itp and ttp in gp practice - igazi foundation · – unnecessary in patients with typical features...
TRANSCRIPT

Willem van Schalkwyk Haematology Pathologist
ITP and TTP in GP practice

Immune thrombocytopenia (ITP)
• Acquired immune-mediated disorder. • Definition:
• Platelet count <100 x 109/L. • Absence of obvious initiation or underlying cause.
Blood, 2010;115: 168-186
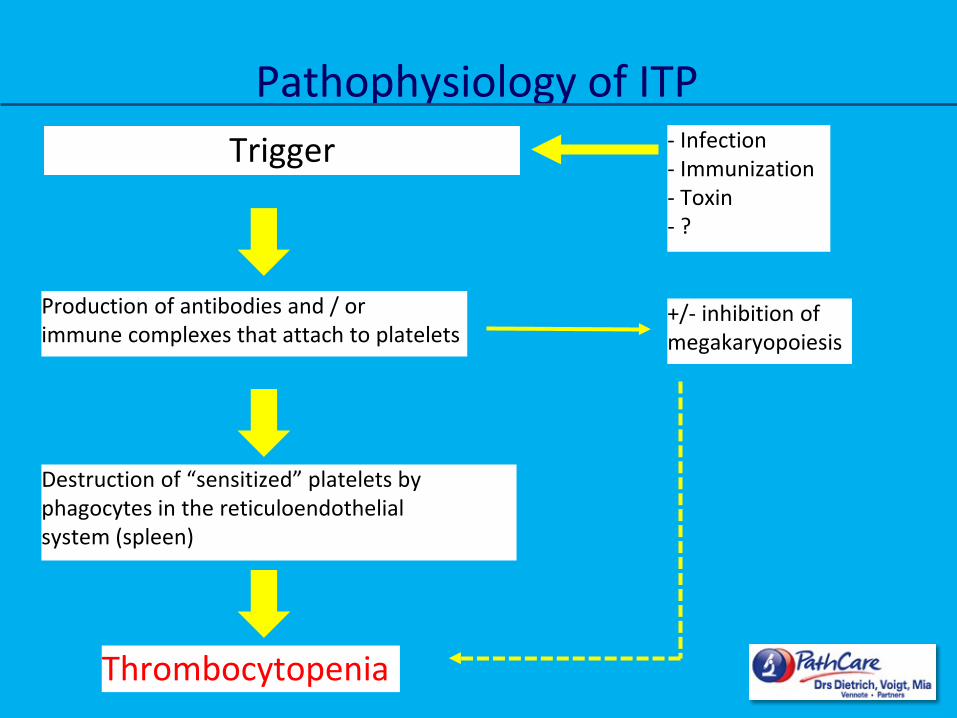
Trigger
Production of antibodies and / or immune complexes that attach to platelets
- Infection - Immunization - Toxin - ?
Destruction of “sensitized” platelets by phagocytes in the reticuloendothelial system (spleen)
+/- inhibition of megakaryopoiesis
Thrombocytopenia
Pathophysiology of ITP

• Primary ITP: – Absence of other causes/disorders that may be associated with ITP.
• Secondary ITP: Underlying disease or to drug exposure.
– HIV, HCV, H Pylori – Auto-immune/immunodeficiency disorders – Lymphoproliferative neoplasms – Recent vaccination – Post transfusion purpura – Drugs
Blood, 2009;113: 2386-2393
Classification


• Usually follows a chronic course. • +/-15% Of patients remit within 1 year. • Muco-cutaneous bleeding most common.
– Petechiae, purpura, epistaxis and gum bleeding.
• Rarely GIT bleeding and intracranial bleeding. • Risk of major bleed increases with:
– Platelet count < 30 x 109/L. – Age >60 years. – Other comorbidities.
Clinical presentation in adults

• No “gold standard” test. • Presumptive diagnosis is made when history, physical examination,
FBC and smear do not suggest other etiologies for the thrombocytopenia.
• Distinction between primary and secondary ITP.
Diagnostic work-up

1. Necessary evaluation: – History:
• Isolated bleeding symptoms consistent with thrombocytopenia without constitutional symptoms. • Review medication usage.
– Physical examination: • Bleeding symptoms in the absence of hepatosplenomegaly, lymphadenopathy, or stigmata of
congenital conditions. – FBC:
• Isolated thrombocytopenia. • Anaemia only if due to significant bleeding. • Normal red cell indices, white cell and differential counts.
– Peripheral blood smear: • Platelets normal to large in size. • Red and white cell morphology normal.
Diagnostic work-up

2. Additional investigations: – Direct coombs test – HIV, HCV – H Pylori – Antinuclear antibodies – Immunoglobulin levels (IgM, IgG and IgA) in children. – Pregnancy test in woman of childbearing age.
3. Bone marrow evaluation: – Unnecessary in patients with typical features of ITP. – Presence of abnormalities in the history, physical examination, or the FBC and smear
should be further investigated before a diagnosis of ITP is made.
Diagnostic work-up

• Therapeutic goals of initial therapy: – Rapidly obtaining a safe platelet count. – Prevent or stop haemorrhage. – To ensure an acceptable quality of life. – Minimize treatment-related toxicity.
• Assessment of disease status:
– Consider treatment for patients with platelet count <30 x 109/L. – Severity of bleeding? – Additional risk factors for bleeding (antithrombotic agents or high risk occupation). – Is a surgical procedure anticipated?
Blood (2010), vol 115, no 2: 168-186
Management in adult patients

• First-line therapy:
Blood (2010), vol 115, no 2: 168-186
Management in adult patients

• Emergency treatments: – Patients needing surgical procedures. – High-risk of bleeding. – Active CNS or GIT haemorrhage.
Combining corticosteroids with IVIg. Platelet transfusions +/- IVIg. Antifibrinolytics (tranexamic acid). Emergency splenectomy.
Management in adult patients

• Durable response in 10 – 30% of patients after first line therapy. • Second line treatment:
– Goal: to attain a sustained increase of the platelet count that is considered hemostatic for the individual patient.
• Splenectomy • Rituximab • Thrombopoietin receptor agonists
American Society of Hematology Educationbook 2013: 276-282
Management in adult patients

• Peak age of presentation between 5 and 6 years. • 50-60% have a preceding febrile illness:
– Rubella – Varicella – Mumps – Rubeola – EBV – Immunization with measles-mumps-rubella vaccine.
• Bleeding manifestations vary from little/none to purpura, petechiae, epistaxis.
• Clinically significant bleeding seen in only 3% of children. • Spontaneous resolution within 6 months (often within 6 weeks) in the
majority of children.
Clinical presentation and natural history of ITP in children

• To treat or not to treat? • To perform a bone marrow aspirate or not? • To hospitalize or not?
Management of ITP in children

• “Watch and wait” policy: – No significant bleeding symptoms. – No abnormalities on peripheral smear or physical examination. – Parents advised to watch for signs of bleeding and contact details of clinician. – Weekly outpatient visits.
• Hospitalization:
– Reserved for those with clinically significant bleeding.
Management of ITP in children

• Beware of children with: – < 6 Months or age and adolescents. – History of systemic symptoms (chronic fevers, bone pain). – Abnormalities on the initial FBC that are not early explained (neutropenia, raised MCV). – Clinically significant hepatosplenomegaly / adenopathy. – Failure to respond to standard “front-line” ITP therapies.
Atypical features of childhood ITP
• Beware of children with: – < 6 Months or age and adolescents. – History of systemic symptoms (chronic fevers, bone pain). – Abnormalities on the initial FBC that are not early explained (neutropenia, raised MCV). – Clinically significant hepatosplenomegaly / adenopathy. – Failure to respond to standard “front-line” ITP therapies.
Consider bone marrow biopsy
Atypical features of childhood ITP

• Initiate treatment only for clinically significant bleeding. • First-line treatment options to raise platelet count:
– Corticosteroids: short courses. – IVIg: if a more rapid increase in platelet count is required. – Anti-D : if corticosteroids contra-indicated.
• Goal: elevate platelet count to a level where risk of severe bleeding is minimized.
Management of ITP in children

• <10% Of cases will present with persistent severe thrombocytopenia after 6 months.
• Second line treatment options for children with persistent or chronic ITP: – Dexamethasone – High-dose methylprednisolone – Splenectomy – Rituximab
Management of ITP in children


• Aggressive form of thrombotic micro-angiopathy. • Deficiency of vWF cleavage protein ADAMTS13. • Multi-organ dysfunction as a consequence of widespread microvascular
ischemia.
• Important diagnosis to make: – Untreated mortality 90% – Half of deaths occur within 24hrs from presentation.
Thrombotic Thrombocytopenic Purpura (TTP)

Pathophysiology of TTP

Pathophysiology of TTP

Pathophysiology of TTP

Pathophysiology of TTP
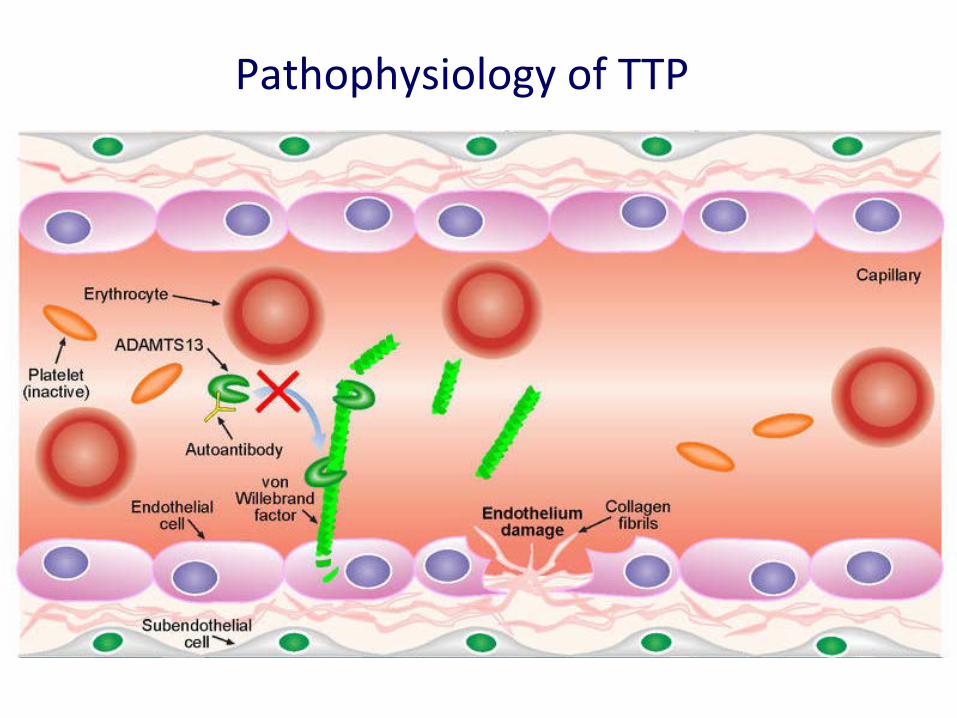
Pathophysiology of TTP

Pathophysiology of TTP

Pathophysiology of TTP

Deficiency in ADAMTS 13
Congenital Acquired
(auto-antibody
mediated) Secondary Idiopathic
Infections: HIV Drugs: Quinine, Ticlopidine, Clopidogrel, Simvastatin,
Trimethoprim Chemotherapy: Cisplatin, Mitomycin C, Cyclosporin Malignancy Connective tissue disorders: SLE. Pregnancy Bone marrow transplant Cardiac surgery
Classification of TTP

• Fever • Neurological symptoms (confusion, visual disturbance, seizures, focal
neurological sings and coma). • Renal dysfunction • Thrombocytopenia • Red cell fragmentation
Clinical presentation

• Confirmation of diagnosis of TTP: • FBC: and smear
– Marked anaemia and thrombocytopenia – Red cell fragments
• Other investigations: – Reticulocyte count, LDH, haptoglobin, unconjugated bilirubin. – DIC screen normal – U&E, serum creatinine
• Decrease in ADAMTS13 enzyme activity (<10%). • Detection of auto-antibodies against ADAMTS13.
Diagnostic work-up


• Identify secondary causes: – HIV, hep B &C serology – Pregnancy test – Auto-immune screen (ANA, RF, Lupus Anticoagulant) – Imaging and investigations to exclude malignancy as directed by clinical history and
examination.
Diagnostic work-up

• Differential diagnosis: – DIC – Pregnancy-associated: Eclampsia, HELLP – Malignant hypertention – Haemolytic uraemic syndrome – Vasculitis – Infections: meningococcus – Catastrophic antiphospholipid syndrome
Diagnostic work-up

• Patients clinically unstable with rapid deterioration. • Resuscitation and stabilization of critical organ dysfunction. • Plasma exchange / plasma infusion most important acute intervention.
Initiate without delay in any patient with suspected TTP.
Management of TTP

• Plasma exchange: – Removal of autoantibody. – Supplementation of ADAMTS13 in exchange plasma. – Replacement fluid: cryoprecipitate-poor plasma, FFP or solvent/detergent-treated pooled
plasma.
• Immunosuppression: – Corticosteroids:
• Methylprednisolone (1g/day for three days) • Oral Prednisolone (1mg/kg/day)
– Rituximab
• Plasma infusion: – Temporary measure if delay in plasma exchange.
Management of TTP

• May be the initial presenting features of HIV disease. • Virological suppression important: ADAMTS13 activity increases as CD4
count recovers and HIV viral load falls. • Novitzky et al (2004):
– Response to plasma infusion significantly better and faster than HIV negative cohort. – Relevant for patients who present to regional hospital that may not have apheresis
facilities. – Prednisone 1mg/kg plus cryoprecipitate-poor FFP 30 ml/kg/day.
HIV-associated TTP
British Journal of Haematology, 128, 373-379

• Recombinant ADAMTS13. • Anti-vWF nanobody: blocking vWF interaction with platelets.
Future directions

• Significant advances in the understanding of the pathological basis of TTP. • Improvement in outcomes. • TTP still has a high mortality rate. • Room for improvement in numerous areas of patient care. • Centralized management of patients in order to concentrate expertise and
experience.
Conclusion
Journal of Blood Medicine 2014: 5, 15-23