jmed genet syndrome ofthe month syndromes with lissencephaly
TRANSCRIPT

JMed Genet 1996;33:319-323
Syndrome of the month
Syndromes with lissencephaly
D T Pilz, 0 W J Quarrell
Institute of MedicalGenetics,University Hospital ofWales,Heath Park,Cardiff CF4 4XN, UKD T Pilz
Centre for HumanGenetics,117 Manchester Road,Sheffield S10 5DN, UK0 W J Quarrell
Correspondence to:Dr Pilz.
Earl Walker's paper in 1942 represents a de-tailed review of early described cases of lis-sencephaly and states that Owen (On theanatomy of vertebrates, vol 3, London: Long-mans, Green & Co, 1868) is said to haveintroduced the term lissencephaly to describean agyric brain, from the Greek words "lissos"(smooth) and "encephalus" (brain).'Further reports of lissencephaly followed by
Miller,2 Dieker et al,3 Warburg,45 and others andtheir contributions are recognised in syndromesnow known as Miller-Dieker syndrome andWalker-Warburg syndrome.
In his detailed analysis in 1984/85, Dobynscategorised lissencephaly into different patho-
logical types, assigned these to previously de-scribed cases or syndromes, and discussedpossible genetic mechanisms.67 The two mainpathological types he described provide a usefulbasis on which to review "syndromes with lis-sencephaly".
Key word: lissencephaly.
Type I or classical lissencephalyPREVALENCEThe only epidemiological data on the pre-valence of type I lissencephaly come from TheNetherlands, with 11-7 per million births.8
Figure 1 Classical lissencephaly (MRI scan). (Above) Axial Tl weighted SLE spinecho MR images showing agyria of the frontal and temporal lobes and pachygyria in theoccipital lobes. Note shallow sylvian fissures, mild colpocephaly (short arrows), andprominent cavum vellum interposition (long arrow). (Below) Coronal Tl weighted spinecho image, showing some temporal pachygyria. Note thick cortex.
PATHOLOGY AND NEUROIMAGINGType I lissencephaly results from a neuro-migrational arrest between 12 and 16 weeks'gestation, and histologically the cortex has fourinstead of six layers. These consist ofa marginal,superficial cellular, cell sparse, and deep cellularlayer.9 The cell sparse layer can appear as ahypodense area on computerised tomography(CT) or hyperintensity on T2 weighted mag-netic resonance (MR) images, especially in theperisylvian region.10Macroscopic abnormalities which can also
be seen on neuroimaging include: agyria, mixedagyria/pachygyria or complete pachygyria, athick cerebral cortex, incomplete oper-cularisation resulting in a shallow sylvian fissureand the typical "figure of eight" appearance ofthe brain, hypoplastic corpus callosum, per-sistent septum cavum pellucidum, dilatation ofthe posterior horns of the lateral ventricles, alsoknown as "colpocephaly", probably because ofincomplete development of the adjacent struc-tures such as the calcarine gyri and the hippo-campi, and heterotopias. Midline calcificationnot associated with infection can be observedin some cases and importantly the cerebellumusually appears normal (fig 1)."'-13
SYNDROMESMiller-Dieker syndrome (MDS)In patients with MDS the cerebral cortex isusually predominantly agyric with few areas ofpachygyria. Perinatal problems include poly-hydramnios and a low birth weight. Feedingproblems and recurrent chest infections arecommon. Patients are generally hypotonic, al-though hypertonia, particularly of the limbs,can ensue. They are profoundly retarded anddevelop seizures in the first few months of life,which can be difficult to control.'2
319
on 12 January 2019 by guest. Protected by copyright.
http://jmg.bm
j.com/
J Med G
enet: first published as 10.1136/jmg.33.4.319 on 1 A
pril 1996. Dow
nloaded from
Pilz, Quarrell
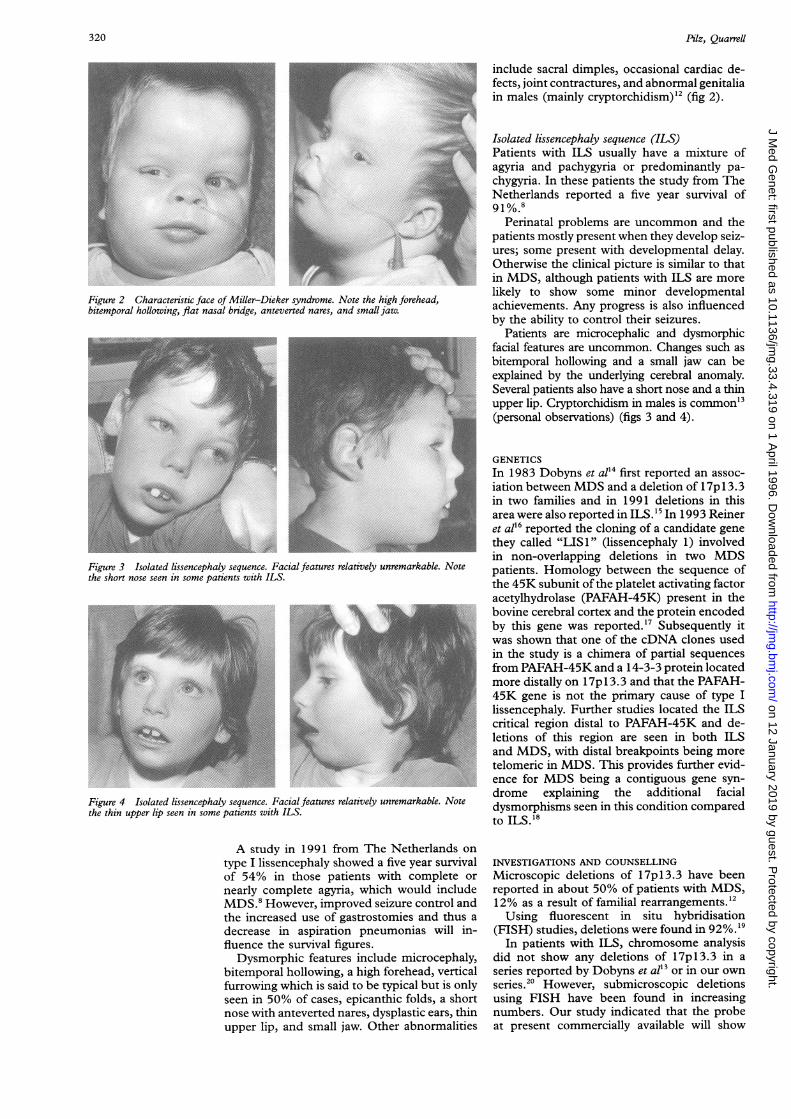
Figure 2 Characteristic face of Miller-Dieker syndrome. Note the high forehead,bitemporal hollowing, flat nasal bridge, anteverted nares, and small jaw.~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~Figure 3 Isolated lissencephaly sequence. Facial features relatively unremarkable. Notethe short nose seen in some patients with ILS.
Figure 4 Isolated lissencephaly sequence. Facial features relatively unremarkable. Notethe thin upper lip seen in some patients with ILS.
A study in 1991 from The Netherlands on
type I lissencephaly showed a five year survivalof 54% in those patients with complete or
nearly complete agyria, which would includeMDS.8 However, improved seizure control andthe increased use of gastrostomies and thus a
decrease in aspiration pneumonias will in-fluence the survival figures.Dysmorphic features include microcephaly,
bitemporal hollowing, a high forehead, verticalfurrowing which is said to be typical but is onlyseen in 50% of cases, epicanthic folds, a shortnose with anteverted nares, dysplastic ears, thinupper lip, and small jaw. Other abnormalities
include sacral dimples, occasional cardiac de-fects, joint contractures, and abnormal genitaliain males (mainly cryptorchidism)'2 (fig 2).
Isolated lissencephaly sequence (ILS)Patients with ILS usually have a mixture ofagyria and pachygyria or predominantly pa-chygyria. In these patients the study from TheNetherlands reported a five year survival of91%.8
Perinatal problems are uncommon and thepatients mostly present when they develop seiz-ures; some present with developmental delay.Otherwise the clinical picture is similar to thatin MDS, although patients with ILS are morelikely to show some minor developmentalachievements. Any progress is also influencedby the ability to control their seizures.
Patients are microcephalic and dysmorphicfacial features are uncommon. Changes such asbitemporal hollowing and a small jaw can beexplained by the underlying cerebral anomaly.Several patients also have a short nose and a thinupper lip. Cryptorchidism in males is common'3(personal observations) (figs 3 and 4).
GENETICSIn 1983 Dobyns et al'4 first reported an assoc-iation between MDS and a deletion of 17p 13.3in two families and in 1991 deletions in thisarea were also reported in ILS.'5 In 1993 Reineret all6 reported the cloning of a candidate genethey called "LIS1" (lissencephaly 1) involvedin non-overlapping deletions in two MDSpatients. Homology between the sequence ofthe 45K subunit of the platelet activating factoracetylhydrolase (PAFAH-45K) present in thebovine cerebral cortex and the protein encodedby this gene was reported. ' Subsequently itwas shown that one of the cDNA clones usedin the study is a chimera of partial sequencesfrom PAFAH-45K and a 14-3-3 protein locatedmore distally on 17p l 3.3 and that the PAFAH-45K gene is not the primary cause of type Ilissencephaly. Further studies located the ILScritical region distal to PAFAH-45K and de-letions of this region are seen in both ILSand MDS, with distal breakpoints being moretelomeric in MDS. This provides further evid-ence for MDS being a contiguous gene syn-drome explaining the additional facialdysmorphisms seen in this condition comparedto ILS.'8
INVESTIGATIONS AND COUNSELLINGMicroscopic deletions of 17pl3.3 have beenreported in about 50% of patients with MDS,12% as a result of familial rearrangements.'2Using fluorescent in situ hybridisation
(FISH) studies, deletions were found in 92%.19In patients with ILS, chromosome analysis
did not show any deletions of 17pl3.3 in a.series reported by- Dobyns et al'3 or in our ownseries."0 However, submicroscopic deletionsusing FISH have been found in increasingnumbers. Our study indicated that the probeat present commercially available will show
320
74
........
on 12 January 2019 by guest. Protected by copyright.
http://jmg.bm
j.com/
J Med G
enet: first published as 10.1136/jmg.33.4.319 on 1 A
pril 1996. Dow
nloaded from

Syndromes with lissencephaly
Figure 5 Cobblestone lissencephaly (US scan). Coronal ultrasound section. Falx (smallarrows), lateral ventricle (LV), cisterna magna (CM). Smooth agyric cortex (mediumarrows), hypoplastic cerebellum (large arrow). Hypoplastic inferior vermis seen on sagittalsections.
Figure 6 Cobblestone lissencephaly (CT scan). Axial CT section. Severe hydrocephalus,absent septum pellucidum. Note agyric cortex (arrows) not as well visualised owing to thebeam hardeninglmisregistration effects of the adjacent skull.
deletions in about 18% of patients with ILS."0Using additional probes and slightly revisedphenotypic criteria (that is, exclusion ofpatients with severe congenital microcephaly)(W B Dobyns, personal communication) de-letions were shown in 44% of patients withILS. 19
In patients with a deletion of 17pl3.3 therecurrence to sibs will be low and prenataldiagnosis can be offered. FISH should alsobe undertaken on the parents in cases withsubmicroscopic deletions to exclude subtle re-arrangements. In ILS cases where a deletionhas not been shown with currently availableprobes the recurrence risk is about 5%.2'
Prenatal ultrasonographic diagnosis of lis-sencephaly has been reported in a few cases ofMDS,"'23 but the absence of gyral formationis unlikely to become apparent before the thirdtrimester. Additional features like colpocephaly
may be more helpful indicators. Overall ultra-sonography is not a reliable method to detecttype I lissencephaly in early pregnancy.
Cases of type I lissencephaly with autosomalrecessive inheritance have been observed.9 21Norman et a124 reported a patient born toconsanguineous parents with two further ap-parently similarly affected sibs. The patient hadclassical lissencephaly, marked microcephaly, alow sloping forehead, and a prominent nasalbridge. Although this has been discussed asa specific syndrome and named Norman-Roberts syndrome,6 further convincing reportshave failed to follow and now the distinctionfrom ILS is unclear.92' At present a 25% re-currence risk should be considered in thosepatients with type I lissencephaly and severecongenital microcephaly in whom a deletionon 17p 13.3 is not detected.
Classical lissencephaly and X linked in-heritance will be discussed later.
Type II or cobblestone lissencephalyPREVALENCEThere is no reliable prevalence data availableon type II lissencephaly. It is most commonlyseen in the United Kingdom as Walker-Warburg syndrome (personal observations).
PATHOLOGY AND NEUROIMAGINGHistologically, type II lissencephaly is char-acterised by a severely disorganised unlayeredcortex with extensive neuronal and glial ectopiain the leptomeninges. These changes are seenin the cerebellum as well as the cerebrum,although to a slightly lesser degree. Macro-scopically, the surface of the brain is pre-dominantly agyric with a somewhat verrucousappearance, hence the name "cobblestone lis-sencephaly". In addition, areas of pachygyriaand polymicrogyria are seen. The cortex isthickened, but to a lesser extent than in type Ilissencephaly. The meninges are also thickenedand adherent to the cortex, resulting inobliteration of the subarachnoid space andconsequently hydrocephalus. Fusion of thecerebral hemispheres is also observed. The sep-tum pellucidum and corpus callosum are hy-poplastic or absent. Myelination is poor. Thecerebellum is often small and the vermishypoplastic. Other abnormalities includeDandy-Walker malformation and occipitalcephaloceles71"25 (figs 5 and 6). The extent ofthe cerebral and cerebellar abnormalities isvariable in the syndromes described below.
SYNDROMESEssentially three syndromes have been outlinedin association with type II lissencephaly:Walker-Warburg syndrome, Muscle-eye-braindisease, and Fukuyama congenital musculardystrophy, eye abnormalities and muscular dys-trophy being the other consistent features.Muscle-eye-brain disease (MEB) has mainlybeen reported in the Finnish population andFukuyama congenital muscular dystrophy(FCMD) has predominantly been seen in the
321
on 12 January 2019 by guest. Protected by copyright.
http://jmg.bm
j.com/
J Med G
enet: first published as 10.1136/jmg.33.4.319 on 1 A
pril 1996. Dow
nloaded from

Pilz, Quarrell
Japanese population. Walker-Warburg syn-drome (WWS) and MEB differ mainly in theseverity of the manifestations, these beingmilder in MEB, and it is suggested that theymay be allelic.212627Type II lissencephaly has also been seen
without ocular changes or muscular dystrophy2'(personal observation).
Walker-Warburg syndromeIn 1989 Dobyns et al28 published an extensivereview of patients with Walker-Warburg syn-drome. They all had type II lissencephaly, withhydrocephalus, cerebellar malformation, andvermian hypoplasia being the other consistentfeatures. Eye abnormalities included retinaldysplasia, microphthalmia, colobomata, andanterior chamber abnormalities (cataracts,corneal clouding commonly secondary to aPeter anomaly, and glaucoma). Congenitalmuscular dystrophy was seen in all patientswith a raised CK, abnormalities on EMG, andpathological changes on muscle histology.
Other abnormalities consisted of cleft lip andpalate, genital anomalies in males (small penis,cryptorchidism), and occasionally congenitalcontractures.
Congenital macrocephaly was common andcongenital microcephaly was observed par-ticularly in patients with a posterior ce-phalocele, leading the authors to postulate thatthe cephalocele might have a "decompressive"effect. Postnatally, microcephaly was also ob-served after the insertion of a shunt.
Pregnancy was commonly complicated bypolyhydramnios and resuscitation was oftenrequired at birth. The median survival was 9months. All patients were profoundly retardedalthough some of those who survived beyondthe first year showed some minor de-velopmental achievements.
Muscle-eye-brain diseaseIn 1989 Santavuori et a129 published a series of19 patients with MEB and recently reviewedtheir findings in 20 patients at a workshop oncongenital muscular dystrophy.30
Cerebral and cerebellar malformations wereconsistent with type II lissencephaly, but oftenless severe than those seen in WWS. Hydro-cephalus was present in 15 out of 20 cases.Eye abnormalities consisted predominantly ofsevere myopia, although glaucoma, retinal dys-trophy, and cataracts were also seen. Very highvisual evoked potentials were common. Con-genital muscular dystrophy was a consistentfeature with CK levels raised 3 to 20 times thenorm, although normal in some cases in thefirst year of life.
Pregnancy was usually uncomplicated withinfants presenting with hypotonia and feedingdifficulties neonatally or in the first few monthsof life. All cases were mentally retarded, usuallyseverely; however, a few did learn to walk and50% developed some speech. Seizures werecommon. Survival was significantly longer thanin WWS. Some of the patients seen were over40 years of age.
Fukuyama congenital muscular dystrophyIn 1981 Fukuyama et al3' published a detailedreview of patients with congenital progressivemuscular dystrophy of the Fukuyama type, alsoknown as FCMD, and in 1994 Yoshioka andKuroki32 reviewed the findings in 48 patients.
Cobblestone lissencephaly was present inthese patients. The cerebral surface showedpachygyria and polymicrogyria. Featuresfurther included mild to moderate ven-triculomegaly, white matter abnormalities, andpolymicrogyria of the cerebellum.Eye abnormalities mainly consisted of my-
opia, although some patients had optic atrophy.All patients had muscular dystrophy. CK
levels ranged from 10 to 50 times normal withdeclining levels after the age of 6 years.A higher rate of threatened miscarriages dur-
ing pregnancy and postmature deliveries werereported. Hypotonia and marked mental re-tardation were predominant features, althoughsome learned to walk and had meaningfulspeech development. Seizures were commonwith a much higher incidence of febrile con-vulsions than seen in the general population.In a recent review of 83 cases, Fukuyama re-ported an average survival of 18 years.30
GENETICSAll three syndromes are considered to be auto-somal recessive with reports of more thanone affected offspring of consanguineousparents.282933 FCMD has recently been linkedto 9q31-33.34 Subsequently the same groupfound linkage to this locus in a family of affectedmale sibs defined as either having FCMD orWWS.35 Other groups have not found linkageof WWS or MEB to 9q31-33.2'A few cases of prenatal ultrasound diagnosis
have been reported.36 37 Features, such as hydro-cephalus, encephaloceles, and microphthalmia,can be detected in the second trimester andultrasound scanning during pregnancy can bea useful diagnostic tool, especially in thosefamilies with a previously affected child.
Other syndromes with defective neuronalmigrationMore than 25 syndromes with lissencephalyand other disorders of neuronal migration havebeen delineated.2' Of particular interest aresome recent reports of apparent X linked in-heritance, which provide evidence of one ormore loci on the X chromosome involved inneuronal migration, and they will therefore bebriefly reviewed as part of this article.
In his review of isolated lissencephaly in1992, Dobyns et al'3 reported a patient withclassical lissencephaly and an apparently bal-anced X;2 translocation with breakpoints atXq22 and 2p25.
In 1994 Pinard et a138 described two veryinteresting families, where the probands, bothboys, had lissencephaly, one with predominentagyria and one with agyria and pachygyria andradiological appearances consistent with type Ilissencephaly. Neither had a deletion of17pl3.3 on high resolution banding. One of
322 on 12 January 2019 by guest. P
rotected by copyright.http://jm
g.bmj.com
/J M
ed Genet: first published as 10.1136/jm
g.33.4.319 on 1 April 1996. D
ownloaded from

Syndromes with lissencephaly
the mothers had a history of epilepsy, the otherof poor school performance. Both had sub-cortical laminar heterotopias on MR imaging.One of the probands had two maternal halfsisters who both had a history of epilepsy andeducational difficulties. MR imaging showedsubcortical laminar heterotopias, similar tothose seen in their mother. The authors sug-gested X linked dominant inheritance in bothfamilies, with the females being less severelyaffected than the males.38
In 1992 Zollino et a139 reported three malesand a female (first cousins) with mental re-tardation born to three healthy sisters. Ad-ditional features variably included character-istic facies, congenital hypotonia, micro-cephaly, talipes equinovarus, and hypo-genitalism. In the female the developmentaldifficulties were less marked than in the males,and both she and one of the males had normalMRI scans. The two other males had pa-chygyria. Again it was postulated that this wasan X linked dominant mental retardation syn-drome.
Finally there is a report by Berry-Kravis andIsrael40 of a family with five affected males intwo generations. Problems included pschy-chomotor retardation, seizures, micrognathia,and micropenis. Imaging studies were availableon two of the patients and showed pre-dominantly pachygyria with some areas of agy-ria, absence of the corpus callosum, andventricular dilatation. An X linked syndromewas discussed.
Conclusion
Lissencephaly is clearly a heterogeneous dis-order with several loci involved in neuronalmigration. The aim of this article is to reviewsome of the well defined syndromes within thecurrent pathological and phenotypic clas-sification and discuss the observed inheritancepatterns and genetic mechanisms, which facili-tate diagnosis of affected patients and coun-selling of their families. The definitivephenotypic range ofthe syndromes will becomeclear as the genes responsible are cloned.
We would like to thank Dr Jaspan for his review of the radio-logical aspects of lissencephaly presented in this article and DrDobyns for his continuing support for our work on lissencephalyand his helpful communication.
1 Walker AE. Lissencephaly. Arch Neurol Psychol 1942;48:13-29.
2 Miller JQ. Lissencephaly in 2 siblings. Neurology 1963;13:841-50.
3 Dieker H, Edwards RH, ZuRhein G, Chou SM, HartmanHA, Opitz JM. The lissencephaly syndrome. Birth Defects1969;5(2):53-64.
4 Warburg M. Hydrocephaly, congenital retinal non-attachment and congenital falciform fold. AmJ7 Ophthalmol1978;85:88-94.
5 Warburg M. The heterogeneity of microphthalmia in thementally retarded. Birth Defects 1971 ;7(3):136-54.
6 Dobyns WB, Stratton RF, Greenberg F. Syndromes withlissencephaly. I.Miller-Dieker and Norman-Roberts syn-dromes and isolated lissencephaly. Am _7 Med Genet 1984;18:509-26.
Dobyns WB, Kirkpatrick JB, Hittner HM, Roberts RM
FL. Syndromes with lissencephaly. II. Walker-
Warburg and Cerebro-oculo-muscular syndromes and a
syndrome with type II lissencephaly. Am_Med Genet
985;22: 157-95.
DeRijk-van Andel JF, Arts WFM, Hofman A, Staal A,
Niermeijer MF. Epidemiology of lissencephaly type I.
Neuroepidemiology 1991;10:200-4.
9 Dobyns WB, Reiner 0, Carrozzo R, Ledbetter DH. Lis-sencephaly. A human brain malformation associated withdeletion of the LIS1 gene located at chromosome l7pl 3.JAAM 1993;270:2838-42.
10 Barkovich AJ. In: Paediatric neuroimaging. New York: RavenPress, 1990:91.
11 Barkovich AJ, Gressens P, Evrard P. Formation, maturation,and disorders of brain neocortex. AJNR 1992;13:423-46.
12 Dobyns WB, Curry CJR, Hoyme HE, Turlington L, Led-better DH. Clinical and molecular diagnosis of Miller-Dieker syndrome. Am 7Hum Genet 199 1;48:584-94.
13 Dobyns WB, Elias ER, Newlin AC, Pagon RA, LedbetterDH. Causal heterogeneity in isolated lissencephaly. Neur-ology 1992;42:1375-88.
14 Dobyns WB, Stratton RF, Parke JT, Greenberg F,Nussbaum RL, Ledbetter DH. Miller-Dieker syndromeand monosomy 17p. J Pediatr 1983;102:552-8.
15 De Rijk-van Andel JF, Catsman-Berrevoets CE, Halley DJJ,Wesby-van Swaay E, Niermeijer M, Oostra BA. Isolatedlissencephaly sequence associated with a microdeletion atchromosome 17pl3. Hum Genet 1991;87:509-10.
16 Reiner 0, Carrozzo R, Shen Y, et al. Isolation of a Miller-Dieker lissencephaly gene containing G protein beta-sub-unit-like repeats. Nature 1993;364:717-21.
17 Hattori M, Adachi H, Tsujimoto M, Arai H, Inoue K.Miller-Dieker lissencephaly gene encodes a subunit ofbrain platelet-activating factor. Nature 1994;370:216-18.
18 Chong SS, Pack S, Tanigami A, Carrozzo R, Dobyns WB,Ledbetter DH. Systematic deletion analysis of MDS andILS patients excludes a candidate gene and delineates thelissencephaly gene locus. Am J Hum Genet Suppl 1995;57:170A.
19 Dobyns WB, Carrozzo R, Ledbetter DH. Frequent deletionsof the LISI gene in classic lissencephaly. Ann Neurol 1994;36:489-90A.
20 Pilz DT, Dalton A, Long A, Jaspan T, Maltby EL, QuarrellOWJ. Detecting deletions in the critical region for lis-sencephaly on 17pl3.3 using fluorescent in situ hy-bridisation and a PCR assay identifying a dinucleotiderepeat polymorphism. J Med Genet 1995;32:275-8A.
21 Dobyns WB, Truwit CL. Lissencephaly and other mal-formations of cortical development: 1995 update. Neuro-paediatrics 1985;26: 132-47.
22 Saltzman DH, Krauss CM, Goldman JM, Benacerraf BR.Prenatal diagnosis of lissencephaly. Prenat Diagn 1991 ;11:139-43.
23 McGahan JP, Grix A, Gersovich EO. Prenatal diagnosis oflissencephaly: Miller-Dieker syndrome. _7 Clin Ultrasound1994;22:560-3.
24 Norman MG, Roberts M, Sirois J, Tremblay LJM. Lis-sencephaly. Can .7 Neurol Sci 1976;3:39-46.
25 Squier MV. Development of the cortical dysplasia of type IIlissencephaly. NeuropatholApplNeurobiol 1993;19:209-13.
26 Santavuori P, Pihko H, Sainio K, et al. Muscle-eye-braindisease and Walker-Warburg syndrome. Am _7 Med Genet1990;36:371-2.
27 Dobyns WB, Pagon RA, Curry CJR, Greenberg F. Responseto Santavuori et al. Regarding Walker-Warburg syndromeand muscle-eye-brain disease. Am . Med Genet 1990;36:373-4.
28 Dobyns WB, Pagon RA, Armstrong D, et al. Diagnosticcriteria for Walker-Warburg syndrome. Am .7 Med Genet1989;32: 195-210.
29 Santavuori P, Somer H, Sainio K, et al. Muscle-eye-braindisease (MEB). Brain Dev 1989;11:147-53.
30 Dubowitz V. Workshop report: 22nd ENMC sponsoredworkshop on congenital muscular dystrophy held in Baarn,The Netherlands, 14-16 May 1993. Neuromusc Disord1994;4:75-81.
31 Fukuyama Y, Osawa M, Susuki H. Congenital progressivemuscular dystrophy of the Fukuyama type - clinical,genetic and pathological considerations. Brain Dev 1981;3:1-29.
32 Yoshioka M, Kuroki S. Clinical spectrum and genetic studiesof Fukuyama congenital muscular dystrophy. Am .7 MedGenet 1994;53:245-50.
33 Fukuyama Y, Ohsawa M. A genetic study of the Fukuyamatype congenital muscular dystrophy. Brain Dev 1984;6:373-90.
34 Toda T, Segawa M, Nomura Y, et al. Localization of thegene for Fukuyama type congenital muscular dystropy tochromosome 9q31-33. Nature Genet 1993;5:283-6.
35 Toda T, Yoshioka M, Nakahori Y, Kanazawa I, NakamuraY, Nakagome Y. Genetic identity of Fukuyama-type con-genital muscular dystrophy and Walker-Warburg syn-drome. Ann Neurol 1995;37:99-101.
36 Crowe C, Jassani M, Dickerman L. The prenatal diagnosisof the Walker-Warburg syndrome. Prenat Diagn 1985;6:177-85.
37 Vohra N, Ghidini A, Alvarez M, Lockwood C. Walker-Warburg syndrome: prenatal ultrasound findings. Prenat_Diagn 1993;13:575-9.
38 PinardJM, Motte J, Chiron C, Brian R, Andermann E,Dulac 0. Subcortical laminar heterotopia and lis-sencephaly in two families: a single X linked dominantgene. .7 NeurolNeurosurg Psychiatry 1994;57:914-20.
39 Zollino M, Mastroiacovo P, Zampino G, Mariotti P,NeriG. New XLMR syndrome with characteristic face, hypo-genitalism, congenital hypotonia and pachygyria. Am.7Med Genet 1992;43:452-7.
40 Berry-Kravis E, Israel J. X-linked pachygyria and agenesisof the corpus callosum: evidence for an X chromosomelissencephaly locus. Ann Neurol 1994;36:229-33.
323
on 12 January 2019 by guest. Protected by copyright.
http://jmg.bm
j.com/
J Med G
enet: first published as 10.1136/jmg.33.4.319 on 1 A
pril 1996. Dow
nloaded from