jennifer glass, krista l lanctot, nathan herrmann, beth a...
TRANSCRIPT

1
Zopiclone
Addition to the List
Note: Commonly prescribed medication.
Literature question
Is zopiclone effective and safe in treating insomnia?
Literature search
Pubmed/Medline: zopiclone AND (insomnia OR Sleep Initiation and Maintenance Disorders); zopiclone AND
(insomnia OR Sleep Initiation and Maintenance Disorders) AND efficacy
e-CPS: Insomnia
Cochrane: Zopiclone
Jennifer Glass, Krista L Lanctot, Nathan Herrmann, Beth A Sproule, and Usoa E Busto. “Sedative hypnotics
in older people with insomnia: meta-analysis of risks and benefits.” BMJ (2005): 1-7
Quality of sleep. Reported sleep quality was significantly better with sedative use (mean effect size 0.14, 0.05 to
0.23; P<0.005). This effect size indicates a difference in mean scores on sleep quality for sedative versus placebo
groups of 0.11. In the most heavily weighted study in the analysis, this would correspond to mean scores of 3.8 in
the placebo group and 3.7 in the sedative group on a seven point scale. Three studies (339 participants) comparing
benzodiazepines with benzodiazepine receptor agonists (zaleplon, zolpidem, and zopiclone) found no significant
difference in sleep quality (mean effect size 0.04, -1.11 to 1.19; test for heterogeneity P=1.0). Two studies (116
participants) that reported sleep quality data for zopiclone versus placebo had a magnitude of effect of 0.41 (-0.76 to
1.58; test for heterogeneity P=0.98).
Amount of sleep. In eight studies (601 participants) with extractable data, the increase in total sleep time with any
sedatives compared with placebo was 25.2 minutes (12.8 to 37.8 minutes; P=0.001; test for heterogeneity P=0.10).
In eight studies (524 participants) that compared benzodiazepines with placebo, the increase in total sleep time was
34.2 minutes (16.2 to 52.8 minutes, P<0.01; test for heterogeneity P=0.13).
Number of awakenings. In six studies (441 participants) with extractable data, the mean number of awakenings
decreased by 0.63 (-0.48 to -0.77, P<0.0001; test for heterogeneity P=0.71). in six studies with benzodiazepines
versus placebo (296 participants) the mean number of awakenings decreased by 0.60 (-0.41 to -0.78, P<0.0001; test
for heterogeneity P=0.58).
Adverse events. The risk of adverse events was higher with sedative treatment. Most adverse events were reported to
be reversible and not severe. Patients who took sedatives had a higher incidence of falls and motor vehicle crashes.
Numbers needed to treat versus numbers needed to harm. The number needed to treat for improved sleep quality
was 13 and the number needed to harm for any adverse event was 6. This ratio indicates that an adverse event is
more than twice as likely as enhanced quality of sleep. This ratio can be used as a rough indicator only, as more than
double the number of participants contributed to the “harm” data than to the “effectiveness” data.
Nina Buscemi, Ben Vandermeer, Carol Friesen, Liza Bialy, Michelle Tubman, Maria Ospina, Terry Klassen,
and Manisha Witmans. “The Efficacy and Safety of Drug Treatments for Chronic Insomnia in Adults: A
Meta-analysis of RCTs.” Journal of General Internal Medicine 22 (2007): 1335-1350
Efficacy. The combined weighted mean difference (WMD) showed that benzodiazepine (BDZ), non-benzodiazepine
(non-BDZ), and anti-depressant (ADP) had significantly shorter sleep onset latency times compared to placebo
when measured by polysomnography (WMD: −10.0 minutes; 95% CI: −16.6, −3.4; WMD: −12.8 minutes; 95% CI:
−16.9, −8.8; WMD: −7.0 minutes; 95% CI: −10.7, −3.3, respectively) or sleep diary (WMD: −19.6 minutes; 95% CI:
−23.9, −15.3; WMD −17.0 minutes; 95% CI: −20.0, −14.0; WMD: −12.2 minutes; 95% CI: −22.3, −2.2, respectively)
(Table 4). The improvements measured by sleep diary were more prominent for all three drug groups. There was
heterogeneity among studies on BDZ and non-BDZ for both measures of sleep onset latency times, but the direction

2
of the estimate was fairly consistent for BDZ. Nine out of 11 comparisons had point estimates that favored BDZ for
polysomnography, while all 26 sleep diary trials had point estimates that favored BDZ (Fig. 2). For non-BDZ, all 12
studies for polysomnography and all 34 studies for sleep diary showed a point estimate that favored non-BDZ (Fig.
3). For ADP, there was moderate heterogeneity among studies in the polysomnography group and negligible
heterogeneity in the sleep diary group (Fig. 4). We conducted meta-analyses for wakefulness after sleep onset, sleep
efficiency, total sleep time, and sleep quality, subcategorized by polysomnography and sleep diary for BDZ, non-
BDZ, and ADP. All results were statistically significant and favored BDZ and non-BDZ with the exception of the
polysomnography studies measuring wakefulness after sleep onset and total sleep time, which were marginally
nonsignificant (Table 4). In contrast, for ADP, polysomnography results significantly favored ADP, but sleep diary
results were fewer and nonsignificantly favored ADP for wakefulness after sleep onset and nonsignificantly favored
placebo for total sleep time (Table 4).
Safety. To analyze the safety of BDZ, non-BDZ, and ADP, there were 34, 27, and 3 studies included, respectively.
The combined risk difference showed that all drug groups had significantly more adverse events than the placebo
group (Table 4). There was substantial heterogeneity among studies in the BDZ and non-BDZ groups, but it was
negligible in the ADP group. The most commonly reported adverse events in studies were somnolence (n=27),
headache (n=18), dizziness (n=16), nausea (n=11), and fatigue (n=11) in the BDZ group. There were no reports of
falls, injury, or death. In the non-BDZ group, the most commonly reported adverse events were headache (n=16),
dizziness (n=14), nausea (n=13), and somnolence (n=13). Accidental injury was reported in one study; however,
there was no significant difference in the frequency of this event between non-BDZ and placebo groups. Finally, for
ADP, the most commonly reported adverse events were somnolence (n=4), headache (n=3), dizziness (n=3), and
nausea (n=3). There were no reports of falls, injury, or death.
Indirect Comparisons of the Three Interventions. Compared to non-BDZ, BDZ showed a larger benefit on sleep
diary measures of sleep onset latency, but non-BDZ was favored when measured by polysomnography – neither
value was statistically significant. Non-BDZ was significantly safer than BDZ. Compared to ADP, the only
significant result was that non-BDZ was significantly more efficacious in terms of sleep onset latency when
measured by polysomnography – the result when measured by sleep diary still favored non-BDZ; but it was not
significant.
Alexander Winkler, Charlotte Auer, Bettina K. Doering, and Winfried Rief. “Drug Treatment of Primary
Insomnia: A Meta-Analysis of Polysomnographic Randomized Controlled Trials.” CNS Drugs 28 (2014): 799-
816.
Efficacy of Different Subgroups of Pharmacotherapy for Primary Insomnia. Comparisons for benzodiazepines
versus placebo were significant for all outcome variables (with the exception of sTST). For comparisons between
benzodiazepine receptor agonists, all effect sizes (with the exception of sWASO and sSE) were significant. With the
exception of sSOL, all effect sizes for comparisons between antidepressants and placebo were significant.
Comparison of Different Drug Classes. The Q test for heterogeneity yielded significant results for the comparison
between benzodiazepines and benzodiazepine receptor agonists with regard to sSOL, and, in the case of SOL, for
the comparisons between benzodiazepine receptor agonists and antidepressants as well as between benzodiazepines
and antidepressants. For outcome variable SOL, benzodiazepines and benzodiazepine receptor agonists are
significantly more effective than antidepressants and, for sSOL, benzodiazepines are more effective than
benzodiazepine receptor agonists.
A principal finding of this meta-analysis is that benzodiazepines and benzodiazepine receptor agonists are
significantly more effective than antidepressants in reducing SOL in patients with primary insomnia. Moreover,
benzodiazepines are significantly more effective than benzodiazepine receptor agonists in reducing sSOL of these
patients. A second main finding is that SOL and TST (which are primary endpoints in most treatment studies
addressing primary insomnia) showed higher effect sizes for objective outcomes in the benzodiazepine subgroup.

3
Naren Gunja. “The Clinical and Forensic Toxicity of Z-drugs.” Journal of Medical Toxicology 9 (2013): 155-
162.
Traditional therapy for insomnia has predominantly involved the use of benzodiazepines for several decades. Since
the 1980s, development of non-benzodiazepine drugs for the management of insomnia has been driven by the
significant adverse effect profile of the former group of drugs. The Z-drugs (zolpidem, zopiclone, zaleplon) have
significant hypnotic effects by reducing sleep latency and improving sleep quality, though their ability to prolong
total sleep time is debatable. While hypnosis and sedation are adequately achieved from oral benzodiazepines, they
invariably alter sleep architecture, reduce deep (stage 3 and 4) sleep, and lead to dependence, tolerance, and
withdrawal. Furthermore, benzodiazepines carry the risk of residual daytime effects such as impairment of cognitive
and psychomotor function. The Z-drugs possess shorter duratioxn of action and half-life, do not disturb overall sleep
architecture, and cause less residual effects during daytime hours, making them more clinically attractive than
benzodiazepines.
Adverse Effects. The Z-drugs are well-tolerated and the most common adverse effects include headache,
gastrointestinal upset, and dizziness. For a given dose, adverse reactions appear to be worse in elderly patients;
hence, lower doses are recommended in this group. The daytime residual effects of hypnotic drugs on cognitive and
psychomotor performance are a major concern in patients regularly taking these medications. Z-drugs have the
potential to cause residual effects post-awakening that related to cognition, memory, parasomnia, and bizarre
behavior. They have a profound effect on nocturnal and next-day psychomotor performance including body balance,
reaction times, and the ability to multi-task. Tolerance, dependence, and withdrawal are all reported with Z-drugs,
though this appears to be less severe and with lower incidence than for traditional benzodiazepines in the treatment
of insomnia.
Insomnia; Jonathan A.E. Fleming, MB, FRCPC, FABPN. FABSM. e-CPS. Date of Revision: July 2015.
Benzodiazepine receptor agonists. Although not a benzodiazepine, the cyclopyrrolone zopiclone acts at the
benzodiazepine receptor and so has similar therapeutic and side effects. Zopliclone can have residual or hangover
effects that could impair morning driving when used with or without alcohol. Warn patients not to drive or operate
machinery until at least 12hours after a bedtime dose of zopiclone. Compared with benzodiazepines, tolerance to
zopiclone’s hypnotic effect may be delayed and rebound insomnia may be reduced. Zolpidem is an imidazopyrine
with preferential affinity to benzodiazepine type I receptors. Memory disturbances and complex sleep behaviours
have been reported in patients using zolpidem. Gender and age-based differences in metabolic clearance of zolpidem
and incidence of complex sleep behaviours have led to lower dosing recommendations for women and elderly
patients.
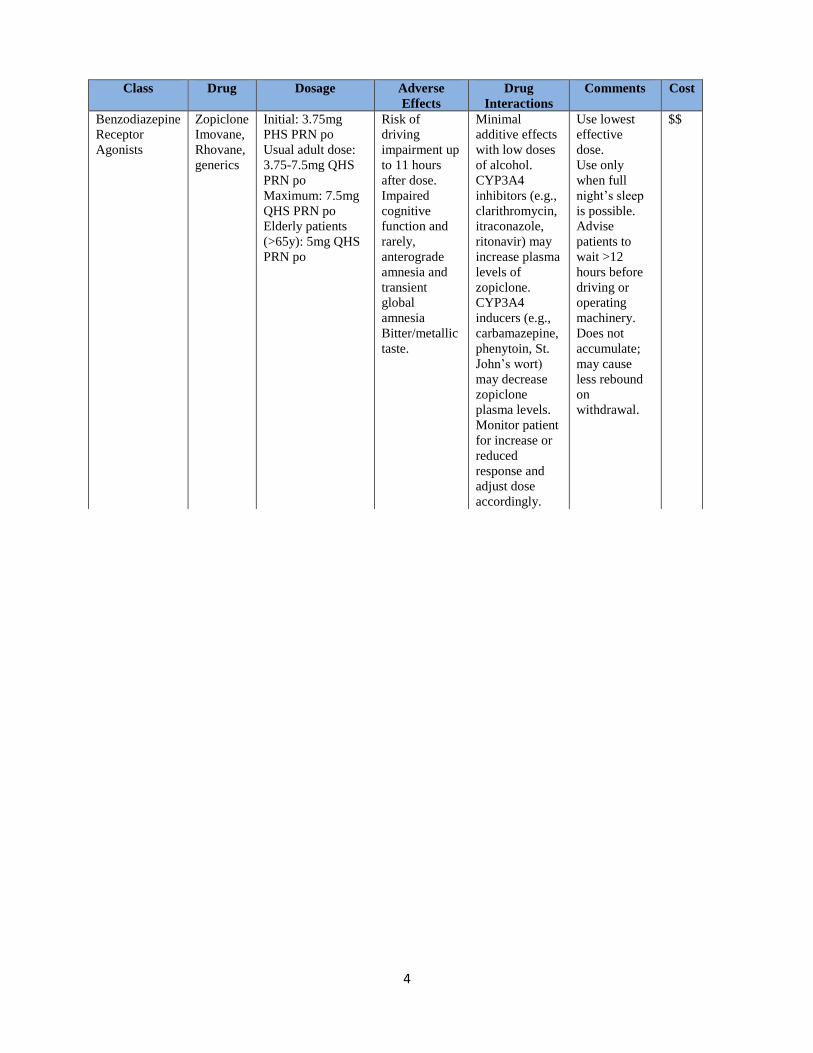
4
Class Drug Dosage Adverse
Effects
Drug
Interactions
Comments Cost
Benzodiazepine
Receptor
Agonists
Zopiclone
Imovane,
Rhovane,
generics
Initial: 3.75mg
PHS PRN po
Usual adult dose:
3.75-7.5mg QHS
PRN po
Maximum: 7.5mg
QHS PRN po
Elderly patients
(>65y): 5mg QHS
PRN po
Risk of
driving
impairment up
to 11 hours
after dose.
Impaired
cognitive
function and
rarely,
anterograde
amnesia and
transient
global
amnesia
Bitter/metallic
taste.
Minimal
additive effects
with low doses
of alcohol.
CYP3A4
inhibitors (e.g.,
clarithromycin,
itraconazole,
ritonavir) may
increase plasma
levels of
zopiclone.
CYP3A4
inducers (e.g.,
carbamazepine,
phenytoin, St.
John’s wort)
may decrease
zopiclone
plasma levels.
Monitor patient
for increase or
reduced
response and
adjust dose
accordingly.
Use lowest
effective
dose.
Use only
when full
night’s sleep
is possible.
Advise
patients to
wait >12
hours before
driving or
operating
machinery.
Does not
accumulate;
may cause
less rebound
on
withdrawal.
$$

5
Varenicline
Addition to the List
Note: Commonly prescribed medication.
Literature question
Is varenicline effective and safe for smoking cessation?
Literature search
Medline: Varenicline AND smoking cessation AND limit to (meta-analysis or review)
e-CPS: Varenicline; smoking cessation
Cochrane: Varenicline AND efficacy
Cahill K, Stevens S, Perera R, and Lancaster T. “Pharmacological interventions for smoking cessation: an
overview and network meta-analysis.” The Cochrane Library (2013).
Summary of main results. This overview identified 12 Cochrane reviews of pharmacotherapies used to assist
smoking cessation. Three treatments (NRT, bupropion and varenicline) are licensed as aids for smoking cessation
in high-income countries and recommended by many national guidelines, and we have concentrated on these
interventions for this overview. Both NRT and bupropion are similarly effective compared with placebo in helping
people to quit. Varenicline is more effective than NRT or bupropion, when compared with placebo. Direct and
indirect comparisons between the three treatments demonstrate no advantage for NRT over bupropion. Varenicline
is shown to be superior to any single type of NRT and to bupropion. Different types of NRT are generally equally
effective. Combinations of NRT outperform single formulations, and may be as effective as varenicline. None of the
network meta-analysis findings show evidence of inconsistency. Varenicline’s superior efficacy must be tempered
by current questions about its safety. Meta-analysis of any SAE while on varenicline compared with placebo finds
no difference between them, and subgroup analyses detect no significant excess of neuropsychiatric or
cardiovascular events. While this finding is supported by a number of observational studies and challenged by
others, the evidence is inconclusive at present, and long-term post-marketing surveillance will continue to inform the
debate.

6
Henri-Jean Aubin, Amandine Luquiens, and Ivan Berlin. “Pharmacotherapy for smoking cessation:
pharmacological principles and clinical practice.” British Journal of Clinical Pharmacology 77 (2013): 324-
336.
Therapeutic efficacy of varenicline. The recommended dosage is 1mg twice daily following a 1 week up-titration.
However, it has been shown that a self-regulated, flexible dosing regimen of varenicline is well tolerated, with
superior effectiveness vs. placebo [64]. Smokers treated with varenicline should normally aim to quit approximately
1 week after the start of the treatment. It has been shown, however, that using a flexible quit-date paradigm had
efficacy and safety similar to those in the previous fixed quit-date paradigm [65]. Meta-analyses have confirmed the
increased efficacy of varenicline on smoking quit rates at the dosage of 2mg day-1 during a 12 week treatment
compared with placebo and also to bupropion (see Table 1) [3, 66, 67]. For instance, the Cochrane review has shown
a risk ratio over placebo of 2.27 (95% CI 2.02, 2.55). Only two published studies have compared varenicline with
NRT [68, 69]. In an open but randomized study, varenicline performed better than the transdermal nicotine patch
[68]. Varenicline has been shown to result in higher abstinence rates than combined or high dose NRT in some [35],
but not all [1], meta-analyses. A 12 week treatment extension yields better cessation rates for smokers who quit
successfully for at least 1 week at the end of the first 12 weeks of treatment [70].The combination of varenicline
with NRT or bupropion seems to be safe [71, 72], but no published, randomized controlled data exist as to the
superiority of this co-administration over one or the other alone. Like NRT and bupropion, varenicline has been
shown to limit post-cessation weight gain during the active treatment phase, an effect that does not persist after
treatment has ended [43]. In addition, some data suggest that varenicline may help reduce alcohol consumption in
smokers who drink heavily [73, 74].
Safety and tolerability of varenicline. Phase I reports have shown that varenicline is tolerated after single doses up to
3mg in smokers and 1mg in nonsmokers. Nausea and vomiting at doses above 3.0 mg in smokers and 1mg in non-
smokers are dose limiting [75]. Recent reviews of the safety profile of varenicline concluded that the most frequent
adverse event was nausea, occurring in 30–40% of users [76–78].The nausea was generally reported as mild to
moderate and diminishing over time, and it was associated with low attributable discontinuation rates. Other
common adverse effects included insomnia, abnormal dreams and headaches. In the randomized, controlled phase
III studies, serious adverse events were rare, with no treatment-related deaths during the treatment or follow-up
phases. There are currently no known contraindications to varenicline [79]. Post-marketing surveillance reports have
suggested an increased risk of reported depression and suicidal/self-injurious behaviour with varenicline (and
bupropion) compared with NRT [58, 80–82]. However, a pooled analysis of more than 5000 smokers without
current psychiatric history who participated in one of 10 randomized, placebo controlled clinical trials found that
there was no significant increase in overall psychiatric adverse events aside from sleep disorders [83]. A study that
assessed neuropsychiatric adverse effects failed to find any difference in measurements of depressive symptoms,
anxiety, or aggression/ hostility between varenicline and placebo among smokers without psychiatric disorders [84].
Furthermore, a retrospective analysis of 80 600 adults prescribed varenicline, using records from the UK General
Practice Research Database, found that the incidence of depression and suicide was not greater with varenicline than
with NRT or bupropion [85].The post-marketing reports led the US Food and Drug Administration (FDA) and the
European Medicine Agency to add a ‘black box warning’ to the product labeling for both varenicline and bupropion
SR. Recently, two FDA-sponsored epidemiological studies that evaluated the risk of neuropsychiatric adverse events
associated with smoking cessation drugs found no difference in risk of neuropsychiatric hospitalizations between
varenicline and NRT [86]. Smoking and/or smoking cessation are frequently associated with neuropsychiatric
symptoms and suicide-related outcomes [87]. Therefore, drug regulatory agencies acknowledge that distinguishing
between drug-related adverse effects/events and the neuropsychiatric effects related to smoking and/or smoking
cessation is difficult, and they strongly recommend that patients be closely monitored for neuropsychiatric
symptoms [88]. A meta-analysis suggested an increased rate of cardiovascular events with varenicline [89].
However, a more recent meta-analysis that accounted for the major biases not taken into account in the previous
work concluded that treatment with varenicline is not associated with increased rates of cardiovascular events
compared with placebo [90].
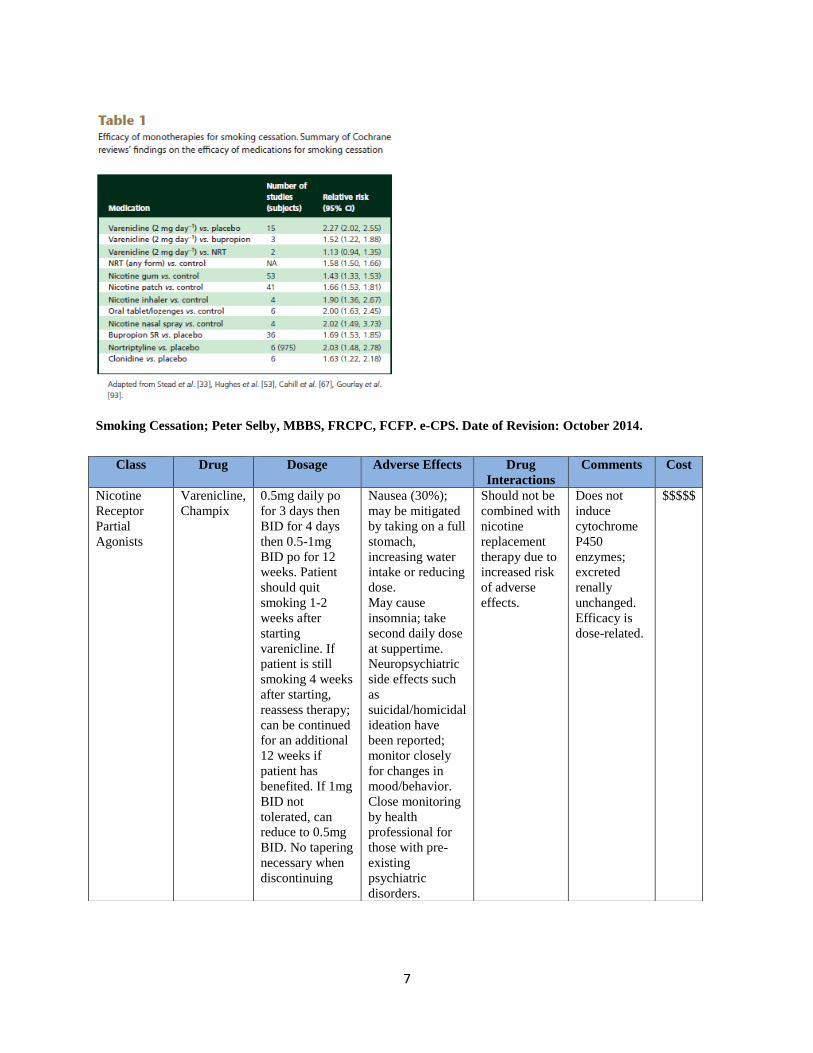
7
Smoking Cessation; Peter Selby, MBBS, FRCPC, FCFP. e-CPS. Date of Revision: October 2014.
Class Drug Dosage Adverse Effects Drug
Interactions
Comments Cost
Nicotine
Receptor
Partial
Agonists
Varenicline,
Champix
0.5mg daily po
for 3 days then
BID for 4 days
then 0.5-1mg
BID po for 12
weeks. Patient
should quit
smoking 1-2
weeks after
starting
varenicline. If
patient is still
smoking 4 weeks
after starting,
reassess therapy;
can be continued
for an additional
12 weeks if
patient has
benefited. If 1mg
BID not
tolerated, can
reduce to 0.5mg
BID. No tapering
necessary when
discontinuing
Nausea (30%);
may be mitigated
by taking on a full
stomach,
increasing water
intake or reducing
dose.
May cause
insomnia; take
second daily dose
at suppertime.
Neuropsychiatric
side effects such
as
suicidal/homicidal
ideation have
been reported;
monitor closely
for changes in
mood/behavior.
Close monitoring
by health
professional for
those with pre-
existing
psychiatric
disorders.
Should not be
combined with
nicotine
replacement
therapy due to
increased risk
of adverse
effects.
Does not
induce
cytochrome
P450
enzymes;
excreted
renally
unchanged.
Efficacy is
dose-related.
$$$$$

8
Vitamin B12
Addition to the List
Note: Commonly prescribed medication.
Literature question
Is vitamin B12 effective and safe?
Literature search
Medline: Vitamin B12 deficiency AND limit to (meta-analysis or review)
e-CPS: Vitamin B12; Nutritional Supplements
Cochrane: Vitamin B12 deficiency
We found no trials comparing B12 to placebo.
Vidal-Alaball J, Butler C, Cannings-John R, Coringe A, Hood K, McCaddon A, McDowell I, and
Papaioannou A. “Oral vitamin B12 versus intramuscular vitamin B12 for vitamin B12 deficiency (Review).”
The Cochrane Library (2009).
Two studies compared oral and intramuscular vitamin B12: Kuzminski and colleagues (Kuzminski 1998) reported
neurologic and haematologic responses. Four of the eighteen participants randomized to receive oral vitamin B12
and four of the fifteen randomized to receive intramuscular vitamin B12 had a neurological response with a marked
improvement or clearing of paresthesias, ataxia, or memory loss. Serum vitamin B12 levels were significantly
higher in the oral (643 +/- 328 pg/mL) compared to the intramuscular group (306 +/- 118 pg/mL) at 2 months
P<0.001). The difference was even greater at four months (1,005 +/- 595 vs. 325 +/- 165 pg/mL; P< 0.0005). Serum
methylmalonic acid concentrations decreased to < 3 SD above the normal range in all participants except one in the
oral and two in the intramuscular group. Mean concentrations of the metabolites were not significantly different
between the oral and the intramuscular groups, except at four months, when the value was higher in the
intramuscular group (P< 0.05). Elevated serum total homocysteine decreased to 3 SD above the normal range in
most participants, but the decrease was over four months in the oral group and during the first month in the
intramuscular group. However, in two patients in each group the response was not optimal. Bolaman and colleagues
(Bolaman 2003) reported neurologic and haematologic responses. Both groups receiving oral or intramuscular
vitamin B12 reported improvements of cognitive function, sensory neuropathy and vibration sense but the difference
between both groups was not statistically significant. Serum vitamin B12 levels increased in those receiving oral and
those receiving intramuscular vitamin B12 for 90 days. The authors reported a statistically significant difference
between day 0 and day 90 within both groups (P< 0.001) but did not analyse differences between both groups.
Vitamin B12 – Dietary Supplement Fact Sheet. National Institutes of Health – Office of Dietary Supplements.
Cardiovascular disease. Elevated homocysteine levels have been identified as an independent risk factor for
cardiovascular disease. Evidence from retrospective, cross-sectional, and prospective studies links elevated
homocysteine levels with coronary heart disease and stroke. Vitamin B12, folate and vitamin B6 are involved in
homocysteine metabolism. In the presence of insufficient vitamin B12, homocysteine levels can rise due to
inadequate function of methionine synthase. Results from several randomized controlled trials indicate that
combinations of vitamin B12 and folic acid supplements with or without vitamin B6 decrease homocysteine levels
in people with vascular disease or diabetes and in young adult women. In another study, older men and women who
took a multivitamin/multimineral supplement for 8 weeks experienced a significant decrease in homocysteine levels.
Evidence supports a role for folic acid and vitamin B12 supplements in lowering homocysteine levels, but results
from several large prospective studies have not shown that these supplements decrease the risk of cardiovascular
disease. In the Women’s Antioxidant and Folic Acid Cardiovascular Study, women at high risk of cardiovascular
disease who took daily supplements containing 1mg vitamin B12, 2.5mg folic acid, and 50mg vitamin B6 for
7.3years did not have a reduced risk of major cardiovascular events, despite lowered homocysteine levels. The Heart
Outcomes Prevention Evaluation (HOPE) 2 trial, which included 5,522 patients older than 54years with vascular
disease or diabetes, found that daily treatment with 2.5mg folic acid, 50mg vitamin B6, and 1mg vitamin B12 for an
average of 5 years reduced homocysteine levels and the risk of stroke, but did not reduce the risk of major

9
cardiovascular events. In the Western Normal B Vitamin Intervention Trial, which included 3,096 patients
undergoing coronary angiography, daily supplements of 0.4mg vitamin B12 and a 0.8mg folic acid with or without
40mg vitamin B6 for 1year reduced homocysteine levels by 30%, but did not affect total mortality or the risk of
major cardiovascular events during 38 months of follow-up. The Norwegian Vitamin (NORVIT) trial and the
Vitamin Intervention for Stroke Prevention trial had similar results. The American Heart Association has concluded
that the available evidence is inadequate to support a role for B vitamins in reducing cardiovascular risk.
Dementia and cognitive function. Observational studies show positive associations between elevated homocysteine
level and the incidence of both Alzheimer’s disease and dementia. Low vitamin B12 status has also been positively
associated with cognitive decline. Despite evidence that vitamin B12 lowers homocysteine levels and correlations
between low vitamin B12 levels and cognitive decline, research has not shown that vitamin B12 has an independent
effect on cognition. In one randomized, double-blind, placebo-controlled trial, 195 subjects aged 70 years or older
with no or moderate cognitive impairment received 1,000mcg vitamin B12, 1,000mcg vitamin B12 plus 400mcg
folic acid, or placebo for 24 weeks. Treatment with vitamin B12 plus folic acid reduced homocysteine
concentrations by 36%, but neither vitamin B12 treatment nor vitamin B12 plus folic acid treatment improved
cognitive function.
Energy and endurance. Due to its role in energy metabolism, vitamin B12 is frequently promoted as an energy
enhancer and an athletic performance and endurance booster. These claims are based on the fact that correcting the
megaloblastic anemia caused by vitamin B12 deficiency should improve the associated symptoms of fatigue and
weakness. However, vitamin B12 supplementation appears to have no beneficial effect on performance in the
absence of a nutritional deficit.
Health Risks from Excessive Vitamin B12. The IOM did not establish a UL for vitamin B12 because of its low
potential for toxicity. In Dietary Reference Intakes: Thiamin, Riboflavin, Niacin, Vitamin B6, Folate, Vitamin B12,
Pantothenic Acid, Biotin, and Choline, the IO states that “no adverse effects have been associated with excess
vitamin B12 intake from food and supplements in healthy individuals”. Findings from intervention trials support
these conclusions. In the NORVIT and HOPE 2 trials, vitamin B12 supplementation (in combination with folic acid
and vitamin B6) did not cause any serious adverse events when administered at doses of 0.4mg for 40 months
(NORVIT trial) and 1.0mg for 5 years (HOPE 2 trial).

10
Tretinoin
Addition to the List
Note: Commonly prescribed medication.
Literature question
Is tretinoin effective and safe?
We also briefly summarize evidence of efficacy in the treatment of other conditions (e.g. rosacea, actinic keratosis).
Literature search
Medline: Tretinoin AND acne AND limit to (meta-analysis or review); Tretinoin AND efficacy AND limit to (meta-
analysis or review)
e-CPS: Tretinoin; Acne
Cochrane: Tretinoin AND efficacy; topical retinoid AND acne AND efficacy
Harald PM Gollnick and Andrea Krautheim. “Topical Treatment in Acne: Current Status and Future
Aspects.” Dermatology 2003 (206): 29-36.
Tretinoin was the first topical retinoid described in the first reports by Stuttgen and by Beer. It significantly reduces
the number of comedones but also of inflammatory acne lesions. It has been shown in several trials that at least
during a 12-week course the reduction of lesion counts ranges between 32-81% for non-inflammatory lesions and
17-71% for inflammatory lesions, i.e. 22-83% for the total lesion count. Comparing tretinoin 0.025% gel or 0.025%
cream with its vehicle in a once daily application manner, tretinoin was significantly more effective than the vehicle
in reducing both inflammatory and non-inflammatory lesions. Currently tretinoin is available in different galenic
formulations: cream (0.025%, 0.1%, gel 0.1%, 0.025%) and as a solution (0.05%). In a topical gel formulation
containing polyolprepolymer-2, tretinoin penetration was shown to be significantly reduced while potentially
enhancing epidermal deposition compared to a commercially available gel preparation at the same concentration.
Polyolprepolymer-2 helps to retain drug molecules on the skin surface and in the upper layers of the skin [19, 20].
Another new formulation is a microsponge delivery system consisting of macroporous beads of 10-25µm in
diameter which are loaded with an active ingredient. After topical application this is gradually released depending
on rubbing, temperature, pH and other factors [21]. Tretinoin 0.1% gel microsponge compared with tretinoin
0.025% gel, tazarotene 0.1% gel or adapalene 0.1% gel in a split face study showed similar facial tolerability for all
retinoids [22]. A formulation of liposomally encapsulated tretinoin 0.01% was in a comparative study equipotent in
clearing acne lesions as tretinoin 0.025 or 0.05% gel after once daily topical application for 10 weeks, but showed a
much better cutaneous tolerability [23]. This was also reproducible on in vitro reconstructed epidermis [24]. With
regard to systemic absorption and risk of embryotoxicity after topical application, the detected fecal and plasma
tretinoin concentrations were much below the endogenous tretinoin levels [25, 26] and did not affect the endogenous
levels of tretinoin or its metabolites or alter the plasma vitamin A levels [27].

11
Sanjiv Jain. “Topical tretinoin or adapalene in acne vulgaris: an overview.” Journal of Dermatological
Treatment 2004 (15): 200-07.
A series of large-scale, multicentre, comparative trials of adapalene 0.1% gel versus tretinoin have been undertaken
(Table III). The outcome of individual primary trials were not completely homogeneous and demonstrated varying
results regarding efficacy. The inconclusive and equivocal outcomes necessitated a meta-analysis that concluded
that adapalene 0.1% gel was equally as efficacious as tretinoin 0.025% gel.32 As it is not rational to extrapolate the
conclusions derived from the comparison of adapalene 0.1% gel with tretinoin 0.025% gel to tretinoin 0.05% gel, it
prompted a scrutiny of the comparative efficacy of 0.1% adapalene gel and tretinoin 0.05% gel. A split-face clinical
and bioinstrumental comparison of 0.1% adapalene gel and 0.05% tretinoin gel in facial acne was undertaken. In
contrast to previous studies, the present trial revealed the greater efficacy of tretinoin 0.05% gel over adapalene
0.1% gel. This may be ascribed to the higher concentration of tretinoin,27 and its higher affinity for gamma
receptors.33 The adverse reactions to adapalene are of the same type, but of significantly lower frequency and lesser
severity than for tretinoin (Table IV). Most studies concluded that adverse effects such as erythema, scaling,
pruritus, and burning could occur in a significant percentage of patients. Overall, 45.7% of the tretinoin-treated
patients experienced some form of irritation, compared with 32.4% of those treated with adapalene.29
A Geng, MA Weinstock, R Hall, D Eilers, M Maylor, and J Kalivas. “Tolerability of high-dose topical
tretinoin: the Veterans Affairs Topical Tretinoin Chemoprevention Trial.” British Journal of Dermatology
2009 (161): 918-24.
This study assessed the long-term tolerability of tretinoin 0.1% cream, and found that while participants using
tretinoin were more likely to have side-effects than the controls, the frequency of these side-effects decreased over
time, and there was no remaining difference between the groups after 2 years. Burning was the most commonly
reported specified side-effect, followed by itching and other local cutaneous reactions. The majority (67%) of
participants on tretinoin tolerated cream application at least once daily (80% tolerated at least every other day), and
the use of tretinoin cream was not associated with an increased drop-out rate from the study. We expected and found
that participants using tretinoin were more likely to have side-effects; this was true even after using the study cream
for 6 months (the first follow-up assessment). While there is a widely held belief in clinical practice that tretinoin
cream at the 0.1% concentration is a poorly tolerated medication, in this study population almost 40% of the
tretinoin group reported no side-effects at 6 months. We expect that the frequency of side-effects would probably
have been higher had the participants been assessed at earlier time points (e.g. at 2 weeks). We also expected and
found that the frequency of side-effects decreased over time, supporting the clinical observation that participants
seem to tolerate tretinoin better with continued usage. Of note, side-effects continued to decrease over 2 years before
the difference between the two groups became non-significant. We examined the frequency of cream application as
a measure of tolerability, as the tretinoin group might have applied the cream less often in response to the known
side-effects. As predicted, participants using tretinoin were less likely to apply their cream twice daily or once daily,
and more likely to apply every other day or ‘not at all’. These frequencies of application persisted over time.
Gupta AK, Paquet M, Villanueva E, and Brintnell W. “Interventions for actinic keratoses (Review).” The
Cochrane Library (2012).
Prescription-based topical treatments – Ro 14-9706 versus 0.05% tretinoin. This intervention was addressed by
only 1 intraindividual study (Misiewicz 1991), which compared the efficacy of 0.05% Ro 14-9706 and 0.05%
tretinoin applied twice-daily for 16 weeks for the treatment of facial actinic keratoses. Assessment was performed at
the end of the 16-week treatment. There was no major source of possible bias. Areas treated with tretinoin cream
showed an initial increase in the number of lesions (weeks 3 to 9), which eventually decreased after week 10. Ro 14-
9706 showed no initial increase in number of actinic keratoses lesions, but a gradual decline over time. The resulting
mean percentage of reduction in lesion counts was significantly higher in the group treated with Ro 14-9706 than the
group treated with tretinoin (MD 7.50, 95% CI 6.57 to 8.43; Analysis 3.1). To summarise, Ro 14-9706 treatment
showed better overall reduction in lesion counts, whereas tretinoin treatment, which showed an initial increase in
lesions, resulted in more participants with complete response.

12
Van Zuuren EJ, Fedorowicz Z, Carter B, van der Linden MMD, and Charland L. “Interventions for rosacea
(Review).” The Cochrane Library (2015).
Topical clindamycin phosphate combined with tretinoin was not considered to be effective compared to placebo
(moderate quality of the evidence)
Acne; Duane Lichtenwald, MD, FRCPC. e-CPS. Date of Revision: June 2015.
Topical retinoids.Topical retinoids are the most powerful comedolytic agents, yet still take months to be effective.
They are also effective for inflammatory lesions. Tretinoin is the most cost effective, but also the most
photosensitizing. Adapalene is the least irritating, tazarotene is the most potent. Topical retinoids (except when
combined in a commercial product) are unstable in the presence of benzoyl peroxide and should be applied at a
different time: usually benzoyl peroxide in the morning and retinoid at bedtime.
Class Drug Dosage Adverse Effects Cost
Retinoids Tretinoin
0.01%,
0.025%,
0.05%, 0.1%
Retin-A,
Retin-A
Micro,
Stieva-A,
Vitamin A
Acid
Crea, gel; once
daily at HS to
limit
photosensitivity
Photosensitivity.
Irritating.
SS

13
Baclofen
Addition to the List
Note: Commonly prescribed medication.
Literature question
Is baclofen effective and safe for the management of low back pain?
Literature search
Medline: : Baclofen AND low back pain AND limit to (meta-analysis or review); Baclofen AND spasticity AND
limit to (meta-analysis or review)
e-CPS: Baclofen; Low back pain
Cochrane: Baclofen AND low back pain; Baclofen AND spasticity AND efficacy
van Tulder MW, Touray T, Furlan AD, Solway S, and Bouter LM. “Muscle relaxants for non-specific low-
back pain (Review).” The Cochrane Library (2008).
Acute LBP. One study compared an antispasticity muscle relaxant (baclofen) with placebo and incorporated bed rest
in the therapeutic regimen (Dapas 1983). In comparison with placebo, there was significant relief of pain and
improvement in terms of global efficacy. Relief of spasm did not reach statistical significance.
Low Back Pain; Eldon Tunks, MD, FRCPC and Paul Stacey, MD, MSc. e-CPS. Date of Revision: February
2014.
Pharmacologic Choices. During the acute phase of low back pain, muscle relaxants may be effective for relief of
pain and spasm during short-term use, but are more likely to cause adverse effects such as drowsiness and
dizziness.5,15 Muscle relaxants might include benzodiazepines, cyclobenzaprine, nonprescription preparations
containing methocarbamol or orphenadrine, or the antispasticity drugs baclofen or tizanidine. A muscle relaxant
may also be combined with an NSAID or COX-2 inhibitor for relief of pain and spasm in acute back pain.
Taricco M, Adone R, Pagliacci C, Telaro E. “Pharmacological interventions for spasticity following spinal
cord injury (Review).” The Cochrane Library (2009).
Oral baclofen versus placebo. Brar 1991 reported significant improvement compared to placebo in angle of flexion
and subjective function report for participants treated with baclofen alone or in combination with stretching
exercises, but no significant added benefit from stretching exercises alone. Significantly more participants improved
Class Drug Dosage Adverse
Effects
Drug
Interactions
Comments Cost
Muscle
Relaxants
Baclofen
Lioresal
Oral,
generics
Start with
5mg TID po;
increase
gradually to
maximum of
20mg TID
Sedation,
weakness,
nausea,
dizziness.
Very rare:
hepatotoxicity
Potential additive
CNS depression
with tricyclic
antidepressants,
opioids,
benzodiazepines
and
antihypertensives
Adjust dose
gradually to
minimize
adverse
effects or
withdrawal
symptoms.
In acute pain,
short-term
use as muscle
relaxant.
Not
recommended
in the elderly.
$$

14
in Ashworth score only with baclofen combined with exercises compared with placebo. Feldman 1978 only reported
the numbers of participants who improved on each treatment. Significantly better improvement was suggested in
passive range of motion, painful spasms and clonus in participants taking baclofen.
Alessandro Dario and Giustino Tomei. “A Benefit-Risk Assessment of Baclofen in Severe Spinal Spasticity.”
Drug Safety 2004 (11): 799-818.
Benefit Evaluation. In open-label studies of oral administration of baclofen, the drug was demonstrated to improve
spasticity in 70-87% of patients; additionally, improvement in spasms was reported in the 75-96% of patients.[37-
40] However, it should be noted that the measures of symptom improvement used differed between the studies.
Spasticity and spasms following spinal cord injury seem to show better clinical improvement with baclofen
treatment than multiple sclerosis-associated symptoms.[39] Despite the reported symptomatic improvements,
baclofen has not yet been clearly shown to produce long-term functional improvement measured by appropriate
scale. [39] In double-blind crossover placebo-controlled trials, [41-44], baclofen was reported to be an effective drug
that produced a statistically significant improvement in symptoms (table II). However, in five patients with
traumatic spinal cord injuries the quantitative effects of baclofen versus placebo treatment failed to provide a
convincing effect of spasticity reduction; [45] of interest, baclofen did appear to have anxiolytic effects in these
patients. [46] The administration of oral baclofen demonstrated an improvement in gait in patients with spasticity
and an improvement in vertical unsteadiness in patients with multiple sclerosis; however, this study only lasted 11
days. [47] However, it should be noted that the clinical measure of improvement was not homogeneous between the
placebo controlled studies. The effective and well-tolerated dosage of oral baclofen lies in the range of 30-80mg
daily, [39] but in multiple sclerosis patients high-dose administration has been suggested. [51] The cost-
effectiveness of managing spasticity using baclofen was calculated as £10.50 per successfully treated day (year 2000
values) in one economic analysis.[22]
Benefit-Risk Evaluation. Oral baclofen appears to be effective in the treatment of spasticity. With regards to
increased tone and spasms, the drug is effective in 55-96% of patients; however, the long-term functional benefit for
patients is not yet certain. Considering the seriousness of the diseases affecting these patients (mainly spinal cord
injury or multiple sclerosis) and their clinical status, this result is favourable. The main risks of oral baclofen
administration are related to withdrawal (ie seizures, psychic symptoms and hyperthermia) [section 1.2]. These
symptoms have been reported to improve after the reintroduction of baclofen therapy, usually without sequelae.
When these symptoms are not related to drug withdrawal, they present mainly only in patients with brain damage
and in the elderly. The limited data available on baclofen toxicity in patients with renal disease suggest that
administration of the drug in these persons may carry an unnecessarily high risk. The interactions between tricyclic
drugs and baclofen are not clearly defined, so the contemporaneous administration of the two drugs should be
cautiously performed. Because the adverse consequences do not exceed the benefits of oral baclofen for the
treatment of patients with spinal spasticity the benefit-risk assessment is favourable.

15
Pramoxine/zinc
Addition to the List
Note: Commonly prescribed medication.
Literature question
Is pramoxine effective and safe in the management of pruritus?
Literature search
Medline: Pramoxine AND (pruritus OR itch*) AND limit to (meta-analysis); (Pruritus OR itch*) AND zinc
Cochrane: Pramoxine AND limit to (meta-analysis or review); Zinc AND limit to (meta-analysis or review)
Trudye A Young, Tejesh S Patel, Fabian Camacho, Adele Clark, Barry I Freedman, Mandeep Kaur, Julie
Fountain, Lisa L Williams, Gil Yosipovitch, and Alan B Fleischer JR. “A pramoxine-based anti-itch lotion is
more effective than a control lotion for the treatment of uremic pruritus in adult hemodialysis patients.”
Journal of Dermatological Treatment 2009 (20): 76-81.
The present study shows that anti-itch lotion containing pramoxine reduces ESRD-associated pruritus to a greater
degree than the control lotion following 4 weeks of topical application. In addition, both the pramoxine lotion and
the control lotion resulted in overall improvements in disease severity, skin hydration and quality of life (in terms of
sleep disturbances and mood) at the end of the 4-week study period. No adverse effects were reported and only one
patient failed to complete the study (owing to death from an unrelated event). Treatment with anti-itch lotion
containing pramoxine was therefore well tolerated. Previous reports on the safety of pramoxine hydrochloride have
described this compound to be well tolerated by tissues in addition to displaying low systemic toxicity and
sensitizing potential (16). Pramoxine lotion and control lotion both resulted in improvements in individual reported
itch intensity. Although disease severity obtained by IGA (in terms of erythema, xerosis and lichenification) and
skin hydration improved following 4 weeks of topical application, because of the small size of this pilot study, the
results were not significant. Xerosis is the most frequent cutaneous manifestation of ESRD and has been suggested
as a cause of pruritus in such patients (17–21). The pramoxine lotion reduces ESRD-associated pruritus to a greater
degree than the control lotion, implying the active ingredients in this preparation independently augment the
reduction of this symptom. Pramoxine, a local anesthetic, has been suggested to affect itch sensation by interfering
with the transmission of impulses along the sensory nerve fibers (23).
Pongcharoen P and Fleischer AB. “An evidence-based review of systemic treatments for itch.” European
Journal of Pain 2016 (20): 24-31.
Zinc sulphate. One RCT (Najafabadi et al., 2012) investigated the effect of zinc sulphate in 40 haemodialysis
participants. Itch was significantly decreased in the treatment group compared with placebo. No severe side effects
were observed. However, due to the small number of sample size and small benefit, the effectiveness of zinc
sulphate cannot be assessed conclusively.

16
Benztropine
Addition to the List
Note: Commonly prescribed medication.
Literature question
Is benztropine effective and safe?
Literature search
Medline: Benztropine AND efficacy AND limit to (meta-analysis)
Cochrane: Benztropine AND efficacy
Soares-Weiser K, Mobsy C, and Holliday E. “Anticholinergic medication for neuroleptic-induced tardive
dyskinesia (Review).” The Cochrane Library (2012).
Implications for practice. Based on the currently available data, this systematic review can provide no reliable
conclusions about the use of anticholinergics (benzhexol, benztropine, biperiden, orphenadrine, procyclidine,
scopolamine or trihexylphenidyl) for the treatment of neuroleptic-induced TD. In addition, there is no evidence to
support the suggestion that the withdrawal of these medications may benefit people with TD. In the absence of
evidence one way or another, the clinician must balance the possible benefits against the potential adverse effects of
the treatment.
Katzenschlager R, Sampaio C, Costa J, and Lees A. “Anticholinergics for symptomatic management of
Parkinson’s disease (Review).” The Cochrane Library (2009).
Main results. Trial duration was between five and 20 weeks and drugs investigated were benzhexol (mean doses: 8
to 20 mg/d), orphenadrine (mean dose not reported), benztropine (mean dose not reported), bornaprine (8 to 8.25
mg/d), benapryzine (200 mg/d), and methixine (45 mg/d). Only one study involved two anticholinergic drugs.
Outcome measures varied widely across studies and in many cases, the scales applied were the authors´ own and
were not defined in detail. Incomplete reporting of methodology and results was frequent. The heterogeneous study
designs as well as incomplete reporting precluded combined statistical analysis. Five studies used both tremor and
other parkinsonian features as outcome measures. Outcome measures in these five studies were too different for a
combined analysis and results varied widely, from a significant improvement in tremor only to significant
improvement in other features but not in tremor. All studies except one (dealing with methixine) found a significant
improvement from baseline on the anticholinergic drug in at least one outcome measure. The difference between
placebo and active drug was reported in four studies and was found to be significant in all cases. No study failed to
show superiority of the anticholinergic over placebo. The occurrence of neuropsychiatric and cognitive adverse
events was reported in all but three studies (in 35 patients on active drug versus 13 on placebo). The most frequently
reported reason for drop-outs from studies was in patients on placebo due to withdrawal from pre-trial
anticholinergic treatment.
Powney MJ, Adams CE, and Jones H. “Haloperidol for psychosis-induced aggression or agitation (rapid
tranquillisation) (Review).” The Cochrane Library (2012).
Adverse events In terms of movement disorders and the emergence of ataxia (1 RCT, n = 66, RR 0.44 CI 0.04 to
4.65), dystonia (1 RCT, n = 66, RR 3.54 CI 0.42 to 30.03), speech disorder (1 RCT,n = 66, RR 1.77 CI 0.35 to 9.01),
rigidity (1 RCT, n = 66, RR 6.22 CI 0.33 to 115.91), tremor (1 RCT, n = 66, RR 1.77 CI 0.17 to 18.60) and the need
for benztropine (1 RCT, n = 66, RR 1.99 CI 0.68 to 5.83). One study (n = 60) reported that significantly more
participants experienced EPS in the haloperidol group compared with the lorazepam group (RR 15.00 CI 2.11 to
106.49, Analysis 13.7).
Walshe M, Smith M, and Pennington L. “Interventions for drooling in children with cerebral palsy
(Review).” The Cochrane Library (2012).

17
Pharmaceutical Interventions. Both studies on pharmaceutical interventions (Camp-Bruno 1989; Mier 2000)
compared intervention versus placebo. They differed in the medications given and outcome measures used. Camp-
Bruno 1989 examined reduction in salivary flow and found a statistically significant difference in salivary flow
between participants (age range 4-44 years) on placebo and those taking benztropine (p<.001) immediately after
intervention. Both studies examined the frequency and severity of drooling albeit using different outcome measures.
Camp-Bruno 1989 defined a ’responder’ as those participants who obtained a mean TDS rating of less than 3. They
also defined ’responsivity’ as a decrease of one baseline SD or greater. Mier 2000 defined an improvement of 4
points or greater in their 9 point scale as a standard for significant ’clinical improvement’. On the Teacher Drool
Scale, Camp-Bruno 1989 found a statistically significant difference between both placebo and intervention in the
frequency and severity of drooling immediately after intervention (p_0.001).Mier 2000 using an adaptation of
Thomas-Stonell and Greenberg Scale also found a statistically significant difference between the placebo and
intervention immediately after intervention (p<0.001). It is unknown how long the effects of these medications
lasted in terms of reducing the quantity of saliva produced and reducing the frequency and severity of drooling.
The adverse effects of benztropine reported were behavior changes such as irritability and listlessness. Medical side
effects reported were insomnia, vomiting, dilated pupils, disorientation, facial flushing, ’glassy eyes’ , stomachache,
and dry mouth. Three children of the 27 (11%) children in Camp-Bruno 1989 were excluded because of adverse
reactions to benztropine. Eight of the 39 (20.5%) children inMier 2000 study dropped out because of the adverse
side effects to glycopyrrolate. As with the trials on BoNT-A, it is difficult to determine whether all adverse effects
reported were directly related to the medication.

18
Fusidic Acid and Mupirocin
Addition to the List
Note: Commonly prescribed medication.
Literature question
Is fusidic acid effective and safe?
Is mupirocin effective and safe?
Literature search
Medline: Fusidic acid AND efficacy AND limit to (meta-analysis); Mupirocin AND efficacy AND limit to (meta-
analysis)
Cochrane: Fusidic acid AND efficacy; Mupirocin AND
Koning S, van der Sande R, Verhagen AP, van Suijlekom-Smit LWA, Morris AD, Butler CC, Berger M, van
der Wouden JC. “Interventions for impetigo (Review).” The Cochrane Library (2011).
Topical antibiotics versus placebo (six studies, four comparisons). Overall topical antibiotics showed better cure
rates or more improvement than placebo (pooled risk ratio (RR) 2.24, 95% CI 1.16 to 3.13 using a random-effects
model, I² = 53%) (see Analysis 1.1). This result was consistent for mupirocin (RR 2.21, 95% CI 1.59 to 3.05; 3
studies - Eells 1986; Gould 1984; Rojas 1985) (see Analysis 1.1), fusidic acid (RR 4.42, 95% CI 2.39 to 8.17; 1
study - Koning 2003) (see Analysis 1.1), and retapamulin (RR 1.64, 95% CI 1.30 to 2.07; 1 study - Koning 2008)
(see Analysis 1.1). In one small study (Ruby 1973), bacitracin did not show a significant difference in cure rate
compared with placebo (RR 3.71, 95% CI 0.16 to 85.29) (see Analysis 1.1).
Topical antibiotics versus oral (systemic) antibiotics (16 studies, 17 comparisons). Pooling 10 studies which
compared mupirocin with oral erythromycin showed significantly better cure rates, or more improvement, with
mupirocin (RR 1.07, 95% CI 1.01 to 1.13) (see Analysis 3.1). However, no significant differences were seen
between mupirocin and dicloxacillin (Arredondo 1987), cephalexin (Bass 1997), or ampicillin (Welsh 1987).
Bacitracin was significantly worse than oral cephalexin in one small study (Bass 1997), but no difference was seen
between bacitracin and erythromycin (Koranyi 1976), or penicillin (Ruby 1973). A sensitivity analysis on the
influence of blinding the outcome assessor on the comparison of mupirocin versus erythromycin (10 studies)
revealed that there was no clear relationship between blinding of the outcome assessor and the outcome. Pooling the
2 studies with observer blinding (Britton 1990;Dagan 1992) showed high heterogeneity (I² statistic = 79%) and
resulted in a non-significant difference between the 2 drugs (random-effects model, RR 1.12, 95% CI 0.86 to 1.46)
(see Analysis 3.2).
Secondary outcomes: Adverse Effects – Topical antibiotics. The trials included in this review usually reported few,
if any, side effects from topical antibiotics (see Table 1). The studies comparing mupirocin, bacitracin, and placebo
reported none (Eells 1986; Ruby 1973). The study that compared fusidic acid to placebo recorded more side-effects
in the placebo group (Koning 2003). Three of 4 studies comparing mupirocin with fusidic acid recorded side-effects:
minor skin side-effects were reported for mupirocin by 10 out of 368 participants (3%) and for fusidic acid by 4 out
of 242 participants (2%). The study that compared retapamulin to placebo found more itching in the group treated
with retapamulin (7%vs 1%; P = 0.17) (Koning 2008). In the other study of retapamulin, this side-effect was
reported in less than 1% of cases (Oranje 2007).Most other trials comparing topical antibiotics reported no side-
effects or reported minor skin side-effects in low numbers (less than 5% of participants).

19

20
George Ajay and Rubin Greg. “A systematic review and meta-analysis of treatments for impetigo.” British
Journal of General Practice 2003 (53): 480-487.
Mupirocin versus fusidic acid. The BNF advises use of mupirocin or fusidic acid as topical treatment for
impetigo.13 Of the four studies comparing these two treatments, one showed 100% cure or improvement in both
groups31 and one favoured fusidic acid,34 while the other two favoured mupirocin35,36 (with only one of these
showing mupirocin to be significantly better than fusidic acid). The three studies that showed differences in
treatment effects were all set in UK general practice. There was no strong evidence of statistical heterogeneity (χ2 =
5.94, df = 2, P = 0.051) and meta-analysis shows no difference between the two treatments, (OR = 1.76, 95% CI =
0.77 to 4.03) (Figure 4).
Kevin B Laupland and John M Conly. “Treatment of Staphylococcus aureus Colonization and Prophylaxis
for Infection with Topical Intranasal Mupirocin: An Evidence-Based Review.” Clinical Infectious Diseases
2003 (37): 933-38.
Several studies conducted with varied populations demonstrated that mupirocin was highly effective in eradicating
nasal colonization with S. aureus when compared with placebo in the short term. The efficacy of mupirocin was also
comparable to a systemic regimen and superior to other topical antibacterials in a few small trials. However, 2–14-
day therapeutic courses of mupirocin did not typically result in long-term clearance of S. aureus colonization.
Repeated application of mupirocin is a potential option for reducing colonization in the long term, but the evolution
of resistance is a risk. Several of the studies included in this review described resistance associated with mupirocin
therapy, but the rates were typically low. Ultimately, the most important goal of intranasal mupirocin application is
to reduce subsequent clinical infection. The body of literature currently does not support routine administration of
prophylactic intranasal mupirocin to patients in an attempt to decrease the rate of clinical infection. Although the
available literature does not support routine use of topical intranasal mupirocin to prevent subsequent infections,
there may be yet-identified patient populations that could benefit. For a significant effect to be seen, one may
speculate as to the characteristics of patients who would benefit the most. Ideally, mupirocin should be used for
patients when the period of risk for infection is acute. Chronic infection risk, such as with dialysis patients, has
demonstrated minimal clinical benefit but increased risk for resistance. Examples of patients with acute disease who
may be candidates for prophylactic mupirocin treatment include patients who have undergone cardiac surgery,
patients with multiple trauma (especially those with head injuries), and, possibly, other selected critically ill patients
[38–41]. The selection of patients with high rates of nasal colonization or documented carriers is also important for
mupirocin therapy to be effective.
21
Olopatadine
Addition to the List
Note: Commonly prescribed medication.
Literature question
Is olopatadine effective and safe?
Literature search
Medline: Olopatadine AND efficacy AND limit to (meta-analysis or review)
e-CPS: Olopatadine
Cochrane: Olopatadine AND efficacy
Castillo M, Scott NW, Mustafa MZ, Mustafa MS, Azuara-Blanco A. “Topical antihistamines and mast cell
stabilisers for treating seasonal and perennial allergic conjunctivitis (Review).” The Cochrane Library (2015).

22
Olopatadine versus placebo or other control. The search identified two eligible studies comparing the antihistamine
olopatadine versus placebo, in Avunduk 2005, or other control, in Lanier 2001. The Avunduk 2005 study was a
three-arm trial comparing both olopatadine and ketotifen with placebo (Avunduk 2005). Duration of treatment was
four weeks, but two-week data were available. The sample size was relatively small, with 16 participants
randomised to olopatadine and 17 participants randomised to artificial tears (placebo). Mean and SD data were
available. Lanier 2001 compared the effect of topical olopatadine in people undergoing treatment with an oral
antihistamine (loratadine). Duration of treatment was one week. A total of 94 participants (49 versus 45) were
randomised but not masked. No SD data were available. Primary outcomes. Avunduk 2005 reported two of the four
symptoms prespecified as primary outcomes (participant-reported itching and watering eyes). Data on mean and SD
were available. Olopatadine had statistically significantly less severe itching (mean 0.76, SD 0.1) and tearing (mean
0.30, SD 0.1) than the placebo group (mean itching 1.85, SD 0.3; mean tearing 1.07, SD 0.2) after two weeks of
treatment. Lanier 2001 included participant-reported itching on a 4-point scale (from 1 to 4). After one week,
reported itching was less in the group treated with olopatadine (mean 2.21 versus 2.74, P = 0.044). Safety outcomes.
There were no adverse events or side effects associated with olopatadine in either study. Overall summary.
There was evidence from two small trials that olopatadine may be effective in improving some ocular symptoms.
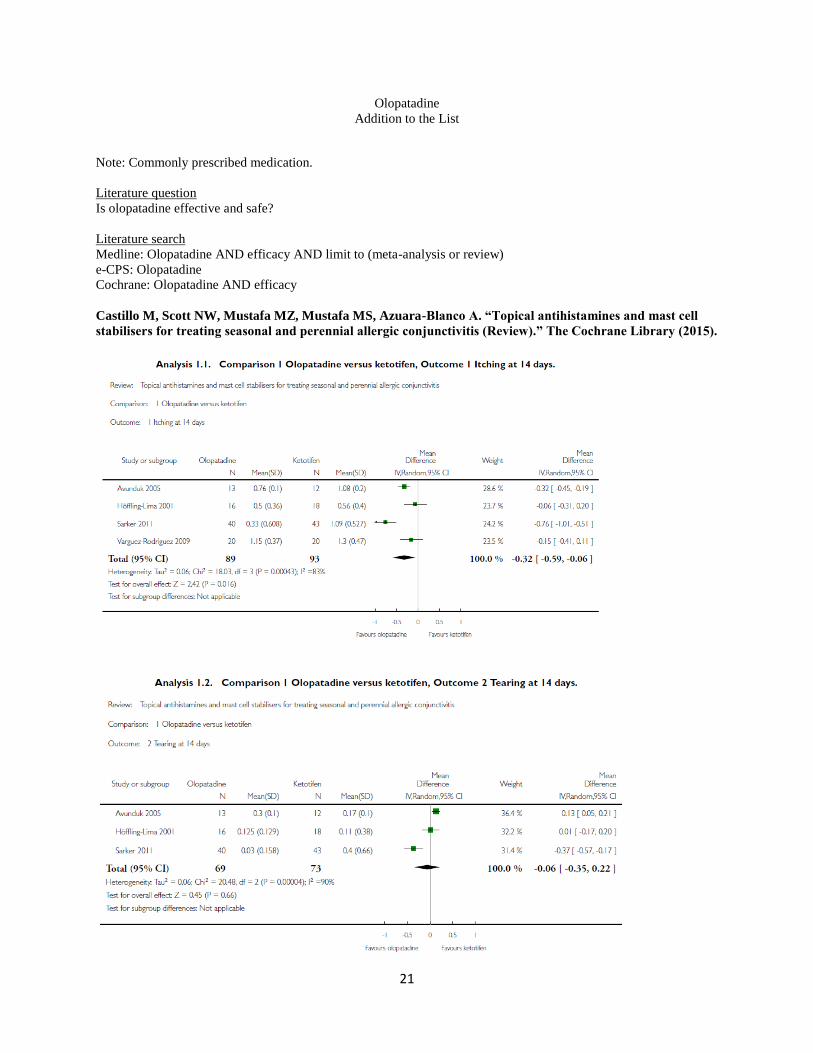
Olopatadine versus ketotifen. The search identified four eligible studies comparing the antihistamines olopatadine
and ketotifen (Avunduk 2005; Höffling- Lima 2001; Sarker 2011; Varguez-Rodriguez 2009). One study was a three-
arm trial comparing both olopatadine and ketotifen with placebo (Avunduk 2005). Drug concentration was the same
in all studies (olopatadine 0.1% and ketotifen 0.025%), except Höffling-Lima 2001, which used ketotifen 0.05%.
Duration of treatment was four weeks in three studies, Avunduk 2005, Höffling-Lima 2001, and Varguez-Rodriguez
2009, and two weeks in one study (Sarker 2011). In all studies the sample size was relatively small. The number of
participants randomised to olopatadine and ketotifen were 16 versus 16 (Avunduk 2005), 20 versus 20 (Höffling-
Lima 2001), 46 versus 46 (Sarker 2011), and 20 versus 20 (Varguez-Rodriguez 2009), respectively. Primary
outcomes. Although four studies reported at least two of the four symptoms prespecified as primary outcomes
(itching and tearing), there was some variation in how these outcomes were reported. All four studies collected data
on participant-reported itching using a 0-3 scale. Two studies reported mean and SD values (Avunduk 2005; Sarker
2011). Two studies did not find any differences between olopatadine and ketotifen in itching reporting (Avunduk
2005;Höffling-Lima 2001), while two studies found a greater reduction in itching with olopatadine than with
ketotifen after two weeks of treatment (Sarker 2011; Varguez-Rodriguez 2009). Sarker 2011 reported two-week
mean scores of 1.09 (SD 0.53) with ketotifen and 0.33 (SD 0.60) with olopatadine. A random-effects meta-analysis
of these four studies showed evidence of a statistically significant difference in favour of olopatadine in the
reduction of itching at 14 days (mean difference (MD) -0.32, 95% confidence interval (CI) -0.59 to -0.06) (Figure
5). However, there was high statistical heterogeneity (I2 = 83%). Two studies presented data for participant-reported
ocular irritation (described in both studies as burning) Höffling-Lima 2001; Varguez-Rodriguez 2009). Höffling-
Lima 2001 did not find any differences between groups, while Varguez-Rodriguez 2009 reported a statistically
significant greater reduction in burning in the lopatadine group (P < 0.05). All four studies reported data on tearing.
Only one study reported differences between groups after two weeks (Sarker 2011),with the group treated with
olopatadine having less tearing (mean 0.03, SD 0.16) than the one treated with ketotifen (mean 0.40, SD 0.66).
Three studies assessed tearing at 14days (Avunduk 2005;Höffling- Lima 2001; Sarker 2011). A random-effects
meta-analysis of these studies found no evidence of a difference between lopatadine and ketotifen (MD -0.06, 95%
CI -0.35 to 0.22). There was no evidence of a difference in the reduction of tearing scores at 14 days between the 2
groups (Figure 6). Once again, there was high statistical heterogeneity between the studies (I2 = 90%).We knew of
two studies that collected data on photophobia (Höffling-Lima 2001; Sarker 2011). There were no differences
between groups. Safety outcomes. No serious adverse events were reported in the four papers. Three studies did not
report any side effects (Avunduk 2005; Höffling- Lima 2001; Varguez-Rodriguez 2009). In one study, ketotifen was
associated with a mild stinging sensation of short duration (less than 30 minutes) in 13 out of 43 participants; no
participants treated with olopatadine reported such discomfort (Sarker 2011). Overall summary. There was some
evidence from individual trials that olopatadine may be more effective than ketotifen in improving some ocular
symptoms such as itching. Both drugs are safe.

23
Atsuki Fukushima and Nobuyuki Ebihara. “Efficacy of Olopatadine versus Epinastine for Treating Allergic
Conjunctivitis Caused by Japanese Cedar Pollen: A Double-Blind Randomized Controlled Trial.” Advances
in Therapeutics 2014 (31): 1045-1058.
We found that administration of olopatadine significantly reduced self-assessed ocular itching at 7 min (the primary
endpoint) and investigator assessed conjunctival hyperemia at 20 min (the main secondary endpoint) compared with
epinastine without apparent safety concerns. These results support the use of olopatadine for the treatment of allergic
conjunctivitis caused by Japanese cedar pollen. In a similarly designed study, Abelson and Greiner [9] performed
CAC tests in 68 subjects and reported that olopatadine 0.1% significantly reduced itching and redness compared
with levocabastine 0.05%. The authors also reported that olopatadine was more tolerable than levocabastine in terms
of reduced discomfort following administration. In another study in which 32 subjects underwent CAC tests, Berdy
et al. [10] reported that olopatadine 0.1% was more effective in reducing ocular itching than ketotifen fumarate
0.025% while causing less ocular discomfort. They also found that olopatadine was preferred over ketotifen by
approximately three times as many patients. In addition, Lanier et al. [8] reported that olopatadine 0.1% was more
effective than epinastine 0.05% in controlling allergic symptoms induced by a CAC test. Taken together, the results
of these studies support the use of olopatadine 0.1% as an effective treatment for preventing allergic conjunctivitis
and other ocular allergic symptoms. However, data should be interpreted with caution because there was no negative
control group treated with physiological saline.

24
Cromolyn
Addition to the List
Note: Commonly prescribed medication.
Literature question
Is cromolyn effective and safe?
Literature search
Medline: Cromolyn AND efficacy AND limit to (meta-analysis)
Cochrane: Cromolyn AND efficacy
Castillo M, Scott NW, Mustafa MZ, Mustafa MS, and Azuara-Blanco A. “Topical antihistamines and mast
cell stabilisers for treating seasonal and perennial allergic conjunctivitis (Review).” The Cochrane Library
(2015)
Nedocromil sodium/sodium cromoglycate versus placebo – Primary outcomes. There was some evidence from
individual trials that nedocromil sodium or sodium cromoglycate is more effective than placebo in improving ocular
symptoms. However, it was not possible to perform formal meta-analyses for this comparison due to variation in
how outcomes were reported and the lack of suitable data (especially standard deviations (SDs)).
Pacharn P and Vichyanond P. “Immunomodulators for conjunctivitis.” Current Opinion in Allergy and
Clinical Immunology. 2013 (13): 550-557
Cromolyn was found to be effective in treating seasonal allergic conjunctivitis [23]. Cromolyn 4% solution was
found to be more effective than 2% solution [24]. However, limitation of ophthalmic cromolyn is that it has to be
applied four times per day. In fact, nedocromil 2% solution applied twice daily was found to be as effective as
cromolyn 2% solution applied four times daily [25]. By increasing concentration of cromolyn to 4%, a satisfactory
effect was obtained with twice-daily application [26]. Both drugs were tried in patients with VKC with more
effective results attained with the use of nedocromil eye drops [27,28]. Moreover, at 22 weeks of treatment,
improvement of severe conjunctivitis sign of VKC such as pannus, papilla and keratitis was improved with
nedocromil treatment only [28]. In an extensive review of experience with nedocromil, it was found to be effective
in seasonal, perennial and VKC with four times daily application being more efficient than twice daily [29].
Surprisingly, the effect of nedocromil was noted to be as rapid as 15 min in this review.
van der Wouden JC, Uijen JHJM, Bernsen RMD, Tasche MJA, de Jongste JC, and Ducharme FM. “Inhaled
sodium cromoglycate for asthma in children (Review).” The Cochrane Library (2011)

25
Effect of interventions. Symptoms. Only four studies provided results for the percentage of symptom-free
days: our primary outcome measure (Figure 2; Table 3). In all but one of the studies (Cogswell 1985), the
confidence interval included the point of no difference. Pooling the results revealed no significant difference
between DSCG and placebo (WMD 6.76% favouring DSCG, 95%CI -2.18 to 15.70), random-effects model. Mean
overall symptom scores favoured DSCG in direction in six out of ten studies (Figure 5;Table 6).The 95%confidence
intervals of four of the studies included the point of no difference. Pooling the results (test of homogeneity rejected,
hence random-effects model) showed an overallmean difference of -0.22 symptom score points (WMD), favouring
the DSCG group (95%CI -0.34 to - 0.09), hence statistically significant.
Sodium Cromoglycate. e-CPS. Date of Revision: December 2011.
Nausea, vomiting, diarrhoea, abdominal discomfort, headache, insomnia, skin rashes sneezing, cough, unpleasant
taste in the mouth, and joint pains have been reported. Hypersensitivity reactions have been reported rarely.
Possible immunologic changes resulting in reactions such as polymyositis, pneumonitis and heart failure, urticaria
and anaphylaxis, have been reported.
Cases of erythema, urticaria or maculopapular rash have been reported and these have cleared within a few days on
withdrawal of the drug.

26
Montelukast
Addition to the List
Note: Commonly prescribed medication.
Literature question
Is montelukast effective and safe?
Literature search
Medline: Montelukast or Leukotriene Antagonists AND efficacy AND limit to (meta-analysis)
Cochrane: Montelukast AND efficacy
Watts K and Chavasse RJPG. “Leukotriene receptor antagonists in addition to usual care for acute asthma
in adults and children (Review).” The Cochrane Library (2012).
27
Oral montelukast or zafirlukast in addition to usual care
Hospitalisation (primary outcome). There was no significant difference between oral leukotriene receptor
antagonists (LTRAs) and control in the risk of admission to hospital in three studies including 194 children
comparing oral montelukast to control in addition to usual care for acute asthma (RR 0.86; 95% CI 0.21 to 3.52;
Figure 3).
Lung function. There was a significant difference in FEV1 favouring LTRAs in the combined results of a single
study on 641 adults on different doses of zafirlukast (MD 0.08; 95% CI 0.01 to 0.14; Figure 4). In Silverman 2004a
20 mg of zafirlukast was administered orally and in Silverman 2004b the dose was 160 mg. Change in predicted
FEV1 did not show a significant effect of LTRAs in a single study ofmontelukast in 26 children (MD-3.10;
95% CI -12.70 to 6.50; Analysis 1.4).
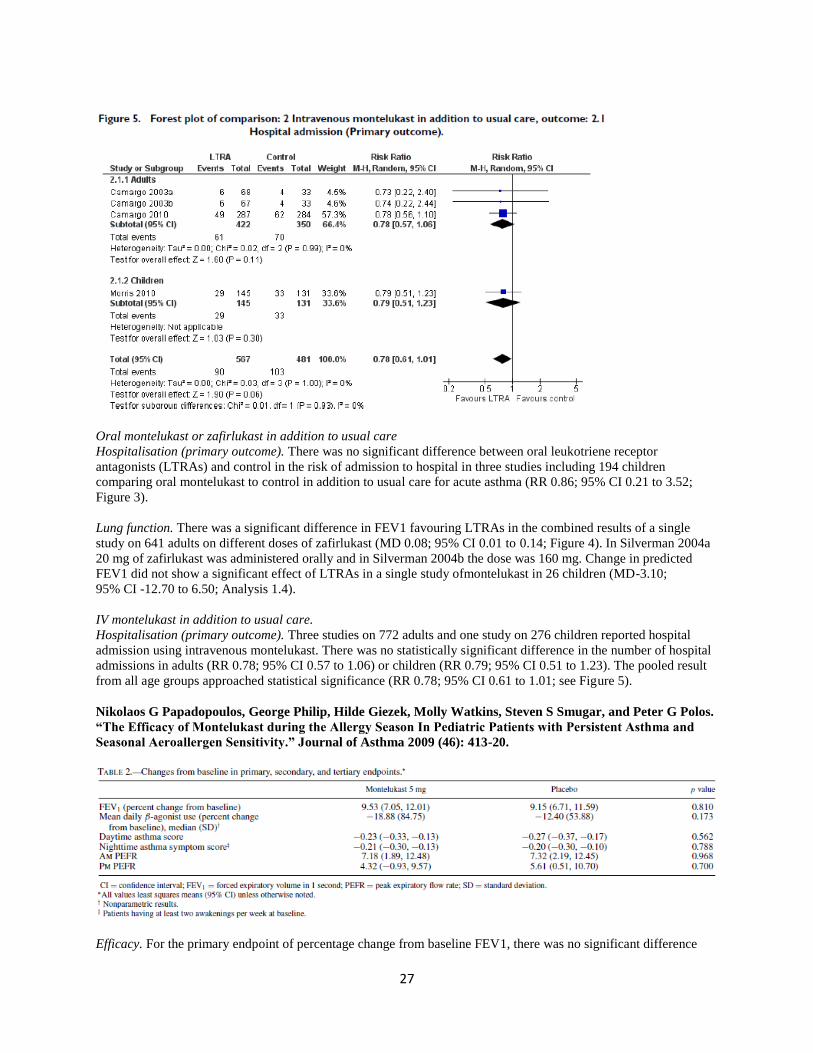
IV montelukast in addition to usual care.
Hospitalisation (primary outcome). Three studies on 772 adults and one study on 276 children reported hospital
admission using intravenous montelukast. There was no statistically significant difference in the number of hospital
admissions in adults (RR 0.78; 95% CI 0.57 to 1.06) or children (RR 0.79; 95% CI 0.51 to 1.23). The pooled result
from all age groups approached statistical significance (RR 0.78; 95% CI 0.61 to 1.01; see Figure 5).
Nikolaos G Papadopoulos, George Philip, Hilde Giezek, Molly Watkins, Steven S Smugar, and Peter G Polos.
“The Efficacy of Montelukast during the Allergy Season In Pediatric Patients with Persistent Asthma and
Seasonal Aeroallergen Sensitivity.” Journal of Asthma 2009 (46): 413-20.
Efficacy. For the primary endpoint of percentage change from baseline FEV1, there was no significant difference

28
between montelukast and placebo (LS mean: 9.53% vs. 9.15%, respectively; difference: 0.38% [95% CI: −2.74,
3.50]; p = 0.810) (Table 2). There were no significant differences between treatment groups in the analyses of the
primary endpoint by prespecified subgroups (Figure 2); treatment difference was consistent among subgroups for
gender, age, race, concomitant inhaled steroid use, asthma severity, and pollen level. A somewhat higher treatment
difference was observed for the subgroup spring 2007 (treatment difference in LS means of 2.52%). Montelukast
was significantly more effective than placebo for the investigator’s global asthma evaluation (LS mean: 2.71 vs.
2.98;p <0.05) (Figure 3A) and parent/guardian global asthma evaluation (LS mean: 2.63 vs. 2.90;p <0.05) (Figure
3B).
Xun Chen, Ke Wang, Min Jiang, and Guang-Min Nong. “Leukotriene receptor antagonists for small-airway
abnormalities in asthmatics: a systematic review and meta-analysis.” Journal of Asthma 2013 (50): 695-704.
LTRAs versus placebo. HRCT One RCT (41) showed that an LTRA was more efficacious than placebo in reducing
regional air-trapping on HRCT (MD 61.00; 95% CI, 26.32–95.68; p50.001). FEF25–75% Three RCTs [41,46,47]
reported changes in FEF25–75% when comparing LTRAs with placebo. There was no heterogeneity among the 3
RCTs (p=0.40, I2=0%). The pooled MD for the treatment effect was 5.29 (95% CI, -4.05 to 14.62; p=0.27),
showing no improvement in the FEF25–75% in participants receiving LTR compared with patients receiving
placebo (Figure 2). RV Three RCTs [41,46,48] reported changes in RV when comparing LTRAs with placebo. One
RCT [48] showed its result for the parameter of residual volume as the percent predicted (RV% predicted), while
other RCTs [41,46] displayed their results as the ratio of the residual volume to the total lung capacity (RV/TLC).
Therefore, the SMD was used as a summary statistic. There was no heterogeneity among the 3 RCTs (p=0.58,
I2=0%). The pooled SMD for the treatment effect was -0.85 (95% CI, -1.29 to -0.42; p<0.001), showing a reduction
in RV in participants receiving LTRAs compared with patients receiving placebo (Figure 3).

29
Drugs Used for Maintenance Therapy for Asthma in Children; Mary Noseworth, MD, FRCPC, and Mark
Montgomery, MD, FRCPC. e-CPS. Date of Revision: April 2015.
Class Drug Dosage Adverse Effects Comments Cost
Leukotriene
Receptor
Antagonists
Montelukast
Singulair,
Montelukast,
generics
>15y: 10mg QHS po
6-14y: 5mg
(chewable tablet)
QHS po
2-5y: 4mg
(chewable tablet or
granules) QHS po
Headache, abdominal
pain, flu-like
symptoms,
hepatotoxicity (rare).
Reports of
neuropsychiatric
effects, e.g. depress,
agitation
Decreased
montelukast levels:
carbamazepine,
rifampin,
phenobarbital,
phenytoin
$$
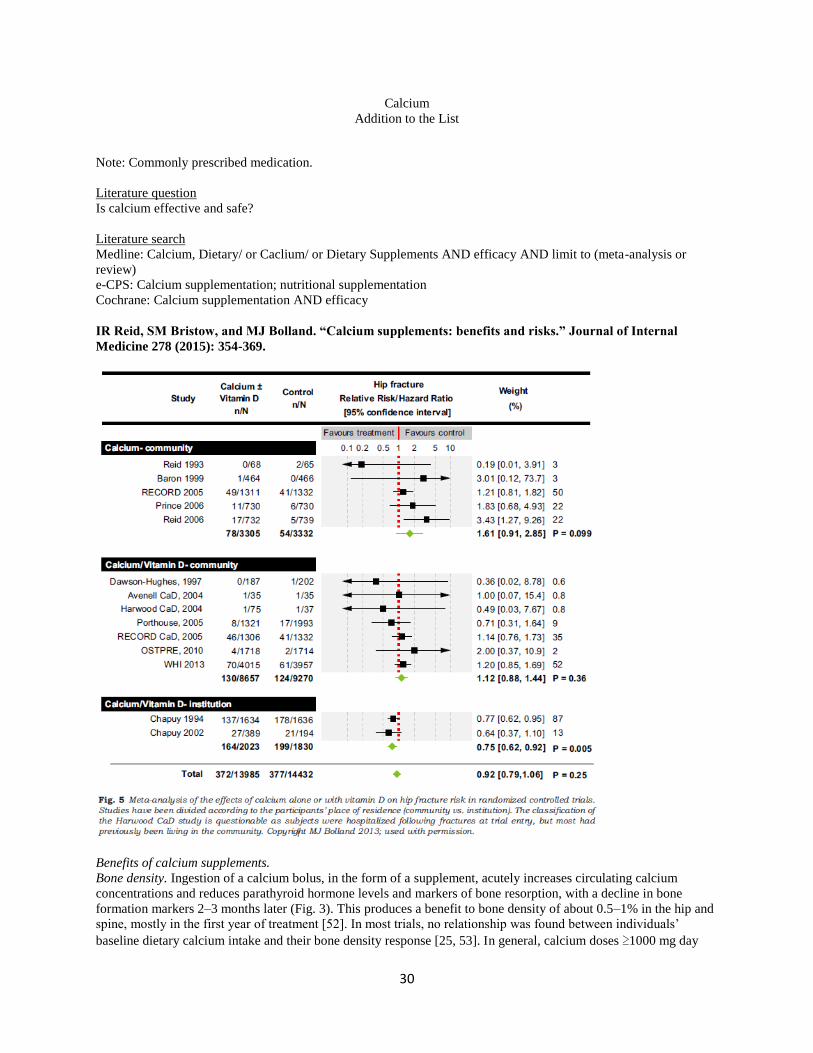
30
Calcium
Addition to the List
Note: Commonly prescribed medication.
Literature question
Is calcium effective and safe?
Literature search
Medline: Calcium, Dietary/ or Caclium/ or Dietary Supplements AND efficacy AND limit to (meta-analysis or
review)
e-CPS: Calcium supplementation; nutritional supplementation
Cochrane: Calcium supplementation AND efficacy
IR Reid, SM Bristow, and MJ Bolland. “Calcium supplements: benefits and risks.” Journal of Internal
Medicine 278 (2015): 354-369.
Benefits of calcium supplements.
Bone density. Ingestion of a calcium bolus, in the form of a supplement, acutely increases circulating calcium
concentrations and reduces parathyroid hormone levels and markers of bone resorption, with a decline in bone
formation markers 2–3 months later (Fig. 3). This produces a benefit to bone density of about 0.5–1% in the hip and
spine, mostly in the first year of treatment [52]. In most trials, no relationship was found between individuals’
baseline dietary calcium intake and their bone density response [25, 53]. In general, calcium doses ≥1000 mg day

31
have been used in supplement studies [54]. Doses of 250–600 mg day have been found to produce no or minimal
effects on BMD [55, 56], except in individuals with very low calcium intakes [12, 57]. Collectively, these findings
suggest that calcium supplements act as weak anti-resorptive agents, reducing bone turnover whatever the baseline
calcium intake, and producing a one-off gain in bone density as a result of filling-in of some of the osteoclastic
resorption sites. This, however, does not produce cumulative benefits in terms of bone mass.
Fractures. In the early 1990s, Chapuy et al. conducted the first study designed to test the anti-fracture efficacy of
calcium (with vitamin D in this case) [14]. At 18 months, there was a 43% decrease in hip fractures in a completers’
analysis, which equated to a 26% decline in fracture rate on an intention-to treat basis. The effect was similar at 36
months [58]. Non-vertebral fracture numbers were decreased by 25% and 17%, at 18 and 36 months, respectively
(intention-to-treat analysis). This led to the conclusion that calcium and vitamin D were an essential part of
osteoporosis management, because of their demonstrable anti-fracture efficacy and their presumed safety. Several
features of this study by Chapuy and colleagues should be mentioned. First, it was carried out in frail elderly women
living in institutions. Secondly, calcium intakes were ~500 mg day_1 and mean serum 25-hydroxyvitamin D
concentration was 25 nmol L_1 in placebo subjects 12 months into the study, equating to 13.7 nmol L_1 after
correction for inaccuracies in assay calibration [59]. Finally, calcium plus vitamin D produced a between-group
difference in total hip bone density of 7.3%, a response larger than reported with any anti-osteoporotic medication.
Such a large effect can only be explained as a response to the treatment of osteomalacia, which is highly likely to
have affected many of these women, considering their marked vitamin D deficiency. In the last 10 years, the results
of five large trials of calcium supplements have been reported; none showed beneficial effects on fractures in their
primary analyses [25, 60–63], although some found beneficial and adverse fracture effects in secondary analyses.
This is reflected in recent meta-analyses, which showed either no effect of calcium on fracture or small effects [54,
64]. The influential nature of the study by Chapuy and colleagues in the first of these meta-analyses, by Tang et al.
[54], is noteworthy. In the subgroup analyses, the group in which Chapuy lies usually is found to have the greatest
therapeutic effect. Thus, total fractures were only reduced by 6% in community- dwelling subjects. Fracture
prevention was reduced with trial duration, reaching no effect at 7 years, and with declining compliance [relative
risk 0_96, 95% confidence interval (CI) 0_91–1_01, with <80% compliance] (Fig. 4). Tang et al. did not consider
hip fracture as a separate endpoint, but at least three meta-analyses have shown upward trends in hip fracture from
the use of calcium alone [51, 64, 65]. The effects of calcium plus vitamin D on hip fracture are dominated by the
results of Chapuy et al., but analyses of community- dwelling subjects have shown no evidence of fracture
prevention. Recently, the investigators from the Women’s Health Initiative (WHI) demonstrated a significant
interaction between randomization to calcium plus vitamin D and to hormones on risk of hip fracture in their study
[66]. When results from the non-hormone-treated WHI subjects were used in the meta-analysis, there was a
trend towards an adverse effect on hip fracture risk from the use of calcium plus vitamin D [65] (Fig. 5).
In summary, calcium supplements clearly have small beneficial effects on bone density, but a cumulative density
benefit has not been demonstrated in most studies. As a result, the anti-fracture efficacy of calcium supplements
remains an open question; there is no evidence to support a role in hip fracture prevention (other than in a cohort
with severe vitamin D deficiency) and total fracture numbers are only reduced by 0–10%, depending on which meta-
analysis is considered. This has led to the US Preventive Services Task Force not recommending their use [67], a
view supported by some journal editorials [68].
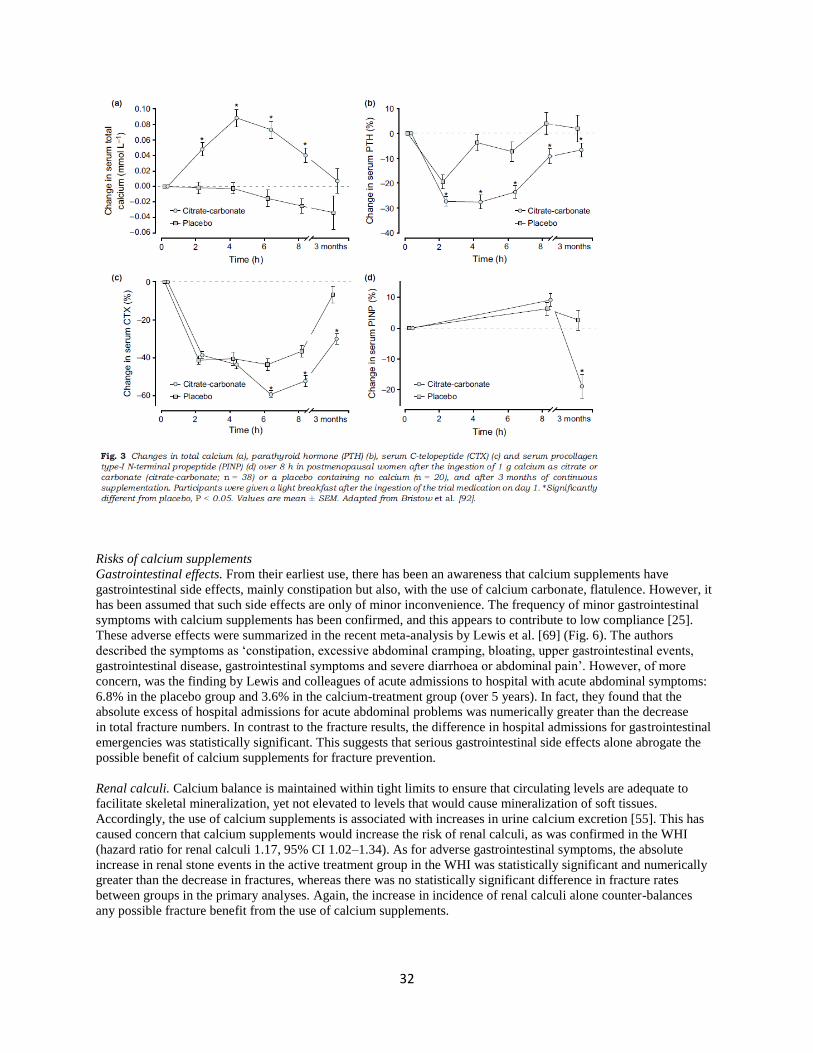
32
Risks of calcium supplements
Gastrointestinal effects. From their earliest use, there has been an awareness that calcium supplements have
gastrointestinal side effects, mainly constipation but also, with the use of calcium carbonate, flatulence. However, it
has been assumed that such side effects are only of minor inconvenience. The frequency of minor gastrointestinal
symptoms with calcium supplements has been confirmed, and this appears to contribute to low compliance [25].
These adverse effects were summarized in the recent meta-analysis by Lewis et al. [69] (Fig. 6). The authors
described the symptoms as ‘constipation, excessive abdominal cramping, bloating, upper gastrointestinal events,
gastrointestinal disease, gastrointestinal symptoms and severe diarrhoea or abdominal pain’. However, of more
concern, was the finding by Lewis and colleagues of acute admissions to hospital with acute abdominal symptoms:
6.8% in the placebo group and 3.6% in the calcium-treatment group (over 5 years). In fact, they found that the
absolute excess of hospital admissions for acute abdominal problems was numerically greater than the decrease
in total fracture numbers. In contrast to the fracture results, the difference in hospital admissions for gastrointestinal
emergencies was statistically significant. This suggests that serious gastrointestinal side effects alone abrogate the
possible benefit of calcium supplements for fracture prevention.
Renal calculi. Calcium balance is maintained within tight limits to ensure that circulating levels are adequate to
facilitate skeletal mineralization, yet not elevated to levels that would cause mineralization of soft tissues.
Accordingly, the use of calcium supplements is associated with increases in urine calcium excretion [55]. This has
caused concern that calcium supplements would increase the risk of renal calculi, as was confirmed in the WHI
(hazard ratio for renal calculi 1.17, 95% CI 1.02–1.34). As for adverse gastrointestinal symptoms, the absolute
increase in renal stone events in the active treatment group in the WHI was statistically significant and numerically
greater than the decrease in fractures, whereas there was no statistically significant difference in fracture rates
between groups in the primary analyses. Again, the increase in incidence of renal calculi alone counter-balances
any possible fracture benefit from the use of calcium supplements.

33
Cardiovascular effects. We have recently reviewed the cardiovascular side effects of calcium supplements in detail
elsewhere [70]. Deposition of calcium into arterial walls is an integral part of the atherosclerotic process, so there
has been concern for some decades that calcium supplementation might increase the risk of cardiovascular disease.
However, this concern has principally been expressed by vascular biologists [71] and often not considered seriously
by those involved in the therapeutic management of osteoporosis. By contrast, there has been long-standing concern
about the use of calcium in patients with chronic renal failure. In pre-dialysis patients, calcium supplements increase
coronary artery calcification [72] and adversely impact on survival [73]. These patients have renal function
comparable to that seen in many older individuals at risk of osteoporosis [74]. This concern has resulted in ‘the
demise of calcium-based phosphate binders’ in many centres [75, 76]. More than a decade ago, we and others
observed that calcium supplements appeared to produce small benefits in terms of blood pressure [77, 78] and
circulating lipid levels [79], although the latter findings have not been consistent. On the basis of these possible
beneficial effects, a number of cardiovascular events were pre-specified as secondary endpoints in the Auckland
Calcium Study. We were surprised to discover that the incidence of myocardial infarction was significantly
increased in this study, and there was an upward trend in stroke incidence [80]. Subsequently, as part of an
international consortium, we conducted a meta-analysis of all trials in older adults randomly assigned to calcium or
placebo for ≥1 year [81]. This analysis confirmed a 27% increase in the incidence of myocardial infarction and again
suggested an adverse effect on the risk of stroke. A quarter of myocardial infarctions in this analysis were self-
reported; the hazard ratio was 1.44 (95% CI 1.08–1.91, P = 0.013) if these events were excluded [82]. A similar
numerical increase in myocardial infarction risk associated with calcium monotherapy has been found in two
subsequent meta-analyses [83, 84], although their statistical significance was marginal because of smaller numbers
in those analyses. Our findings with calcium monotherapy appeared to contradict those of the WHI, which
concluded that there was no adverse effect of calcium plus vitamin D on cardiovascular health [85]. However, their
analyses did demonstrate an interaction between body mass index and cardiovascular disease risk, such that the
incidence of myocardial infarction increased by 17% in non-obese subjects. In addition, an almost significant hazard
ratio of 1.08 (95% CI 0.99–1.19) was reported for a composite endpoint that included myocardial infarction and
coronary artery revascularization, which is not reassuring from a safety perspective. The WHI differed in several
important respects from those studies included in the first meta-analysis led by Bolland [80]. The subjects were, on
average, 10 years younger, the active treatment group received vitamin D in addition to calcium, and participants
were accepted into the trial even if they were already taking calcium supplements, and were permitted to continue
these self-administered calcium supplements throughout the trial. Thus, at randomization, 54% of subjects were self-
administering calcium, and this proportion rose to 69% at trial end. We hypothesized that contamination with self-
administration of calcium might have obscured an adverse effect of supplements on cardiovascular disease risk and
designed a protocol to address this issue. Following approval of the analysis plan by the NIH, we obtained the
publicly available WHI data set. We found a significant interaction between self-administration of calcium
supplements and the effect of the calcium/vitamin D intervention on cardiovascular disease risk [86]. There were no
adverse effects from the addition of further calcium in those already taking a supplement, but, in calcium-naive
subjects, an increase in risk very similar to that found in our first meta-analysis was demonstrated. The adverse
trends applied to both myocardial infarction and stroke. As shown in Fig. 7, the time courses of the effects of
calcium supplementation on myocardial infarction and stroke are very similar between our meta-analysis of calcium
monotherapy and the analysis of the calcium-naive subjects in the WHI. It is also noteworthy that the onset of
adverse effect is more rapid for myocardial infarction than for stroke and this difference is consistent between the
two separate data sets. Thus, there is no evidence that the addition of vitamin D abrogates the adverse effects
of calcium on cardiovascular disease risk, and this is consistent with the much larger body of clinical trial evidence
suggesting that vitamin D is not cardioprotective [87, 88]. Recently, Lewis et al. have performed another meta-
analysis of these data, but excluding men and self-reported events [89]. For calcium monotherapy and myocardial
infarction, their results were very similar to those of Bolland and colleagues [81] (relative risk 1.37, 95% CI 0.98–
1.92); the lack of statistical significance was accounted for by the smaller numbers they included (6333 vs. 10 210).
For their analysis of calcium plus vitamin D, Lewis and colleagues added two groups of women not included in the
meta-analysis by Bolland et al.: 20 000 participants from the WHI who were already taking calcium at the time of
randomization, and 6000 women from the study of Larsen et al. [90], which was not a randomized, controlled trial.
Because we have shown that self-administration of calcium significantly influenced the cardiovascular outcomes of
the WHI [86], the first of these additions is not appropriate. In the Larsen study [90], the authors divided the
residential area into district ‘clusters’, with one ‘cluster’ per intervention. Prospective participants knew what their
intervention would be before agreeing to participate, and there was a higher participation rate among those offered
calcium plus vitamin D. The use of cardiovascular medications, sedatives and analgesics was lower in this group
compared with those agreeing to act as controls, suggesting a difference in cardiovascular disease risk and other

34
comorbidities at baseline, which would bias the study against finding an adverse effect of calcium supplements.
Thus, this study does not qualify for inclusion in a meta-analysis of randomized, controlled trials. In the analysis by
Lewis et al., no effect of calcium plus vitamin D supplements on total coronary heart disease was found, but 82% of
the weight in this analysis was from the WHI, so it is essentially a re-publication of those data. As for
gastrointestinal and renal adverse effects, it is instructive to compare the absolute increase in numbers of
cardiovascular events with the absolute decrease in number of fractures. For calcium monotherapy, treatment of
1000 persons for 5 years will cause 14 myocardial infarctions, 10 strokes and 13 deaths, while preventing 26
fractures [81]. Thus, consideration of the cardiovascular adverse effects in isolation suggests that calcium
supplements produce no net benefit. When these cardiovascular adverse events are considered alongside
gastrointestinal events and renal calculi, it is apparent that a negative impact of calcium supplement use is likely.
Benjamin MP Tang, Guy D Eslick, Caryl Nowson, Caroline Smith, and Alan Bensoussan. “Use of calcium or
calcium in combination with vitamin D supplementation to prevent fractures and bone loss in people aged 50
years and older: a meta-analysis.” The Lancet 2007 (370):657-66.
Discussion. Our meta-analysis has shown that calcium supplementation, alone or in combination with vitamin D, is
effective in the preventive treatment of osteoporotic fracture. Over an average treatment duration of 3・5 years,
the risk of fracture was reduced and was accompanied by a reduction of bone loss at the hip and spine. The fracture
risk reduction was greater in individuals who were elderly, lived in institutions, had a low bodyweight, had a low
calcium intake, or were at a high baseline risk than it was in others. The treatment effect was consistent irrespective
of sex, fracture sites, or history of previous fracture. Moreover, the treatment was similarly effective whether the
person used calcium or calcium in combination with vitamin D supplementation. However, the treatment was less
effective if compliance was poor. For calcium-only supplementation, a minimum dose of 1200 mg is needed for best
therapeutic effect. For calcium in combination of vitamin D supplementation, a minimum dose of 800 IU of vitamin
D is recommended. Although addition of vitamin D supplementation was not shown to offer additional risk
reduction over and above the use of calcium alone, a significant difference was observed between the effects of
different vitamin D doses. This discrepancy could be due to statistical artifact. However, we would like to point out
that our analysis was limited by the scarcity of data for vitamin D doses higher than 800 IU. It is possible that
vitamin D does have a beneficial effect when the dose is large enough (ie, >800 IU). In the absence of such data, we
recommend that if vitamin D is to be used as an adjunct supplementation to calcium, its dose should be at least
800 IU or more. Poor compliance is a major obstacle to obtaining the full benefit of calcium supplementation. When
we analysed trials with a compliance rate of at least 80% separately, the risk reduction was doubled. We noted a
greater treatment effect in individuals with low dietary calcium intake than in those whose dietary intake was high.
This result is important since inadequate dietary calcium is prevalent throughout the world, especially in elderly
people and women.47,48 It is also consistent with what is understood about the pathophysiology of osteoporosis.
After midlife there is an age-related yearly loss of bone in both sexes of about 1%,49 which is accelerated to 2% for
up to 14 years in women around the age of menopause.50 This bone loss is characterised by the loss of calcium from
bone. To keep bone loss to a minimum, increased dietary calcium is needed to offset the continuing loss. It is
possible that the role of supplementation might diminish with increased intake. It can be difficult, however, for
many elderly people to maintain a calcium intake above 1000 mg per day, especially when energy intake falls with
increasing age. Our study has implications for both clinicians and policymakers. The estimated number needed to
treat shows that 63 patients will need to be treated over 3・5 years to prevent one fracture. This result is comparable
to other preventive treatments such as statins, in which 40 people would need to be treated for 5 years to prevent one
major cardiovascular event,52 and it is substantially better than aspirin treatment, in which more than 270
participants would need to be treated for 6 years to prevent one cardiovascular event.53 Furthermore, the number
needed to treat decreased to 30 or fewer in individuals who were elderly, had low dietary calcium intake, or were
compliant with calcium supplementation. On the basis of our recommended minimum dose of 1200 mg of calcium
or 800 IU of vitamin D, many formulations of calcium or combined calcium with vitamin D tablets that are available
contain insufficient quantities of the active ingredients. In view of the large number of calcium supplementation
tablets sold worldwide, adequate dosage is an important issue to address if best possible public-health benefits are to
be realised. Our study also has implications for future studies of cost-effectiveness. Although our findings confirmed
that therapeutic effect generally increased with age, it also suggested that the effect becomes much greater and
clinically significant after the age of 70 years. The cost-effectiveness of selecting a specific age group, such as
people aged 70 years or older, will therefore need to be addressed in future studies. Our study has several strengths.

35
e-CPS – Calcium Salts: Oral
Serious Warnings and Precautions. A 2010 meta-analysis showed an association between high-dose calcium
supplementation (taken without supplemental vitamin D) and an increased risk of myocardial infarction [BMJ 2010;
341:c3691]. Since then, cohort studies, randomized controlled trials and systematic reviews have shown conflicting
results, ranging from positive, neutral and negative associations between calcium and increased cardio vascular
disease risk [JAMA 2013; 173(8):639-46]. Important factors to consider are the dietary versus supplemental sources
of calcium, low versus high dose and benefit versus risk in the particular patient. Further study is warranted to
clarify the effect of calcium on cardiovascular disease. Until further safety information is available, high doses of
calcium supplements should not be used in those who do not need extra calcium. Those who have inadequate
calcium intake should consider first increasing dietary intake before taking supplements. Health Canada
recommends that patients not exceed 1500 mg/day from supplements alone; some experts recommend a lower
maximum of 500 mg/day from supplements alone.

36
Vaginal ring eluting etonogestrel and ethinyl estradiol
Addition to the List
Note: Commonly prescribed medication.
Literature question
Is the combined hormonal vaginal ring effective and safe?
Literature search
Medline: combined hormonal vaginal ring AND efficacy AND limit to (meta-analysis)
Cochrane: combined hormonal vaginal ring AND efficacy And limit to (meta-analysis or review)
Lopez LM, Grimes DA, Gallo MF, Stockton LL, and Schulz KF. “Skin patch and vaginal ring versus
combined oral contraceptives for contraception (Review).” The Cochrane Library (2013).
Vaginal ring versus COC. Effectiveness Contraceptive effectiveness was not significantly different for the vaginal
ring and comparison COCs in the eight trials that reported pregnancy. Compliance (adherence) Compliance results
varied across the four vaginal ring studies with those data. One found poorer compliance among the ring users, two
showed no difference, and one showed ‘perfect’ use was more common for the vaginal ring users compared to COC
users. Cycle control The ring trials with bleeding data generally showed fewer problems for the ring group
compared to the COC group. In three of seven trials with such data, the ring group had fewer episodes of
breakthrough bleeding and spotting. One study showed the ring group had a greater mean for breakthrough bleeding
and spotting days. Fewer problems may relate to steady-state hormone levels, in contrast to peaks and troughs with
oral contraceptives. The better bleeding patterns did not translate into less discontinuation, though. Adverse events
For adverse events, five patch trials and seven ring trials reported data. Ring users had more vaginal irritation and
discharge than COC users in several trials, but less nausea and acne. One trial showed ring users had fewer reports
of worsening body weight, headaches, or sex drive. Another found less irritability, depression, and vaginal dryness
among the ring users than the COC users. However, that trial investigated certain tolerability issues, presumably
actively, whereas other researchers usually summarized participants’ reports of adverse events. Participants may
report more problems if prompted, but the effect should be similar in both groups. Since these trials were not
blinded, the results could be biased. For the etonogestrel ring users, the risk of deep venous thrombosis was 149
(95% xCI 18 to 538) per 100,000 women, but one woman with deep venous thrombosis had an additional risk
factor. Because of the rarity of venous thromboembolism, these randomized controlled trials are not informative
concerning comparative risk.

37
Estrogen (conjugated estrogens & ethinyl estradiol)
Addition to the List
Note: Commonly prescribed medication.
Literature question
Is estrogen effective and safe?
Are conjugated estrogens effective and safe?
Literature search
Medline: Conjugated estrogens AND efficacy AND limit to (meta-analysis or review); Ethinyl estrogen AND
efficacy AND limit to (meta-analysis or review)
Cochrane: Conjugated estrogens AND efficacy And limit to (meta-analysis or review); Ethinyl estrogen AND
efficacy And limit to (meta-analysis or review)
Boardman HMP, Hartley L, Eisinga A, Main C, Roque I Figuls M, Bonfill Cosp X, Gabriel Sanchez R,
Knight B. “Hormone therapy for preventing cardiovascular disease in post-menopausal women (Review).”
The Cochrane Library (2015).
We found high quality evidence that hormone therapy in both primary and secondary prevention conferred no
protective effects for all-cause mortality, cardiovascular death, non-fatal myocardial infarction, angina, or re-
vascularisation. However, there was an increased risk of stroke in those in the hormone therapy arm for combined
primary and secondary prevention (RR 1.24, 95% CI 1.10 to 1.41). Venous thromboembolic events were increased
(RR 1.92, 95% CI 1.36 to 2.69), as were pulmonary emboli (RR 1.81, 95% CI 1.32 to 2.48) on hormone therapy
relative to placebo.

38
Heidi D Nelson. “Commonly Used Types of Postmenopausal Estrogen for Treatment of Hot Flashes.” JAMA
2004 (291): 1610-1620.
All 8 trials of oral CEE reported statistically significant improvements in hot flash frequency, severity, or both
compared with placebo (Table 1, Table 7, Table 8).20,42-48 Three trials included treatment groups with
concomitant progestin or progesterone use (cyclic and continuous medroxyprogesterone acetate, cyclic micronized
progesterone).42,47,48 One trial compared 3 doses of CEE alone (0.3, 0.45, and 0.625 mg/d) and reported bigger
treatment effects with 0.625mg than 0.45 mg or 0.3 mg (P_.05).48 Differences between estrogen doses were not
found in patients provided with CEE (0.3, 0.45, and 0.625 mg/d) and continuous medroxyprogesterone acetate
(1.5 or 2.5 mg/d) in this trial.48
Lethaby A, Hogervorst E, Richards M, Yesufu A, and Yaffe K. “Hormone replacement therapy for cognitive
function in postmenopausal women (Review).” The Cochrane Library (2015).
Meta-analyses showed no effects of either ERT or HRT on prevention of cognitive impairment after five and four
years of treatment, respectively (odds ratio 1.34, 95% CI 0.95 to 1.9; odds ratio 1.05, 95% CI 0.72 to 1.54
respectively) (trend favouring control in both instances). Analyses assessing the effects of treatment over time found
that both ERT and HRT did not maintain or improve cognitive function and may even adversely affect this outcome
(WMD = -0.45, 95% CI -0.99 to 0.09;WMD = -0.16, 95% CI -0.58 to 0.26, respectively at maximum follow up).
Negative effects were found for ERT after one year and HRT after three and four years of therapy. Results from
smaller trials assessing effects onindividual cognitive domains mostly reported no evidence of benefit.
Suckling JA, Kennedy R, Lethaby A, Roberts H. “Local oestrogen for vaginal atrophy in postmenopausal
women (Review).” The Cochrane Library (2010).
The vaginal oestrogen ring was significantly more effective than placebo in reducing the following symptoms of
vaginal atrophy: dyspareunia, pallor, friability, pruritis and dryness. The oestrogen vaginal tablets, when compared
to the ring and placebo, were significantly more effective for reducing the symptoms of dyspareunia, frequency,
dryness and burning and itching. Vaginal oestrogen cream was significantly effective for reducing vaginal dryness,
vaginal moisture, vaginal fluid volume and elasticity when compared to moisturizing gel. When the ring was
compared to the cream there were no noted significant differences. The cream (conjugated equine oestrogen), when
39
compared with the tablets (17B-oestradiol), was the only treatment with any reported significant adverse effects
(uterine bleeding, breast pain and perineal pain). These results should be viewed with caution as they are based on
the results of two trials. Although not statistically significant, there were cases of hyperplasia and endometrial
overstimulation with the ring, cream and tablets (17B-oestradiol). This raises the question of whether women need
progestogenic protection from possible vaginal absorption of oestrogen from the ring, cream or tablets when used
beyond six months.
Menopause; Amie J. Cullimore, Bed, MD, MSc, FRCSC. e-CPS. Date of Revision: July 2015.
Class Drug Dosage Adverse Effects Comments Cost
Estrogens, oral Conjugated estrogens
Premarin
0.3-1.25mg daily Bloating, headache,
nausea, chloasma,
breast tenderness,
breakthrough
bleeding/spotting.
Increased risk of VTE,
CVD, breast cancer
Administer with a
progestogen in
patients with
intact uterus to
prevent
endometrial
hyperplasia or
cancer.
Consider vaginal
estrogen for
patients with
vaginal
symptoms only.
$