isolation and characterization of exosomes
TRANSCRIPT

ISOLATION AND
CHARACTERIZATION OF EXOSOMES
BY
BOW JESSE TAURO
SUBMITTED IN TOTAL FULFILMENT OF THE REQUIREMENTS OF THE DEGREE
OF DOCTOR OF PHILOSOPHY
AUGUST 2013
DEPARTMENT OF BIOCHEMISTRY AND MOLECULAR BIOLOGY
THE UNIVERSITY OF MELBOURNE
LUDWIG INSTITUTE FOR CANCER RESEARCH LTD.
PARKVILLE BRANCH
&
LA TROBE INSTITUTE FOR MOLECULAR SCIENCE
LA TROBE UNIVERSITY


iii
“It is the theory which decides what we can observe.”
Albert Einstein
“At the heart of science is an essential balance between two seemingly contradictory
attitudes - an openness to new ideas, no matter how bizarre or counterintuitive they may
be, and the most ruthless skeptical scrutiny of all ideas, old and new. This is how deep
truths are winnowed from deep nonsense.”
Carl Sagan

iv
Abstract
Cell-cell communication is an integral physiological process that relies on the sending and
receiving of signals. Communication may involve direct contact between adjoining cells, or
require the release of secreted molecules to facilitate the interaction. Recently, extracellular
vesicles (EVs) secreted from cells have been recognized to be involved in cell-cell
communication. EVs comprise shed microvesicles (sMVs), apoptotic bodies and exosomes
which differ based on their mechanism of biogenesis and size; of these, exosomes have
been most widely studied. Exosomes are ~40-100 nm EVs released from a multitude of cell
types that perform pleiotropic extracellular functions within the cellular microenvironment.
These functions include autocrine and paracrine signaling, immunological modulation, and
horizontal transfer of proteins, lipids and genetic material (miRNA/mRNA) to recipient
cells. When investigating exosome functionality, sample homogeneity is of critical
importance, a caveat which, thus far, has been ostensibly overlooked. Although numerous
functions have been ascribed to exosomes, the interpretation of the majority of these studies
has been confounded by sample heterogeneity with other secreted vesicles.
This thesis aims to address the concern of exosome purity by evaluating widely-used
exosome isolation procedures, using the human colorectal cancer (CRC) cell line LIM1863
as a model. The information gained from this comparative purification study will be used to
provide insights into the isolation and characterization of two exosome sub-populations
released from this highly-polarized CRC cell line. Additionally, using an epithelial-
mesenchymal transition (EMT) cell model, exosomes will be isolated and characterized for
the purpose of identifying their contribution to the EMT process.
The first experimental chapter compared and evaluated three strategies for exosome
isolation: differential ultracentrifugation, density gradient centrifugation using iodixanol
(OptiPrep™) and EpCAM immunoaffinity capture. All exosome preparations contained 40-
100 nm diameter vesicles based on electron microscopy, and were positive for stereotypical
exosome markers Alix, TSG101, HSP70 using Western blotting. Using MS–based
proteomic profiling, the protein composition of exosomes isolated from each of these

v
procedures was investigated. The effectiveness of each method was assessed by
quantitating the number of MS/MS spectra identified for exosome markers and proteins
associated with key exosome processes including their biogenesis, intracellular trafficking,
and exocytic release. This study revealed that proteins in all aforementioned categories
were significantly enriched (at least 2-fold) in immunoaffinity isolated exosomes when
compared to density gradient- or differential ultracentrifugation-derived exosomes. Overall,
this study concluded that immunoaffinity capture was the most effective method for
isolating exosomes.
The second experimental chapter investigated sub-populations of exosomes from highly-
polarized LIM1863 cells. In this study, a sequential immunoaffinity capture strategy was
developed using anti-A33- and anti-EpCAM-coupled magnetic beads to isolate A33- and
EpCAM-Exos. A key finding of this study was the exclusive identification in EpCAM-
Exos of the classical intracellular apical trafficking molecules CD63, mucin 13, and apical
intestinal enzyme sucrase isomaltase. The increased expression of dipeptidyl peptidase IV
and the apically-restricted pentaspan membrane glycoprotein prominin 1 was also
observed. In comparison, A33-Exos were enriched with intracellular basolateral trafficking
molecules including early endosome antigen 1 (EEA1), the Golgi membrane protein ADP-
ribosylation factor (ARF1) and clathrin. Collectively, these data are consistent with
EpCAM- and A33-Exos being released from the apical and basolateral surfaces,
respectively. Intriguingly, several members of the MHC class I family of antigen
presentation molecules were exclusively observed in A33-Exos. Additionally, EpCAM-
Exos contained molecules known to complex together to promote tumor progression
including, EpCAM, claudin-7 and CD44. This study is the first robust characterization of
two distinct populations of exosomes from highly-polarized epithelial cells.
A proteome analysis of LIM1863-derived sMVs (also referred to as plasma membrane
blebs, microvesicles and oncosomes) was also performed to reveal that sMVs are clearly
distinguishable from A33- and EpCAM-Exos. Interestingly, sMVs were shown to be
significantly enriched with members of the human ATP-binding cassette (ABC) transporter
superfamily, which act to shuttle substrates (e.g., drugs) across cellular membranes.

vi
The third experimental chapter analyzed exosomes released from cells following EMT.
EMT is a highly conserved morphogenic process defined by the loss of epithelial
characteristics and the acquisition of a mesenchymal phenotype. This process is associated
with increased aggressiveness, invasiveness, and metastatic potential in carcinoma cells.
Using an EMT cell model consisting of Madin-Darby canine kidney cells (MDCK) and its
oncogenic H-Ras-induced variant (21D1) established by Dr. Zhu (The University of
Melbourne), MDCK- and 21D1- exosomes (MDCK- and 21D1-Exos) were isolated using
density gradient centrifugation (OptiPrep™). Proteomic profiling revealed that typical
cellular EMT marker proteins are present in MDCK- and 21D1-Exos. These include
reduction of a characteristic inhibitor of angiogenesis (e.g., thrombospondin-1) and
epithelial markers (e.g., E-cadherin and EpCAM), with a concomitant upregulation of a
mesenchymal marker (e.g., vimentin). Further, this study revealed that 21D1-Exos were
enriched with several proteases and integrins that have been recently implicated in
regulating the tumor microenvironment to promote metastatic progression. A salient
finding, however, was the unique enrichment of key transcriptional regulators (e.g., the
master transcriptional regulator YBX1) and core splicing complex components in
mesenchymal 21D1 cell-derived exosomes. Overall, these findings demonstrate that
exosomes from Ras-transformed MDCK cells are reprogrammed with factors which may
be capable of inducing EMT in recipient cells.
In summary, this thesis evaluated three strategies for purifying exosomes. The findings of
this study revealed that immunoaffinity capture was significantly more effective at
enriching exosomes from cellular media than density gradient centrifugation or differential
centrifugation. This purification knowledge was used to isolate and characterize two
populations of exosomes secreted from the colon carcinoma cell line LIM1863 as well as to
evaluate the contribution of exosomes in the epithelial-mesenchymal transition process.

vii
Declaration
This is to certify that:
(i) this thesis comprises only my original work towards the PhD except where
indicated in the Preface.
(ii) due acknowledgement has been made in the text to all other material used.
(iii) the thesis is less than 100,000 words in length, exclusive of tables, maps,
bibliographies, appendices and footnotes.
Signed: _________________________
Bow Tauro

viii
Preface
Publications during candidature not included in this thesis:
1. Mathivanan, S., Lim, J. W. E., Tauro, B. J., Ji, H., Moritz, R. L., and Simpson, R. J.
(2010) Proteomics analysis of A33 immunoaffinity-purified exosomes released from the
human colon tumor cell line LIM1215 reveals a tissue-specific protein signature. Molecular
& Cellular Proteomics 9 (2), 197-208. PMID: 19837982.
2. Mathivanan, S., Ji, H., Tauro, B. J., Chen, Y. S., and Simpson, R. J. (2012) Identifying
mutated proteins secreted by colon cancer cell lines using mass spectrometry. Journal of
Proteomics 5 (76), 141-149. PMID: 22796352.
3. Ji, H., Greening, D. W., Barnes, T. W., Lim, J. W. E., Tauro, B. J., Adda, C.,
Mathivanan, S., Zhao, W., Xue, Y., Xu, T., and Simpson, R. J. (2013) Proteome profiling
of exosomes derived from human primary and metastatic colorectal cells reveal differential
expression of key metastatic factors and signal transduction components. Proteomics 13
(10-11), 1672-1686. PMID: 23585443.

ix
Publications during candidature included in this thesis:
1. Tauro, B. J., Greening, D. W., Mathias, R. A., Ji, H., Mathivanan, S., Scott, A. M., and
Simpson, R. J. (2012) Comparison of ultracentrifugation, density gradient separation, and
immunoaffinity capture methods for isolating human colon cancer cell line LIM1863-
derived exosomes. Methods 56, 293-304. PMID: 22285593. Chapter 2 of this thesis.
2. Tauro, B. J., Greening, D. W., Mathias, R. A., Mathivanan, S., Ji, H., and Simpson, R. J.
(2012) Two distinct populations of exosomes are released from LIM1863 colon carcinoma
cell-derived organoids. Molecular & Cellular Proteomics 12, 587-598 PMID: 23230278.
Chapter 3 of this thesis.
3. Tauro, B. J., Mathias, R. A., Greening, D. W., Gopal, S. K., Ji, H, Kapp, E. A.,
Coleman, B. M., Hill, A. F., Kusebauch, U., Hallows, J. L., Shteynberg, D., Moritz, R. L.,
Zhu, H. J., and Simpson R. J. (2013) Oncogenic H-Ras reprograms Madin-Darby canine
kidney (MDCK) cell-derived exosomal proteins during epithelial-mesenchymal transition.
Molecular & Cellular Proteomics 12, 2148-2159. PMID: 23645497. Chapter 4 of this
thesis.

x
The work described in the manuscripts included this thesis was performed entirely by
myself except for the following collaborations: Chapters 2 and 3, Dr. David Greening and
Dr. Rommel Mathias (assistance with manuscript editing), Dr. Suresh Mathivanan
(performed initial bioinformatic analysis), Chapter 4, Dr. Rommel Mathias (assistance with
manuscript writing), Dr. David Greening (assistance with manuscript editing), Mr. Shashi
Kumar Gopal Krishnan (assistance with cell culture for Figure 2), Mr. Eugene Kapp
(performed initial bioinformatic analysis), Dr. Bradley Coleman (cryo-transmission
electron microscopy imaging), A/Prof. Robert Moritz (MS analysis of a second (replicate)
dataset). I estimate that my contribution to Chapter 2 and 3 to be greater than 90% and my
contribution to Chapter 4 to be greater than 80% therefore achieving an overall contribution
of my work in the thesis to be greater than 85%.

xi
Acknowledgements
This thesis is an account of several years of devoted work while enrolled through the
Department of Biochemistry and Molecular Biology at The University of Melbourne.
Experimental work was performed at the Ludwig Institute for Cancer Research (LICR) and
the La Trobe Institute for Molecular Science (LIMS).
First, I would like to express my deep appreciation to my primary supervisor, Prof. Richard
Simpson, for allowing me to undertake my PhD. He is incredibly accomplished within the
scientific field and his level of success is something to aspire to. His ability to continually
enthuse and inspire me, even in the toughest of times, has been amazing. I have genuinely
enjoyed spending these years with him and value the friendship that has evolved further
than a supervisor/student relationship. Attending conferences together, particularly the
International Society for Extracellular Vesicles meeting in Sweden, have been a great
highlight with many fond memories.
Next I would like to thank my co-supervisors, Dr. Hong Ji and Dr. Hong-Jian Zhu. From
the first day I stepped into the laboratory, a friendship with Ji Hong grew like the orchids
she would nurture; we were instantly a great team. Her skills within the laboratory are
second to none. Her experimental design and execution are impeccable; I can only hope
that my skills will continue to grow to one day reach her level. Hong-Jian’s guidance as an
external supervisor has been amazing; his knowledge regarding key concepts including
epithelial-mesenchymal transition has been invaluable.
I would also like to thank the members of my PhD Committee, Prof. Andrew Hill and
A/Prof. Matthew Perugini, who both provided critical review and guidance during my PhD
research. Additionally, I would also like to acknowledge Prof. Antony Burgess and Prof.
Nick Hoogenraad who were the directors of LICR and LIMS, respectively, during my PhD
candidature.

xii
I would also like to thank staff and students, past and present, who have given me unending
support. In particular, Dr. David Greening and Dr. Rommel Mathias whose clear thinking
and focused attitudes I admire greatly. With these qualities they assisted me to distill my
vast amounts of experimental data into a publishable form for which I am deeply greatful.
Additionally, I would like to thank Dr. Thomas Barnes, Mr. Robert Goode, Dr. Yuan-Shou
Chen, Dr. Justin Lim, Dr. Nicole Harris, Mr. Robert Speirs, Mr. Shashi Kumar Gopal
Krishnan, Mr. Alin Rai, and Mr. Rong Xu. I thank them all for the support, laughter and the
great times we shared together throughout my PhD.
Thanks must also go to A/Prof. Robert Moritz, Dr. Suresh Mathivanan and Dr. Eugene
Kapp. Their collective knowledge of proteomics and mass spectrometry has been vital to
my learning and the completion of this thesis.
I would also like to give thanks to my parents, Geraldine and Peter, my sister Eve, and
brothers Rory and Tristan, I love and thank them all. My family have given me unending
support in all of my endeavors and helped me to grow to become who I am today. I would
also like to acknowledge my influential and motivating chemistry teacher, Mr. Robert
Timoney, who taught me many key scientific concepts that I still reflect on to this day.
Finally, I would like to thank my fiancée, Jess Williamson. Literally since day one, Jess has
been by my side, encouraging and supporting me along on the way, I could not have done it
without her. My love continues to grow for this inspiring person and I look forward to
tackling the next chapter with her in Ghana.

xiii
Table of Contents
Abstract .................................................................................................................................... iv
Declaration............................................................................................................................. vii
Preface .................................................................................................................................. viii
Acknowledgements ................................................................................................................. xi
List of Tables ......................................................................................................................... xvi
List of Figures and Illustrations ........................................................................................... xvii
Abbreviations ........................................................................................................................ xix
Chapter 1: Background and literature review ........................................................................... 1
1.1 Preface ................................................................................................................... 1
1.2 Secretome .............................................................................................................. 2
1.3 Extracellular vesicles ............................................................................................. 2
1.3.1 Shed microvesicles ................................................................................................ 5
1.3.2 Apoptotic blebs ...................................................................................................... 6
1.4 Exosomes ............................................................................................................... 7
1.4.1 Exosome biogenesis and cargo selection ............................................................... 8
1.4.2 Endosome motility and exosome release ............................................................. 11
1.4.2.1 Exosome biogenesis and release from polarized cells ......................................... 12
1.4.3 Exosome sizing and morphology ........................................................................ 13
1.4.4 Exosomal proteins ............................................................................................... 14
1.4.5 Exosomal lipids ................................................................................................... 16
1.4.6 Exosomal RNA .................................................................................................... 18
1.4.7 Exosome targeting and uptake ............................................................................. 20
1.4.8 Exosome function in the tumor microenvironment and cancer progression ....... 23

xiv
1.4.9 Exosome therapeutics .......................................................................................... 27
1.4.10 Exosome and microvesicle diagnostics ............................................................... 32
1.4.11 Exosome isolation strategies................................................................................ 32
1.5 Research models used in this thesis ..................................................................... 34
1.5.1 Colorectal cancer model ...................................................................................... 34
1.5.1.1 Colorectal cancer ................................................................................................. 34
1.5.1.2 CRC statistics and subtypes ................................................................................. 35
1.5.1.3 Colorectal cancer cell line LIM1863 ................................................................... 37
1.5.2 Epithelial-mesenchymal transition (EMT) model ............................................... 38
1.5.2.1 EMT during cancer progression .......................................................................... 38
1.5.2.2 Molecular markers of EMT ................................................................................. 41
1.5.2.3 Signaling pathways and inducers of EMT ........................................................... 42
1.5.2.4 Madin-Darby canine kidney (MDCK) and 21D1 cell lines................................. 43
1.5.2.5 Proteomic analysis of the secretome from MDCK cells following EMT ........... 43
1.5.2.6 Exosomes and EMT ............................................................................................. 45
1.6 Thesis aims .......................................................................................................... 45
Chapter 2: Comparison of methods for isolating exosomes ................................................... 49
2.1 Overview.............................................................................................................. 49
Chapter 3: Use of sequential immunoaffinity capture to isolate and characterize two distinct
populations of exosomes released from LIM1863 colon carcinoma cells ............................. 65
3.1 Overview.............................................................................................................. 65
Chapter 4: Characterization of Madin-Darby canine kidney (MDCK) cell-derived exosomal
proteins following oncogenic H-Ras induced epithelial-mesenchymal transition ................. 81

xv
4.1 Overview.............................................................................................................. 81
Chapter 5: Summary and future directions ............................................................................. 95
5.1 Evaluation of exosome isolation methods and the importance of purity............. 95
5.2 Analysis of two exosome sub-populations released from LIM1863 cells ........... 96
5.3 Analysis of exosomes following epithelial-mesenchymal transition (EMT) ...... 98
5.4 Conclusion ......................................................................................................... 100
References ............................................................................................................................ 102
Appendix .............................................................................................................................. 139
Appendix A-1 Isolation and characterization of cell line-derived exosomes ............... 140
Appendix A-2 Isolation and characterization of body fluid-derived exosomes............ 146
Appendix A-3 Human colon cancer LIM1863 cells ..................................................... 149
Appendix A-4 Distinguishing features of EMT ............................................................ 150
Appendix A-5 Secreted extracellular modulators of EMT ........................................... 151
Appendix A-6 Supplemental information for manuscripts included in this thesis ....... 152
Appendix A-7 Distribution of proteins identified in UC-, DG-, and IAC-Exos ........... 154
Appendix A-8 Plasma membrane proteins identified in the exosome datasets ............ 155
Appendix A-9 21D1 cells require their own culture medium to maintain a
mesenchymal phenotype ....................................................................... 156
Appendix A-10 Exosome characterization following OptiPrep™ density gradient
separation .............................................................................................. 157
Appendix A-11 Experimental procedures for replicate GeLC-MS/MS analysis of
MDCK- and 21D1-Exos ........................................................................ 158

xvi
List of Tables
Chapter 1
Table 1-1 Properties of extracellular vesicles ................................................................... 4
Table 1-2 A summary of in vitro studies of miRNAs identified in cancer cell
exosomes ........................................................................................................ 19
Table 1-3 Examples of EV-mediated transfer of oncogenic/transforming molecules .... 25
Table 1-4 Therapeutic applications of exosomes ............................................................ 31
Chapter 2
Table 2-1 Relative quantitation of selected exosome proteins by label-free spectral
counting .......................................................................................................... 61
Chapter 3
Table 3-1 Representative list of proteins either uniquely localized or significantly
enriched in EpCAM-Exos .............................................................................. 74
Table 3-2 Representative list of proteins either uniquely localized or significantly
enriched in A33-Exos ..................................................................................... 75
Table 3-3 List of colon cancer-related and normal colon tissue specific membrane
proteins in LIM1863-derived exosomes ......................................................... 76
Chapter 4
Table 4-1 EMT hallmark proteins identified in MDCK- and 21D1-Exos .......................... 88
Table 4-2 Exosomal factors involved in metastatic niche formation and metastasis .......... 89
Table 4-3 Splicing factors and transcription factors enriched in 21D1-Exos ...................... 90

xvii
List of Figures and Illustrations
Chapter 1
Figure 1-1 Extracellular membranous vesicles released from cells ................................... 3
Figure 1-2 Exosomal cargo selection involving ESCRT complexes.................................. 9
Figure 1-3 Exosome targeting and uptake ........................................................................ 21
Figure 1-4 Extracellular vesicles (EVs) are components of the intra- and intercellular
oncogenic signaling circuitry ......................................................................... 24
Figure 1-5 Functional roles and therapeutic targeting of extracellular vesicles (EVs) .... 28
Figure 1-6 Advantages and drawbacks of siRNA delivery by exosomes, viruses and
lipid nanoparticles .......................................................................................... 29
Figure 1-7 Model of intestinal cancer initiation and progression ..................................... 36
Figure 1-8 Epithelial-mesenchymal transition.................................................................. 39
Figure 1-9 EMT during tumor progression and metastasis .............................................. 40
Figure 1-10 MDCK and Ras-transformed MDCK cells (21D1) morphology .................... 44
Chapter 2
Figure 2-1 Exosome isolation .......................................................................................... 56
Figure 2-2 Characterization of exosomes ........................................................................ 57
Figure 2-3 Semi-quantitative normalized spectral count ratios of selected exosome
proteins ........................................................................................................... 58
Chapter 3
Figure 3-1 Isolation of exosomes and shed microvesicles from the colon carcinoma cell
line LIM1863 .................................................................................................. 72
Figure 3-2 Morphological characterization and proteome analysis of LIM1863 cell-
derived A33- and EpCAM-Exos .................................................................... 73
Figure 3-3 A confocal optical section though a preparation of LIM1863 organoids ...... 77
Figure 3-4 A model depicting the molecular structure of two discreet populations of
LIM1863-derived exosomes ........................................................................... 70

xviii
Chapter 4
Figure 4-1 Isolation of exosomes from MDCK and 21D1 cells ...................................... 86
Figure 4-2 Characterization of MDCK- and 21D1-Exos................................................. 87
Figure 4-3 Proteomic analysis of exosomes .................................................................... 87

xix
Abbreviations
ADAM a disintegrin and metalloproteinase
A33-Exos exosomes isolated using anti-A33 immunoaffinity beads
BMDC bone marrow-derived hematopoietic progenitor cells
BMP bone morphogenic protein
CCM concentrated culture medium
CM culture medium
CRC colorectal cancer
DMEM Dulbecco’s modified eagle medium
EGF epidermal growth factor
EGFR epidermal growth factor receptor
EM electron microscopy
EMMPRIN extracellular matrix metalloproteinase inducer
EMT epithelial-mesenchymal transition
eMVs extracellular microvesicles
EpCAM epithelial cell adhesion molecule
EpCAM-Exos exosomes isolated using anti-EpCAM immunoaffinity beads
ESCRT endosomal sorting complex required for transport
FACS fluorescence-activated cell sorting
FCS fetal calf serum
FGF fibroblast growth factor
FITC fluorescein isothiocyanate
FOXC2 forkhead box protein C2
GI gastrointestinal
GSK-3β glycogen synthase kinase 3 β
HMGB high mobility group box
HPLC high-performance liquid chromatography
HSP heat shock protein
HUVEC human umbilical vein endothelial cells
ILV intraluminal vesicle
ITS insulin-transferrin-selenium
LDH lactate dehydrogenase

xx
LEF lymphoid enhancer factor
MACS magnetic activated cell sorting
MAPK mitogen-activated protein kinase
MDCK Madin-Darby canine kidney
MMP matrix metalloproteinase
MTT 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide
MVB multivesicular body
MVs microvesicles
NSF N-Ethylmaleimide-sensitive fusion protein
PDCD6IP/Alix programmed cell death 6 interacting protein
PI(3)K phosphatidylinositol 3-kinase
PM plasma membrane
Rsc relative spectral count fold change ratios
SIP smad interacting protein
SMART simple modular architecture research tool
sMVs shed microvesicles
SNARE soluble N-ethylmaleimide-sensitive factor attachment protein receptor
SSM solid support magnet
TEM tetraspanin enriched microdomains
TGN trans-Golgi network
TMHMM transmembrane hidden Markov model
TF tissue factor
TGFβ transforming growth factor β
TNFα tumor necrosis factor α
TPP trans-proteomic pipeline
TNTs tunneling nanotubes
TSG101 tumor susceptibility gene 101
VEGF-A vascular endothelial growth factor A
VEGFR-1 vascular endothelial growth factor receptor 1
VAMP vesicle-associated membrane protein
WCL whole cell lysate
YBX1 Y-box binding protein 1
ZEB1 zinc finger E-box binding homeobox 1
21D1 Ras-transformed MDCK cells

1
Chapter 1: Background and literature review
1.1 Preface
Exosomes are 40-100 nm diameter membraneous vesicles released from most cell types.
They function in extracellular communication by transferring a variety of bioactive
molecules (e.g., proteins, lipids, RNA species such as mRNA/miRNAs, and possibly DNA)
between cells [1, 2]. Over the past decade they have gained much attention for their role in
cancer progression [3]. Although exosomes were first described almost thirty years ago [4-
6], their purification to homogeneity has proven a difficult task. This is due to their inherent
biophysical properties (e.g., size and buoyant density) being similar to extracellular vesicles
(EVs) (e.g., shed microvesicles and apoptotic blebs), the problem of co-purification with
protein oligomers (e.g., proteasome complexes), and the possible contamination with
cellular pathogens (e.g., adenoviruses and prions) [7]. Obtaining homogeneous exosomes is
a pre-requisite for in-depth biophysical and functional analyses.
Using semi-quantitative MS-based proteomic techniques, such as label-free spectral
counting [8], this thesis first compares widely used exosome isolation strategies to assess
their capability for enriching several classes of proteins involved in exosome biogenesis,
trafficking, release and uptake. Secondly, a sequential immunoaffinity capture technique
was developed to isolate exosome sub-populations released from apical and basolateral
surfaces of highly-polarized cells. Finally, in the absence of an immunoaffinity capture
strategy, density gradient centrifugation (OptiPrep™) was used to isolate and characterize
exosomes, for the first time, from an epithelial-mesenchymal transition (EMT) cell model.

2
1.2 Secretome
The term ‘secretome’ refers to all soluble proteins and extracellular membranous vesicles
that are secreted or shed into the extracellular space [9-15]. The secretome is a tightly
regulated and sensitive feature required for cell-cell communication and normal
physiological function [9]. Secreted proteins can constitute structural components of the
extracellular matrix (ECM), such as collagens and laminins, while others are able to trigger
intercellular responses in target cells [16, 17]. Secreted proteins are of particular importance
as aberrant protein secretion is associated with pathological events within diseases such as
cancer [13, 14], including pre-metastatic niche formation and metastasis [18-21]. In
addition to soluble-secreted proteins, the secretome contains several types of EVs including
shed microvesicles (sMVs), apoptotic blebs and exosomes (Figure 1-1, Table 1-1) [1]. It is
now recognized that these vesicles are also important regulators of cell-cell communication,
particularly within the tumor microenvironment [19, 22].
1.3 Extracellular vesicles
The release of EVs was first described in 1967 when Wolf and colleagues recognized pro-
coagulant particulate matter around activated platelets (‘platelet dust’) [23]. Platelets were
later identified to release microvesicles and exosomes [24]. In addition to normal cellular
functioning, EV release can be caused by diverse biological mechanisms that are triggered
during oncogenic transformation, cellular activation by stimuli in the microenvironment,
stress or apoptosis [25]. The formation of vesicles may occur as budding events at the
plasma membrane (e.g., sMVs) or within late endosomal compartments such as
multivesicular bodies (e.g., exosomes), or as a consequence of apoptosis (e.g., apoptotic
blebs). Following release, different EVs may have distinct functions. It is understood that
isolating EVs to homogeneity is the first step towards obtaining a clear understanding of
their biophysical properties and biological functions. However, such an undertaking has
been impeded by the inherent technical difficulties – due largely to the similarity of their
physiochemical characteristics (Table 1-1) [26].

3
Figure 1-1. Extracellular membranous vesicles released from cells
Shed microvesicles (sMVs) range in size between 100-1000 nm in diameter and are formed
by direct outward budding of the plasma membrane (PM). During endocytosis, a receptor is
internalized and sorted in early endosomes, where cargo may be recycled to the PM via
recycling endosomes [27, 28]. Intraluminal vesicles (ILVs) are generated by inward
budding from the limiting membrane of late endosomes, forming multivesicular bodies
(MVBs). MVBs may be (i) sorted to the lysosome and become degradative or (ii) fuse with
the PM to release 40-100 nm ILVs, now termed exosomes [29]. Exosomes display the same
surface topology as the cell with extracellular domains of proteins at the surface, and
enclosing cytoplasmic contents (cargo) in the lumen. Apoptotic blebs (50-5000 nm in
diameter) are formed from cells undergoing programmed cell death.
4
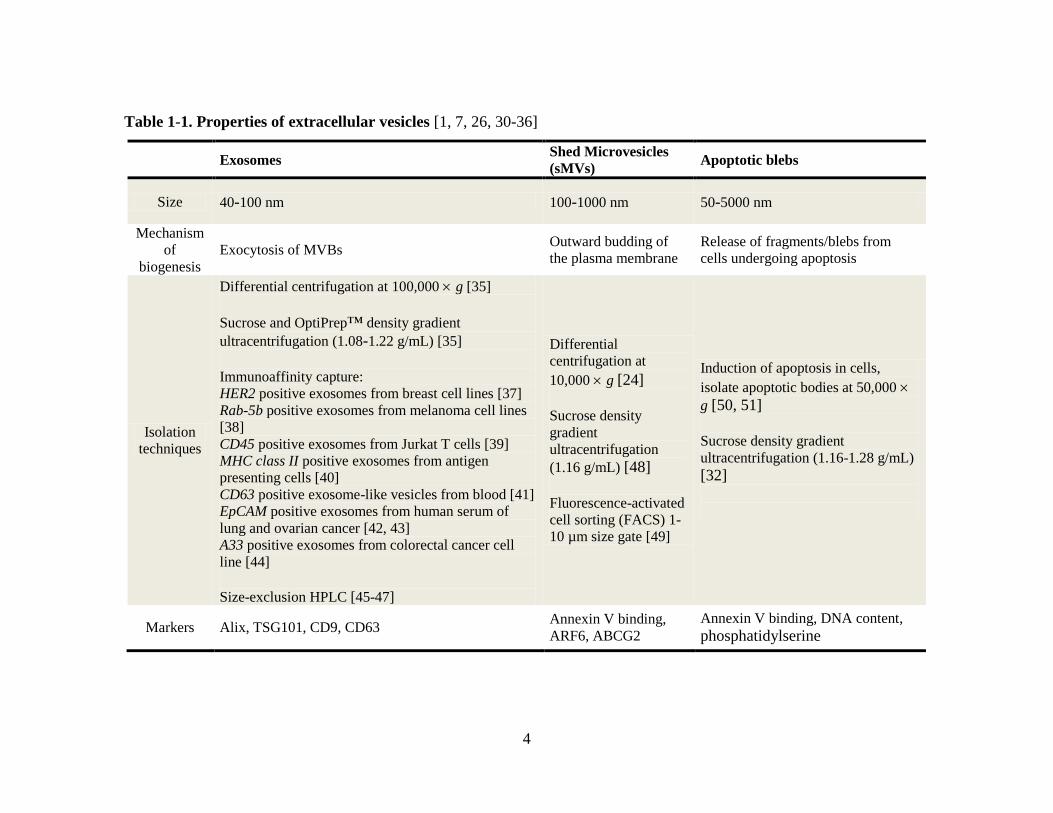
Table 1-1. Properties of extracellular vesicles [1, 7, 26, 30-36]
Exosomes Shed Microvesicles
(sMVs) Apoptotic blebs
Size 40-100 nm 100-1000 nm 50-5000 nm
Mechanism
of
biogenesis Exocytosis of MVBs
Outward budding of
the plasma membrane Release of fragments/blebs from
cells undergoing apoptosis
Isolation
techniques
Differential centrifugation at 100,000 g [35]
Sucrose and OptiPrep™ density gradient
ultracentrifugation (1.08-1.22 g/mL) [35]
Immunoaffinity capture: HER2 positive exosomes from breast cell lines [37] Rab-5b positive exosomes from melanoma cell lines
[38] CD45 positive exosomes from Jurkat T cells [39] MHC class II positive exosomes from antigen
presenting cells [40] CD63 positive exosome-like vesicles from blood [41] EpCAM positive exosomes from human serum of
lung and ovarian cancer [42, 43] A33 positive exosomes from colorectal cancer cell
line [44] Size-exclusion HPLC [45-47]
Differential
centrifugation at
10,000 g [24]
Sucrose density
gradient
ultracentrifugation
(1.16 g/mL) [48]
Fluorescence-activated
cell sorting (FACS) 1-
10 µm size gate [49]
Induction of apoptosis in cells,
isolate apoptotic bodies at 50,000
g [50, 51]
Sucrose density gradient
ultracentrifugation (1.16-1.28 g/mL) [32]
Markers Alix, TSG101, CD9, CD63 Annexin V binding,
ARF6, ABCG2 Annexin V binding, DNA content,
phosphatidylserine

5
A recent initiative to assist in the cataloguing of EVs is the database Vesiclepedia
(http://www.microvesicles.org/) [52]. Vesiclepedia is a manually-curated compendium of
molecular information (protein, lipid and RNA) identified in different classes of EVs.
Vesiclepedia comprises 35,264 protein, 18,718 mRNA, 1,772 miRNA and 342 lipid entries
comprised from 341 independent studies. A summary of EVs (sMVs, apoptotic blebs and
exosomes) and their biophysical properties is outlined below.
1.3.1 Shed microvesicles
Shed microvesicles (sMVs) range in diameter between 100-1000 nm and are formed by
direct budding of cytoplasmic protrusions from the cell membrane upon activation of
various internal and external stimuli (Figure 1-1) [7, 26]. Nomenclature of these vesicles
has been defined depending on their parent cell; for example, microvesicles released by
platelets are referred to as ‘microparticles’ while microvesicles released by leukocytes are
called ‘ectosomes’ [31]. Prostate cancer xenograft released vesicles are termed
‘oncosomes’ [49]. They are collectively referred to as sMVs in this thesis. The biogenesis
of sMVs is reported to involve phospholipid redistribution caused by flippases and
floppases translocating phospholipids from the outer-to-inner and inner-to-outer membrane
leaflets, respectively [36]. Following this rearrangement, contraction of cytoskeletal
structures by actin-myosin interactions, caused by ADP-ribosylation factor 6 (ARF6),
initiates a signaling cascade that culminates in the phosphorylation and activation of
myosin light chain, facilitating membrane budding and sMV release [36]. Shedding of
vesicles occurs in resting cells, however, Ca2+
is known to increase the rate of the process
dramatically [53]. Interestingly, despite being generated by the analogous process of
budding from the cell surface, sMVs isolated at rest and following stimulation can be
molecularly different. For example, it was shown that vesicles shed from stimulated
neutrophils express increased levels of integrin αMβ2 [54].
sMVs are recognized to participate in important biological events including coagulation by
mediating the contribution of platelets, macrophages and neutrophils, and horizontal

6
trafficking of protein and RNA between cells [26] (for an excellent review see [55]). It is
well recognized that various classes of sMVs are rich in ATP-Binding Cassette (ABC)
transporters which form a defense network against a range of chemotherapeutics [56]. This
network can result in multidrug resistance, a major impediment to curative cancer
chemotherapy [57-60]. Following release into the extracellular space, sMVs may be
degraded locally or move by diffusion to appear in biological fluids including cerebrospinal
fluid, blood and urine [26]. When investigating sMVs released from tumor cells, it was
demonstrated that they may rapidly break down to release cargo. This may include
extracellular matrix metalloproteinase inducer (EMMPRIN), a transmembrane glycoprotein
expressed at high levels by tumor cells that can stimulate matrix metalloproteinase
expression in fibroblasts to facilitate tumor invasion and metastasis [61]. Additionally,
sMVs (oncosomes) isolated by FACS from prostate cancer xenograft mouse blood were
shown to stimulate the migration of normal endothelial cells indicating their ability to
modify the microvasculature [49]. Methods used to isolate sMVs from culture medium or
platelet-poor plasma include density gradient centrifugation (buoyant density of 1.16 g/mL)
on sucrose gradients [24, 48, 49, 62]. sMVs can also be isolated by FACS using size-gating
determined by polymeric bead standards [49]. Although separation by size-exclusion
chromatography has been performed to isolate sMVs of varying size from urine, this
process resulted in contamination with other EVs and proteins [63]. To date, little is known
about sMVs structure and specific information about their biogenesis is limited.
1.3.2 Apoptotic blebs
Apoptotic blebs (and larger ‘apoptotic bodies’) are heterogeneous fragments of the cell (50-
5000 nm) formed during programmed cell death (Figure 1-1) [36]. The occurrence of cell
shrinkage and condensation of nuclear chromatin, hallmarks of apoptosis, leads to the
generation of apoptotic blebs. These blebs contain remnants of the cell degradation
processes including cytoplasmic components and DNA [64]. It is understood that during
apoptosis, phosphatidylserine (PS) is relocalized from the inner leaflet of the PM to the
outer leaflet where it triggers recognition for phagocytosis [65]. In addition to PS, other

7
phagocytic surface signals including integrins αVβ3/αVβ5 and CD36 are displayed [33, 34,
66]. Interestingly, from a functional aspect, phagocytosis of apoptotic blebs by dendritic
cells results in efficient processing and presentation of apoptotic cell antigens on CD4+ and
CD8+ T lymphocytes [67]. Further, apoptotic bodies derived from H-RasV12 or human c-
myc transfected cells were up taken by murine fibroblasts resulting in loss of contact
inhibition in vitro and a tumorigenic phenotype in vivo [68]. Protocols for the biogenesis
and isolation of apoptotic blebs involve induction of cell apoptosis, commonly by UV
irradiation or ethanol induction, and subsequent cell culture for up to 24 h. At different
times after apoptotic induction, cells are collected and stained with FITC-labeled annexin
V, an early marker of apoptosis. Blebs are then isolated by centrifugation at 50,000 g [50,
51] or by sucrose density gradients at a buoyant density of 1.16-1.28 g/mL [32]. In contrast
to sMVs, which are formed and released at the cell membrane, and apoptotic blebs which
are formed by cell shrinkage during programmed cell death, exosomes are formed by
inward budding of late endosomes. These MVBs subsequently traffic to and fuse with the
plasma membrane to release exosomes into the extracellular environment.
1.4 Exosomes
Exosomes were first described in the 1980s by Johnstone and colleagues while culturing
maturing reticulocytes and were originally thought to be a mechanism for removal of
obsolete cellular proteins [5, 6]. Using transmission electron microscopy they observed the
internalization and release of immunogold labeled transferrin receptors associated with
small (50 nm diameter) vesicles [4]. Exosomes are now recognized to be released from
most cell types including B lymphocytes [69], dendritic cells [70], neurons [71], intestinal
epithelial cells [72] and tumor cell lines [37, 44, 73]. A current definition describes
exosomes as a discrete population of small 40-100 nm diameter membranous vesicles that
are formed within endosomes and released into the extracellular space following fusion of
MVBs with the plasma membrane (Figure 1-1) [74, 75]. This mechanism of biogenesis
differs from that of sMVs and apoptotic blebs and led to the hypothesis that exosomes
might have specialized functions that differ from other EVs [76].

8
1.4.1 Exosome biogenesis and cargo selection
The biogenesis of exosomes occurs within the endosomal network, a membranous
compartment used to sort various intraluminal vesicles and proteins within the cell [77].
Endosomes are separated into three distinct compartments including early, late and
recycling endosomes, each of which are defined by the association of specific proteins on
their cytosolic surface [27, 78]. Early endosomes are characterized by the presence of Rab5
together with its effector VPS34/p150, a phosphatidylinositol 3-kinase (PI(3)K) complex
[78]. These endosomes patrol the peripheral cytoplasm close to the plasma membrane by
movement along microtubules [79] and are the major entry site for endocytosed material
that arrives by clathrin-mediated-, caveolar- and ARF6-dependent pathways [80]. It is
reported that endosomes accept content for approximately 10 min and cargo destined for
recycling is sorted, via syntenin and syndecan heparin sulphate proteoglycans, into
recycling endosomes, which fuse with the plasma membrane [27, 28]. The remaining
material accumulates and is retained in the early endosomes which then undergo maturation
into late endosomes [27].
Maturation of early to late endosomes occurs to reveal a series of transformations including
increased size, a decrease in pH from (6.8-5.9) to (6.0-4.9) and a gradual change in markers
including the loss of Rab5 and acquisition of Rab7 [81, 82]. Following this transformation,
inward budding occurs to form intraluminal vesicles (ILVs/exosomes) in the late endosome
lumen (also termed MVB). This process is governed by endosomal sorting complex
required for transport complexes (ESCRT-0, -I, -II, -III) which are also reported to be
involved in cargo selection [83-85]. The molecular mechanisms regulating cargo selection
into exosomes remain to be fully characterized, however, mono-ubiquitination of the
cytoplasmic tail of membrane proteins has been reported as one of the methods for cargo
selection and sorting [84, 85] (Figure. 1-2). This process requires at least 18 conserved
ESCRT complex proteins that were originally identified in yeast Saccharomyces cerevisiae
[85-87]. ESCRT components are individually recruited from the cytosol to the surface of
endosomes to segregate cargo into domains within the limiting membrane. Specifically,
ESCRT-0, -I, and -II complexes function in sorting of ubiquitin-tagged proteins into the
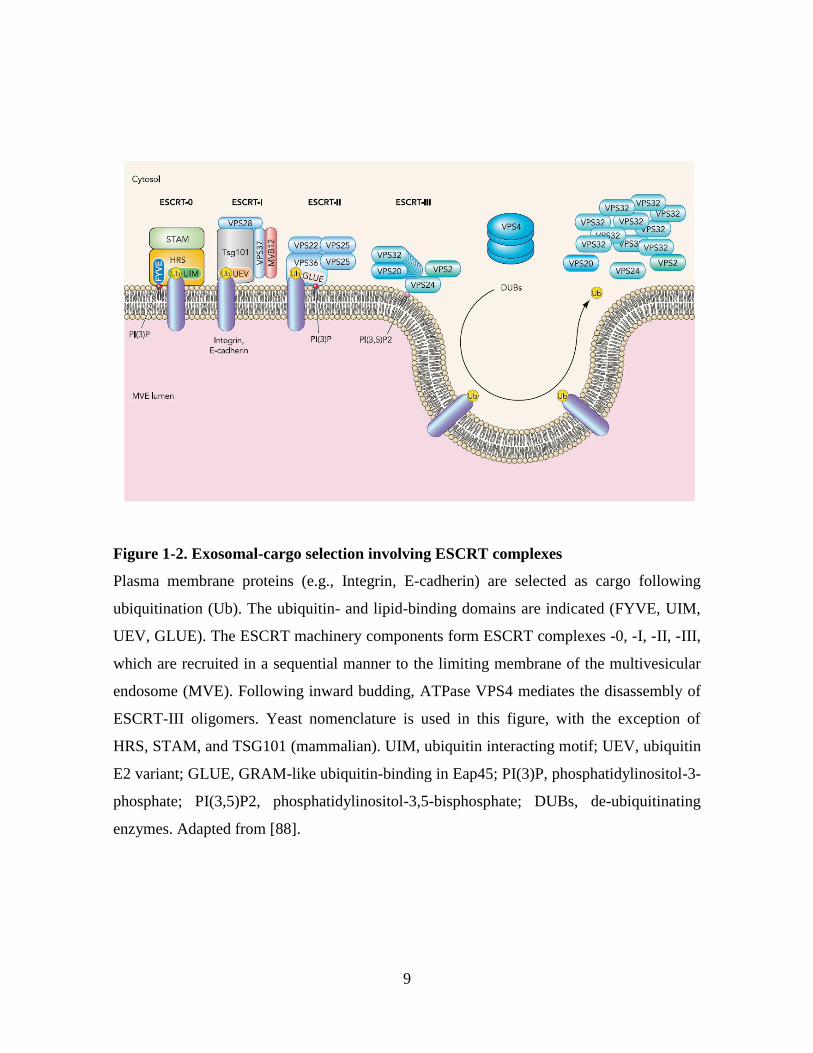
9
Figure 1-2. Exosomal-cargo selection involving ESCRT complexes
Plasma membrane proteins (e.g., Integrin, E-cadherin) are selected as cargo following
ubiquitination (Ub). The ubiquitin- and lipid-binding domains are indicated (FYVE, UIM,
UEV, GLUE). The ESCRT machinery components form ESCRT complexes -0, -I, -II, -III,
which are recruited in a sequential manner to the limiting membrane of the multivesicular
endosome (MVE). Following inward budding, ATPase VPS4 mediates the disassembly of
ESCRT-III oligomers. Yeast nomenclature is used in this figure, with the exception of
HRS, STAM, and TSG101 (mammalian). UIM, ubiquitin interacting motif; UEV, ubiquitin
E2 variant; GLUE, GRAM-like ubiquitin-binding in Eap45; PI(3)P, phosphatidylinositol-3-
phosphate; PI(3,5)P2, phosphatidylinositol-3,5-bisphosphate; DUBs, de-ubiquitinating
enzymes. Adapted from [88].

10
endosomal delimiting membrane [84, 89]. After sorting, the ESCRT-III complex
sequentially associates and recruits the de-ubiquitinating enzyme Doa4 to remove the
ubiquitin tag from the protein cargo, prior to incorporation into ILVs [90]. The ESCRT-III
complex is also responsible for membrane budding and scission in a ‘purse string’ model
[84, 89, 91]. It is reported that depletion of ESCRT machinery components results in fewer
ILVs, and accumulation of cargo in endosomes with abnormal morphology. This indicates
their importance in this process [92].
Conversely, it has been reported that cargo incorporation into exosomes does not depend on
ubiquitination and ESCRT machinery [1]. This is exemplified by the trafficking of pre-
melanosome protein 17 (Pmel17), which through association with CD63, relies on a
luminal domain enabling its sorting and inclusion into exosomes [93, 94]. Exosome
biogenesis also appears to be dependent on cytoplasmic adaptors syntenin and syndecan
[95] and sphingolipid ceramide [96]. In a study by Trajkovic and colleagues, ceramide was
shown to be enriched in exosomes; they demonstrated that the release of exosomes was
reduced following inhibition of neutral sphingomyelinase (nSMase), suggesting the lateral
segregation of lipids contributes to exosome formation and subsequent release, a process
devoid of ESCRT machinery [96].
Cargo selection is also dependent on adaptor proteins. For example, phosphatase and tensin
homolog deleted on chromosome 10 (PTEN), a tumor suppressor protein localized to the
cytoplasm and nucleus, has been demonstrated to be secreted in exosomes [97]. It was
found that PTEN secretion in exosomes requires Ndfip1, an adaptor protein for members of
the Nedd4 family of E3 ubiquity ligases [97]. To clearly define the exosome formation
pathway, further examination into ESCRT-dependent and ESCRT-independent formation
mechanisms is required.

11
1.4.2 Endosome motility and exosome release
Following vesicle formation, motility of endosomes is dependent on dynein and kinesin
motors that migrate endosomes in opposite directions along microtubules [98]. Rab
GTPases are also integral in ensuring endosome cargo is delivered to the correct
destination. Through sorting adaptors and tethering factors, Rab GTPases control
membrane identity and vesicle budding, motility and fusion. Following endosome
maturation (see section 1.4.1), late endosomes containing ILVs may be targeted to
lysosomes for degradation via the Rab7 effector Rab-interacting lysosomal protein (RILP),
which mediates minus-end-directed trafficking [99]. Late endosomes move to the
perinuclear area of the cell, fuse together to form larger bodies, undergo transient ‘kiss-and-
run’ interactions and eventually full fusions with lysosomes [100].
Alternative to the lysosome pathway, late endosomes may traffic to, and fuse with, the
plasma membrane to release ILVs; ILVs released into the extracellular space are referred to
as exosomes. This process requires various proteins involved in docking, tethering and
fusion. Previous studies have reported the involvement of Rab35 in regulating exosome
secretion by assisting docking and tethering of MVBs to the plasma membrane [101].
Additionally, Rab27a/b and their effectors Slp4 and Slac2b are reported to be involved
exosome release [102, 103]. Soluble N-Ethylmaleimide-sensitive factor attachment protein
receptor (SNARE) molecules are also involved in recognition and fusion of endosomal
membranes to the plasma membrane by forming tetrahelical bundles on opposing
membranes [104]. In a recent study, it was demonstrated that the SNARE Ykt6 was integral
in the secretion of exosomes carrying the morphogen Wnt [105]. Further, vesicle-associated
membrane proteins (VAMPs or synaptobrevins) are anchored in the vesicular membrane to
mediate intracellular vesicle fusion [106, 107]. Finally, VAMP7 and ATPase NSF, a
protein required for SNARE disassembly, are shown to mediate the fusion between MVBs
with the plasma membrane to release exosomes into the extracellular space [108].

12
1.4.2.1 Exosome biogenesis and release from polarized cells
Investigation into exosome biogenesis and release in polarized cell systems has received
little attention to date. In polarized cells, proteins are sorted into different sub-populations
of carrier vesicles, a process mediated by sorting signals (e.g., tyrosine motifs, GPI anchors
and N-glycans) and signal-decoding machinery (e.g., clathrin adaptors and lipid rafts)
[109]. In these cells, endosomes are strategically located between the biosynthetic and
endocytic routes. This allows newly synthesized and endocytosed proteins to be directed to
the appropriate membrane for interaction with different extracellular environments at their
apical and basolateral surfaces [110]. When considering the biogenesis, trafficking and
release of exosomes in polarized cells, it is interesting to note that apical and basolateral
endocytic routes have been shown to converge at the level of late endosomes in Madin-
Darby canine kidney (MDCK) cells [111]. This has led to a hypothesis that exosomes
destined for apical or basolateral membrane release might be generated within a common
late endosomal pool [110]. However, it has also been suggested that epithelial cells could
have distinct MVBs into which apical and basolateral proteins are differentially sorted and
exosomes are formed [110].
Studies investigating these hypotheses have been quite limited. To date, only a single
experimental study has investigated exosomes released from apical and basolateral
surfaces. It was observed that polarized intestinal epithelial cells (T84), grown as a
monolayer on microporous filters, allowed for collection of apical or basolateral exosomes.
These exosomes contained proteins localized to corresponding plasma membrane surfaces
including dipeptidyl peptidase IV on apical exosomes and glycoprotein A33 on basolateral
exosomes [72]. Interestingly, using gel filtration, another study identified two populations
of exosomes in human saliva, however it cannot be claimed that these are apical/basolateral
exosomes due to the potential of multiple cell types contributing to the saliva sample [47].
Further investigation is required to isolate and characterize the contents of apical and
basolateral exosomes. This may lead to an understanding of their formation and trafficking
within polarized cells and potentially identify their varying functions following release.

13
1.4.3 Exosome sizing and morphology
Exosome are characterized by their size of 40-100 nm in diameter [74, 75], however some
studies have reported the identification of larger vesicles up to 150 nm [112]. Various
methods have been used to define vesicle size including transmission electron microscopy
(TEM) which originally identified exosomes to have ‘cup shaped’ morphology [35]. It was
later realized that this phenotype may be a consequence of fixation and dehydration during
sample preparation [1]. In contrast, cryo-TEM preparations, where samples are snap frozen
in liquid ethane, revealed spherical-shaped exosomes that visibly contained ultra-structural
features [113, 114].
Another method used to characterize exosomes is atomic force microscopy (AFM). AFM
provides topographical imaging of exosomes by scanning the surface of deposited samples
with a tipped cantilever and translating tip deflection into a three-dimensional (lateral and
vertical) image of the surface [115]. This method can reach sub-nanometer resolution of
polydisperse samples. However, surface binding may affect the morphology of vesicles and
can hamper the determination of their true diameter [116].
Size-based analysis may also be accomplished by examining the light-scattering properties
of vesicles within a fluid medium that is based upon Brownian motion [117]. Dynamic light
scattering (DLS) is a fast and accurate measurement tool for size distribution and
electrokinetic potential (zeta potential) analysis of monosized particles in suspension.
However, it does have limitations when analyzing polydisperse samples where large
fluctuations in intensity of scattered light are measured [118, 119]. This limitation occurs as
the size distribution analysis is highly influenced by the presence of small amounts of large
particles which scatter more light than small vesicles, causing the distribution to be
weighted towards larger vesicles [116]. Alternatively, it is reported that nanoparticle
tracking analysis (NTA) can analyze polydisperse samples of vesicles between 50-1000 nm
by measuring the absolute size distribution of particles in a mixture [116]. Particles in a
fluid are illuminated by a laser and viewed as small bright spots through a conventional
optical microscope. The Brownian motion of individual particles is recorded through live

14
imaging video and the mean velocity of each particle is calculated with image analysis
software [116]. Although individual particles may be tracked, some limitations of NTA in
comparison to DLS include a limited concentration range (107-10
9 vs. 10
8-10
12
particles/mL) and increased acquisition time (2-5 min vs. 5-60 min). Additionally in NTA,
large aggregates have been reported to mask smaller aggregates, which may enter and leave
the viewing area, making size measurement impossible [120].
Finally, a novel high resolution flow cytometry technique has been developed to quantitate
immunolabeled nano-sized vesicles. Using a new custom-made Influx™ flow cytometer,
small-particle detection has been improved by a wider forward scatter angler and a
photomultiplier tube to allow detection below the previously limiting 300 nm level [121].
This technological advance in the physical characterization of exosomes and other released
vesicles awaits commercialization before becoming widely applicable.
1.4.4 Exosomal proteins
Exosomes have been extensively characterized with respect to their proteome content [35,
52, 122, 123]. Proteomics describes the unbiased and global analysis of all proteins in a
given system [124]. Needless to say, for an accurate protein representation, a homogeneous
exosome sample is essential. Several proteomic techniques, including 1-D sodium dodecyl
sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and 2-D differential in gel
electrophoresis (DIGE) coupled with mass spectrometry (MS) have been used to identify
between ~30 to ~900 proteins from exosome samples [114, 125]. In these workflows, SDS-
PAGE bands/spots are cut, reduced, alkylated and trypsinised. Generated tryptic peptides
are extracted and separated by reversed-phase high-performance liquid chromatography
(HPLC) that is coupled online to a mass spectrometer for analysis [126]. MS tryptic peptide
spectra are then searched against databases using programs including SEQUEST [127],
PROWL [128], and MASCOT [129] to identify proteins. Additionally, Trans-Proteomic
Pipeline (TPP) is an integrated software platform that encompasses most of the steps in a
proteomic data analysis workflow [130]. In addition to identifying proteins, techniques can

15
be used to define the abundance of proteins between samples. Stable isotope labeling by
amino acids in cell culture (SILAC) experimentally measures relative-abundance ratio of
peptides by comparing heavy/light peptide pairs [131]. Other examples of chemical
derivatization techniques for quantitative proteomics include isotope-coded affinity tags
(ICAT), used for the labeling of free cysteine, and isobaric tags for relative and absolute
quantification (iTRAQ), used for the labeling of free primary amine groups [132]. Label-
free quantitation includes either spectral counting or peptide signal intensity to estimate
abundance of proteins [133]. Additionally, semi-quantitative spectral counting using
significant spectral count fold change ratios (RSC) can also be used to quantitate protein
levels between samples [8, 134]. Alternatively, absolute quantitation using selected reaction
monitoring (SRM) [135], also referred to as multiple reaction monitoring (MRM), is a
targeted approach requiring individually designed assays. This involves the selection of a
‘quantotypic’ peptide that is unique to the protein of interest and directly proportional to the
amount of protein present [136]. These isotopically labeled peptides become the internal
calibrant and therefore the unknown abundance of a specific protein can be determined by
comparing its peptide signal intensity with the known calibrant standards [135, 137].
Proteomic studies have been performed on exosomes derived from multiple cell lines and
body fluid sources [122]. Exosomes derived from hematopoietic cells including mast [138],
B and T cells [139, 140], and dendritic cells [141]. Tumor cell lines derived from breast
adenocarcinoma [37], melanoma [142], mesothelioma [143], medulloblastoma [144],
bladder [145], and colorectal cancer [114] are used to characterize cancer-related exosomes
which may assist in defining cell specific markers for disease. Proteomic studies have also
been performed on exosomes derived from body fluids including breast milk [146],
malignant pleural effusions [147], semen [148], urine [149], saliva [47] and blood [41]. An
extensive review of proteomic studies of exosomes isolated from cell lines and body fluids
has been included in Appendix A1 and A2. For a comprehensive account of exosome
studies, including unpublished datasets, see ExoCarta (http://www.exocarta.org/) [122, 123]
and Vesiclepedia (http://www.microvesicles.org/) [52].

16
A conserved set of proteins are observed in exosomes including those involved in their
biogenesis, these include, Alix, TSG101 and other ESCRT complex proteins. Tetraspanin
superfamily proteins CD9, CD63, CD81 and CD82 are commonly found in exosomes; they
are reported to contribute to the protein sorting pathway [74, 150, 151]. Cytosolic proteins
actin and tubulin, and chaperone proteins HSP70 and HSP90 are also often identified in
exosomes. Additionally, exosomes are enriched in proteins that associate with lipid rafts,
including glycosylphosphatidylinositol (GPI) -anchored proteins and flotillin [139] and Rab
GTPases involved in trafficking and endosome fusion events [152]. Exosomes also contain
lipid and nucleic acid (mRNA/miRNA) species [138, 153] (see section 1.4.5 and 1.4.6).
In addition to a conserved set of proteins generally identified in exosomes, a seminal study
comparing proteomic analyses of exosomes revealed that proteins harbored by exosomes
may be cell or tissue specific. This was demonstrated by a comparison of urine, mast cell
and colorectal carcinoma-derived exosomes by Mathivanan and colleagues [44]. A
proteomic analysis was performed on exosomes isolated from the LIM1215 colorectal
carcinoma cell model using immunoaffinity targeting anti-A33, a marker of the basolateral
surface in intestinal epithelial cells [154-156]. This study identified 394 exosomal proteins.
A comparative protein profiling analysis of this study with murine mast cell and human
urine-derived exosomes revealed a common set of 31 proteins including Alix, Rab5 and
annexins A6 and A11. Interestingly, a protein signature reflecting the colorectal cell origin
(LIM1215) was identified. This signature included A33, cadherin-17, carcinoembryonic
antigen (CEA) and epithelial cell adhesion molecule (EpCAM).
1.4.5 Exosomal lipids
In addition to the protein content of exosomes, the lipid components of exosomes have also
been investigated. Characterizing the lipid content of exosomes may assist in our
understanding of their biogenesis and cell-mediated uptake, and in the longer term, the
development of exosome (liposome) encapsulated drugs [153, 157].

17
The process of inward budding of MVBs and exosome formation requires critical lipid
sorting [153]. During inward budding, as two thirds of the lipids required for ILV
formation are located within the inner leaflet of the endosomal membrane, careful
rearrangement and equilibration of lipids from inner and outer leaflets is required [153]. It
is reported that lysobisphosphatidic acid (LBPA) accumulates in the MVB membrane and
interacts strongly with Alix during exosome biogenesis [153]. Interestingly, addition of
LBPA to a lipid composition similar to that of MVBs caused internal budding of small
vesicles within liposomes when adjusted to pH 5.5 (pH of MVBs) [158]. A lipidomic study
of exosomes in comparison to parental cells revealed enrichment in sphingomyelins by 1.3
times in erythrocytes and 2.3 times in B lymphocytes. It was also observed that cholesterol
was enriched in B-lymphocyte-derived exosomes while phosphatidylserine was enriched in
dendritic cell-derived exosomes [153]. Further, it was recognized that phosphatidylcholines
and phosphatidylethanolamines present in exosomes are enriched in saturated fatty acids
comparatively to parental cells [153]. Interestingly, bioactive lipids such as prostaglandins,
and lipid mediators that display multiple biological effects related to inflammation, are
sorted into exosomes [159]. Lipid raft-like domains have also been reported in exosome
membranes. These domains consist of glycerophospholipids with long saturated fatty acyl
chains, cholesterol, flotillin, stomatin, high amounts of sphingolipids and low levels of
phosphatidylcholine [160].
A recent comprehensive lipid profiling study of isogenic primary and metastatic colon cell
lines (SW480 and SW620) revealed differential expression of lipids identified as being
associated with cancer progression [161]. These included increased plasmanylcholine and
triglyceride lipid levels, decreased plasmenylethanolamine and ceramide lipid levels, and a
significant increase of total cholesterol ester and triglyceride lipids in the SW620 cells
compared to those in SW480 cells [161]. It is interesting to hypothesize if similar lipid
contributions would be observed in exosomes. Understanding the lipid content of
exosomes, through comprehensive lipidomic studies of exosomes from various sources,
may shed light on their fate and physiological function.

18
1.4.6 Exosomal RNA
A major breakthrough in characterizing exosomes was the finding of functional mRNAs
and miRNAs, small 22-25 nucleotide non-coding RNAs that suppress the translation of
target mRNAs [162]. This finding highlighted exosomes as prospective vehicles for the
horizontal transfer of biologically important information [163, 164]. Valadi and colleagues
demonstrated that mast cells secreted exosomes containing mRNAs from approximately
1300 genes and more than 100 different miRNAs [138]. Further, an in vitro translational
assay demonstrated that exosomal mRNAs from mouse mast cell line MC/9 are functional
in human mast cell line HMC-1 cells [138]. In a separate study, glioblastoma cell-derived
exosomes containing a reporter gene were incubated with recipient human brain
microvascular endothelial cells (HBMVECs) to reveal mRNA was delivered and translated
[165]. A summary of miRNAs identified from in vitro studies of cancer cell exosomes is
outlined (Table 1-2).
In-depth RNA analysis has been performed to characterize multiple RNA species contained
in exosomes. For example, a small RNA deep sequencing study revealed RNA profiles in
exosomes released from prion-infected neuronal cells [166]. Bellingham and colleagues
demonstrated that neuronal exosomes contain a diverse range of RNA species including
retroviral RNA repeat regions, messenger RNA fragments, transfer RNA fragments, non-
coding RNA, small nuclear RNA, small nucleolar RNA, small cytoplasmic RNA, silencing
RNA as well as known and novel candidate miRNAs. The authors concluded that
exosomes released from prion-infected neuronal cells have a distinct mRNA/miRNA
signature that can be utilized for diagnosis of prion disease [166]. Further work is required
to establish a link between these exosomal RNA species and prion disease pathogenesis.
In another study, deep sequencing technology was used to characterize the RNA content of
exosomes isolated from breast milk [167]. This study revealed 602 unique miRNAs. Of
these, 59 were well characterized immune-relate miRNAs [167]. The longevity of miRNAs
contained within exosomes was increased when compared with circulating RNAs. It was
speculated that due to the protection and durability of exosomal miRNAs, they may assist
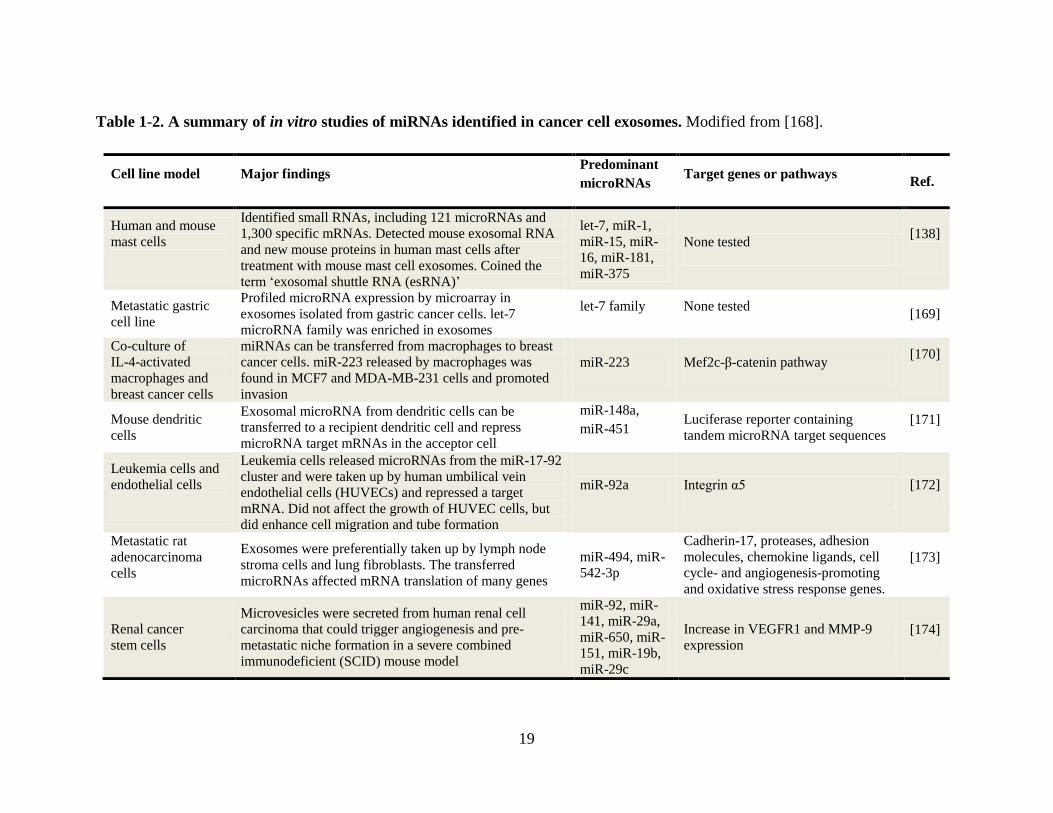
19
Table 1-2. A summary of in vitro studies of miRNAs identified in cancer cell exosomes. Modified from [168].
Cell line model Major findings Predominant
microRNAs Target genes or pathways
Ref.
Human and mouse
mast cells
Identified small RNAs, including 121 microRNAs and
1,300 specific mRNAs. Detected mouse exosomal RNA
and new mouse proteins in human mast cells after
treatment with mouse mast cell exosomes. Coined the
term ‘exosomal shuttle RNA (esRNA)’
let-7, miR-1,
miR-15, miR-
16, miR-181,
miR-375
None tested [138]
Metastatic gastric
cell line
Profiled microRNA expression by microarray in
exosomes isolated from gastric cancer cells. let-7
microRNA family was enriched in exosomes
let-7 family None tested [169]
Co-culture of
IL-4-activated
macrophages and
breast cancer cells
miRNAs can be transferred from macrophages to breast
cancer cells. miR-223 released by macrophages was
found in MCF7 and MDA-MB-231 cells and promoted
invasion
miR-223 Mef2c-β-catenin pathway [170]
Mouse dendritic
cells
Exosomal microRNA from dendritic cells can be
transferred to a recipient dendritic cell and repress
microRNA target mRNAs in the acceptor cell
miR-148a,
miR-451 Luciferase reporter containing
tandem microRNA target sequences [171]
Leukemia cells and
endothelial cells
Leukemia cells released microRNAs from the miR-17-92
cluster and were taken up by human umbilical vein
endothelial cells (HUVECs) and repressed a target
mRNA. Did not affect the growth of HUVEC cells, but
did enhance cell migration and tube formation
miR-92a Integrin α5 [172]
Metastatic rat
adenocarcinoma
cells
Exosomes were preferentially taken up by lymph node
stroma cells and lung fibroblasts. The transferred
microRNAs affected mRNA translation of many genes
miR-494, miR-
542-3p
Cadherin-17, proteases, adhesion
molecules, chemokine ligands, cell
cycle- and angiogenesis-promoting
and oxidative stress response genes.
[173]
Renal cancer
stem cells
Microvesicles were secreted from human renal cell
carcinoma that could trigger angiogenesis and pre-
metastatic niche formation in a severe combined
immunodeficient (SCID) mouse model
miR-92, miR-
141, miR-29a,
miR-650, miR-
151, miR-19b,
miR-29c
Increase in VEGFR1 and MMP-9
expression [174]

20
the development of the infant immune system [167]. Further studies are required to
determine whether specific intracellular miRNAs are enriched in exosomes. Recent
evidence supporting specific miRNA loading of exosomes was the identification that
different miRNA content depended on the maturation stage of the DCs [171], and that cis-
acting elements may target specific RNAs into exosomes [175]. Interestingly, the
identification of a ‘zipcode’ contained within the 3’ region of mRNA has been identified
that leads to incorporation of specific mRNAs into EVs [176]. In-depth characterization of
mRNA and miRNA signatures within exosomes from cancer cells may lead to their use as
clinical biomarkers and give insight into their contribution in disease progression.
1.4.7 Exosome targeting and uptake
It is understood that exosomes are key mediators of intercellular communication and,
therefore, the molecular mechanisms governing exosome recognition and uptake by
recipient cells have gained much attention. An overview of possible modes of exosome
interaction and uptake by recipient cells is given in (Figure 1-3) [32].
Differences in exosomal tetraspanin complexes appear to influence target cell interaction in
vitro and in vivo, possibly by modulating the functions of associated integrin adhesion
molecules [177]. For example, exosome capture by dendritic cells was reduced by 5-30%
by co-incubation with blocking antibodies specific for various integrins, adhesion
molecules or tetraspanins [178]. Other membrane proteins also reported to be important in
targeting exosomes to recipient cells include intercellular adhesion molecule 1 (ICAM-1)
and milk fat globule-epidermal growth factor VIII protein (MFGE-8) [179, 180]. In
addition to membrane proteins, the delivery efficiency of exosomes to cells is reported to
be directly related to rigidity of exosomes that contain lipids including sphingomyelin and
N-acetylneuraminyl-galactosylglucosylceramide (GM3) [181].
In addition to receptor-mediated interactions leading to activation of cell signaling
pathways [25], exosomes may also be internalized. Escrevente and colleagues
demonstrated that fluorescently-labeled ovarian cancer cell-derived exosomes were

21
Figure 1-3. Exosome targeting and uptake
Following release by parent cells, exosomes are shown to communicate and elicit an effect
with recipient cells by (i) receptor-mediated interactions involving tetraspanins and
integrins [177], (ii) membrane fusion [181], and (iii) endocytic uptake [182] by
phagocytosis [183] and macropinocytosis [184].

22
internalized by clathrin-mediated endocytosis in ovarian cancer cells [182], while Feng and
colleagues reported exosomes being internalized via phagocytosis in monocyte-derived
macrophages [183]. Fitzner and co-workers also showed that selective transfer of exosomes
from oligodendrocytes to microglia could occur by macropinocytosis [184], a process
whereby bending of single surface lamellipodia gives rise to circular curved ruffles which
are finally sealed to form discrete endocytic vacuoles [184]. Exosome uptake has been
reported to be a temperature dependent process. For example, exosomes from rat
pheochromocytoma (PC12 cells) were internalized by resting PC12 cells by actin-
dependent endocytosis [185]. Exosomes were shown to be internalized by lipid raft
mediated endocytosis in HUVECs – this process was subsequently negatively regulated by
caveolin-1 [186, 187].
In another study, exosomes were shown to fuse with the plasma membrane in a pH
dependent manner [181]. For example, exosomes purified from metastatic melanoma cell
culture medium were labeled with a self-quenching lipid fluorescent probe, R18, and shown
to fuse at the plasma membrane of recipient melanoma cells [181]. Interestingly, fusion
efficiency was higher with exosomes released from metastatic cells compared to those
derived from primary melanoma cell tumors or normal peripheral blood mononuclear cells
[181]. A mechanism of exosome uptake involving a combination of fusion and endocytosis
has also been reported [171]. Montecalvo and colleagues demonstrated that dendritic cell-
derived exosomes were shown to fuse in ‘2-step event’ consisting of exosome hemifusion
with the cell membrane, followed by endocytosis and complete fusion of the exosomes with
the limiting membrane of the phagosome [171].
Although there are numerous studies demonstrating exosome uptake by recipient cells, very
little is known about the specific target recognition motifs in exosomes. This highlights the
need for in-depth characterization of exosome membranes to identify barcode signatures
that allow and signal for their recognition and uptake by specific recipient cells. Following
exosome uptake there is limited data showing how exosomes traffic intracellularly to exert
their biological effect.

23
1.4.8 Exosome function in the tumor microenvironment and cancer progression
In addition to cancer cells, the tumor microenvironment comprises endothelial cells,
supporting pericytes, fibroblasts, and both innate and adaptive infiltrating immune cells
[188]. Interestingly, as cancer cells accumulate genetic mutations, they can secrete
molecules (and vesicles) that can reprogram normal stromal cells to serve the budding
neoplasm; this process ultimately enables cancer cells to invade adjacent tissues and
disseminate to distant loci [189]. Recently, there has been significant interest in
understanding the involvement of exosomes in the tumor microenvironment and cancer
progression [190]. For example, exosomes have been reported to be involved in multiple
aspects of cancer progression including causing immune suppression [191], stimulating
angiogenesis [192], modulating stromal cells [193], promoting metastatic ability [194], and
establishing the pre-metastatic niche [195] through the transfer of oncogenic material in an
intra- and inter-cellular circuitry [196] (Figure 1-4, Table 1-3).
It has been reported that tumor-derived exosomes bear immunosuppressive molecules and
are implicated in immune suppression [191]. Further, it was demonstrated that exosomes
can inactivate T lymphocytes, or inhibit the differentiation of precursors to mature antigen
presenting cells to suppress immune surveillance, thereby assisting cancer progression
[191]. Angiogenesis can also be influenced by exosomes [192]. For example, melanoma
exosomes were observed to rapidly stimulate the production of endothelial spheroids and
sprouts in a dose-dependent manner [192]. These exosomes elicited paracrine endothelial
signaling by regulation of inflammatory cytokines, including IL-1α, TNFα and VEGF,
suggesting that exosomes can promote endothelial angiogenic responses and contribute to
tumor metastatic potential [192]. Tumor-derived exosomes were also shown to express
membrane TGF-β and activate the SMAD-dependent signaling pathway in stromal
fibroblasts, resulting in myofibroblast differentiation [193]. Myofibroblasts are enriched in
solid tumors and represent an altered stroma that usually supports tumor growth,
vascularization, and metastasis [193].

24
Figure 1-4. Extracellular vesicles (EVs) are components of the intra- and intercellular
oncogenic signaling circuitry
Oncogenic mutations (onc) and the resulting synthesis of altered mRNA and oncoproteins
(ONC) result in perturbations of the intracellular signaling circuitry (SIGNAL) in a primary
tumor. EVs containing onc are released into the extracellular space, and sometimes further
into circulation, enabling exposure of stromal or indolent cells to transforming onc
molecules. Upon uptake of tumor-derived EVs, oncogenic mutations may affect cellular
signaling which can contribute to increased proliferation [197] and invasiveness [198].
Within the circulation, EVs can educate bone marrow-derived cells (BMDC onc) resulting
in mobilization of BMDC onc to lymph nodes to assist pre-metastatic niche formation
[195]. EVs alone can also home to the lymph node for pre-metastatic niche formation
[119]. Figure adapted from [196] using Sevier Medical Art.
25
Table 1-3. Examples of EV-mediated transfer of oncogenic/transforming molecules. Adapted and updated from [196].
Transforming
molecule Vesicle Observations Ref.
Protein/EGFRvIII EVs Glioblastoma EVs transfer EGFRvIII protein to indolent cancer cells causing oncogenic activity
including activation of transforming signaling pathways MAPK and Akt [199]
mRNA/EGFRvIII EVs Glioblastoma EVs containing EGFRvIII oncogene transfer functional mRNA causing stimulation
of tubule formation by endothelial cells [165]
Protein/EGFR EVs Cancer EVs transfer oncogenic EGFR to endothelial cells causing autocrine reprogramming of
VEGF [200]
Protein/MyrAkt1 EVs Cancer EVs transfer oncogenic Akt to recipient cells causing rapid phosphor-tyrosine and Akt
pathway signaling stimulating proliferation and migration [62]
Protein/tTG and FNa EVs Cancer EVs transfer tissue transglutaminase and fibronectin to NIH3T3 fibroblasts causing
tumorigenic conversion [201]
DNA/retrotransposona EVs Cancer cell-derived EVs transfer retrotransposons to endothelial cells [202]
Protein/LMP1 Exosomes Cancer exosomes transfer Epstein-Barr virus latent membrane protein 1 causing activation of
ERK and AKT signaling pathways in recipient epithelial cells [203]
Mutant KRAS Exosomes Exosomes containing mutant KRAS can cause enhanced three-dimensional growth of wild-type
KRAS-expressing non-transformed cells [197]
Wnt11 Exosomes Fibroblast-derived exosomes mobilize autocrine Wnt-PCP signaling in breast cancer cells to
stimulate invasive behavior and metastasis in animal models [198]
Wnt3a Exosomes Exosomes containing Wnt3a can induce Wnt signaling activity in target cells [105]
Tyrosine kinase MET Exosomes Exosomes expressing tyrosine kinase MET increased the metastatic capability of bone marrow
cells [195]
a Oncogenic activity not directly demonstrated

26
The transfer of metastatic activity has also been attributed to exosomes. Melanoma-derived
exosomes were shown to promote epigenetic transfer of metastatic activity in murine
models. Highly metastatic BL6-10-derived exosomes were transferred to poorly metastatic
melanoma F1 cells to express Met 72 tumor antigen [194]. The subsequent Met 72-positive
F1 tumor cells showed increased metastatic activity by growing numerous lung tumor
colonies following their intravenous injection into mice [194]. In addition to exosomes,
microvesicles (MVs) derived from platelets are also capable of enhancing the invasive
potential of breast cancer and induce angiogenesis and metastasis in lung cancer [204, 205].
In a further study, transfer of oncogenic potential to a recipient cell through activation of
MAPK and Akt signaling pathways highlights new mechanisms of intercellular
communication of oncogenic material via MVs in the tumor microenvironment [165, 200].
It is well recognized that tumors preferentially home to specific metastatic sites. For
example, metastasis of melanoma often homes to the lung and brain [206], while colorectal
cancer to the liver [207]. Interestingly, it has recently been shown that malignant cancers
use secreted signals to effectively prime a specific site for metastasis in a ‘seed-soil’
manner by preparing a fertile environment [19, 208]. Melanoma-derived exosomes injected
into the foot pads of mice preferentially colonized and conditioned sentinel lymph nodes by
the introduction of genetic molecular signals for cell recruitment, extracellular matrix
deposition, and vascular proliferation [119]. Further, studies by Peinado and colleagues
have demonstrated that tumor exosomes can horizontally transfer genetic material to bone
marrow-derived cells (BMDCs) resulting in upregulation of the MET oncoprotein. This led
to mobilization of BMDCs to home to the lung and lymph node to support tumor
vasculogenesis, invasion and metastasis [195] (Figure 1-4).
Interestingly, the ability to interfere with molecular pathways responsible for exosome
formation and release may in turn lessen their contribution to disease progression [209].
This could potentially involve the modulation of a key protein required for their formation
(i.e., Rab27a) [103, 210]. Additionally, it has previously been demonstrated that regulation
of Rab GTPase-activating proteins (GAPs) can alter Rab35 activity which leads to the
accumulation of endosomal vesicles and impairment of exosome secretion [101].

27
Attenuation of exosome release from cancer cells can also be accomplished by
administration of neutral sphingomyelinase inhibitors [96], or treatment with the blood-
pressure-lowering drug amiloride [211]. Alternatively, the inhibition of EV uptake by
recipient cells can be altered using diannexin to block phosphatidylserine, which is an
important molecule required for cell adhesion [200, 212]. This experimental process was
shown to reduce the growth rate of human glioma xenografts in mice [200].
1.4.9 Exosome therapeutics
The ability of exosomes to transport bioactive molecules has driven research to improve
their large-scale isolation and characterization for use in translational nano-medicine [213]
(Figure 1-5). Current drug delivery vehicles, including viral and bacterial vectors, have
limited efficacy following repeat administration. These vectors may also elicit cellular
toxicity by causing insertional mutagenesis in host cells [214] (Figure 1-6). Alternatively,
exosomes may have advantageous properties over other drug delivery vehicles as they have
limited immunogenicity when derived from ‘self’, increased stability and efficient delivery
of cargo delivery abilities.
A groundbreaking discovery in exosome therapeutics was the ability of exosomes to
function as natural cell-derived nano-carriers that can cross biological barriers [215, 216].
For example, exosomes may be used to encapsulate and protect exogenous RNA
interference (RNAi) and miRNA regulatory molecules in addition to other single stranded
oligonucleotides [216]. Exciting new work by Alvarez-Erviti and colleagues have
demonstrated that following systemic injections, neuron-specific peptide targeting allowed
effective delivery of exosomes loaded with siRNA into the mouse brain [216]. As a result
of siRNA contained within exosomes, they observed the knock-down of BACE-1, a
therapeutic target in Alzheimer’s disease. Additionally, injections of exosomes (150 µg)
appear to be well-tolerated with little or no immunogenicity or toxicity, even following
repeat (n=3) administration [210, 216].
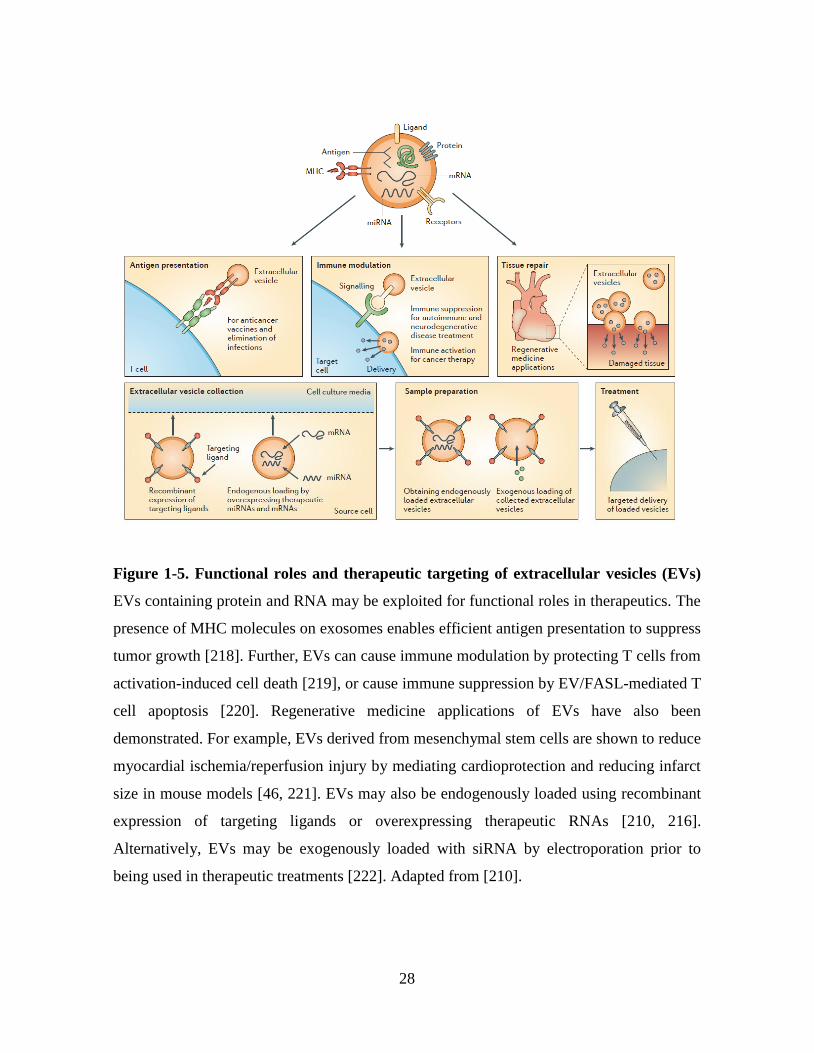
28
Figure 1-5. Functional roles and therapeutic targeting of extracellular vesicles (EVs)
EVs containing protein and RNA may be exploited for functional roles in therapeutics. The
presence of MHC molecules on exosomes enables efficient antigen presentation to suppress
tumor growth [218]. Further, EVs can cause immune modulation by protecting T cells from
activation-induced cell death [219], or cause immune suppression by EV/FASL-mediated T
cell apoptosis [220]. Regenerative medicine applications of EVs have also been
demonstrated. For example, EVs derived from mesenchymal stem cells are shown to reduce
myocardial ischemia/reperfusion injury by mediating cardioprotection and reducing infarct
size in mouse models [46, 221]. EVs may also be endogenously loaded using recombinant
expression of targeting ligands or overexpressing therapeutic RNAs [210, 216].
Alternatively, EVs may be exogenously loaded with siRNA by electroporation prior to
being used in therapeutic treatments [222]. Adapted from [210].

29
Figure 1-6. Advantages and drawbacks of siRNA delivery by exosomes, viruses and
lipid nanoparticles
Major advantages are indicated in green, disadvantages in red. (a) Dendritic cell-derived
exosomes can directly fuse with the plasma membrane through CD9 tetraspanin
interactions with surface glycoproteins on the target cell to ensure direct cytosolic delivery
of siRNA [223]. (b) Viruses mediate direct delivery of vectors encoding short hairpin (sh)
RNA to the cytosol. However, viruses are prone to clearance by opsonins, complement,
coagulation factors and virus-specific antibodies in the systemic circulation. Although
genomic insertion of the shRNA-encoding vectors allows long-term RNAi, a potential risk
of this strategy is genetic deregulation and oncogene activation by insertional mutagenesis
[214]. (c) Internalization of lipid nanoparticles occurs by endocytosis. Endosomal escape is
required for cytoplasmic delivery by fusion with, or rupture of, the endosomal membrane
using viral or bacterial proteins. This escape is often hampered by the man-made nature of
lipid nanoparticles The direct delivery of RNA into endosomes can lead to unwanted
TLR7/8 activation [224]. Adapted from [225].

30
The ability of exosomes to activate T cells has prompted their potential use as vaccines
[217]. Chaput and colleagues demonstrated that peptide loaded dendritic cell-derived
exosomes could mediate tumor rejection in mice [217]. Further, exosomes secreted from
Gm-CSF/IL-4 bone marrow dendritic cells were shown to eradicate established murine
tumors in mammary TSA carcinoma, P815 mastocytoma and MCA205 sarcoma models
[217]. These immunogenic properties have allowed the first phase I clinical trial of
dendritic cell-derived exosomes to treat metastatic-tumor-bearing patients [226]. Good
manufacturing practice (GMP) was used to isolate large-scale pharmaceutical-grade
exosomes and directly load peptides on functional MHC complexes. Autologous exosomes
were inserted into patients to observe an increase in NK cells, thus highlighting their
potential use as a vaccine [226]. It was also observed that increased levels of hsa-let-7b and
hsa-let-7g miRNAs in mesenchymal stem cell-derived exosomes were able to treat
myocardial infarction [227, 228] and provide protection against myocardial ischemia [46].
A list of other therapeutic applications of exosomes is outlined in (Table 1-4).
An alternative to tailored biological exosomes are artificial exosome ‘mimetics’, which are
being developed for clinical immunology [229]. It is hypothesized that the incorporation of
only key exosomal lipids, protein and nucleic acids will lead to the creation of a viable
therapeutic vehicle [229]. Using nanotechnology-based approaches, targeted and traceable
in vivo super paramagnetic artificial exosomes (termed liposomes) have been developed
[230, 231]. Liposomes coated with a MHC class I/peptide complexes and a range of ligands
for adhesion were tracked in vivo using fluorescence and magnetic resonance imaging and
shown to activate and expand functional T cells [232]. Interestingly, Delcayre and
colleagues have coined ‘exosome display technology’ which utilizes the fusion of antigens
onto the C1C2 domain of lactadherin [233], allowing for presentation of antigens to the
immune system. Further, this technology could also be incorporated into liposomes.
Inclusion of tetraspanin proteins in exosome mimetics has been proposed due to their
ability to functionally mediate fusion and cell-cell adhesion [234]. Challenges associated
with liposome creation and usability include their sensitiveness to clearance by opsonins,
complement and coagulation factors in the blood [225] (Figure 1-6). Additionally,
although lipid assembly is understood, incorporation of functional membrane proteins into

31
Table 1-4. Therapeutic applications of exosomes
Application Therapeutic observation Ref.
Antigen
presentation
Dendritic cells pulsed with tumor-related peptides are release on exosomes to suppress tumor
growth [218]
Immune
modulation
Exosomes cause immune activation by protecting T cells from activation-induced cell death [219]
Immune suppression by exosome/FASL-mediated T cell apoptosis [220]
Tissue repair Manufacturing of therapeutic exosomes following immortalizing of mesenchymal stem cells [236]
Exosomes secreted by mesenchymal stem cells reduces myocardial ischemia/reperfusion injury [46, 221]
Gene therapy
Transfection of miR-150 in monocytes released miR-150 containing functional exosomes [237]
siRNA loaded into exosomes by electroporation is shown to be functional by knocking down
BACE1 in mouse brain, a therapeutic target in Alzheimer’s disease
[210,
216]
Human plasma-derived exosomes were electroporated with siRNA to deliver a functional RNAi
response in human monocytes and lymphocytes in vitro [222]
Adendo-associated viruses (AAVs) displaying viral caspid proteins contained within ‘vexosomes’
can deliver cargo to recipient cells [238]
Tissue targeting
Plasmid expressing exosomal proteins fused to brain specific peptide (rabies virus glycoprotein,
RVG) is displayed on exosomes and delivered across blood brain barrier [216]
Exosomes containing the transmembrane domain of platelet-derived growth factor receptor fused
to the GE11 peptide homed to EGFR-expressing tumor cells to deliver let-7a anti-tumor miRNA [239]

32
liposomes has proven to be a more difficult process [235]. Overall, it is hypothesized that
these challenges can be overcome, and that liposomes may be produced on a large scale to
act as pharmaceutical-grade delivery vehicles that carry similar therapeutic properties to
exosomes [229].
1.4.10 Exosome and microvesicle diagnostics
The capacity of exosomes and other EVs to protect genomic material makes them a
promising source of potential biomarkers. One assay reaching clinical utility is the use of
mRNA profiling of serum-derived EVs to identify glioblastoma-bearing patients [240]. It
has also been reported by Mitchell and colleagues that miR-141 levels from serum,
proposed to be contained within exosomes, can robustly distinguish prostate cancer patients
from healthy controls [241]. Additionally, there are several companies that have begun
development of exosome based diagnostics. New York based biotechnology company,
Exosome Diagnostics (http://www.exosomedx.com/), is performing the characterization of
mRNA and miRNA contained within exosomes (and other EVs) to develop biofluid-based
molecular diagnostic tests for use in personalized medicine. Caris Life Sciences
(http://www.carislifesciences.com/) is also developing methods for isolating and
characterizing blood based circulating microvesicles. Finally, HansaBioMed
(http://www.hansabiomed.eu/) has developed an ELISA based platform called Exotest
which utilizes CD63, CD9 and CD81 antibodies immobilized on 96-well plates in an effort
to capture, quantify and characterize exosomes. It is evident that there is significant interest
in this field of research, with several companies having initiated programs that exploit the
diagnostic potential of exosomes.
1.4.11 Exosome isolation strategies
A major challenge in the field is to improve and standardize methods for exosome isolation
and characterization. Current strategies for isolating exosomes differ significantly based

33
upon their various physical attributes. These strategies include ultracentrifugation [242],
sucrose or OptiPrep™ gradients relying on vesicle density [35, 243] and immunoaffinity
approaches relying on a specific exosome surface protein target (Table 1-1). This diversity
in purification methodology has led to variations in sample homogeneity and confusion
with respect to nomenclature within the field (e.g., exosomes vs. exosome-like vesicles). As
different isolation strategies may have varying fractionation capabilities, it is critical to
evaluate these techniques and their abilities to isolate a homogeneous population exosomes
These isolation strategies are described in turn:
Differential ultracentrifugation involves removal of intact cells and bulky cell debris by low
speed centrifugation (e.g., 500 g, 2,000 g) followed by high-speed centrifugation (e.g.,
100,000 g) to sediment exosomes [242]. This widely-used methodology, however, can
potentially result in co-sedimentation of protein aggregates and/or other membranous
vesicles which may be pelleted during ultracentrifugation [7].
Ultracentrifugation using a linear sucrose gradient has been developed to isolate exosomes
based on their density of 1.08-1.22 g/cm3 [35, 242]. For clinical purposes, the preparation
of GMP-grade exosomes can be prepared using a combination of ultrafiltration,
ultracentrifugation and a 30% sucrose/deuterium (D2O) (98%) cushion (1.21 g/cm3) [244].
Interestingly, sucrose gradients have been inefficient in separating exosomes from HIV-1
particles due to similarities in their size and buoyant density [245]. To overcome this
problem, Cantin and colleagues describe the use of iodixanol (OptiPrep™) 6-18% gradients
to separate HIV-1 particles and apoptotic vesicles from exosomes [243]. Iodixanol is a non-
ionic, water-soluble radiographic contrast medium [246] that may be beneficial for future
clinical use with exosomes due to its physical tolerance and low incidence of adverse
effects when injected and used as an x-ray contrast medium [247].
Immunoaffinity capture (IAC) is another widely used method for exosome isolation. This
strategy is based on the use of specific exosome-surface protein directed antibodies coupled
to magnetic beads (Table 1-1). IAC can be a powerful tool for exosome isolation, provided
a specific exosomal cell surface protein can be identified that discriminates the exosome of

34
interest from other EVs present in the biological matrix. Immuno-isolation of exosomes has
been performed to purifying antigen presenting cell-derived exosomes [40], CD45-
exosomes from Jurkat T cells [39], HER2-positive exosomes from breast adenocarcinoma
cell lines and ovarian cancer patient-derived ascites [37], A33-positive exosomes released
from colon carcinoma cancer cells [44], and EpCAM-positive exosomes from the sera of
lung cancer [42] and ovarian cancer [43] patients. This technique is limited by the number
of known cell, and thus, specific exosome membrane proteins. A final strategy, size-
exclusion HPLC, has also been successfully employed for the large-scale production of
microparticles from human mesenchymal stem cells [45, 46], and human saliva [47].
1.5 Research models used in this thesis
Cell lines that are readily cultured in vitro are often used as surrogates for tumors as they
can reflect their tissue of origin [248]. Cell lines have an advantage over cancer biopsy
tissue in that they are homogeneous as compared to heterogenic solid tumors which, in
addition to cancer cells, contain multiple other cell types [249]. Cell lines also provide an
unlimited auto-replicative source that can be easily handled, may contain specific known
mutations, or can be genetically manipulated [250]. In this thesis, two different cell line
models were utilized: the colorectal cancer (CRC) cell line LIM1863 [251] and an EMT
cell model created from epithelial Madin-Darby canine kidney (MDCK) cells [250].
1.5.1 Colorectal cancer model
1.5.1.1 Colorectal cancer
The structure of the adult colon, or large intestine, is composed of three concentric tissue
layers. The outer layer consists of several sheets of smooth muscle that execute the
rhythmic peristaltic movements of the intestine [252]. The space between the outer muscle
and the inner epithelial layer is filled by connective stromal tissue that contains numerous

35
blood and lymph vessels, nerve fibers, and various immune cells. The inner luminal surface
consists of a single-cell layer comprising differentiated epithelial cells that mediate
functions of the intestinal epithelium [252]. Finally, finger like invaginations of this
epithelial sheet into the underlying connective tissue form ‘colonic crypts’, the basic
functional unit of the colon [253]. CRC is defined as the development of malignant tissue
in the wall of the colon or rectum and is caused by a series of stepwise genetic changes
(Figure 1-7) [254].
1.5.1.2 CRC statistics and subtypes
About 608,000 deaths from colorectal cancer are estimated to occur worldwide each year.
This accounts for 8% of all cancer deaths, making it the fourth most common cause of
cancer-related death [255]. According to the Australian Institute of Health and Welfare,
CRC was the most commonly registered form of cancer in Australia in 2009 with 14,410
new cases and 3,982 deaths, making up 9.3% of all cancer deaths in Australia [256]. The
Dukes’ staging system (A-D) is commonly employed to stage progression and it has been
shown that CRC can be effectively treated if detected early [257, 258]. Based on 5-year
survival rates, patients presenting with Dukes’ stage A CRC have >90% chance of survival,
while more commonly, patients presenting with late stage Dukes’ stage D CRC have 5-year
survival rates <10% [259]. A lack of early detection occurs due to low patient compliance
with current testing modalities coupled with the limited sensitivity of current detection
techniques [260].
Typically there are two forms of CRC, termed sporadic and hereditary. Sporadic CRC
refers to random occurrences in which family history is absent [261]. This accounts for
approximately 70% of all CRCs [262]. Sporadic CRC arises from the accumulation of
multiple genetic mutations in somatic cells over an extended period of time and therefore a
late age onset is commonly observed [263]. Hereditary CRC is divided into two classes,
familial adenomatous polyposis (FAP) and hereditary non-polyposis colorectal cancer

36
Figure 1-7. Model of intestinal cancer initiation and progression
Crypt base columnar cells (green) and adenoma cell (red) are located at the base of the
crypt. Loss of adenomatous polyposis coli (APC) is known to be an initiating event behind
early adenoma formation (red-hued) where cells begin to migrate up the transit amplifying
compartment (blue). The accumulation of additional oncogenic mutations (shown in italics)
drives tumor progression from a benign intermediate and late adenoma to an invasive
carcinoma. Other cell types include entero-endocrine cell (yellow), goblet cell (orange), and
tuft cell (purple). Adapted from [254].

37
(HNPCC), which together make up ~10% of all colon cancers [262]. FAP is characterized
by germline mutations in the adenomatous polyposis coli (APC) gene, resulting in the
presence of numerous adenomatous polyps [264]. The APC gene encodes a ~300 kDa
protein which selectively localizes to the nucleus and cytoplasm [265]. Under normal
circumstances, APC functions as a tumor suppressor by mediating the downregulation of β-
catenin. When APC is mutated, β-catenin accumulates in the nucleus and binds to
transcription factors to promote cellular proliferation [266]. HNPCC is caused by mutations
in one or more DNA mismatch repair genes, most commonly hMSH2 and hMLH1 [267].
Other less frequently mutated genes are hMSH6 and hPMS2 [267]. These mutations are
autosomal dominant, resulting in aggressive tumor growth in the right side colon in 70% of
cases [268].
1.5.1.3 Colorectal cancer cell line LIM1863
The human LIM1863 (Ludwig Institute Melbourne) cell line was derived from a tumor
sample taken from the ileocecal valve, between the ileum of the small intestine and the
caecum of the large intestine, from a 74-year-old Caucasian female [251]. This sporadic
carcinoma extended through the full thickness of the muscle wall and mutational analysis
revealed truncated APC (stop at aa 1506) and mutated p53 (aa 234 Y to H) genes. In vitro,
the LIM1863 cell line grows as a suspension culture of floating organoids, clusters of cells
resembling crypt like structures, which are morphologically and functionally organized.
LIM1863 organoids comprise two polarized cell types, columnar and goblet cells
surrounding a central lumen (Appendix A-3). Along with continuous cell proliferation and
organoid formation, dead cells are continuously shed. This organization is very similar to
that of in vivo, where intestinal epithelial cells form intestinal crypts [251]. Significantly,
single cell cloning was used to isolate 29 subclones of the LIM1863 cell line, all of which
grew to display the same phenotype and percentage of morphological cell types as the
parent line, thus indicating ‘stemness’ like properties of the cells [269].

38
Isolating exosomes from CRC cell lines may allow insights into their contribution to the
disease. As the LIM1863 cell line grows as floating organoids in suspension, it can be
cultured on a large scale, this is beneficial for characterization studies as large amounts of
exosomes can be isolated. Further, as LIM1863 cells are polarized, the cell model can be
used to understand exosomes in the context of apical and basolateral trafficking and release.
1.5.2 Epithelial-mesenchymal transition (EMT) model
Epithelial-mesenchymal transition is a process by which epithelial cells lose polarity and
cell adhesion characteristics and acquire migratory and invasive properties of mesenchymal
cells [270, 271]. For many decades, EMT has been recognized as a cell phenotypic switch
in developmental biology, embryogenesis, tissue remodeling and wound healing [272,
273]. During these processes, a series of molecular and cellular changes occur allowing
epithelial cells to escape from their rigid structural constraints to accommodate a phenotype
more amenable to cell migration and invasion (Figure 1-8) [274]. These changes include
reduced cell-cell adhesion, loss of cell polarity, elongated spindle-shaped morphology, an
invasive phenotype, induction of mesenchymal-specific proteins, ECM alterations, and
increased migratory capacity (Appendix A-4) [275]. The activation of an EMT program
has also been proposed as the critical mechanism for the acquisition of malignant
phenotypes by epithelial cancer cells [189, 276, 277] (Figure 1-9).
1.5.2.1 EMT during cancer progression
Early growth of primary epithelial cancers is characterized by excessive cell proliferation
and angiogenesis [189]. Following these changes, local invasiveness, initially through the
basement membrane, a dense specialized ECM barrier between the epithelial cell layer and
underlying stroma, is thought to begin the multi-step process that leads to metastatic
dissemination [270]. The ability of cells to penetrate through the basement membrane may
be attributed to the increased expression of secreted proteases [278].

39
Figure 1-8. Epithelial-mesenchymal transition
Epithelial-mesenchymal transition (EMT) is a morphogenetic process whereby immotile
polarized epithelial cells become migratory mesenchymal cells. This highly conserved
cellular process involves dissolution of cell-cell junctions, i.e., tight junctions (dark blue),
adherens junctions (light blue) and desmosomes (green), and loss of apical-basolateral
polarity. Cells will then acquire a mesenchymal phenotype, this is characterized by actin
reorganization, stress fiber formation (red), a ‘leading-edge’ polarity, migration and
invasion phenotypes. Modified from [274].

40
Figure 1-9. EMT during tumor progression and metastasis
Normal epithelia proliferate to form a local adenoma. Genetic alterations and epigenetic
events produce a carcinoma in situ. Loss of cell-cell adhesion and increased invasiveness,
possibly by EMT, drives cells through a fragmented basement membrane where they can
intravasate into lymph or blood vessels. It is proposed that the invasive tumor cells are
passaged to distant sites where they repopulate and form secondary metastases. Modified
from [276].

41
These proteases include matrix metalloproteinases (MMPs) and a disintegrin and
metalloproteinases (ADAMs) that function at cells invasive leading edge to proteolyse
collagens and laminins to assist cell migration [278, 279]. Cells expressing a mesenchymal
phenotype are typically at the invasive tumor front and are considered to ultimately follow
an invasion-metastasis cascade [270]. Interestingly, at distant sites colonized mesenchymal
cells are reported to revert back and resemble their originating tumor phenotype. This
process is known as mesenchymal-epithelial transition (MET), whereby mesenchymal
characteristics are lost in favor of epithelial traits [270]. It is proposed that this may occur
as signals originally required for driving and maintaining the mesenchymal phenotype in
the primary tumor environment are no longer apparent [270, 280].
1.5.2.2 Molecular markers of EMT
The architecture of epithelial cells is controlled by the expression of various proteins
including E-Cadherin, which are associated with cell-cell contact [281]. E-cadherin is a
transmembrane protein required for the establishment and maintenance of adherens
junctions and is fundamental to the epithelial phenotype [282-284]. The suppression of E-
cadherin diminishes epithelial cell adhesion, alters polarity and is associated with increased
cell invasiveness during cancer metastasis [285]. Following E-cadherin suppression, the
mesenchymal cell-cell adhesion molecule N-cadherin becomes highly expressed, a process
also known as the ‘cadherin switch’ [284].
In addition to E-cadherin, the modulation of many other components has been reported
during EMT [286]. These alterations include downregulation of epithelial adhesion
molecules (e.g., claudins, occludin and the ZO-family) and upregulation of mesenchymal
molecules (e.g., vimentin, fibronectin and α5β1 integrin) [287]. Additionally, during EMT
cytokeratin networks are replaced by vimentin-rich intermediate filaments, leading to
enhanced cell migration and invasiveness [288].

42
1.5.2.3 Signaling pathways and inducers of EMT
The induction of EMT can occur via a complex array of extracellular signaling
components, cytoplasmic molecules and nuclear effectors of transcription [289]. The EMT
process may be initiated by growth factors such as fibroblast growth factor (FGF),
epidermal growth factor (EGF), TGF-β, TNF-α, and bone morphogenic protein (BMP)
[270, 290, 291]. These initiating events can activate signaling pathways of cytoplasmic
effectors including SMADs, glycogen synthase kinase 3 β (GSK-3β), mitogen-activated
protein kinase (MAPK), and reactive oxygen species leading to activation of transcriptional
regulators [289]. It is understood that once expressed and activated, EMT-inducing
transcription factors, notably, smad interacting protein (SIP-1), Lymphoid enhancer factor
(LEF), Snail, Slug, Zinc finger E-box binding homeobox 1 (ZEB1), Twist, Goosecoid, and
Forkhead box protein C2 (FOXC2) can act pleiotropically to choreograph the complex
EMT program [276, 289, 292].
In addition to extracellular signaling components, the transfection of oncogenes into
epithelial cells can also induce EMT [293]. Ras is a small GTPase that is involved in many
signaling pathways that regulate cellular processes including cell proliferation,
differentiation, and cell motility [294]. Active Ras-GTP initiates a MAPK phosphorylation
cascade that facilitates the altered expression of transcription factors including Jun, Fos and
Slug, which can induce EMT [282]. Additionally, it was found that oncogenic Ras
signaling downregulated Rac activity and upregulated Rho activity, leading to
rearrangement of the actin cytoskeleton during EMT [295]. Further, the Ras effector
pathway, Raf/MAPK, was shown to transform Madin-Darby canine kidney (MDCK) cells
towards a mesenchymal phenotype [296]. Examination of MDCK cells following
transformation with oncogenic H-RasV12 demonstrated a loss of polarity, detachment from
the substratum, and multilayer growth [297].

43
1.5.2.4 Madin-Darby canine kidney (MDCK) and 21D1 cell lines
The MDCK cell line was derived from the kidney of a normal male cocker spaniel in 1958
[298] and is one of the most-widely used and best characterized epithelial cell lines
available [299]. MDCK cells appear round and resemble cobble-stone like structures. They
grow as a polarized monolayer, with strong cell-cell contact and show expression of
epithelial cell junction markers E-cadherin and ZO-1 [250].
MDCK cells and oncogenic H-Ras-transformed MDCK cells, termed 21D1 cells, are
together recognized as an epithelial-mesenchymal cell model [250]. 21D1 cells were
generated by transfecting pcDNA3 containing v-Ha-Ras under the control of a CMV
promoter and a SV40-driven neomycin resistance plasmid gene into MDCK cells [250]. In
this study, 21D1 cells were shown to have reduced cell-cell contact and display spindle
shaped morphology following transformation [250] (Figure 1-10). Confocal microscopy
images of mesenchymal 21D1 cells showed reduced expression of E-cadherin and ZO-1
between cell junctions, and increased expression of the mesenchymal marker vimentin.
Wound healing and migration assays also revealed that 21D1 cells have enhanced motility
and invasive capabilities when compared to MDCK cells [300].
1.5.2.5 Proteomic analysis of the secretome from MDCK cells following EMT
Using the EMT model previously described, the secretome of MDCK cells following Ras-
induced EMT was analyzed in the Simpson laboratory to identify differentially expressed
secreted proteins which may influence tumor cell state and invasive potential [250, 300].
Using a semi-quantitative label-free spectral counting method, common constituents of the
basement membrane including laminins and collagens were downregulated, indicating
ECM remodeling following EMT. Several proteases such as MMP-1 and kallikrein 6 were
identified to be highly upregulated after EMT (Appendix A5). The precise function of
MMP-1 in EMT was further studied by an MMP-1 siRNA knock-down assay in 21D1 cells
which led to reduced cell migration. Collectively, it is hypothesized that secreted effectors

44
Figure 1-10. MDCK and H-Ras-transformed MDCK cells (21D1) morphology
(A) MDCK cells grow as round cobblestone-like sheets. (B) 21D1 cells exhibit elongated
and spindle shaped cell morphology.
A B

45
might co-ordinate cell migration and EMT in a hierarchical manner [300].
1.5.2.6 Exosomes and EMT
While the direct involvement of exosomes in the process of EMT has not yet been studied,
pioneering work investigating exosome-like vesicles following EMT has been undertaken
[301]. It was found that A431 epithelial cancer cells adopt mesenchymal-like features upon
activation of epidermal growth factor receptor (EGFR) and repression of E-cadherin.
Exosomes were isolated from these mesenchymal-like cells and shown to contain increased
levels of tissue factor (TF) that caused pro-coagulant conversion of endothelial cells. This
indicated that cells with mesenchymal-like features release vesicles capable of altering cell
interactions within the vasculature [301]. Further, there are a significant number of studies
identifying that exosomes are able to transfer ‘oncogenic’ material to assist cancer
progression (see section 1.4.8) [196]. Although exosomes are identified to be released from
mesenchymal cells [301], a detailed proteomic characterization has not yet been performed.
It is therefore hypothesized that an in-depth proteomic analysis of exosomes isolated from
MDCK and 21D1 cell lines may define extracellular modulators contained in exosomes and
lead to a better understanding of their funtional roles.
1.6 Thesis aims
Exosomes are 40-100 nm diameter membrane-derived nanovesicles of endocytic origin that
are released by most cell types. They are reported to have pleiotropic biological roles
including involvement in cell-cell communication. The majority of functional studies
performed on exosomes to date have involved heterogeneous or crude vesicle populations.
It is anticipated that by developing robust exosome purification strategies, and studying
precise protein profiles in homogeneous exosome populations, valuable biological insights
will be gained.

46
The overarching aim of this thesis is to undertake in-depth proteomic analyses on highly-
purified exosomes in order to gain insights into the role of exosomes in cancer biology and
the epithelial-mesenchymal transition process.
The specific aims of this thesis are:
1. To evaluate exosome purification strategies
Our capacity to gain meaningful biochemical insights from exosomes is impeded by the
fundamental difficulty in isolating exosomes to homogeneity. Chapter 2 describes a
comparative evaluation of three widely-used methods for isolating exosomes: differential
ultracentrifugation, OptiPrep™ density gradient centrifugation and immunoaffinity capture
using EpCAM monoclonal antibody bound to magnetic beads. In this study the human
colorectal cancer cell line LIM1863 [251] was used as a model. In order to compare
proteome profiles between the three isolation methods, GeLC-MS/MS and label-free MS
spectral counting was employed. Following isolation of exosomse, sample homogeneity
was based on the expression of defined stereotypical exosomal proteins reported in the
literature.
This aim is addressed in Chapter 2 and incorporates - Tauro, B. J., Greening, D. W.,
Mathias, R. A., Ji, H., Mathivanan, S., Scott, A. M., and Simpson, R. J. (2012) Comparison
of ultracentrifugation, density gradient separation, and immunoaffinity capture methods for
isolating human colon cancer cell line LIM1863-derived exosomes. Methods 56, 293-304.
2. To analyze exosome sub-populations released from LIM1863 cells
The colon cancer cell line LIM1863 [251] grows as crypt-like ‘organoid’ structures with
highly-polarized columnar and goblet cells organized around a central lumen. Chapter 3
describes a strategy that involves the sequential use of two immunoaffinity target reagents

47
(anti-A33 antibody and anti-EpCAM antibody coated magnetic beads) for isolating
different sub-populations of exosomes. This study was directed at characterizing protein
cargo contained in apical- (e.g., positive for epithelial cell marker EpCAM) and basolateral-
(e.g., positive for basolateral marker A33) derived exosome populations. A proteomic
analysis of sMVs was also undertaken to assess whether these vesicles differ from
exosomes.
This aim is addressed in Chapter 3 and incorporates - Tauro, B. J., Greening, D. W.,
Mathias, R. A., Mathivanan, S., Ji, H., and Simpson, R. J. (2013) Two distinct populations
of exosomes are released from LIM1863 colon carcinoma cell-derived organoids.
Molecular & Cellular Proteomics 12, 587-598.
3. To analyze exosomes following epithelial-mesenchymal transition
While it is becoming increasingly evident that epithelial-mesenchymal transition (EMT)
plays a critical role in cancer cell plasticity [277], it is not known whether exosomes
contribute to the EMT process. Chapter 4 describes the in-depth proteome analysis of
exosomes released from Madin-Darby canine kidney (MDCK) cells and upon oncogenic H-
Ras induced EMT (21D1 cells). This study identified exosomal proteins that were
significantly dysregulated following oncogenic H-Ras induced EMT. This finding indicates
potential functions of exosomes associated with the EMT process.
This aim is addressed in Chapter 4 and incorporates - Tauro, B. J., Mathias, R. A.,
Greening, D. W., Gopal, S. K., Ji, H, Kapp, E. A., Coleman, B. M., Hill, A. F., Kusebauch,
U., Hallows, J. L., Shteynberg, D., Moritz, R. L., Zhu, H. J., and Simpson R. J. (2013)
Oncogenic H-Ras reprograms Madin-Darby canine kidney (MDCK) cell-derived exosomal
proteins during epithelial-mesenchymal transition. Molecular & Cellular Proteomics 12,
2148-2159.

48

49
Chapter 2: Comparison of methods for isolating
exosomes
2.1 Overview
The secretome, a rich complex subset of macromolecules secreted by a cell, is a vital aspect
of cell-cell communication in eukaryotes [9]. There has been much interest in the secretome
from cancer cells due to the recognition that secreted molecules could spawn new therapies
[9, 13, 14, 302]. In addition to soluble proteins, the secretome also contains a multitude of
extracellular nano-membraneous vesicles (EVs) [303] (see section 1.2); of these, 40-100
nm diameter exosomes have been the most widely studied from a functional perspective.
However, one of the setbacks in the exosome field is that most functional studies have been
conducted using impure preparations, typically containing a mixture of EVs. Since this
situation confounds the interpretation of such studies there is a fundamental need to purify
exosomes to homogeneity. For these reasons, I have performed, and describe in this
chapter, a detailed comparison of typical exosome isolation methods currently used in the
field. Due to the scope of this chapter and instrument limitations, we were unable to include
HPLC purification of microparticles in this comparison. Size-exclusion HPLC has been
successfully employed for the large-scale production of cardioprotective microparticles
from human fetal mesenchymal stem cells for clinical purposes [45, 46], and for isolating
two distinct exosome populations from human saliva [47].
Commonly used exosome isolation methods include: (i) differential ultracentrifugation to
‘pellet’ exosomes, (ii) density-gradient centrifugation to fractionate exosomes from other
EVs by exploiting their buoyant density, and (iii) immunoaffinity capture on magnetic
beads using antibodies directed towards known exosomal membrane proteins (see section
1.4.11) [35].

50
Differential centrifugation utilizes multiple centrifugation and filtration stages to separate
organelles [304], intracellular vesicles [305], and exosomes [50] based on differences in
density. Typically, this method involves serial low speed centrifugation (500 g, 2000 g)
to remove cells and cellular debris, followed by 0.1 µm [306] or 0.22 µm [50] membrane
filtration, and ultracentrifugation between 60-100,000 g to pellet exosomes [307]. Due to
its limited resolving ability, the differential ultracentrifugation procedure can result in
exosome preparations being significantly contaminated by other classes of EVs, protein
oligomers and pathogens (e.g., adenoviruses, prions etc) [7]. For example, it has been
shown that shed microvesicles (sMVs, also referred to as oncosomes) and exosomes are
morphologically distinct, the former being heterogeneous in size (100 nm-1000 nm) and
sedimenting at lower speeds relative to exosomes [308]. Thus it is likely that EVs that
sediment at 100,000 g may contain a mixture of sMVs and exosomes.
The biogenesis of sMVs and exosomes are quite distinct. sMVs are formed by outward
budding of the plasma membrane, however, it should be noted that not all plasma
membrane proteins are incorporated into shed microvesicles, yet the topology of the
membrane remains intact. Oncogenic membrane proteins and other growth factor receptor
proteins, integrin receptors and soluble proteins such as proteases, cytokines and nucleic
acids are found in microvesicles, however, it is not clear how specific cargo proteins are
trafficked to microvesicles [49]. In contrast, exosomes are formed by endocytic
invagination of late endosomes to form multivesicular bodies (MVBs) which then traffic to,
and fuse with, the plasma membrane, releasing their cargo of exosomes (see section 1.4.1 –
1.4.2). Like sMVs, exosomes contain bioactive molecules such as oncogenic proteins,
mRNAs and miRNAs that can be horizontally transferred to recipient cells and are thought
to be implicated in tumor invasion and metastasis [165]. Whether biological differences
exist between sMVs and exosomes remains to be determined. For these reasons it is
important to purify exosomes to homogeneity (free of sMVs) in order to understand their
biological function [49, 308].
Isolation of exosomes by density gradient centrifugation involves their fractionation across
a continuous or discontinuous density gradient, i.e., sucrose or iodixanol (OptiPrep).

51
Exosomes can be purified by floatation on linear sucrose gradients (2.0-0.25 M) at densities
of 1.08-1.22 g/cm3 [35, 242]. However, sucrose gradients have been shown to be inefficient
in separating exosomes and HIV-1 particles due to their similarity in size and buoyant
densities [245]. Recently, OptiPrep™ gradients have been shown to be an alternative to
sucrose gradients due to their ability to separate exosomes from HIV-1 particles [243].
Interestingly, iodixanol (sold under the FDA approved Visipaque™ UCM198414) is
commonly used as a radiopaque contrast agent for intravascular use to diagnose coronary
angiopathy, brain, blood vessel and kidney disorders. For this reason, OptiPrep prepared
exosomes are compatible with in vivo functional studies.
Immunoaffinity capture can be used to isolate exosomes from a variety of cellular sources
(Table 1-1), however, this method is dependent on the availability of monoclonal
antibodies directed towards tissue-specific exosomal membrane markers [44]. For example,
immunoaffinity capture has been used to isolate exosomes containing A33 from epithelial
colorectal cancer cells [44], HER2-tumor exosomes from breast adenocarcinoma cell lines
[37] and ascites of ovarian cancer patients [37], Rab5b-positive exosomes from melanoma
cell lines [38], CD45-positive exosomes from Jurkat T cells [39], MHC class II-exosomes
from antigen presenting cells [40], CD63-positive exosome-like vesicles from blood [41],
and EpCAM-positive exosomes from human sera of lung cancer [42] and ovarian cancer
patients [43].
In this chapter, label-free MS spectral counting [8, 134] was used to quantitate the relative
levels of proteins in exosomes prepared by the three commonly-used exosomal isolation
strategies described above. Using the LIM1863 colorectal carcinoma cell line as a model,
exosomes were isolated from concentrated culture medium by differential
ultracentrifugation (UC-Exos), OptiPrep density gradient centrifugation (DG-Exos), and
EpCAM-magnetic microbead immunoaffinity capture (IAC-Exos). This study revealed that
stereotypical exosome markers Alix, TSG101, and HSP70 were significantly enriched in
the IAC-Exos dataset when compared with differential centrifugation and density gradient
centrifugation (OptiPrep) prepared samples. Detailed examination of the IAC-Exos
dataset also revealed the enrichment of other ESCRT complex subunit proteins involved in

52
exosome biogenesis, and Rab proteins involved in exosome trafficking and release. Taken
together, the findings from this study indicate that EpCAM-based immunoaffinity capture
is more effective than density gradient centrifugation, which in turn is more effective than
differential centrifugation, for enriching exosomes.
For IAC to be more generally applicable there is a need, first, to identify tissue- and cell-
type specific membrane proteins and, secondly, to generate mAbs directed to such proteins
that allow for the selective capture of exosomes using mAb-loaded magnetic beads. In
addition, there is a need to fully evaluate the effectiveness of different immunoaffinity
capture beads, since in my studies (used in Chapter 3), the background contaminants
associated with Protein G Dynabeads® and magnetic assisted cell sorting (MACS)
microbeads varied significantly. Further studies need to be performed to discriminate
whether this is due to mAb isotype or the chemistry of the magnetic beads employed.
Advances in the identification of tissue-specific exosome surface proteins, the development
of corresponding mAbs directed toward these proteins, and improvement of magnetic bead
chemistry will lead to better understanding of exosome biochemistry and may also allow
them to be exploited for clinical use (e.g., biomarker diagnostic), vaccines [309], and
delivery vehicles for therapeutic cargo [215].
Following is the manuscript: Comparison of ultracentrifugation, density gradient
separation, and immunoaffinity capture methods for isolating human colon cancer cell line
LIM1863-derived exosomes. Tauro, B. J., Greening, D. W., Mathias, R. A., Ji, H.,
Mathivanan, S., Scott, A. M., and Simpson, R. J. (2012) Methods 56, 293-304.
Supplemental Information located in Appendix A-6 and online at
doi:10.1016/j.ymeth.2012.01.002.

53
SEE CHAPTER 2 MANUSCRIPT - PAGE 1

54
SEE CHAPTER 2 MANUSCRIPT - PAGE 2

55
SEE CHAPTER 2 MANUSCRIPT - PAGE 3

56
SEE CHAPTER 2 MANUSCRIPT - PAGE 4

57
SEE CHAPTER 2 MANUSCRIPT - PAGE 5

58
SEE CHAPTER 2 MANUSCRIPT - PAGE 6

59
SEE CHAPTER 2 MANUSCRIPT - PAGE 7

60
SEE CHAPTER 2 MANUSCRIPT - PAGE 8

61
SEE CHAPTER 2 MANUSCRIPT - PAGE 9

62
SEE CHAPTER 2 MANUSCRIPT - PAGE 10

63
SEE CHAPTER 2 MANUSCRIPT - PAGE 11

64
SEE CHAPTER 2 MANUSCRIPT - PAGE 12

65
Chapter 3: Use of sequential immunoaffinity capture to
isolate and characterize two distinct populations of
exosomes released from LIM1863 colon carcinoma cells
3.1 Overview
To date, the content and function of exosomes released from highly-polarized epithelial
cells has received limited attention. In polarized cells, the apical and basolateral surfaces
are separated by adherens and tight junctions [310], and endosomes are used to recycle
apical and basolateral proteins to their respective plasma membrane surfaces in order to
maintain cell polarity. It has been reported that apical and basolateral exosomes, each
containing different cargos, may traffic to and be released from apical and basolateral
plasma membranes, respectively [110]. Van Neil and colleagues have hypothesized that
basolateral exosomes and their cargo may be able to cross the basement membrane and
convey immune information to non-contiguous immune cells [72]. To test this hypothesis,
polarized T84 colorectal cancer cells were grown as a monolayer on microporous filters to
allow for apical and basolateral exosome collection. This group subsequently performed a
limited proteomic analysis of exosomes to identify 31 proteins. A key finding was the
differential protein expression of apical trafficking molecule syntaxin 3 in apical exosomes
and basolateral transmembrane protein A33 in basolateral exosomes [72].
To further investigate apical and basolateral trafficking of exosomes in the context of colon
cancer biology, I performed an in-depth proteomic analysis of these exosome sub-
populations from a polarized colon cancer cell line. Having established in Chapter 2 that
immunoaffinity capture (IAC) is the preferred method to enrich exosomes from cell culture
medium, I used this approach to isolate highly-purified apical and basolateral exosomes
using the human colon carcinoma cell line LIM1863 as an experimental model (see section
1.5.1.3). I designed a sequential immunoaffinity method utilizing magnetic beads coated

66
with mAbs directed towards A33 antigen, a definitive marker of basolateral surfaces in
intestinal epithelial cells [154-156], followed by anti-EpCAM loaded magnetic beads, a
ubiquitously expressed epithelial cancer marker [311]. This strategy allowed for the
isolation of two sub-populations of exosomes, A33-Exos and EpCAM-Exos. Proteomic-
profiling revealed the unique identification in EpCAM-Exos of classical apical trafficking
molecules, apical intestinal enzymes and the apically-restricted membrane proteins. By
comparison, A33-Exos were selectively enriched with basolateral trafficking molecules,
such as early endosome antigen 1 (EEA1), ADP-ribosylation factor (ARF1) and clathrin.
Intriguingly, several members of the MHC class I family of antigen presentation molecules
were exclusively observed in A33-Exos, while neither MHC class I or II molecules were
observed in EpCAM-Exos. Findings from this study also showed the co-localization of
EpCAM, claudin-7 and CD44 in EpCAM-Exos. When complexed together, but not alone,
these proteins promote tumor progression [312]. Further investigation is required to
ascertain whether EpCAM/claudin-7/CD44 do in fact complex together in this study, and
whether this complex confers tumor promoting activity in recipient cells.
The findings in this chapter also shed light on the membrane composition of A33- and
EpCAM-Exos. For example, a total of 119 transmembrane-containing proteins were
identified, of which 13 have been previously implicated in colon cancer (e.g., ADAM10
and tumor-associated calcium signal transducer 2). An interesting finding was the
expression of three cell surface proteins specifically localized on normal colon epithelium,
MUC13, TSPAN8 and A33, as indicated following examination of the Human Protein
Atlas (http://www.proteinatlas.org/). Although these three membrane proteins are
specifically expressed in normal colon epithelium, they are over-expressed in many other
cancer tissues. Ongoing studies are required to identify specific membrane proteins in colon
cancer cell-derived exosomes that will morphologically distinguish these exosomes in
blood and other body fluids from exosomes generated from other tissue/cell types. In
addition, two other integral membrane proteins (CD46 and CD59) identified in EpCAM-
Exos, but not A33-Exos, may have the potential to increase the half-life of these exosomes
by protecting them from complement mediated lysis. CD59 prevents activation of the
membrane attack complex by inhibition of C9 incorporation in C5b-9 [313], whereas CD46

67
has cofactor activity for inactivation of complement components C3b and C46 by serum
factor 1, which protects cells from damage by the complement system [314]. Further,
EpCAM-Exos also uniquely expressed high-mobility group box proteins HMGB2 and
HMGB3 which act as sensors to distinct types of nucleic acid and activate innate immune
responses [315]. Studies aimed at determining the functional significance of apical and
basolateral exosomes in colon cancer biology were limited due to time constraints.
In order to eliminate the possibility that A33- and EpCAM-Exos were actually shed
microvesicles (sMVs) (see section 1.3.1), I isolated sMVs and examined their protein
content. This analysis identified 1392 proteins, of which 426 were unique to sMVs. This
showed that A33- and EpCAM-Exos were clearly distinguishable from sMVs. A key
finding was the enrichment of ATP binding cassette transporter proteins, which suggests
that sMVs may be implicated in multidrug resistance [316, 317]. Ongoing studies are
underway in the Simpson laboratory to establish whether LIM1863-derived sMVs are
capable of transferring resistance to drug sensitive colon cancer cells.
Following is the manuscript: Tauro, B. J., Greening, D. W., Mathias, R. A., Mathivanan,
S., Ji, H., and Simpson, R. J. (2013) Two distinct populations of exosomes are released
from LIM1863 colon carcinoma cell-derived organoids. Molecular & Cellular Proteomics
12, 587-598. Supplemental Information located in Appendix A-6 and online at
http://www.mcponline.org/content/suppl/2013/07/11/M112.021303.DC1.

68

69
SEE CHAPTER 3 MANUSCRIPT - PAGE 1

70
SEE CHAPTER 3 MANUSCRIPT - PAGE 2

71
SEE CHAPTER 3 MANUSCRIPT - PAGE 3

72
SEE CHAPTER 3 MANUSCRIPT - PAGE 4

73
SEE CHAPTER 3 MANUSCRIPT - PAGE 5

74
SEE CHAPTER 3 MANUSCRIPT - PAGE 6

75
SEE CHAPTER 3 MANUSCRIPT - PAGE 7

76
SEE CHAPTER 3 MANUSCRIPT - PAGE 8

77
SEE CHAPTER 3 MANUSCRIPT - PAGE 9

78
SEE CHAPTER 3 MANUSCRIPT - PAGE 10

79
SEE CHAPTER 3 MANUSCRIPT - PAGE 11

80
SEE CHAPTER 3 MANUSCRIPT - PAGE 12

81
Chapter 4: Characterization of Madin-Darby canine
kidney (MDCK) cell-derived exosomal proteins following
oncogenic H-Ras induced epithelial-mesenchymal
transition
4.1 Overview
Epithelial-mesenchymal transition (EMT) occurs during embryonic morphogenesis and has
recently been implicated in cancer invasion and metastasis [277, 318-320]. In previous
studies, Mathias and colleagues in the Simpson laboratory examined the components of the
secretome in EMT using oncogenic H-Ras transformed Madin-Darby canine kidney
(MDCK) cells as a model (see sections 1.5.2.4 and 1.5.2.5). These studies, which focused
on the proteomic profiles of soluble-secreted proteins, revealed that following EMT,
secreted components mediating cell-cell and cell-matrix adhesion (e.g., collagen IV, XVII,
and laminin 5) were decreased in abundance, with the concordant upregulation of proteases
and ECM constituents promoting cell motility and invasion (e.g., MMP-1, TIMP-1
kallikrein-6, -7) [250, 300].
In this chapter I extended the secretome studies of Mathias and colleagues [250, 300] by
examining the contribution of exosomes in oncogenic H-Ras-induced EMT in MDCK cells.
As this was the first study of the role of exosomes in EMT using the MDCK model, and
given that there are no known specific MDCK-derived exosomal membrane marker
proteins, immunoaffinity capture isolation was not a viable purification option. Instead I
employed density gradient centrifugation using iodixanol (OptiPrep™) described in
Chapter 2. Proteomic profiling of MDCK-derived exosomes (MDCK-Exos) and oncogenic
H-Ras transformed MDCK-derived exosomes (21D1-Exos) was performed using GeLC-
MS/MS as described in Chapter 2 and 3.
Findings from this study showed that purified MDCK- and 21D1-Exos were
morphologically similar by cryo-electron microscopy and contained stereotypical exosome

82
marker proteins such as TSG101, Alix and CD63. Typical cellular EMT hallmark protein
changes are also mirrored in MDCK and 21D1-Exos, including decreased expression of E-
Cadherin and increased expression of vimentin. This study also revealed that 21D1-Exos
are enriched with several metalloproteinases (e.g., MMP-1, -14, -19, ADAM10,
ADAMTS1), and integrins (e.g., ITGB1, ITGA3, and ITGA6) that have been recently
implicated in regulating the tumor microenvironment to promote metastatic progression
[321-323]. A significant finding of this study was the observation that upon EMT,
mesenchymal 21D1-Exos are extensively reprogrammed to be enriched with key
transcriptional regulators (e.g., master transcriptional regulator YBX1) and core splicing
complex components (e.g., SF3B1, SF3B3, and SFRS1). These findings suggest that Ras-
transformed MDCK cell-derived exosomes may be reprogrammed with factors capable of
inducing EMT in recipient cells. In this regard, preliminary experiments showed that 21D1-
Exos were capable of maintaining the mesenchymal morphology of 21D1 cells. Further
experiments are required to reveal whether 21D1-Exos are capable of inducing EMT in
other recipient cell types, and whether over-expression of YBX1 alone in epithelial cells, or
in conjunction with soluble secreted proteins, is capable of inducing tumorigenesis in
‘normal’ epithelial cells.
Following is the manuscript: Tauro, B. J., Mathias, R. A., Greening, D. W., Gopal, S. K.,
Ji, H, Kapp, E. A., Coleman, B. M., Hill, A. F., Kusebauch, U., Hallows, J. L., Shteynberg,
D., Moritz, R. L., Zhu, H. J., and Simpson R. J. (2013) Oncogenic H-Ras reprograms
Madin-Darby canine kidney (MDCK) cell-derived exosomal proteins during epithelial-
mesenchymal transition. Molecular & Cellular Proteomics 12, 2148-2159. Supplemental
Information located in Appendix A-6 and online at
http://www.mcponline.org/content/early/2013/05/03/mcp.M112.027086/suppl/DC1.

83
SEE CHAPTER 4 MANUSCRIPT - PAGE 1

84
SEE CHAPTER 4 MANUSCRIPT - PAGE 2

85
SEE CHAPTER 4 MANUSCRIPT - PAGE 3

86
SEE CHAPTER 4 MANUSCRIPT - PAGE 4

87
SEE CHAPTER 4 MANUSCRIPT - PAGE 5

88
SEE CHAPTER 4 MANUSCRIPT - PAGE 6

89
SEE CHAPTER 4 MANUSCRIPT - PAGE 7

90
SEE CHAPTER 4 MANUSCRIPT - PAGE 8

91
SEE CHAPTER 4 MANUSCRIPT - PAGE 9

92
SEE CHAPTER 4 MANUSCRIPT - PAGE 10

93
SEE CHAPTER 4 MANUSCRIPT - PAGE 11

94
SEE CHAPTER 4 MANUSCRIPT - PAGE 12

95
Chapter 5: Summary and future directions
5.1 Evaluation of exosome isolation methods and the importance of purity
As microvesicles, 100-1000 nm in diameter, and exosomes, 40-100 nm in diameter, are
morphologically distinct and may have different biological functions, there is a pressing
need to evaluate methods for obtaining homogeneous vesicle preparations in order to
characterise their bioactive content (e.g., proteins, lipids and RNA species). This is
particularly important when considering exosomes for use as clinical agents in therapeutics
(see section 1.4.9). A clear understanding of all exosome components may also lead to the
design of exosome mimetics for personalized medicine [229]. Recently, as circulating
exosomes have been identified to contain disease-specific miRNA signatures, there is also
evidence for the use of exosomes as diagnostic biomarkers [241]. Exosomes also display
the capabilities to be therapeutic agents, these include immune modulation, tissue repair,
and drug delivery due to their biocompatibility, immunological inertness, and ability to
cross the blood brain barrier [209, 210]. Before the full potential of exosomes use in drug
delivery can be realized, large-scale production protocols must be standardized [209].
Currently, detailed biochemical and functional analyses of exosomes are confounded by
technical difficulties in their isolation from cell culture medium and body fluids. Without
stringent purification, exosome samples are typically contaminated with other membranous
vesicles including shed plasma membrane vesicles (e.g., microparticles and oncosomes)
and apoptotic blebs. As these other secreted vesicles share similar physiochemical
characteristics (size, buoyant density) to exosomes, Chapter 2 compared the effectiveness
of three widely-used isolation strategies (ultracentrifugation, density gradient separation
and immunoaffinity capture) to obtain exosomes from colorectal cancer cells. Further, an
in-depth proteomic analysis was performed to establish the purity of secreted exosomes.
The findings from this chapter showed that immunoaffinity capture (IAC) using anti-

96
EpCAM antibody was the optimum method for purifying exosomes. In this study the
assessment of exosome sample homogeneity was based upon selective enrichment of
proteins involved in exosome biogenesis, trafficking, and release. To further this research,
investigation into the isolation of exosomes from other cell-derived sources and biofluids
(e.g., blood, urine, and malignant ascites) needs to be performed to validate the
compatability of the IAC approach.
Further, it is noted that the potential for exosomes to act as a blood-based biomarker
diagnostic source (e.g., using specific disease miRNA signatures) is confounded by the
challenge in isolating exosomes that are characteristic of specific disease cell types, such as
cancer. Such cancer-derived exosomes may be in low abundance when compared to the
general population of exosomes and other microvesicles in circulation. Additionally,
because these disease specific miRNA signatures may be masked by analyzing low
abundance disease-derived exosomes in blood, there is a need to isolate highly pure
exosomes (e.g., using IAC) for diagnostic purposes. In future studies it is therefore essential
to identify tissue-specific membrane protein markers for IAC isolation of disease-related
exosomes.
It remains to be seen whether a large-scale IAC approach will emerge as an alternative to
the filtration-based/size-exclusion HPLC approach currently used for preparing clinical
grade exosomes for regenerative medical purposes [45, 221]. Needless to say, such
evaluation of exosome purification methods is required, as pre-clinical and clinical trials
(http://www.australianclinicaltrials.gov.au/) require large-scale cost-effective standardized
protocols for the production of GMP exosomes.
5.2 Analysis of two exosome sub-populations released from LIM1863 cells
In Chapter 3, a sequential immunoaffinity capture strategy using magnetic beads coupled
with antibodies towards A33 and EpCAM was developed to isolate exosome sub-
populations released from apical and basolateral surfaces of polarized human colon

97
carcinoma cells. An in-depth proteomic characterization of exosome sub-populations
revealed distinct apical and basolateral trafficking proteins, consistent with release of
exosomes from the respective surfaces of polarized epithelial cells. Proteomic analysis
revealed 684 proteins were common to both exosomal datasets, while 340 and 214 proteins
were unique to A33- and EpCAM-Exos, respectively. Interestingly, the key components of
a metastasis-promoting complex, EpCAM/claudin-7/CD44, were uniquely identified in
apically-derived exosomes, while MHC-I components were unique to basolaterally-derived
exosomes.
The finding of selectively enriched proteins in apical and basolateral exosomes raises
questions into the functional roles of these two distinct exosome populations, especially in
the context of tumor biology. To examine their biological function, studies investigating the
abilities of the metastasis-promoting complex, EpCAM/claudin-7/CD44, may be
performed. Although all three components were uniquely identified in EpCAM-Exos, it
could not be claimed that they form a functional complex within exosomes. Further studies
are required to determine whether exosomes contain active signaling complexes involving
oncogenic proteins and, if so, whether such complexes may remain intact upon exosome
uptake in recipient cells and contribute to functional signaling pathways in target cells.
Ongoing efforts using Blue-Native PAGE and co-immunoprecipitation are underway in the
Simpson laboratory to assess the presence of this protein complex and functional validation
of its constituents. Following identification of this complex, experiments involving its
modification (e.g., the knock-down of one or more of the components from the originating
cell line) could be performed. To assess the ability of the complex to act as a metastasis
promoter, experiments involving the application of exosomes, with or without the intact
complex, to recipient cells may be performed. Functional readouts assessing cell
proliferation, migration, invasion or resistance to apoptosis may indicate the hypothesized
metastatic effect of exosomes on recipient cells.
Interestingly, the presence of MHC-I components were only observed in A33-Exos. Further
experiments are required to establish whether these components also form a functional
complex in exosomes. It would be exciting to determine whether the MHC complexes carry

98
peptides, or are capable of carrying peptides, that can elicit a stimulatory or inhibitory
immune cell response.
To further differentiate apical and basolateral exosomes and understand their biological
functions, characterization of their RNA content (miRNA and mRNA) may be investigated.
The first report of exosome-mediated transfer of exogenous nucleic acids was published in
2010, when it was shown that monocytic miR-150 was functionally delivered to endothelial
cells [237]. Since then, numerous other studies demonstrating the RNA transporting
capabilities of exosomes have been reported [171, 173, 324, 325]. Although at an early
stage, these findings collectively illustrate the potential of exosomes for therapeutic
delivery of nucleic acids – for reviews see El-Andaloussi et al., Kooijmans et al., and Rak
et al. [196, 210, 229].
In addition to the characterization of apical and basolateral-derived exosomes, shed
microvesicles (sMVs) were also isolated using differential centrifugation (10,000 g) to
differentiate these vesicles from exosomes. A proteomic characterization revealed 1392
proteins in sMVs with 462 proteins unique to these vesicles. This characterization revealed
another distinct vesicle population and therefore, the potential of different functional
capabilities to that of exosomes. One intriguing finding was the enrichment of ATP binding
cassette transporter proteins, which suggests that sMVs may be involved in multidrug
resistance [316, 317]. Functional studies are planned to establish whether LIM1863-derived
sMVs are capable of transferring resistance to drug sensitive colon cancer cells. Further
investigation into the isolation of sMVs is also being performed in the Simpson laboratory
to identify if sup-populations of sMVs exist within their large size range of 100-1000 nm
diameter.
5.3 Analysis of exosomes following epithelial-mesenchymal transition (EMT)
In Chapter 4 of this thesis, I investigated whether exosomes are modified during the EMT
process. I studied the EMT process using the epithelial MDCK cell line and its oncogenic

99
H-Ras-transformed mesenchymal variant (21D1) as an experimental model. Exosomes
released into the culture media from both cell lines were isolated by density gradient
centrifugation (as described in Chapter 2) and their protein content characterized by MS-
based profiling, ultimately to gain insight into any functional roles of exosomes in EMT.
Proteomic data revealed profound changes in 21D1 cell-derived exosomes. Foremost, these
studies revealed that cellular hallmarks typically identified in the epithelial-mesenchymal
transition (e.g., downregulation of E-cadherin and upregulation of vimentin in
mesenchymal cells) were mirrored in exosomes. These studies also showed that oncogenic
H-Ras reprograms MDCK cell-derived exosomal proteins. For example, mesenchymal
exosomes were enriched with several proteases (e.g., MMP-1, -14, -19, ADAM10 and
ADAMTS1) and integrins (e.g., ITGB1, ITGA3 and ITGA6) which are implicated in
promoting metastasis. A key finding in mesenchymal exosomes was the unique expression
of numerous splicing factors (e.g., SF3B1, SF3B3, and SFRS1) and master transcriptional
regulator, YBX1. Whether these proteins are functional in recipient cells remains to be
investigated.
It is interesting to speculate that exosomes from 21D1 cells may induce a mesenchymal
phenotype in recipient cells. If so, it would be important to identify the specific cargo (e.g.,
proteins, transcription factors or RNAs) that can cause such a transformation. To assess
this, initial experiments involving the addition of exosomes to recipient cells are planned to
be performed to assess changes in proliferation, invasion and migration abilities. Following
an observed morphological change, identification of the specific factors contributing to
these effects could be undertaken. For example, using an RNAi based system to create
stable knock-down cell lines; the expression of selected target genes that govern key
cellular processes may be altered in 21D1 cells. For these studies, target gene selection will
be based on proteomic data identified in 21D1-Exos. For example, highly upregulated
protein and inducer of EMT (e.g., transcription factor YBX1), or characteristic proteins of
the mesenchymal phenotype (e.g., MMP-1) will be selected first for generating knock-
down lines from 21D1 cells. Following gene knock-down, exosomes will be isolated and
characterized to assess that the corresponding protein expression is also reduced. Exosomes
isolated from knock-down and wild-type cell lines will then be applied to recipient cells to

100
identify the contribution of these modified constituents (i.e., YBX1 and MMP-1) in
exosome functionality and recipient cell modulation.
Additionally, the characterization of exosomal miRNA may also allow functional insights
into epithelial and mesenchymal cell-derived exosomes. Rana and colleagues have recently
demonstrated that miRNA contained within exosomes from metastatic rat adenocarcinoma
cells can alter mRNA transcription of proteases, adhesion molecules, chemokine ligands,
cell cycle- and angiogenesis-promoting genes, and genes engaged in oxidative stress
responses [173]. It is hypothesized that similarly to the proteomic analysis of exosomes,
miRNA content may also be dysregulated following EMT and therefore cause different
mRNA transcription in target cells which may result in altered biological functions. To
further support this theory, it has been demonstrated that exosomes can also deliver
functional miRNA to dysregulate mRNA in dendritic cells [171, 324]. Additionally it is
reported that mRNA and miRNA within melanoma exosomes may be actively transported
into normal melanocytes enabling increased invasive properties of these cells [325]. Based
on these recent observations, it would be exciting to examine the RNA species that are
contained within exosomes derived from MDCK to 21D1 cells. This may allow the
identification of bioactive molecules (RNAs) that have functional capabilities associated
with the EMT process.
5.4 Conclusion
Exosomes are important extracellular conveyors of information between cells. This may
occur through the transmission of bioactive molecules such as oncogenic proteins, tumor
suppressors, transcription factors, bioactive lipids and genetic information such as
mRNAs/miRNAs that alter the phenotypic and biological function of target cells. One of
the key obstacles in ascribing any biochemical or functional characteristics to exosomes is
that of sample homogeneity. To date, the majority of functional exosome studies reported
in the literature have been performed on impure samples that are significantly contaminated
with other EV types such as microvesicles and possibly apoptotic bodies.

101
In this thesis I carefully evaluated three commonly-used exosome purification protocols
(differential ultracentrifugation, density gradient centrifugation and immunoaffinity
capture) to establish that immunoaffinity capture was the superior method for the
enrichment of exosomes. With this knowledge, I then developed a sequential
immunoaffinity capture method to isolate and characterize, for the first time, two distinct
populations of exosomes from LIM1863 organoids. In a further application, I used density
gradient centrifugation (OptiPrep™) to investigate, for the first time, exosomes in
oncogenic H-Ras MDCK induced EMT. This study showed that exosomes were
significantly reprogrammed during the EMT process, being selectively enriched with the
master transcriptional regulator, transcription factor YBX1, and components of protein
splicing machinery.
In summary, the findings of this thesis are the first step towards standardizing protocols for
the purification of exosomes. Already, the applications of these purification strategies have
paved the way for a better understanding of the roles of exosomes in colon cancer biology
and their contribution to the oncogenic H-Ras induced EMT process.

102
References
1. Raposo, G. and W. Stoorvogel. (2013) Extracellular vesicles: Exosomes,
microvesicles, and friends. J Cell Biol. 200(4). 373-383
2. Thery, C. (2011) Exosomes: secreted vesicles and intercellular communications.
F1000 Biol Rep. 3. 15
3. Kahlert, C. and R. Kalluri. (2013) Exosomes in tumor microenvironment influence
cancer progression and metastasis. J Mol Med (Berl). 91(4). 431-437
4. Pan, B.T., K. Teng, C. Wu, M. Adam, and R.M. Johnstone. (1985) Electron
microscopic evidence for externalization of the transferrin receptor in vesicular
form in sheep reticulocytes. J Cell Biol. 101(3). 942-948
5. Pan, B.T. and R.M. Johnstone. (1983) Fate of the transferrin receptor during
maturation of sheep reticulocytes in vitro: selective externalization of the receptor.
Cell. 33(3). 967-978
6. Johnstone, R.M., M. Adam, J.R. Hammond, L. Orr, and C. Turbide. (1987) Vesicle
formation during reticulocyte maturation. Association of plasma membrane
activities with released vesicles (exosomes). J Biol Chem. 262(19). 9412-9420
7. Gyorgy, B., T.G. Szabo, M. Pasztoi, Z. Pal, P. Misjak, B. Aradi, V. Laszlo, E.
Pallinger, E. Pap, A. Kittel, G. Nagy, A. Falus, and E.I. Buzas. (2011) Membrane
vesicles, current state-of-the-art: emerging role of extracellular vesicles. Cell Mol
Life Sci. 68(16). 2667-2688
8. Vogel, C. and E.M. Marcotte. (2012) Label-free protein quantitation using weighted
spectral counting. Methods Mol Biol. 893. 321-341
9. Paltridge, J.L., L. Belle, and Y. Khew-Goodall. (2013) The secretome in cancer
progression. Biochim Biophys Acta. Epub ahead of print
10. Greening, D.W., E.A. Kapp, H. Ji, T.P. Speed, and R.J. Simpson. (2013) Colon
tumour secretopeptidome: Insights into endogenous proteolytic cleavage events in
the colon tumour microenvironment. Biochim Biophys Acta. Epub ahead of print
11. Greening, D. and R. Simpson. (2013) An updated secretome. Biochim Biophys Acta,
12. Makridakis, M. and A. Vlahou. (2010) Secretome proteomics for discovery of
cancer biomarkers. J Proteomics. 73(12). 2291-2305

103
13. Pavlou, M.P. and E.P. Diamandis. (2010) The cancer cell secretome: a good source
for discovering biomarkers? J Proteomics. 73(10). 1896-1906
14. Karagiannis, G.S., M.P. Pavlou, and E.P. Diamandis. (2010) Cancer secretomics
reveal pathophysiological pathways in cancer molecular oncology. Mol Oncol. 4(6).
496-510
15. Ji, H., R.J. Goode, F. Vaillant, S. Mathivanan, E.A. Kapp, R.A. Mathias, G.J.
Lindeman, J.E. Visvader, and R.J. Simpson. (2011) Proteomic profiling of
secretome and adherent plasma membranes from distinct mammary epithelial cell
subpopulations. Proteomics. 11(20). 4029-4039
16. Dumortier, J., D.N. Streblow, A.V. Moses, J.M. Jacobs, C.N. Kreklywich, D.
Camp, R.D. Smith, S.L. Orloff, and J.A. Nelson. (2008) Human cytomegalovirus
secretome contains factors that induce angiogenesis and wound healing. J Virol.
82(13). 6524-6535
17. Formolo, C.A., R. Williams, H. Gordish-Dressman, T.J. MacDonald, N.H. Lee, and
Y. Hathout. (2011) Secretome signature of invasive glioblastoma multiforme. J
Proteome Res. 10(7). 3149-3159
18. Hathout, Y. (2007) Approaches to the study of the cell secretome. Expert Rev
Proteomics. 4(2). 239-248
19. Peinado, H., S. Lavotshkin, and D. Lyden. (2011) The secreted factors responsible
for pre-metastatic niche formation: old sayings and new thoughts. Semin Cancer
Biol. 21(2). 139-146
20. Gadea, B.B. and J.A. Joyce. (2006) Tumour-host interactions: implications for
developing anti-cancer therapies. Expert Rev Mol Med. 8(30). 1-32
21. Joyce, J.A. (2005) Therapeutic targeting of the tumor microenvironment. Cancer
Cell. 7(6). 513-520
22. Al-Nedawi, K., B. Meehan, and J. Rak. (2009) Microvesicles: messengers and
mediators of tumor progression. Cell Cycle. 8(13). 2014-2018
23. Wolf, P. (1967) The nature and significance of platelet products in human plasma.
Br J Haematol. 13(3). 269-288

104
24. Heijnen, H.F., A.E. Schiel, R. Fijnheer, H.J. Geuze, and J.J. Sixma. (1999)
Activated platelets release two types of membrane vesicles: microvesicles by
surface shedding and exosomes derived from exocytosis of multivesicular bodies
and alpha-granules. Blood. 94(11). 3791-3799
25. Lee, T.H., E. D'Asti, N. Magnus, K. Al-Nedawi, B. Meehan, and J. Rak. (2011)
Microvesicles as mediators of intercellular communication in cancer--the emerging
science of cellular 'debris'. Semin Immunopathol. 33(5). 455-467
26. Cocucci, E., G. Racchetti, and J. Meldolesi. (2009) Shedding microvesicles:
artefacts no more. Trends Cell Biol. 19(2). 43-51
27. Huotari, J. and A. Helenius. (2011) Endosome maturation. Embo J. 30(17). 3481-
3500
28. Beekman, J.M. and P.J. Coffer. (2008) The ins and outs of syntenin, a
multifunctional intracellular adaptor protein. J Cell Sci. 121(Pt 9). 1349-1355
29. Camussi, G., M.C. Deregibus, S. Bruno, V. Cantaluppi, and L. Biancone. (2010)
Exosomes/microvesicles as a mechanism of cell-to-cell communication. Kidney Int.
78(9). 838-848
30. Breakefield, X.O., R.M. Frederickson, and R.J. Simpson. (2011) Gesicles:
Microvesicle "Cookies" for Transient Information Transfer Between Cells. Mol
Ther. 19(9). 1574-1576
31. Gasser, O., C. Hess, S. Miot, C. Deon, J.C. Sanchez, and J.A. Schifferli. (2003)
Characterisation and properties of ectosomes released by human polymorphonuclear
neutrophils. Exp Cell Res. 285(2). 243-257
32. Thery, C., M. Ostrowski, and E. Segura. (2009) Membrane vesicles as conveyors of
immune responses. Nat Rev Immunol. 9(8). 581-593
33. Beyer, C. and D.S. Pisetsky. (2010) The role of microparticles in the pathogenesis
of rheumatic diseases. Nat Rev Rheumatol. 6(1). 21-29
34. Gregory, C.D. and A. Devitt. (2004) The macrophage and the apoptotic cell: an
innate immune interaction viewed simplistically? Immunology. 113(1). 1-14
35. Thery, C., S. Amigorena, G. Raposo, and A. Clayton. (2006) Isolation and
characterization of exosomes from cell culture supernatants and biological fluids.
Curr Protoc Cell Biol. Chapter 3. Unit 3 22

105
36. Akers, J.C., D. Gonda, R. Kim, B.S. Carter, and C.C. Chen. (2013) Biogenesis of
extracellular vesicles (EV): exosomes, microvesicles, retrovirus-like vesicles, and
apoptotic bodies. J Neurooncol. 113(1). 1-11
37. Koga, K., K. Matsumoto, T. Akiyoshi, M. Kubo, N. Yamanaka, A. Tasaki, H.
Nakashima, M. Nakamura, S. Kuroki, M. Tanaka, and M. Katano. (2005)
Purification, characterization and biological significance of tumor-derived
exosomes. Anticancer Res. 25(6A). 3703-3707
38. Logozzi, M., A. De Milito, L. Lugini, M. Borghi, L. Calabro, M. Spada, M.
Perdicchio, M.L. Marino, C. Federici, E. Iessi, D. Brambilla, G. Venturi, F.
Lozupone, M. Santinami, V. Huber, M. Maio, L. Rivoltini, and S. Fais. (2009) High
levels of exosomes expressing CD63 and caveolin-1 in plasma of melanoma
patients. PLoS One. 4(4). e5219
39. Coren, L.V., T. Shatzer, and D.E. Ott. (2008) CD45 immunoaffinity depletion of
vesicles from Jurkat T cells demonstrates that exosomes contain CD45: no evidence
for a distinct exosome/HIV-1 budding pathway. Retrovirology. 5. 64
40. Clayton, A., J. Court, H. Navabi, M. Adams, M.D. Mason, J.A. Hobot, G.R.
Newman, and B. Jasani. (2001) Analysis of antigen presenting cell derived
exosomes, based on immuno-magnetic isolation and flow cytometry. J Immunol
Methods. 247(1-2). 163-174
41. Caby, M.P., D. Lankar, C. Vincendeau-Scherrer, G. Raposo, and C. Bonnerot.
(2005) Exosomal-like vesicles are present in human blood plasma. Int Immunol.
17(7). 879-887
42. Rabinowits, G., C. Gercel-Taylor, J.M. Day, D.D. Taylor, and G.H. Kloecker.
(2009) Exosomal microRNA: a diagnostic marker for lung cancer. Clin Lung
Cancer. 10(1). 42-46
43. Taylor, D.D. and C. Gercel-Taylor. (2008) MicroRNA signatures of tumor-derived
exosomes as diagnostic biomarkers of ovarian cancer. Gynecol Oncol. 110(1). 13-
21

106
44. Mathivanan, S., J.W. Lim, B.J. Tauro, H. Ji, R.L. Moritz, and R.J. Simpson. (2010)
Proteomics analysis of A33 immunoaffinity-purified exosomes released from the
human colon tumor cell line LIM1215 reveals a tissue-specific protein signature.
Mol Cell Proteomics. 9(2). 197-208
45. Lai, R.C., F. Arslan, S.S. Tan, B. Tan, A. Choo, M.M. Lee, T.S. Chen, B.J. Teh,
J.K. Eng, H. Sidik, V. Tanavde, W.S. Hwang, C.N. Lee, R.M. El Oakley, G.
Pasterkamp, D.P. de Kleijn, K.H. Tan, and S.K. Lim. (2010) Derivation and
characterization of human fetal MSCs: an alternative cell source for large-scale
production of cardioprotective microparticles. J Mol Cell Cardiol. 48(6). 1215-1224
46. Lai, R.C., F. Arslan, M.M. Lee, N.S. Sze, A. Choo, T.S. Chen, M. Salto-Tellez, L.
Timmers, C.N. Lee, R.M. El Oakley, G. Pasterkamp, D.P. de Kleijn, and S.K. Lim.
(2010) Exosome secreted by MSC reduces myocardial ischemia/reperfusion injury.
Stem Cell Res. 4(3). 214-222
47. Ogawa, Y., Y. Miura, A. Harazono, M. Kanai-Azuma, Y. Akimoto, H. Kawakami,
T. Yamaguchi, T. Toda, T. Endo, M. Tsubuki, and R. Yanoshita. (2011) Proteomic
analysis of two types of exosomes in human whole saliva. Biol Pharm Bull. 34(1).
13-23
48. Choi, D.S., J.M. Lee, G.W. Park, H.W. Lim, J.Y. Bang, Y.K. Kim, K.H. Kwon, H.J.
Kwon, K.P. Kim, and Y.S. Gho. (2007) Proteomic analysis of microvesicles derived
from human colorectal cancer cells. J Proteome Res. 6(12). 4646-4655
49. Di Vizio, D., M. Morello, A.C. Dudley, P.W. Schow, R.M. Adam, S. Morley, D.
Mulholland, M. Rotinen, M.H. Hager, L. Insabato, M.A. Moses, F. Demichelis,
M.P. Lisanti, H. Wu, M. Klagsbrun, N.A. Bhowmick, M.A. Rubin, C. D'Souza-
Schorey, and M.R. Freeman. (2012) Large oncosomes in human prostate cancer
tissues and in the circulation of mice with metastatic disease. Am J Pathol. 181(5).
1573-1584
50. Thery, C., M. Boussac, P. Veron, P. Ricciardi-Castagnoli, G. Raposo, J. Garin, and
S. Amigorena. (2001) Proteomic analysis of dendritic cell-derived exosomes: a
secreted subcellular compartment distinct from apoptotic vesicles. J Immunol.
166(12). 7309-7318

107
51. Zhou, Z., X. Sun, and Y.J. Kang. (2001) Ethanol-induced apoptosis in mouse liver:
Fas- and cytochrome c-mediated caspase-3 activation pathway. Am J Pathol.
159(1). 329-338
52. Kalra, H., R.J. Simpson, H. Ji, E. Aikawa, P. Altevogt, P. Askenase, V.C. Bond,
F.E. Borras, X. Breakefield, V. Budnik, E. Buzas, G. Camussi, A. Clayton, E.
Cocucci, J.M. Falcon-Perez, S. Gabrielsson, Y.S. Gho, D. Gupta, H.C. Harsha, A.
Hendrix, A.F. Hill, J.M. Inal, G. Jenster, E.M. Kramer-Albers, S.K. Lim, A.
Llorente, J. Lotvall, A. Marcilla, L. Mincheva-Nilsson, I. Nazarenko, R. Nieuwland,
E.N. Nolte-'t Hoen, A. Pandey, T. Patel, M.G. Piper, S. Pluchino, T.S. Prasad, L.
Rajendran, G. Raposo, M. Record, G.E. Reid, F. Sanchez-Madrid, R.M. Schiffelers,
P. Siljander, A. Stensballe, W. Stoorvogel, D. Taylor, C. Thery, H. Valadi, B.W.
van Balkom, J. Vazquez, M. Vidal, M.H. Wauben, M. Yanez-Mo, M. Zoeller, and
S. Mathivanan. (2012) Vesiclepedia: a compendium for extracellular vesicles with
continuous community annotation. PLoS Biol. 10(12). e1001450
53. Gonzalez, L.J., E. Gibbons, R.W. Bailey, J. Fairbourn, T. Nguyen, S.K. Smith, K.B.
Best, J. Nelson, A.M. Judd, and J.D. Bell. (2009) The influence of membrane
physical properties on microvesicle release in human erythrocytes. PMC Biophys.
2(1). 7
54. Pluskota, E., N.M. Woody, D. Szpak, C.M. Ballantyne, D.A. Soloviev, D.I. Simon,
and E.F. Plow. (2008) Expression, activation, and function of integrin alphaMbeta2
(Mac-1) on neutrophil-derived microparticles. Blood. 112(6). 2327-2335
55. Muralidharan-Chari, V., J.W. Clancy, A. Sedgwick, and C. D'Souza-Schorey.
(2010) Microvesicles: mediators of extracellular communication during cancer
progression. J Cell Sci. 123(Pt 10). 1603-1611
56. Goler-Baron, V. and Y.G. Assaraf. (2011) Structure and function of ABCG2-rich
extracellular vesicles mediating multidrug resistance. PLoS One. 6(1). e16007
57. Han, B. and J.T. Zhang. (2004) Multidrug resistance in cancer chemotherapy and
xenobiotic protection mediated by the half ATP-binding cassette transporter
ABCG2. Curr Med Chem Anticancer Agents. 4(1). 31-42
58. Ejendal, K.F. and C.A. Hrycyna. (2002) Multidrug resistance and cancer: the role of
the human ABC transporter ABCG2. Curr Protein Pept Sci. 3(5). 503-511

108
59. Robey, R.W., O. Polgar, J. Deeken, K.W. To, and S.E. Bates. (2007) ABCG2:
determining its relevance in clinical drug resistance. Cancer Metastasis Rev. 26(1).
39-57
60. Mealey, K.L. (2012) ABCG2 transporter: therapeutic and physiologic implications
in veterinary species. J Vet Pharmacol Ther. 35(2). 105-112
61. Sidhu, S.S., A.T. Mengistab, A.N. Tauscher, J. LaVail, and C. Basbaum. (2004)
The microvesicle as a vehicle for EMMPRIN in tumor-stromal interactions.
Oncogene. 23(4). 956-963
62. Di Vizio, D., J. Kim, M.H. Hager, M. Morello, W. Yang, C.J. Lafargue, L.D. True,
M.A. Rubin, R.M. Adam, R. Beroukhim, F. Demichelis, and M.R. Freeman. (2009)
Oncosome formation in prostate cancer: association with a region of frequent
chromosomal deletion in metastatic disease. Cancer Res. 69(13). 5601-5609
63. Rood, I.M., J.K. Deegens, M.L. Merchant, W.P. Tamboer, D.W. Wilkey, J.F.
Wetzels, and J.B. Klein. (2010) Comparison of three methods for isolation of
urinary microvesicles to identify biomarkers of nephrotic syndrome. Kidney Int.
78(8). 810-816
64. Elmore, S. (2007) Apoptosis: a review of programmed cell death. Toxicol Pathol.
35(4). 495-516
65. Bratton, D.L., V.A. Fadok, D.A. Richter, J.M. Kailey, S.C. Frasch, T. Nakamura,
and P.M. Henson. (1999) Polyamine regulation of plasma membrane phospholipid
flip-flop during apoptosis. J Biol Chem. 274(40). 28113-28120
66. Albert, M.L., S.F. Pearce, L.M. Francisco, B. Sauter, P. Roy, R.L. Silverstein, and
N. Bhardwaj. (1998) Immature dendritic cells phagocytose apoptotic cells via
alphavbeta5 and CD36, and cross-present antigens to cytotoxic T lymphocytes. J
Exp Med. 188(7). 1359-1368
67. Albert, M.L., B. Sauter, and N. Bhardwaj. (1998) Dendritic cells acquire antigen
from apoptotic cells and induce class I-restricted CTLs. Nature. 392(6671). 86-89
68. Bergsmedh, A., A. Szeles, M. Henriksson, A. Bratt, M.J. Folkman, A.L. Spetz, and
L. Holmgren. (2001) Horizontal transfer of oncogenes by uptake of apoptotic
bodies. Proc Natl Acad Sci U S A. 98(11). 6407-6411

109
69. McLellan, A.D. (2009) Exosome release by primary B cells. Crit Rev Immunol.
29(3). 203-217
70. Buschow, S.I., E.N. Nolte-'t Hoen, G. van Niel, M.S. Pols, T. ten Broeke, M.
Lauwen, F. Ossendorp, C.J. Melief, G. Raposo, R. Wubbolts, M.H. Wauben, and
W. Stoorvogel. (2009) MHC II in dendritic cells is targeted to lysosomes or T cell-
induced exosomes via distinct multivesicular body pathways. Traffic. 10(10). 1528-
1542
71. Ghidoni, R., A. Paterlini, V. Albertini, M. Glionna, E. Monti, L. Schiaffonati, L.
Benussi, E. Levy, and G. Binetti. (2009) Cystatin C is released in association with
exosomes: A new tool of neuronal communication which is unbalanced in
Alzheimer's disease. Neurobiol Aging,
72. van Niel, G., G. Raposo, C. Candalh, M. Boussac, R. Hershberg, N. Cerf-
Bensussan, and M. Heyman. (2001) Intestinal epithelial cells secrete exosome-like
vesicles. Gastroenterology. 121(2). 337-349
73. Wolfers, J., A. Lozier, G. Raposo, A. Regnault, C. Thery, C. Masurier, C. Flament,
S. Pouzieux, F. Faure, T. Tursz, E. Angevin, S. Amigorena, and L. Zitvogel. (2001)
Tumor-derived exosomes are a source of shared tumor rejection antigens for CTL
cross-priming. Nat Med. 7(3). 297-303
74. Simpson, R.J., J.W. Lim, R.L. Moritz, and S. Mathivanan. (2009) Exosomes:
proteomic insights and diagnostic potential. Expert Rev Proteomics. 6(3). 267-283
75. Bobrie, A., M. Colombo, G. Raposo, and C. Thery. (2011) Exosome secretion:
molecular mechanisms and roles in immune responses. Traffic. 12(12). 1659-1668
76. van Niel, G., I. Porto-Carreiro, S. Simoes, and G. Raposo. (2006) Exosomes: a
common pathway for a specialized function. J Biochem. 140(1). 13-21
77. Simons, M. and G. Raposo. (2009) Exosomes--vesicular carriers for intercellular
communication. Curr Opin Cell Biol. 21(4). 575-581
78. Christoforidis, S., M. Miaczynska, K. Ashman, M. Wilm, L. Zhao, S.C. Yip, M.D.
Waterfield, J.M. Backer, and M. Zerial. (1999) Phosphatidylinositol-3-OH kinases
are Rab5 effectors. Nat Cell Biol. 1(4). 249-252
79. Nielsen, E., F. Severin, J.M. Backer, A.A. Hyman, and M. Zerial. (1999) Rab5
regulates motility of early endosomes on microtubules. Nat Cell Biol. 1(6). 376-382

110
80. Mayor, S. and R.E. Pagano. (2007) Pathways of clathrin-independent endocytosis.
Nat Rev Mol Cell Biol. 8(8). 603-612
81. Maxfield, F.R. and D.J. Yamashiro. (1987) Endosome acidification and the
pathways of receptor-mediated endocytosis. Adv Exp Med Biol. 225. 189-198
82. Poteryaev, D., S. Datta, K. Ackema, M. Zerial, and A. Spang. (2010) Identification
of the switch in early-to-late endosome transition. Cell. 141(3). 497-508
83. Hurley, J.H. and G. Odorizzi. (2012) Get on the exosome bus with ALIX. Nat Cell
Biol. 14(7). 654-655
84. Raiborg, C. and H. Stenmark. (2009) The ESCRT machinery in endosomal sorting
of ubiquitylated membrane proteins. Nature. 458(7237). 445-452
85. Babst, M. (2005) A protein's final ESCRT. Traffic. 6(1). 2-9
86. Raiborg, C., T.E. Rusten, and H. Stenmark. (2003) Protein sorting into
multivesicular endosomes. Curr Opin Cell Biol. 15(4). 446-455
87. Katzmann, D.J., G. Odorizzi, and S.D. Emr. (2002) Receptor downregulation and
multivesicular-body sorting. Nat Rev Mol Cell Biol. 3(12). 893-905
88. Lobert, V.H. and H. Stenmark. (2011) Cell polarity and migration: emerging role
for the endosomal sorting machinery. Physiology (Bethesda). 26(3). 171-180
89. Hurley, J.H. (2010) The ESCRT complexes. Crit Rev Biochem Mol Biol. 45(6).
463-487
90. Amerik, A.Y., J. Nowak, S. Swaminathan, and M. Hochstrasser. (2000) The Doa4
deubiquitinating enzyme is functionally linked to the vacuolar protein-sorting and
endocytic pathways. Mol Biol Cell. 11(10). 3365-3380
91. Saksena, S., J. Wahlman, D. Teis, A.E. Johnson, and S.D. Emr. (2009) Functional
reconstitution of ESCRT-III assembly and disassembly. Cell. 136(1). 97-109
92. Doyotte, A., M.R. Russell, C.R. Hopkins, and P.G. Woodman. (2005) Depletion of
TSG101 forms a mammalian "Class E" compartment: a multicisternal early
endosome with multiple sorting defects. J Cell Sci. 118(Pt 14). 3003-3017
93. Theos, A.C., S.T. Truschel, D. Tenza, I. Hurbain, D.C. Harper, J.F. Berson, P.C.
Thomas, G. Raposo, and M.S. Marks. (2006) A lumenal domain-dependent
pathway for sorting to intralumenal vesicles of multivesicular endosomes involved
in organelle morphogenesis. Dev Cell. 10(3). 343-354

111
94. van Niel, G., S. Charrin, S. Simoes, M. Romao, L. Rochin, P. Saftig, M.S. Marks,
E. Rubinstein, and G. Raposo. (2011) The tetraspanin CD63 regulates ESCRT-
independent and -dependent endosomal sorting during melanogenesis. Dev Cell.
21(4). 708-721
95. Baietti, M.F., Z. Zhang, E. Mortier, A. Melchior, G. Degeest, A. Geeraerts, Y.
Ivarsson, F. Depoortere, C. Coomans, E. Vermeiren, P. Zimmermann, and G.
David. (2012) Syndecan-syntenin-ALIX regulates the biogenesis of exosomes. Nat
Cell Biol. 14(7). 677-685
96. Trajkovic, K., C. Hsu, S. Chiantia, L. Rajendran, D. Wenzel, F. Wieland, P.
Schwille, B. Brugger, and M. Simons. (2008) Ceramide triggers budding of
exosome vesicles into multivesicular endosomes. Science. 319(5867). 1244-1247
97. Putz, U., J. Howitt, A. Doan, C.P. Goh, L.H. Low, J. Silke, and S.S. Tan. (2012)
The tumor suppressor PTEN is exported in exosomes and has phosphatase activity
in recipient cells. Sci Signal. 5(243). ra70
98. Loubery, S., C. Wilhelm, I. Hurbain, S. Neveu, D. Louvard, and E. Coudrier. (2008)
Different microtubule motors move early and late endocytic compartments. Traffic.
9(4). 492-509
99. Jordens, I., M. Fernandez-Borja, M. Marsman, S. Dusseljee, L. Janssen, J. Calafat,
H. Janssen, R. Wubbolts, and J. Neefjes. (2001) The Rab7 effector protein RILP
controls lysosomal transport by inducing the recruitment of dynein-dynactin motors.
Curr Biol. 11(21). 1680-1685
100. Luzio, J.P., P.R. Pryor, and N.A. Bright. (2007) Lysosomes: fusion and function.
Nat Rev Mol Cell Biol. 8(8). 622-632
101. Hsu, C., Y. Morohashi, S. Yoshimura, N. Manrique-Hoyos, S. Jung, M.A.
Lauterbach, M. Bakhti, M. Gronborg, W. Mobius, J. Rhee, F.A. Barr, and M.
Simons. (2010) Regulation of exosome secretion by Rab35 and its GTPase-
activating proteins TBC1D10A-C. J Cell Biol. 189(2). 223-232
102. Savina, A., M. Vidal, and M.I. Colombo. (2002) The exosome pathway in K562
cells is regulated by Rab11. J Cell Sci. 115(Pt 12). 2505-2515

112
103. Ostrowski, M., N.B. Carmo, S. Krumeich, I. Fanget, G. Raposo, A. Savina, C.F.
Moita, K. Schauer, A.N. Hume, R.P. Freitas, B. Goud, P. Benaroch, N. Hacohen,
M. Fukuda, C. Desnos, M.C. Seabra, F. Darchen, S. Amigorena, L.F. Moita, and C.
Thery. (2010) Rab27a and Rab27b control different steps of the exosome secretion
pathway. Nat Cell Biol. 12(1). 19-30; sup pp 11-13
104. Hong, W. (2005) SNAREs and traffic. Biochim Biophys Acta. 1744(3). 493-517
105. Gross, J.C., V. Chaudhary, K. Bartscherer, and M. Boutros. (2012) Active Wnt
proteins are secreted on exosomes. Nat Cell Biol. 14(10). 1036-1045
106. Hasan, N. and C. Hu. (2010) Vesicle-associated membrane protein 2 mediates
trafficking of alpha5beta1 integrin to the plasma membrane. Exp Cell Res. 316(1).
12-23
107. Jahn, R. and R.H. Scheller. (2006) SNAREs--engines for membrane fusion. Nat Rev
Mol Cell Biol. 7(9). 631-643
108. Fader, C.M., D.G. Sanchez, M.B. Mestre, and M.I. Colombo. (2009) TI-
VAMP/VAMP7 and VAMP3/cellubrevin: two v-SNARE proteins involved in
specific steps of the autophagy/multivesicular body pathways. Biochim Biophys
Acta. 1793(12). 1901-1916
109. Rodriguez-Boulan, E., G. Kreitzer, and A. Musch. (2005) Organization of vesicular
trafficking in epithelia. Nat Rev Mol Cell Biol. 6(3). 233-247
110. Lakkaraju, A. and E. Rodriguez-Boulan. (2008) Itinerant exosomes: emerging roles
in cell and tissue polarity. Trends Cell Biol. 18(5). 199-209
111. Parton, R.G., K. Prydz, M. Bomsel, K. Simons, and G. Griffiths. (1989) Meeting of
the apical and basolateral endocytic pathways of the Madin-Darby canine kidney
cell in late endosomes. J Cell Biol. 109(6 Pt 2). 3259-3272
112. Ng, Y.H., S. Rome, A. Jalabert, A. Forterre, H. Singh, C.L. Hincks, and L.A.
Salamonsen. (2013) Endometrial exosomes/microvesicles in the uterine
microenvironment: a new paradigm for embryo-endometrial cross talk at
implantation. PLoS One. 8(3). e58502
113. Coleman, B.M., E. Hanssen, V.A. Lawson, and A.F. Hill. (2012) Prion-infected
cells regulate the release of exosomes with distinct ultrastructural features. Faseb J.
26(10). 4160-4173

113
114. Ji, H., D.W. Greening, T.W. Barnes, J.W. Lim, B.J. Tauro, A. Rai, R. Xu, C. Adda,
S. Mathivanan, W. Zhao, Y. Xue, T. Xu, H.J. Zhu, and R.J. Simpson. (2013)
Proteome profiling of exosomes derived from human primary and metastatic
colorectal cells reveal differential expression of key metastatic factors and signal
transduction components. Proteomics. 10-11. 1672-1686
115. Binnig, G., C.F. Quate, and C. Gerber. (1986) Atomic force microscope. Phys Rev
Lett. 56(9). 930-933
116. van der Pol, E., A.G. Hoekstra, A. Sturk, C. Otto, T.G. van Leeuwen, and R.
Nieuwland. (2010) Optical and non-optical methods for detection and
characterization of microparticles and exosomes. J Thromb Haemost. 8(12). 2596-
2607
117. D'Souza-Schorey, C. and J.W. Clancy. (2012) Tumor-derived microvesicles:
shedding light on novel microenvironment modulators and prospective cancer
biomarkers. Genes Dev. 26(12). 1287-1299
118. Lawrie, A.S., A. Albanyan, R.A. Cardigan, I.J. Mackie, and P. Harrison. (2009)
Microparticle sizing by dynamic light scattering in fresh-frozen plasma. Vox Sang.
96(3). 206-212
119. Hood, J.L., R.S. San, and S.A. Wickline. (2011) Exosomes released by melanoma
cells prepare sentinel lymph nodes for tumor metastasis. Cancer Res. 71(11). 3792-
3801
120. Filipe, V., A. Hawe, and W. Jiskoot. (2010) Critical evaluation of Nanoparticle
Tracking Analysis (NTA) by NanoSight for the measurement of nanoparticles and
protein aggregates. Pharm Res. 27(5). 796-810
121. Nolte-'t Hoen, E.N., E.J. van der Vlist, M. Aalberts, H.C. Mertens, B.J. Bosch, W.
Bartelink, E. Mastrobattista, E.V. van Gaal, W. Stoorvogel, G.J. Arkesteijn, and
M.H. Wauben. (2012) Quantitative and qualitative flow cytometric analysis of
nanosized cell-derived membrane vesicles. Nanomedicine. 8(5). 712-720
122. Mathivanan, S., C.J. Fahner, G.E. Reid, and R.J. Simpson. (2012) ExoCarta 2012:
database of exosomal proteins, RNA and lipids. Nucleic Acids Res. 40(1). D1241-
1244

114
123. Mathivanan, S. and R.J. Simpson. (2009) ExoCarta: A compendium of exosomal
proteins and RNA. Proteomics. 9(21). 4997-5000
124. Anderson, N.L. and N.G. Anderson. (1998) Proteome and proteomics: new
technologies, new concepts, and new words. Electrophoresis. 19(11). 1853-1861
125. Graner, M.W., O. Alzate, A.M. Dechkovskaia, J.D. Keene, J.H. Sampson, D.A.
Mitchell, and D.D. Bigner. (2008) Proteomic and immunologic analyses of brain
tumor exosomes. Faseb J. 5. 1541-1557
126. Raimondo, F., L. Morosi, C. Chinello, F. Magni, and M. Pitto. (2011) Advances in
membranous vesicle and exosome proteomics improving biological understanding
and biomarker discovery. Proteomics. 11(4). 709-720
127. Ducret, A., I. Van Oostveen, J.K. Eng, J.R. Yates, 3rd, and R. Aebersold. (1998)
High throughput protein characterization by automated reverse-phase
chromatography/electrospray tandem mass spectrometry. Protein Sci. 7(3). 706-719
128. Qin, J., D. Fenyo, Y. Zhao, W.W. Hall, D.M. Chao, C.J. Wilson, R.A. Young, and
B.T. Chait. (1997) A strategy for rapid, high-confidence protein identification. Anal
Chem. 69(19). 3995-4001
129. Perkins, D.N., D.J. Pappin, D.M. Creasy, and J.S. Cottrell. (1999) Probability-based
protein identification by searching sequence databases using mass spectrometry
data. Electrophoresis. 20(18). 3551-3567
130. Deutsch, E.W., L. Mendoza, D. Shteynberg, T. Farrah, H. Lam, N. Tasman, Z. Sun,
E. Nilsson, B. Pratt, B. Prazen, J.K. Eng, D.B. Martin, A.I. Nesvizhskii, and R.
Aebersold. (2010) A guided tour of the Trans-Proteomic Pipeline. Proteomics.
10(6). 1150-1159
131. Ong, S.E., B. Blagoev, I. Kratchmarova, D.B. Kristensen, H. Steen, A. Pandey, and
M. Mann. (2002) Stable isotope labeling by amino acids in cell culture, SILAC, as a
simple and accurate approach to expression proteomics. Mol Cell Proteomics. 1(5).
376-386
132. Chong, P.K., C.S. Gan, T.K. Pham, and P.C. Wright. (2006) Isobaric tags for
relative and absolute quantitation (iTRAQ) reproducibility: Implication of multiple
injections. J Proteome Res. 5(5). 1232-1240

115
133. Domon, B. and R. Aebersold. (2006) Mass spectrometry and protein analysis.
Science. 312(5771). 212-217
134. Beissbarth, T., L. Hyde, G.K. Smyth, C. Job, W.M. Boon, S.S. Tan, H.S. Scott, and
T.P. Speed. (2004) Statistical modeling of sequencing errors in SAGE libraries.
Bioinformatics. 20 Suppl 1. i31-39
135. Holman, S.W., P.F. Sims, and C.E. Eyers. (2012) The use of selected reaction
monitoring in quantitative proteomics. Bioanalysis. 4(14). 1763-1786
136. Brownridge, P., S.W. Holman, S.J. Gaskell, C.M. Grant, V.M. Harman, S.J.
Hubbard, K. Lanthaler, C. Lawless, R. O'Cualain, P. Sims, R. Watkins, and R.J.
Beynon. (2011) Global absolute quantification of a proteome: Challenges in the
deployment of a QconCAT strategy. Proteomics. 11(15). 2957-2970
137. Schmidt, C. and H. Urlaub. (2012) Absolute quantification of proteins using
standard peptides and multiple reaction monitoring. Methods Mol Biol. 893. 249-
265
138. Valadi, H., K. Ekstrom, A. Bossios, M. Sjostrand, J.J. Lee, and J.O. Lotvall. (2007)
Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of
genetic exchange between cells. Nat Cell Biol. 9(6). 654-659
139. Wubbolts, R., R.S. Leckie, P.T. Veenhuizen, G. Schwarzmann, W. Mobius, J.
Hoernschemeyer, J.W. Slot, H.J. Geuze, and W. Stoorvogel. (2003) Proteomic and
biochemical analyses of human B cell-derived exosomes. Potential implications for
their function and multivesicular body formation. J Biol Chem. 278(13). 10963-
10972
140. Blanchard, N., D. Lankar, F. Faure, A. Regnault, C. Dumont, G. Raposo, and C.
Hivroz. (2002) TCR activation of human T cells induces the production of
exosomes bearing the TCR/CD3/zeta complex. J Immunol. 168(7). 3235-3241
141. Segura, E., C. Nicco, B. Lombard, P. Veron, G. Raposo, F. Batteux, S. Amigorena,
and C. Thery. (2005) ICAM-1 on exosomes from mature dendritic cells is critical
for efficient naive T-cell priming. Blood. 106(1). 216-223

116
142. Mears, R., R.A. Craven, S. Hanrahan, N. Totty, C. Upton, S.L. Young, P. Patel, P.J.
Selby, and R.E. Banks. (2004) Proteomic analysis of melanoma-derived exosomes
by two-dimensional polyacrylamide gel electrophoresis and mass spectrometry.
Proteomics. 4(12). 4019-4031
143. Hegmans, J.P., M.P. Bard, A. Hemmes, T.M. Luider, M.J. Kleijmeer, J.B. Prins, L.
Zitvogel, S.A. Burgers, H.C. Hoogsteden, and B.N. Lambrecht. (2004) Proteomic
analysis of exosomes secreted by human mesothelioma cells. Am J Pathol. 164(5).
1807-1815
144. Epple, L.M., S.G. Griffiths, A.M. Dechkovskaia, N.L. Dusto, J. White, R.J.
Ouellette, T.J. Anchordoquy, L.T. Bemis, and M.W. Graner. (2012)
Medulloblastoma exosome proteomics yield functional roles for extracellular
vesicles. PLoS One. 7(7). e42064
145. Welton, J.L., S. Khanna, P.J. Giles, P. Brennan, I.A. Brewis, J. Staffurth, M.D.
Mason, and A. Clayton. (2010) Proteomics analysis of bladder cancer exosomes.
Mol Cell Proteomics. 9(6). 1324-1338
146. Admyre, C., S.M. Johansson, K.R. Qazi, J.J. Filen, R. Lahesmaa, M. Norman, E.P.
Neve, A. Scheynius, and S. Gabrielsson. (2007) Exosomes with immune
modulatory features are present in human breast milk. J Immunol. 179(3). 1969-
1978
147. Bard, M.P., J.P. Hegmans, A. Hemmes, T.M. Luider, R. Willemsen, L.A.
Severijnen, J.P. van Meerbeeck, S.A. Burgers, H.C. Hoogsteden, and B.N.
Lambrecht. (2004) Proteomic analysis of exosomes isolated from human malignant
pleural effusions. Am J Respir Cell Mol Biol. 31(1). 114-121
148. Poliakov, A., M. Spilman, T. Dokland, C.L. Amling, and J.A. Mobley. (2009)
Structural heterogeneity and protein composition of exosome-like vesicles
(prostasomes) in human semen. Prostate. 69(2). 159-167
149. Raimondo, F., S. Corbetta, L. Morosi, C. Chinello, E. Gianazza, G. Castoldi, C. Di
Gioia, C. Bombardi, A. Stella, C. Battaglia, C. Bianchi, F. Magni, and M. Pitto.
(2013) Urinary exosomes and diabetic nephropathy: a proteomic approach. Mol
Biosyst. 9(6). 1139-1146

117
150. Hurley, J.H. (2008) ESCRT complexes and the biogenesis of multivesicular bodies.
Curr Opin Cell Biol. 20(1). 4-11
151. Fehon, R.G., A.I. McClatchey, and A. Bretscher. (2010) Organizing the cell cortex:
the role of ERM proteins. Nat Rev Mol Cell Biol. 11(4). 276-287
152. Savina, A., C.M. Fader, M.T. Damiani, and M.I. Colombo. (2005) Rab11 promotes
docking and fusion of multivesicular bodies in a calcium-dependent manner.
Traffic. 6(2). 131-143
153. Subra, C., K. Laulagnier, B. Perret, and M. Record. (2007) Exosome lipidomics
unravels lipid sorting at the level of multivesicular bodies. Biochimie. 89(2). 205-
212
154. Heath, J.K., S.J. White, C.N. Johnstone, B. Catimel, R.J. Simpson, R.L. Moritz,
G.F. Tu, H. Ji, R.H. Whitehead, L.C. Groenen, A.M. Scott, G. Ritter, L. Cohen, S.
Welt, L.J. Old, E.C. Nice, and A.W. Burgess. (1997) The human A33 antigen is a
transmembrane glycoprotein and a novel member of the immunoglobulin
superfamily. Proc Natl Acad Sci U S A. 94(2). 469-474
155. Ji, H., R.L. Moritz, G.E. Reid, G. Ritter, B. Catimel, E. Nice, J.K. Heath, S.J.
White, S. Welt, L.J. Old, A.W. Burgess, and R.J. Simpson. (1997) Electrophoretic
analysis of the novel antigen for the gastrointestinal-specific monoclonal antibody,
A33. Electrophoresis. 18(3-4). 614-621
156. Ritter, G., L.S. Cohen, E.C. Nice, B. Catimel, A.W. Burgess, R.L. Moritz, H. Ji,
J.K. Heath, S.J. White, S. Welt, L.J. Old, and R.J. Simpson. (1997) Characterization
of posttranslational modifications of human A33 antigen, a novel palmitoylated
surface glycoprotein of human gastrointestinal epithelium. Biochem Biophys Res
Commun. 236(3). 682-686
157. Record, M., C. Subra, S. Silvente-Poirot, and M. Poirot. (2011) Exosomes as
intercellular signalosomes and pharmacological effectors. Biochem Pharmacol.
81(10). 1171-1182
158. Matsuo, H., J. Chevallier, N. Mayran, I. Le Blanc, C. Ferguson, J. Faure, N.S.
Blanc, S. Matile, J. Dubochet, R. Sadoul, R.G. Parton, F. Vilbois, and J. Gruenberg.
(2004) Role of LBPA and Alix in multivesicular liposome formation and endosome
organization. Science. 303(5657). 531-534

118
159. Subra, C., D. Grand, K. Laulagnier, A. Stella, G. Lambeau, M. Paillasse, P. De
Medina, B. Monsarrat, B. Perret, S. Silvente-Poirot, M. Poirot, and M. Record.
(2010) Exosomes account for vesicle-mediated transcellular transport of activatable
phospholipases and prostaglandins. J Lipid Res. 51(8). 2105-2120
160. Laulagnier, K., C. Motta, S. Hamdi, S. Roy, F. Fauvelle, J.F. Pageaux, T.
Kobayashi, J.P. Salles, B. Perret, C. Bonnerot, and M. Record. (2004) Mast cell-
and dendritic cell-derived exosomes display a specific lipid composition and an
unusual membrane organization. Biochem J. 380(Pt 1). 161-171
161. Fhaner, C.J., S. Liu, H. Ji, R.J. Simpson, and G.E. Reid. (2012) Comprehensive
lipidome profiling of isogenic primary and metastatic colon adenocarcinoma cell
lines. Anal Chem. 84(21). 8917-8926
162. Calin, G.A. and C.M. Croce. (2006) MicroRNA signatures in human cancers. Nat
Rev Cancer. 6(11). 857-866
163. Baj-Krzyworzeka, M., R. Szatanek, K. Weglarczyk, J. Baran, B. Urbanowicz, P.
Branski, M.Z. Ratajczak, and M. Zembala. (2006) Tumour-derived microvesicles
carry several surface determinants and mRNA of tumour cells and transfer some of
these determinants to monocytes. Cancer Immunol Immunother. 55(7). 808-818
164. Ratajczak, J., K. Miekus, M. Kucia, J. Zhang, R. Reca, P. Dvorak, and M.Z.
Ratajczak. (2006) Embryonic stem cell-derived microvesicles reprogram
hematopoietic progenitors: evidence for horizontal transfer of mRNA and protein
delivery. Leukemia. 20(5). 847-856
165. Skog, J., T. Wurdinger, S. van Rijn, D.H. Meijer, L. Gainche, M. Sena-Esteves,
W.T. Curry, Jr., B.S. Carter, A.M. Krichevsky, and X.O. Breakefield. (2008)
Glioblastoma microvesicles transport RNA and proteins that promote tumour
growth and provide diagnostic biomarkers. Nat Cell Biol. 10(12). 1470-1476
166. Bellingham, S.A., B.M. Coleman, and A.F. Hill. (2012) Small RNA deep
sequencing reveals a distinct miRNA signature released in exosomes from prion-
infected neuronal cells. Nucleic Acids Res. 40(21). 10937-10949
167. Zhou, Q., M. Li, X. Wang, Q. Li, T. Wang, Q. Zhu, X. Zhou, X. Gao, and X. Li.
(2012) Immune-related microRNAs are abundant in breast milk exosomes. Int J
Biol Sci. 8(1). 118-123

119
168. Hannafon, B.N. and W.Q. Ding. (2013) Intercellular Communication by Exosome-
Derived microRNAs in Cancer. Int J Mol Sci. 14(7). 14240-14269
169. Ohshima, K., K. Inoue, A. Fujiwara, K. Hatakeyama, K. Kanto, Y. Watanabe, K.
Muramatsu, Y. Fukuda, S. Ogura, K. Yamaguchi, and T. Mochizuki. (2010) Let-7
microRNA family is selectively secreted into the extracellular environment via
exosomes in a metastatic gastric cancer cell line. PLoS One. 5(10). e13247
170. Yang, M., J. Chen, F. Su, B. Yu, L. Lin, Y. Liu, J.D. Huang, and E. Song. (2011)
Microvesicles secreted by macrophages shuttle invasion-potentiating microRNAs
into breast cancer cells. Mol Cancer. 10. 117
171. Montecalvo, A., A.T. Larregina, W.J. Shufesky, D.B. Stolz, M.L. Sullivan, J.M.
Karlsson, C.J. Baty, G.A. Gibson, G. Erdos, Z. Wang, J. Milosevic, O.A. Tkacheva,
S.J. Divito, R. Jordan, J. Lyons-Weiler, S.C. Watkins, and A.E. Morelli. (2012)
Mechanism of transfer of functional microRNAs between mouse dendritic cells via
exosomes. Blood. 119(3). 756-766
172. Umezu, T., K. Ohyashiki, M. Kuroda, and J.H. Ohyashiki. (2013) Leukemia cell to
endothelial cell communication via exosomal miRNAs. Oncogene. 32(22). 2747-
2755
173. Rana, S., K. Malinowska, and M. Zoller. (2013) Exosomal tumor microRNA
modulates premetastatic organ cells. Neoplasia. 15(3). 281-295
174. Grange, C., M. Tapparo, F. Collino, L. Vitillo, C. Damasco, M.C. Deregibus, C.
Tetta, B. Bussolati, and G. Camussi. (2011) Microvesicles released from human
renal cancer stem cells stimulate angiogenesis and formation of lung premetastatic
niche. Cancer Res. 71(15). 5346-5356
175. Batagov, A.O., V.A. Kuznetsov, and I.V. Kurochkin. (2011) Identification of
nucleotide patterns enriched in secreted RNAs as putative cis-acting elements
targeting them to exosome nano-vesicles. BMC Genomics. 12 Suppl 3. S18
176. Bolukbasi, M.F., A. Mizrak, G.B. Ozdener, S. Madlener, T. Strobel, E.P. Erkan,
J.B. Fan, X.O. Breakefield, and O. Saydam. (2012) miR-1289 and "Zipcode"-like
Sequence Enrich mRNAs in Microvesicles. Mol Ther Nucleic Acids. 1. e10

120
177. Rana, S., S. Yue, D. Stadel, and M. Zoller. (2012) Toward tailored exosomes: the
exosomal tetraspanin web contributes to target cell selection. Int J Biochem Cell
Biol. 44(9). 1574-1584
178. Morelli, A.E., A.T. Larregina, W.J. Shufesky, M.L. Sullivan, D.B. Stolz, G.D.
Papworth, A.F. Zahorchak, A.J. Logar, Z. Wang, S.C. Watkins, L.D. Falo, Jr., and
A.W. Thomson. (2004) Endocytosis, intracellular sorting, and processing of
exosomes by dendritic cells. Blood. 104(10). 3257-3266
179. Segura, E., C. Guerin, N. Hogg, S. Amigorena, and C. Thery. (2007) CD8+
dendritic cells use LFA-1 to capture MHC-peptide complexes from exosomes in
vivo. J Immunol. 179(3). 1489-1496
180. Hanayama, R., M. Tanaka, K. Miwa, A. Shinohara, A. Iwamatsu, and S. Nagata.
(2002) Identification of a factor that links apoptotic cells to phagocytes. Nature.
417(6885). 182-187
181. Parolini, I., C. Federici, C. Raggi, L. Lugini, S. Palleschi, A. De Milito, C. Coscia,
E. Iessi, M. Logozzi, A. Molinari, M. Colone, M. Tatti, M. Sargiacomo, and S. Fais.
(2009) Microenvironmental pH is a key factor for exosome traffic in tumor cells. J
Biol Chem. 284(49). 34211-34222
182. Escrevente, C., S. Keller, P. Altevogt, and J. Costa. (2011) Interaction and uptake of
exosomes by ovarian cancer cells. BMC Cancer. 11. 108
183. Feng, D., W.L. Zhao, Y.Y. Ye, X.C. Bai, R.Q. Liu, L.F. Chang, Q. Zhou, and S.F.
Sui. (2010) Cellular internalization of exosomes occurs through phagocytosis.
Traffic. 11(5). 675-687
184. Fitzner, D., M. Schnaars, D. van Rossum, G. Krishnamoorthy, P. Dibaj, M. Bakhti,
T. Regen, U.K. Hanisch, and M. Simons. (2011) Selective transfer of exosomes
from oligodendrocytes to microglia by macropinocytosis. J Cell Sci. 124(Pt 3). 447-
458
185. Tian, T., Y. Wang, H. Wang, Z. Zhu, and Z. Xiao. (2010) Visualizing of the cellular
uptake and intracellular trafficking of exosomes by live-cell microscopy. J Cell
Biochem. 111(2). 488-496
186. Ewers, H. and A. Helenius. (2011) Lipid-mediated endocytosis. Cold Spring Harb
Perspect Biol. 3(8). a004721

121
187. Svensson, K.J., H.C. Christianson, A. Wittrup, E. Bourseau-Guilmain, E. Lindqvist,
L.M. Svensson, M. Morgelin, and M. Belting. (2013) Exosome uptake depends on
ERK1/2-heat shock protein 27 signalling and lipid raft-mediated endocytosis
negatively regulated by caveolin-1. J Biol Chem,
188. Kucerova, L. and S. Skolekova. (2013) Tumor microenvironment and the role of
mesenchymal stromal cells. Neoplasma. 60(1). 1-10
189. Hanahan, D. and R.A. Weinberg. (2011) Hallmarks of cancer: the next generation.
Cell. 144(5). 646-674
190. Ge, R., E. Tan, S. Sharghi-Namini, and H.H. Asada. (2012) Exosomes in Cancer
Microenvironment and Beyond: have we Overlooked these Extracellular
Messengers? Cancer Microenviron. 5(3). 323-332
191. Zhang, H.G. and W.E. Grizzle. (2011) Exosomes and cancer: a newly described
pathway of immune suppression. Clin Cancer Res. 17(5). 959-964
192. Hood, J.L., H. Pan, G.M. Lanza, and S.A. Wickline. (2009) Paracrine induction of
endothelium by tumor exosomes. Lab Invest. 89(11). 1317-1328
193. Webber, J., R. Steadman, M.D. Mason, Z. Tabi, and A. Clayton. (2010) Cancer
exosomes trigger fibroblast to myofibroblast differentiation. Cancer Res. 70(23).
9621-9630
194. Hao, S., Z. Ye, F. Li, Q. Meng, M. Qureshi, J. Yang, and J. Xiang. (2006)
Epigenetic transfer of metastatic activity by uptake of highly metastatic B16
melanoma cell-released exosomes. Exp Oncol. 28(2). 126-131
195. Peinado, H., M. Aleckovic, S. Lavotshkin, I. Matei, B. Costa-Silva, G. Moreno-
Bueno, M. Hergueta-Redondo, C. Williams, G. Garcia-Santos, C.M. Ghajar, A.
Nitadori-Hoshino, C. Hoffman, K. Badal, B.A. Garcia, M.K. Callahan, J. Yuan,
V.R. Martins, J. Skog, R.N. Kaplan, M.S. Brady, J.D. Wolchok, P.B. Chapman, Y.
Kang, J. Bromberg, and D. Lyden. (2012) Melanoma exosomes educate bone
marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat Med.
18(6). 883-891
196. Rak, J. and A. Guha. (2012) Extracellular vesicles--vehicles that spread cancer
genes. Bioessays. 34(6). 489-497

122
197. Demory Beckler, M., J.N. Higginbotham, J.L. Franklin, A.J. Ham, P.J. Halvey, I.E.
Imasuen, C. Whitwell, M. Li, D.C. Liebler, and R.J. Coffey. (2013) Proteomic
analysis of exosomes from mutant KRAS colon cancer cells identifies intercellular
transfer of mutant KRAS. Mol Cell Proteomics. 12(2). 343-355
198. Luga, V., L. Zhang, A.M. Viloria-Petit, A.A. Ogunjimi, M.R. Inanlou, E. Chiu, M.
Buchanan, A.N. Hosein, M. Basik, and J.L. Wrana. (2012) Exosomes mediate
stromal mobilization of autocrine Wnt-PCP signaling in breast cancer cell
migration. Cell. 151(7). 1542-1556
199. Al-Nedawi, K., B. Meehan, J. Micallef, V. Lhotak, L. May, A. Guha, and J. Rak.
(2008) Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles
derived from tumour cells. Nat Cell Biol. 10(5). 619-624
200. Al-Nedawi, K., B. Meehan, R.S. Kerbel, A.C. Allison, and J. Rak. (2009)
Endothelial expression of autocrine VEGF upon the uptake of tumor-derived
microvesicles containing oncogenic EGFR. Proc Natl Acad Sci U S A. 106(10).
3794-3799
201. Antonyak, M.A., B. Li, L.K. Boroughs, J.L. Johnson, J.E. Druso, K.L. Bryant, D.A.
Holowka, and R.A. Cerione. (2011) Cancer cell-derived microvesicles induce
transformation by transferring tissue transglutaminase and fibronectin to recipient
cells. Proc Natl Acad Sci U S A. 108(12). 4852-4857
202. Balaj, L., R. Lessard, L. Dai, Y.J. Cho, S.L. Pomeroy, X.O. Breakefield, and J.
Skog. (2011) Tumour microvesicles contain retrotransposon elements and amplified
oncogene sequences. Nat Commun. 2. 180
203. Meckes, D.G., Jr., K.H. Shair, A.R. Marquitz, C.P. Kung, R.H. Edwards, and N.
Raab-Traub. (2010) Human tumor virus utilizes exosomes for intercellular
communication. Proc Natl Acad Sci U S A. 107(47). 20370-20375
204. Janowska-Wieczorek, A., L.A. Marquez-Curtis, M. Wysoczynski, and M.Z.
Ratajczak. (2006) Enhancing effect of platelet-derived microvesicles on the invasive
potential of breast cancer cells. Transfusion. 46(7). 1199-1209

123
205. Janowska-Wieczorek, A., M. Wysoczynski, J. Kijowski, L. Marquez-Curtis, B.
Machalinski, J. Ratajczak, and M.Z. Ratajczak. (2005) Microvesicles derived from
activated platelets induce metastasis and angiogenesis in lung cancer. Int J Cancer.
113(5). 752-760
206. Fidler, I.J., S. Yano, R.D. Zhang, T. Fujimaki, and C.D. Bucana. (2002) The seed
and soil hypothesis: vascularisation and brain metastases. Lancet Oncol. 3(1). 53-57
207. Schluter, K., P. Gassmann, A. Enns, T. Korb, A. Hemping-Bovenkerk, J. Holzen,
and J. Haier. (2006) Organ-specific metastatic tumor cell adhesion and
extravasation of colon carcinoma cells with different metastatic potential. Am J
Pathol. 169(3). 1064-1073
208. Kaplan, R.N., S. Rafii, and D. Lyden. (2006) Preparing the "soil": the premetastatic
niche. Cancer Res. 66(23). 11089-11093
209. S, E.L.A., I. Mager, X.O. Breakefield, and M.J. Wood. (2013) Extracellular
vesicles: biology and emerging therapeutic opportunities. Nat Rev Drug Discov.
12(5). 347-357
210. El-Andaloussi, S., Y. Lee, S. Lakhal-Littleton, J. Li, Y. Seow, C. Gardiner, L.
Alvarez-Erviti, I.L. Sargent, and M.J. Wood. (2012) Exosome-mediated delivery of
siRNA in vitro and in vivo. Nat Protoc. 7(12). 2112-2126
211. Chalmin, F., S. Ladoire, G. Mignot, J. Vincent, M. Bruchard, J.P. Remy-Martin, W.
Boireau, A. Rouleau, B. Simon, D. Lanneau, A. De Thonel, G. Multhoff, A.
Hamman, F. Martin, B. Chauffert, E. Solary, L. Zitvogel, C. Garrido, B. Ryffel, C.
Borg, L. Apetoh, C. Rebe, and F. Ghiringhelli. (2010) Membrane-associated Hsp72
from tumor-derived exosomes mediates STAT3-dependent immunosuppressive
function of mouse and human myeloid-derived suppressor cells. J Clin Invest.
120(2). 457-471
212. Lima, L.G., R. Chammas, R.Q. Monteiro, M.E. Moreira, and M.A. Barcinski.
(2009) Tumor-derived microvesicles modulate the establishment of metastatic
melanoma in a phosphatidylserine-dependent manner. Cancer Lett. 283(2). 168-175
213. Hood, J.L. and S.A. Wickline. (2012) A systematic approach to exosome-based
translational nanomedicine. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 4(4).
458-467

124
214. Seow, Y. and M.J. Wood. (2009) Biological gene delivery vehicles: beyond viral
vectors. Mol Ther. 17(5). 767-777
215. O'Loughlin, A.J., C.A. Woffindale, and M.J. Wood. (2012) Exosomes and the
emerging field of exosome-based gene therapy. Curr Gene Ther. 12(4). 262-274
216. Alvarez-Erviti, L., Y. Seow, H. Yin, C. Betts, S. Lakhal, and M.J. Wood. (2011)
Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes.
Nat Biotechnol. 29(4). 341-345
217. Chaput, N., J. Taieb, N. Schartz, C. Flament, S. Novault, F. Andre, and L. Zitvogel.
(2005) The potential of exosomes in immunotherapy of cancer. Blood Cells Mol
Dis. 35(2). 111-115
218. Zitvogel, L., A. Regnault, A. Lozier, J. Wolfers, C. Flament, D. Tenza, P. Ricciardi-
Castagnoli, G. Raposo, and S. Amigorena. (1998) Eradication of established murine
tumors using a novel cell-free vaccine: dendritic cell-derived exosomes. Nat Med.
4(5). 594-600
219. Monleon, I., M.J. Martinez-Lorenzo, L. Monteagudo, P. Lasierra, M. Taules, M.
Iturralde, A. Pineiro, L. Larrad, M.A. Alava, J. Naval, and A. Anel. (2001)
Differential secretion of Fas ligand- or APO2 ligand/TNF-related apoptosis-
inducing ligand-carrying microvesicles during activation-induced death of human T
cells. J Immunol. 167(12). 6736-6744
220. Kim, S.H., E.R. Lechman, N. Bianco, R. Menon, A. Keravala, J. Nash, Z. Mi, S.C.
Watkins, A. Gambotto, and P.D. Robbins. (2005) Exosomes derived from IL-10-
treated dendritic cells can suppress inflammation and collagen-induced arthritis. J
Immunol. 174(10). 6440-6448
221. Lai, R.C., R.W. Yeo, K.H. Tan, and S.K. Lim. (2013) Exosomes for drug delivery -
a novel application for the mesenchymal stem cell. Biotechnol Adv. 31(5). 543-551
222. Wahlgren, J., L.K.T. De, M. Brisslert, F. Vaziri Sani, E. Telemo, P. Sunnerhagen,
and H. Valadi. (2012) Plasma exosomes can deliver exogenous short interfering
RNA to monocytes and lymphocytes. Nucleic Acids Res. 40(17). e130

125
223. Thery, C., A. Regnault, J. Garin, J. Wolfers, L. Zitvogel, P. Ricciardi-Castagnoli, G.
Raposo, and S. Amigorena. (1999) Molecular characterization of dendritic cell-
derived exosomes. Selective accumulation of the heat shock protein hsc73. J Cell
Biol. 147(3). 599-610
224. Dobrovolskaia, M.A., P. Aggarwal, J.B. Hall, and S.E. McNeil. (2008) Preclinical
studies to understand nanoparticle interaction with the immune system and its
potential effects on nanoparticle biodistribution. Mol Pharm. 5(4). 487-495
225. van den Boorn, J.G., M. Schlee, C. Coch, and G. Hartmann. (2011) SiRNA delivery
with exosome nanoparticles. Nat Biotechnol. 29(4). 325-326
226. Escudier, B., T. Dorval, N. Chaput, F. Andre, M.P. Caby, S. Novault, C. Flament,
C. Leboulaire, C. Borg, S. Amigorena, C. Boccaccio, C. Bonnerot, O. Dhellin, M.
Movassagh, S. Piperno, C. Robert, V. Serra, N. Valente, J.B. Le Pecq, A. Spatz, O.
Lantz, T. Tursz, E. Angevin, and L. Zitvogel. (2005) Vaccination of metastatic
melanoma patients with autologous dendritic cell (DC) derived-exosomes: results of
the first phase I clinical trial. J Transl Med. 3(1). 10
227. Chen, T.S., R.C. Lai, M.M. Lee, A.B. Choo, C.N. Lee, and S.K. Lim. (2010)
Mesenchymal stem cell secretes microparticles enriched in pre-microRNAs. Nucleic
Acids Res. 38(1). 215-224
228. Lai, R.C., T.S. Chen, and S.K. Lim. (2011) Mesenchymal stem cell exosome: a
novel stem cell-based therapy for cardiovascular disease. Regen Med. 6(4). 481-492
229. Kooijmans, S.A., P. Vader, S.M. van Dommelen, W.W. van Solinge, and R.M.
Schiffelers. (2012) Exosome mimetics: a novel class of drug delivery systems. Int J
Nanomedicine. 7. 1525-1541
230. Puri, A., K. Loomis, B. Smith, J.H. Lee, A. Yavlovich, E. Heldman, and R.
Blumenthal. (2009) Lipid-based nanoparticles as pharmaceutical drug carriers: from
concepts to clinic. Crit Rev Ther Drug Carrier Syst. 26(6). 523-580
231. Fenske, D.B. and P.R. Cullis. (2008) Liposomal nanomedicines. Expert Opin Drug
Deliv. 5(1). 25-44
232. De La Pena, H., J.A. Madrigal, S. Rusakiewicz, M. Bencsik, G.W. Cave, A.
Selman, R.C. Rees, P.J. Travers, and I.A. Dodi. (2009) Artificial exosomes as tools
for basic and clinical immunology. J Immunol Methods. 344(2). 121-132

126
233. Delcayre, A., A. Estelles, J. Sperinde, T. Roulon, P. Paz, B. Aguilar, J. Villanueva,
S. Khine, and J.B. Le Pecq. (2005) Exosome Display technology: applications to the
development of new diagnostics and therapeutics. Blood Cells Mol Dis. 35(2). 158-
168
234. Hemler, M.E. (2005) Tetraspanin functions and associated microdomains. Nat Rev
Mol Cell Biol. 6(10). 801-811
235. Varnier, A., F. Kermarrec, I. Blesneac, C. Moreau, L. Liguori, J.L. Lenormand, and
N. Picollet-D'hahan. (2010) A simple method for the reconstitution of membrane
proteins into giant unilamellar vesicles. J Membr Biol. 233(1-3). 85-92
236. Chen, T.S., F. Arslan, Y. Yin, S.S. Tan, R.C. Lai, A.B. Choo, J. Padmanabhan, C.N.
Lee, D.P. de Kleijn, and S.K. Lim. (2011) Enabling a robust scalable manufacturing
process for therapeutic exosomes through oncogenic immortalization of human
ESC-derived MSCs. J Transl Med. 9. 47
237. Zhang, Y., D. Liu, X. Chen, J. Li, L. Li, Z. Bian, F. Sun, J. Lu, Y. Yin, X. Cai, Q.
Sun, K. Wang, Y. Ba, Q. Wang, D. Wang, J. Yang, P. Liu, T. Xu, Q. Yan, J. Zhang,
K. Zen, and C.Y. Zhang. (2010) Secreted monocytic miR-150 enhances targeted
endothelial cell migration. Mol Cell. 39(1). 133-144
238. Maguire, C.A., L. Balaj, S. Sivaraman, M.H. Crommentuijn, M. Ericsson, L.
Mincheva-Nilsson, V. Baranov, D. Gianni, B.A. Tannous, M. Sena-Esteves, X.O.
Breakefield, and J. Skog. (2012) Microvesicle-associated AAV vector as a novel
gene delivery system. Mol Ther. 20(5). 960-971
239. Ohno, S., M. Takanashi, K. Sudo, S. Ueda, A. Ishikawa, N. Matsuyama, K. Fujita,
T. Mizutani, T. Ohgi, T. Ochiya, N. Gotoh, and M. Kuroda. (2013) Systemically
injected exosomes targeted to EGFR deliver antitumor microRNA to breast cancer
cells. Mol Ther. 21(1). 185-191
240. Noerholm, M., L. Balaj, T. Limperg, A. Salehi, L.D. Zhu, F.H. Hochberg, X.O.
Breakefield, B.S. Carter, and J. Skog. (2012) RNA expression patterns in serum
microvesicles from patients with glioblastoma multiforme and controls. BMC
Cancer. 12(22). 1-11

127
241. Mitchell, P.S., R.K. Parkin, E.M. Kroh, B.R. Fritz, S.K. Wyman, E.L. Pogosova-
Agadjanyan, A. Peterson, J. Noteboom, K.C. O'Briant, A. Allen, D.W. Lin, N.
Urban, C.W. Drescher, B.S. Knudsen, D.L. Stirewalt, R. Gentleman, R.L. Vessella,
P.S. Nelson, D.B. Martin, and M. Tewari. (2008) Circulating microRNAs as stable
blood-based markers for cancer detection. Proc Natl Acad Sci U S A. 105(30).
10513-10518
242. Raposo, G., H.W. Nijman, W. Stoorvogel, R. Liejendekker, C.V. Harding, C.J.
Melief, and H.J. Geuze. (1996) B lymphocytes secrete antigen-presenting vesicles. J
Exp Med. 183(3). 1161-1172
243. Cantin, R., J. Diou, D. Belanger, A.M. Tremblay, and C. Gilbert. (2008)
Discrimination between exosomes and HIV-1: purification of both vesicles from
cell-free supernatants. J Immunol Methods. 338(1-2). 21-30
244. Lamparski, H.G., A. Metha-Damani, J.Y. Yao, S. Patel, D.H. Hsu, C. Ruegg, and
J.B. Le Pecq. (2002) Production and characterization of clinical grade exosomes
derived from dendritic cells. J Immunol Methods. 270(2). 211-226
245. Nguyen, D.G., A. Booth, S.J. Gould, and J.E. Hildreth. (2003) Evidence that HIV
budding in primary macrophages occurs through the exosome release pathway. J
Biol Chem. 278(52). 52347-52354
246. Andersen, R., J. Hosaka, T.S. Halstensen, A. Stordahl, A. Tverdal, L. Aabakken,
and F. Laerum. (1996) The X-ray contrast medium iodixanol detects increased
colonic permeability equally well as 51Cr-labeled ethylenediaminetetraacetic acid
in experimental colitis of rats. Scand J Gastroenterol. 31(2). 140-146
247. Spencer, C.M. and K.L. Goa. (1996) Iodixanol. A review of its pharmacodynamic
and pharmacokinetic properties and diagnostic use as an x-ray contrast medium.
Drugs. 52(6). 899-927
248. Borrell, B. (2010) How accurate are cancer cell lines? Nature. 463(7283). 858
249. Anglard, P., E. Trahan, S. Liu, F. Latif, M.J. Merino, M.I. Lerman, B. Zbar, and
W.M. Linehan. (1992) Molecular and cellular characterization of human renal cell
carcinoma cell lines. Cancer Res. 52(2). 348-356

128
250. Mathias, R.A., B. Wang, H. Ji, E.A. Kapp, R.L. Moritz, H.J. Zhu, and R.J. Simpson.
(2009) Secretome-based proteomic profiling of Ras-transformed MDCK cells
reveals extracellular modulators of epithelial-mesenchymal transition. J Proteome
Res. 8(6). 2827-2837
251. Whitehead, R.H., J.K. Jones, A. Gabriel, and R.E. Lukies. (1987) A new colon
carcinoma cell line (LIM1863) that grows as organoids with spontaneous
differentiation into crypt-like structures in vitro. Cancer Res. 47(10). 2683-2689
252. Sancho, E., E. Batlle, and H. Clevers. (2003) Live and let die in the intestinal
epithelium. Curr Opin Cell Biol. 15(6). 763-770
253. Humphries, A. and N.A. Wright. (2008) Colonic crypt organization and
tumorigenesis. Nat Rev Cancer. 8(6). 415-424
254. Rizk, P. and N. Barker. (2012) Gut stem cells in tissue renewal and disease:
methods, markers, and myths. Wiley Interdiscip Rev Syst Biol Med. 4(5). 475-496
255. Ferlay, J., H.R. Shin, F. Bray, D. Forman, C. Mathers, and D.M. Parkin. (2010)
Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J
Cancer. 127(12). 2893-2917
256. McDermid, I. (2005) Cancer incidence projections Australia 2002 to 2011.
Australian Institute of Health and Welfare. 1-166
257. Gomez-Dominguez, E., M. Trapero-Marugan, A.J. del Pozo, J. Cantero, J.P.
Gisbert, and J. Mate. (2006) The colorectal carcinoma prognosis factors.
Significance of diagnosis delay. Rev Esp Enferm Dig. 98(5). 322-329
258. Cserni, G. (2003) Nodal staging of colorectal carcinomas and sentinel nodes. J Clin
Pathol. 56(5). 327-335
259. Steele, G.D., Jr. (1994) The National Cancer Data Base report on colorectal cancer.
Cancer. 74(7). 1979-1989
260. Whitlock, E.P., J. Lin, E. Liles, T. Beil, R. Fu, E. O'Connor, R.N. Thompson, and T.
Cardenas, in Screening for Colorectal Cancer: An Updated Systematic Review2008:
Rockville (MD).
261. Hamilton, S.R. (1992) The adenoma-adenocarcinoma sequence in the large bowel:
variations on a theme. J Cell Biochem Suppl. 16G. 41-46

129
262. Calvert, P.M. and H. Frucht. (2002) The genetics of colorectal cancer. Ann Intern
Med. 137(7). 603-612
263. Fearon, E.R. and B. Vogelstein. (1990) A genetic model for colorectal
tumorigenesis. Cell. 61(5). 759-767
264. Debinski, H.S., S. Love, A.D. Spigelman, and R.K. Phillips. (1996) Colorectal
polyp counts and cancer risk in familial adenomatous polyposis. Gastroenterology.
110(4). 1028-1030
265. Akiyama, T. (2000) Wnt/beta-catenin signaling. Cytokine Growth Factor Rev.
11(4). 273-282
266. MacDonald, B.T., K. Tamai, and X. He. (2009) Wnt/beta-catenin signaling:
components, mechanisms, and diseases. Dev Cell. 17(1). 9-26
267. Fishel, R., M.K. Lescoe, M.R. Rao, N.G. Copeland, N.A. Jenkins, J. Garber, M.
Kane, and R. Kolodner. (1993) The human mutator gene homolog MSH2 and its
association with hereditary nonpolyposis colon cancer. Cell. 75(5). 1027-1038
268. Muller, A. and R. Fishel. (2002) Mismatch repair and the hereditary non-polyposis
colorectal cancer syndrome (HNPCC). Cancer Invest. 20(1). 102-109
269. Hayward, I.P. and R.H. Whitehead. (1992) Patterns of growth and differentiation in
the colon carcinoma cell line LIM 1863. Int J Cancer. 50(5). 752-759
270. Kalluri, R. and R.A. Weinberg. (2009) The basics of epithelial-mesenchymal
transition. J Clin Invest. 119(6). 1420-1428
271. Thiery, J.P. and J.P. Sleeman. (2006) Complex networks orchestrate epithelial-
mesenchymal transitions. Nat Rev Mol Cell Biol. 7(2). 131-142
272. Greenburg, G. and E.D. Hay. (1982) Epithelia suspended in collagen gels can lose
polarity and express characteristics of migrating mesenchymal cells. J Cell Biol.
95(1). 333-339
273. Thiery, J.P. (2003) Epithelial-mesenchymal transitions in development and
pathologies. Curr Opin Cell Biol. 15(6). 740-746
274. Xu, J., S. Lamouille, and R. Derynck. (2009) TGF-beta-induced epithelial to
mesenchymal transition. Cell Res. 19(2). 156-172

130
275. Grunert, S., M. Jechlinger, and H. Beug. (2003) Diverse cellular and molecular
mechanisms contribute to epithelial plasticity and metastasis. Nat Rev Mol Cell
Biol. 4(8). 657-665
276. Thiery, J.P. (2002) Epithelial-mesenchymal transitions in tumour progression. Nat
Rev Cancer. 2(6). 442-454
277. Thompson, E.W., D.F. Newgreen, and D. Tarin. (2005) Carcinoma invasion and
metastasis: a role for epithelial-mesenchymal transition? Cancer Res. 65(14). 5991-
5995
278. Egeblad, M. and Z. Werb. (2002) New functions for the matrix metalloproteinases
in cancer progression. Nat Rev Cancer. 2(3). 161-174
279. Chang, C. and Z. Werb. (2001) The many faces of metalloproteases: cell growth,
invasion, angiogenesis and metastasis. Trends Cell Biol. 11(11). S37-S43
280. Bissell, M.J., D.C. Radisky, A. Rizki, V.M. Weaver, and O.W. Petersen. (2002) The
organizing principle: microenvironmental influences in the normal and malignant
breast. Differentiation. 70(9-10). 537-546
281. Yang, J. and R.A. Weinberg. (2008) Epithelial-mesenchymal transition: at the
crossroads of development and tumor metastasis. Dev Cell. 14(6). 818-829
282. Guarino, M., B. Rubino, and G. Ballabio. (2007) The role of epithelial-
mesenchymal transition in cancer pathology. Pathology. 39(3). 305-318
283. Thiery, J.P. (2002) Epithelial-mesenchymal transitions in tumour progression. Nat
Rev Cancer. 2(6). 442-454
284. Yilmaz, M. and G. Christofori. (2009) EMT, the cytoskeleton, and cancer cell
invasion. Cancer Metastasis Rev. 28(1-2). 15-33
285. Canel, M., A. Serrels, M.C. Frame, and V.G. Brunton. (2013) E-cadherin-integrin
crosstalk in cancer invasion and metastasis. J Cell Sci. 126(Pt 2). 393-401
286. Savagner, P. (2010) The epithelial-mesenchymal transition (EMT) phenomenon.
Ann Oncol. 21 Suppl 7. vii89-92
287. Moustakas, A. and C.H. Heldin. (2007) Signaling networks guiding epithelial-
mesenchymal transitions during embryogenesis and cancer progression. Cancer Sci.
98(10). 1512-1520

131
288. Kokkinos, M.I., R. Wafai, M.K. Wong, D.F. Newgreen, E.W. Thompson, and M.
Waltham. (2007) Vimentin and epithelial-mesenchymal transition in human breast
cancer--observations in vitro and in vivo. Cells Tissues Organs. 185(1-3). 191-203
289. Radisky, D.C. (2005) Epithelial-mesenchymal transition. J Cell Sci. 118(Pt 19).
4325-4326
290. Brattain, M.G., G. Howell, L.Z. Sun, and J.K. Willson. (1994) Growth factor
balance and tumor progression. Curr Opin Oncol. 6(1). 77-81
291. Strutz, F., M. Zeisberg, F.N. Ziyadeh, C.Q. Yang, R. Kalluri, G.A. Muller, and E.G.
Neilson. (2002) Role of basic fibroblast growth factor-2 in epithelial-mesenchymal
transformation. Kidney Int. 61(5). 1714-1728
292. Niessen, K., Y. Fu, L. Chang, P.A. Hoodless, D. McFadden, and A. Karsan. (2008)
Slug is a direct Notch target required for initiation of cardiac cushion
cellularization. J Cell Biol. 182(2). 315-325
293. Pelaez, I.M., M. Kalogeropoulou, A. Ferraro, A. Voulgari, T. Pankotai, I. Boros,
and A. Pintzas. (2010) Oncogenic RAS alters the global and gene-specific histone
modification pattern during epithelial-mesenchymal transition in colorectal
carcinoma cells. Int J Biochem Cell Biol. 42(6). 911-920
294. Giehl, K. (2005) Oncogenic Ras in tumour progression and metastasis. Biol Chem.
386(3). 193-205
295. Zondag, G.C., E.E. Evers, J.P. ten Klooster, L. Janssen, R.A. van der Kammen, and
J.G. Collard. (2000) Oncogenic Ras downregulates Rac activity, which leads to
increased Rho activity and epithelial-mesenchymal transition. J Cell Biol. 149(4).
775-782
296. Schramek, H., E. Feifel, I. Marschitz, N. Golochtchapova, G. Gstraunthaler, and R.
Montesano. (2003) Loss of active MEK1-ERK1/2 restores epithelial phenotype and
morphogenesis in transdifferentiated MDCK cells. Am J Physiol Cell Physiol.
285(3). C652-661
297. Hogan, C., S. Dupre-Crochet, M. Norman, M. Kajita, C. Zimmermann, A.E.
Pelling, E. Piddini, L.A. Baena-Lopez, J.P. Vincent, Y. Itoh, H. Hosoya, F. Pichaud,
and Y. Fujita. (2009) Characterization of the interface between normal and
transformed epithelial cells. Nat Cell Biol. 11(4). 460-467

132
298. Madin, S.H., P.C. Andriese, and N.B. Darby. (1957) The in vitro cultivation of
tissues of domestic and laboratory animals. Am J Vet Res. 18(69). 932-941
299. Gaush, C.R., W.L. Hard, and T.F. Smith. (1966) Characterization of an established
line of canine kidney cells (MDCK). Proc Soc Exp Biol Med. 122(3). 931-935
300. Mathias, R.A., Y.S. Chen, B. Wang, H. Ji, E.A. Kapp, R.L. Moritz, H.J. Zhu, and
R.J. Simpson. (2010) Extracellular remodelling during oncogenic Ras-induced
epithelial-mesenchymal transition facilitates MDCK cell migration. J Proteome Res.
9(2). 1007-1019
301. Garnier, D., N. Magnus, T.H. Lee, V. Bentley, B. Meehan, C. Milsom, L.
Montermini, T. Kislinger, and J. Rak. (2012) Cancer cells induced to express
mesenchymal phenotype release exosome-like extracellular vesicles carrying tissue
factor. J Biol Chem. 287(52). 43565-43572
302. Clark, H.F., A.L. Gurney, E. Abaya, K. Baker, D. Baldwin, J. Brush, J. Chen, B.
Chow, C. Chui, C. Crowley, B. Currell, B. Deuel, P. Dowd, D. Eaton, J. Foster, C.
Grimaldi, Q. Gu, P.E. Hass, S. Heldens, A. Huang, H.S. Kim, L. Klimowski, Y. Jin,
S. Johnson, J. Lee, L. Lewis, D. Liao, M. Mark, E. Robbie, C. Sanchez, J.
Schoenfeld, S. Seshagiri, L. Simmons, J. Singh, V. Smith, J. Stinson, A. Vagts, R.
Vandlen, C. Watanabe, D. Wieand, K. Woods, M.H. Xie, D. Yansura, S. Yi, G. Yu,
J. Yuan, M. Zhang, Z. Zhang, A. Goddard, W.I. Wood, P. Godowski, and A. Gray.
(2003) The secreted protein discovery initiative (SPDI), a large-scale effort to
identify novel human secreted and transmembrane proteins: a bioinformatics
assessment. Genome Res. 13(10). 2265-2270
303. Mathivanan, S., H. Ji, and R.J. Simpson. (2010) Exosomes: extracellular organelles
important in intercellular communication. J Proteomics. 73(10). 1907-1920
304. Bronfman, M., G. Loyola, and C.S. Koenig. (1998) Isolation of intact organelles by
differential centrifugation of digitonin-treated hepatocytes using a table Eppendorf
centrifuge. Anal Biochem. 255(2). 252-256
305. Girard, M., P.D. Allaire, F. Blondeau, and P.S. McPherson. (2005) Isolation of
clathrin-coated vesicles by differential and density gradient centrifugation. Curr
Protoc Cell Biol. Chapter 3. Unit 3 13

133
306. Ji, H., N. Erfani, B.J. Tauro, E.A. Kapp, H.J. Zhu, R.L. Moritz, J.W. Lim, and R.J.
Simpson. (2008) Difference gel electrophoresis analysis of Ras-transformed
fibroblast cell-derived exosomes. Electrophoresis. 29(12). 2660-2671
307. Mathias, R.A., J.W. Lim, H. Ji, and R.J. Simpson. (2009) Isolation of extracellular
membranous vesicles for proteomic analysis. Methods Mol Biol. 528. 227-242
308. Muralidharan-Chari, V., J. Clancy, C. Plou, M. Romao, P. Chavrier, G. Raposo, and
C. D'Souza-Schorey. (2009) ARF6-regulated shedding of tumor cell-derived plasma
membrane microvesicles. Curr Biol. 19(22). 1875-1885
309. Tan, A., H. De La Pena, and A.M. Seifalian. (2010) The application of exosomes as
a nanoscale cancer vaccine. Int J Nanomedicine. 5. 889-900
310. Martin-Belmonte, F. and M. Perez-Moreno. (2012) Epithelial cell polarity, stem
cells and cancer. Nat Rev Cancer. 12(1). 23-38
311. Went, P., M. Vasei, L. Bubendorf, L. Terracciano, L. Tornillo, U. Riede, J.
Kononen, R. Simon, G. Sauter, and P.A. Baeuerle. (2006) Frequent high-level
expression of the immunotherapeutic target Ep-CAM in colon, stomach, prostate
and lung cancers. Br J Cancer. 94(1). 128-135
312. Kuhn, S., M. Koch, T. Nubel, M. Ladwein, D. Antolovic, P. Klingbeil, D.
Hildebrand, G. Moldenhauer, L. Langbein, W.W. Franke, J. Weitz, and M. Zoller.
(2007) A complex of EpCAM, claudin-7, CD44 variant isoforms, and tetraspanins
promotes colorectal cancer progression. Mol Cancer Res. 5(6). 553-567
313. Rollins, S.A. and P.J. Sims. (1990) The complement-inhibitory activity of CD59
resides in its capacity to block incorporation of C9 into membrane C5b-9. J
Immunol. 144(9). 3478-3483
314. Riley-Vargas, R.C., D.B. Gill, C. Kemper, M.K. Liszewski, and J.P. Atkinson.
(2004) CD46: expanding beyond complement regulation. Trends Immunol. 25(9).
496-503
315. Yanai, H., T. Ban, Z. Wang, M.K. Choi, T. Kawamura, H. Negishi, M. Nakasato, Y.
Lu, S. Hangai, R. Koshiba, D. Savitsky, L. Ronfani, S. Akira, M.E. Bianchi, K.
Honda, T. Tamura, T. Kodama, and T. Taniguchi. (2009) HMGB proteins function
as universal sentinels for nucleic-acid-mediated innate immune responses. Nature.
462(7269). 99-103

134
316. Jaiswal, R., F. Luk, P.V. Dalla, G.E. Grau, and M. Bebawy. (2013) Breast cancer-
derived microparticles display tissue selectivity in the transfer of resistance proteins
to cells. PLoS One. 8(4). e61515
317. Sarkadi, B., C. Ozvegy-Laczka, K. Nemet, and A. Varadi. (2004) ABCG2 -- a
transporter for all seasons. FEBS Lett. 567(1). 116-120
318. Huber, M.A., N. Kraut, and H. Beug. (2005) Molecular requirements for epithelial-
mesenchymal transition during tumor progression. Curr Opin Cell Biol. 17(5). 548-
558
319. Nistico, P., M.J. Bissell, and D.C. Radisky. (2012) Epithelial-mesenchymal
transition: general principles and pathological relevance with special emphasis on
the role of matrix metalloproteinases. Cold Spring Harb Perspect Biol. 4(2). pii:
a011908
320. De Craene, B. and G. Berx. (2013) Regulatory networks defining EMT during
cancer initiation and progression. Nat Rev Cancer. 13(2). 97-110
321. Pulukuri, S.M. and J.S. Rao. (2008) Matrix metalloproteinase-1 promotes prostate
tumor growth and metastasis. Int J Oncol. 32(4). 757-765
322. Guise, T.A. (2009) Breaking down bone: new insight into site-specific mechanisms
of breast cancer osteolysis mediated by metalloproteinases. Genes Dev. 23(18).
2117-2123
323. Guo, W. and F.G. Giancotti. (2004) Integrin signalling during tumour progression.
Nat Rev Mol Cell Biol. 5(10). 816-826
324. Pegtel, D.M., K. Cosmopoulos, D.A. Thorley-Lawson, M.A. van Eijndhoven, E.S.
Hopmans, J.L. Lindenberg, T.D. de Gruijl, T. Wurdinger, and J.M. Middeldorp.
(2010) Functional delivery of viral miRNAs via exosomes. Proc Natl Acad Sci U S
A. 107(14). 6328-6333
325. Xiao, D., J. Ohlendorf, Y. Chen, D.D. Taylor, S.N. Rai, S. Waigel, W. Zacharias, H.
Hao, and K.M. McMasters. (2012) Identifying mRNA, microRNA and protein
profiles of melanoma exosomes. PLoS One. 7(10). e46874

135
326. Escola, J.M., M.J. Kleijmeer, W. Stoorvogel, J.M. Griffith, O. Yoshie, and H.J.
Geuze. (1998) Selective enrichment of tetraspan proteins on the internal vesicles of
multivesicular endosomes and on exosomes secreted by human B-lymphocytes. J
Biol Chem. 273(32). 20121-20127
327. Chaput, N., C. Flament, S. Viaud, J. Taieb, S. Roux, A. Spatz, F. Andre, J.B.
LePecq, M. Boussac, J. Garin, S. Amigorena, C. Thery, and L. Zitvogel. (2006)
Dendritic cell derived-exosomes: biology and clinical implementations. J Leukoc
Biol. 80(3). 471-478
328. Skokos, D., S. Le Panse, I. Villa, J.C. Rousselle, R. Peronet, B. David, A. Namane,
and S. Mecheri. (2001) Mast cell-dependent B and T lymphocyte activation is
mediated by the secretion of immunologically active exosomes. J Immunol. 166(2).
868-876
329. Huber, V., S. Fais, M. Iero, L. Lugini, P. Canese, P. Squarcina, A. Zaccheddu, M.
Colone, G. Arancia, M. Gentile, E. Seregni, R. Valenti, G. Ballabio, F. Belli, E.
Leo, G. Parmiani, and L. Rivoltini. (2005) Human colorectal cancer cells induce T-
cell death through release of proapoptotic microvesicles: role in immune escape.
Gastroenterology. 128(7). 1796-1804
330. Lim, J.W., R.A. Mathias, E.A. Kapp, M.J. Layton, M.C. Faux, A.W. Burgess, H. Ji,
and R.J. Simpson. (2012) Restoration of full-length APC protein in SW480 colon
cancer cells induces exosome-mediated secretion of DKK-4. Electrophoresis.
33(12). 1873-1880
331. Malik, Z.A., K.S. Kott, A.J. Poe, T. Kuo, L. Chen, K.W. Ferrara, and A.A.
Knowlton. (2013) Cardiac myocyte exosomes: stability, HSP60, and proteomics.
Am J Physiol Heart Circ Physiol. 304(7). H954-965
332. Faure, J., G. Lachenal, M. Court, J. Hirrlinger, C. Chatellard-Causse, B. Blot, J.
Grange, G. Schoehn, Y. Goldberg, V. Boyer, F. Kirchhoff, G. Raposo, J. Garin, and
R. Sadoul. (2006) Exosomes are released by cultured cortical neurones. Mol Cell
Neurosci. 31(4). 642-648

136
333. Potolicchio, I., G.J. Carven, X. Xu, C. Stipp, R.J. Riese, L.J. Stern, and L.
Santambrogio. (2005) Proteomic analysis of microglia-derived exosomes: metabolic
role of the aminopeptidase CD13 in neuropeptide catabolism. J Immunol. 175(4).
2237-2243
334. Krämer-Albers, E.-M., N. Bretz, S. Tenzer, C. Winterstein, W. Möbius, H. Berger,
K.-A. Nave, H. Schild, and J. Trotter. (2007) Oligodendrocytes secrete exosomes
containing major myelin and stress-protective proteins: Trophic support for axons?
PROTEOMICS - CLINICAL APPLICATIONS. 1(11). 1446-1461
335. Kesimer, M., M. Scull, B. Brighton, G. Demaria, K. Burns, W. O'Neal, R.J. Pickles,
and J.K. Sheehan. (2009) Characterization of exosome-like vesicles released from
human tracheobronchial ciliated epithelium: a possible role in innate defense. Faseb
J. 23(6). 1858-1868
336. Conde-Vancells, J., E. Rodriguez-Suarez, N. Embade, D. Gil, R. Matthiesen, M.
Valle, F. Elortza, S.C. Lu, J.M. Mato, and J.M. Falcon-Perez. (2008)
Characterization and Comprehensive Proteome Profiling of Exosomes Secreted by
Hepatocytes. J Proteome Res. 7(12). 5157-5166
337. Chavez-Munoz, C., J. Morse, R. Kilani, and A. Ghahary. (2008) Primary human
keratinocytes externalize stratifin protein via exosomes. J Cell Biochem. 104(6).
2165-2173
338. Perez-Hernandez, D., C. Gutierrez-Vazquez, I. Jorge, S. Lopez-Martin, A. Ursa, F.
Sanchez-Madrid, J. Vazquez, and M. Yanez-Mo. (2013) The intracellular
interactome of tetraspanin-enriched microdomains reveals their function as sorting
machineries to exosomes. J Biol Chem. 288(17). 11649-11661
339. Fevrier, B., D. Vilette, F. Archer, D. Loew, W. Faigle, M. Vidal, H. Laude, and G.
Raposo. (2004) Cells release prions in association with exosomes. Proc Natl Acad
Sci U S A. 101(26). 9683-9688
340. Dukers, D.F., P. Meij, M.B. Vervoort, W. Vos, R.J. Scheper, C.J. Meijer, E.
Bloemena, and J.M. Middeldorp. (2000) Direct immunosuppressive effects of EBV-
encoded latent membrane protein 1. J Immunol. 165(2). 663-670

137
341. Admyre, C., J. Grunewald, J. Thyberg, S. Gripenback, G. Tornling, A. Eklund, A.
Scheynius, and S. Gabrielsson. (2003) Exosomes with major histocompatibility
complex class II and co-stimulatory molecules are present in human BAL fluid. Eur
Respir J. 22(4). 578-583
342. Andre, F., N.E. Schartz, M. Movassagh, C. Flament, P. Pautier, P. Morice, C.
Pomel, C. Lhomme, B. Escudier, T. Le Chevalier, T. Tursz, S. Amigorena, G.
Raposo, E. Angevin, and L. Zitvogel. (2002) Malignant effusions and immunogenic
tumour-derived exosomes. Lancet. 360(9329). 295-305
343. Ogawa, Y., M. Kanai-Azuma, Y. Akimoto, H. Kawakami, and R. Yanoshita. (2008)
Exosome-like vesicles with dipeptidyl peptidase IV in human saliva. Biol Pharm
Bull. 31(6). 1059-1062
344. Skriner, K., K. Adolph, P.R. Jungblut, and G.R. Burmester. (2006) Association of
citrullinated proteins with synovial exosomes. Arthritis Rheum. 54(12). 3809-3814
345. Keller, S., C. Rupp, A. Stoeck, S. Runz, M. Fogel, S. Lugert, H.D. Hager, M.S.
Abdel-Bakky, P. Gutwein, and P. Altevogt. (2007) CD24 is a marker of exosomes
secreted into urine and amniotic fluid. Kidney Int. 72(9). 1095-1102
346. Pisitkun, T., R.F. Shen, and M.A. Knepper. (2004) Identification and proteomic
profiling of exosomes in human urine. Proc Natl Acad Sci U S A. 101(36). 13368-
13373
347. Gonzales, P.A., T. Pisitkun, J.D. Hoffert, D. Tchapyjnikov, R.A. Star, R. Kleta,
N.S. Wang, and M.A. Knepper. (2009) Large-scale proteomics and
phosphoproteomics of urinary exosomes. J Am Soc Nephrol. 20(2). 363-379
348. Principe, S., E.E. Jones, Y. Kim, A. Sinha, J.O. Nyalwidhe, J. Brooks, O.J.
Semmes, D.A. Troyer, R.S. Lance, T. Kislinger, and R.R. Drake. (2013) In-depth
proteomic analyses of exosomes isolated from expressed prostatic secretions in
urine. Proteomics. 13(10-11). 1667-1671
349. Raimondo, F., L. Morosi, S. Corbetta, C. Chinello, P. Brambilla, P. Della Mina, A.
Villa, G. Albo, C. Battaglia, S. Bosari, F. Magni, and M. Pitto. (2013) Differential
protein profiling of renal cell carcinoma urinary exosomes. Mol Biosyst. 9(6). 1220-
1233

138
350. Wang, Z., S. Hill, J.M. Luther, D.L. Hachey, and K.L. Schey. (2012) Proteomic
analysis of urine exosomes by multidimensional protein identification technology
(MudPIT). Proteomics. 12(2). 329-338
351. Saleem, R.A., R.S. Rogers, A.V. Ratushny, D.J. Dilworth, P.T. Shannon, D.
Shteynberg, Y. Wan, R.L. Moritz, A.I. Nesvizhskii, R.A. Rachubinski, and J.D.
Aitchison. (2010) Integrated phosphoproteomics analysis of a signaling network
governing nutrient response and peroxisome induction. Mol Cell Proteomics. 9(9).
2076-2088
352. Deutsch, E.W., H. Lam, and R. Aebersold. (2008) PeptideAtlas: a resource for
target selection for emerging targeted proteomics workflows. EMBO Rep. 9(5). 429-
434
353. Desiere, F., E.W. Deutsch, N.L. King, A.I. Nesvizhskii, P. Mallick, J. Eng, S. Chen,
J. Eddes, S.N. Loevenich, and R. Aebersold. (2006) The PeptideAtlas project.
Nucleic Acids Res. 34(Database issue). D655-658
354. Desiere, F., E.W. Deutsch, A.I. Nesvizhskii, P. Mallick, N.L. King, J.K. Eng, A.
Aderem, R. Boyle, E. Brunner, S. Donohoe, N. Fausto, E. Hafen, L. Hood, M.G.
Katze, K.A. Kennedy, F. Kregenow, H. Lee, B. Lin, D. Martin, J.A. Ranish, D.J.
Rawlings, L.E. Samelson, Y. Shiio, J.D. Watts, B. Wollscheid, M.E. Wright, W.
Yan, L. Yang, E.C. Yi, H. Zhang, and R. Aebersold. (2005) Integration with the
human genome of peptide sequences obtained by high-throughput mass
spectrometry. Genome Biol. 6(1). R9

139
Appendix
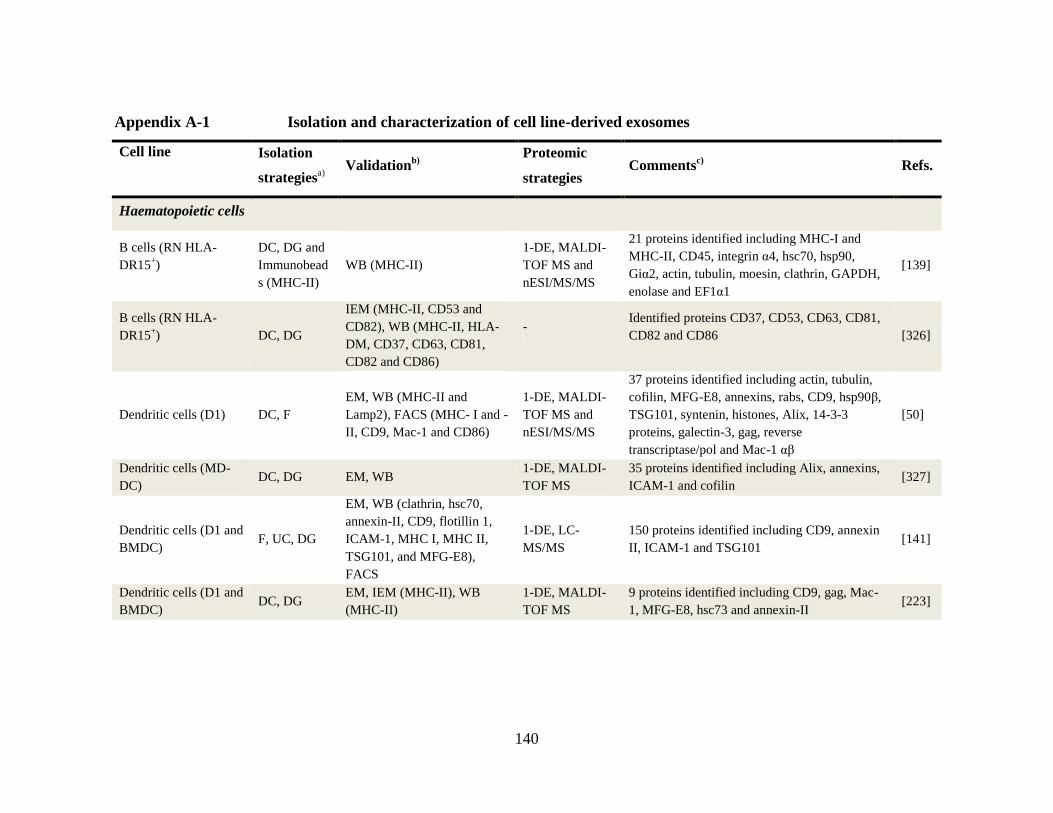
140
Appendix A-1 Isolation and characterization of cell line-derived exosomes
Cell line
Isolation
strategiesa)
Validationb)
Proteomic
strategies Comments
c) Refs.
Haematopoietic cells
B cells (RN HLA-
DR15+)
DC, DG and
Immunobead
s (MHC-II)
WB (MHC-II)
1-DE, MALDI-
TOF MS and
nESI/MS/MS
21 proteins identified including MHC-I and
MHC-II, CD45, integrin α4, hsc70, hsp90,
Giα2, actin, tubulin, moesin, clathrin, GAPDH,
enolase and EF1α1
[139]
B cells (RN HLA-
DR15+)
DC, DG
IEM (MHC-II, CD53 and
CD82), WB (MHC-II, HLA-
DM, CD37, CD63, CD81,
CD82 and CD86)
-
Identified proteins CD37, CD53, CD63, CD81,
CD82 and CD86
[326]
Dendritic cells (D1) DC, F
EM, WB (MHC-II and
Lamp2), FACS (MHC- I and -
II, CD9, Mac-1 and CD86)
1-DE, MALDI-
TOF MS and
nESI/MS/MS
37 proteins identified including actin, tubulin,
cofilin, MFG-E8, annexins, rabs, CD9, hsp90β,
TSG101, syntenin, histones, Alix, 14-3-3
proteins, galectin-3, gag, reverse
transcriptase/pol and Mac-1 αβ
[50]
Dendritic cells (MD-
DC) DC, DG EM, WB
1-DE, MALDI-
TOF MS
35 proteins identified including Alix, annexins,
ICAM-1 and cofilin [327]
Dendritic cells (D1 and
BMDC) F, UC, DG
EM, WB (clathrin, hsc70,
annexin-II, CD9, flotillin 1,
ICAM-1, MHC I, MHC II,
TSG101, and MFG-E8),
FACS
1-DE, LC-
MS/MS
150 proteins identified including CD9, annexin
II, ICAM-1 and TSG101 [141]
Dendritic cells (D1 and
BMDC) DC, DG
EM, IEM (MHC-II), WB
(MHC-II)
1-DE, MALDI-
TOF MS
9 proteins identified including CD9, gag, Mac-
1, MFG-E8, hsc73 and annexin-II [223]

141
Mast cell (MC/9,
HMC-1, BMMC) DC, F, DG EM, FACS (CD63)
1-DE, LC-
MS/MS
271 proteins identified including mast
carboxypeptidase A, tubulins, TCP proteins,
ezrin, moesin, 40S ribosomal proteins, 14-3-3
proteins,CD43, CD63, CD97, annexins, MHC-I,
histones, hsc70 and integrin-α6
[138]
Mast cell (MC/9 and
BMMC) and
mastocytoma (P815)
DC EM, IEM
1-DE, MALDI-
TOF MS,
ELISA
7 proteins identified including MHC-II, CD40,
CD40L, CD86, LFA-1, ICAM-1, CD13,
annexin-VI, actins and CDC25
[328]
T cells (Jurkat cells, T
cell blasts, E+ cells and
MART-1+ T cell)
F, UC
EM, IEM (TCR β and CD3ε),
WB (TCR β and CD3ε),
FACS (CD63, TCR β, CD3ε,
MHC-I and -II)
WB, FACS
Proteins identified including TCR β, CD3ε and
ζ (phosphorylated ζ), MHC-I and -II, CD2,
CD18, chemokine receptor CXCR4, c-Cbl,
tyrosine kinases Fyn and Lck
[140]
Tumor cells
Bladder cancer cells
(HT1376) DC, SC
WB (TSG101, CD9 and
CD81)
LC-MALDI-
TOF/TOF MS
353 proteins identified including basigin,
galectin-3, trophoblast glycoprotein (5T4),
LAMP2, CD73 HLA-G and β-Catenin
[145]
Brain tumor
(EGFRvIII-transfected
SMA560)
DC, DG
(OptiPrep™)
EM, WB (Alix, GAPDH, α1-
anti-trypsin, CD9, PDI, CRT,
transferrin, GPNMB, TGF-β1
and EGFRvIII),
acetylcholinesterase assay
2-DE, MALDI-
TOF/TOF
36 proteins identified including HSP70, α-
enolase, MVP, ARRDC2, CENP-P, GAPDH,
syntenin, PCNA, CRMP-2 and sialic acid
synthase
[125]
Breast adenocarcinoma
(BT-474 and MDA-
MB-231)
DC, F, DG,
Immunobead
s (HER2)
EM, FACS (HER2), WB
(HER2 and actin) WB, FACS Protein HER2 identified [37]
Colorectal cancer
(HT29)
DC,
diafiltration
(100K), DG
EM, WB (CD63 and CD81) 1-DE, LC-
MS/MS
547 proteins identified including annexins,
ARFs, rabs, ADAM10, CD44, NG2, ephrin-B1,
MIF, β-catenin, junction plakoglobin, galectin-
4, RACK1 and tetraspanin-8
[48]

142
Colon carcinoma cell
lines (SW403, 1869col
and CRC28462)
DC
EM, IEM (CD63, FasL and
TRAIL), WB and FACS
(CD63, FasL, TRAIL, CEA
and MHC-I)
WB FasL and TRAIL identified [329]
Colorectal cancer
(LIM1215)
F,
diafiltration
(5K), UC
EM, IEM (A33), WB (CD9,
A33, TSG101, Rab7 and
hsc70)
1-DE, LC-
MS/MS
400 proteins identified including A33, CEA,
EGFR, ADAM10, dipeptidase 1, ephrin-B1,
hsc70, tetraspanins, ESCRT proteins, integrins,
annexins, rabs and GTPases
[44]
Colorectal carcinoma
cell lines SW480 and
SW620
DG
(OptiPrep™)
Cryo-EM, WB (Alix,
TSG101, CD9, HSP70,
Flotillin-1, and EGFR
1-DE, LC-
MS/MS
941 and 796 proteins identified in SW480 and
SW620 exosomes, respectively, including MET,
S100A8, S100A9, TNC, EFNB2, EGFR, JAG1,
SRC and TNIK
[114]
Colorectal carcinoma
cell lines SW480 and
SW480APC
F, DC EM, WB (Alix, TSG101, CD9
and HSP70)
1-DE, LC-
MS/MS
563 and 497 proteins were identified in SW480
and SW480APC exosomes respectively
including DKK4, Alix, TSG101, VPS28,
VPS25, CHMP2A and tetraspanins CD9, CD63,
CD81 and CD82
[330]
Mammary
adenocarcinoma
(TS/A, H-2d), P815
mastocytoma (H-2d),
melanoma (Fon and
Mel-888)
DC, DG EM, WB (hsc70 and MHC-I) WB, IEM Proteins MHC-I, hsp70, MART-1 and TRP
identified [73]
Melanoma (MeWo and
SK-MEL-28) F, UC
WB (MHC-I, MART-1, Mel-
CAM and annexin II), DG
2-DE, MALDI-
TOF/TOF MS
41 proteins identified including Alix, hsp70,
Giβ2, Giα, moesin, GAPDH, malate
dehydrogenase, p120 catenin, PGRL, syntaxin-
binding protein 1 and 2, septin-2 and WD
repeat-containing protein 1
[142]

143
Mesothelioma (PMR-
MM7 and 8) DC IEM (CD63)
1-DE, MALDI-
TOF MS
30 proteins identified including annexins,
actins, actinin-4 tubulins, hsc70, hsp90,
integrins, fibronectin, GAPDH, MHC-I,
PLVAP and DEL-1
[143]
Medulloblastoma cells
(D283MED) F, DG
NTA, WB (CD9 and HSP70)
acetylcholinesterase assay
1-DE, LC-
MS/MS
148 proteins proteins identified including PDI,
HPX, HNF4A and GPNMB [144]
Primary and normal cells
Cardiac myocytes
(Sprague-Dawly rats) DC, EQ EM
1-DE LC-
MS/MS
57 proteins identified including tropomyosin-
1α, myomesin 2 (M-band), α-crystallin B,
cardiac α-actin, GAPDH and long-chain
specific acyl-CoA dehydrogenase.
[331]
Cortical neurons (8-
day primary culture) DC, DG
EM, WB (Alix, TSG101 and
flotillin)
1-DE, LC-
MS/MS
19 proteins identified including GLAST1,
brain-specific ceruloplasmin, L1 cell adhesion
molecule, GPI-anchored prion protein and
GluR2/3
[332]
Intestinal epithelial
(HT29-19A and T84-
DRB1*0401/CIITA)
DC, DG
EM, IEM (CD26, CD63,
MHC-I and -IIα), WB
(MHC-I and -IIα, CD26,
CD63, TfR and Na+K
+-
ATPase)
1-DE, MALDI-
TOF MS
28 proteins identified including syntaxin-3,
syntaxin-binding protein 2, EPS8, microsomal
dipeptidase in AM and A33, epithelial cell
surface antigen and major vault protein
[72]
Microglia (N9 and
primary culture from
SJL/J mice)
DC, DG
EM, WB (CD9, CD63,
syntaxin-8, Rab7, Rab11,
clathrin, Lamp-1 and -2, Vti-
1A and -1B)
1-DE, LC-
MS/MS
59 proteins identified including cathepsin S,
aminopeptidase N (CD13), MCT-1, MHC class
II-associated chaperone Ii, CD14, NAP-22, FcR
for IgE and GP42
[333]
Oligodendrocytes
(primary culture and
Oli-neu)
DC, DG EM, WB (Alix, PLP, CNP
and TSG101) LC-MS/MS
143 proteins identified including CD81, 14-3-3
proteins, actins, tubulins, histones, EF1 and 2,
hsp90, hsc70, Na+K
+ATPase α chains, PLP,
CNP, MBP and MOG
[334]
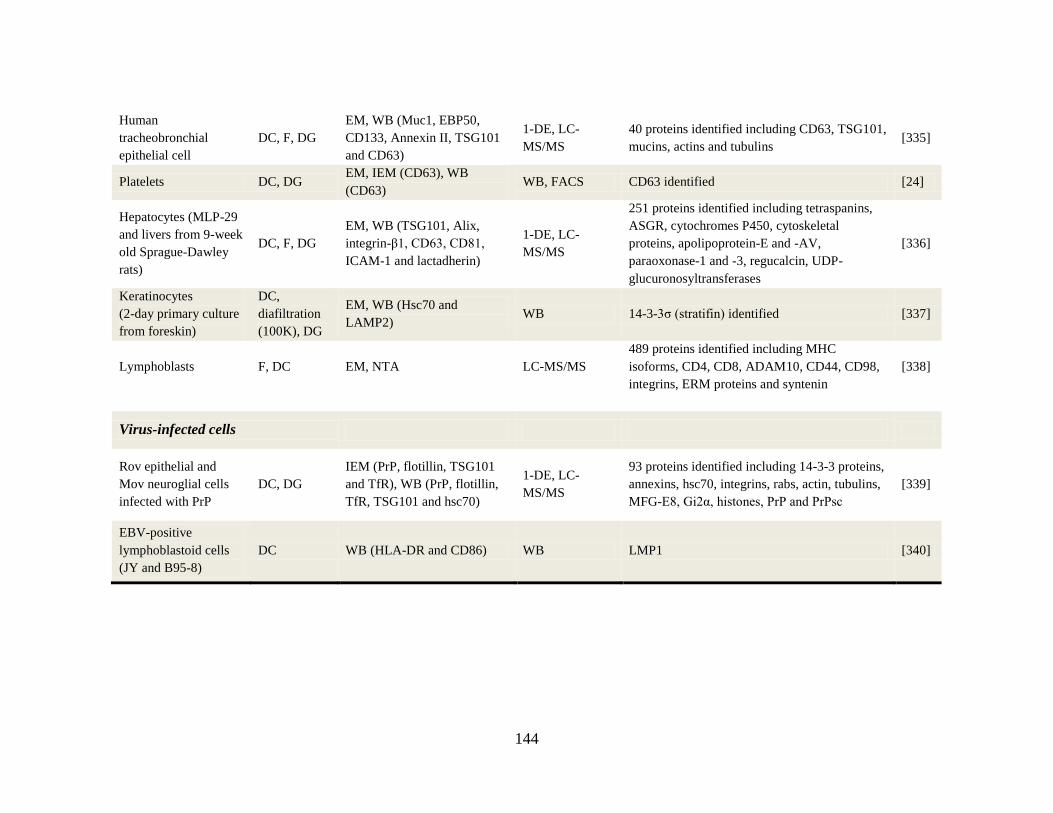
144
Human
tracheobronchial
epithelial cell
DC, F, DG
EM, WB (Muc1, EBP50,
CD133, Annexin II, TSG101
and CD63)
1-DE, LC-
MS/MS
40 proteins identified including CD63, TSG101,
mucins, actins and tubulins [335]
Platelets DC, DG EM, IEM (CD63), WB
(CD63) WB, FACS CD63 identified [24]
Hepatocytes (MLP-29
and livers from 9-week
old Sprague-Dawley
rats)
DC, F, DG
EM, WB (TSG101, Alix,
integrin-β1, CD63, CD81,
ICAM-1 and lactadherin)
1-DE, LC-
MS/MS
251 proteins identified including tetraspanins,
ASGR, cytochromes P450, cytoskeletal
proteins, apolipoprotein-E and -AV,
paraoxonase-1 and -3, regucalcin, UDP-
glucuronosyltransferases
[336]
Keratinocytes
(2-day primary culture
from foreskin)
DC,
diafiltration
(100K), DG
EM, WB (Hsc70 and
LAMP2) WB 14-3-3σ (stratifin) identified [337]
Lymphoblasts F, DC EM, NTA LC-MS/MS
489 proteins identified including MHC
isoforms, CD4, CD8, ADAM10, CD44, CD98,
integrins, ERM proteins and syntenin
[338]
Virus-infected cells
Rov epithelial and
Mov neuroglial cells
infected with PrP
DC, DG
IEM (PrP, flotillin, TSG101
and TfR), WB (PrP, flotillin,
TfR, TSG101 and hsc70)
1-DE, LC-
MS/MS
93 proteins identified including 14-3-3 proteins,
annexins, hsc70, integrins, rabs, actin, tubulins,
MFG-E8, Gi2α, histones, PrP and PrPsc
[339]
EBV-positive
lymphoblastoid cells
(JY and B95-8)
DC WB (HLA-DR and CD86) WB LMP1 [340]

145
a) DC: Differential centrifugation; DG: Sucrose density gradient or otherwise stated; EQ: Exoquick; F: Filtration; GF: Gel filtration; SC: Sucrose
cushion; UC: Ultracentrifugation
b) CRT: Calreticulin; EM: Electron microscopy; EGFRvIII: Epidermal growth factor receptor variant III; FACS: Fluorescence–activated cell sorting;
GPNMB: Glycoprotein nonmetastatic B; IEM: Immunoelectron microscopy; NTA: Nanoparticle tracking analysis; PDI: Protein disulfide isomerase;
TGF-β1: Transforming growth factor beta 1; TfR: Transferrin receptor; WB: Western blot.
c) ADAM10: A Disintegrin and metalloproteinase domain-containing protein 10; AM: Apical membrane; ARF: ADP-ribosylation factor; ARRDC2:
Arrestin domain-containing protein 2; ASGR: Asiaglycoprotein receptor; BM: Basolateral membrane; CENP-P: Centromere protein P; CHMP2A:
Charged multivesicular body protein 2A; CNP: 2',3'-cyclic-nucleotide 3'-phosphodiesterase; CRMP-2: Collapsing response mediator protein 2; DEL-
1: developmental endothelial locus-1; DKK4: Dickkopf 4; Di EBV: Epstein-Barr virus; EFNB2: Ephrin-B2; EGFR: Epidermal growth factor
receptor; ERM proteins (ezrin, radixin, and moesin); ESCRT: Endosomal sorting complex required for transport; FasL: Fas Ligand; GAPDH:
glyceraldehyde-3-phosphate dehydrogenase; GPNMB: glycoprotein non-metastatic B; HNF4A: hepatocyte nuclear factor alpha; HPX: Hemopexin;
JAG1: Jagged 1; LAMP1: Lysosome associated membrane protein 1; LMP1: Latent membrane protein 1; MART-1:Melanoma antigen recognized by
T cells 1; MBP: Myelin basic protein; MET: Met proto-oncogene (hepatocyte growth factor receptor); MIF: Macrophage migration inhibitory factor;
MOG: Myelin oligodendrocyte glycoprotein; MHC: Major histocompatibility complex; MVP: Major vault protein; NG2: Chondroitin sulfate
proteoglycan; PDI: Protein disulfide isomerase; PLP: Myelin proteolipid protein; PrP: Prion protein; PrPsc: Prion protein scrapie; RACK1: Lung
cancer oncogene 7; S100A8: S100 calcium binding protein A8; S100A9: S100 calcium binding protein A9; SRC: v-src sarcoma; TNC: tenascin C;
TRAIL: TNF-related apoptosis-inducing ligand; TNIK; TRAF2 and NCK interacting kinase; TRP: 5,6-dihydroxyindole-2-carboxylic acid oxidase;
VPS proteins: Class E Vacuolar protein-sorting proteins. Reproduced and updated from [74].

146
Appendix A-2 Isolation and characterization of body fluid-derived exosomes
Body fluid Isolation
strategiesa)
Validationb)
Proteomic
strategies Comments
c) Refs.
Breast milk DC, F, DG
IEM (CD63 and HLA-DR), WB
(HLA-DR, CD81 and HSC70),
FACS (HLA-DR, CD63, CD81 and
mucin-1)
In-solution
trypsinization,
scx-LC-MS/MS
73 proteins identified including MFG-
E8, MUC1, hsp70, ARF-1, EH
domain-containing protein 1, CD36,
butyrophilin and polymeric-Ig
receptor
[146]
Bronchoalveolar
lavage fluid
DC, immuno-
beads (MHC-II)
IEM (HLA-DR and CD63), FACS
(HLA-DR, CD54, CD63 and CD86)
-
-
[341]
Malignant pleural
effusions DC, DG EM
1-DE, MALDI-
TOF MS
50 proteins identified including MHC-I,
actin, G protein, hsp90, BTG1, Bamacan,
PEDF, BTG-1, TSG14 and TSP2
[147]
Malignant pleural
effusions and
malignant ascites
DC, DG
EM, IEM (MHC-I and -II, TRP,
gp100 and CD81), WB (MHC-I and
-II ,MART-1, HER2 and hsc70)
WB Proteins including MART-1, TRP, gp100
and HER2 identified [342]
Saliva GF EM WB (Alix, TSG101, HSP70 and
CD63)
2-DE LC-
MS/MS
68 proteins were identified including
GW182, immunoglobulin A, polymeric
immunoglobulin receptor, Mucin-5B, Alpha
–amylase 1, Dipeptidyl peptidase 4 (CD26).
[47]
Saliva F, diafiltration
(100K), GF EM
N-terminal
amino acid
sequencing, WB
Dipeptidyl peptidase IV (CD26), polymeric-
Ig receptor, actin, galectin-3 and Ig chains [343]
Semen DG EM 1-DE, LC-
MS/MS 440 proteins identified [148]
Synovial fluid
(RA, OA and
reactive arthritis)
DC, DG EM, IEM (IgG)
2-DE, WB,
MALDI-TOF
MS
Citrullinated fibrin α-chain, CD5 antigen-
like, fibrinogen fragment D and β-chain [344]
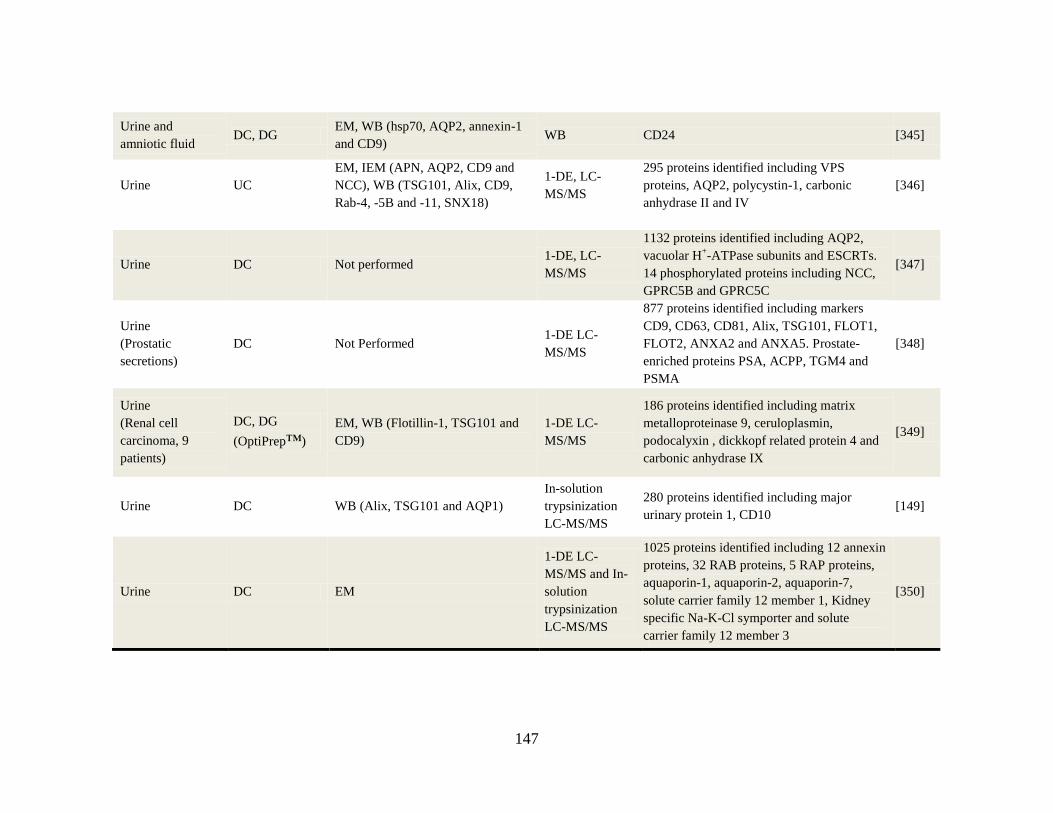
147
Urine and
amniotic fluid DC, DG
EM, WB (hsp70, AQP2, annexin-1
and CD9) WB CD24 [345]
Urine UC
EM, IEM (APN, AQP2, CD9 and
NCC), WB (TSG101, Alix, CD9,
Rab-4, -5B and -11, SNX18)
1-DE, LC-
MS/MS
295 proteins identified including VPS
proteins, AQP2, polycystin-1, carbonic
anhydrase II and IV
[346]
Urine DC Not performed 1-DE, LC-
MS/MS
1132 proteins identified including AQP2,
vacuolar H+-ATPase subunits and ESCRTs.
14 phosphorylated proteins including NCC,
GPRC5B and GPRC5C
[347]
Urine
(Prostatic
secretions)
DC Not Performed 1-DE LC-
MS/MS
877 proteins identified including markers
CD9, CD63, CD81, Alix, TSG101, FLOT1,
FLOT2, ANXA2 and ANXA5. Prostate-
enriched proteins PSA, ACPP, TGM4 and
PSMA
[348]
Urine
(Renal cell
carcinoma, 9
patients)
DC, DG
(OptiPrep™)
EM, WB (Flotillin-1, TSG101 and
CD9)
1-DE LC-
MS/MS
186 proteins identified including matrix
metalloproteinase 9, ceruloplasmin,
podocalyxin , dickkopf related protein 4 and
carbonic anhydrase IX
[349]
Urine DC WB (Alix, TSG101 and AQP1)
In-solution
trypsinization
LC-MS/MS
280 proteins identified including major
urinary protein 1, CD10 [149]
Urine DC EM
1-DE LC-
MS/MS and In-
solution
trypsinization
LC-MS/MS
1025 proteins identified including 12 annexin
proteins, 32 RAB proteins, 5 RAP proteins,
aquaporin-1, aquaporin-2, aquaporin-7,
solute carrier family 12 member 1, Kidney
specific Na-K-Cl symporter and solute
carrier family 12 member 3
[350]

148
a) DC: Differential centrifugations; DG: Sucrose density gradient or otherwise stated; F: Filtration; GF: Gel filtration; UC: Ultracentrifugation.
b) APN: Aminopeptidase N; EM: Electron microscopy; FACS: Fluorescence–activated cell sorter; IEM: Immunoelectron microscopy; SNX18:
Sorting nexin-18; WB: Western blot.
c) ACPP: Prostatic Acid Phosphatase; ANXA2: Annexin A2; ANXA5: Annexin A5; AQP1: Aquaporin-1; AQP2: Aquaporin-2; ARF: ADP-
ribosylation factor; Bamacan: Basement membrane chrondroitin sulfate proteoglycan; BTG1: B-cell translocation gene 1; FLOT: Flotillin GP100:
Melanocyte protein Pmel 17; GPRC5B and GPRC5C: G-protein coupled receptor family C group 5 member B and C; MART-1: Melanoma antigen
recognized by T cells 1; NCC: Na-Cl Cotransporter; OA: osteoarthritis; PEDF: Pigment epithelium- derived factor; PSA: Prostate specific antigen;
PSMA: Prostate specific membrane antigen; RA: Rheumatoid arthritis; TGM4; Transglutaminase 4; TRP: 5,6-dihydroxyindole-2-carboxylic acid
oxidase; TSG14: Tumor necrosis factor-stimulated gene 14; TSP2: Thrombospondin-2; VPS proteins: Class E Vacuolar protein-sorting proteins.
Reproduced and updated from [74].

149
Appendix A-3 Human colon cancer LIM1863 cells
(A) Phase contrast microscopy of LIM1863 organoids Scale 20 μm. (B) A confocal
microscopy cross-section through a LIM1863 organoid highlights concentrated syntaxin 3
staining of the apical ring (red), and A33 staining at the basolateral cell periphery (green).
Scale 20 μm. (C) LIM1863 cell metabolic activity was measured using the MTT assay
following 24 h culture in RPMI containing either 5% FCS or 0.6% ITS. (D) Cell viability
of LIM1863 cells was measured using the lactate dehydrogenase (LDH) assay following 24
h culture in RPMI containing either 5% FCS or 0.6% ITS. Data represents the mean ± S.D
of three independent experiments performed in triplicate.

150
Appendix A-4 Distinguishing features of EMT
References [270, 286, 289].
Feature
observed Epithelial cells Mesenchymal cells
Morphology
• Round cobblestone growth in culture
• Strong cell-cell contact
• Elongated, spindle shaped, scattered
growth in culture
• Weak cell-cell contact
Polarity
• Well maintained apical basolateral
polarity
• Intact tight junctions (ZO-1), adherens
junctions (E-cadherin) and desmosomes
•Leading edge front-back polarity
• No tight junction, adherens junction or
desmosomes. Diminished E-cadherin and
ZO-1 expression
Motility • Little cell migration due to intact
epithelial sheets
• Increased cell migration of individual
cells
Invasiveness • Minimal, spherical cyst growth in
collagen gel
• Increased, penetrating growth into
collagen gel
Cytoskeleton
• Apical actin ring and actin stress fibers
• Cytokeratin intermediate filament
network inside cell border
• No vimentin intermediate filaments
• No apical actin ring and dissolved actin
stress fibers
• Loss of cytokeratin intermediate filaments
• Vimentin intermediate filaments across
cell body

151
Appendix A-5 Secreted extracellular modulators of EMT
Secretome analysis of MDCK cells reduced expression of cell-cell and cell-matrix
attachment proteins following oncogenic Ras-transformed EMT, possibly to enable greater
mobility. Basement membrane constituents were downregulated, and increased protease
expression was observed following transformation. Production of factors associated with
migration and invasion were also elevated in the 21D1 cell secretome. Adapted from [250].

152
Appendix A-6 Supplemental information for manuscripts included in this thesis
Chapter 2. Tauro, B. J., Greening, D. W., Mathias, R. A., Ji, H., Mathivanan, S., Scott, A.
M., and Simpson, R. J. (2012) Comparison of ultracentrifugation, density gradient
separation, and immunoaffinity capture methods for isolating human colon cancer cell line
LIM1863-derived exosomes. Methods 56, 293-304. PMID: 22285593.
Supplemental data associated with this article can be found at
doi:10.1016/j.ymeth.2012.01.002.
Supplemental Table 1 - Proteins identified in UC-Exos, DG-Exos, and IAC-Exos.
Supplemental Table 2 - Relative quantification of cancer related proteins using label-free-
based spectral counting.
Supplemental Figure 1 - Human colon cancer LIM1863 cells (Appendix A-3).
Supplemental Figure 2 - Distribution of proteins identified in UC-, DG-, and IAC-Exos
(Appendix A-7).
Supplemental Figure 3 - Plasma membrane proteins identified in the exosome datasets
(Appendix A-8).
Chapter 3 Tauro, B. J., Greening, D. W., Mathias, R. A., Mathivanan, S., Ji, H., and
Simpson, R. J. (2012) Two distinct populations of exosomes are released from LIM1863
colon carcinoma cell-derived organoids. Molecular & Cellular Proteomics 12, 587-598
PMID: 23230278.
Supplemental data associated with this article can be found at
http://www.mcponline.org/content/suppl/2013/07/11/M112.021303.DC1.
Supplemental Table 1 - A33- and EpCAM-Exos Protein Datasets.
Supplemental Table 2 - sMVs Protein Dataset.

153
Supplemental Table 3 - A33-Exos Peptide Identifications.
Supplemental Table 4 - EpCAM-Exos Peptide Identifications.
Supplemental Table 5 - sMVs Peptide Identifications.
Chapter 4 Tauro, B. J., Mathias, R. A., Greening, D. W., Gopal, S. K., Ji, H, Kapp, E. A.,
Coleman, B. M., Hill, A. F., Kusebauch, U., Hallows, J. L., Shteynberg, D., Moritz, R. L.,
Zhu, H. J., and Simpson R. J. (2013) Oncogenic H-Ras reprograms Madin-Darby canine
kidney (MDCK) cell-derived exosomal proteins during epithelial-mesenchymal transition.
Molecular & Cellular Proteomics 12, 2148-2159. PMID: 23645497.
Supplemental data associated with this article can be found at
http://www.mcponline.org/content/early/2013/05/03/mcp.M112.027086/suppl/DC1.
Supplemental Table 1- Proteins identified in exosome proteome profiling of H-Ras-induced
EMT (21D1) in MDCK cells.
Supplemental Table 2 - Peptides identified in MDCK-Exos.
Supplemental Table 3 - Peptides identified in 21D1-Exos.
Supplemental Table 4 - 139 proteins identified in MDCK- and 21D1-Exos not reported in
ExoCarta database.
Supplemental Figure 1 - 21D1 cells require their own culture medium to maintain a
mesenchymal phenotype (Appendix A-9).
Supplemental Figure 2 - Exosome characterization following OptiPrep™ density gradient
separation (Appendix A-10).
Supplemental Data - Experimental procedures for replicate GeLC-MS/MS analysis of
MDCK- and 21D1-Exos (Appendix A-11).

154
Appendix A-7 Distribution of proteins identified in UC-, DG-, and IAC-Exos
A three-way Venn diagram depicting number of proteins commonly observed in all three
datasets (265), and those that are unique to each purification strategy; ultracentrifugation
(UC-Exos), density gradient centrifugation (DG-Exos), and immunoaffinity capture (IAC-
Exos).

155
Appendix A-8 Plasma membrane proteins identified in the exosome datasets
Proteins were annotated as plasma membrane based on HPRD classification (GO:
0005886). Proteins containing one or more transmembrane domains were predicted using
TMHMM (v2.0). IAC-Exos contained the highest proportion of proteins annotated as
plasma membrane (32.1%), and predicted to contain at least one TM spanning domain
(19.8%).
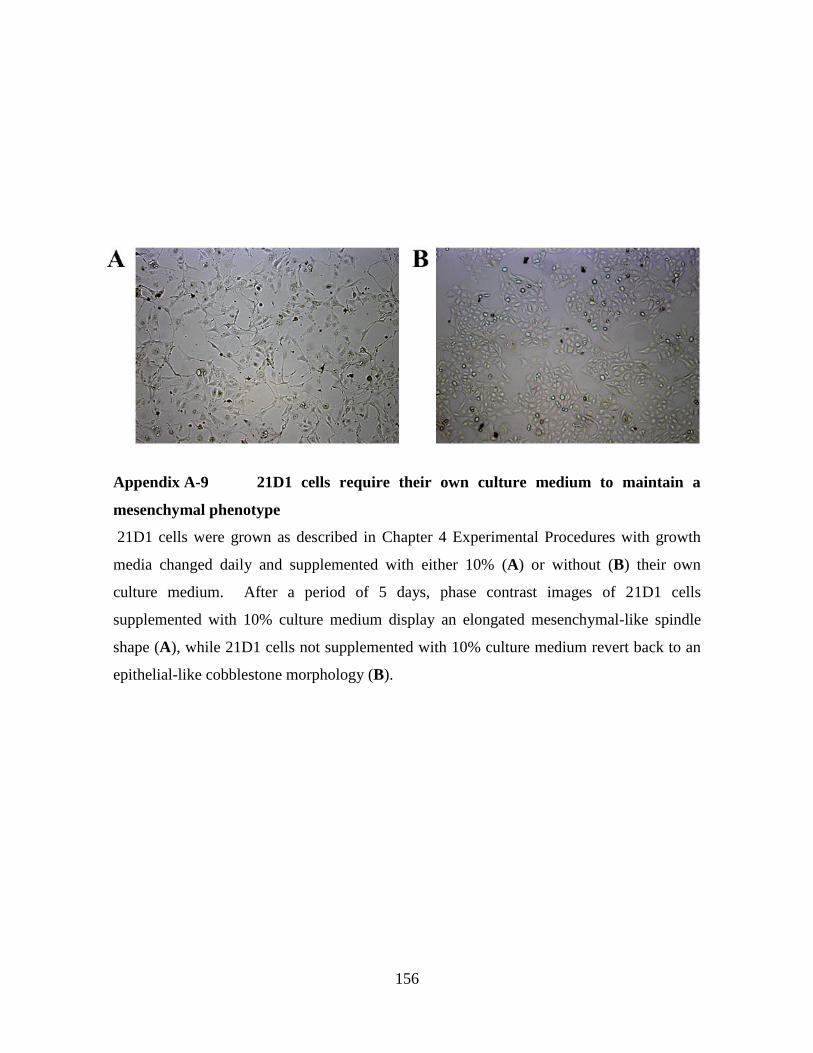
156
Appendix A-9 21D1 cells require their own culture medium to maintain a
mesenchymal phenotype
21D1 cells were grown as described in Chapter 4 Experimental Procedures with growth
media changed daily and supplemented with either 10% (A) or without (B) their own
culture medium. After a period of 5 days, phase contrast images of 21D1 cells
supplemented with 10% culture medium display an elongated mesenchymal-like spindle
shape (A), while 21D1 cells not supplemented with 10% culture medium revert back to an
epithelial-like cobblestone morphology (B).

157
Appendix A-10 Exosome characterization following OptiPrep™ density gradient
separation
Concentrated culture medium from MDCK and 21D1 cells was isolated as described in
Chapter 4 Experimental Procedures (Cell culture and CCM preparation) and separated
using OptiPrep™ density gradient. Protein fractions (1.01-1.30 g/mL) from MDCK (A) and
21D1 (B) were stained with SYPRO® Ruby protein stain. Western blot analysis of MDCK
(C) and 21D1 (D) OptiPrep™ density gradient fractions (10 µg) revealed the exosomal
marker Alix predominantly detected in fraction 1.09 g/mL. This fraction was selected for
proteomic analysis.

158
Appendix A-11 Experimental procedures for replicate GeLC-MS/MS analysis of
MDCK- and 21D1-Exos
MDCK- and 21D1-Exos samples (20 μg) were lysed in SDS sample buffer, and proteins
separated by SDS-PAGE and visualized by Imperial™ Protein Stain (Thermo Fisher
Scientific), according to manufacturer’s instructions. Gel lanes were cut into 20 × 2 mm
bands using a GridCutter (The Gel Company, San Francisco, CA) and individual bands
subjected to automated in-gel reduction, alkylation and trypsinization using a Tecan EVO
liquid handler robotic system. Briefly, gel bands were reduced with 10 mM DTT
(Calbiochem, San Diego, USA) for 20 min, washed and alkylated for 30 min with 50 mM
iodoacetic acid (Fluka, St. Louis, USA), and digested with 100 ng trypsin (Promega
Sequencing Grade Product V5111X, Wisconsin, USA) for 8 h at 35 °C with automated
agitation. Tryptic peptides were extracted with 3 50 μL 50% (v/v) acetonitrile, 50 mM
ammonium bicarbonate, concentrated to ~10 μL by centrifugal lyophilisation, pooled (2
bands pooled as 1 fraction) and analyzed by LC-MS/MS. RP-HPLC was performed on a
Dr. Maisch 3 μm ReproSil-Pur C18 AQ (Dr Maisch GmbH, Germany) 150 × 0.075-mm-
internal diameter reversed phase column with an integrated KASIL Frit using an Agilent
1100 nanoHPLC coupled online to an LTQ-Orbitrap mass spectrometer equipped with a
nanoelectrospray ion source (Thermo Fisher Scientific, San Jose, USA). The column was
developed with a linear 60 min gradient with a flow rate of 0.3 μL/min from 0-60% solvent
B, 60-80% solvent B in 2 min and held for 10 min and then returned to 2% solvent B for
15 min equilibration where solvent A was 0.1% (v/v) aqueous formic acid and solvent B
was 0.1% (v/v) aqueous formic acid/100% acetonitrile. Survey MS scans were acquired
with the resolution set to a value of 30,000. Real-time recalibration was performed using a
background ion from ambient air in the C-trap. Up to 10 selected target ions were
fragmented and then were dynamically excluded from further analysis for 120 secs from
sample analysis [351]. Raw mass spectrometry data is deposited in the PeptideAtlas and
can be accessed at http://www.peptideatlas.org/PASS/PASS00225 [352-354].

159

Minerva Access is the Institutional Repository of The University of Melbourne
Author/s:
Tauro, Bow Jesse
Title:
The isolation and characterization of exosomes
Date:
2013
Citation:
Tauro, B. J. (2013). The isolation and characterization of exosomes. PhD thesis, Ludwig
Institute for Cancer Research Ltd. (Parkville Branch), La Trobe Institute for Molecular
Science, La Trobe University & Department of Biochemistry and Molecular Biology, The
University of Melbourne.
Persistent Link:
http://hdl.handle.net/11343/38509
File Description:
Thesis text