johns hopkins university - fall 2003 phylogenetics & computational genomics - 410.640.71 lecture...
TRANSCRIPT

Johns Hopkins University - Fall 2003 Phylogenetics & Computational Genomics - 410.640.71
Lecture #6 Page 1
Week6: Intro to Phylogenetic Reconstruction& Distance Based Methods
• introduction to phylogenies• distance based methods• phylogeny exercises
Phylogeny Objectives
1 - understand the essence of phylogenies (definition of terms)2 - understand distance based methods of phylogenetic reconstruction3 - should be able to use various software packages to reconstruct and view phylogenies: ClustalX, MEGA, DAMBE, Treeview

study of relationships among organisms (molecular systematics), proteins or genes using molecular biology techniques
Molecular phylogeny
• Darwin - thesis #1 - organisms descend with modification from common ancestors (CA)
internal node - common ancestor (CA)external node - operational taxonomic unit (OTU)
order of branches define the relationships (topology)branch length defines the number of changes
• relationships among organisms, proteins, genes are illustrated by a phylogenetic tree
Lecture #6 Page 2
Johns Hopkins University - Fall 2003 Phylogenetics & Computational Genomics - 410.640.71

A brief history of molecular phylogeny
Lecture #6 Page 3
Johns Hopkins University - Fall 2003 Phylogenetics & Computational Genomics - 410.640.71
• phylogenetic inference is old (for Biology)
Ernst Haeckel “Tree of life” (1891)
Charles Darwin – Orgin of Species (1859)Illustration of ‘descent with modification’

A brief history of molecular phylogeny
• Nuttall (1904) found that the strength of serological cross reactions was correlated with the level of relatedness between animals - applied to primate phylogeny
Lecture #6 Page 4
Johns Hopkins University - Fall 2003 Phylogenetics & Computational Genomics - 410.640.71
Molecular phylogeny
Algorithmic approaches
more modern developments …
• starting 1950’s many more sources of molecular information become avaiable: e.g. amino acid sequences, allozyme frequencies, DNA hybridization
• these data stimulated the development of quantitative ‘numerical taxonomy’ techniques for phylogenetic analysis
• first numerical approach to phylogeny based on phenetic approach – i.e. similarity of morphological characters (Michener and Sokal 1957)
• phylogenetic studies of human populations based on blood allele frequencies led to the introduction of distance, parsimony & likelihood methods (Edwards & Cavalli-Sforza 1963, 1964)

A brief history of molecular phylogeny …
Lecture #6 Page 5
Johns Hopkins University - Fall 2003 Phylogenetics & Computational Genomics - 410.640.71
emergence of sequence based methods:
accumulation of amino acid sequences stimulates development of sequence based phylogenetic methods
these soon emerge as the most powerful methods (see slide #6 for reasons)
Parsimony, Distance & Maximum likelihood methods (see slide # 10)
Eck and Dayhoff (1966) – working of Atlas of Protein Sequence and Structure – publish first method for phylogenetic analysis of sequences based on parsimony
Fitch and Margoliash (1967) publish first distance based method – weighted least squares – for sequence based (cytochrome c) phylogenetic inference
Statistician Neyman (1971) publishes first maximum likelihood method for phylogenetic analysis of sequence data

Benefits of using molecular sequences for phylogenetics
1 - sequences evolve in a much more regular manner than morphological characters
2 - less prone to confusion between homology and analogy, homoplasies
3 - vast abundance of characters to analyze
4 - molecular data more amenable to quantitative treatments
5 - molecular data ubiquitous - can be used for microorganisms
6 - can be used to study relationships at many different evolutionary levels faster evolving genes - mitochondrial DNA - closely related species slower evolving genes - ribosomal RNA genes - distantly related species
Some success stories ….
• primate evolution - who are humans closest relatives ?
• origin of Cetacea mammals (whales, dolphins, porpoises)
• revising deep taxonomic classification scheme - 3 domains of life
Lecture #6 Page 6
Johns Hopkins University - Fall 2003 Phylogenetics & Computational Genomics - 410.640.71
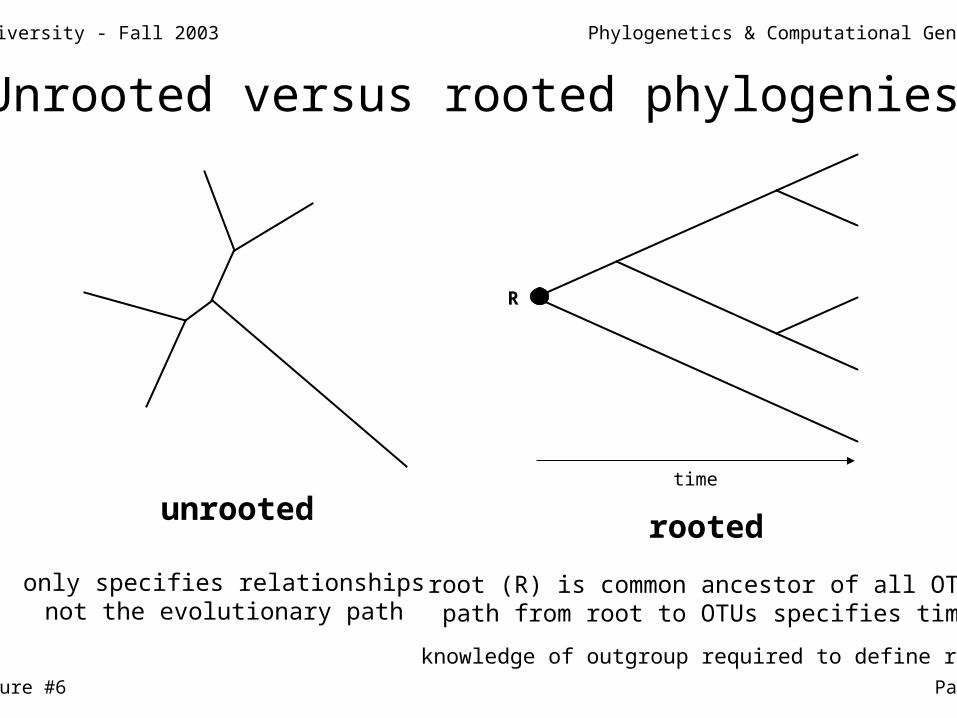
unrootedrooted
R
only specifies relationshipsnot the evolutionary path
root (R) is common ancestor of all OTUspath from root to OTUs specifies time
knowledge of outgroup required to define root
time
Unrooted versus rooted phylogenies
Lecture #6 Page 7
Johns Hopkins University - Fall 2003 Phylogenetics & Computational Genomics - 410.640.71
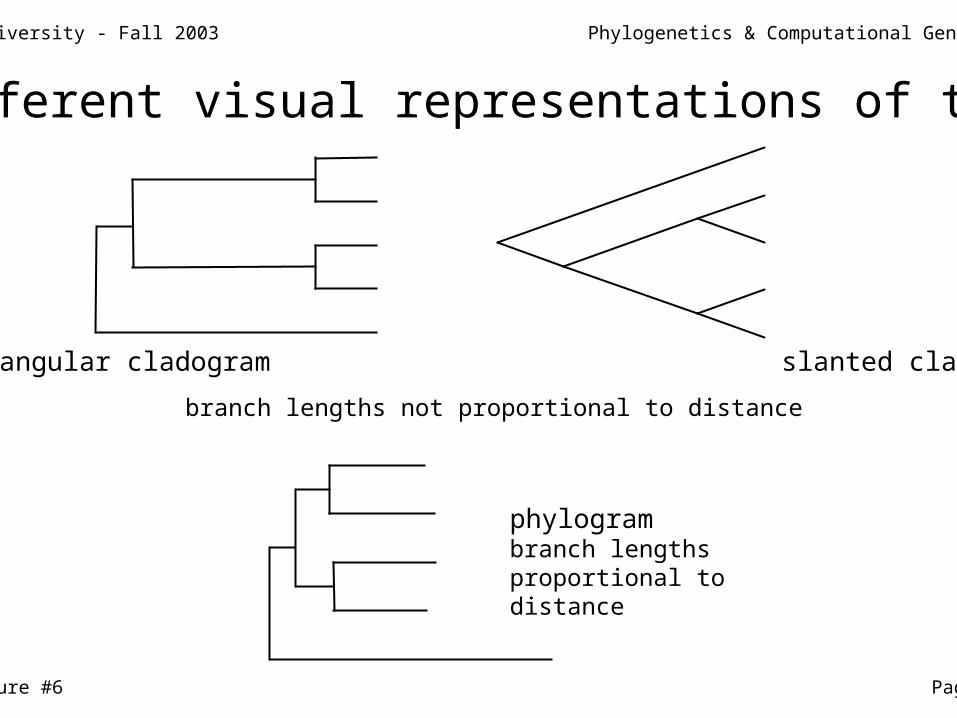
Different visual representations of trees
phylogrambranch lengthsproportional todistance
rectangular cladogram slanted cladogram
branch lengths not proportional to distance
Lecture #6 Page 8
Johns Hopkins University - Fall 2003 Phylogenetics & Computational Genomics - 410.640.71

Species tree versus gene tree
species tree - represents evolutionary relationships among species
gene tree - represents evolutionary relationship among genes
species trees and genes trees can (and often do!) differ
Reasons for this ??• comparison of orthologous versus paralogous genes• horizontal (later) transfer of genes
the concept of an accurate species tree is notoriously difficult to pin downin this class we will deal almost exclusively with genes trees
more on these important concepts later in course
Lecture #6 Page 9
Johns Hopkins University - Fall 2003 Phylogenetics & Computational Genomics - 410.640.71

Distance based• pairwise evolutionary distances computed for all taxa• tree constructed using algorithm based on relationships between distances
Maximum parsimony• nucleotides or amino acids are considered as character states• best phylogeny is chosen as the one that minimizes the number of changes between character states
Maximum likelihood• statistical method of phylogeny reconstruction• explicit model for how data set generated - nucleotide or amino acid substitution• find topology that maximizes the probability of the data given the model and the parameter values (estimated from data)
Methods of phylogenetic reconstruction
Lecture #6 Page 10
Johns Hopkins University - Fall 2003 Phylogenetics & Computational Genomics - 410.640.71

Lecture #6 Page 11
Johns Hopkins University - Fall 2003 Phylogenetics & Computational Genomics - 410.640.71
Phylogenetic inference
1 – sequences change as they evolve from a common ancestor over time
2 – a group of related sequences retains information (incomplete) about the evolutionary history that unites them – based on the pattern of changes
3 – phylogeny is estimation, make the best estimate about evolutionary historygiven the incomplete information in the sequences being analyzed
4 – information about the past is not available, only extant sequences
5 – therefore any evolutionary scenario (i.e. phylogeny) can be postulated to explain the changes in the sequences being analyzed
6 – must have some way to discriminate among the (many!) possible phylogenies

n
(2i-5) (2n-3) i=3
Number of OTUs and number of possible trees
# rooted trees
2 1 1 3 1 3 4 3 15 5 15 105 6 105 954 7 954 10,395 8 10,395 135,135 9 135,135 2,027,02510 2,027,025 34,459,425
true tree - true evolutionary history is one of many possibilitiesdifficult to infer true tree when # OTUs is large
inferred tree - obtained using data and reconstruction methodnot necessarily the same as the true tree - a hypothesis
Lecture #6 Page 12
n
(2i-5) i=3
# OTUs (n)# unrooted trees
Johns Hopkins University - Fall 2003 Phylogenetics & Computational Genomics - 410.640.71

Lecture #6 Page 13
Johns Hopkins University - Fall 2003 Phylogenetics & Computational Genomics - 410.640.71
Alogrithms & Optimality CriteriaTwo ways for selecting preferred phylogenies:
Algorithms – sequence of steps that leads to the selection of a phylogeny - combine phylogeny inference and criterion definition into single step - move directly to toward the best tree without evaluating many different trees e.g. UPGMA & Neighbor-joining
Optimality criteria – a criteria is defined whereby different phylogenies are - compared to one another to determine which is better - two steps involved: 1 – define criteria (objective function) 2 – use algorithm to compute objective function on different trees - this method is much slower – must evaluate many trees (shorcuts often necessary) - may be more robust because scores are assigned to every phylogeny and then they are ranked – yields information about how well specified the tree is e.g. Least squares & Minimum evolution
Compromise – define starting tree with algorithm approach and then search nearby tree-space using optimality criteria approach

Lecture #6 Page 14
Johns Hopkins University - Fall 2003 Phylogenetics & Computational Genomics - 410.640.71
Least squares method First distance based method developed; Cavalli-Sforza & Edwards (1967) Fitch & Margoliash (1967)
Optimality criterion = minimize the residual sum of squares (RS) between the observed distances (dij - based on distance matrix) and the patristic differences (eij – based on the branch lengths of the inferred phylogeny)
RS = (dij – eij)2
i<j
A B C DA 0 17 21 27B 17 0 12 18C 21 12 0 14D 27 18 14 0
e.g.dBD = 18 eBD = 6 + 2 + 8 = 16RS-BD = (18 – 16)2 = 4

Lecture #6 Page 15
Johns Hopkins University - Fall 2003 Phylogenetics & Computational Genomics - 410.640.71
Least squares method … Statisitcally very sound method since based on Least squares
Logically challenged since it formally estimates branch lengths and not topologies
In principle RS is computed for all possible topologies but in practice this quickly becomes impracticable (see slide #12) – short cuts are available to minimize search space (see lecture week7)
Fitch & Margoliash (1967) introduced weighted least squares that corrects for the bias introduced by long distances
Negative branch length estimates can confound method – constraint of non-negative branch lengths results in substantial improvement
RS = [(dij – eij)2 / dij] i<j
Lecture #6 Page 16
Johns Hopkins University - Fall 2003 Phylogenetics & Computational Genomics - 410.640.71
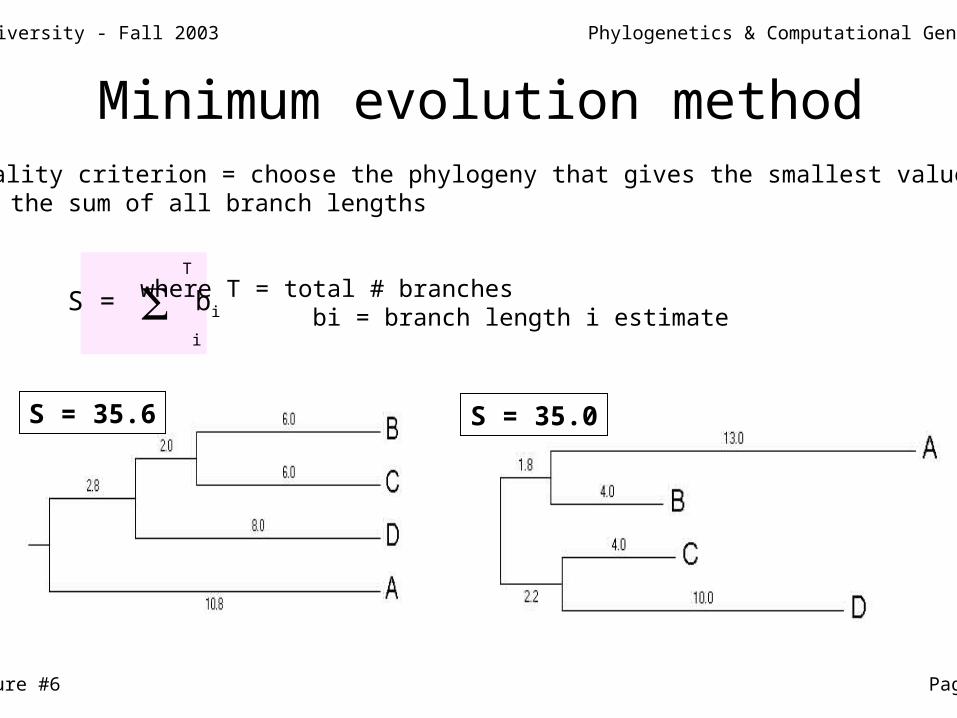
Minimum evolution method Optimality criterion = choose the phylogeny that gives the smallest value of S - the sum of all branch lengths
T
S = bi
i
where T = total # branches bi = branch length i estimate
S = 35.6 S = 35.0

Lecture #6 Page 17
Johns Hopkins University - Fall 2003 Phylogenetics & Computational Genomics - 410.640.71
Minimum evolution method …
As with least squares, S should ideally be computed for all possible trees but this is impossible with many taxa
One shortcut is to start search with neighbor-joining tree and then evaluate closely related trees to find the best one
Close neighbor interchange (CNI) start with temporary ME tree (e.g. NJ tree for first step) and evaluate all trees that differ by one or two topological changes
This approach may be more robust than using neighbor-joining alone because it can result in an ordered list of trees, if many trees represent the data almost equally well then the best tree may not be so well supported

UPGMA method(unweighted pair group method with arithmetic mean)
simplest method - uses sequential clustering algorithm
results in ‘ultrameric’ trees – equal distances from root to all tips
based on assumption of strict rate constancy among lineages – this is often violated and so method often gives erroneous trees (not reccomended)
Lecture #6 Page 18
Johns Hopkins University - Fall 2003 Phylogenetics & Computational Genomics - 410.640.71
A BB dABC dAC dBC
(AB)C d(AB)C d(AB)C = (dAC + dAB) / 2Distance matrix
Tree
dAB / 2
A
B
A
d(AB)C / 2
B
C
step 1 step 2
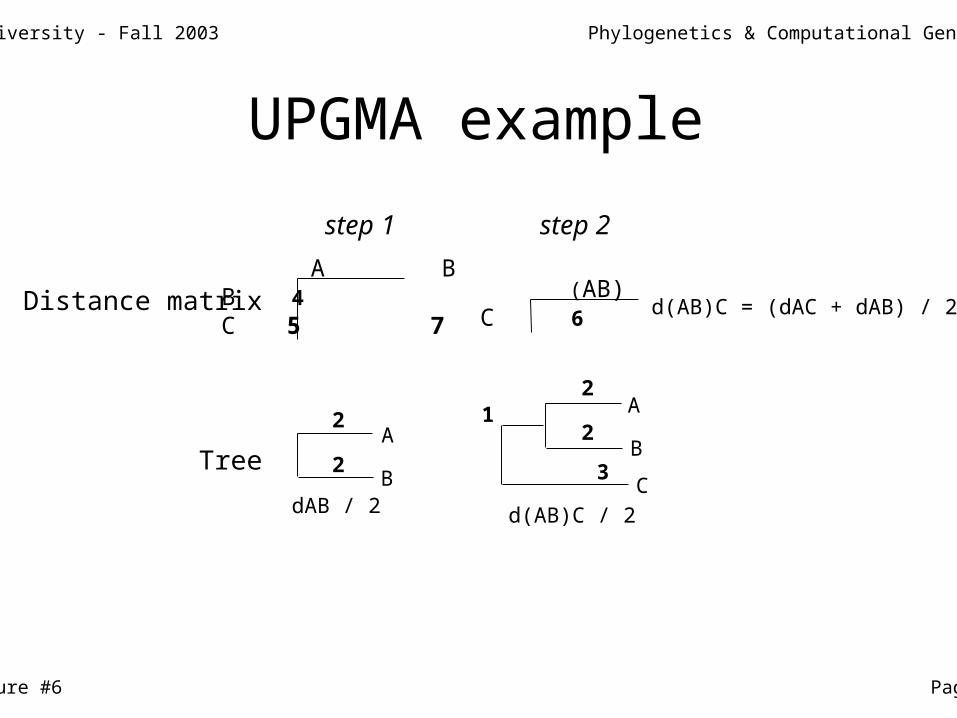
UPGMA example
Lecture #6 Page 19
Johns Hopkins University - Fall 2003 Phylogenetics & Computational Genomics - 410.640.71
A BB 4C 5 7
(AB)C 6 d(AB)C = (dAC + dAB) / 2Distance matrix
Tree
dAB / 2
A
B
A
d(AB)C / 2
B
C
step 1 step 2
2
2
2
21
3
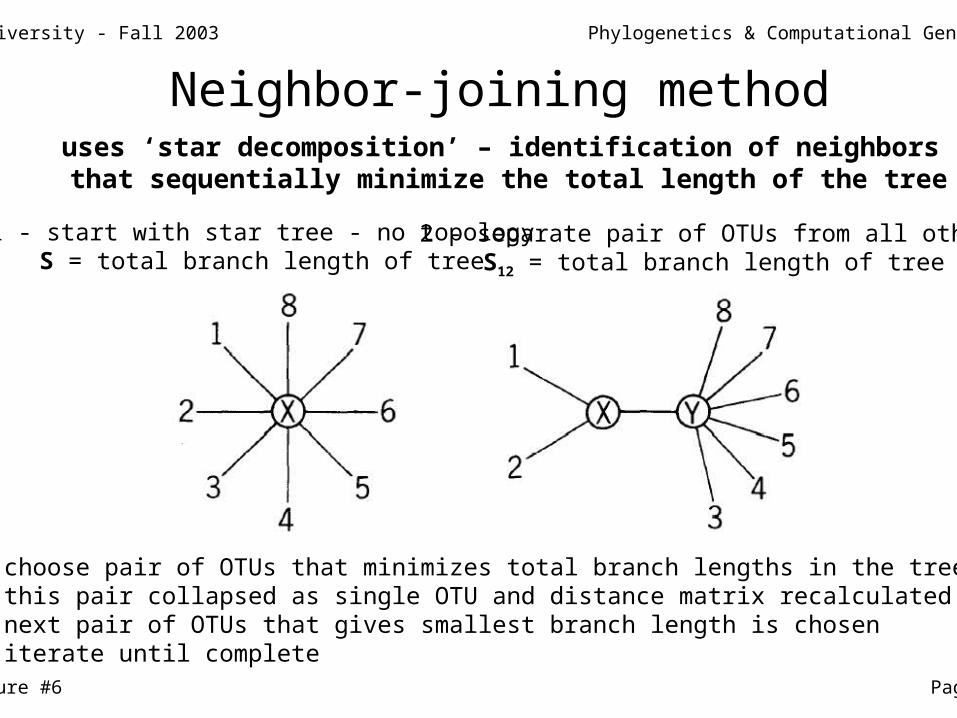
3 - choose pair of OTUs that minimizes total branch lengths in the tree4 - this pair collapsed as single OTU and distance matrix recalculated5 - next pair of OTUs that gives smallest branch length is chosen6 - iterate until complete
1 - start with star tree - no topologyS = total branch length of tree
2 - separate pair of OTUs from all othersS12 = total branch length of tree
uses ‘star decomposition’ – identification of neighbors that sequentially minimize the total length of the tree
Neighbor-joining method
Lecture #6 Page 20
Johns Hopkins University - Fall 2003 Phylogenetics & Computational Genomics - 410.640.71
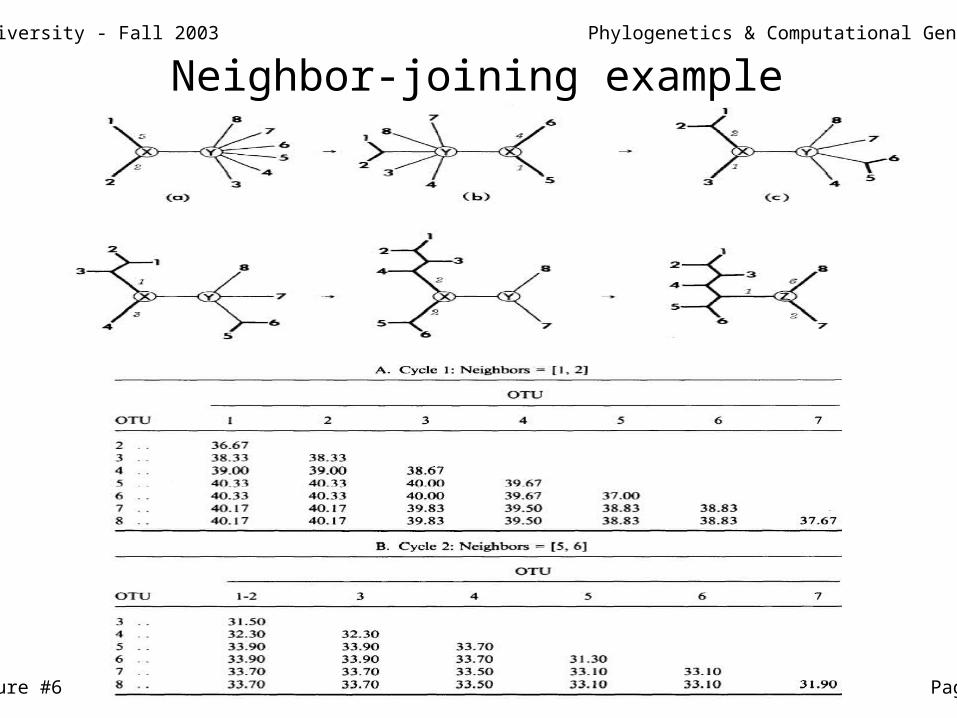
Neighbor-joining example
Lecture #6 Page 21
Johns Hopkins University - Fall 2003 Phylogenetics & Computational Genomics - 410.640.71

Neighbor-joining method …
Lecture #6 Page 22
Johns Hopkins University - Fall 2003 Phylogenetics & Computational Genomics - 410.640.71
Extremely fast and efficient method, widely used & found in numerous publications
Tends to perform fairly well in simulation studies
May produce tie trees from data set but this appears to be rare
Algorithm is ‘greedy’ and so can get stuck in local optima
Main criticism is that it produces only one tree and does not give any idea of how many other trees are equally well or almost as supported by the data
For this reason, neighbor-joining is often used as a method to find a starting tree that other methods (e.g. minimum evolution) will evaluate to find the best tree

Exercises1 - choose some alignment to work on
2 - load alignment into Clustal and build neighbor-joining tree
3 - open tree in Treeview and view, manipulate and save tree
4 - load alignment into DAMBE and into MEGA and reconstruct and view trees using all distance methods available – look for differences in results
5 - manually reconstruct UPGMA tree for the distance matrix on slide #14
6 - open the MEGA formatted version of this same distance matrix http://jhunix.hcf.jhu.edu/~kjordan6/distances.meg in MEGA and reconstruct distance based trees using all 3 methods available (check UPGMA result against manually reconstructed UPGMA tree)
7 - calculate RS for all three distance based trees from #6 and pick best tree
8 - calculate S for all three distance based trees from #6 and pick best tree
Lecture #6 Page 23
Johns Hopkins University - Fall 2003 Phylogenetics & Computational Genomics - 410.640.71