isotope effects in nucleophilic substitution reactions. vii. the effect of ion pairing on the...
TRANSCRIPT

Isotope effects in nucleophilic substitution reactions. VII. The effect of ion pairing on the substituent effects on S,2 transition state structure
ZHU-GEN LAI AND KENNETH CHARLES WESTAWAY' Chemistry Department, Laurerltian lir~iversity, Sudbury, Ont., Carlada P3E 2C6
Received October 6, 19872
ZHU-GEN LAI and KENNETH CHARLES WESTAWAY. Can. J . Chem. 67, 21 (1989). The secondary a-deuterium kinetic isotope effects and substituent effect found in the SN2 reactions between a series of
para-substituted sodium thiophenoxides and benzyldimethylphenylammonium ion are significantly larger when the reacting nucleophile is a free ion than when it is a solvent-separated ion pair complex. Tighter transition states are found when a poorer nucleophile is used in both the free ion and ion pair reactions. Also, the transition states for all but one substituent are tighter for the reactions with the solvent-separated ion pair complex than with the free ion. Hammett p values found by changing the substi- tuent on the nucleophile do not appear to be useful for determining the length of the sulfur-a-carbon bond in the ion pair and free ion transition states.
Key words: Isotope effects, ion pairing, nucleophilic substitution, SN2 reactions, transition states.
ZHU-GEN LAI et KENNETH CHARLES WESTAWAY. Can. J. Chem. 67, 21 (1989). On a trouvt que, lorsque le nucltophile est un ion libre, les effets isotopiques cinttiques secondaires des atomes deuterium
en a ainsi que I'effet de substituant qui ont Ctt observes lors de reactions SN2 d'une strie de thioph6nolates de sodium substituts en pa ra avec l'ion benzyldimtthylphtnylammonium sont beaucoup plus importants que lorsque ce nuclCophile est une paire d'ions complexe stparte par le solvant. On a trouvt que les ttats de transition sont beaucoup plus compacts lorsqu'on utilise un mauvais nuclCophile tant dans des reactions d'ions libres que dans des rtactions de paires d'ions. Par comparaison avec les rtactions se produisant avec I'ion libre, les ttats de transition de tous les substituants, 2 I'exception d'un, sont plus compacts pour les reactions avec la paire d'ions complexe stparte par le solvant. I1 ne semble pas que les valeurs p de Hammett que l'on trouve en faisant varier la nature du substituant attach6 au nucleophile soient utiles pour determiner la longueur de la liaison entre le soufre et le carbone en a , tant dans la paire d'ions que dans les etats de transition de l'ion libre.
Mots cl6s : Effets isotopiques, couplage ionique, substitution nucltophile, rtactions SN2, ttats de transition. [Traduit par la revue]
Introduction Recent work by Westaway and Lai (1) has shown that the
form of the reacting anion has a significant effect on the magnitude of the secondary a-deuterium kinetic isotope effect and transition state structure of an SN2 reaction. For example, changing the form of the reacting anion from a solvent- separated ion pair complex (ref. 1, vide infra) to a free ion in the SN2 reaction between n-butyl chloride and sodium thiophenox- ide in DMSO at 20°C, eq. [I], changes the secondary a-deu- terium kinetic isotope effect from 1.034 t. 0.003 to 1.118 +. 0.009 (1). In fact, this change in the form of the reacting nucleo- phile seems to be a general phenomenon because the secondary a-deuterium kinetic isotope effect changes in at least three other SN2 reactions when the form of the nucleophile is altered ( I ) . ~ These results clearly demonstrate that the magnitude of the secondary a-deuterium kinetic isotope effect and transition state structure can be strongly dependent on the form of the reacting anion in an SN2 reaction. Our interest in the substituent effects on the structure of S N ~ transitions states (3-5) and the many cases where the substituent effects found experimentally do not show a consistent pattern (6) made it important to determine how the form of the reacting anion in an SN2 reaction affects the substituent effects on transition state structure.
'Author to whom correspondence may be addressed. 2Revision received September 2, 1988. 3 ~ l s o , K. C. Westaway and Z.-g. Lai. Unpublished results.
Results and discussion The effect of ion pairing (the form of the reacting anion) on the
substituent effect on transition state structure has been assessed by measuring the secondary a-deuterium kinetic isotope effects for the SN2 reactions of a series of para-substituted sodium thiophenoxides and benzyldimethylphenylammonium ion in methanol at 20°C, eq. [ 2 ] .
[2] ( ~ a ' ) - s - @ Z + c~H~cL~&(cH, )~c~H, NO,- +
L = H , D Z = CH30, CH3, H, and Cl
Obviously, the first requirement for this study of the effect of ion pairing on substituent effects was to demonstrate that the form of the reacting thiophenoxide ion could be changed from a free ion to a solvent-separated ion pair complex under the reaction conditions. Several results indicate that sodium thio- phenoxide exists as ion pairs in methanol at high concentrations (1). First, the slope of the conductance versus concentration plot decreases sharply at a sodium thiophenoxide concentration of approximately 2 X M. In addition, there is a rapid change in the uv spectrum when the sodium thiophenoxide concentra- tion is 1 X M. Finally, the addition of 15-crown-5 ether causes the same changes as dilution in the uv spectrum of sodium thiophenoxide solutions.
The cation independent A,,, of the uv spectra of lithium, sodium, and cesium thiophenoxide solutions, at concentrations where the salts exist as ion pairs in methanol, indicates that the nucleophilic reactant is a solvent-separated ion pair (7). Finally, the extremely sharp change in the uv spectrum and in the
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
Uni
vers
ity o
f W
ater
loo
on 0
6/01
/14
For
pers
onal
use
onl
y.

22 CAN. J. CHEM. VOL. 67. 1989
TABLE 1. The rate constants and secondary a-deuterium kinetic isotope effects for the SN2 reaction between benzyldimethylphenylammonium ion and a series of para-substituted thiophenoxide ions in methanol
at 20°C
FIG. 1. The ultraviolet spectrum of a 1.54 x M solution of the solvent-separated sodium thiophenoxide ion pair complex (-) and the free thiophenoxide ion (---) in methanol at 20°C. The free thiophenoxide ion was generated by adding 15-crown-5 ether.
secondary a-deuterium kinetic isotope effects with concentra- tion (1) indicate that the solvent-separated ion pair must exist as a complex consisting of several solvent-separated sodium thiophenoxide ion pairs, eq. [3].
All of the experimental findings are consistent with such an equilibrium. First, the form of the reacting anion would only change rapidly with concentration if this equilibrium existed. As a result, this equilibrium will produce the rapid changes in the uv spectra and in the secondary a-deuterium kinetic isotope effects with concentration that are observed. The conductivity data are also consistent with the formation of the ion pair complex because the conductance of the ion pair complex is lower than that of the free ions (8). Finally, when 15-crown-5 ether is present, the secondary a-deuterium kinetic isotope effect for the SN2 reaction between n-butyl chloride and sodium thiophenoxide at concentrations where the salt exists as a solvent-separated ion pair complex is identical to those found in the free ion reactions. This occurs because the 15-crown-5 ether complexes the sodium ions (9), preventing the formation of the ion pair complex.
The above experiments indicate that the sodium thiophenox- ide exists as a solvent-separated ion pair complex at concentra- tions greater than 1 X loF4 M in methanol at 20°C. However,
para kH x lo3 k, x lo3 Substituent (L mol-I S-I) (L mol-' s-') ( k ~ / k ~ ) a
T h e errors are the standard deviation from at least three different kinetic mns. ?he error is l /kD[(AkH)2 + ( k H / k ~ ) ~ X ( ~ k , ) ~ ] ' / ~ where AkH and Ak, are
the standard deviations for the rate constants for the undeuterated and deuterated substrates, respectively ( 4 ) .
TABLE 2. The rate constants and secondary a-deuterium kinetic isotope effects for the SN2 reaction between benzyldimethylphenylammonium ion and a series of solvent-separated para-substituted sodium thiophen-
oxide ion pair complexes in methanol at 20°C
para kH x lo3 kD x lo3 Substituent (L mol-' S-') (L mol-' S-') ( k ~ I k ~ ) a
T h e errors are the standard deviation from at least three different kinetic runs. b ~ h e error is l / k D [ ( A k H ) 2 + ( k ~ / k , ) ~ x ( ~ k , ) ~ ] ' / ~ where A ~ H and A k , are
the standard deviations for the rate constants for the undeuterated and deuterated substrates, respectively ( 4 ) .
the uv spectra in Fig. 1 show that the addition of 15-crown-5 ether clearly changes the form of the thiophenoxide ion. When the sodium thiophenoxide concentration is 3 x M, the concentration used throughout this study, the uv spectrum shows a strong maximum at 267 nm, where the solvent- separated sodium thiophenoxide ion pair complex absorbs (I). When 15-crown-5 ether is added, the absorption at 267 nm almost completely disappears and a new absorption at 235 nm, where the free thiophenoxide anion absorbs, is found (1). Thus, the form of the reacting thiophenoxide ion can be changed from the solvent-separated ion pair complex to the free anion by adding 15-crown-5 ether, and a study of the effect of ion pairing on the substituent effects in SN2 reactions is f e a ~ i b l e . ~
The secondary a-deuterium kinetic isotope effects found for the SN2 reactions between benzyldimethylphenylammonium ion and a series of para-substituted free thiophenoxide ions and solvent-separated sodium thiophenoxide ion pair complexes, Tables 1 and 2, differ in several ways. First, the substituent effects on the secondary a-deuterium kinetic isotope effects cover a much larger range in the free ion reactions, i.e., the isotope effects range from 1.27 1 to 1.12 1 when the nucleophile is a free ion but only from 1.216 to 1.150 when the solvent- separated sodium thiophenoxide ion-pair complex is the react- ing species. This observation is important because it illustrates
4Neither adding benzyldimethylphenylammonium nitrate nor chang- ing the para substituent on the thiophenoxide ion affects the concen- tration at which the solvent-separated ion pair complexes begin to form in methanol.
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
Uni
vers
ity o
f W
ater
loo
on 0
6/01
/14
For
pers
onal
use
onl
y.

LA1 AND WESTAWAY
TABLE 3. The C N D 0 / 2 calculated charge densities on the sulfur atom of the partially solvated thiophenoxide ion and sodium thiophenoxide ion pair in methanol
para Substituent
Charge density on Charge density on the sulfur atom of the sulfur atom of
HOCHl HOCH,
C H 3 0 CH3
H C1
Change in the charge density on sulfur
Change in the ( k ~ / k ~ ) a
that the substituent effect on SN2 transition state structure is strongly dependent on the form of the ionic nucleophile.
One possible explanation for the different secondary a-deu- terium kinetic isotope effects in the free ion and ion pair complex reactions is that the sodium ion alters the reduced mass of the nucleophile sufficiently to affect the isotope effect. This explanation seems unlikely for several reasons. First, the ion pair is a solvent-separated ion pair so the change in mass does not occur near the nucleophilic atom, i.e., the sodium is replacing a methanol molecule in the second or perhaps third solvent shell around the free ion and should have little or no effect on the reduced mass of the nucleophile. In any case, the mass of the methanol molecule and a sodium ion are similar. Also, theoretical calculations (10) indicate that the secondary a-deuterium kinetic isotope effect in the SN2 reaction between benzyl chloride and an alkoxide ion only changes by 0.0002 when the developing chloride ion is solvated with three hydrogen- bonding solvent molecules. Since this change in the reduced mass of the chloride ion is much larger than that associated with changing a solvent molecule (methanol) to a sodium ion, it is unlikely that the mass of the sodium ion is responsible for the difference in the isotope effect.
A more plausible explanation for the change in the secondary a-deuterium kinetic isotope effect is that the sodium ion of the solvent-separated ion pair complex reduces the charge on the sulfur atom of the thiophenoxide ion. If the charge on the sulfur of the solvent-separated ion pair complex were indeed smaller than that on the sulfur of the free ion, one would expect that the nucleophile would have to approach closer to the a carbon to distort the a-carbon-chlorine bond and cause reaction. This would result in a tighter transition state and the smaller secondary a-deuterium kinetic isotope effect that is observed for all but one of the ion pair complex reactions (vide infra).
The sodium ion could also reduce the magnitude of the substituent effect resulting from a change in the para substituent on the nucleophile, by reducing the charge on the sulfur atom of the thiophenoxide ion. If the charge on the sulfur of the solvent-separated ion pair complex were smaller than that on the sulfur of the free ion, a change in the para substituent might have a smaller effect on the charge on the sulfur atom of the sodium thiophenoxide ion pair. If this occurred, the nucleophi- licity of the thiophenoxide ion would not be altered as much by a
change in substituent, and the change in transition state structure with para substituent (secondary a-deuterium kinetic isotope effect (substituent effect)) should also be smaller in the ion pair complex reactions.
This rationalization is supported by the results from CND0/2 calculations of the charge on the sulfur atom of the partially solvated free thiophenoxide ion and the contact sodium thio- phenoxide ion pair, Table 3. The CND0/2 calculations were carried out on the thiophenoxide ion solvated by two methanol molecules and on the contact ion pail5 solvated by only one methanol molecule because one of the lone pairs of electrons on the sulfur is involved in bond formation in the SN2 transition state and cannot be solvated. As expected, the calculations indicate that the negative charge on the sulfur atom of the nucleophile is much smaller when the positive sodium ion is present. Although the substituent changes the amount of negative charge on the sulfur atom by the same relative amount, i.e., changing a hydrogen to a para-chloro substituent reduces the charge on the sulfur atom by the same percentage in both the free ion and the ion pair, it is important to note that the absolute change in charge on the sulfur ion with substituent is greater for the free ion. Since the substituent effect on the charge on the nucleophile is greater for the free ion than for the ion pair complex, the change in the nucleophilicity should be greater in the free ion reactions. As a result, one would expect the substituent effect on the secondary a-deuterium kinetic isotope effect (transition state structure) to be greater for the free ion than for the ion pair complex reactions. This is, in fact, what is observed.
Although CND0/2 calculations predict charges in molecules very well (1 l ) , the calculated charges on the sulfur atom of the free ion and ion pair complex transition states are only representative of the changes in charge that occur in the actual reactions. However, the same trends in the charge on the sulfur ion with substituent were found for both the unsolvated and fully solvated ion pairs and free ions in methan01.~ Moreover, the same trends were also found when the position of the sodium ion in the ion pair was varied. Since the same trends in the charge
he calculations were not done on the solvent-separated ion pair complex because neither the structure nor the number of solvent molecules between the sodium and sulfur ions is known.
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
Uni
vers
ity o
f W
ater
loo
on 0
6/01
/14
For
pers
onal
use
onl
y.
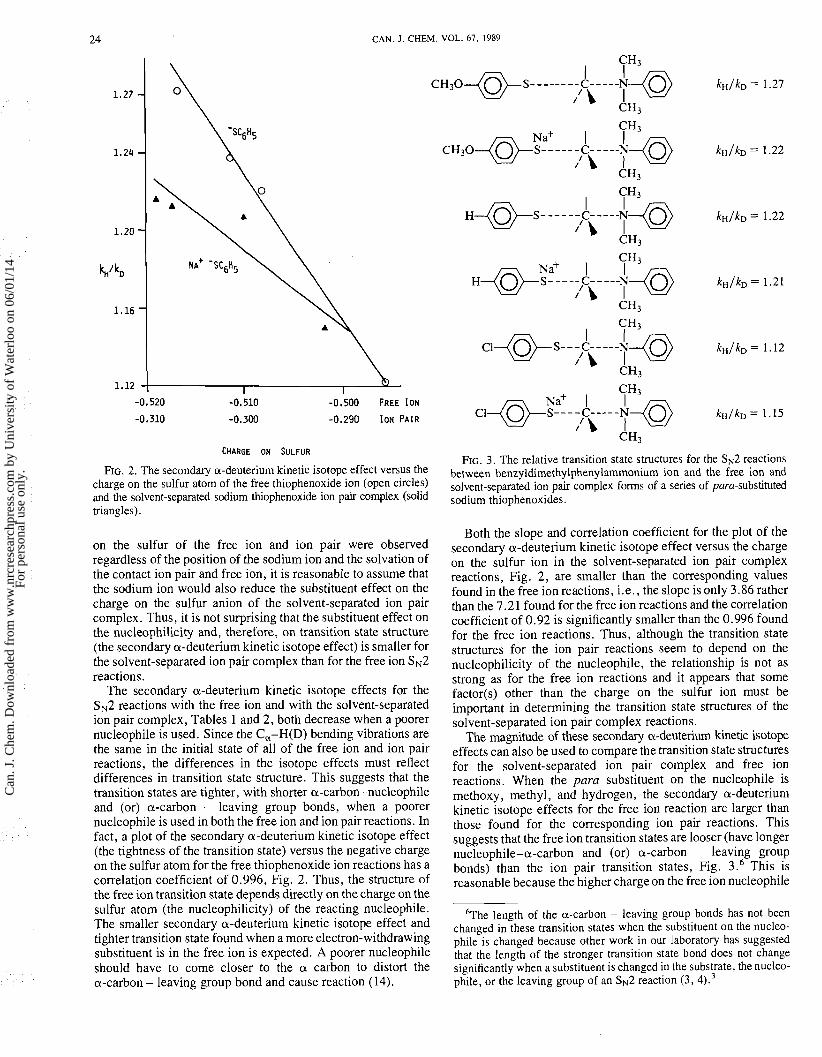
CAN. J. CHEM. VOL. 67, 1989
FIG. 2. The secondary a-deuterium kinetic isotope effect versus the charge on the sulfur atom of the free thiophenoxide ion (open circles) and the solvent-separated sodium thiophenoxide ion pair complex (solid triangles).
on the sulfur of the free ion and ion pair were observed regardless of the position of the sodium ion and the solvation of the contact ion pair and free ion, it is reasonable to assume that the sodium ion would also reduce the substituent effect on the charge on the sulfur anion of the solvent-separated ion pair complex. Thus, it is not surprising that the substituent effect on the nucleophilicity and, therefore, on transition state structure (the secondary a-deuterium kinetic isotope effect) is smaller for the solvent-separated ion pair complex than for the free ion SN2 reactions.
The secondary a-deuterium kinetic isotope effects for the SN2 reactions with the free ion and with the solvent-separated ion pair complex, Tables 1 and 2, both decrease when a poorer nucleophile is used. Since the C,-H(D) bending vibrations are the same in the initial state of all of the free ion and ion pair reactions, the differences in the isotope effects must reflect differences in transition state structure. This suggests that the transition states are tighter, with shorter a-carbon-nucleophile and (or) a-carbon - leaving group bonds, when a poorer nucleophile is used in both the free ion and ion pair reactions. In fact, a plot of the secondary a-deuterium kinetic isotope effect (the tightness of the transition state) versus the negative charge on the sulfur atom for the free thiophenoxide ion reactions has a correlation coefficient of 0.996, Fig. 2. Thus, the structure of the free ion transition state depends directly on the charge on the sulfur atom (the nucleophilicity) of the reacting nucleophile. The smaller secondary a-deuterium kinetic isotope effect and tighter transition state found when a more electron-withdrawing substituent is in the free ion is expected. A poorer nucleophile should have to come closer to the a carbon to distort the a-carbon - leaving group bond and cause reaction ( 14).
FIG. 3. The relative transition state structures for the SN2 reactions between benzyldimethylphenylammonium ion and the free ion and solvent-separated ion pair complex forms of a series of para-substituted sodium thiophenoxides.
Both the slope and correlation coefficient for the plot of the secondary a-deuterium kinetic isotope effect versus the charge on the sulfur ion in the solvent-separated ion pair complex reactions, Fig. 2, are smaller than the corresponding values found in the free ion reactions, i.e., the slope is only 3.86 rather than the 7.21 found for the free ion reactions and the correlation coefficient of 0.92 is significantly smaller than the 0.996 found for the free ion reactions. Thus, although the transition state structures for the ion pair reactions seem to depend on the nucleophilicity of the nucleophile, the relationship is not as strong as for the free ion reactions and it appears that some factor(s) other than the charge on the sulfur ion must be important in determining the transition state structures of the solvent-separated ion pair complex reactions.
The magnitude of these secondary a-deuterium kinetic isotope effects can also be used to compare the transition state structures for the solvent-separated ion pair complex and free ion reactions. When the para substituent on the nucleophile is methoxy, methyl, and hydrogen, the secondary a-deuterium kinetic isotope effects for the free ion reaction are larger than those found for the corresponding ion pair reactions. This suggests that the free ion transition states are looser (have longer nucleophile-a-carbon and (or) a-carbon - leaving group bonds) than the ion pair transition states, Fig. 3.6 This is reasonable because the higher charge on the free ion nucleophile
q h e length of the a-carbon - leaving group bonds has not been changed in these transition states when the substituent on the nucleo- phile is changed because other work in our laboratory has suggested that the length of the stronger transition state bond does not change significantly when a substituent is changed in the substrate, the nucleo- phile, or the leaving group of an SN2 reaction (3, 4).3
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
Uni
vers
ity o
f W
ater
loo
on 0
6/01
/14
For
pers
onal
use
onl
y.

LA1 AND WESTAWAY 25
TABLE 4. The rate constants and Harnmett p values for the free ion and the solvent-separated ion pair complex SN2 reactions between benzyl- dimethylphenylammonium ion and a series of para-substituted sodium
thiophenoxides in methanol at 20°C
para-Substituent kree ion X lo3 kion pair X lo3 on the nucleophile (L mol-I s- l ) ( ~ m o l - I s - I ' )
CH30 10.33 k 0.11 10.09 k 0.10 ' 3 3 6.87 ? 0.02 7.06 k 0.05
H 5.14 k 0.03 5.18 k 0.03 C1 3.66 + 0.03 3.69 -t 0.03
P -0.85 + 0.14 -0.84 -t 0.11 Correlation coefficient 0.98 0.98
would distort the a-carbon - leaving group bond and cause reaction at a greater distance from the a carbon ( 14).
Unfortunately, not all of the free ion transition states are looser than the ion pair transition states. The secondary a-deuterium kinetic isotope effect for the reaction with the para-chlorothiophenoxide ion pair complex is greater7 (1.150 2 0.009 versus 1.12 1 ? 0.014) than the isotope effect for the corresponding free ion reaction. Although other factors, such as a change in the amount of conjugation to the phenyl ring on the a carbon ( 10, 15), changes in the reaction coordinate motion ( 16, 17), tunneling (16, 17), and changes in the product composition (18), can affect the magnitude of secondary a-deuterium kinetic isotope effects, the simplest explanation is that the free ion transition state is tighter than the ion pair transition state when the nucleophile is para-chlorothiophenoxide ion, Fig. 3, and that some factor(s) other than the charge on the sulfur of the nucleophile is important in determining transition state structure.
The unexpected changes in the secondary a-deuterium kinetic isotope effect found when the para-chlorothiophenoxide ion is the nucleophile are not caused by changes l'n product composition (18) for two reasons. First, quantitative yields of the SN2 products, benzyl phenyl sulfide and N,N-dimethyl- aniline, are found in these reactions. Secondly, the isotope effects in the side reaction would be very similar to that in the major reaction. Thus, even if a small amount of the side reaction occurred, the effect on the observed secondary a-deuterium kinetic isotope effect would be very small. Thus product composition cannot be responsible for the unexpected changes in the isotope effect.
Conjugation between the a carbon and the phenyl ring, which reduces the magnitude of secondary a-deuterium kinetic isotope effects, only appears to be important when strongly electron- donating groups are on the benzene ring on the a carbon (15). Thus, changes in conjugation would not seem to be the cause of the unexpected isotope effects.
Finally, the secondary a-deuterium kinetic isotope effects found in this study are less than or equal to those found in the SN2 reactions of other quaternary ammonium salts (5, 19, 20). ~ i n ~ e large secondary a-deuterium kinetic isotope effects, significantly greater than the equilibrium isotope effect, are found when changes in the reaction coordinate motion and (or) tunneling are important in determining the magnitude of secondary a-deuterium isotope effects (16, 17), it seems unlikely that this factor is important in affecting the magnitude
'TWO different statistical tests, the Wilcoxin test and the Student's T test (12, 13) indicate that the secondary a-deuterium kinetic isotope effects for the ion pair and free ion reactions with pora-chlorothio- phenoxide ion are significantly different at the 99% confidence level.
of these isotope effects. As a result, it would appear that the more likely explanation is the simple one, i.e., that the magnitude of the isotope effects is related to the structure of the transition state. In any case, it would seem unlikely that changing a para-hydrogen to a para-chloro substituent, which only changes the rate constant by 40%, would change the reaction coordinate motion and (or) tunneling enough to cause the different behaviour of the isotope effect.
The Hammett p values, obtained by changing the para substituent on the nucleophile, were determined for both the solvent-separated ion pair complex and the free ion reactions, Table 4 , in an effort to learn more about the relative SN2 transition state structures for these reactions. The identical Hammett p values for the two types of reactions show that this technique is not useful in distinguishing between the lengths of the a-carbon-sulfur bonds in the free ion and solvent-separated ion pair complex transition states (1).
Finally, the difference between the secondary a-deuterium kinetic isotope effects for the free ion and the ion pair complex reactions is large when the nucleophile is para-methoxy- thiophenoxide ion but becomes smaller when a more electron- withdrawing substituent is on the thiophenoxide ion. In fact, the difference in the isotope effects is almost zero when the nucleophile is thiophenoxide ion and is inverse when the reactant is the para-chlorothiophenoxide ion. Although we do not understand these observations, these results are interesting because they suggest that the effect of ion pairing is largest when strongly electron-donating and electron-withdrawing sub- stituents are present.
It is interesting that the difference between the rate constants for the free ion and solvent-separated ion pair complex reactions is very small. Although we do not understand why this is so, similar differences in rate have been found in several other SN2 reactions we have studied ( I ) . ~
Conclusion This work has shown that the secondary a-deuterium kinetic
isotope effect (transition state structure) is altered significantly when the form of the reacting ionic nucleophile is altered from a free ion to a solvent-separated ion pair complex in the SN2 reactions between benzyldimethylphenylammonium ion and a series of para-substituted sodium thiophenoxides in methanol. Also, the magnitude of the substituent effect found by changing a para substituent on the ionic nucleophile is significantly larger when the nucleophile is a free ion rather than a solvent-separated ion pair complex. The transition states are tighter when a poorer nucleophile is used in both the free ion and ion pair reactions. Also, with one exception, the SN2 reaction between para- chlorothiophenoxide ion and benzyldimethylphenylammonium ion, the ion pair transition states are tighter than the free ion transition states. Finally, the Hammett p values found by changing the substituent on the nucleophile do not appear to be useful in determining the relative lengths of the a-carbon-sulfur bonds in the ion pair complex and free ion transition states.
Experimental Preparation of reagents
The preparation of the benzyldimethylphenylammonium nitrate and the para-substituted sodium thiophenoxides is described elsewhere (2, 19). The purification of the solvent is described in ref. 1. The 15-crown-5 ether was purchased from Parrish Chemical Co.
Product analysis Approximately 0.12 g of benzyldimethylphenylammonium nitrate
was reacted with 0.12 g sodium thiophenoxide in 85 mL of purified
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
Uni
vers
ity o
f W
ater
loo
on 0
6/01
/14
For
pers
onal
use
onl
y.

26 CAN. J. CHEM. VOL. 67, 1989
methanol for 5500 min (approximately 14 half-lives) at 20°C. Then, the reaction mixture was poured into 1400 mL of pH 12 water and the products were extracted once with 200 mL, twice with 150 mL, and twice with 100 mL of ether. The combined ether layers wre extracted twice with40 mL, and once with 30 mL of a 10% sulfuric acid solution. Evaporation of the ether layer under reduced pressure left a solid whose nmr spectrum was consistent with that of the expected SN2 product, benzyl phenyl sulfide. No peaks associated with the other possible product, methyl phenyl sulfide, were apparent even at a high spectrum amplitude.
Most of the water was removedfrom the combined 10% sulfuric acid solutions on a rotary evaporator. A Kjeldahl analysis (2) of the concentrate was consistent with a 98% yield of N,N-dimethylaniline (the expected SN2 product). If even a small amount of the other possible product, N-benzyl-N-methylaniline, had been present, a much lower yield of N,N-dimethylaniline would have been obtained.
An identical experiment, except that 2 mL of 15-crown-5 ether was present in the reaction mixture, gave the same results. The only substitution product visible in the nmr spectrum was benzyl phenyl sulfide, and the Kjeldahl analysis indicated the yield of N,N- dimethylaniline was 99%.
Kinetic method After all of the apparatus had been cleaned and dried at 140°C, it was
sealed while hot or cooled in a desiccator. Six 125-mL Erlenmeyer flasks with ground glass stoppers were filled with extra dry nitrogen and weighed accurately. Then, these flasks, samples of the appropriate para-substituted sodium thiophenoxide and benzyldimethylphenylam- monium nitrate, a crude balance, and some spatulas were placed in an I R ~ glove bag that was sealed, filled, evacuated, and refilled with extra-dry nitrogen. About 520 mg (3.9 X mol) of the para- substituted sodium thiophenoxide was placed in three of the Erlen- meyer flasks and approximately 59 mg (2.2 X mol) of benzyldi- methylphenylammonium nitrate was placed in the other Erlenmeyer flasks. These flasks were removed from the glove bag and the amount of each salt determined by weighing them accurately.
The Erlenmeyers containing the para-substituted sodium thiophen- oxide, the Erlenmeyers containing the quaternary ammonium salt, adaptors fitted with a serum cap, a bottle of purified methanol (vide supra), a graduated cylinder, three glass-stoppered 50-mL Erlenmeyer flasks for each para-substituted sodium thiophenoxide sample, three 1-mL and one 75-mL pipettes, and arubber pipetting bulb, were placed in the glove bag under a nitrogen atmosphere (vide supra). Approxi- mately 40 mL of methanol was added to each Erlenmeyer containing the para-substituted sodium thiophenoxide and 75 mL of methanol was pipetted into the Erlenmeyers containing the quaternary ammonium salt. Finally, three 1-mL samples of each sodium thiophenoxide solution were transferred to the three 50-mL Erlenmeyers.
The reaction flasks containing the quaternary ammonium salt and the sodium thiophenoxide solutions were temperature equilibrated at 20.00 * 0.02"C for at least 45 min. The purity of the thiophenoxide salt was determined by titrating the samples in the 50-mL Erlenmeyers with standard mercury I1 nitrate (2). Finally, the kinetic run was begun by
injecting 5 mL of the para-substituted sodium thiophenoxide solution into the reaction vessel with a 5-mL Hamilton air-tight syringe.
The kinetic data were obtained by withdrawing 5-mL aliquots of the reaction mixture with a 5-mL Hamilton gas-tight syringe fitted with a 6-in. needle. Each sample was quenched by adding it to 15 mL of a solution prepared by diluting 2.3 mL of a 25% solution of nitric acid to 1 L with methanol. The unreacted thiophenoxide ion was determined in a titration using a standard mercury I1 nitrate (2). The rate constants were calculated using standard methods (3).
Acknowledgement The authors gratefully acknowledge the financial support
provided by the Natural Sciences and Engineering Research Council of Canada.
1. K. C. WESTAWAY and Z.-G. LAI. Can. J . Chem. 66, 1263 (1988). 2. K. C. WESTAWAY and R. A. POIRIER. Can. J. Chem. 53, 3216,
(1975). 3. K. C. WESTAWAY and S. F. ALI. Can. J. Chem. 57,1354 (1979). 4. K. C. WESTAWAY and Z. WASZCZYLO. Can. J. Chem. 60, 2500
(1 982). 5. H. A. JOLY and K. C. WESTAWAY. Can. J. Chem. 64, 1206
(1986). 6. K. C. WESTAWAY. In Isotopes in organic chemistry. Vol. 7.
Edited by E. Buncel and C. C. Lee. Elsevier, New York, NY. 1987. p. 275.
7. E. BUNCEL, B. C. MENON, and J. P. COLPA. Can. J. Chem. 57, 999 (1979).
8. R. J. WILLIAMS, J. N. PHILLIPS, and K. J. MYSELS. Trans. Faraday Soc. 51, 728 (1955).
9. J. J . CHRISTENSEN, D. J. EATOUGH, and R. M. IZATT. Chem. Rev. 74, 351 (1974).
10. D. M. BRUBAKER. Ph. D. dissertation. University of Arkansas, Fayetteville, AR. 1978.
11. J. A. POPLE and D. L. BEVERIDGE. Approximate molecular orbital theory. McGraw-Hill, New York, NY. 1970.
12. R. D. G. STEEL and J . H. TORRIE. Principles and procedures of statistics with special reference to biological sciences. McGraw- Hill, Toronto, Ont. 1960. pp. 404 and 470.
13. C. WHITE. Biometrics, 8, 33 (1950). 14. J. E. GORDON. The organic chemistry of electrolyte solutions. J.
Wiley and Sons, Toronto, Ont. 1975. p. 372. 15. Z. WASZCZYLO and K. C. WESTAWAY. Tetrahedron Lett. 23, 143
(1982). 16. W. P. HUSKEY and R. L. SCHOWEN. J. Am. Chem. Soc. 105,
5704 (1983). 17. W. H. SAUNDERS, JR. J. Am. Chem. Soc. 106, 2223 (1984). 18. D. J. MCLENNAN and P. M. W. GILL. Isr. J. Chem. 26, 378
(1985). 19. K. C. WESTAWAY and S. F. ALI. Can. J. Chem. 57, 1089 (1979). 20. E. C. F. KO and K. T. LEFFEK. Can. J. Chem. 49, 129 (1971).
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
Uni
vers
ity o
f W
ater
loo
on 0
6/01
/14
For
pers
onal
use
onl
y.