keratin biomaterial treatments for burn injury and mechanisms of tissue … · ·...
TRANSCRIPT

______________________________________________________________________________
KERATIN BIOMATERIAL TREATMENTS FOR BURN INJURY AND
MECHANISMS OF TISSUE SURVIVAL
BY
DEEPIKA RANI PORANKI
A Dissertation Submitted to the Graduate Faculty of
WAKE FOREST UNIVERSITY GRADUATE SCHOOL OF ARTS AND SCIENCES
in Partial Fulfillment of the Requirements
for the Degree of
DOCTOR OF PHILOSOPHY
in the Molecular Genetics and Genomics Program
August 2012
Winston-Salem, North Carolina
Approved by
Mark Van Dyke, PhD., Advisor
Michael Tytell, PhD., Chair
Mark O. Lively, PhD
Joseph Molnar, M.D., Ph.D., F.A.C.S
Mark Willingham, PhD

______________________________________________________________________________
ii
ACKNOWLEDGEMENTS
My time at Wake Forest had been an incredible learning experience. This thesis is the
culmination of my work at Wake Forest but the experiences and knowledge I gained here will
stay with me for a lifetime. My work and my personal transformation would not have been
possible without the invaluable support of my excellent mentors and amazing colleagues.
First and foremost, I am indebted to my mentor, Dr. Mark Van Dyke, for his constant help,
expert guidance, enormous support and committed mentorship. I feel extremely fortunate to have
worked with the “best mentor“, anyone can hope for. My gratitude goes to him for always
putting the students first. Mark’s personal attributes of dedication, commitment and poise will
always be something that I wish to strive for.
As program director and committee member, Dr. Mark Lively was always very helpful with his
meticulous nature and timely advice. I am also grateful to my other committee members: Dr.
Michael Tytell, Dr. Joseph Molnar and Dr. Mark Willingham for their guidance, advice and
willingness to serve on my committee amidst their busy schedules.
I am thankful to Carmen Gaines for her support and friendship at WFIRM and after. It was a
great experience working with her on the wound healing projects. Her detail-oriented nature and
thoroughness have been a great source of help for our project(s). It seemed like yesterday that
Heather Coan taught me the nuances of tissue culture work, when I just started working in the
lab. She moved on but the techniques that I learnt from her helped me for the major part of my
thesis.
Thanks to many others, who helped me at different stages of this work. I cannot forget the help
of Christina Ross and Mary Ellenberg during the hectic animal study. Renae Hall, Cynthia

______________________________________________________________________________
iii
Miller, Sandy Sink and Tammy Cockerham from the surgery core helped us complete the project
without any issues. I am appreciative of Dr. Richard Clark for hosting us in his lab at Stony
Brook and training us in the intricacies of histological analysis; Dr. Wei Du for continuing on
that training at Wake Forest and answering the questions we had in histology and Bridgette Jones
for helping with the sectioning. If it was not for Barbara Summerlin, Deborah McLaughlin and
Susan Pierce working behind the scenes, my graduate student life would have been so much
tougher.
The past and present members of the Keratin group: Paulina Hill, Mária Bahawdory, Bailey
Fearing, Lauren Pace, Julie Gupton, Fiesky Nunez, Roche de Guzman, Jill Richter, Olga
Roberts, Matt Stern, Alana Sampson and Arthur J.Allen have been a constant source of
knowledge and support to me.
I am very grateful to my parents, Satyaprasad Poranki and Annapurna Poranki, for teaching the
value of honesty and integrity and always being there for me –no matter what. I am also grateful
to my in-laws, Prabhakara Raju Vinukonda and Sri Lakshmi Vinukonda, for their commitment to
higher education and wholeheartedly encouraging me. Thanks to other family members: Uday,
Priya, Ravi, Rama, Suresh, Sruthi and their kids for all their love and support. There was also a
great deal of support we received from family friends in Winston-Salem.
Last, thanks to my husband, Naveen Vinukonda and daughter, Asmithaa Vinukonda, without
whom, I wouldn’t have come this far. My loving, encouraging and patient husband has always
been the strongest source of support, right from the beginning of this worthwhile journey. My
daughter was a few-month old baby, when I started preparing for GRE. Today she is a 6 yr. old
getting ready to graduate from kindergarten. If not for her bringing joy and happiness, this

______________________________________________________________________________
iv
journey would have been so much more strenuous. All along my daughter saw her mom struggle
to fulfill a dream and I hope this stays with her as an inspiration.

______________________________________________________________________________
v
DEDICATION
Dedicated to
Daddy & Amma

______________________________________________________________________________
vi
TABLE OF CONTENTS
LIST OF FIGURES AND TABLES .......................................................................................... vii
ABBREVIATIONS ....................................................................................................................... x
ABSTRACT ................................................................................................................................ xiii
CHAPTER 1 - INTRODUCTION ............................................................................................... 1
CHAPTER 2- A KERATIN BIOMATERIAL PROMOTES SKIN REGENERATION
AFTER BURNS IN VIVO AND RESCUES CELLS AFTER THERMAL
STRESS IN VITRO ............................................................................................... 37
CHAPTER 3 - GAMMA KERATOSE MAINTAINS CELL VIABILITY IN VITRO
AFTER THERMAL STRESS BY REGULATING THE EXPRESSION OF
CELL DEATH PATHWAY SPECIFIC GENES ................................................ 78
CHAPTER 4 - DEVELOPMENT OF A PORCINE DEEP PARTIAL THICKNESS BURN
MODEL ................................................................................................................. 119
CHAPTER 5 - ASSESSMENT OF DEEP PARTIAL THICKNESS BURN TREATMENT
WITH KERATIN BIOMATERIAL HYDROGELS IN A SWINE MODEL 146
CHAPTER 6 - DELAYED TREATMENT OF PORCINE DEEP PARTIAL THICKNESS
BURNS SUBJECT TO THERAPEUTIC KERATIN BIOMATERIALS ...... 170
CHAPTER 7 - SUMMARY, CONCLUSIONS AND PROSPECTIVE RESEARCH ........ 200
SCHOLASTIC VITA ................................................................................................................ 208

______________________________________________________________________________
vii
LIST OF FIGURES AND TABLES
CHAPTER 1 - INTRODUCTION ............................................................................................... 1
Figure 1: Three Zones in Burn Wounds: ..................................................................................... 3
Figure 2: Schematic diagram of Wool fiber ................................................................................ 7
Figure 3: Apoptotic Pathways ................................................................................................... 14
Figure 4: Autophagy and its inhibitors ...................................................................................... 16
Table 1: Gene Glossary ............................................................................................................. 18
CHAPTER 2 - A KERATIN BIOMATERIAL PROMOTES SKIN REGENERATION
AFTER BURNS IN VIVO AND RESCUES CELLS AFTER THERMAL
STRESS IN VITRO ............................................................................................... 37
Figure 1. Mouse Chemical Burn Wound Area .......................................................................... 65
Figure 2. Digital Images from Mice Chemical burn study ........................................................ 66
Figure 3. Swine Thermal Burn Wound Area ............................................................................. 67
Figure 4. Digital Images from Swine thermal burn study ......................................................... 68
Figure 5. In Vitro Thermal Stress Model .................................................................................. 69
Figure 6. Percent Cell Viability Curve ..................................................................................... 70
Figure 7. Keratose SDS PAGE .................................................................................................. 71
Supplementary Table 1: Band # 1, Molecular Weight ~110 KDa ............................................ 72
Supplementary Table 2: Band # 2, Molecular Weight ~60 KDa .............................................. 73
Supplementary Table 3: Band # 3, Molecular Weight ~50-40 KDa ......................................... 74
Supplementary Table 4: Band # 4, Molecular Weight ~30 Kda ............................................... 75
Supplementary Table 5: Band # 5, Molecular Weight ~20-15 Kda .......................................... 76
Supplementary Table 6: Band # 6, Molecular Weight ~8-10 KDa ........................................... 77
CHAPTER 3 - GAMMA KERATOSE MAINTAINS CELL VIABILITY IN VITRO
AFTER THERMAL STRESS BY REGULATING THE EXPRESSION OF
CELL DEATH PATHWAY SPECIFIC GENES ................................................ 78

______________________________________________________________________________
viii
Figure 1: In vitro Heat shock Model ....................................................................................... 105
Table 1: RNA Quantification .................................................................................................. 106
Table 2: Gene expression of gamma keratose treated cells at 12hr......................................... 107
Table 3: Gene expression of gamma keratose treated cells at 18hr......................................... 109
Table 4: Gene expression of growth media treated cells at 18hr............................................. 110
Table 5: Gene expression of gamma keratose treated cells at 24hr......................................... 112
Table 6: Gene expression of growth media treated cells at 24 hr............................................ 113
Table 7: Total number of genes significantly expressed in gamma keratose treated cells and
growth media treated cells with p<0.05 .................................................................................. 114
Supplementary Table 1: Genes included in the PCR array ..................................................... 115
CHAPTER 4 - DEVELOPMENT OF A PORCINE DEEP PARTIAL THICKNESS BURN
MODEL ................................................................................................................. 119
Figure 1. Differences in healing of deep partial thickness scald burns due to inconsistent
burning. .................................................................................................................................... 135
Figure 2. Burns with non-uniform severity created with brass block heated in boiling water
along the right flank of the animal .......................................................................................... 136
Figure 3. 360 grade, 3 cm diameter brass block used for burn wound creation ...................... 137
Figure 4. Burn device operation. ............................................................................................. 138
Figure 5. Heated brass blocks with temperature sensors. ........................................................ 139
Figure 6. Contact angle analysis using water and PEG:H2O azeotropic mixture to determine
surface wetting. ........................................................................................................................ 140
Figure 7. Healing progression of wounds with and without islands ....................................... 141
Figure 8. Visually consistent burn wounds ............................................................................. 142
Figure 9. Histological scoring data from day 1 wounds .......................................................... 143
Figure 10. Tissue sections stained with Gomori trichrome stain ............................................ 144
Figure 11. Percent dermal tissue damage ................................................................................ 145
CHAPTER 5 - ASSESSMENT OF DEEP PARTIAL THICKNESS BURN TREATMENT
WITH KERATIN BIOMATERIAL HYDROGELS IN A SWINE MODEL 146
Figure 1: Visual wound assessment ........................................................................................ 164

______________________________________________________________________________
ix
Figure 2: Burn depth ................................................................................................................ 165
Figure 3: Early time points rates of re-epithelialization .......................................................... 166
Figure 4. Rates of re-epithelialization during later stages of wound healing .......................... 167
Figure 5: Thickness of granulation tissue ................................................................................ 168
Table 1: Percent increase in burn depth from day 3 to 6 ......................................................... 168
Table 2. Rates of re-epithelialization at day 3 and day 6 ....................................................... 169
Table 3. Rates of re-epithelialization between days 9 and 15. ................................................ 169
Table 4. Days to wound closure .............................................................................................. 169
CHAPTER 6 - DELAYED TREATMENT OF PORCINE DEEP PARTIAL THICKNESS
BURNS SUBJECT TO THERAPEUTIC KERATIN BIOMATERIALS ...... 170
Figure 1. Schematic of randomized placement of treatments ................................................ 191
Figure 2. Extent of burn wound size reduction over time ...................................................... 192
Figure 3. Degree of wound contracture as a function of time ................................................ 193
Figure 4. H&E images of deep partial thickness burn depth at Day 1 ................................... 194
Figure 5. H&E images of deep partial thickness burns at Day 6............................................ 194
Figure 6. H&E images of deep partial thickness burns at Day 12.......................................... 195
Figure 7. Initial rates of re-epithelialization in deep partial thickness burns ......................... 196
Figure 8. Rates of re-epithelialization in second degree burns during late stages of healing 197
Figure 9. H&E images of deep partial thickness epithelium at Day 12 .................................. 198
Figure 10. Degree of reepithelialization at Day 30 post-burn ................................................. 198
Table 1. Initial rates of reepithelialization between Days 0 and 6 ......................................... 199
Table 2. Rates of reepithelialization between Days 9 and 15 ................................................. 199

______________________________________________________________________________
x
ABBREVIATIONS
µm: Micrometer
ACUC: Animal Care and Use Committee
APC: Activated protein C
cm: Centimeter
DE: Dermal-epidermal
DI: Deionized water
DoD: Department of Defense
EGF: Epidermal growth factor
FGF2: fibroblast growth factor 2
GM-CSF: Granulocyte monocyte colony stimulating factor
H&E: Hemotoxylin and Eosin
h: Hour
H2O: Water
HBSS: Hanks balanced salt solution
hr: hour
IPA: Isopropyl alcohol

______________________________________________________________________________
xi
KAP: Keratin associated proteins
KDa: Kilo dalton
kg: Kilograms
KOS: Keratose
mcg/h: Micrograms per hour
mg: Milligrams
min: Minutes
MKH: Modified keratose hydrogel
mL: Milliliters
MRad: Mega radiation absorbed dose
ºC: degrees celsius
PBS: Phosphate buffer saline
PCR: Polymerase chain reaction
PEG: Polyethylene glycol
RCF: Relative centrifugal force
rpm: Revolutions per minute
r-TPA: Recombinant tissue type plasminogen

______________________________________________________________________________
xii
s: Seconds
SD: Standard Deviation
sec: Second
SIRC: Statens Seruminstitut Rabbit Cornea
SSD: Silver sulfadiazine
TBSA: Total body surface area
TCP: Tissue culture plastic
w/v: weight/volume
WFIRM: Wake Forest Institute for Regenerative Medicine
WFU IACUC: Wake Forest University Institutional Animal Care and Use Committee

______________________________________________________________________________
xiii
ABSTRACT
Deepika Rani Poranki
KERATIN BIOMATERIAL TREATMENTS FOR BURN INJURY AND
MECHANISMS OF TISSUE SURVIVAL
Dissertation under the direction of
Mark Van Dyke, Ph.D., Assistant Professor
Wake Forest Institute for Regenerative Medicine
Thermal burns typically display an injury pattern dictated by the transfer of the thermal energy
into the skin and underlying tissues. There are often three zones of injury represented by a
necrotic zone of disrupted cells and tissue, an intermediate zone of injured and dying cells, and a
distant zone of stressed cells that will recover with proper treatment. There are currently no
approved clinical therapies that target the zone of stasis and can reduce the need for excision and
grafting by salvaging potentially viable cells and tissue and thereby contributing to spontaneous
healing. In this project, we endeavored to investigate the potential of keratin biomaterials to
mediate such healing. Pilot studies testing treatment of chemical (mouse model) and thermal
(swine model) burns showed that a native mixture (termed “crude keratose” throughout this
thesis) of keratose was able to promote wound healing by stabilizing the size of the burn,
suggesting that cell survival was being facilitated in the zone of stasis. These results led us to
hypothesize that the interaction of the keratose biomaterial with cells and tissue promoted
survival and reduced the total body surface area burned. As crude keratose is a heterogeneous
mixture of proteins representing alpha and gamma protein fractions, we determined the
differential activity on cell survival associated with these fractions by using an in vitro thermal
stress model and showed that gamma keratose contributed to cell survival while alpha keratose
did not. In additional studies, the specific mechanism of cell survival associated with the gamma
keratose fraction was investigated. These results showed that gamma keratose was able to down
regulate the genes involved in cell death pathways. Later, an in vivo porcine deep partial
thickness burn model was developed to study the wound healing properties of a modified
keratose hydrogel (MKH) that contained a reduced amount of gamma keratose, reflecting the
same concentrations used in in vitro thermal stress model. The MKH showed a faster re-
epithelialization rate and wound closure compared to other treatments. Finally, to investigate the
potential clinical relevance to actual burn treatment, a delayed treatment thermal burn study was
performed. These results showed that MKH was able to promote faster wound healing compared
to other treatments but not with the same efficacy compared to more immediate post-burn
treatment. This work suggested the potential benefit of using keratin biomaterial in burn therapy
and provided informative data for developing second generation keratin biomaterial treatments.

______________________________________________________________________________
CHAPTER 1
_____________________________________________________________________________________
INTRODUCTION

______________________________________________________________________________
2
Introduction:
Skin is the largest organ of the body and acts primarily as a homeostatic tissue
maintaining body temperature and fluid balance. There are two main layers of the skin:
epidermis and dermis. Cells in the basal layer of the epidermis give rise to proliferating
keratinocytes that differentiate as they move up the strata [1, 2]. As they reach the surface of the
skin, they undergo programmed cell death [3] and these flattened, enucleated cells form the outer
stratum corneum, a heavily keratinized layer of highly cross-linked proteins that provide a
structural barrier to keep pathogens out and fluid in. This entire process is relatively fast and the
epidermis turns over every 2 weeks. The dermis consists of blood vessels, nerves, hair follicles,
smooth muscle, lymphatic tissue, and elastin fibers, along with loose connective tissue collagen.
Fibroblasts, adipocytes and macrophages are the main cell types found in dermis. The renewing
property of the epidermis helps in burn healing or any other skin injury.
Burns:
Burns are defined as injuries caused by heat, friction, electricity, radiation or chemicals.
They are classified into first degree (1°), second degree (2°), third degree (3°) and fourth degree
(4°) depending on the depth of the burn. First degree burns mainly involve the epidermal layer of
the skin. Second degree burns are further divided to superficial or deep partial thickness burns.
Superficial partial thickness burns do not extend entirely through the dermis and leave behind
epithelial-lined dermal appendages including sweat glands, hair follicles and sebaceous glands.
Deep partial thickness burns extend to the lower layers of dermis and usually heal in 3-9 weeks.
Third degree burns are full thickness burns that involve all layers of the dermis and subcutaneous

______________________________________________________________________________
3
adipose tissue. Fourth degree burns involve all the layers of dermis, subcutaneous tissue and the
underlying fascia or muscle.
According to Douglas Mac G Jackson, intensity of the burn is characterized by 3
concentric zones – the Zones of hyperemia, stasis and coagulation [4] (Figure 1). The outer zone
of hyperemia is characterized by the presence of blood circulation and active metabolism. The
intermediate zone of stasis is where the blood circulation is not reduced but metabolism is
diminished. If proper treatment is provided, the viable cells in this zone will survive. The central
zone of coagulation is the zone where injury occurred and is characterized by complete
destruction of the blood vessels. The cells in this zone are completely necrosed [4].
Figure 1: Three Zones in Burn Wounds: The central zone of coagulation is where injury
occurred and cells are completely necrosed, intermediate zone of stasis is where the blood
circulation is not reduced but metabolism is diminished and outer zone of hyperemia is
characterized by the presence of blood circulation and active metabolism.
(www.indiasurgeons.com)

______________________________________________________________________________
4
The majority of burn wounds are healed either by re-epithelialization, scar formation,
contraction, skin grafting, or combinations thereof. Re-epithelialization occurs mainly in first and
second degree burns where remnants of epithelium are present. Epithelial cells migrate from the
wound edges and regions around hair follicles to the wound bed and help in resurfacing the
residual dermis [4-7].
In scar formation, collagen deposition occurs in the wounded region. Scarring can be
divided into 3 phases – 1) Inflammatory phase where high number of inflammatory cells are
present in the wounded region, 2) Proliferative/collagen phase where there is rapid increase in
the collagen content, and 3) Maturation phase where the tensile strength of the skin increases but
there is no increase in the collagen content [8]. Contraction of the wound is mainly due to
myofibroblasts that express α-actin. The last form of healing is skin grafting which can be
divided into 3 phases – 1) Phase of imbibition where skin absorbs nutrients from the wound bed,
2) Phase of neovascularization where blood vessels invade the graft, and 3) Phase of maturation
where collagen bridges are formed between the wound bed and the graft [9].
Some burns are superficial at presentation and do not require extensive treatment and re-
epithelialize on their own, while some are full thickness burns and are best treated with excision
and grafting. However, many burns are indeterminate with mixed burn depth which may require
grafting. A typical protocol for burn care is to allow the wound to re-epithelialize if it can occur
by itself, preventing further tissue injury and avoiding infection of the wounded area.
As a standard of care, the patient is topically treated with antimicrobial agents like silver
sulfadiazine cream or silver impregnated dressings [10]. However, silver sulfadiazine has been
shown to inhibit re-epithelialization [11, 12]. In burns that are superficial and re-epithelializing,
other antimicrobial agents like topical antibiotic creams are preferred [13].

______________________________________________________________________________
5
Skin grafts are used in treating partial thickness and full thickness burns. Autografts are
the best skin grafts and are harvested from appropriate areas of the patient’s unburned skin.
When large areas of the patient are burned, harvesting sufficient autograft can be a major
challenge, particularly when unburned areas are inaccessible or undesirable (e.g. soles of the feet
or palms). In these cases there are temporary skin substitutes or permanent skin substitutes
available in the market.
Temporary skin substitutes provide transient physiologic wound coverage, help to
control pain and can absorb modest amounts of wound exudate. These include human allograft,
human amnion, xenografts and synthetic membranes. These are usually applied on a cleaned,
debrided wound. Other than human allograft, temporary skin substitutes do not vascularize but
provide effective temporary wound coverage and control pain. Human allograft is the most
often used temporary biological dressing. Allograft can either be viable with live cells
(keratinocytes, fibroblasts, endothelial cells and langerhans cells) or non-viable without any live
cells and usually is glycerolized or gamma irradiated, freeze dried or ethylene oxide treated [14-
16]. Human amnion is derived from the amniotic membrane. It acts as a temporary dressing for
clean wounds like partial thickness burns, donor sites or full thickness wounds [17-19]. One of
the main drawbacks of amnion is the difficulty of screening for viral diseases. Xenograft is
porcine derived and is used as temporary coverage for clean superficial second degree burns and
donor sites [20]. Synthetic membranes are semi-permeable membrane dressings that provide a
barrier for bacteria, vapor and also control pain until the underlying wound heals [21, 22].
Biobrane is a two layer nylon mesh that helps in fibrovascular in-growth and has an outer silastic
layer that act as vapor and bacterial barriers. One type of hydrocolloid dressings is a three
layered structure that has a porous inner layer, absorbent methyl cellulose middle layer, and semi

______________________________________________________________________________
6
permeable outer layer. This provides a moist environment for the wound to heal and absorbs
wound exudate [23, 24].
Permanent skin substitutes are also available but are not ideal. Epicel, Integra and
Alloderm are marketed as permanent skin substitutes because some or all of their components
become integrated into the healing wound. Epicel is cultured epidermal autograft. Epithelial cells
are isolated from a small skin biopsy and are cultured in vitro until there are enough cells to form
a dressing of useful size [25, 26]. These epithelial cell sheets are attached to petroleum coated
gauze which acts as a carrier and then applied on burn wounds [27]. Integra is the first dermal
substitute approved by the US Food and Drug Administration. Integra is also known as artificial
skin [28]. It contains an inner bovine collagen fibrous layer and outer silicone layer with vapor
transmission characteristics similar to normal epithelium. Once full thickness wounds are
covered with Integra and become vascularized, the outer silicone layer is replaced with thin
epithelial autograft 2-3 weeks later [29]. Alloderm is an acellular human dermal allograft. It is
devoid of epidermis and must be covered by a split thickness autograft at the time of initial
operation. It helps in reducing post operative scarring [30, 31].
Keratin Biomaterial:
Keratins are intermediate filament proteins that provide mechanical stability and integrity
for epithelial cells and tissues [32]. Mammalian keratins are classified into two groups – hard
keratins and soft keratins based on their structure, function and regulation. Hard or trichocytic
keratins, have high sulfur content and are found in a number of external appendages such as
wool and hair fibers. These have ordered arrays of intermediate filaments embedded in a matrix
of cysteine rich proteins (Figure 1). Soft or cyto keratins, are characteristic of epithelial cells
with low sulfur content [33-36]. Hair keratins and cyto keratins consist of obligate heterodimers:

______________________________________________________________________________
7
type I and type II proteins with non-helical N-terminal and C-terminal domains and a central
coiled rod domain. Type I and II chains differ in their molecular weight [36].
The hair follicle is a remarkably proliferative and regenerative organelle. To produce new
hair, the follicle undergoes a series of cycles – anagen (growth), catagen (regression) and telogen
(rest) [37]. During anagen follicles produce the entire hair shaft. During catagen and telogen,
stem cells are activated; the lower cycling portion of the hair follicle regresses in growth and is
in a resting phase. Stem cells activated in this phase trigger the new hair growth cycle again.
More than 30 growth factors and cytokines are involved in the hair cycling process, and have
also been shown to play a role in the regeneration of other tissues [38].
Figure 2: Schematic diagram of Wool fiber: Human hair has a similar superstructure to wool
fiber with 80% alpha- to 20% gamma-keratin. Alpha-keratins are the high molecular weight (40-
60kDa), low sulfur containing proteins that are mainly composed of the right-handed, alpha-
helical proteins. Gamma-keratins are the low molecular weight (10-25kDa), high sulfur

______________________________________________________________________________
8
containing proteins that hold the keratin intermediate filaments together (Used with permission
from Elsevier, Plowman JE, Proteomic database of wool fibers. J Chromatogr B Analyt Technol
Biomed Life Sci. 2003 Apr 5;787(1):63-76) .
Hair fiber consists of 3 components – cuticle, cortex and medulla (Figure 2). Cuticle is
the thin outer surface of the fiber and consists of β-keratins that help to protect the hair fiber from
external physical and chemical damage. Cortex is composed of spindle shaped cells with keratin
filaments contained within them. Type I (acidic) and Type II (basic to neutral) intermediate
filament (IF) keratins form coiled coil obligatory heterodimers through the interaction of their α-
helical rod domains [39]. Two dimers assemble in a staggered anti-parallel fashion to make a
tetramer. Two tetramers form an octamer and four octamers join to form a cylindrical unit length
filament (ULF). Individual ULF join to form short filaments which fuse end to end followed by a
compaction phase that leads to the formation of 10nm diameter IF [40]. The hair keratins have
high sulfur content and are further reinforced by highly cross linked matrix proteins called
keratin associated protein (KAPs) [32, 41].Medulla is present occasionally in the center of the
hair fiber. The many different structures and keratin sub-types lead to the complexity of the hard
keratin protein family.
Keratins Classification: Keratins constitute two classes of the intermediate filament (IF)
family proteins – type I acidic keratins and type II basic keratins [42, 43]. Type I keratins are
smaller in size with acidic isoelectric point (pI) and type II keratins are larger in size with neutral
to slightly basic pI. Genome analyses demonstrated that there are a total of 54 functional keratin
genes in humans – 28 type I keratins on chr 17q21.2 and 26 type II keratins on chr 12q13.13 [44-
46]. In 2006, Schweizer et al., published a new consensus nomenclature for mammalian keratins

______________________________________________________________________________
9
by dividing the keratins into 3 categories: 1) epithelial keratins/genes 2) hair keratins/genes, and
3) keratin pseudogenes..
The cortical proteins in the hair fiber can be divided into two groups – a cystine poor
fraction called α-keratin and cystine rich fraction called γ-keratins. The α-keratins are the main
structural component of the hair with average molecular weight 40-60KDa [47]. In the
terminology of past wool literature, the cystine rich fraction is termed γ-keratins with a
molecular weight range of 10-25KDa [47]. However, more recent publications state that the
cysteine rich fraction is more appropriately termed keratin associated proteins (KAP) and that
these matrix proteins in human hair are coded by 85 genes [41]. To add to the nomenclature
confusion, γ-keratin is often used to describe an acid-soluble fraction isolated after extraction of
oxidized keratin proteins, or so-called “keratose”. KAP can account for as much as 20-30% of
hair fiber proteins. The α-keratins assemble together to form keratin intermediate filaments
(KIF’s) that impart toughness to the fiber. The matrix proteins functions as disulfide cross linkers
that hold the KIF’s together. In humans, there are total 17 hair keratin genes – 11 type I and 6
type II [46] and more than 85 KAP genes [41].
Using chemical methods, soluble keratin proteins are extracted from the hair fiber by
breaking the disulfide crosslinks. If an oxidant is used cystine is converted to cysteic acid
derivatives and the product is termed “keratoses”, and if a reductant is used cystine is converted
to cysteine and the product is termed “kerateines”. Keratoses are hygroscopic, non-disulfide
cross linkable, water soluble, and susceptible to hydrolytic degradation at extreme of pH due to
polarization of the backbone caused by the electron withdrawing properties of cysteic acid.
These characteristics cause relatively quick degradation (days to weeks) of keratoses in vivo.

______________________________________________________________________________
10
Kerateines are less polar, more stable at extremes of pH and can be re-crosslinked through
oxidative coupling of cysteine groups. These can persist in vivo for weeks to months.
Hydrogel Treatments for Burns:
A burn dressing should serve three principle functions: comfort, metabolic and protective
[48]. They act as a protective barrier, absorb excess wound exudate, and maintain a moist
environment, which in turn helps in pain reduction. A moist wound environment is known to
accelerate wound healing [49-51], and there is abundant literature on various hydrogel
treatments. Collagen and chitosan hydrogels, and a combination of these two with other
hydrogels like alginate or dextran are the commonly used treatments. Collagen sheets were
introduced commercially in the 1960’s for the covering of large excised wound areas[52].
Research and variations soon followed, with one study showing that modified collagen
membrane was permeable to topical antibiotics with no significant antigenicity and was superior
to homograft and heterograft in autograft uptake [53]. In vitro experiments have shown that
human keratinocytes were able to grow extensively on human dermal collagen and differentiate
within a few days into columnar conformation [54]. Porous collagen sponges were able to
provide three dimensional matrix for tissue infiltration in vivo and also cell growth in vitro [55].
Collagen sheets were shown to promote proliferation and attachment of neonatal rat
keratinocytes in vitro and promote faster re-epithelialization when these sheets were used to
cover full thickness burns on the dorsum of rats [56]. More recently, collagen hydrogel has been
used synergistically with other hydrogel systems in the treatment of burns. Collagen and chitosan
porous scaffolds cross linked with glutaraldehyde have shown an increase in proliferation in
vitro, and when tested in animals have shown that the scaffold was able to support and accelerate

______________________________________________________________________________
11
fibroblast cell infiltration from surrounding tissue, suggesting its use as a potential dermal
equivalent [57]. Chitosan –collagen hydrogel incorporated with lysostaphin was shown to control
methicillin-resistant Staphylococcus aureus infection in third degree burn patients and promoted
healing [58].
Chitosan is a de-acetylated derivative of chitin that has been shown to facilitate burn
wound healing in vivo [59-67] and cell adhesion and viability in vitro in primary rat fibroblast
cells [59]. Full thickness burn wounds that were treated with high molecular weight, high de-
acetylated chitosan showed faster re-epithelialization and wound closure in rats compared to 2%
Fucidin treatment [68]. Chitosan hydrogel was also used as a carrier for biopharmaceuticals,
antimicrobials and growth factors. A chitosan gel formulation with epidermal growth factor
(EGF) was shown to significantly increase cell proliferation and had a faster re-epithelialization
rate in second degree burns in rats [60, 67]. It was also used as a fibroblast growth factor 2
(FGF2) carrier that induced angiogenesis and collateral blood circulation in impaired diabetic
mice [69]. In Wistar-Albino rats, Chitosan gels with controlled slow release granulocyte-
monocyte colony stimulating factor (GM-CSF) assisted burn wound healing with better collagen
fibril organization [70]. Chitosan acetate bandages were shown to control the growth of
Pseudomonas aeruginosa and Pseudomonas mirabilis bacteria, and control the development of
systemic sepsis in third degree burns in mice [64]. A hydrogel sheet composed of alginate,
chitosan and fucoidan was shown to promote wound healing in healing impaired rats [71].
Dextran hydrogel scaffolds were shown to promote early inflammatory cell infiltration in full
thickness burns, which led to the degradation of the scaffold and in turn promoted the infiltration
of endothelial cells into the wounded region. This enhanced neo-vascularization and restoration
of hair fibers and epidermal morphology similar to normal mouse skin [72].

______________________________________________________________________________
12
Alginates are polysaccharides derived from sea weed and there are a wide variety
available commercially that have been shown to promote wound healing [73-78]. Alginate
dressings are typically made from calcium alginate, which has cytotoxicity and immune
reactions associated with it. [79]. Development of alginate dressing with less calcium ions
reduced the cytotoxicity and foreign body reaction in both in vitro and in vivo experiments [79,
80]. Hydrocolloid dressings are used in partial thickness and small burns where the wound
exudate is comparatively less [81, 82]. Compared to hydrocolloid dressings, alginate dressings
remain gelled longer as the presence of calcium ion crosslinks decreases the biodegradability of
alginate gels [83]. The antimicrobial properties of the above mentioned alginate dressings have
been increased by the addition of silver ions [84-90].
Cell Death Pathways:
In burns, one of the main reasons for the increase in total body surface area (TBSA)
burned is the increase in cell death after the thermal insult. Apoptosis and Necrosis are the two
main modes of cell death that occur when the tissues are exposed to heat. There are two
apoptotic pathways – extrinsic or death receptor pathway, and intrinsic or mitochondrial pathway
(Figure 3). In both these pathways, cysteine aspartyl-specific proteases (caspases) are activated,
which further leads to the morphological and biochemical changes associated with apoptosis.
The death receptors usually belong to the tumor necrosis factor super family, comprising a
subfamily with a characteristic intracellular death domain [91]. When the ligands of the tissue
necrosis factor (TNF) family bind to their respective death receptors such as CD-95 and TRAIL-
R1, they are activated and attract intracellular Fas-associated death domain (FADD), which
further recruits caspases [92]. Caspase 8 and 10 function as initiator caspases that are recruited to
the death inducing signaling complex (DISC) [93]. The DISC cleaves the procaspase 8 and 10

______________________________________________________________________________
13
and yield active initiator caspases 8 and 10 [94]. Active caspase 8 cleaves BH3 interacting
domain (BID), a BCL2 family protein which translocates to mitochondria and initiates the
intrinsic pathway [95, 96]. Activation of mitochondria leads to the release of Cyt C into the
cytosol, which binds to apoptotic protease activating factor 1(APAF1) to form the apoptosome.
Apoptosome, along with TP, cleave procaspase 9 and activate it. Any of the BCL2 family
proteins: BAD, BID, BIM can activate the intrinsic pathway of apoptosis. Once the initiator
caspases are activated, they cleave and activate executioner caspases 3, 6 and 7, which cleave the
cellular substrates downstream [92]. Once the phosphatudylserine are exposed on the cell
membrane, the remains of the cell are engulfed by phagocytosis [97].

______________________________________________________________________________
14
Figure 3: Apoptotic Pathways: The 2 major pathways of apoptosis:the extrinsic (Fas and other
TNFR superfamily members and ligands) and the intrinsic (mitochondria-associated) pathways.
Both pathways lead to activation of caspase-3, giving rise to apoptotic cell death.(Used with
permission from Seminars from Arthritis and Rheumatism, Schultz DZ, Apoptosis: Programmed
cell death at a molecular level. Semin Arthritis Rheum. 2003 Jun;32(6):345-69)
There are some other cell death pathways that are not well defined and do not require caspase
activation [98-100]. Apoptotic pathways can be regulated at the initial receptor level, by
inhibiting caspase activation, or by influencing the permeability of the mitochondrial membrane.
Receptor level regulation is performed by FLIPs (FADD-like interleukin-1 -converting
enzyme-like protease (FLICE/caspase-8)-inhibitory proteins) [101]. The members of BCL2
family proteins regulate apoptosis at the mitochondrial level and may be either pro-apoptotic or
anti-apoptotic [95, 96]. Inhibitor of Apoptotic Proteins (IAPs) bind and inhibit activation of
caspases [102]. IAPs are inhibited by SMAC/DIABLO (second mitochondria-derived activator
of caspase/direct IAP binding protein with low pI), which is released from mitochondria along
with cyt c [103, 104].
Necrosis used to be considered a disorganized mode of cell death, especially when the
cells are exposed to severe physiochemical conditions. But recently it was shown that necrosis
occurs during normal physiological development [105, 106]. There are published in vitro studies
showing that even though apoptosis is completely inhibited, cell death still occurs and is usually
independent of caspases, following a specific signaling pathway where the cell morphology
looks more like necrotic cell death [107-111]. FADD (Fas-associated death domain) plays a
crucial role in propagating apoptotic or necrotic signals as it has both a death domain that can
trigger necrosis and a death effector domain that can propagate apoptosis [112, 113]. When

______________________________________________________________________________
15
caspase activation is impeded, FADD is not recruited when the TNF ligand binds to the death
receptor TRAIL-R1 [114-116]. Instead, a complex 1 is formed at the plasma membrane
consisting of TNF-R1, TRAF2 and RIP1 that activates NF-κB and MAPKs [117, 118]. After
receptor endocytosis, a second complex with TRADD that recruits FADD and procaspase 8 is
formed [119, 120]. If complex 1 is not able to induce sufficient anti-apoptotic proteins, then
caspase 8 is activated leading to caspase induced apoptosis [121]. If the anti-apoptotic proteins
block the caspases, then necrosis occurs through the caspase independent pathway [109]. Holler
et al. have shown that RIP is required for necrotic cell death induced by TNF and TRAIL, and
that it has its own kinase activity that activates the cell death pathway when phosphorylated
[122]. In Rip null (Rip–/–) mouse fibroblast cells, it is shown that RIP is required for TNF-
induced activation of the MAPKs extracellular-signal-related kinase (ERK), p38 and c-Jun
amino-terminal kinase (JNK) [123].
Autophagy refers to a catabolic process involving degradation of the cell’s own
components and is usually activated by oxidative, nutritional or toxic stresses. Macroautophagy
is an evolutionarily conserved, genetically controlled multi-step process in which intracellular
organelles are sequestered within characteristic double or multi-membrane autophagic vacuoles
termed autophagosome, and finally delivered to lysosomes for bulk degradation [124] (Figure 4).
Embryonic fibroblasts from double knock out Bax-/-
Bak-/-
mice are resistant to apoptotic
inducers. When treated with etoposide, the cells failed to undergo apoptosis and instead
manifested a massive autophagy followed by cell death [125]. But knocking down the autophagy
related gene (Atg) products beclin-1 and Atg5 reduced etoposide induced cell death [125]. Using
other apoptotic inhibitors did not trigger the autophagy cell death pathway, indicating that Bax
and Bak absence played an important role in autophagy activation [125] . In other cell types like

______________________________________________________________________________
16
L929 mouse fibrosarcome, Human Jurkat T lymphoma and U937 monocytoid cells, inhibition of
cystein proteases (caspases) was shown to induce autophagy [126, 127]. In stress conditions like
nutrition depletion or loss of growth factors, autophagy gets activated by the inhibition of
apoptosis and promotes cell survival [128, 129]. If autophagy is inhibited in cell stress conditions
then the cells usually die through apoptosis. This cell death can be postponed by inhibiting BAX
or BAK or caspases [130]. In vitro and in vivo experiments have shown induced apoptotic cell
death by the inactivation of Atg genes – Atg 5 or 7, beclin-1[131-134].
Figure 4: Autophagy and its inhibitors: Autophagy starts with the stepwise engulfment of
cytoplasmic material (cytosol and/or organelles) by the phagophore (also called isolation
membrane), which sequesters material in double-membraned vesicles named autophagosomes
(also called autophagic vacuoles). There are 4 regulatory process: (1)De‑repression of the

______________________________________________________________________________
17
mTOR Ser/Thr kinase (2)Activation of mammalian Vps34 in the initial steps of vesicle
nucleation (3)Two ubiquitin-like conjugation systems that are part of the vesicle elongation
process (4)Autophagosomes maturation by the fusion with lysosomes to create autolysosomes.
(Used with permission from Nature Publishing Group, Maiuri MC, Zalckvar E, Kimchi A,
Kroemer G Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev
Mol Cell Biol. 2007 Sep;8(9):741-52)
Bcl2 and Bcl-XL are regulators of beclin-1. Lamp2,lysosome-associated membrane
glycoprotein-2. Autophagy may develop as a primary response to stress stimuli, which then
triggers either apoptosis or necrosis [135, 136]. There is crosstalk between all the cell death
pathways, apoptosis, necrosis and autophagy, depending on the external and internal conditions
to which the cells are exposed.

______________________________________________________________________________
18
Table 1: Gene Glossary
Abbreviation Full Form Function
CD-95 Fas ligand
Cytokine that binds to TNFRSF6/FAS, a receptor
that transduces the apoptotic signal into cells.
TRAIL-r1
tumor necrosis
factor receptor
superfamily Receptor for the cytotoxic ligand TNFSF10/TRAIL.
FADD
Fas-associated death
domain
Apoptotic adaptor molecule that recruits caspase-8
or caspase-10 to the activated Fas (CD95) or TNFR-
1 receptors.
BID
BH3-interacting
domain death
agonist
This gene encodes a death agonist that
heterodimerizes with either agonist BAX or
antagonist BCL2.
Cyt C Cytochrome C
Electron carrier protein. Plays a role in apoptosis.
Suppression of the anti-apoptotic members or
activation of the pro-apoptotic members of the Bcl-2
family leads to altered mitochondrial membrane
permeability resulting in release of cytochrome c
into the cytosol. Binding of cytochrome c to Apaf-1
triggers the activation of caspase-9, which then
accelerates apoptosis by activating other caspases
APAF1
Apoptotic protease
activating factor 1
This gene encodes a cytoplasmic protein that
initiates apoptosis. This protein contains several
copies of the WD-40 (beta transducin)domain, a
caspase recruitment domain (CARD), and an
ATPase domain (NB-ARC).
BAD
BCL2-associated
agonist of cell death
Promotes cell death. Successfully competes for the
binding to Bcl-X(L), Bcl-2 and Bcl-W, thereby
affecting the level of heterodimerization of these
proteins with BAX. Can reverse the death repressor
activity of Bcl-X(L), but not that of Bcl-2 (By
similarity). Appears to act as a link between growth
factor receptor signaling and the apoptotic pathways
BIM BCL2-like 11 Induces apoptosis.
Caspases
3,6,7,8,10
cysteinyl aspartate
proteases
Caspases are involved in the signal transduction
pathways of apoptosis, necrosis and inflammation.
Initiator caspases include Capases 1,-4,-5,-8,-9,-10,-
11,-12 and effector caspases include Caspase -3,-6,-7

______________________________________________________________________________
19
FLIP
FADD-like
interleukin-1 -
converting enzyme-
like protease
(FLICE/caspase-8)-
inhibitory proteins
Apoptosis regulator protein which functions as a
crucial link between cell survival and cell death
pathways in mammalian cells.
SMAC,
DIABLO
second
mitochondria-
derived activator of
caspase/direct IAP
binding protein with
low pI
Promotes apoptosis by activating caspases in the
cytochrome c/Apaf-1/caspase-9 pathway.
TRAF2
TNF receptor-
associated factor 2
Regulates activation of NF-kappa-B and JNK and
plays a central role in the regulation of cell survival
and apoptosis.
RIP1
receptor (TNFRSF)-
interacting serine-
threonine kinase
Serine-threonine kinase which transduces
inflammatory and cell-death signals (necroptosis)
following death receptors ligation.
NF-kB
nuclear factor
kappa-light-chain-
enhancer of
activated B cells
NF-kappaB (nuclear factor-kappa B) is a rapidly
acting primary transcription factor involved in
cellular responses to stimuli such as cytokines and
stress and plays a key role in regulating the immune
response to infection.
TRADD
TNFRSF1A-
associated via death
domain
Adapter molecule for TNFRSF1A/TNFR1 that
specifically associates with the cytoplasmic domain
of activated TNFRSF1A/TNFR1 mediating its
interaction with FADD. Overexpression of TRADD
leads to two major TNF-induced responses,
apoptosis and activation of NF-kappa-B
TNF
Tumor Necrosis
Factor
Cytokine that binds to TNFRSF1A/TNFR1 and
TNFRSF1B/TNFBR.
ERK
MAPKs
extracellular-signal-
related kinase
Involved in both the initiation and regulation of
meiosis, mitosis, and postmitotic functions in
differentiated cells by phosphorylating of
transcription factors
P38, JNK,
MAPK9
p38, c-Jun amino
terminal kinase,
Mitogen activated
rotein kinase
Three major MAPKs include ERKs (Extracellular
signal-Regulated Kinases), JNKs (c-Jun NH(2)-
terminal protein Kinases), and p38 Kinases. These
are activated by environmental stresses and
inflammatory cytokines
Beclin-1 autophagy-related Plays a central role in autophagy.

______________________________________________________________________________
20
ATG5
autophagy related 5
homolog
Required for autophagy. Its expression is a relatively
late event in the apoptotic process, occurring
downstream of caspase activity.
ATG7
autophagy related 7
homolog
Functions as an E1 enzyme essential for multi
substrates such as ATG8-like proteins and ATG12.
BAX, BAK1
Bcl-2-associated X
protein
Accelerates programmed cell death by binding to,
and antagonizing the apoptosis repressor BCL2.
Bcl2 B-cell lymphoma-2
Potent inhibitor of cell death. Inhibits activation of
caspases.
Bcl-XL
B-cell lymphoma-
extra large Positive regulator of apoptosis
Lamp2
Lysosomal-
associated
membrane protein 2 Implicated in tumor cell metastasis.

______________________________________________________________________________
21
Rationale for the hypothesis that the interaction of keratin biomaterial with burned tissue
promotes cell survival and will reduce total body surface area (TBSA) burned:
Previous work in our group has focused on testing the wound healing capacities of keratin
biomaterials in animals. Two wound healing burn studies were performed – a mouse chemical
burn study and a swine thermal burn study. Both of the animal studies have shown that keratin
biomaterials promote tissue salvage after burn injury and speed healing. Generally, hydrogel
treatments are able to provide a moist environment, help control exudation of wound fluid, which
in turn reduces pain. As previously discussed, there are many hydrogel treatments available for
burn patients, but none of those treatments were able to show that they facilitate cell survival and
tissue salvage by any of the aforementioned pathways. From our previous wound healing studies,
it was shown that a hydrogel made from a “crude” preparation of keratose (i.e. not otherwise
refined or purified) was able to reduce TBSA burned and accelerate wound healing. As crude
keratose is a heterogeneous mixture of alpha, gamma and KAPs, we hypothesized that separating
the different fractions and testing their cell salvage abilities would be helpful in the development
of better keratin formulations for the treatment of burns. To test this hypothesis, three specific
aims were undertaken and form the basis of this thesis project:
Specific Aim 1: To develop an in vitro thermal injury model that mimics the Jackson model [4]
of burn injury and determine the effect of keratin subtype on cell survival and recovery from
thermal injury.
Specific Aim 2: To understand the molecular mechanism by which keratose biomaterial
promotes cell survival.
Specific Aim 3: To test the wound repair capabilities of meta-keratose in a partial thickness pig
burn injury model.

______________________________________________________________________________
22
References:
[1] Alonso L, Fuchs E. Stem cells in the skin: waste not, Wnt not. Genes Dev. 2003;17:1189-
200.
[2] Fuchs E. Epidermal differentiation: the bare essentials. J Cell Biol. 1990;111:2807-14.
[3] Lippens S, Denecker G, Ovaere P, Vandenabeele P, Declercq W. Death penalty for
keratinocytes: apoptosis versus cornification. Cell Death Differ. 2005;12 Suppl 2:1497-508.
[4] Jackson DM. [The diagnosis of the depth of burning]. Br J Surg. 1953;40:588-96.
[5] Woodley DT, O'Keefe EJ, Prunieras M. Cutaneous wound healing: a model for cell-matrix
interactions. J Am Acad Dermatol. 1985;12:420-33.
[6] Barrandon Y, Green H. Cell migration is essential for sustained growth of keratinocyte
colonies: the roles of transforming growth factor-alpha and epidermal growth factor. Cell.
1987;50:1131-7.
[7] Cooper ML, Hansbrough JF, Foreman TJ, Sakabu SA, Laxer JA. The effects of epidermal
growth factor and basic fibroblast growth factor on epithelialization of meshed skin graft
interstices. Prog Clin Biol Res. 1991;365:429-42.
[8] Staiano-Coico L, Krueger JG, Rubin JS, D'Limi S, Vallat VP, Valentino L, et al. Human
keratinocyte growth factor effects in a porcine model of epidermal wound healing. J Exp Med.
1993;178:865-78.
[9] N.Herndon D. Total Burn Care. Third ed: Saunders Elsevier; 2007.
[10] Hussain S, Ferguson C. Best evidence topic report. Silver sulphadiazine cream in burns.
Emerg Med J. 2006;23:929-32.

______________________________________________________________________________
23
[11] Geronemus RG, Mertz PM, Eaglstein WH. Wound healing. The effects of topical
antimicrobial agents. Arch Dermatol. 1979;115:1311-4.
[12] Muller MJ, Hollyoak MA, Moaveni Z, Brown TL, Herndon DN, Heggers JP. Retardation of
wound healing by silver sulfadiazine is reversed by Aloe vera and nystatin. Burns. 2003;29:834-
6.
[13] Atiyeh BS, Costagliola M, Hayek SN, Dibo SA. Effect of silver on burn wound infection
control and healing: review of the literature. Burns. 2007;33:139-48.
[14] Bondoc CC, Burke JF. Clinical experience with viable frozen human skin and a frozen skin
bank. Ann Surg. 1971;174:371-82.
[15] Herndon DN. Perspectives in the use of allograft. J Burn Care Rehabil. 1997;18:S6.
[16] Kagan RJ, Robb EC, Plessinger RT. Human skin banking. Clin Lab Med. 2005;25:587-605.
[17] Ramakrishnan KM, Jayaraman V. Management of partial-thickness burn wounds by
amniotic membrane: a cost-effective treatment in developing countries. Burns. 1997;23 Suppl
1:S33-6.
[18] Sawhney CP. Amniotic membrane as a biological dressing in the management of burns.
Burns. 1989;15:339-42.
[19] Subrahmanyam M. Amniotic membrane as a cover for microskin grafts. Br J Plast Surg.
1995;48:477-8.
[20] Elliott RA, Jr., Hoehn JG. Use of commercial porcine skin for wound dressings. Plast
Reconstr Surg. 1973;52:401-5.
[21] Salisbury RE, Carnes RW, Enterline D. Biological dressings and evaporative water loss
from burn wounds. Ann Plast Surg. 1980;5:270-2.

______________________________________________________________________________
24
[22] Salisbury RE, Wilmore DW, Silverstein P, Pruitt BA, Jr. Biological dressings for skin graft
donor sites. Arch Surg. 1973;106:705-6.
[23] Hermans MH. HydroColloid dressing (Duoderm) for the treatment of superficial and deep
partial thickness burns. Scand J Plast Reconstr Surg Hand Surg. 1987;21:283-5.
[24] Nangia A, Hung CT. Design of a new hydrocolloid dressing. Burns. 1989;15:385-8.
[25] Gallico GG, 3rd, O'Connor NE, Compton CC, Kehinde O, Green H. Permanent coverage of
large burn wounds with autologous cultured human epithelium. N Engl J Med. 1984;311:448-51.
[26] Green H, Kehinde O, Thomas J. Growth of cultured human epidermal cells into multiple
epithelia suitable for grafting. Proc Natl Acad Sci U S A. 1979;76:5665-8.
[27] Sheridan RL, Tompkins RG. Cultured autologous epithelium in patients with burns of
ninety percent or more of the body surface. J Trauma. 1995;38:48-50.
[28] Tompkins RG, Burke JF. Progress in burn treatment and the use of artificial skin. World J
Surg. 1990;14:819-24.
[29] Tompkins RG, Hilton JF, Burke JF, Schoenfeld DA, Hegarty MT, Bondoc CC, et al.
Increased survival after massive thermal injuries in adults: preliminary report using artificial
skin. Crit Care Med. 1989;17:734-40.
[30] Wainwright D, Madden M, Luterman A, Hunt J, Monafo W, Heimbach D, et al. Clinical
evaluation of an acellular allograft dermal matrix in full-thickness burns. J Burn Care Rehabil.
1996;17:124-36.
[31] Wainwright DJ. Use of an acellular allograft dermal matrix (AlloDerm) in the management
of full-thickness burns. Burns. 1995;21:243-8.
[32] Moll R, Divo M, Langbein L. The human keratins: biology and pathology. Histochem Cell
Biol. 2008;129:705-33.

______________________________________________________________________________
25
[33] Heid HW, Moll I, Franke WW. Patterns of expression of trichocytic and epithelial
cytokeratins in mammalian tissues. II. Concomitant and mutually exclusive synthesis of
trichocytic and epithelial cytokeratins in diverse human and bovine tissues (hair follicle, nail bed
and matrix, lingual papilla, thymic reticulum). Differentiation. 1988;37:215-30.
[34] Langbein L, Rogers MA, Winter H, Praetzel S, Schweizer J. The catalog of human hair
keratins. II. Expression of the six type II members in the hair follicle and the combined catalog
of human type I and II keratins. J Biol Chem. 2001;276:35123-32.
[35] Moll R, Franke WW, Schiller DL, Geiger B, Krepler R. The catalog of human cytokeratins:
patterns of expression in normal epithelia, tumors and cultured cells. Cell. 1982;31:11-24.
[36] Fraser RD, MacRae TP, Parry DA, Suzuki E. Intermediate filaments in alpha-keratins. Proc
Natl Acad Sci U S A. 1986;83:1179-83.
[37] Alonso L, Fuchs E. The hair cycle. J Cell Sci. 2006;119:391-3.
[38] Stenn KS, Paus R. What controls hair follicle cycling? Exp Dermatol. 1999;8:229-33;
discussion 33-6.
[39] Popescu C, Hocker H. Hair--the most sophisticated biological composite material. Chem
Soc Rev. 2007;36:1282-91.
[40] Goldman RD, Grin B, Mendez MG, Kuczmarski ER. Intermediate filaments: versatile
building blocks of cell structure. Curr Opin Cell Biol. 2008;20:28-34.
[41] Rogers MA, Langbein L, Praetzel-Wunder S, Winter H, Schweizer J. Human hair keratin-
associated proteins (KAPs). Int Rev Cytol. 2006;251:209-63.
[42] Yu J, Yu DW, Checkla DM, Freedberg IM, Bertolino AP. Human hair keratins. J Invest
Dermatol. 1993;101:56S-9S.

______________________________________________________________________________
26
[43] Fuchs EV, Coppock SM, Green H, Cleveland DW. Two distinct classes of keratin genes and
their evolutionary significance. Cell. 1981;27:75-84.
[44] Hesse M, Magin TM, Weber K. Genes for intermediate filament proteins and the draft
sequence of the human genome: novel keratin genes and a surprisingly high number of
pseudogenes related to keratin genes 8 and 18. J Cell Sci. 2001;114:2569-75.
[45] Rogers MA, Winter H, Langbein L, Bleiler R, Schweizer J. The human type I keratin gene
family: characterization of new hair follicle specific members and evaluation of the chromosome
17q21.2 gene domain. Differentiation. 2004;72:527-40.
[46] Rogers MA, Edler L, Winter H, Langbein L, Beckmann I, Schweizer J. Characterization of
new members of the human type II keratin gene family and a general evaluation of the keratin
gene domain on chromosome 12q13.13. J Invest Dermatol. 2005;124:536-44.
[47] Buchanan JH. A cystine-rich protein fraction from oxidized alpha-keratin. Biochem J.
1977;167:489-91.
[48] Herndon DN. Total Burn Care. Third ed: Saunders Elsevier; 2007.
[49] Alvarez O. Moist environment for healing: matching the dressing to the wound. Ostomy
Wound Manage. 1988;21:64-83.
[50] Bryan J. Moist wound healing: a concept that changed our practice. J Wound Care.
2004;13:227-8.
[51] Chang H, Wind S, Kerstein MD. Moist wound healing. Dermatol Nurs. 1996;8:174-6, 204.
[52] Abbenhaus JI, MacMahon RA, Rosenkrantz JG, Paton BC. Collagen sheets as a dressing for
large excised areas. Surg Forum. 1965;16:477-8.
[53] Tavis MJ, Harney JH, Thornton JW, Bartlett RH. Modified collagen membrane as a skin
substitute: preliminary studies. J Biomed Mater Res. 1975;9:285-301.

______________________________________________________________________________
27
[54] Shakespeare VA, Shakespeare PG. Growth of cultured human keratinocytes on fibrous
dermal collagen: a scanning electron microscope study. Burns Incl Therm Inj. 1987;13:343-8.
[55] Doillon CJ. Porous collagen sponge wound dressings: in vivo and in vitro studies. J
Biomater Appl. 1988;2:562-78.
[56] Morykwas MJ, Stevenson TR, Marcelo CL, Thornton JW, Smith DJ, Jr. In vitro and in vivo
testing of a collagen sheet to support keratinocyte growth for use as a burn wound covering. J
Trauma. 1989;29:1163-6; discussion 6-7.
[57] Ma L, Gao C, Mao Z, Zhou J, Shen J, Hu X, et al. Collagen/chitosan porous scaffolds with
improved biostability for skin tissue engineering. Biomaterials. 2003;24:4833-41.
[58] Cui F, Li G, Huang J, Zhang J, Lu M, Lu W, et al. Development of chitosan-collagen
hydrogel incorporated with lysostaphin (CCHL) burn dressing with anti-methicillin-resistant
Staphylococcus aureus and promotion wound healing properties. Drug Deliv. 2011;18:173-80.
[59] Ribeiro MP, Espiga A, Silva D, Baptista P, Henriques J, Ferreira C, et al. Development of a
new chitosan hydrogel for wound dressing. Wound Repair Regen. 2009;17:817-24.
[60] Alemdaroglu C, Degim Z, Celebi N, Zor F, Ozturk S, Erdogan D. An investigation on burn
wound healing in rats with chitosan gel formulation containing epidermal growth factor. Burns.
2006;32:319-27.
[61] Boucard N, Viton C, Agay D, Mari E, Roger T, Chancerelle Y, et al. The use of physical
hydrogels of chitosan for skin regeneration following third-degree burns. Biomaterials.
2007;28:3478-88.
[62] Jin Y, Ling PX, He YL, Zhang TM. Effects of chitosan and heparin on early extension of
burns. Burns. 2007;33:1027-31.

______________________________________________________________________________
28
[63] Sezer AD, Cevher E, Hatipoglu F, Ogurtan Z, Bas AL, Akbuga J. Preparation of fucoidan-
chitosan hydrogel and its application as burn healing accelerator on rabbits. Biol Pharm Bull.
2008;31:2326-33.
[64] Dai T, Tegos GP, Burkatovskaya M, Castano AP, Hamblin MR. Chitosan acetate bandage
as a topical antimicrobial dressing for infected burns. Antimicrob Agents Chemother.
2009;53:393-400.
[65] Nascimento EG, Sampaio TB, Medeiros AC, Azevedo EP. Evaluation of chitosan gel with
1% silver sulfadiazine as an alternative for burn wound treatment in rats. Acta Cir Bras.
2009;24:460-5.
[66] Dantas MD, Cavalcante DR, Araujo FE, Barretto SR, Aciole GT, Pinheiro AL, et al.
Improvement of dermal burn healing by combining sodium alginate/chitosan-based films and
low level laser therapy. J Photochem Photobiol B. 2011;105:51-9.
[67] Degim Z, Celebi N, Alemdaroglu C, Deveci M, Ozturk S, Ozogul C. Evaluation of chitosan
gel containing liposome-loaded epidermal growth factor on burn wound healing. Int Wound J.
2011;8:343-54.
[68] Alsarra IA. Chitosan topical gel formulation in the management of burn wounds. Int J Biol
Macromol. 2009;45:16-21.
[69] Ishihara M, Obara K, Nakamura S, Fujita M, Masuoka K, Kanatani Y, et al. Chitosan
hydrogel as a drug delivery carrier to control angiogenesis. J Artif Organs. 2006;9:8-16.
[70] Simsek S, Canter HI, Konas E, Korkusuz P, Demir D, Oner F, et al. A new concept in
treatment of burn injury: controlled slow-release granulocyte-monocyte colony-stimulating
factor chitosan gel system. Ann Plast Surg. 2011;67:583-8.

______________________________________________________________________________
29
[71] Murakami K, Aoki H, Nakamura S, Takikawa M, Hanzawa M, Kishimoto S, et al. Hydrogel
blends of chitin/chitosan, fucoidan and alginate as healing-impaired wound dressings.
Biomaterials. 2010;31:83-90.
[72] Sun G, Zhang X, Shen YI, Sebastian R, Dickinson LE, Fox-Talbot K, et al. Dextran
hydrogel scaffolds enhance angiogenic responses and promote complete skin regeneration during
burn wound healing. Proc Natl Acad Sci U S A. 2011;108:20976-81.
[73] Piacquadio D, Nelson DB. Alginates. A "new" dressing alternative. J Dermatol Surg Oncol.
1992;18:992-5.
[74] Agren MS. Four alginate dressings in the treatment of partial thickness wounds: a
comparative experimental study. Br J Plast Surg. 1996;49:129-34.
[75] Williams C. Melgisorb: a highly absorbent calcium/sodium alginate dressing. Br J Nurs.
1998;7:975-6.
[76] Williams C. Tegagen alginate dressing for moderate to heavily exuding wounds. Br J Nurs.
1998;7:550-2.
[77] Williams C. Algosteril calcium alginate dressing for moderate/high exudate. Br J Nurs.
1999;8:313-7.
[78] Kammerlander G, Eberlein T. An assessment of the wound healing properties of Algisite M
dressings. Nurs Times. 2003;99:54-6.
[79] Suzuki Y, Nishimura Y, Tanihara M, Suzuki K, Nakamura T, Shimizu Y, et al. Evaluation
of a novel alginate gel dressing: cytotoxicity to fibroblasts in vitro and foreign-body reaction in
pig skin in vivo. J Biomed Mater Res. 1998;39:317-22.
[80] Suzuki Y, Tanihara M, Nishimura Y, Suzuki K, Yamawaki Y, Kudo H, et al. In vivo
evaluation of a novel alginate dressing. J Biomed Mater Res. 1999;48:522-7.

______________________________________________________________________________
30
[81] Vloemans AF, Soesman AM, Suijker M, Kreis RW, Middelkoop E. A randomised clinical
trial comparing a hydrocolloid-derived dressing and glycerol preserved allograft skin in the
management of partial thickness burns. Burns. 2003;29:702-10.
[82] Thomas S. Hydrocolloid dressings in the management of acute wounds: a review of the
literature. Int Wound J. 2008;5:602-13.
[83] Ichioka S, Harii K, Nakahara M, Sato Y. An experimental comparison of hydrocolloid and
alginate dressings, and the effect of calcium ions on the behaviour of alginate gel. Scand J Plast
Reconstr Surg Hand Surg. 1998;32:311-6.
[84] Qin Y. Silver-containing alginate fibres and dressings. Int Wound J. 2005;2:172-6.
[85] Muangman P, Muangman S, Opasanon S, Keorochana K, Chuntrasakul C. Benefit of
hydrocolloid SSD dressing in the outpatient management of partial thickness burns. J Med Assoc
Thai. 2009;92:1300-5.
[86] Opasanon S, Muangman P, Namviriyachote N. Clinical effectiveness of alginate silver
dressing in outpatient management of partial-thickness burns. Int Wound J. 2010;7:467-71.
[87] Lu S, Gao W, Gu HY. Construction, application and biosafety of silver nanocrystalline
chitosan wound dressing. Burns. 2008;34:623-8.
[88] Ong SY, Wu J, Moochhala SM, Tan MH, Lu J. Development of a chitosan-based wound
dressing with improved hemostatic and antimicrobial properties. Biomaterials. 2008;29:4323-32.
[89] Huang L, Dai T, Xuan Y, Tegos GP, Hamblin MR. Synergistic combination of chitosan
acetate with nanoparticle silver as a topical antimicrobial: efficacy against bacterial burn
infections. Antimicrob Agents Chemother. 2011;55:3432-8.

______________________________________________________________________________
31
[90] Li D, Diao J, Zhang J, Liu J. Fabrication of new chitosan-based composite sponge
containing silver nanoparticles and its antibacterial properties for wound dressing. J Nanosci
Nanotechnol. 2011;11:4733-8.
[91] Yu KY, Kwon B, Ni J, Zhai Y, Ebner R, Kwon BS. A newly identified member of tumor
necrosis factor receptor superfamily (TR6) suppresses LIGHT-mediated apoptosis. J Biol Chem.
1999;274:13733-6.
[92] Igney FH, Krammer PH. Death and anti-death: tumour resistance to apoptosis. Nat Rev
Cancer. 2002;2:277-88.
[93] Kischkel FC, Hellbardt S, Behrmann I, Germer M, Pawlita M, Krammer PH, et al.
Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling
complex (DISC) with the receptor. EMBO J. 1995;14:5579-88.
[94] Kischkel FC, Lawrence DA, Tinel A, LeBlanc H, Virmani A, Schow P, et al. Death receptor
recruitment of endogenous caspase-10 and apoptosis initiation in the absence of caspase-8. J Biol
Chem. 2001;276:46639-46.
[95] Zamzami N, Kroemer G. The mitochondrion in apoptosis: how Pandora's box opens. Nat
Rev Mol Cell Biol. 2001;2:67-71.
[96] Martinou JC, Green DR. Breaking the mitochondrial barrier. Nat Rev Mol Cell Biol.
2001;2:63-7.
[97] Savill J, Fadok V. Corpse clearance defines the meaning of cell death. Nature.
2000;407:784-8.
[98] Borner C, Monney L. Apoptosis without caspases: an inefficient molecular guillotine? Cell
Death Differ. 1999;6:497-507.

______________________________________________________________________________
32
[99] Xiang J, Chao DT, Korsmeyer SJ. BAX-induced cell death may not require interleukin 1
beta-converting enzyme-like proteases. Proc Natl Acad Sci U S A. 1996;93:14559-63.
[100] Sperandio S, de Belle I, Bredesen DE. An alternative, nonapoptotic form of programmed
cell death. Proc Natl Acad Sci U S A. 2000;97:14376-81.
[101] Krueger A, Baumann S, Krammer PH, Kirchhoff S. FLICE-inhibitory proteins: regulators
of death receptor-mediated apoptosis. Mol Cell Biol. 2001;21:8247-54.
[102] Deveraux QL, Reed JC. IAP family proteins--suppressors of apoptosis. Genes Dev.
1999;13:239-52.
[103] Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes
cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102:33-42.
[104] Verhagen AM, Ekert PG, Pakusch M, Silke J, Connolly LM, Reid GE, et al. Identification
of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP
proteins. Cell. 2000;102:43-53.
[105] Chautan M, Chazal G, Cecconi F, Gruss P, Golstein P. Interdigital cell death can occur
through a necrotic and caspase-independent pathway. Curr Biol. 1999;9:967-70.
[106] Kitanaka C, Kuchino Y. Caspase-independent programmed cell death with necrotic
morphology. Cell Death Differ. 1999;6:508-15.
[107] Goossens V, Stange G, Moens K, Pipeleers D, Grooten J. Regulation of tumor necrosis
factor-induced, mitochondria- and reactive oxygen species-dependent cell death by the electron
flux through the electron transport chain complex I. Antioxid Redox Signal. 1999;1:285-95.
[108] Kawahara A, Ohsawa Y, Matsumura H, Uchiyama Y, Nagata S. Caspase-independent cell
killing by Fas-associated protein with death domain. J Cell Biol. 1998;143:1353-60.

______________________________________________________________________________
33
[109] Vercammen D, Beyaert R, Denecker G, Goossens V, Van Loo G, Declercq W, et al.
Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor
necrosis factor. J Exp Med. 1998;187:1477-85.
[110] Vercammen D, Brouckaert G, Denecker G, Van de Craen M, Declercq W, Fiers W, et al.
Dual signaling of the Fas receptor: initiation of both apoptotic and necrotic cell death pathways. J
Exp Med. 1998;188:919-30.
[111] Vercammen D, Vandenabeele P, Beyaert R, Declercq W, Fiers W. Tumour necrosis factor-
induced necrosis versus anti-Fas-induced apoptosis in L929 cells. Cytokine. 1997;9:801-8.
[112] Vanden Berghe T, van Loo G, Saelens X, Van Gurp M, Brouckaert G, Kalai M, et al.
Differential signaling to apoptotic and necrotic cell death by Fas-associated death domain protein
FADD. J Biol Chem. 2004;279:7925-33.
[113] Boone E, Vanden Berghe T, Van Loo G, De Wilde G, De Wael N, Vercammen D, et al.
Structure/Function analysis of p55 tumor necrosis factor receptor and fas-associated death
domain. Effect on necrosis in L929sA cells. J Biol Chem. 2000;275:37596-603.
[114] Schneider-Brachert W, Tchikov V, Neumeyer J, Jakob M, Winoto-Morbach S, Held-
Feindt J, et al. Compartmentalization of TNF receptor 1 signaling: internalized TNF
receptosomes as death signaling vesicles. Immunity. 2004;21:415-28.
[115] Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential
signaling complexes. Cell. 2003;114:181-90.
[116] Harper N, Hughes M, MacFarlane M, Cohen GM. Fas-associated death domain protein
and caspase-8 are not recruited to the tumor necrosis factor receptor 1 signaling complex during
tumor necrosis factor-induced apoptosis. J Biol Chem. 2003;278:25534-41.

______________________________________________________________________________
34
[117] Devin A, Cook A, Lin Y, Rodriguez Y, Kelliher M, Liu Z. The distinct roles of TRAF2
and RIP in IKK activation by TNF-R1: TRAF2 recruits IKK to TNF-R1 while RIP mediates
IKK activation. Immunity. 2000;12:419-29.
[118] Ting AT, Pimentel-Muinos FX, Seed B. RIP mediates tumor necrosis factor receptor 1
activation of NF-kappaB but not Fas/APO-1-initiated apoptosis. EMBO J. 1996;15:6189-96.
[119] Hsu H, Shu HB, Pan MG, Goeddel DV. TRADD-TRAF2 and TRADD-FADD interactions
define two distinct TNF receptor 1 signal transduction pathways. Cell. 1996;84:299-308.
[120] Thomas LR, Henson A, Reed JC, Salsbury FR, Thorburn A. Direct binding of Fas-
associated death domain (FADD) to the tumor necrosis factor-related apoptosis-inducing ligand
receptor DR5 is regulated by the death effector domain of FADD. J Biol Chem. 2004;279:32780-
5.
[121] Lin Y, Devin A, Rodriguez Y, Liu ZG. Cleavage of the death domain kinase RIP by
caspase-8 prompts TNF-induced apoptosis. Genes Dev. 1999;13:2514-26.
[122] Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, et al. Fas triggers an
alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule.
Nat Immunol. 2000;1:489-95.
[123] Devin A, Lin Y, Liu ZG. The role of the death-domain kinase RIP in tumour-necrosis-
factor-induced activation of mitogen-activated protein kinases. EMBO Rep. 2003;4:623-7.
[124] Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk
between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8:741-52.
[125] Shimizu S, Kanaseki T, Mizushima N, Mizuta T, Arakawa-Kobayashi S, Thompson CB, et
al. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on
autophagy genes. Nat Cell Biol. 2004;6:1221-8.

______________________________________________________________________________
35
[126] Madden DT, Egger L, Bredesen DE. A calpain-like protease inhibits autophagic cell death.
Autophagy. 2007;3:519-22.
[127] Xu Y, Kim SO, Li Y, Han J. Autophagy contributes to caspase-independent macrophage
cell death. J Biol Chem. 2006;281:19179-87.
[128] Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, et al. Growth factor regulation
of autophagy and cell survival in the absence of apoptosis. Cell. 2005;120:237-48.
[129] Lum JJ, DeBerardinis RJ, Thompson CB. Autophagy in metazoans: cell survival in the
land of plenty. Nat Rev Mol Cell Biol. 2005;6:439-48.
[130] Gonzalez-Polo RA, Boya P, Pauleau AL, Jalil A, Larochette N, Souquere S, et al. The
apoptosis/autophagy paradox: autophagic vacuolization before apoptotic death. J Cell Sci.
2005;118:3091-102.
[131] Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, et al.
Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature.
2006;441:885-9.
[132] Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, et al. Loss of autophagy in
the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880-4.
[133] Pua HH, Dzhagalov I, Chuck M, Mizushima N, He YW. A critical role for the autophagy
gene Atg5 in T cell survival and proliferation. J Exp Med. 2007;204:25-31.
[134] Takacs-Vellai K, Vellai T, Puoti A, Passannante M, Wicky C, Streit A, et al. Inactivation
of the autophagy gene bec-1 triggers apoptotic cell death in C. elegans. Curr Biol. 2005;15:1513-
7.

______________________________________________________________________________
36
[135] Espert L, Denizot M, Grimaldi M, Robert-Hebmann V, Gay B, Varbanov M, et al.
Autophagy is involved in T cell death after binding of HIV-1 envelope proteins to CXCR4. J
Clin Invest. 2006;116:2161-72.
[136] Yu L, Wan F, Dutta S, Welsh S, Liu Z, Freundt E, et al. Autophagic programmed cell
death by selective catalase degradation. Proc Natl Acad Sci U S A. 2006;103:4952-7.

______________________________________________________________________________
37
CHAPTER 2
_____________________________________________________________________________________
A KERATIN BIOMATERIAL PROMOTES SKIN REGENERATION
AFTER BURNS IN VIVO AND RESCUES CELLS AFTER THERMAL
STRESS IN VITRO
Poranki Da, Whitener W
a, Howse S
a, Mesen T
a, Howse B
a, Burnell J
a, Greengauz-Roberts O
a,
Molnar Jb, Van Dyke M
a
aWake Forest School of Medicine, Wake Forest Institute for Regenerative Medicine
bDepartment of Plastic and Reconstructive Surgery
Wake Forest School of Medicine, Medical Center Boulevard, Winston Salem, NC 27157
The following manuscript has not yet been submitted for publication. Deepika Poranki prepared
the manuscript. Dr. Mark Van Dyke acted in an advisory and editorial capacity.

______________________________________________________________________________
38
Abstract:
Thermal burns typically display an injury pattern dictated by the transfer of the thermal
energy into the skin and underlying tissues. There are often three zones of injury represented by
a necrotic zone of disrupted cells and tissue, an intermediate zone of injured and dying cells, and
a distant zone of stressed cells that will recover with proper treatment. Burn treatment is directed
toward removal of clearly necrotic skin and therapeutic support for remaining tissue that may
spontaneously heal without complication. Excised areas of the wound then require grafting.
Decisions regarding when and how much damaged tissue to excise are largely determined
empirically, and there is substantial debate about the relative merits of early or late excision and
grafting. Early grafting carries the risk of removing too little tissue, as cells in the intermediate
zone continue to die up to 10 days after injury. Late excision can expose the patient to many
inflammatory mediators produced by the dead and dying tissue. Strategies to facilitate the
survival of cells and tissue in the intermediate zone of burn injury may be capable of mediating
spontaneous healing, or reducing the overall extent of excision and grafting, but to date no such
therapies are available for clinical use.
The wound healing capabilities of a keratin biomaterial hydrogel were studied in two
pilot studies, one using a chemical burn model in mice and the other a thermal burn model in
swine. In both studies, an oxidized form of keratin biomaterial called “keratose” was shown to
prevent enlargement of the initial wound area and promote faster wound closure. This keratose
hydrogel was provided as a heterogeneous mixture of proteins representing an “alpha” fraction
and a “gamma” fraction. These different fractions of keratin have disparate properties including
molecular weights, amino acid content, and solubility characteristics. They can be easily

______________________________________________________________________________
39
separated by techniques such as dialysis or isoelectric precipitation. To determine if there was a
differential wound healing activity associated with these two keratose fractions, an in vitro heat
stress model mimicking the different zones of burn injury was developed. Treating thermally
stressed dermal fibroblast with growth media containing alpha, gamma or the native mixture
(termed “crude keratose” throughout this manuscript) showed that gamma keratose was able to
maintain cell viability and promote cell proliferation. By the third day of treatment, the cell
viability for gamma keratose-treated cells was 125% ± 15.6, whereas with other treatments the
cell viability was 65% ± 20.7 (crude), 46% ± 32.5 (alpha) and 37% ± 20 (fibroblast media).
Electrophoretic separation of the gamma keratose fraction revealed sub-fractions of
different molecular weights ranging from 110kDa to 8kDa. Mass spectrometry analysis
identified keratins 81, 83, 85, 86, 31, 33A, 33B, 34 in the excised bands. Epithelial keratins 1 and
10 were identified in the excised bands with molecular weights ~60 kDa and ~8 kDa,
respectively. Keratins 81, 83, 85, 86, 31, 33A, 33B and 34 were also identified with lower than
expected molecular weights, suggesting that these were degraded alpha keratose peptides.
Keratin-associated protein 19-5, galectin-7, calmodulin-like protein 3, thioredoxin, and cystatin
A were also identified in the ~8 kDa excised band.
These results suggest that the gamma fraction of keratose is comprised essentially of
degraded alpha keratose proteins, and that these peptides may facilitate the rescue of cells from
thermal injury. Treatment of burns with gamma keratose may therefore represent a potential
therapy for wounds with an intermediate zone of damaged tissue that has the potential to
contribute to spontaneous healing.

______________________________________________________________________________
40
Keywords: keratin, biomaterial, burn, wound, heat shock, hydrogel, keratose, alpha keratin,
gamma keratin, proteomics, mass spectrometry

______________________________________________________________________________
41
1. Introduction:
Burn wound repair is a complex process with a series of overlapping phases:
Inflammation; matrix deposition; and tissue remodeling. The duration of these individual phases
varies depending on the intensity and depth of the burn. Most burn dressing treatments aim to
facilitate these stages of wound healing by providing a moist environment, control excessive
exudate buildup, and protect against infection that would perturb normal healing. It has been
suggested that a burn dressing should serve three principle functions: Provide comfort by
protecting the wound surface from external air currents; absorb wound secretions and reduce
water loss from exposed burns; and protect from microorganisms [1]. In recent years, hydrogel
wound dressings have drawn increased attention as they serve all of these purposes. They act as a
protective barrier, absorb excess wound exudate, and maintain a moist environment, which in
turn helps in pain reduction. A moist wound environment is known to accelerate wound healing
[2-4].
There is abundant literature on various hydrogel treatments, among them chitosan,
alginate, collagen and hydro-colloid dressings that have been shown to promote burn wound
healing. Chitosan is a de-acetylated derivative of chitin that has been shown to promote burn
wound healing in vivo [5-13]. Chitosan did not appear to have a detrimental effect on primary rat
fibroblast viability in vitro [5]. In another study, full thickness burn wounds treated with high
molecular weight, highly de-acetylated chitosan showed faster re-epithelialization and wound
closure in rats compared to 2% Fucidin treatment. [14]. Chitosan hydrogel has also been used as
a carrier for biopharmaceuticals, antimicrobials and growth factors. Chitosan gel formulation
with epidermal growth factor (EGF) was shown to significantly increase cell proliferation and to

______________________________________________________________________________
42
promote faster re-epithelialization in second degree burns in rats [6, 13]. It was also used as a
fibroblast growth factor 2 (FGF2) carrier that induced angiogenesis and collateral blood
circulation in impaired diabetic mice [15]. Another study using pluronic/chitosan hydrogels with
recombinant human EGF enhanced epidermal cell differentiation in second degree burns in mice
with impaired wound healing [16]. Also, chitosan gels with controlled slow release granulocyte-
monocyte colony stimulating factor (GM-CSF) assisted in better collagen fibril organization in
Wistar-Albino rats [17]. Chitosan acetate bandages were shown to control the growth of
Pseudomonas aeruginosa and Pseudomonas mirabilis and to prevent the development of
systemic sepsis in mice with third degree burns [10]. Despite these positive findings, chitosan
has no apparent utility beyond providing a moist wound healing environment and a physical
barrier, and typically is combined with another therapeutic to enhance its efficacy.
Wound dressings containing alginate, a polysaccharide derived from sea weed, have also
been investigated. In 1987, Barnett and coworkers showed that a calcium alginate dressing was
an effective hemostat and facilitated wound repair [18]. A wide variety of alginate dressings are
available commercially that promote wound healing by providing a moist environment [19-24].
However, like chitosan, alginates have no intrinsic capacity for facilitating wound healing
beyond their physical attributes. Moreover, a disadvantage of alginate dressings containing
calcium alginate is that some cytotoxicity and immune reactions have been noted [25].
Development of alginate dressings containing less calcium has reduced the cytotoxicity and
foreign body reaction in experiments in vitro and in vivo [25, 26], but there are no market-
leading burn dressings based on alginate biomaterials.

______________________________________________________________________________
43
Dressings containing collagen have also been used in the treatment of burns. They
provide a moist wound environment and act as a barrier to microorganisms. Collagen dressings
have been shown to promote faster re-epithelialization, early granulation tissue formation [27-
29], and when used as a composite wound dressing along with other biomaterials or antibiotics,
were shown to promote faster wound healing with a reduced rate of infection [30-32].
Lyophilized bovine collagen has been shown to help in the management of pain and faster
epithelialization of skin graft donor sites [33]. However, these findings do not differ substantially
than those for other hydrogel biomaterials and have no specific efficacy for burn wound healing.
An additional limitation to the use of collagen dressings for burn wounds is that the environment
of a burn is highly proteolytic, and protein-based dressings such as collagen can breakdown
rapidly [34].
Hydrocolloid dressings are primarily used in partial thickness burns and small burns with
less wound exudate [35, 36]. In their intact state hydrocolloid dressings are impermeable to water
vapor and provide an effective barrier to transepidermal moisture loss, whereas in presence of
wound exudate the dressings absorb the liquid and form gel, making them permeable to vapor
[36]. Hydrocolloid dressings along with silver sulfadiazine have been shown to promote faster
wound closure and also help in pain management [37]. The principal benefit of using these
dressings for burns is pain management.
Recognizing the limited functionality of the above mentioned dressings, additional
enhancements have been made by commercial vendors of these products. Prominent among them
is the addition of antimicrobial properties by the addition of silver ions [37-43]. However, the
underlying limitation of all of these products is that they have no particular efficacy for burn

______________________________________________________________________________
44
wounds and the complex physiology of skin that has been damaged by the application of external
energy (thermal and electrical) or agents (chemical). The opportunity therefore exists to develop
new treatments that can specifically address the pathophysiology of burns.
Keratin biomaterial hydrogels may represent a burn-specific wound dressing technology.
In addition to possessing the physical attributes for a wound dressing (facilitates a moist
environment, protects the wound, absorbs excess wound fluid, etc.), keratins may facilitate the
survival of thermally and chemically damaged cells. Keratins represent a class of biomaterials
that have been used in a variety of biomedical applications, but the understanding of how keratin
proteins function in these applications is only at its nascent stages.
The major proteins contained in human hair fibers are classified as alpha keratins [44],
beta keratins and keratin-associated proteins (KAP) [45]. They can be extracted using oxidative
or reductive chemical techniques, and separated using various methods into purified fractions
with different molecular weights, amino acid content, and solubility characteristics [46]. Another
fraction derived from the oxidative extraction of these proteins from hair fibers is often referred
to in the literature as “gamma” keratin [47].
The general biocompatibility and utility of keratin biomaterials in potential medical and
biotechnology applications has been demonstrated in numerous studies showing proof of
principle for cell culture, wound healing, bone and nerve regeneration, drug delivery, hemostasis,
corneal tissue engineering, cardiac repair, and wound healing [48-61]. Recently, keratin
dressings were shown to enhance wound healing by the activation of keratinocytes in a porcine
partial thickness burn model [54]. However, no data was provided on the type of keratin used in
these dressings, or the underlying mechanism of wound repair. Given this body of evidence, we

______________________________________________________________________________
45
hypothesized that keratose hydrogels would facilitate both chemical and thermal burn repair, and
that there may be differential efficacy among the different keratose fractions present in a mixed
protein hydrogel. To test this postulate, we utilized a mouse chemical burn model and a swine
thermal burn model to conduct pilot studies using a hydrogel prepared from an un-purified
sample of crude keratose extract. We then investigated the differential effect of alpha and gamma
keratose fractions on cell survival in an in vitro model of thermal insult.
2. Materials and Methods:
2.1. Keratin Biomaterial Extraction: Keratin biomaterial was extracted from hair using an
oxidative protocol described by de Guzman et al. [62]. This heterogeneous mixture of
cortical proteins is referred to as crude keratose. For crude keratin hydrogel preparations,
the extract solution was dialyzed against endotoxin free water using a 5kDa spiral wound
cartridge (Millipore, Billerica, MA) for 5 volume washes. Finally, the solution was
concentrated to minimal volume, adjusted to pH 7.4 and lyophilized.
2.2. Separation of Alpha and Gamma Keratose fractions: Crude keratose was further
separated into alpha and gamma keratose fractions by isoelectric precipitation.
Concentrated hydrochloric acid was added to the crude keratose solution with stirring
until the pH dropped to 4.2, facilitating the precipitation of the insoluble alpha keratose.
The soluble gamma keratose was separated by fixed angle centrifugation at 1,500 rpm
(252 RCF) for 15min at 4ºC and filtered. After neutralizing the gamma solution to pH
8.5, it was dialyzed against endotoxin free water using a 5kDa spiral wound cartridge
(Millipore) for 5 volume washes. Finally, the solution was concentrated to minimal
volume, adjusted to pH 7.4 and lyophilized. The precipitated alpha keratose was

______________________________________________________________________________
46
reconstituted to a concentrated solution using a minimum of 0.1M sodium hydroxide and
then dialyzed against endotoxin free water using a 30kDa spiral wound cartridge
(Millipore) for 5 volume washes. Finally the solution was concentrated to minimal
volume, adjusted to pH 7.4 and lyophilized.
2.3. Hydrogel Preparation: Crude keratin hydrogel was prepared by reconstituting the
lyophilized powder at 15 percent by weight with normal saline. It was not sterilized
prior to use. Chitosan (Sigma-Aldrich) was similarly prepared by reconstituting the dry
powder at 9 weight percent with saline. Chitosan also was not sterilized prior to use.
Gels were loaded into syringes so that they could be accurately applied to the wounds.
2.4. Mouse Chemical Burn Model: Forty eight CD1 mice were used for this study under a
protocol approved by the Wake Forest University Institutional Animal Care and Use
Committee (WFU IACUC). A few minutes prior to the surgery and under general
anesthesia (2-3% halothane by nose cone inhalation), hair at the dorsal mid-line and
below the shoulders of the mice was removed with a commercial depilatory cream. The
area of exposed skin was washed with soap and water, then disinfected with iodine and
cleaned with saline soaked gauze. Chemical burns were created with 90% phenol in a 1
cm x 2 cm area of skin exposed for 60 sec. Phenol was removed with copious amounts
of phosphate buffered saline. Twenty minutes after creation of the phenol burns, the
treatments were applied, covered with a commercial adhesive bandage, and the animals
placed in a protective jacket to keep the dressings in place. Treatments included crude
keratose hydrogel, chitosan hydrogel, and saline only. Dressings were changed every 2
days for 20 days. Digital pictures were taken with a ruler in the field and wound size
measurements made using digital image analysis software (ImageJ, NIH, Bethesda,

______________________________________________________________________________
47
MD). Wound size values for each treatment group were averaged (e.g. n=12 on day 0,
n=11 on day 2, n=10 on day 4, etc.) and reported as mean ± standard error of the mean
(SEM).
2.5. Swine Thermal Burn Model: Four Yorkshire swine were used for this study under a
protocol approved by the WFU IACUC. Anesthesia was introduced intramuscularly with
ketamine (20mg/kg), xylazine (2.2mg/kg), acepromazine (1mg/kg) and then maintained
by nose cone with 2-3% halothane throughout the surgery and during dressing changes.
Several minutes prior to producing the burn, the animals were shaved and washed with
soap and water. Thermal burns were created using 3.5 cm diameter heated brass blocks
with a contact time of 12 sec [63]. Sixteen such wounds were created, eight on each side
of the dorsal mid-line between the shoulders and hip. Treatments were applied within 30
minutes after burn creation and included crude keratose hydrogel, chitosan hydrogel or
saline soaked gauze. Treatment areas were covered with dry gauze followed by an
occlusive dressing (adhesive Ioban), a cotton stocking, a plastic saddle over the back,
and a nylon jacket to keep all dressings in place. Dressing changes were performed on
anesthetized swine every 3 days. Digital pictures were taken with a ruler in view and
wound size measurements made using digital image analysis software (ImageJ). Wound
size values for each treatment group were averaged (e.g. n=16 on days 0, 3, 6, and 9;
n=12 on days 12 and 15; n=8 on day 18; n=4 on days 21, 24, and 27) and reported as
mean ± SEM.
2.6. In Vitro Thermal Injury Model:
2.6.1. Primary Mouse Skin Fibroblast Isolation: Primary mouse fibroblasts were
isolated from adult CD1 mouse ear pads. The ear pads were incubated in Dispase II

______________________________________________________________________________
48
overnight and then the dermis was peeled off using forceps and minced before
incubating in 0.35% type I collagenase reconstituted in DMEM high glucose for 30
min at 37˚C on a shaker. The suspension was filtered using a 100 micron mesh filter
and cells pelleted by centrifugation. The cell pellet was washed once with Hank’s
balanced salt solution (HBSS) to remove the residual collagenase. Finally, the
fibroblasts were seeded in a 150 mm tissue culture plastic (TCP) dish at 2.5 x 106
cells/mL in fibroblast media [DMEM high glucose plus 10% Fetal Bovine Serum
(Hyclone® Laboratories, Inc., Logan,Utah) and 1% Antibiotic/Antimycotic solution
(Hyclone® Laboratories, Inc.)].
2.6.2. In Vitro Thermal Stress Method: Primary mouse fibroblasts from passages 3 to
6 were used for these experiments. Fibroblasts were seeded in the range of 1.83 x
105
to 2.36 x 105cells/mL in a 100 mm TCP dish. The cells were grown in fibroblast
media until 95% confluent. Media changes were performed every 3 days. For a
thermal injury experiment, a media change was performed one day prior to heating
the cells. On the day of the experiment, fibroblasts were moved from a 37˚C, 5%
CO2 incubator to a 44˚C, 5% CO2 incubator for 60 minutes. After the thermal stress,
culture dishes were moved back to a 37˚C, 5% CO2 incubator to allow for
reattachment of the cells and for the media temperature to equilibrate. Six hours
after heat exposure, various treatments were applied in fresh media. Treatments
included 0.01mg/mL crude keratose, 0.001mg/ml gamma keratose, 0.001mg/ml
alpha keratose and fibroblast media. All keratose samples were sterilized by gamma
irradiation at 1 MRad before adding to the media. This experiment was performed

______________________________________________________________________________
49
with three replicates. All the data were normalized to 0 hours (before heat shock)
and reported as mean ± standard error of the mean (SEM).
2.6.3. Trypan Blue Assay: Equal amounts of cell suspension and 0.4% trypan blue
(Invitrogen, Gibco, Grand Island, NY) were mixed together and stained cells were
counted using a hemocytometer. Live cells with intact cell membrane excluded the
trypan blue dye whereas dead cells retained the dye and were blue.
2.7. Detection and Identification of Gamma Keratose proteins: Proteins associated with
the gamma keratose fraction were separated by sodium dodecyl sulfate polyacrylamide
gel electrophoresis (SDS-PAGE) and stained protein bands were recovered and analyzed
by mass spectrometry as previously described [62].
2.8. Statistical Analysis: Two-way analysis of variance (ANOVA) and Bonferroni post-hoc
analyses were performed using Graph pad Prism at 95% confidence intervals for both
the in vivo and in vitro studies.
3. Results:
3.1. Mouse Chemical Burn Study: Compared to the other treatments, keratose hydrogel
treated burns showed no increase in wound size over the first four days post-injury and
faster wound closure. There was a statistically significant reduction in wound area with
keratose treatment relative to either chitosan or saline at days 4 through 16 (Figure 1).
Digital pictures of representative burn wounds at day 0, 8 and 16 showed faster wound
healing progression and closure (Figure 2). A well-defined eschar (sloughed skin) was
observed early in the keratose treated wound at day 8 compared to day 16 for the
chitosan and saline soaked gauze treatments (occlusive dressing).

______________________________________________________________________________
50
3.2. Swine Thermal Burn Pilot Study: Compared to the other treatments, keratose hydrogel
treatment again showed no increase in burn wound size and resulted in faster wound
closure (Figure 3). There was a statistically significant reduction in wound area at days
3, 6, and 12 in the keratose treated group compared to both chitosan and saline. Digital
pictures of representative burn wounds at day 3, 9 and 24 shows wound healing
progression and faster wound closure (Figure 4).
3.3. In Vitro Thermal Stress Model: Control cultures were examined for their ability to
mimic the phases of heat induced cell damage. Immediately after heat stress, 40-50%
cell death was noted by cell counting using trypan blue staining. Further cell death was
evident in the following 24 hours if proper treatment was not provided. After 7 days,
cultures grew back to a confluent state (Figure 5). Using this in vitro heat injury model,
different keratose treatments were compared to each other and to control treatment (i.e.
fibroblast growth media containing serum). Fibroblasts that received a single treatment
of gamma keratose at 0.001mg/mL dissolved in the fibroblast media and were able to
maintain their viability, whereas all other treatments resulted in additional cell death
between the time of treatment (6 hours) and 24 hours. Even by day 3, the other
treatments were only able to maintain their post-treatment cell numbers [65% ± 20.7
(crude), 46% ± 32.5 (alpha) and 37% ± 20 (fibroblast media)], while the gamma keratose
treated fibroblasts had 125% ± 15.6 cell viability (Figure 6). The gamma keratose
treatment resulted in statistically significant cell growth compare to all other treatments
at day 3, and compared to the alpha keratose treatment at day 5.
3.4. Identification and separation of gamma keratose proteins: Electrophoretic separation
of gamma keratose revealed proteins of different molecular weights ranging from

______________________________________________________________________________
51
110kDa to 8kDa. Protein bands at 110 kDa, 40-60 kDa, ~30 kDa, ~15-20 kDa, and ~8-
10 kDa that were excised for mass spectrometry analysis were identified as containing
keratins 81, 83, 85, 86, 31, 33A, 33B, 34 in all the excised regions. Epithelial keratins 1
and 10 were identified in the excised regions with molecular weight 60kDa and 8kDa.
The excised region at ~110 kDa band was identified as alpha keratin heterodimers,
which were previously reported by de Guzman et al [62]. Keratin-associated protein 19-
5, galectin-7, calmodulin-like protein 3, thioredoxin, and cystatin A were identified in
the band at ~8 kDa (Figure 7). Mass spectrometry analysis of the excised bands with
protein identities, molecular weights and protein scores is provided as supplementary
data (Tables 1-6).
4. Discussion:
Hydrogels are gaining more interest as wound dressings, especially for the treatment of
burns, ulcers, and other necrotic wounds [64]. They have inherent properties such as the ability
to absorb wound exudate and provide a moist wound environment that enhances healing [3, 64].
Keratin proteins provide a readily available, inexpensive biomaterial hydrogel that has been
shown to be beneficial for regenerating tissues [51, 54, 56-58, 65-67]. Recently, a keratin
biomaterial extracted from wool was shown to promote faster re-epithelialization in deep partial
thickness burns [54]. Moreover, keratose, the oxidized form of keratin protein extracted from
hair and wool fibers, was previously purported to be beneficial for wound healing [68].
However, neither of these accounts attempts to describe a plausible mechanism for improved
wound healing, nor do they describe the keratin in sufficient detail to provide much insight into
its structure or composition. The ability of a hydrogel made from a structural protein such as

______________________________________________________________________________
52
keratin to provide beneficial wound healing is not surprising, given the general nature of
hydrogels and the proven efficacy of providing a protected wound environment where moisture
can be controlled. However, in this study, we sought to demonstrate the particular efficacy of a
keratose biomaterial hydrogel for the treatment of chemical and thermal burns, then investigate a
potential mechanism of cell rescue that was tied to a specific fraction of keratin biomaterial that
has until now, been uncharacterized in the literature. In both pilot animal studies, a crude form of
keratose was used that contained both gamma and alpha fractions. While this mixture of proteins
was efficacious in both models, we were not able to determine if either of these fractions
individually or synergistically produced differential effects on the damaged tissues. To better
understand the relative contributions of these different forms of keratin biomaterial, a heat stress
model that mimics thermal injury in vivo was developed.
The injury to tissue that results from thermal insult was first described by Jackson [69].
According to Jackson’s burn model, burn injury is characterized by three zones: 1) the inner zone
of coagulation, which is characterized by necrotic tissue, 2) an intermediate zone of stasis, which
is characterized by damaged cells that may live or die depending on treatment and other factors,
and 3) an outer zone of hyperemia where cells are stressed but will likely survive if the wound
does not get infected and standard treatment is given [69]. Similar to Jackson’s model, the heat
stress culture system was optimized to produce a population of cells that died from necrosis
during the heat treatment phase, a second population of cells that died from apoptosis in the
hours following thermal injury, and a third population that recovered and could re-establish the
culture. Using this model, we were able to show that gamma keratose was better able to maintain

______________________________________________________________________________
53
cell viability compared to crude keratose, alpha keratose, and fibroblast growth media. By day 5,
gamma keratose treated fibroblast growth was so robust that the culture was over-confluent.
Heat stress models have been used previously to isolate cells into a more simplistic
system and study molecular mechanisms of damage and survival [70]. In this study we not only
showed that a biomaterial made from oxidized keratin protein can improve fibroblast survival,
but that this phenomenon is associated only with the gamma keratose fraction. As this is the first
study to show that this keratose fraction has particular efficacy for cell salvage after heat stress,
we performed mass spectrometry analysis to understand the composition of proteins present in
the gamma keratose fraction. Mass spectrometry analysis of the excised bands showed that they
contain keratins 81, 83, 85, 86, 31, 33A, 33B, and 34. Because molecular weights between 26
and 46 kDa do not correspond to any known, intact human hair alpha keratin or KAP, and
because there are no alpha keratin proteins with molecular weights below 46kDa, we conclude
that all proteins identified by mass spectroscopy in the 30kDa, 15-20kDa, and 8kDa
electrophoretic bands must represent fragments of degraded alpha keratose proteins. In much of
the early published literature on keratins, the gamma fraction has been described as being
derived from the cortical matrix [71], and this concept has become dogma in the current keratin
biomaterials field. However, given the oxidation and extraction conditions employed in our
protein isolation method, it is not surprising that the generation of peptide fragments occurs,
especially when considering recent evidence that shows a number of weak bonds in alpha keratin
proteins [72].
The concept that fragments of larger proteins have a biological function different than
that of the parent molecule is not new. Extra cellular matrix (ECM) derived from various tissues

______________________________________________________________________________
54
contain growth factors that promote tissue remodeling and angiogenesis [73]. Badylak et al.
showed that bone marrow derived stem cells actively participated in the remodeling of ECM
scaffolds in vivo. They also suggested that portion of these bone marrow derived stem cells
might have participated in angiogenic response [74]. In another study, it was shown that a low
molecular weight peptide fraction of ECM (5-16kDa) has chemoattractant properties for mature
endothelial cells both in in vitro and in vivo assays. This low molecular weight peptide fraction
was derived from small intestine sub mucosa ECM. The ECM containing structural proteins
(collagens, proteoglycans, glycoproteins, glycosylaminoglycans) and growth factors was reduced
to particulate form and further separated into seven fractions based on molecular weight. These
biologically active peptides were not purified or characterized completely but have been shown
to attract endothelial cells that might have promoted angiogenesis when ECM scaffolds were
implanted in vivo [75].
It has been shown that proteolytic degradation exposes cryptic sites on resulting peptide
fragments, imparting biological function that was not possessed by the intact protein [76].
Understanding more about the composition and structural nature of gamma keratose and the
active peptide region(s) that may be responsible for promoting survival in thermally stressed
cells may assist in the development of better keratin-based treatments for burn injuries.
5. Conclusions:
Pilot burn studies in mice and pigs have identified the potential for a keratose biomaterial
to spare burned tissue and facilitate skin regeneration. A closer examination of this observation
has revealed that gamma keratose, identified through proteomic techniques as peptide fragments
cleaved from the structural alpha keratins during extraction, were primarily responsible for this

______________________________________________________________________________
55
phenomenon. The gamma keratose fraction was better able to rescue cells after thermal insult in
an in vitro model system and therefore may represent a potential therapy for burn injury.
Conflict of interest statement
Mark Van Dyke, Ph.D. holds stock and is an officer in the company, KeraNetics LLC, which has
provided partial funding for this research. Wake Forest Health Sciences has a potential financial
interest in KeraNetics, LLC through licensing agreements.

______________________________________________________________________________
56
References:
[1] Herndon DN. Total Burn Care. Third ed: Saunders Elsevier; 2007.
[2] Alvarez O. Moist environment for healing: matching the dressing to the wound. Ostomy
Wound Manage. 1988;21:64-83.
[3] Bryan J. Moist wound healing: a concept that changed our practice. J Wound Care.
2004;13:227-8.
[4] Chang H, Wind S, Kerstein MD. Moist wound healing. Dermatol Nurs. 1996;8:174-6, 204.
[5] Ribeiro MP, Espiga A, Silva D, Baptista P, Henriques J, Ferreira C, et al. Development of a
new chitosan hydrogel for wound dressing. Wound Repair Regen. 2009;17:817-24.
[6] Alemdaroglu C, Degim Z, Celebi N, Zor F, Ozturk S, Erdogan D. An investigation on burn
wound healing in rats with chitosan gel formulation containing epidermal growth factor. Burns.
2006;32:319-27.
[7] Boucard N, Viton C, Agay D, Mari E, Roger T, Chancerelle Y, et al. The use of physical
hydrogels of chitosan for skin regeneration following third-degree burns. Biomaterials.
2007;28:3478-88.
[8] Jin Y, Ling PX, He YL, Zhang TM. Effects of chitosan and heparin on early extension of
burns. Burns. 2007;33:1027-31.
[9] Sezer AD, Cevher E, Hatipoglu F, Ogurtan Z, Bas AL, Akbuga J. Preparation of fucoidan-
chitosan hydrogel and its application as burn healing accelerator on rabbits. Biol Pharm Bull.
2008;31:2326-33.

______________________________________________________________________________
57
[10] Dai T, Tegos GP, Burkatovskaya M, Castano AP, Hamblin MR. Chitosan acetate bandage
as a topical antimicrobial dressing for infected burns. Antimicrob Agents Chemother.
2009;53:393-400.
[11] Nascimento EG, Sampaio TB, Medeiros AC, Azevedo EP. Evaluation of chitosan gel with
1% silver sulfadiazine as an alternative for burn wound treatment in rats. Acta Cir Bras.
2009;24:460-5.
[12] Dantas MD, Cavalcante DR, Araujo FE, Barretto SR, Aciole GT, Pinheiro AL, et al.
Improvement of dermal burn healing by combining sodium alginate/chitosan-based films and
low level laser therapy. J Photochem Photobiol B. 2011;105:51-9.
[13] Degim Z, Celebi N, Alemdaroglu C, Deveci M, Ozturk S, Ozogul C. Evaluation of chitosan
gel containing liposome-loaded epidermal growth factor on burn wound healing. Int Wound J.
2011;8:343-54.
[14] Alsarra IA. Chitosan topical gel formulation in the management of burn wounds. Int J Biol
Macromol. 2009;45:16-21.
[15] Ishihara M, Obara K, Nakamura S, Fujita M, Masuoka K, Kanatani Y, et al. Chitosan
hydrogel as a drug delivery carrier to control angiogenesis. J Artif Organs. 2006;9:8-16.
[16] Choi JS, Yoo HS. Pluronic/chitosan hydrogels containing epidermal growth factor with
wound-adhesive and photo-crosslinkable properties. J Biomed Mater Res A. 2010;95:564-73.
[17] Simsek S, Canter HI, Konas E, Korkusuz P, Demir D, Oner F, et al. A new concept in
treatment of burn injury: controlled slow-release granulocyte-monocyte colony-stimulating
factor chitosan gel system. Ann Plast Surg. 2011;67:583-8.
[18] Barnett SE, Varley SJ. The effects of calcium alginate on wound healing. Ann R Coll Surg
Engl. 1987;69:153-5.

______________________________________________________________________________
58
[19] Piacquadio D, Nelson DB. Alginates. A "new" dressing alternative. J Dermatol Surg Oncol.
1992;18:992-5.
[20] Agren MS. Four alginate dressings in the treatment of partial thickness wounds: a
comparative experimental study. Br J Plast Surg. 1996;49:129-34.
[21] Williams C. Melgisorb: a highly absorbent calcium/sodium alginate dressing. Br J Nurs.
1998;7:975-6.
[22] Williams C. Tegagen alginate dressing for moderate to heavily exuding wounds. Br J Nurs.
1998;7:550-2.
[23] Williams C. Algosteril calcium alginate dressing for moderate/high exudate. Br J Nurs.
1999;8:313-7.
[24] Kammerlander G, Eberlein T. An assessment of the wound healing properties of Algisite M
dressings. Nurs Times. 2003;99:54-6.
[25] Suzuki Y, Nishimura Y, Tanihara M, Suzuki K, Nakamura T, Shimizu Y, et al. Evaluation
of a novel alginate gel dressing: cytotoxicity to fibroblasts in vitro and foreign-body reaction in
pig skin in vivo. J Biomed Mater Res. 1998;39:317-22.
[26] Suzuki Y, Tanihara M, Nishimura Y, Suzuki K, Yamawaki Y, Kudo H, et al. In vivo
evaluation of a novel alginate dressing. J Biomed Mater Res. 1999;48:522-7.
[27] Horch RE, Stark GB. Comparison of the effect of a collagen dressing and a polyurethane
dressing on the healing of split thickness skin graft (STSG) donor sites. Scand J Plast Reconstr
Surg Hand Surg. 1998;32:407-13.
[28] Landsman A, Taft D, Riemer K. The role of collagen bioscaffolds, foamed collagen, and
living skin equivalents in wound healing. Clin Podiatr Med Surg. 2009;26:525-33.

______________________________________________________________________________
59
[29] Singh O, Gupta SS, Soni M, Moses S, Shukla S, Mathur RK. Collagen dressing versus
conventional dressings in burn and chronic wounds: a retrospective study. J Cutan Aesthet Surg.
2011;4:12-6.
[30] Adhirajan N, Shanmugasundaram N, Shanmuganathan S, Babu M. Collagen-based wound
dressing for doxycycline delivery: in-vivo evaluation in an infected excisional wound model in
rats. J Pharm Pharmacol. 2009;61:1617-23.
[31] Kondo S, Niiyama H, Yu A, Kuroyanagi Y. Evaluation of a Wound Dressing Composed of
Hyaluronic Acid and Collagen Sponge Containing Epidermal Growth Factor in Diabetic Mice. J
Biomater Sci Polym Ed. 2011.
[32] Nunes PS, Albuquerque RL, Jr., Cavalcante DR, Dantas MD, Cardoso JC, Bezerra MS, et
al. Collagen-based films containing liposome-loaded usnic acid as dressing for dermal burn
healing. J Biomed Biotechnol. 2011;2011:761593.
[33] Uygur F, Evinc R, Ulkur E, Celikoz B. Use of lyophilized bovine collagen for split-
thickness skin graft donor site management. Burns. 2008;34:1011-4.
[34] Neely AN, Brown RL, Clendening CE, Orloff MM, Gardner J, Greenhalgh DG. Proteolytic
activity in human burn wounds. Wound Repair Regen. 1997;5:302-9.
[35] Vloemans AF, Soesman AM, Suijker M, Kreis RW, Middelkoop E. A randomised clinical
trial comparing a hydrocolloid-derived dressing and glycerol preserved allograft skin in the
management of partial thickness burns. Burns. 2003;29:702-10.
[36] Thomas S. Hydrocolloid dressings in the management of acute wounds: a review of the
literature. Int Wound J. 2008;5:602-13.

______________________________________________________________________________
60
[37] Muangman P, Muangman S, Opasanon S, Keorochana K, Chuntrasakul C. Benefit of
hydrocolloid SSD dressing in the outpatient management of partial thickness burns. J Med Assoc
Thai. 2009;92:1300-5.
[38] Qin Y. Silver-containing alginate fibres and dressings. Int Wound J. 2005;2:172-6.
[39] Opasanon S, Muangman P, Namviriyachote N. Clinical effectiveness of alginate silver
dressing in outpatient management of partial-thickness burns. Int Wound J. 2010;7:467-71.
[40] Lu S, Gao W, Gu HY. Construction, application and biosafety of silver nanocrystalline
chitosan wound dressing. Burns. 2008;34:623-8.
[41] Ong SY, Wu J, Moochhala SM, Tan MH, Lu J. Development of a chitosan-based wound
dressing with improved hemostatic and antimicrobial properties. Biomaterials. 2008;29:4323-32.
[42] Huang L, Dai T, Xuan Y, Tegos GP, Hamblin MR. Synergistic combination of chitosan
acetate with nanoparticle silver as a topical antimicrobial: efficacy against bacterial burn
infections. Antimicrob Agents Chemother. 2011;55:3432-8.
[43] Li D, Diao J, Zhang J, Liu J. Fabrication of new chitosan-based composite sponge
containing silver nanoparticles and its antibacterial properties for wound dressing. J Nanosci
Nanotechnol. 2011;11:4733-8.
[44] Schweizer J, Bowden PE, Coulombe PA, Langbein L, Lane EB, Magin TM, et al. New
consensus nomenclature for mammalian keratins. J Cell Biol. 2006;174:169-74.
[45] Rogers MA, Langbein L, Praetzel-Wunder S, Winter H, Schweizer J. Human hair keratin-
associated proteins (KAPs). Int Rev Cytol. 2006;251:209-63.
[46] Singer AJ, Taira BR, Lin F, Lim T, Anderson R, McClain SA, et al. Curcumin reduces
injury progression in a rat comb burn model. J Burn Care Res. 2011;32:135-42.

______________________________________________________________________________
61
[47] Buchanan JH. A cystine-rich protein fraction from oxidized alpha-keratin. Biochem J.
1977;167:489-91.
[48] Yamauchi K, Maniwa M, Mori T. Cultivation of fibroblast cells on keratin-coated substrata.
J Biomater Sci Polym Ed. 1998;9:259-70.
[49] Tachibana A, Kaneko S, Tanabe T, Yamauchi K. Rapid fabrication of keratin-
hydroxyapatite hybrid sponges toward osteoblast cultivation and differentiation. Biomaterials.
2005;26:297-302.
[50] Sierpinski P, Garrett J, Ma J, Apel P, Klorig D, Smith T, et al. The use of keratin
biomaterials derived from human hair for the promotion of rapid regeneration of peripheral
nerves. Biomaterials. 2008;29:118-28.
[51] Shen D, Wang X, Zhang L, Zhao X, Li J, Cheng K, et al. The amelioration of cardiac
dysfunction after myocardial infarction by the injection of keratin biomaterials derived from
human hair. Biomaterials. 2011;32:9290-9.
[52] Saul JM, Ellenburg MD, de Guzman RC, Van Dyke M. Keratin hydrogels support the
sustained release of bioactive ciprofloxacin. J Biomed Mater Res A. 2011;98:544-53.
[53] Reichl S. Films based on human hair keratin as substrates for cell culture and tissue
engineering. Biomaterials. 2009;30:6854-66.
[54] Pechter PM, Gil J, Valdes J, Tomic-Canic M, Pastar I, Stojadinovic O, et al. Keratin
dressings speed epithelialization of deep partial-thickness wounds. Wound Repair Regen. 2012
Mar-Apr;20(2):236-42.
[55] Lee KY, Kong SJ, Park WH, Ha WS, Kwon IC. Effect of surface properties on the
antithrombogenicity of silk fibroin/S-carboxymethyl kerateine blend films. J Biomater Sci Polym
Ed. 1998;9:905-14.

______________________________________________________________________________
62
[56] Dias GJ, Peplow PV, McLaughlin A, Teixeira F, Kelly RJ. Biocompatibility and
osseointegration of reconstituted keratin in an ovine model. J Biomed Mater Res A.
2010;92:513-20.
[57] Dias GJ, Mahoney P, Swain M, Kelly RJ, Smith RA, Ali MA. Keratin-hydroxyapatite
composites: biocompatibility, osseointegration, and physical properties in an ovine model. J
Biomed Mater Res A. 2010;95:1084-95.
[58] Belcarz A, Ginalska G, Zalewska J, Rzeski W, Slosarczyk A, Kowalczuk D, et al. Covalent
coating of hydroxyapatite by keratin stabilizes gentamicin release. J Biomed Mater Res B Appl
Biomater. 2009;89:102-13.
[59] Thilagar S, Jothi NA, Omar AR, Kamaruddin MY, Ganabadi S. Effect of keratin-gelatin and
bFGF-gelatin composite film as a sandwich layer for full-thickness skin mesh graft in
experimental dogs. J Biomed Mater Res B Appl Biomater. 2009;88:12-6.
[60] Reichl S, Borrelli M, Geerling G. Keratin films for ocular surface reconstruction.
Biomaterials. 2011;32:3375-86.
[61] Hill PS, Apel PJ, Barnwell J, Smith T, Koman LA, Atala A, et al. Repair of peripheral nerve
defects in rabbits using keratin hydrogel scaffolds. Tissue Eng Part A. 2011;17:1499-505.
[62] de Guzman RC, Merrill MR, Richter JR, Hamzi RI, Greengauz-Roberts OK, Van Dyke ME.
Mechanical and biological properties of keratose biomaterials. Biomaterials. 2011;32:8205-17.
[63] Papp A, Kiraly K, Harma M, Lahtinen T, Uusaro A, Alhava E. The progression of burn
depth in experimental burns: a histological and methodological study. Burns. 2004;30:684-90.
[64] Edwards J. Hydrogels and their potential uses in burn wound management. Br J Nurs.
2010;19:S12, S4-6.

______________________________________________________________________________
63
[65] Tachibana A, Nishikawa Y, Nishino M, Kaneko S, Tanabe T, Yamauchi K. Modified
keratin sponge: binding of bone morphogenetic protein-2 and osteoblast differentiation. J Biosci
Bioeng. 2006;102:425-9.
[66] Nunez F, Trach S, Burnett L, Handa R, Van Dyke M, Callahan M, et al. Vasoactive
properties of keratin-derived compounds. Microcirculation. 2011;18:663-9.
[67] Richter JR, de Guzman RC, Van Dyke ME. Mechanisms of hepatocyte attachment to
keratin biomaterials. Biomaterials. 2011;32:7555-61.
[68] Widra A. Hydrophilic biopolymeric copolyelectrolytes, and biodegradable wound dressing
comprising same. . In: Office Up, editor. US: University of Illinois Foundation; 1986.
[69] Jackson DM. [The diagnosis of the depth of burning]. Br J Surg. 1953;40:588-96.
[70] Park CH, Lee MJ, Ahn J, Kim S, Kim HH, Kim KH, et al. Heat shock-induced matrix
metalloproteinase (MMP)-1 and MMP-3 are mediated through ERK and JNK activation and via
an autocrine interleukin-6 loop. J Invest Dermatol. 2004;123:1012-9.
[71] Crewther WG, Fraser RD, Lennox FG, Lindley H. The chemistry of keratins. Adv Protein
Chem. 1965;20:191-346.
[72] Barthelemy NR, Bednarczyk A, Schaeffer-Reiss C, Jullien D, Van Dorsselaer A, Cavusoglu
N. Proteomic tools for the investigation of human hair structural proteins and evidence of
weakness sites on hair keratin coil segments. Anal Biochem. 2012;421:43-55.
[73] Vorotnikova E, McIntosh D, Dewilde A, Zhang J, Reing JE, Zhang L, et al. Extracellular
matrix-derived products modulate endothelial and progenitor cell migration and proliferation in
vitro and stimulate regenerative healing in vivo. Matrix Biol. 2010;29:690-700.
[74] Badylak SF, Park K, Peppas N, McCabe G, Yoder M. Marrow-derived cells populate
scaffolds composed of xenogeneic extracellular matrix. Exp Hematol. 2001;29:1310-8.

______________________________________________________________________________
64
[75] Li F, Li W, Johnson S, Ingram D, Yoder M, Badylak S. Low-molecular-weight peptides
derived from extracellular matrix as chemoattractants for primary endothelial cells. Endothelium.
2004;11:199-206.
[76] Xu J, Rodriguez D, Petitclerc E, Kim JJ, Hangai M, Moon YS, et al. Proteolytic exposure of
a cryptic site within collagen type IV is required for angiogenesis and tumor growth in vivo. J
Cell Biol. 2001;154:1069-79.

______________________________________________________________________________
65
Figures:
Figure 1. Mouse Chemical Burn Wound Area
Digital Image analysis shows that crude keratose hydrogel treatment of chemical burns results in
a significant difference in wound area compared to saline and chitosan hydrogel control
treatments at days 4 through 16 (indicated by asterisks).
0 2 4 6 8 10 12 14 16 18 200.0
0.2
0.4
0.6
0.8
1.0
1.2
Saline
Chitosan
Keratose* ****
Day
Norm
alized W
ound S
ize

______________________________________________________________________________
66
Figure 2. Digital Images from Mice Chemical burn study
Digital pictures of representative burn wounds at day 0, 8 and 16 show enhanced wound healing
and closure with keratose treatment compared to chitosan or saline (occlusive dressing).

______________________________________________________________________________
67
Figure 3. Swine Thermal Burn Wound Area
Digital Image analysis shows that crude keratose hydrogel treatment of thermal burns results in a
significant difference in wound area compared to saline and chitosan hydrogel control at days 3,
6 and 12.
0 2 4 6 8 10 12 14 160.0
0.2
0.4
0.6
0.8
1.0
1.2
Saline
Chitosan
Keratose* *
*
Day
Norm
alized W
ound S
ize
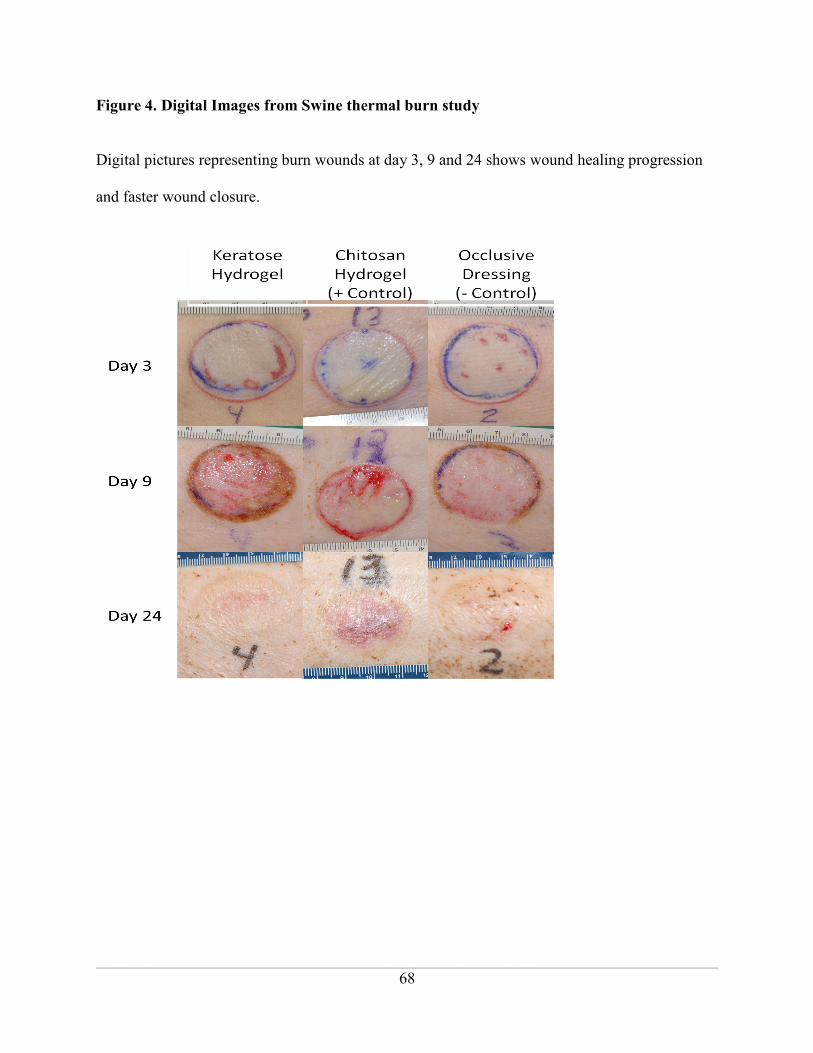
______________________________________________________________________________
68
Figure 4. Digital Images from Swine thermal burn study
Digital pictures representing burn wounds at day 3, 9 and 24 shows wound healing progression
and faster wound closure.

______________________________________________________________________________
69
Figure 5. In Vitro Thermal Stress Model
An in-vitro heat shock model was developed that produced three populations of cells: necrotic,
apoptotic, and survivable. This graph represents the growth curve of the survivable cells treated
with fibroblast growth media after heat treatment at 44oC for 60 minutes.
0
20
40
60
80
100
120
140
160
0hr 8hr 1d 3d 5d 7d
% C
ell
Via
bil
ity
Time Point
% Cell Viability
Average
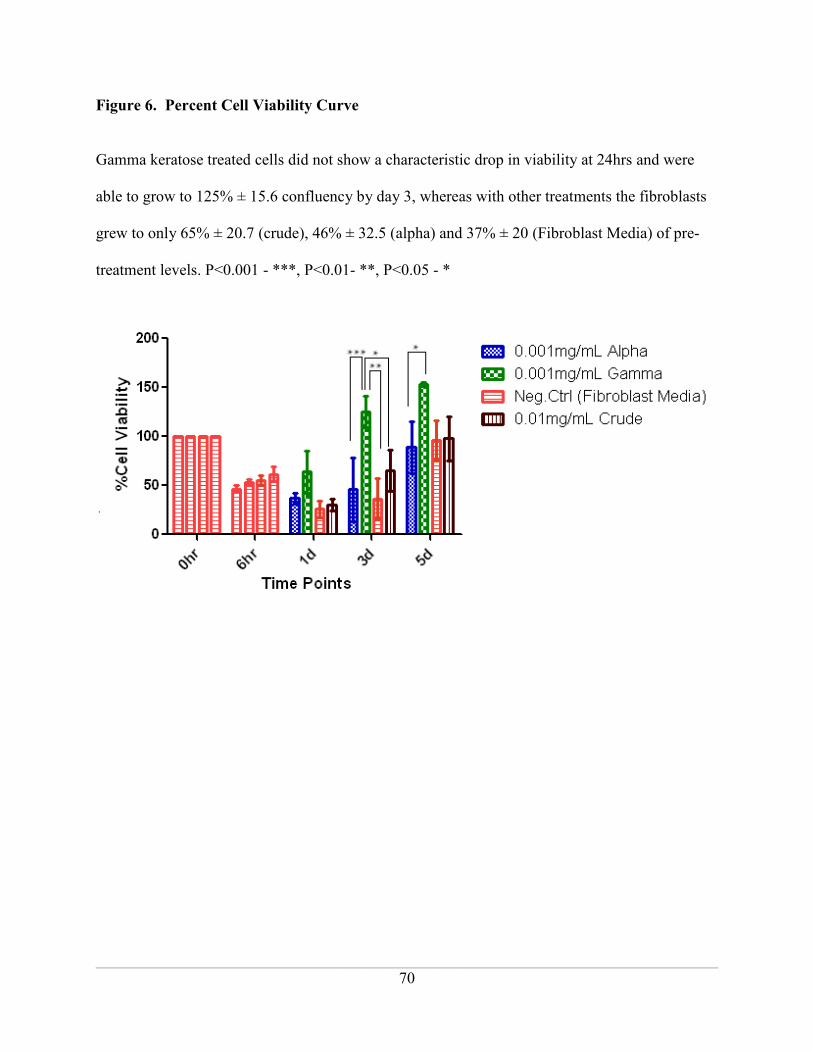
______________________________________________________________________________
70
Figure 6. Percent Cell Viability Curve
Gamma keratose treated cells did not show a characteristic drop in viability at 24hrs and were
able to grow to 125% ± 15.6 confluency by day 3, whereas with other treatments the fibroblasts
grew to only 65% ± 20.7 (crude), 46% ± 32.5 (alpha) and 37% ± 20 (Fibroblast Media) of pre-
treatment levels. P<0.001 - ***, P<0.01- **, P<0.05 - *

______________________________________________________________________________
71
Figure 7. Keratose SDS PAGE
Mass spectrometry analysis of gamma keratose identified keratin 81, 83, 85, 86, 31, 33A, 33B,
34 in all the excised regions. Epithelial keratins – 1 and 10 were identified in the excised regions
with molecular weight 60 kDa and 8 kDa. Keratin-associated protein 19-5, galectin-7,
calmodulin-like protein 3, thioredoxin, and cystatin A were also identified in the ~8 kDa excised
band.
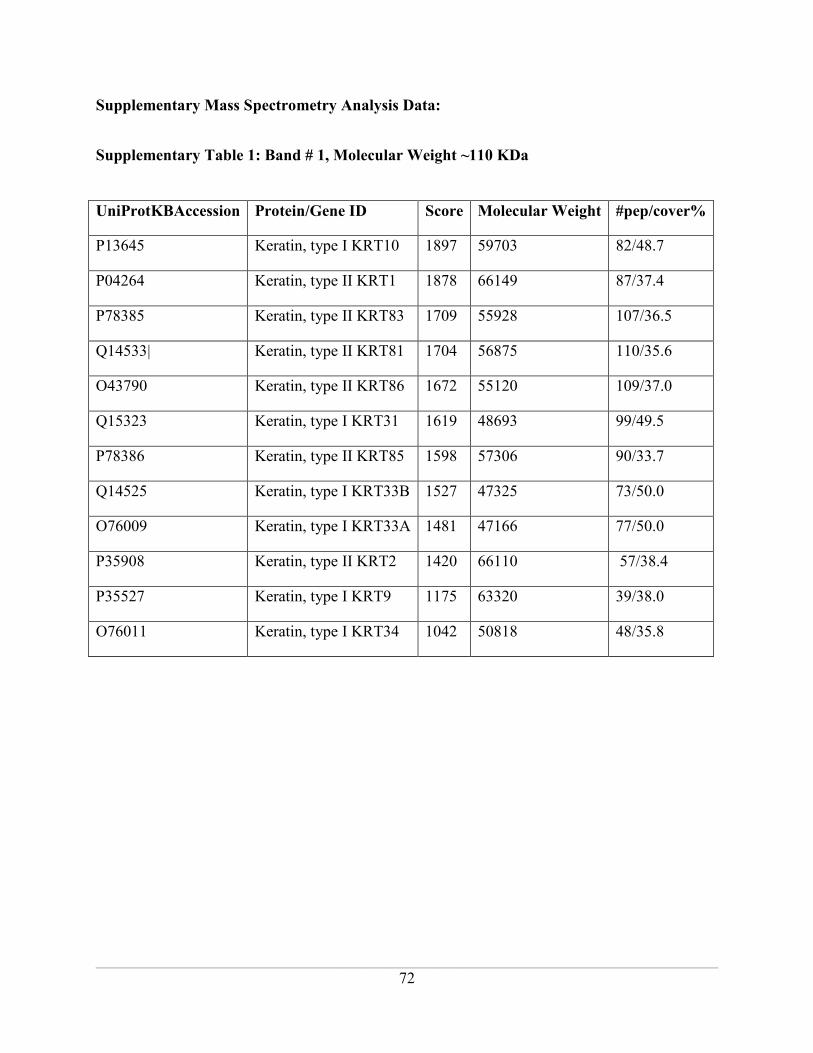
______________________________________________________________________________
72
Supplementary Mass Spectrometry Analysis Data:
Supplementary Table 1: Band # 1, Molecular Weight ~110 KDa
UniProtKBAccession Protein/Gene ID Score Molecular Weight #pep/cover%
P13645 Keratin, type I KRT10 1897 59703 82/48.7
P04264 Keratin, type II KRT1 1878 66149 87/37.4
P78385 Keratin, type II KRT83 1709 55928 107/36.5
Q14533| Keratin, type II KRT81 1704 56875 110/35.6
O43790 Keratin, type II KRT86 1672 55120 109/37.0
Q15323 Keratin, type I KRT31 1619 48693 99/49.5
P78386 Keratin, type II KRT85 1598 57306 90/33.7
Q14525 Keratin, type I KRT33B 1527 47325 73/50.0
O76009 Keratin, type I KRT33A 1481 47166 77/50.0
P35908 Keratin, type II KRT2 1420 66110 57/38.4
P35527 Keratin, type I KRT9 1175 63320 39/38.0
O76011 Keratin, type I KRT34 1042 50818 48/35.8
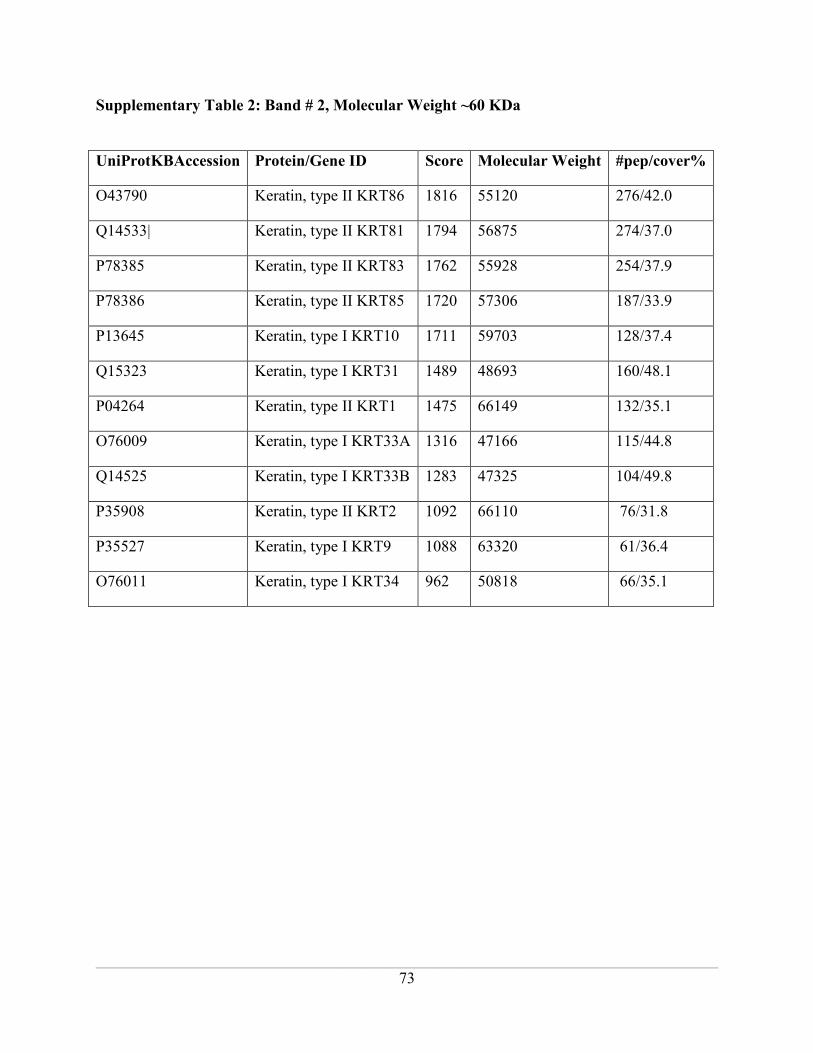
______________________________________________________________________________
73
Supplementary Table 2: Band # 2, Molecular Weight ~60 KDa
UniProtKBAccession Protein/Gene ID Score Molecular Weight #pep/cover%
O43790 Keratin, type II KRT86 1816 55120 276/42.0
Q14533| Keratin, type II KRT81 1794 56875 274/37.0
P78385 Keratin, type II KRT83 1762 55928 254/37.9
P78386 Keratin, type II KRT85 1720 57306 187/33.9
P13645 Keratin, type I KRT10 1711 59703 128/37.4
Q15323 Keratin, type I KRT31 1489 48693 160/48.1
P04264 Keratin, type II KRT1 1475 66149 132/35.1
O76009 Keratin, type I KRT33A 1316 47166 115/44.8
Q14525 Keratin, type I KRT33B 1283 47325 104/49.8
P35908 Keratin, type II KRT2 1092 66110 76/31.8
P35527 Keratin, type I KRT9 1088 63320 61/36.4
O76011 Keratin, type I KRT34 962 50818 66/35.1
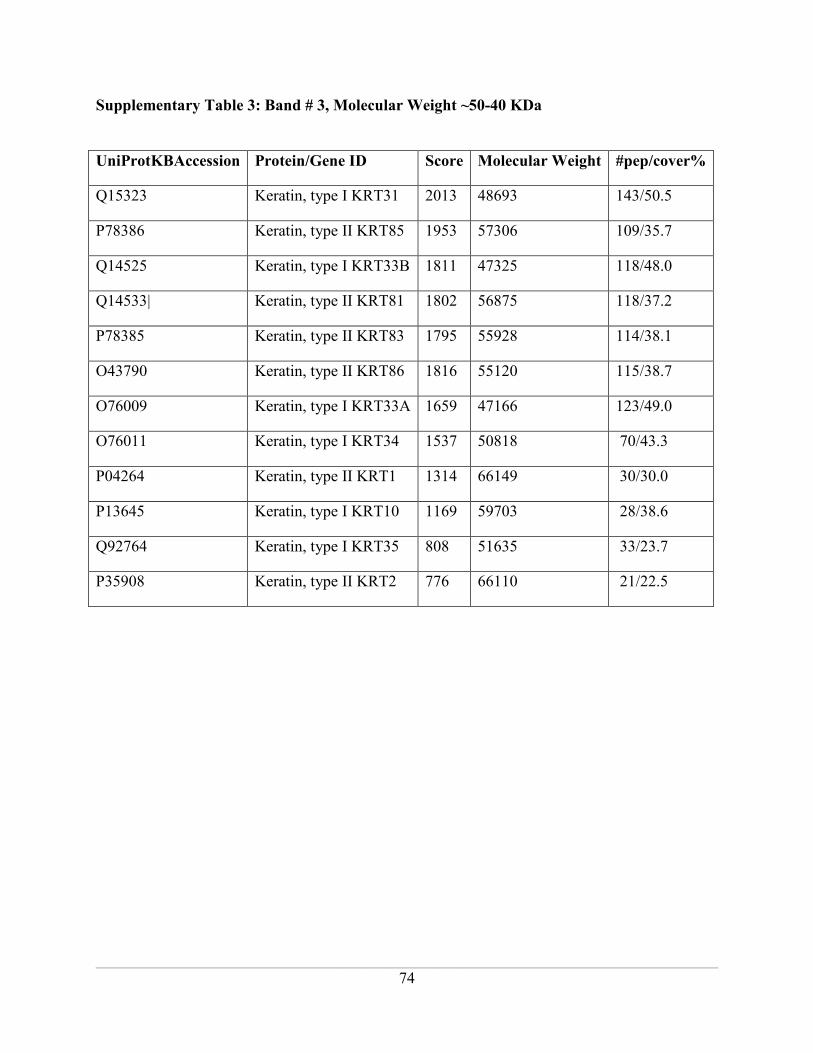
______________________________________________________________________________
74
Supplementary Table 3: Band # 3, Molecular Weight ~50-40 KDa
UniProtKBAccession Protein/Gene ID Score Molecular Weight #pep/cover%
Q15323 Keratin, type I KRT31 2013 48693 143/50.5
P78386 Keratin, type II KRT85 1953 57306 109/35.7
Q14525 Keratin, type I KRT33B 1811 47325 118/48.0
Q14533| Keratin, type II KRT81 1802 56875 118/37.2
P78385 Keratin, type II KRT83 1795 55928 114/38.1
O43790 Keratin, type II KRT86 1816 55120 115/38.7
O76009 Keratin, type I KRT33A 1659 47166 123/49.0
O76011 Keratin, type I KRT34 1537 50818 70/43.3
P04264 Keratin, type II KRT1 1314 66149 30/30.0
P13645 Keratin, type I KRT10 1169 59703 28/38.6
Q92764 Keratin, type I KRT35 808 51635 33/23.7
P35908 Keratin, type II KRT2 776 66110 21/22.5

______________________________________________________________________________
75
Supplementary Table 4: Band # 4, Molecular Weight ~30 Kda
UniProtKBAccession Protein/Gene ID Score Molecular Weight #pep/cover%
P78386 Keratin, type II KRT85 2038 57306 113/42.2
Q15323 Keratin, type I KRT31 1810 48693 108/48.1
Q14533| Keratin, type II KRT81 1761 56875 121/42.8
Q14525 Keratin, type I KRT33B 1758 47325 78/48.0
O43790 Keratin, type II KRT86 1757 55120 118/44.4
P78385 Keratin, type II KRT83 1709 55928 116/41.4
P04264 Keratin, type II KRT1 1467 66149 44/40.2
O76011 Keratin, type I KRT34 1448 50818 61/41.1
O76009 Keratin, type I KRT33A 1399 47166 82/42.8
P13645 Keratin, type I KRT10 1248 59703 44/41.0
P35527 Keratin, type I KRT9 771 63320 18/37.2
O76015 Keratin, type I KRT38 690 52054 22/19.7

______________________________________________________________________________
76
Supplementary Table 5: Band # 5, Molecular Weight ~20-15 Kda
UniProtKBAccession Protein/Gene ID Score Molecular Weight #pep/cover%
P78386 Keratin, type II KRT85 2252 57306 171/42.2
O43790 Keratin, type II KRT86 2132 55120 199/44.2
Q14533| Keratin, type II KRT81 2088 56875 194/39.2
P78385 Keratin, type II KRT83 2080 55928 194/40.2
Q15323 Keratin, type I KRT31 1847 48693 146/49.0
Q14525 Keratin, type I KRT33B 1587 47325 107/50.5
O76009 Keratin,type I KRT33A 1570 47166 126/48.0
O76011 Keratin, type I KRT34 1413 50818 86/42.2
P04264 Keratin, type II KRT1 959 66149 38/28.9
P13645 Keratin, type I KRT10 723 59703 23/19.4
P35908 Keratin, type II KRT2 600 66110 35/25.6
O76015 Keratin, type I KRT38 579 52054 18/18.9
P47929 Galectin-7 331 15123 8/36.0
P27482 Calmodulin-like protein 3 310 16937 15/30.2
P15090 Fatty acid-binding protein 273 14824 19/34.1
P10599 Thioredoxin 221 12015 8/31.4
P01040| Cystatin A 220 11000 5/48.0

______________________________________________________________________________
77
Supplementary Table 6: Band # 6, Molecular Weight ~8-10 KDa
UniProtKBAccession Protein/Gene ID Score Molecular
Weight
#pep/cover%
P78385 Keratin, type II KRT83 1301 55928 35/31.0
Q14533| Keratin, type II KRT81 1271 56875 36/32.3
O43790 Keratin, type II KRT86 1271 55120 37/33.5
P04264 Keratin, type II KRT1 1240 66149 31/38.7
Q15323 Keratin, type I KRT31 1167 48693 45/40.6
P13645 Keratin, type I KRT10 1157 59703 29/32.9
P78386 Keratin, type II KRT85 1150 57306 29/29.0
O76009 Keratin, type I KRT33A 1117 47166 41/36.4
P35908 Keratin, type II KRT2 1001 66110 20/30.9
Q14525 Keratin, type I KRT33B 990 47325 31/35.1
O76011 Keratin, type I KRT34 955 50818 21/39.2
P35527 Keratin, type I KRT9 735 62320 19/36.8
P47929 Galectin-7 228 15123 5/36.8
Q3LI72 Keratin-associated protein
19-5
122 7847 1/30.6
P27482 Calmodulin-like protein 3 102 16937 1/11.4
P10599 Thioredoxin 88 12015 1/12.4

______________________________________________________________________________
78
CHAPTER 3
_____________________________________________________________________________________
GAMMA KERATOSE MAINTAINS CELL VIABILITY IN VITRO AFTER THERMAL
STRESS BY REGULATING THE EXPRESSION OF CELL DEATH PATHWAY
SPECIFIC GENES
Poranki D, Van Dyke M
Wake Forest Institute for Regenerative Medicine
Wake Forest School of Medicine, Medical Center Boulevard, Winston Salem, NC 27157
The following manuscript has not yet been submitted for publication. Deepika Poranki prepared
the manuscript. Dr. Mark Van Dyke acted in an advisory and editorial capacity.

______________________________________________________________________________
79
Abstract:
When skin is thermally burned, transfer of heat energy into the skin results in the destruction of
cells. Some of these skin cells are damaged but may be capable of self-repair and survival,
thereby contributing to spontaneous healing of the wound. This phenomenon can be replicated in
cell cultures through models that utilize mild heating, where cells are thermally stressed and their
response to injury studied in the presence and absence of therapeutic treatments. Keratin protein-
based biomaterials have been suggested as potential treatments for burn injury. Isolation of
cortical proteins from hair fibers results in an acid soluble fraction of keratin proteins referred to
as “gamma” keratose. In the present study, treatment with this fraction dissolved in media was
able to maintain cell viability after thermal stress in an in vitro model using primary mouse
dermal fibroblasts. PCR array analysis identified that at 12hr compared to the pre-treatment
control (6hr), gamma keratose treated cells has a significant (p<0.01) up regulation of Ulk1
(autophagy) and Casp3 (apoptosis and autophagy). Whereas in the growth media treated cells
although there is relatively high expression (fold change) of the genes, significant difference was
not observed. At 18hr, necrotic (Parp2) and autophagic (Atg16l2) genes were highly expressed in
growth media treated cells with statistically significant (p<0.01) difference compared to the pre-
treatment control (6hr), whereas none of the cell death related genes were highly expressed in the
gamma keratose treated cells. Moreover, gamma keratose treatment significantly (p<0.01) down
regulated autophagic (Esr1) and necrotic (Parp2) genes compared to the pre-treatment control
(6hr). By 24 hours, there was similar expression in both the treatment groups. These results
suggest that gamma keratose treatment may assist in the survival and salvage of thermally
stressed cells, maintaining their viability by regulating cell death pathway related genes. Gamma

______________________________________________________________________________
80
keratose may be a promising material for burn treatment that aids in spontaneous wound healing
from viable tissue surrounding the burn.
Keywords: gamma keratose, thermal stress, PCR array, apoptosis, necrosis, autophagy

______________________________________________________________________________
81
1. Introduction
When skin is thermally burned, transfer of the heat into the tissue results in cell
destruction. Some damaged cells may be capable of self-repair and survival, if properly treated,
thereby contributing to spontaneous healing of the wound. Complete tissue necrosis is often
observed in the region where the injury occurs, beginning with the surface of the skin and
radiating downward into the tissue. The tissue surrounding this necrotic zone can be termed a
“thermally stressed zone”, which has received the energy as an initiating factor but is susceptible
to several further insults that result in cell death [1]. This phenomenon can be replicated in cell
culture by models that utilize mild heating (i.e. 42-48oC), resulting in a heterogeneous population
of cells, some of which die as a direct result of the thermal injury and others that die secondary to
the thermal insult due to the inflammatory cytokines released into the media. There are also some
cells that survive both insults and are capable of self repair. Heating cells has been shown to
induce various cell death pathways [2-8]. Depending on the intensity and duration of the
exposure, apoptosis, necrosis or autophagy pathways are triggered. Apoptosis is a genetically
programmed cell death mechanism that is associated with the activation of cysteine-dependent
aspartate-specific proteases called caspases and is involved in the development of homeostasis
[9]. Necrosis is morphologically characterized by cellular swelling, swelling of organelles,
plasma membrane rupture and loss of intracellular contents [10]. Until recently, it was
considered as non-programmed and unregulated cell death, but recent evidence indicates that
programmed necrosis occurs through the activation of death receptors in a caspase independent
manner termed “necroptosis”[11]. Autophagy is a catabolic process involving degradation of the
cell’s own components and is usually activated by oxidative, nutritional or toxic stresses [12]. In

______________________________________________________________________________
82
contrast to other cell death pathways, autophagy reflects a cell survival mechanism by
maintaining normal cellular function during nutrient deficient conditions or by removing
damaged organelles and aggregated proteins [8, 13].
Numerous studies have been published in which in vitro models have been used to
investigate thermally-induced cell death. In one study the log-phase cultures of mouse
mastocytoma cells exposed to heat from 42ºC - 44ºC showed an increase in apoptosis; at 45ºC
both necrosis and apoptosis were observed, and at 46º and 47ºC only necrosis was observed [2].
In another study, prolonged energy deprivation followed by thermal stress (43ºC for 10 min) of
murine P3 O1 myeloma and Ehrlich ascites carcinoma cells induced high expression levels of
Hsp70 that protected the cells from necrosis [4]. A third study used osteoblasts exposed to 48ºC
for 10 min and showed a 15-20% increase in necrotic cells within minutes after heating. After a
12 hour recovery at 37ºC, there was a 10% increase in apoptotic cells compared to control cells
maintained at 37ºC, indicating that thermal stress induced necrosis in the early phase and
apoptosis in the late phase [5]. These studies have shown that mild heating of cells in culture can
induce both necrosis and apoptosis at different stages, thus serving as a basis for further
investigation of these pathways at the cell and molecular level.
More extensive studies have been published in which pathway analysis at the gene level
has been conducted. For example, exposure of NIH3T3 cells to 45ºC for 15 min induced the
activation of Akt mediated by PI3Kinase. Phosphotidylinositol 3 kinase (PI3 kinase) activates
Akt (a serine threonine kinase) downstream in the signaling pathway of growth factors and
cytokines [14-16]. Akt is also activated by PI3Kinase in cell stress conditions such as hydrogen
peroxide (H2O2) and heat shock [17]. Phosphorylated Akt gradually decreased to basal levels in

______________________________________________________________________________
83
9 hours after insult and a high rate of cell proliferation was observed within 24 hours. Whereas
when the cells were exposed to prolonged heating at 45ºC for 60min, apoptosis was increased by
about 70% compared to untreated cells at 37ºC. This treatment failed to activate Akt, indicating
that cells might be irreversibly damaged when exposed to high temperatures for prolonged
periods[6]. In another study, heat treatment of Statens Seruminstitut Rabbit Cornea (SIRC) cells
at 42ºC for 30 min triggered a mitochondrial apoptotic pathway indicated by a change in
Bax/Bcl-2 ratios, resulting in caspase-9 activity [7]. Thermal stress to cells was also shown to
trigger death receptor apoptotic pathways by a change in Fas ligand expression, which in turn
resulted in the activation of caspase 8. Addition of an antioxidant decreased heat mediated
apoptosis, suggesting that ROS plays a critical role in the mitochondrial and death receptor
pathways of these cells [7].
The aforementioned studies shed further light on the time/temperature dependence on cell
death pathways, and begin to elucidate some of the molecular pathways involved in necrosis and
apoptosis. However, autophagy has been less widely studied. There are published in vitro studies
showing that in spite of apoptosis being completely inhibited, cell death still occurs and is
usually independent of caspases, following a specific signaling pathway where the cell
morphology resembles necrotic cell death [18-22]. If anti-apoptotic proteins block the caspases,
then necrosis occurs through the caspase independent pathway [20]. Embryonic fibroblasts from
double knock out Bax/Bak-/-
mice are resistant to apoptotic inducers. Bax and Bak regulate the
intrinsic pathway of apoptosis by controlling the activation of caspases [23]. Bax/Bak-/-
double
knockouts are used in research to study the different mechanisms of cell death (autophagy and
necrosis) that occur when apoptosis is completely inhibited. When treated with etoposide, the

______________________________________________________________________________
84
cells failed to undergo apoptosis and instead manifested a massive autophagy response followed
by cell death, but knocking down the autophagy related gene (Atg) products beclin-1 and Atg5,
reduced etoposide induced cell death [24]. Using other apoptotic inhibitors did not trigger the
autophagic cell death pathway, indicating that Bax and Bak absence played an important role in
autophagy activation [24]. In other cell types like L929 mouse fibrosarcoma, human Jurkat T
lymphoma, and U937 monocytoid cells, inhibition of cystein proteases (caspases) was shown to
induce autophagy [25, 26]. In stress conditions like nutrition depletion or loss of growth factors,
autophagy gets activated by the inhibition of apoptosis and promotes cell survival [27, 28]. If
autophagy is inhibited in cell stress conditions, the cells usually die through apoptosis. This cell
death can be postponed by inhibiting Bax or Bak or caspases [29]. In vitro and in vivo
experiments have shown induced apoptotic cell death by the inactivation of Atg genes Atg 5 or
7, and beclin-1[30-33]. Autophagy may develop as a primary response to stress stimuli, which
then triggers either apoptosis or necrosis [34, 35].
Previous studies (Chapter 2) have shown that gamma keratose was able to maintain cell
viability post thermal stress compared to the other treatments in an in vitro model using primary
mouse fibroblasts. It was evident in these studies that a large number of cells died from the initial
heat treatment, additional cells died within hours after heating, and surviving cells re-established
the culture. Thus, we concluded that all three pathways of cell death may be present in our cell
culture model, and that keratin treatment may affect one or more of these pathways. In the
present study, we hypothesize that treating primary dermal fibroblast cells with gamma keratose
six hours post thermal stress will induce differential expression of genes related to the cell death
pathways. The RT2 Profiler
TM PCR array was used to investigate cell death pathway related gene

______________________________________________________________________________
85
expression in thermally stressed cells treated with gamma keratose dissolved in the media
compared to cells treated with fibroblast growth media only.
2. Methods
2.1. Fibroblast cell Isolation: Primary mouse fibroblasts were isolated from adult CD1
mouse ear pads. The ear pads were incubated in Dispace II overnight and then the
dermis was peeled off using forceps and the remaining tissue minced before incubating
in 0.35% type I collagenase reconstituted in DMEM high glucose for 30 min at 37˚C on
a shaker. The suspension was filtered using a 100 micron meshed filter and the cells
were pelleted by centrifugation. The cell pellet was washed once with Hank’s balanced
salt solution (HBSS) to remove the residual collagenase. Finally, the fibroblast cells
were seeded in a 150 mm tissue culture plastic (TCP) dish at 2.5 x 106 cells/mL in
fibroblast growth media [DMEM high glucose plus 10% Fetal Bovine Serum (Hyclone®
Laboratories,Inc. Logan,Utah)] and 1% Antibiotic/Antimycotic solution (Hyclone®
Laboratories,Inc. Logan,Utah)).
2.2. Gamma Keratose Extraction: Keratin biomaterial was extracted from human hair
using the oxidative protocol specified by de Guzman et al. [36]. The extracted keratin
biomaterial was termed crude keratose. The gamma keratose fraction was further
separated from crude keratose by isoelectric precipitation. Concentrated hydrochloric
acid was added dropwise to the crude keratose solution with stirring until a pH 4.2 is
reached. At this pH, the alpha keratose precipitates leaving the acid-soluble gamma
fraction in solution. The insoluble alpha keratose was separated by fixed angle
centrifugation at 1500 rpm (252 RCF) for 15min at 4ºC. After neutralizing the gamma-

______________________________________________________________________________
86
containing supernatant to pH 8.4, it was dialyzed against endotoxin free water using a
5kDa spiral wound cartridge (Millipore, Billerica, MA) for 5 volume washes. Finally,
the solution was concentrated to minimal volume, pH adjusted to 7.4, lyophilized, and
sterilized by gamma irradiation at 1 MRad prior to use.
2.3. In vitro thermal stress model: For this experiment, primary mouse fibroblasts between
passages 3 to 6 were seeded at approximately 8x105cells/mL in 100mm TCP dishes. The
cells were allowed to grow in fibroblast growth media until they were 95% confluent.
Media changes were performed every 3 days. For a thermal stress experimental run, a
media change was performed a day prior to heating the cells. On the day of the
experiment, cultures were moved from a 37˚C, 5% CO2 incubator to a 44˚C, 5% CO2
incubator and heated for 150 minutes. After the heat treatment, culture dishes were
moved back to a 37˚C, 5% CO2 incubator to allow re-attachment of cells and the media
temperature to equilibrate. Six hours post heat treatment, cultures were randomized and
treatment was applied in fresh media. Treatments included 0.1mg/mL gamma keratose
supplemented to fibroblast growth media and fibroblast growth media alone (control
containing 10% FBS). The former will be termed gamma keratose treatment and the
latter growth media treatment in the rest of the chapter.
2.4. Trypan Blue Assay: Cell counting with a hemocytometer was performed using 0.4%
trypan blue reagent (Invitrogen, Gibco, Grand Island, NY), which stains cells with a
disrupted plasma membrane. Cells were counted at 6hr, 12hr, 18hr and 24 hr post heat
treatment. Out of two separate in vitro experiments performed, three samples showing
the highest percent cell viability after gamma keratose treatment and three samples
showing the lowest percent cell viability with growth media treatment were selected for

______________________________________________________________________________
87
further analysis using PCR arrays. Cell viability data from these samples was presented
as mean ± SEM (standard error of the mean).
2.5. RNA extraction & quantification: RNA extraction was performed using 5 prime
perfect pure RNA cultured cell kit (5 Prime Inc., Gaithersburg, MD 20878). After
trypsinization and cell counting, the cell pellet obtained after centrifugation was lysed
with a solution provided in the kit. The suspension was loaded onto a column with a
filter and centrifuged, followed by two washes with wash solution and RNA elution
from the column filter was performed using an elution buffer. Extracted RNA was
quantified using a Thermoscientific Nanodrop 2000 spectrophotometer. Out of the two
separate thermal stress experiments that were performed, RNA was extracted from three
individual (10cm) tissue culture plates for each treatment that showed the highest
percent cell viability after gamma keratose treatment and the least percent cell viability
after growth media treatment.
2.6. PCR arrays: cDNA synthesis was performed with the RT2
First Strand Kit
(SABiosciences) using an AB Applied Biosystems Veit 96 well thermocycler (850
Lincoln Center Drive, Foster City, CA) following the protocol specified by the
manufacturer. After the cDNA synthesis was performed, RT2 Profiler
TM PCR
microarray mouse cell death pathway finder (SABiosciences) was used to determine the
expression of genes that are involved in cell death. RT2 Sybr green ROX qPCR master
mix (SABiosciences) was used to perform the gene array experiment using an AB
Applied Biosystems 7300 Real time PCR system (850 Lincoln center drive, Foster City,
CA) following the protocol specified by the manufacturer. RT2 Profiler
TM PCR array
mouse cell death pathway finder analyzes the real-time expression of 86 genes

______________________________________________________________________________
88
(Supplementary Table 1) related to cell death pathways, which includes apoptosis,
necrosis and autophagy.
3. Data Analysis: Data was analyzed using the Auto Ct settings provided by the 7300 Real
Time PCR system software. Further data analysis was done using RT2
profiler PCR array
web based software provided by the manufacturer that follows ∆∆Ct method of calculation
[37]. This provided a comparison of post-treatment (12hr, 18hr, and 24hr) to the pre-
treatment control (6hr). To compare the relative up or down regulation between the gamma
keratose and fibroblast growth media treatment groups, the ratio of gene expression (fold
change) for gamma keratose treatment to growth media treatment was calculated. Using this
calculation, if the ratio is less than 1, it indicates that the gene expression is higher in growth
media treated cells, if it is equal to 1, the expression is the same in both the treatments, and if
it is greater than 1, the expression is greater in the gamma keratose treated cells.
4. Results:
4.1. In vitro thermal stress model: Six hours after heating, the average percent cell viability
was 53.7%±4.35. At 12hr, there is a significant difference (p<0.05) in the percent cell
viability between gamma keratose treated cells and growth media only treated cells. The
average percent cell viability for gamma keratose treated cells was 37.8%±0.85, whereas
growth media treated cells was 24.41%±2.20. At 18 hr, gamma keratose treated cells
were able to maintain cell viability at 39.83%±5.54, whereas in growth media treated
cells, additional cell death was noted with an average percent cell viability of only
20.18%±4.58. This difference was statistically significant (p<0.01). At 24 hr, the average
percent cell viability for gamma keratose treated cells was 36.9%±3.74 compared to

______________________________________________________________________________
89
growth media treated cells at 24.83%±0.69. This difference was not statistically
significant (p>0.05) (Figure 1).
4.2. RNA quantification: The total amount of RNA extracted from all the 24 samples is
listed in Table 2. The A260:280 and A260:230 ratios were approximately 2.1 and 2.0
indicating the purity of extracted RNA (Table 1).
4.3. PCR array data analysis: Gene expression of gamma keratose and growth media
treated cells at 12hr, 18hr and 24hr was compared to that of post thermal stress control
(i.e. cells at 6 hours post-heating, but immediately prior to treatment with fresh growth
media or gamma keratose supplemented media).
At 12hr compared to the pre-treatment control (6hr), the growth media treated
cells have no genes that were statistically significant even though their gene expression
(fold change) was high. In the gamma keratose treated cells, 29 genes were significantly
(p<0.05) up-regulated compared to the pre-treatment control. Among these 29 genes, 19
genes were down-regulated, six genes were up-regulated in gamma keratose treated cells
and four genes had a similar expression when compared to growth media treated cells.
The 19 genes that were down-regulated were involved in necrosis ( 9430015G10Rik,
Bmf, Txnl4b, Parp2, Pvr, Jph3), autophagy (Atg7, Atg5, Atg12, Map1lc3a), both
apoptosis and autophagy (Akt1, Bcl2, Bax) and apoptosis (anti-apoptotic [Traf2, Xiap]
and pro-apoptotic [Dffa, Fasl, Apaf1, Birc2]).The six up-regulated genes were involved
in autophagy (Htt, Ulk1, Atg16l1, Irgm1), both apoptosis and autophagy (Casp3) and
apoptosis (anti-apoptotic [Mcl1]). The genes that had similar expression between both

______________________________________________________________________________
90
growth media and gamma keratose treated cells were involved in autophagy (App,
Mapk8, Rps6kb1) and apoptosis (pro-apoptotic [Cflar]) (Table 2).
At 18hr, compared to the pre-treatment control (6hr), 34 genes in the growth
media treated cells were statistically significant (p<0.05). Of them seven genes were
down-regulated and 27 genes were up-regulated in the growth media treated cells. In the
gamma keratose treated cells, 18 genes had a gene expression that was statistically
significant (p<0.05) compared to pre-treatment control (6hr). Of these, six genes were
down-regulated and 12 genes were up-regulated in the gamma keratose treated cells.
When the 34 genes that were significantly expressed in growth media treated cells were
compared to the gamma keratose treated cells, six genes involved in necrosis (Galbt5,
Foxi1, Olfr1404), autophagy (Ins2, Esr1) and apoptosis (anti-apoptotic [Tnfrsf11b]) were
down-regulated and six genes involved in necrosis (Parp2, Rab25, Bmf, S100a7a),
autophagy (Atg16l1) and apoptosis (pro-apoptotic [Fasl]) were up-regulated in growth
media treated cells. Twenty two genes involved in necrosis (Commd4, Txnl4b,
9430015g10Rik), autophagy (Htt, Atg7, Pik3c3, Atg5, Ulk1, Mapk8, Map1lc3a),
apoptosis (anti-apoptotic [Xiap, Birc3] and pro-apoptotic [Dffa, Birc2, Casp1, Apaf1,
Gadd45a]) and both apoptosis and autophagy (Casp3, Bcl2l1, Bcl2, Bax, Akt1) had a
similar expression in both the groups. Comparison of the 18 genes that were expressed in
gamma keratose treated cells to growth media have shown that three genes involved in
necrosis (Rab25, Parp2) and autophagy (Atg16l1) were down-regulated and three genes
involved in necrosis (Galnt5), autophagy (Esr1) and anti-apoptosis (Sqstm1) were up-
regulated in gamma keratose treated cells, The 12 remaining genes involved in necrosis

______________________________________________________________________________
91
(Txnl4b), autophagy (Htt, Atg7, Atg5, Ulk1, Mapk8, Map1lca), apoptosis (anti-apoptotic
[Xiap, Birc3] and pro-apoptotic [Birc2]) and both apoptosis and autophagy (Casp3, Bax)
had a similar expression between both the groups (Table 3 & 4).
At 24hr compared to the pre-treatment control (6hr), 21 genes were expressed in
growth media treated cells with statistical significance (p<0.05). Of them, 10 genes were
down-regulated and 11 genes were up-regulated in the growth media treated cells.
Similar gene expression was noticed in the gamma keratose treated cells. Twenty one
genes in gamma keratose treated cells had significant difference (p<0.05) compared to
pre-treatment control. Of them, 10 genes were down-regulated and 11 genes were up-
regulated in the gamma keratose treated cells. When the 21 genes that were significantly
expressed in growth media treated cells were compared to gamma keratose treated cells,
seven genes involved in necrosis (Galnt5, Bmf, Txnl4b), autophagy (Esr1), apotosis (pro-
apoptotic [Gadd45a] and anti-apoptotic [Tnfrsf11b]), apoptosis and necrosis (Sycp2)
were up-regulated. The remaining 14 genes involved in necrosis (Olfr1404, Commd4,
S100a7a, Parp2), autophagy (Ifng, Ctsb, Atg16l1, Gaa), apoptosis (anti-apoptotic [Xiap]
and pro-apoptotic [Birc2, Nol3, Casp1]) and both apoptosis and autophagy (Bcl2l1, Bax)
were expressed similarly between the two groups. Comparison of the 21 genes that were
significantly expressed in gamma keratose treated cells to the growth media treated cells
have shown that six genes involved in necrosis (Galnt5), autophagy (Esr1, Ins2),
apoptosis (anti-apoptotic [Tnfrsf11b] and pro-apoptotic [Gadd45a]) and both apoptosis
and necrosis (Sycp2) were down-regulated and one gene involved in apoptosis (pro-
apoptotic [Abl1]) were up-regulated in gamma keratose treated cells. The remaining 14

______________________________________________________________________________
92
genes involved in necrosis (Rab25, Olfr1404, Commd4, S100a7a, Parp2), autophagy
(Ifng, Ctsb, Atg16l1, Gaa), apoptosis (anti-apoptotic [Xiap] and pro-apoptotic [Birc2,
Nol3, Casp1]) and both apoptosis and autophagy (Bcl2l1) were similarly expressed
between the groups (Table 5 & 6).
5. Discussion
Thermal burns typically display an injury pattern dictated by the transfer of the thermal
energy into the skin and underlying tissues. According to Jackson’s burn model, the zone of
stasis contains viable cells initially but due to the inflammatory cytokines released by the
surrounding necrotic tissue, more cell death occurs [38]. Necrosis is the major mechanism of cell
death that occurs as thermally damaged tissue converts to dead tissue and the wound progresses
from second degree to third degree. This process has sometimes been described as burn wound
progression [39]. This concept was shown by Clark et al. using a validated porcine hot comb
model in which high mobility group box 1 (HMGB-1) protein was used as a marker for cellular
necrosis and activated cleaved caspase-3 as a marker for apoptosis. The authors showed that
cellular necrosis in the zone of stasis occurred between one and four hours and apoptosis was
responsible for cell death only after 24 hr. They also suggested that an early intervention within
the first four hours is necessary to limit the irreversible cellular changes that account for burn
injury progression. However, there have been relatively few therapies that specifically target
cessation of burn injury progression by treating the thermally stressed tissue immediately
following injury. In one rare example, c-jun amino terminal kinase, a member of the mitogen-
activated protein kinase family involved in both apoptosis and necrotic pathways was
investigated as a potential target [40, 41]. In a mouse thermal injury burn model, a peptide

______________________________________________________________________________
93
inhibitor of c-jun was shown to promote re-epithelialization faster and also reduced cell death
surrounding the wounded region by 24 hr post injury [42]. Clearly, treatments that can arrest the
death of potentially viable cells are feasible and represent a novel therapy that may provide tissue
sparring treatment, but surprisingly little research has been conducted in this field.
Previous studies (Chapter 2) showed that gamma keratose was able to reduce the TBSA
in in vivo studies and maintain cell viability in an in vitro thermal stress model. Using the same
thermal stress model, we attempted to understand whether gamma keratose induces differential
expression of cell death pathway genes as a possible mechanism of its apparent tissue-sparing
activity. There are four main findings from the present experiments:
1. As expected, heat treatment of mouse dermal fibroblasts up regulates cell death
pathway related genes.
2. Gene expression was more consistent (i.e. smaller p values) in cells treated
with gamma keratose compared to cells treated with fibroblast growth media at
the early time point post thermal stress (12hr).
3. A single gamma keratose treatment at 0.1mg/mL appeared to influence gene
expression at 12 and 18 hr post-thermal stress (6 and 12 hr post-treatment,
respectively), but this effect was diminished by 24 hr.
4. In general, treating these thermally stressed cells with gamma keratose
substantially diminishes the gene up regulation compared to treatment with
fresh fibroblast growth media.
To further explain the first point, real time PCR array data analysis showed that there was
an overall up regulation of the genes involved in cell death pathways (necrosis, autophagy,

______________________________________________________________________________
94
apoptosis) six hours post thermal stress, and the cell viability data from the in vitro thermal stress
model correlates with this point. After the 6hr post thermal stress interval, there was a drop in the
cell viability from 100% to 53.7%±4.35 (Figure 1).
To further explain the second point, at 12hr post thermal stress, gamma keratose treated
cells showed statistically significant (p<0.05) alterations in the expression of 29 genes compared
to the pre-treatment control (6hr). Whereas in the growth media treated cells although there was
high expression (fold change) of the genes compared to the pre-treatment control, no statistical
significance was observed (p>0.05) due to the variability in the sample replicates for this
treatment group (Table 2).
As mentioned in point three, treating the thermally stressed cells once with 0.1mg/mL
gamma keratose was able to maintain cell viability by regulating the cell death related genes at
12hr and 18hr. At 12hr, 29 genes had a significant (p<0.05) gene expression in gamma keratose
treated cells compared to the pre-treatment control (6hr). Of these, 19 genes were highly
expressed by the growth media treated cells. By 18hr, only 18 genes were significantly (p<0.05)
expressed in gamma keratose treated cells, whereas 34 genes were significantly (p<0.05)
expressed in growth media treated cells compared to the pre-treatment control (6hr). The
regulation of cell death related genes by gamma keratose strongly correlates with the in vitro
thermal stress model cell viability results. In the in vitro model at 12hr post thermal stress,
gamma keratose treated cells were able to maintain cell viability at 37.8%±0.85 compared to
growth media treated cells at 24.41%±2.20 and the difference was statistically significant
(p<0.05). At 18 hr, gamma keratose treated cells were able to maintain cell viability at
39.83%±5.54, whereas in growth media treated cells, additional cell death was observed with an

______________________________________________________________________________
95
average percent cell viability of only 20.18%±4.58. This difference was statistically significant
(p<0.01). By 24hr, there was no significant difference in the cell viability and the gene
expression was similar between the two treatment groups (Figure 1 and Table 7 respectively).
To further explain the fourth point, at 12hr, 10 genes in gamma keratose treated cells
showed a statistically significant difference (p<0.01) compared to the pre-treatment control.
Among these 10 genes, two genes involved in autophagy (Ulk1) and both apoptosis and
autophagy (Casp 3) were up-regulated, 8 genes involved in necrosis (Parp2, Pvr), autophagy
(Atg5, Map1lc3a, Atg7), apoptosis (pro-apoptotic[Fas1]) and both apoptosis and autophagy
(Bax, Akt1) were down-regulated in the gamma keratose treated group compared to the growth
media treated cells (Table 2).
Tozhe et al. screened embryonic kidney 293 cells by kinase specific si-RNA library using
autophagy assay and identified Ulk1 as a novel candidate for autophagy [43]. In mammalian
cells, Ulk1/2 forms a complex with Atg13, Atg101 and FIP200 that was positively regulated by
AMPK (AMP-activated protein kinase) and negatively regulated by mTOR (mammalian target
of rapamycin) [44, 45]. AMPK is an evolutionarily conserved energy sensing kinase that is
activated by metabolic stress [45]. mTOR regulates growth, proliferation and protein synthesis of
cells in response to growth inducing stimuli [46]. Caspase 3 (Casp 3) is involved in the cleavage
of Atg4D (regulates autophagosome formation) and the truncated Atg4D is highly toxic and
couples both apoptosis and autophagy [47]. Fasl is a member of tumor necrosis family (TNF) of
type II transmembrane proteins. It is prototypic death factor that induces apoptosis when it binds
to its receptor Fas [48]. Atg5 and Atg7are involved in the formation of autophagosome [49].
Starved mouse embryonic fibroblasts lacking the autophagy gene Atg7 failed to undergo cell

______________________________________________________________________________
96
cycle arrest. Whereas prolonged metabolic stress resulted in augmented DNA damage and
increased p-53 mediated apoptotic cell death [50].
The gene expression data suggests that gamma keratose treatment was able to control the
up regulation of most of the cell death genes at 12hr compared to the growth media. The genes
that were up regulated in the gamma keratose treatment are involved in the autophagy pathway,
suggesting that the cell death observed in the gamma keratose treatment from 6 to 12 hrs post
thermal stress was due to autophagy. In vitro studies have shown that in stressed conditions like
starvation and energy deprivation, cell death occurs through autophagy when other cell death
pathways (like apoptosis) were inhibited [24-26]. At 18hr compared to the pre-treatment control,
gamma keratose treated cells had a significant (p<0.01) expression of 4 genes. Of the four genes,
two genes involved in autophagy (Esr1) and necrosis (Parp2) were down-regulated compared to
growth media treated cells (Table 3). The remaining two genes had a similar expression between
both the groups. Even though down regulation of Esr1 was observed in our thermal stress model,
no studies were published that suggest that Esr1 expression regulates autophagy when the cells
are exposed to stress conditions. Parp2 was shown to be involved in programmed necrosis (also
called necroptosis). Hitomi et al. treated L929 cells with TNF-α (tumor necrosis receptor family
involved in apoptosis by the activation of caspase 3) and zVAD.fmk (caspase inhibitor) and
performed a genome wide siRNA screen to identify the regulators of necroptosis. They identified
Parp-2 as a key regulator of necroptosis and showed that knockdown of Parp-2 inhibits
necroptosis [51]. This suggests that gamma keratose treated cells were able to maintain their cell
viability (39.83%±5.54) at 18hr when compared to 12hr (20.18%±4.58) by controlling the down
regulation of genes involved in autophagy and necrosis. By 24 hrs, the gene expression between

______________________________________________________________________________
97
the two treatment groups was similar, suggesting that the effect of the gamma keratose treatment
had diminished (Table 5 & 6). Although the mechanism of interaction of the gamma keratose
with the cell was not investigated in this study, the metabolic demand of thermally stressed cells
is known to be pathologically high [52-55], so it would not be surprising if the keratose were
consumed rapidly and its effects short lived.
6. Conclusion:
This study has shown that a single treatment of gamma keratose at 0.1mg/mL after
thermal stress can mediate survival of mouse dermal fibroblasts and faster recovery of the
culture. At 12hr compared to pre-treatment control (6hr), there was approximately 15% decrease
in cell viability and an increase in expression of autophagy related genes in the gamma keratose
treated cells. However, by 18hr gamma keratose was able to maintain cell viability similar to that
observed at 12 hrs and also down regulated the genes related to autophagy. This study suggests
that along with apoptosis and necrosis, autophagy plays an important role in the viability of
thermally stressed cells. Importantly, gamma keratose treatment was able to maintain cell
viability by down regulating the expression of the genes involved in apoptosis, necrosis and
autophagy. Gamma keratose, as a component in a keratin biomaterial hydrogel, may therefore
have been beneficial as a burn treatment as shown in earlier studies due to this mechanism.
Further detailed studies are necessary to determine if this is indeed the case, as the processes of
cell death are complex and any potential role of gamma keratose would need to be examined in
more detail, particularly with regard to how the cell interacts with the gamma keratose
molecules.

______________________________________________________________________________
98
Conflict of interest statement
Mark Van Dyke, Ph.D. holds stock and is an officer in the company, KeraNetics LLC, which has
provided partial funding for this research. Wake Forest Health Sciences has a potential financial
interest in KeraNetics, LLC through licensing agreements.

______________________________________________________________________________
99
References:
[1] Harada T, Kuroda T, Tsutsumi H, Kobayashi M. Heat shock zone of the burn wound. Burns.
1996;22:578-9.
[2] Harmon BV, Corder AM, Collins RJ, Gobe GC, Allen J, Allan DJ, et al. Cell death induced
in a murine mastocytoma by 42-47 degrees C heating in vitro: evidence that the form of death
changes from apoptosis to necrosis above a critical heat load. Int J Radiat Biol. 1990;58:845-58.
[3] Mailhos C, Howard MK, Latchman DS. Heat shock proteins hsp90 and hsp70 protect
neuronal cells from thermal stress but not from programmed cell death. J Neurochem.
1994;63:1787-95.
[4] Kabakov AE, Gabai VL. Heat shock-induced accumulation of 70-kDa stress protein (HSP70)
can protect ATP-depleted tumor cells from necrosis. Exp Cell Res. 1995;217:15-21.
[5] Li S, Chien S, Branemark PI. Heat shock-induced necrosis and apoptosis in osteoblasts. J
Orthop Res. 1999;17:891-9.
[6] Bang OS, Ha BG, Park EK, Kang SS. Activation of Akt is induced by heat shock and
involved in suppression of heat-shock-induced apoptosis of NIH3T3 cells. Biochem Biophys Res
Commun. 2000;278:306-11.
[7] Hsu YL, Yu HS, Lin HC, Wu KY, Yang RC, Kuo PL. Heat shock induces apoptosis through
reactive oxygen species involving mitochondrial and death receptor pathways in corneal cells.
Exp Eye Res. 2011;93:405-12.
[8] Zhang Y, Calderwood SK. Autophagy, protein aggregation and hyperthermia: a mini-review.
Int J Hyperthermia. 2011;27:409-14.
[9] Bortner CD, Cidlowski JA. Cellular mechanisms for the repression of apoptosis. Annu Rev
Pharmacol Toxicol. 2002;42:259-81.

______________________________________________________________________________
100
[10] Kitanaka C, Kuchino Y. Caspase-independent programmed cell death with necrotic
morphology. Cell Death Differ. 1999;6:508-15.
[11] Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, et al. Chemical inhibitor of
nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol.
2005;1:112-9.
[12] Uchiyama Y, Shibata M, Koike M, Yoshimura K, Sasaki M. Autophagy-physiology and
pathophysiology. Histochem Cell Biol. 2008;129:407-20.
[13] Nishida K, Kyoi S, Yamaguchi O, Sadoshima J, Otsu K. The role of autophagy in the heart.
Cell Death Differ. 2009;16:31-8.
[14] del Peso L, Gonzalez-Garcia M, Page C, Herrera R, Nunez G. Interleukin-3-induced
phosphorylation of BAD through the protein kinase Akt. Science. 1997;278:687-9.
[15] Burgering BM, Coffer PJ. Protein kinase B (c-Akt) in phosphatidylinositol-3-OH kinase
signal transduction. Nature. 1995;376:599-602.
[16] Ahmed NN, Grimes HL, Bellacosa A, Chan TO, Tsichlis PN. Transduction of interleukin-2
antiapoptotic and proliferative signals via Akt protein kinase. Proc Natl Acad Sci U S A.
1997;94:3627-32.
[17] Shaw M, Cohen P, Alessi DR. The activation of protein kinase B by H2O2 or heat shock is
mediated by phosphoinositide 3-kinase and not by mitogen-activated protein kinase-activated
protein kinase-2. Biochem J. 1998;336 ( Pt 1):241-6.
[18] Goossens V, Stange G, Moens K, Pipeleers D, Grooten J. Regulation of tumor necrosis
factor-induced, mitochondria- and reactive oxygen species-dependent cell death by the electron
flux through the electron transport chain complex I. Antioxid Redox Signal. 1999;1:285-95.

______________________________________________________________________________
101
[19] Kawahara A, Ohsawa Y, Matsumura H, Uchiyama Y, Nagata S. Caspase-independent cell
killing by Fas-associated protein with death domain. J Cell Biol. 1998;143:1353-60.
[20] Vercammen D, Beyaert R, Denecker G, Goossens V, Van Loo G, Declercq W, et al.
Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor
necrosis factor. J Exp Med. 1998;187:1477-85.
[21] Vercammen D, Brouckaert G, Denecker G, Van de Craen M, Declercq W, Fiers W, et al.
Dual signaling of the Fas receptor: initiation of both apoptotic and necrotic cell death pathways. J
Exp Med. 1998;188:919-30.
[22] Vercammen D, Vandenabeele P, Beyaert R, Declercq W, Fiers W. Tumour necrosis factor-
induced necrosis versus anti-Fas-induced apoptosis in L929 cells. Cytokine. 1997;9:801-8.
[23] Ruiz-Vela A, Opferman JT, Cheng EH, Korsmeyer SJ. Proapoptotic BAX and BAK control
multiple initiator caspases. EMBO Rep. 2005;6:379-85.
[24] Shimizu S, Kanaseki T, Mizushima N, Mizuta T, Arakawa-Kobayashi S, Thompson CB, et
al. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on
autophagy genes. Nat Cell Biol. 2004;6:1221-8.
[25] Madden DT, Egger L, Bredesen DE. A calpain-like protease inhibits autophagic cell death.
Autophagy. 2007;3:519-22.
[26] Xu Y, Kim SO, Li Y, Han J. Autophagy contributes to caspase-independent macrophage
cell death. J Biol Chem. 2006;281:19179-87.
[27] Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, et al. Growth factor regulation of
autophagy and cell survival in the absence of apoptosis. Cell. 2005;120:237-48.
[28] Lum JJ, DeBerardinis RJ, Thompson CB. Autophagy in metazoans: cell survival in the land
of plenty. Nat Rev Mol Cell Biol. 2005;6:439-48.

______________________________________________________________________________
102
[29] Gonzalez-Polo RA, Boya P, Pauleau AL, Jalil A, Larochette N, Souquere S, et al. The
apoptosis/autophagy paradox: autophagic vacuolization before apoptotic death. J Cell Sci.
2005;118:3091-102.
[30] Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, et al.
Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature.
2006;441:885-9.
[31] Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, et al. Loss of autophagy in the
central nervous system causes neurodegeneration in mice. Nature. 2006;441:880-4.
[32] Pua HH, Dzhagalov I, Chuck M, Mizushima N, He YW. A critical role for the autophagy
gene Atg5 in T cell survival and proliferation. J Exp Med. 2007;204:25-31.
[33] Takacs-Vellai K, Vellai T, Puoti A, Passannante M, Wicky C, Streit A, et al. Inactivation of
the autophagy gene bec-1 triggers apoptotic cell death in C. elegans. Curr Biol. 2005;15:1513-7.
[34] Espert L, Denizot M, Grimaldi M, Robert-Hebmann V, Gay B, Varbanov M, et al.
Autophagy is involved in T cell death after binding of HIV-1 envelope proteins to CXCR4. J
Clin Invest. 2006;116:2161-72.
[35] Yu L, Wan F, Dutta S, Welsh S, Liu Z, Freundt E, et al. Autophagic programmed cell death
by selective catalase degradation. Proc Natl Acad Sci U S A. 2006;103:4952-7.
[36] de Guzman RC, Merrill MR, Richter JR, Hamzi RI, Greengauz-Roberts OK, Van Dyke ME.
Mechanical and biological properties of keratose biomaterials. Biomaterials. 2011;32:8205-17.
[37] Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method.
Nat Protoc. 2008;3:1101-8.
[38] Jackson DM. [The diagnosis of the depth of burning]. Br J Surg. 1953;40:588-96.

______________________________________________________________________________
103
[39] Lanier ST, McClain SA, Lin F, Singer AJ, Clark RA. Spatiotemporal progression of cell
death in the zone of ischemia surrounding burns. Wound Repair Regen. 2011;19:622-32.
[40] Devin A, Lin Y, Liu ZG. The role of the death-domain kinase RIP in tumour-necrosis-
factor-induced activation of mitogen-activated protein kinases. EMBO Rep. 2003;4:623-7.
[41] Kuranaga E, Miura M. Molecular genetic control of caspases and JNK-mediated neural cell
death. Arch Histol Cytol. 2002;65:291-300.
[42] Giles N, Rea S, Beer T, Wood FM, Fear MW. A peptide inhibitor of c-Jun promotes wound
healing in a mouse full-thickness burn model. Wound Repair Regen. 2008;16:58-64.
[43] Chan EY, Kir S, Tooze SA. siRNA screening of the kinome identifies ULK1 as a
multidomain modulator of autophagy. J Biol Chem. 2007;282:25464-74.
[44] Alers S, Loffler AS, Wesselborg S, Stork B. The incredible ULKs. Cell Commun Signal.
2012;10:7.
[45] Alers S, Loffler AS, Wesselborg S, Stork B. Role of AMPK-mTOR-Ulk1/2 in the regulation
of autophagy: cross talk, shortcuts, and feedbacks. Mol Cell Biol. 2012;32:2-11.
[46] Kim DH, Sabatini DM. Raptor and mTOR: subunits of a nutrient-sensitive complex. Curr
Top Microbiol Immunol. 2004;279:259-70.
[47] Betin VM, Lane JD. Atg4D at the interface between autophagy and apoptosis. Autophagy.
2009;5:1057-9.
[48] Suda T, Takahashi T, Golstein P, Nagata S. Molecular cloning and expression of the Fas
ligand, a novel member of the tumor necrosis factor family. Cell. 1993;75:1169-78.
[49] Heath RJ, Xavier RJ. Autophagy, immunity and human disease. Curr Opin Gastroenterol.
2009;25:512-20.

______________________________________________________________________________
104
[50] Lee IH, Kawai Y, Fergusson MM, Rovira, II, Bishop AJ, Motoyama N, et al. Atg7
modulates p53 activity to regulate cell cycle and survival during metabolic stress. Science.
2012;336:225-8.
[51] Hitomi J, Christofferson DE, Ng A, Yao J, Degterev A, Xavier RJ, et al. Identification of a
molecular signaling network that regulates a cellular necrotic cell death pathway. Cell.
2008;135:1311-23.
[52] Yu YM, Tompkins RG, Ryan CM, Young VR. The metabolic basis of the increase of the
increase in energy expenditure in severely burned patients. JPEN J Parenter Enteral Nutr.
1999;23:160-8.
[53] Herndon DN, Tompkins RG. Support of the metabolic response to burn injury. Lancet.
2004;363:1895-902.
[54] Pereira CT, Murphy KD, Herndon DN. Altering metabolism. J Burn Care Rehabil.
2005;26:194-9.
[55] Mendonca Machado N, Gragnani A, Masako Ferreira L. Burns, metabolism and nutritional
requirements. Nutr Hosp. 2011;26:692-700.

______________________________________________________________________________
105
Figures & Tables:
Figure 1: In vitro Heat shock Model
Gamma keratose treated cells were able to maintain cell viability at 12hr, 18hr post-thermal
stress compared to the fibroblast growth media treated cells with statistical significance. P<0.01-
**, P<0.05*
* **
P<0.05 *
P<0.01 **

______________________________________________________________________________
106
Table 1: RNA Quantification
The 260/280 and 260/230 ratios were close to 2.1 and 2.0 indicating the purity of the extracted
RNA
# Sample ID
Nucleic Acid
Conc.(ng/µl) A260 A280 260/280 260/230
1 0h/N1 2210.9 55.3 26.1 2.1 2.2
2 0h/N2 3974.9 99.4 47.2 2.1 2.2
3 0h/N3 1973.9 49.3 23.1 2.1 2.2
4 6h/N1 835.6 20.9 9.8 2.1 2.2
5 6h/N2 1742.7 43.6 20.5 2.1 2.2
6 6h/N3 1270.6 31.8 14.8 2.2 2.2
7 12h/N1 486.7 12.2 5.6 2.2 2.2
8 12h/N2 477.9 11.9 5.5 2.2 2.1
9 12h/N3 574.5 14.4 6.6 2.2 2.2
10 12h/N1G 682.8 17.1 7.9 2.2 2.2
11 12h/N2G 385 9.6 4.6 2.1 1.9
12 12h/N3G 383.7 9.6 4.6 2.1 2.2
13 18h/N1 203.8 5.1 2.4 2.1 2.2
14 18h/N2 285.1 7.1 3.4 2.1 1.9
15 18h/N3 279.8 7.0 3.3 2.1 2.0
16 18h/N1G 285.4 7.1 3.4 2.1 1.8
17 18h/N2G 334.1 8.4 4.0 2.1 2.2
18 18h/N3G 732.9 18.3 8.7 2.1 2.2
19 24h/N1 114.9 2.9 1.4 2.1 2.2
20 24h/N2 185.6 4.6 2.2 2.1 2.1
21 24h/N3 226.1 5.7 2.7 2.1 2.1
22 24h/N1G 241.3 6.0 2.9 2.1 2.2
23 24h/N2G 324.8 8.1 3.9 2.1 2.1
24 24h/N3G 180.7 4.5 2.1 2.1 2.2

______________________________________________________________________________
107
Table 2: Gene expression of gamma keratose treated cells at 12hr
These are list of genes that were having significant p-value <0.05 for the gamma keratose treated
cells at 12hrs compared to the post heat shock pre-treated cells. Red color represents the up
regulated genes and blue color represents down regulated genes.
Gene
Symbol
Gamma
keratose
treated
cells p-
value
<0.05
Growth
media
treated
cells
fold
change
Gamma
keratos
e
treated
cells
fold
change
Gamma
keratose
treated cells
fold
change/Growt
h media
treated cells
fold change Pathway Involved
Traf2 0.02 20.63 2.14 0.1 Anti-apoptotic
Xiap 0.02 5.41 3.85 0.71 Anti-apoptotic
Mcl1 0.04 1.28 2.46 1.93 Anti-apoptotic
Akt1 0.01 21.75 2.86 0.13 Apoptosis and Autophagy
Bcl2 0.04 10.64 2.68 0.25 Apoptosis and Autophagy
Bax 0 4.65 2.84 0.61 Apoptosis and Autophagy
Casp3 0.01 0.49 3.03 6.17 Apoptosis and Autophagy
Atg7 0.01 41.19 2.63 0.06 Autophagy
Atg5 0 15.49 2.44 0.16 Autophagy
Atg12 0.02 9.09 3 0.33 Autophagy
Map1lc3a 0 6.8 2.91 0.43 Autophagy
App 0.03 2.65 2.31 0.87 Autophagy
Mapk8 0.05 2.32 2.1 0.91 Autophagy
Rps6kb1 0.04 2.68 2.64 0.98 Autophagy
Htt 0.02 2.87 5.06 1.77 Autophagy
Ulk1 0.01 3.76 8.02 2.13 Autophagy
Atg16l1 0.02 0.85 4.62 5.43 Autophagy
Irgm1 0.05 0.39 2.15 5.46 Autophagy
9430015G1
0Rik 0.02 70.88 3.39 0.05 Necrosis
Bmf 0.03 44.63 7.14 0.16 Necrosis
Txnl4b 0.04 15.37 3.61 0.24 Necrosis

______________________________________________________________________________
108
Parp2 0 9.51 2.62 0.28 Necrosis
Pvr 0 5.87 2.14 0.36 Necrosis
Jph3 0.02 13.64 9.91 0.73 Necrosis
Dffa 0.01 15.23 2.67 0.18 Pro-Apoptotic
Fasl 0 38.16 8.48 0.22 Pro-Apoptotic
Apaf1 0.01 43.33 9.91 0.23 Pro-Apoptotic
Birc2 0.05 15.78 3.81 0.24 Pro-Apoptotic
Cflar 0.02 4.55 3.92 0.86 Pro-Apoptotic
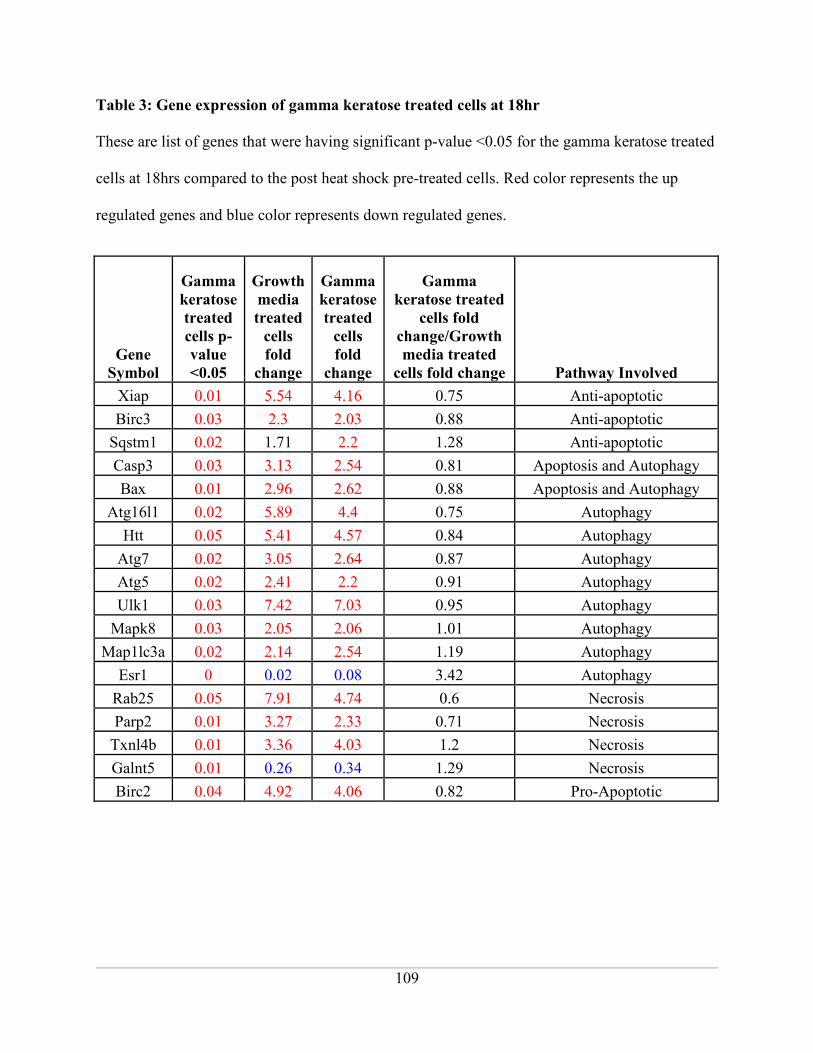
______________________________________________________________________________
109
Table 3: Gene expression of gamma keratose treated cells at 18hr
These are list of genes that were having significant p-value <0.05 for the gamma keratose treated
cells at 18hrs compared to the post heat shock pre-treated cells. Red color represents the up
regulated genes and blue color represents down regulated genes.
Gene
Symbol
Gamma
keratose
treated
cells p-
value
<0.05
Growth
media
treated
cells
fold
change
Gamma
keratose
treated
cells
fold
change
Gamma
keratose treated
cells fold
change/Growth
media treated
cells fold change Pathway Involved
Xiap 0.01 5.54 4.16 0.75 Anti-apoptotic
Birc3 0.03 2.3 2.03 0.88 Anti-apoptotic
Sqstm1 0.02 1.71 2.2 1.28 Anti-apoptotic
Casp3 0.03 3.13 2.54 0.81 Apoptosis and Autophagy
Bax 0.01 2.96 2.62 0.88 Apoptosis and Autophagy
Atg16l1 0.02 5.89 4.4 0.75 Autophagy
Htt 0.05 5.41 4.57 0.84 Autophagy
Atg7 0.02 3.05 2.64 0.87 Autophagy
Atg5 0.02 2.41 2.2 0.91 Autophagy
Ulk1 0.03 7.42 7.03 0.95 Autophagy
Mapk8 0.03 2.05 2.06 1.01 Autophagy
Map1lc3a 0.02 2.14 2.54 1.19 Autophagy
Esr1 0 0.02 0.08 3.42 Autophagy
Rab25 0.05 7.91 4.74 0.6 Necrosis
Parp2 0.01 3.27 2.33 0.71 Necrosis
Txnl4b 0.01 3.36 4.03 1.2 Necrosis
Galnt5 0.01 0.26 0.34 1.29 Necrosis
Birc2 0.04 4.92 4.06 0.82 Pro-Apoptotic

______________________________________________________________________________
110
Table 4: Gene expression of growth media treated cells at 18hr
These are list of genes that were having significant p-value <0.05 for the growth media treated
cells at 18hrs compared to the post heat shock pre-treated cells. Red color represents the up
regulated genes and blue color represents down regulated genes.
Gene Symbol
Growt
h
media
treated
cells p-
value
<0.05
Fibroblas
t media
treated
cells fold
change
Gamma
keratos
e
treated
cells
fold
change
Gamma keratose
treated cells fold
change/Fibroblas
t Media treated
cells fold change Pathway Involved
Xiap 0.00 5.54 4.16 0.75 Anti-apoptotic
Birc3 0.00 2.30 2.03 0.88 Anti-apoptotic
Tnfrsf11b 0.01 0.40 0.51 1.26 Anti-apoptotic
Casp3 0.01 3.13 2.54 0.81
Apoptosis and
Autophagy
Bcl2l1 0.00 3.11 2.55 0.82
Apoptosis and
Autophagy
Bcl2 0.03 2.59 2.17 0.84
Apoptosis and
Autophagy
Bax 0.00 2.96 2.62 0.88
Apoptosis and
Autophagy
Akt1 0.02 2.30 2.32 1.01
Apoptosis and
Autophagy
Atg16l1 0.00 5.89 4.40 0.75 Autophagy
Htt 0.04 5.41 4.57 0.84 Autophagy
Atg7 0.00 3.05 2.64 0.87 Autophagy
Pik3c3 0.02 2.10 1.85 0.88 Autophagy
Atg5 0.00 2.41 2.20 0.91 Autophagy
Ulk1 0.00 7.42 7.03 0.95 Autophagy
Mapk8 0.03 2.05 2.06 1.00 Autophagy
Map1lc3a 0.03 2.14 2.54 1.19 Autophagy
Ins2 0.05 0.23 0.37 1.60 Autophagy
Esr1 0.00 0.02 0.08 3.42 Autophagy

______________________________________________________________________________
111
Rab25 0.02 7.91 4.74 0.60 Necrosis
Bmf 0.01 7.75 4.73 0.61 Necrosis
S100a7a 0.02 9.00 6.27 0.70 Necrosis
Parp2 0.00 3.27 2.33 0.71 Necrosis
Commd4 0.01 2.01 1.60 0.80 Necrosis
9430015G10Ri
k 0.05 3.40 3.04 0.90 Necrosis
Txnl4b 0.02 3.35 4.03 1.20 Necrosis
Galnt5 0.00 0.26 0.34 1.29 Necrosis
Foxi1 0.05 0.23 0.37 1.60 Necrosis
Olfr1404 0.05 0.23 0.37 1.60 Necrosis
Fasl 0.04 7.88 5.65 0.72 Pro-Apoptotic
Dffa 0.00 2.87 2.26 0.79 Pro-Apoptotic
Birc2 0.00 4.92 4.06 0.82 Pro-Apoptotic
Casp1 0.01 2.61 2.22 0.85 Pro-Apoptotic
Apaf1 0.02 6.15 5.25 0.85 Pro-Apoptotic
Gadd45a 0.00 0.43 0.47 1.10 Pro-Apoptotic

______________________________________________________________________________
112
Table 5: Gene expression of gamma keratose treated cells at 24hr
These are list of genes that were having significant p-value <0.05 for the gamma keratose treated
cells at 24hrs compared to the post heat shock pre-treated cells. Red color represents the up
regulated genes and blue color represents down regulated genes.
Gene
Symbol
Gamma
keratose
treated
cells p-
value
<0.05
Growth
media
treated
cells
fold
change
Gamma
keratose
treated
cells
fold
change
Gamma keratose
treated cells fold
change/Growth
media treated cells
fold change Pathway Involved
Tnfrsf11b 0.00 0.34 0.24 0.73 Anti Apoptotic
Xiap 0.02 3.71 3.59 0.97 Anti Apoptotic
Sycp2 0.03 0.23 0.13 0.58 Apoptosis and Necrosis
Bcl2l1 0.00 2.93 3.09 1.06 Apotosis and Autophagy
Esr1 0.00 0.10 0.04 0.41 Autophagy
Ins2 0.03 0.21 0.13 0.64 Autophagy
Ifng 0.03 0.14 0.13 0.95 Autophagy
Ctsb 0.00 0.46 0.45 0.96 Autophagy
Atg16l1 0.04 3.74 3.84 1.03 Autophagy
Gaa 0.03 0.47 0.57 1.20 Autophagy
Galnt5 0.00 0.24 0.13 0.51 Necrosis
Rab25 0.01 6.96 5.51 0.79 Necrosis
Olfr1404 0.03 0.14 0.13 0.95 Necrosis
Commd4 0.01 2.18 2.21 1.01 Necrosis
S100a7a 0.01 7.87 8.18 1.04 Necrosis
Parp2 0.01 2.04 2.50 1.23 Necrosis
Gadd45a 0.00 0.46 0.33 0.71 Pro Apoptotic
Birc2 0.00 4.22 3.21 0.76 Pro Apoptotic
Nol3 0.04 0.43 0.44 1.02 Pro Apoptotic
Casp1 0.01 2.44 2.55 1.05 Pro Apoptotic
Abl1 0.01 3.76 5.23 1.39 Pro Apoptotic

______________________________________________________________________________
113
Table 6: Gene expression of growth media treated cells at 24 hr
These are list of genes that were having significant p-value <0.05 for the growth media treated
cells at 24hrs compared to the post heat shock pre-treated cells. Red color represents the up
regulated genes and blue color represents down regulated genes.
Gene
Symbol
Growth
media
treated
cells p-
value
<0.05
Growth
media
treated
cells
fold
change
Gamma
keratose
treated
cells fold
change
Gamma keratose
treated cells fold
change/Growth
media treated
cells fold change Pathway Involved
Tnfrsf11b 0.00 0.34 0.24 0.73 Anti Apoptotic
Xiap 0.01 3.71 3.59 0.97 Anti Apoptotic
Bcl2l1 0.00 2.93 3.09 1.06
Apoptosis and
Autophagy
Bax 0.03 2.14 2.29 1.07
Apoptosis and
Autophagy
Sycp2 0.04 0.23 0.13 0.58 Apoptosis and Necrosis
Esr1 0.00 0.10 0.04 0.41 Autophagy
Ifng 0.03 0.14 0.13 0.95 Autophagy
Ctsb 0.00 0.46 0.45 0.96 Autophagy
Atg16l1 0.01 3.74 3.84 1.03 Autophagy
Gaa 0.01 0.47 0.57 1.20 Autophagy
Galnt5 0.01 0.24 0.13 0.51 Necrosis
Bmf 0.03 3.55 2.30 0.65 Necrosis
Txnl4b 0.02 2.91 2.14 0.73 Necrosis
Olfr1404 0.03 0.14 0.13 0.95 Necrosis
Commd4 0.04 2.18 2.21 1.01 Necrosis
S100a7a 0.02 7.87 8.18 1.04 Necrosis
Parp2 0.05 2.04 2.50 1.23 Necrosis
Gadd45a 0.01 0.46 0.33 0.71 Pro Apoptotic
Birc2 0.01 4.22 3.21 0.76 Pro Apoptotic
Nol3 0.03 0.43 0.44 1.02 Pro Apoptotic
Casp1 0.00 2.44 2.55 1.05 Pro Apoptotic
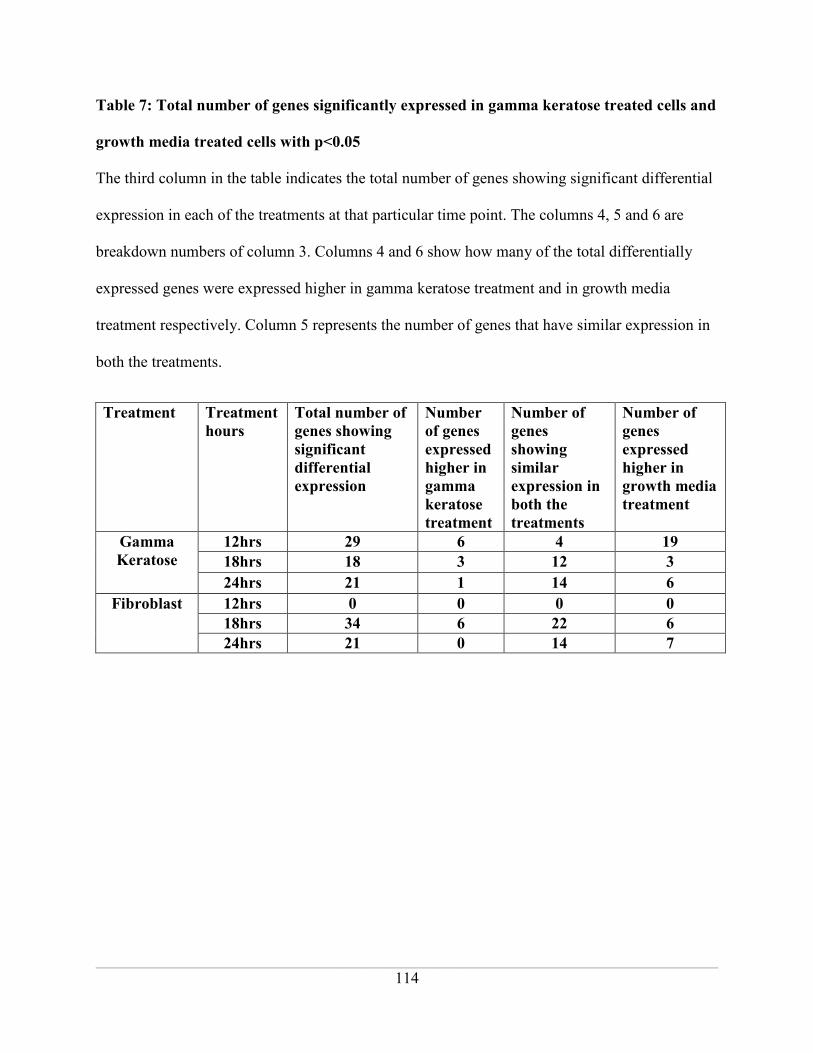
______________________________________________________________________________
114
Table 7: Total number of genes significantly expressed in gamma keratose treated cells and
growth media treated cells with p<0.05
The third column in the table indicates the total number of genes showing significant differential
expression in each of the treatments at that particular time point. The columns 4, 5 and 6 are
breakdown numbers of column 3. Columns 4 and 6 show how many of the total differentially
expressed genes were expressed higher in gamma keratose treatment and in growth media
treatment respectively. Column 5 represents the number of genes that have similar expression in
both the treatments.
Treatment Treatment
hours
Total number of
genes showing
significant
differential
expression
Number
of genes
expressed
higher in
gamma
keratose
treatment
Number of
genes
showing
similar
expression in
both the
treatments
Number of
genes
expressed
higher in
growth media
treatment
Gamma
Keratose
12hrs 29 6 4 19
18hrs 18 3 12 3
24hrs 21 1 14 6
Fibroblast 12hrs 0 0 0 0
18hrs 34 6 22 6
24hrs 21 0 14 7

______________________________________________________________________________
115
Supplementary Table 1: Genes included in the PCR array
Gene Symbol Gene Name
Cell death
Pathway
Associated
9430015G10Rik RIKEN cDNA 9430015G10 gene Necrosis
Abl1 C-abl oncogene 1, non-receptor tyrosine kinase Pro-Apoptotic
Akt1 Thymoma viral proto-oncogene 1 Apoptosis and
Autophagy
Apaf1 Apoptotic peptidase activating factor 1 Pro-Apoptotic
App Amyloid beta (A4) precursor protein Autophagy
Atg12 Autophagy-related 12 (yeast) Autophagy
Atg16l1 Autophagy-related 16-like 1 (yeast) Autophagy
Atg3 Autophagy-related 3 (yeast) Autophagy
Atg5 Autophagy-related 5 (yeast) Autophagy
Atg7 Autophagy-related 7 (yeast) Autophagy
Atp6v1g2
ATPase, H+ transporting, lysosomal V1 subunit
G2
Apoptosis and
Necrosis
Bax Bcl2-associated X protein Apoptosis and
Autophagy
Bcl2 B-cell leukemia/lymphoma 2 Apoptosis and
Autophagy
Bcl2a1a B-cell leukemia/lymphoma 2 related protein A1a Anti-apoptotic
Bcl2l1 Bcl2-like 1 Apoptosis and
Autophagy
Bcl2l11 BCL2-like 11 (apoptosis facilitator) Pro-Apoptotic
Becn1 Beclin 1, autophagy related Autophagy
Birc2 Baculoviral IAP repeat-containing 2 Pro-apoptotic
Birc3 Baculoviral IAP repeat-containing 3 Anti-apoptotic

______________________________________________________________________________
116
Bmf Bcl2 modifying factor Necrosis
Casp1 Caspase 1 Pro-apoptotic
Casp2 Caspase 2 Anti-apoptotic
Casp3 Caspase 3 Apoptosis and
Autophagy
Casp6 Caspase 6 Pro-apoptotic
Casp7 Caspase 7 Pro-apoptotic
Casp9 Caspase 9 Pro-apoptotic
Ccdc103 Coiled-coil domain containing 103 Necrosis
Cd40 CD40 antigen Pro-apoptotic
Cd40lg CD40 ligand Pro-apoptotic
Cflar CASP8 and FADD-like apoptosis regulator Pro-apoptotic
Commd4 COMM domain containing 4 Necrosis
Ctsb Cathepsin B Autophagy
Ctss Cathepsin S Autophagy
Cyld Cylindromatosis (turban tumor syndrome) Apoptosis and
Necrosis
Defb1 Defensin beta 1 Necrosis
Dennd4a DENN/MADD domain containing 4A Necrosis
Dffa DNA fragmentation factor, alpha subunit Pro-apoptotic
Dpysl4 Dihydropyrimidinase-like 4 Necrosis
Eif5b Eukaryotic translation initiation factor 5B Necrosis
Esr1 Estrogen receptor 1 (alpha) Autophagy
Fas Fas (TNF receptor superfamily member 6) Apoptosis and
Autophagy

______________________________________________________________________________
117
Fasl Fas ligand (TNF superfamily, member 6) Pro-apoptotic
Foxi1 Forkhead box I1 Necrosis
Gaa Glucosidase, alpha, acid Autophagy
Gadd45a
Growth arrest and DNA-damage-inducible 45
alpha
Pro-apoptotic
Galnt5 UDP-N-acetyl-alpha-D-galactosamine:polypeptide
N-acetylgalactosaminyltransferase 5
Necrosis
Grb2 Growth factor receptor bound protein 2 Necrosis
Hspbap1 Hspb associated protein 1 Necrosis
Htt Huntingtin Autophagy
Ifng Interferon gamma Autophagy
Igf1 Insulin-like growth factor 1 Autophagy
Igf1r Insulin-like growth factor I receptor Anti-apoptotic
Ins2 Insulin II Autophagy
Irgm1 Immunity-related GTPase family M member 1 Autophagy
Jph3 Junctophilin 3 Necrosis
Kcnip1 Kv channel-interacting protein 1 Necrosis
Mag Myelin-associated glycoprotein Necrosis
Map1lc3a Microtubule-associated protein 1 light chain 3
alpha
Autophagy
Mapk8 Mitogen-activated protein kinase 8 Autophagy
Mcl1 Myeloid cell leukemia sequence 1 Anti-apoptotic
Nfkb1 Nuclear factor of kappa light polypeptide gene
enhancer in B-cells 1, p105
Autophagy
Nol3 Nucleolar protein 3 (apoptosis repressor with
CARD domain)
Pro-apoptotic
______________________________________________________________________________
118
Olfr1404 Olfactory receptor 1404 Necrosis
Parp1 Poly (ADP-ribose) polymerase family, member 1 Necrosis
Parp2 Poly (ADP-ribose) polymerase family, member 2 Necrosis
Pik3c3 Phosphoinositide-3-kinase, class 3 Autophagy
Pvr Poliovirus receptor Necrosis
Rab25 RAB25, member RAS oncogene family Necrosis
Rps6kb1 Ribosomal protein S6 kinase, polypeptide 1 Autophagy
S100a7a S100 calcium binding protein A7A Necrosis
Snca Synuclein, alpha Autophagy
Spata2 Spermatogenesis associated 2 Apoptosis and
Necrosis
Sqstm1 Sequestosome 1 Autophagy
Sycp2 Synaptonemal complex protein 2 Apoptosis and
Necrosis
Tmem57 Transmembrane protein 57 Necrosis
Tnf
Tumor necrosis factor
Apoptosis and
Autophagy
Tnfrsf10b Tumor necrosis factor receptor superfamily,
member 10b
Pro-apoptotic
Tnfrsf11b Tumor necrosis factor receptor superfamily,
member 11b (osteoprotegerin)
Anti-apoptotic
Tnfrsf1a Tumor necrosis factor receptor superfamily,
member 1a
Apoptosis and
Necrosis
Traf2
Tnf receptor-associated factor 2
Anti-apoptotic
Trp53 Transformation related protein 53 Apoptosis and
Autophagy
Txnl4b Thioredoxin-like 4B Necrosis
Ulk1 Unc-51 like kinase 1 (C. elegans) Autophagy
Xiap X-linked inhibitor of apoptosis Anti-apoptotic

______________________________________________________________________________
119
CHAPTER 4
_____________________________________________________________________________________
DEVELOPMENT OF A PORCINE DEEP PARTIAL THICKNESS BURN MODEL
Carmen Gaines*a, Deepika Poranki*
a, Wei Du
b, Richard A. F. Clark
c, Mark Van Dyke
a
a Institute for Regenerative Medicine, Wake Forest School of Medicine, Winston-Salem, NC
27157, USA
b Department of Pathology-Tumor Biology, Wake Forest School of Medicine, Winston-Salem,
NC 27157, USA
c Department of Biomedical Engineering, Stony Brook University, Stony Brook, NY 11794,
USA
* Both Authors contributed equally to this work
This manuscript was submitted for publication to Burns Journal. Stylistic variations are due to
requirements of the journal. Deepika Poranki and Carmen Gaines prepared the manuscript. Mark
Van Dyke acted in an advisory and editorial capacity.

______________________________________________________________________________
120
Abstract:
Swine are the preferred animal models to study the effects of burns on dermal wound healing.
Various studies have been published in which little emphasis was placed on minimizing burn
variability and inconsistency. We developed a novel method to create deep partial thickness
burns that are highly consistent.
A custom‐made burn device was fabricated to control the pressure applied on the swine skin
during burn creation. Cylindrical brass blocks, measuring 3 cm in diameter, are used to create the
burns. A stainless steel post extends from the block for insertion into the device holder. In this
study, burns were created in four female Yorkshire swine. Heating of the brass blocks was
conducted using a boiling azeotropic solution of 80% polyethylene glycol (PEG) and 20% water
and boiling water alone. Contact times ranging from 12 to 20s were used. At 24h and 7d
post‐injury, two swine were euthanized and tissues collected for histological assessment using
Gomori trichrome staining. Histological analysis showed that burns created using boiling water
were superficial compared to those created using the boiling PEG:H2O solution. With a burn
contact time of 20s, 48.5% ± 5.7 tissue damage was demonstrated at 24h when the PEG:H2O
solution was used, whereas only 11.9% ± 1.3 was observed with boiling water.
Keywords: contact burn, polyethylene glycol, azeotropic mixture, deep partial thickness burn,
swine, skin

______________________________________________________________________________
121
1. Introduction
The structural similarities of swine and human skin render swine as preferred subjects for in vivo
burn therapy research [1, 2]. Additionally, the large bodies of swine allow for ample surface area
for multiple wound creation and clinical evaluation [3]. Of the many contact injury models
published [4-6], few studies have considered the incidence of burn wound variability and
inconsistency as a function of burn technique. The use of models with such inherent sources of
error may give an inaccurate picture of the treatment being employed in studies where
differences between treatments is being quantified and statistically compared.
Kempf et al. [7] reported the occurrence of variable deep partial thickness scald burn wounds
upon creation, despite full adherence to a consistent burn procedure. Consequently, reddened,
superficially-burned regions within the wounds healed faster than the surrounding eschar-
covered regions by 14 days post-injury (Figure 1). Papp et al. [8] reported similar difficulties in
burn wound creation, displaying obvious signs of inconsistency in a representative photo (Figure
2). In the aforementioned studies, a single operator created all burn wounds with only the weight
of gravity on the heat source serving as applied pressure. Singer et al. [9] have shown that the
amount of pressure applied on the animal has a direct effect on the intensity and depth of burn.
As such, maintaining a constant pressure every time is important in creating consistent burns.
The observed incidence of burn wound variability suggests that better control over the pressure
applied by the heat source on the skin combined with a more effective heat transfer technique
may produce wound uniformity. A large portion of the published literature describes the use of
boiling water to heat the brass blocks employed in wound creation [8, 10-12]. Boiling water

______________________________________________________________________________
122
produces air bubbles that adhere to the brass blocks and can create non-uniform heating (i.e.
“cold spots”). Uniform thermal energy transfer is then compromised when the brass block is in
contact with the skin of the animal. The cold spots can produce superficially-injured skin regions
that, over time, develop into regenerated ‘islands’ of new skin within the wound at earlier time
points than the rest of the burn. As migration of healthy cells from the periphery of normal skin
to the wounded area is part of the wound healing process, the wounds that have these islands of
less damaged, faster regenerating skin heal faster compared to wounds without islands [13-15].
Our lab has developed a model in which a custom-made, spring-loaded burn device was
fabricated to standardize the pressure applied on the swine skin during burn creation,
independent of the operator. Cylindrical brass blocks served as our heat source for wound
creation. Preliminary evaluations of the brass block temperature upon immersion in boiling water
indicated that the blocks did not thermally equilibrate in the water bath and reach an optimal
temperature. Use of a boiling azeotropic mixture of deionized water and polyethylene glycol
raised the equilibration temperature of the brass blocks and also appeared to eliminate the
formation of cold spots. This allowed for the creation of highly consistent burn wounds and
facilitated the determination of an optimal contact time to achieve the desired burn depth.
2. Materials and methods
0.1. Animal model: This swine study was approved by the Wake Forest University Animal
Care and Use Committee. Six female, Yorkshire swine (Baux-Mountain, Winston-
Salem, NC) weighing 20-25 kg at the time of surgery were used in this study. The
animals were housed in individual pens upon their arrival and allowed to become

______________________________________________________________________________
123
acclimated for at least 7 days. The animals were washed and shaved one day prior to
surgery, at which time they were pretreated with a transdermal fentanyl patch (50mcg/h)
for pain management. While on study, the animals were fasted each night prior to the
administration of anesthesia.
0.2. General anesthesia and surgical monitoring: Anesthesia was induced intramuscularly
with ketamine (10mg/kg) and dexmedetomidine (0.05mg/kg), and was later continued
with isoflurane during surgery. The previously applied fentanyl patch was removed and
a new patch (75mcg/h) applied during surgery, which was replaced every 3 days. Blood
pressure, heart rate and body temperature were monitored during surgery for any
complications. Ten burn wounds were to be created with 3 cm diameter cylindrical brass
blocks, 5 on each side of the mid-dorsal line between the shoulder and the hip.
0.3. Wound creation
2.3.1 Pressure-controlled burn device: Cylindrical brass blocks (360 grade), measuring 3 cm
in diameter and 5 cm in height, were used to create the burn wounds (Figure 3). The
blocks were heated in a boiling solution until used. A stainless steel post protruded from
the top of each block to allow for easy insertion into the burn device holder, made of
insulative Delrin®. An adjustable spring was housed within the otherwise hollow body of
the holder to enable a consistent force response when the block was pressed onto the
animal’s body (Figure 4). During burn creation, the operator positioned the device holder
over the brass block in solution, dislodging a pin connector to allow for insertion of the
steel post into the holder. Release of the pin connector ensured secure locking of the
block within the holder. The holder was then used to remove the block from solution,
residual liquid was quickly removed with a towel, and the block placed onto the animal’s

______________________________________________________________________________
124
skin in a randomly assigned location. Enough force was applied so that the bottom of the
device holder was flush with the brass block, allowing for a consistent block pressure
controlled by the spring. Prior to use on animal subjects, the burn device was tested for
operational consistency by two separate researchers using pressure-sensitive film (Sensor
Products Inc., Madison, NJ). This was further verified under experimental conditions
through visual observation within the current study. A total of 3 identical brass blocks
were rotated for use during the burn surgery to assure temperature equilibration and
minimize the amount of time needed to maintain the animal under general anesthesia.
2.3.2 Metal block surface temperature validation: Initial studies conducted by our lab have
shown that the brass blocks reached a temperature range of only 88‐92°C when
equilibrated in boiling water. A potential solution was to raise the temperature of the
water through the addition of PEG. An 80:20 ratio of PEG:H2O produced a negative
azeotropic mixture that raised the boiling temperature to 115-125°C. Temperature of the
bottom surface of the brass blocks after equilibration in the boiling liquids was tested
using temperature sensing strips (Omega Engineering, Stamford, CT). An unanticipated
effect of the use of this azeotropic liquid was its wetting characteristics and the potential
to reduce the prevalence and adherence of vapor bubbles on the under side of the brass
blocks. As the azeotropic solution contained mostly PEG, we evaluated the difference in
surface tension compared to water alone. A contact angle meter (KSV Cam100
Instruments Ltd, Helsinki, Finland) was used to determine the average contact angle
measurements of water and the azeotropic PEG:H2O mixture on polished brass metal
coupons. The coupons were of the same composition and surface finish as the brass
blocks used with the burn device. Contact angles were measured in 5 places along the

______________________________________________________________________________
125
diameter of the metal coupons and the difference between average values was evaluated
using a Student’s T-test with p<0.05 considered significant.
2.3.3 Burn wound creation: Two Yorkshire swine were used for each boiling solution - water
alone and the 80:20 PEG:H2O mixture. Ten burn wounds were created on each animal
using the custom-made device. Brass block contact times from 12-20s, in 2s increments,
were used in wound creation. Two replicate burns were created for each contact time and
were randomly assigned locations on the animal. Sixty minutes after burning, the wounds
were covered with saline soaked non-adhesive gauze (Telfa, Tyco Healthcare, Mansfield,
MA) followed by an occlusive adhesive dressing (IobanTM
2, 3M, St.Paul, MN), a
protective plastic shield and a nylon jacket to keep the dressings in place. Atipamezole
(0.05mg/kg) was administered intramuscularly to reverse the effect of the
dexmedetomidine post-surgery. Two animals were euthanized after 24h and the wounds
were harvested for further histological analysis. The other two animals were euthanized
after 7days. Dressing changes were performed every 3 days for the 7 day animals.
2.4 Data acquisition
2.4.1 Digital wound capture: Digital pictures were taken at 0, 1, 3 and 7 days post-surgery
(Nikon-D90, Nikon Inc., Melville, NY) to document the presence or absence of islands of
normal tissue. A ruler and color wheel were located within each photograph for later
processing of the image.
2.4.2 Histology: The entire wound was excised from the animal at the designated time point
and half of the tissue was fixed in 10% neutral buffered formalin for 2 days. After
fixation, a tissue cross section closest to the center of the wound was cut and processed in
70% isopropyl alcohol (IPA), 80% IPA, 95% IPA, 100% IPA and xylene (Leica

______________________________________________________________________________
126
ASP300S Tissue processor, Buffalo Grove, IL) overnight. The tissue was embedded in
paraffin and cut in 5 �m sections using a microtome (Leica CM 1850, Leica
Microsystems Inc., Buffalo Grove, IL) and stained with Gomori trichrome.
Representative sections of each wound were evaluated by an experimental pathologist
blinded to the protocol. At pre-determined distance intervals from the edge of the wound,
tissue morphology was scored according to the following scale:
Epidermis
0 Normal
1 Flattening of the dermal-epidermal (DE) junction
2 Flattening and disruption of the DE junction
3 Absence of epidermis
Dermis
0 Normal
1 Loss of collagen architecture (papillary dermis)
2 Loss of collagen architecture and dye intensity color change
3 Loss of collagen architecture in reticular dermis (mid dermis)
4 Loss of collagen (full thickness)
Digital images of stained tissue sections were captured (Axio Imager M1 Microscope,
Carl Zeiss USA, Thornwood, NY) and the burn areas (i.e. demonstrating infiltration of
inflammatory cells, edema, and damaged collagen) and total area of the skin from the
epidermis down to the dermis-fat junction were measured (ImageJ, National Institutes of
Health). Total percent tissue loss was determined by calculating the area of burn (as
indicated by a change from green to red color and denaturation of the collagen bundles

______________________________________________________________________________
127
[16]) divided by the total area of the skin. Average percent tissue loss was calculated
from two replicate wounds and reported as mean ± standard deviation.
3 Results
3.1 Brass block temperature validation and surface chemistry: The brass blocks heated in
boiling azeotropic mixture of 80:20 PEG:H2O reached a temperature range of 99‐103°C
(Figure 5B), whereas the blocks heated with water only reach 88-92oC (Figure 5A). The
contact angle analysis showed a relatively lower average contact angle for the PEG:H2O
mixture compared to water alone (Figure 6). During heating, it was observed that vapor
bubbles did not adhere to the surface of the brass blocks.
3.2 Visual differences due to heating method: When water alone was used, burn wounds can
be uneven as shown in other studies. Islands of superficially-burned skin are evident,
causing an unwanted increase in burn edges that would be expected to affect the rate of
healing. These islands presumably arise from the presence of vapor bubbles that adhere to
the bottom of the brass blocks, thereby insulating the surface and creating cold spots. The
PEG:H2O azeotropic mixture has low surface tension compared to water alone, creating a
continuous liquid interface along the bottom of the block surface. In addition, the PEG:H2O
mixture does not boil as vigorously as water alone, resulting in noticeably fewer vapor
bubbles. The combination of fewer bubbles and better wetting mitigated the formation of
cold spots and created uniform heating when the PEG:H2O azeotrope was used. This
difference can be seen visually with digital pictures of wounds taken on days 0, 3 and 7
post-surgery (Figure 7). Wounds created by two separate operators and using the PEG:H2O
azeotropic mixture showed greater consistency (Figure 8), as verified by an initial
experiment using pressure-sensitive film (data not shown).

______________________________________________________________________________
128
3.3 Histological analysis
3.3.1 Wound variability evaluation: Wound scoring data for day 1 is shown graphically in
Figure 9. Each bar represents the mean value for contact time with the burning device,
and the error bars provide a measure of the variance seen between the two replicate burns
(i.e. large relative error bars indicate larger variance). As can be seen in the graphs, the
damage created by the water heated blocks (Figure 9A) was relatively less severe than
those created by the PEG:H2O heated blocks (Figure 9B) in the epidermis. In addition,
the PEG:H2O burns were more consistent as suggested by the small error bars. Evaluation
of the dermis showed similar results (Figures 9C and 9D) in that the PEG:H2O burns
were relatively more severe and more consistent across replicates and contact times. By
day 7, the epidermis in both groups was necrotic and had been completely removed by
debridement during dressing changes. In the dermis, the extent of damage in the
PEG:H2O burns was extensive as these wounds had progressed to nearly full thickness in
most cases and were not scored, while the water burns showed only modest damage and
continued variability (scoring data not shown).
3.3.2 Percent dermal tissue damage: Gomori trichrome staining was used to measure the
burn depth. When water alone was used for wound creation, the burns were more
superficial, whereas the PEG:H2O heating system created a more severe partial thickness
burn (Figure 10). Importantly, there was no correlation of burn depth with the contact
time as has been reported in earlier studies [8]. When the PEG:H2O mixture was used, the
burns were more consistent and deep and there was a measurable correlation between the
contact time and intensity of the burn. For a 20s contact time, 48.5% ± 5.7 of tissue loss

______________________________________________________________________________
129
was observed with the PEG:H2O boiling solution but only 11.9% ± 1.3 tissue loss was
observed with boiling water alone (Figure 11).
4. Discussion
The creation of highly-reproducible burn wounds is essential in the assessment of healing
progression in therapeutic models. Researchers have recognized the need to create consistent
burn wounds, but have cited the change in underlying tissue structure of the animal as a
hindrance. While we acknowledge the inherent variability encountered in working with animal
subjects, the present model offers control over variability through two simple means, a burn
device that creates operator-independent pressure, and a heating system that ensures consistent
and uniform heating of the brass blocks. Several other groups have limited their attempts at
controlling variability to designating a single operator to create burn wounds during surgery.
While this method has led to slightly better control, this study demonstrates that a higher level of
control can be achieved. Through the customized design of our spring-loaded device, any
operator can create the burn wounds without concern for uneven pressure when applied
appropriately. Pressure applied from the operator is only transferred along the periphery of the
device holder, where the device holder hits the animal’s skin along its insulated bottom surface.
This portion of the holder does not directly encounter the hot bath and does not achieve a
temperature as high as the actual brass block. In addition, the device and mode of operation is
potentially less hazardous to users than some other methods such as scalding [7].
The use of the PEG:H2O azeotrope improved the heat transfer efficiency within our
system. Moreover, the azeotropic mixture appears to maintain a contiguous coating along the
contact surface of the brass blocks, even in the presence of boiling fluid. Water vapor bubbles
adhere more strongly to the brass surface and are more difficult to dislodge through wetting

______________________________________________________________________________
130
because of the hydrophobicity of the brass block. The lower surface tension of the PEG:H2O-
brass interface minimizes the insulating effects of adhered vapor bubbles and thus the formation
of cold spots. If such uneven heating is present, a more superficial burn is created in this area,
accounting for the ‘islands’ that many researchers have encountered. The use of a non-toxic,
biocompatible polymer such as PEG for thermal burn models not only increases overall
maximum temperature, but provides an added benefit of ensuring more even distribution of heat
to allow for more consistent burning and the elimination of lateral and depth temperature
gradients. PEG is used in a variety of applications such as coating the surfaces of nanoparticles
to increase the amount of surface active reactive groups [17], and in many cosmetic products
[18]. It has also been used to neutralize the residual phenol left in a chemical burn [19-21].
Therefore it is unlikely that residual PEG on the brass block surface would induce a chemical
burn or toxicity tissue reaction.
Histological analysis with Gomori trichrome staining showed that using PEG:H2O as a
heating solution created more consistent and deep burns compared to boiling water alone. For a
20s burn, more tissue loss was observed with PEG:H2O compared to water after 24 hours. One of
the possible reasons might be that the brass blocks are not being heated to 100oC with water
alone, as shown by other investigators [8]. Also, given the dimensions of the brass blocks used in
this study, they did not provide an adequate heat sink for 20s when the temperature reached
using water alone was only 88-92oC. This is demonstrated by the fact that we expected to see
more superficial burns with a contact time of 12s compared to 20s. However, when boiling water
was used the burns were more variable overall and the 20s burns were more superficial
compared to the 12s burns.

______________________________________________________________________________
131
There are many variables that need to be considered when creating a burn wound, and
controlling those variables will help in creating a consistent burn. Several prior studies have
discussed the use of animals of similar size/age, controlling contact time, and using the same
operator. Randomization and the use of a large number of replicate wounds is also advocated.
The need for randomization is evident in the data for the 18s contact time in the water only group
where the percent burn was unusually high. Despite randomly locating all wounds, these two
burns ended up over the abdomen, which likely contributed to a high value for this test condition.
Heating brass blocks to a consistently high temperature and avoiding the formation of
cold spots are also important considerations in developing a burn model with consistent, efficient
thermal energy transfer. This study demonstrates that these aspects can be addressed through the
application of simple surface chemistry and thermodynamic principles.
Conflict of interest statement
Mark Van Dyke, Ph.D. holds stock and is an officer in the company, KeraNetics LLC, which has
provided partial funding for this research. Wake Forest Health Sciences has a potential financial
interest in KeraNetics, LLC through licensing agreements.
Acknowledgements
This work is supported by the Armed Forces Institute for Regenerative Medicine (DoD Contract
# W81XWH-08-2-0032) and KeraNetics LLC.
The authors would like to thank Christina Ross and Mary Ellenburg (WFIRM) for their
assistance with the burn surgeries.

______________________________________________________________________________
132
References:
1. Meyer W, Schwarz R, Neurand K. The skin of domestic mammals as a model for the
human skin, with special reference to the domestic pig. Curr Prob Dermatol 1978; 7:39-52.
2. Singer AJ, McClain SA. A porcine burn model. In: DiPietro LA, Burns, AL, editors.
Methods in Molecular Medicine, New Jersey: Humana Press Inc; 2003, p. 107-119.
3. E.Middelkoop, A.J. van den Bogaerdt, E.N. Lamme, M.J. Hoekstra, K.Brandsma,
M.M.W.Ulrich. Porcine wound models for skin substitution and burn treatment.
Biomaterials 25 (2004) 1559–1567.
4. Liu P, Deng Z, Han S, Liu T, Wen N, Lu W, Geng X, Huang S, Jin Y. Tissue-engineered
skin containing mesenchymal stem cells improves burn wounds. Artif Organs 2008;
32(12):925-931.
5. Branski L, Mittermeyr R, Herndon DN, Norbury WB, Masters OE, Hofmann M, Traber
DL, Redl H, Jeschke MG. A porcine model of full-thickness burn excision and skin
autografting. Burns 2008; 34:1119-1127.
6. Shi L, Ermis R, Lam K, Cowart J, Attar P, Aust D. Study on the debridement efficacy of
formulated enzymatic wound debriding agents by in vitro assessment using artificial wound
eschar and by in vivo pig model. Wound Rep Reg 2009; 17:853-862.
7. Kempf M, Cuttle L, Liu P-Y, Wang X-Q, Kimble RM. Important improvements to porcine
skin burn models, in search of the perfect burn. Burns 2009; 35:454-455.
8. Papp A, Kiraly K, Harma M, Lahtinen T, Uusaro A, Alhava E. The progression of burn
depth in experimental burns: a histological and methodological study. Burns 2004; 30:684-
690.

______________________________________________________________________________
133
9. Adam J. Singer, Breena R. Taira, Ryon Anderson, Steve A. McClain, Lior Rosenberg.
Does Pressure Matter in Creating Burns in a Porcine Model? Journal of Burn Care &
Research July/August 2010.
10. White J, Maass DL, Giroir B, Horton JW. Development of an acute burn model in adult
mice for studies of cardiac function and cardiomyocyte cellular function. Shock. 2001
Aug;16(2):122-9.
11. Singer AJ, Taira BR, Anderson R, McClain SA, Rosenberg L. The effects of rapid
enzymatic debridement of deep partial-thickness burns with Debrase on wound
reepithelialization in swine. J Burn Care Res. 2010 Sep-Oct;31(5):795-802.
12. Simonetti O, Cirioni O, Lucarini G, Orlando F, Ghiselli R, Silvestri C, Brescini L, Rocchi
M, Provinciali M, Guerrieri M, Di Primio R, Giacometti A, Offidani A. Tigecycline
accelerates staphylococcal-infected burn wound healing through matrix metalloproteinase-
9 modulation. J Antimicrob Chemother. 2012 Jan;67(1):191-201. Epub 2011 Nov 6.
13. Vogot PM, Ericksson E. Current aspects of epidermal healing. Handchir Mikrochir Plast
Chir. 1992 Sep;24(5):259-66.
14. Fan X, Liang HP. Circulating fibrocytes: A potent cell population in antigen-presenting and
wound healing. Chin J Traumatol. 2010 Apr 1;13(2):111-6.
15. Li L, Jiang J. Regulatory factors of mesenchymal stem cell migration into injured tissues and
their signal transduction mechanisms. Front Med. 2011 Mar;5(1):33-9. Epub 2011 Mar 17.
16. Chvapil M, Speer DP, Owen JA, Chvapil TA. Identification of the depth of burn injury by
collagen stainability. Plast Reconstr Surg. 1984 Mar;73(3):438-41.

______________________________________________________________________________
134
17. Tong S, Hou S, Ren B, Zheng Z, Bao G. Self-assembly of phospholipid-PEG coating on
nanoparticles through dual solvent exchange. Nano Lett. 2011 Sep 14;11(9):3720-6. Epub
2011 Aug 1.
18. Claudia Fruijtier-Pölloth. Safety assessment on polyethylene glycols (PEGs) and their
derivatives as used in cosmetic products. Toxicology Volume 214, Issues 1-2, 15 October
2005.
19. Horch R, Spilker G, Stark GB. Phenol burns and intoxications. Burns. 1994 Feb;20(1):45-
50
20. Brown VK, Box VL, Simpson BJ. Decontamination procedures for skin exposed to
phenolic substances. Arch Environ Health. 1975 Jan;30(1):1-6.
21. Monteiro-Riviere NA, Inman AO, Jackson H, Dunn B, Dimond S. Efficacy of topical
phenol decontamination strategies on severity of acute phenol chemical burns and dermal
absorption: in vitro and in vivo studies in pig skin. Toxicol Ind Health. 2001 May;17(4):95-
104.

______________________________________________________________________________
135
Figure 1. Differences in healing of deep partial thickness scald burns due to inconsistent
burning. Consistent, deep partial thickness burn wound (A) compared to a wound containing red,
superficially-burned areas (inside dashed lines; B) will heal more uniformly (C) compared to the
superficially-burned areas which were healed by day 14 (inside dashed lines; D). Used with
permission [7].

______________________________________________________________________________
136
Figure 2. Burns with non-uniform severity created with brass block heated in boiling water
along the right flank of the animal. A burn wound created with a contact time of 12s is indicated
by the arrow. Used with permission [8].

______________________________________________________________________________
137
Figure 3. 360 grade, 3 cm diameter brass block used for burn wound creation. Notched stainless
steel post extends from block for easy insertion into the device holder.

______________________________________________________________________________
138
Figure 4. Burn device operation. The brass block is held within the device holder by engaging
a pin connector at the notched end. Upon depression of the fully-extended assembly, the spring
contracts exerting an even force onto the block with no extraneous forces from the operator.

______________________________________________________________________________
139
Figure 5. Heated brass blocks with temperature sensors. Equilibrium temperatures of the
block contact surfaces upon heating in boiling water alone (A; up to 88-92oC; arrow) and the
PEG:H2O azeotropic mixture (B; 99-103oC; arrow).
H2O Only PEG:H2O

______________________________________________________________________________
140
Figure 6. Contact angle analysis using water and PEG:H2O azeotropic mixture to determine
surface wetting. The addition of PEG caused a significant drop in surface tension and
consequently, static contact angle (n=5, p<0.05, Student’s T-test).
Water Only 80:20 PEG:H2O0
20
40
60
80
100**
Mean Conta
ct Angle

______________________________________________________________________________
141
Figure 7. Healing progression of wounds with and without islands. The presence of
superficially burned skin in the lower left of the water heated block would be expected to heal
more non-uniformly. The PEG:H2O azeotropic mixture avoided the island formation and created
a more uniform burn.

______________________________________________________________________________
142
Figure 8. Visually consistent burn wounds created by using boiling PEG:H2O azeotropic
mixture. Each row of wounds was created by a different operator.

______________________________________________________________________________
143
Water Only
PEG:H2O
Figure 9. Histological scoring data from day 1 wounds. Wounds created using water heating
caused less severe damage to the epidermis and were more variable (A) compared to wounds
created using PEG:H2O (B). This trend continued in the dermis (C and D, respectively).
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
0‐3mm 3‐6mm 6‐9mm 9‐12mm 12‐15mm 15‐18mm
His
tolo
gy S
co
re o
f E
pid
erm
is
Distance from Center of Wound
12 sec
14 sec
16 sec
18 sec
20 sec
A B
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
0‐3mm 3‐6mm 6‐9mm 9‐12mm 12‐15mm 15‐18mm
His
tolo
gy S
core
of
Ep
ide
rmis
Distance from Center of Wound
12 sec
14 sec
16 sec
18 sec
20 sec
C
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
0‐3mm 3‐6mm 6‐9mm 9‐12mm 12‐15mm 15‐18mm
His
tolo
gy S
core
of
De
rmis
Distance from Center of Wound
12 sec
14 sec
16 sec
18 sec
20 sec
D
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
0‐3mm 3‐6mm 6‐9mm 9‐12mm 12‐15mm 15‐18mm
His
tolo
gy S
core
of
De
rmis
Distance from Center of Wound
12 sec
14 sec
16 sec
18 sec
20 sec

______________________________________________________________________________
144
Figure 10. Tissue sections stained with Gomori trichrome stain showing a variation in the
burn depth for a 20s contact burn after 24h when PEG:H2O azeotropic mixture and boiling water
alone were used – Boiling water alone has created superficial burn compared to PEG:H2O

______________________________________________________________________________
145
Figure 11. Percent dermal tissue damage. For a 20s contact time, 48.5% ± 5.7 tissue loss was
observed using PEG:H2O boiling solution for heating whereas only 11.9% ± 1.3 tissue loss was
observed with boiling water after 24h.
PEG
Water
0
20
40
6020sec
18sec
16sec
14sec
12sec
% Tissue Loss
Time
% T
issue Loss

______________________________________________________________________________
146
CHAPTER 5
_____________________________________________________________________________________
ASSESSMENT OF DEEP PARTIAL THICKNESS BURN TREATMENT WITH
KERATIN BIOMATERIAL HYDROGELS IN A SWINE MODEL
Poranki Da, Gaines C
a, Van Dyke M
a
aWake Forest School of Medicine, Wake Forest Institute for Regenerative Medicine
Wake Forest School of Medicine, Medical Center Boulevard, Winston Salem, NC 27157
The following manuscript has not yet been submitted for publication. Deepika Poranki prepared
the manuscript. Dr. Mark Van Dyke acted in an advisory and editorial capacity.

______________________________________________________________________________
147
Abstract:
Partial thickness burns can advance to full thickness burns due to inadequate tissue
perfusion and increased production of inflammatory cytokines, even with proper hospitalization
and treatment. This advancement of wound severity in the first several days after injury has been
referred to as burn wound progression and is the result of a cascade of events that results in
further tissue loss in the periphery of the original burn. There are currently no commercially
available treatments that can control burn wound progression. In our previous work (Chapters 2
and 3), we demonstrated that a keratin biomaterial hydrogel placed over a burn wound appeared
to reduce progression and that this may be due to a particular fraction of keratin, gamma
keratose, that showed the ability to regulate cell death pathway genes.
In the present thermal burn study, we tested the hypothesis that a modified formulation of
keratin, hereafter referred to as modified keratose hydrogel or MKH, which contained a reduced
amount of the gamma keratose fraction would heal wounds faster compared to the original
formulation of crude keratose. Crude keratose was used in the original pilot studies and was
included here as a control, as well as a collagen hydrogel (Coloplast®) and silver sulfadiazine
cream (SSD; clinical standard of care).
Fourteen female Yorkshire swine weighing 20-25 kg were used in the study.
Standardized burn wounds were created and treated within 60 minutes. Two animals were
euthanized on days 1, 3, 6, 9, 12, 15 and 30; their tissues collected. Digital images of the wounds
immediately prior to sacrifice were captured and analyzed using ImageJ software. The tissue
sections were stained and analyzed for burn depth, granulation of tissue thickness and re-
epithelialization.

______________________________________________________________________________
148
Wound assessment from digital images showed a significant difference between the
MKH and SSD on days 9 (p<0.05), 12 (p<0.001) and 15(p<0.05). Significant differences were
also found between crude keratose and SSD on days 12 (p<0.001) and 15 (p<0.05), and between
Coloplast and SSD on days 9 and 12(p<0.01). Rates of re-epithelialization at early time points
showed a 220% increase for the MKH compared to SSD. A Linear regression model predicted
that MKH and crude keratose had a time to wound closure of 19 days while Coloplast and SSD
treatments took 24 days. However, these data were not statistically significant due to high
variability in the SSD treated wounds. Measurements of burn depth and granulation tissue
thickness showed that by day 9, vertical burn progression had occurred in all wounds. Moreover,
crude keratose treatment appeared to result in a relatively smaller increase in burn depth (5.56%)
compared to other treatments (SSD - 9.81%, MKH - 20.01%, Coloplast - 22.5%). SSD treatment
appeared to show a higher difference (1.115cm) in the thickness of granulation tissue from days
9 to 15 compared to other treatments, while crude keratose appeared to show the least difference
(0.746cm). Again, no statistically significant differences between treatment groups were noted.
These results did demonstrate a smaller wound area with treatment using the MKH at mid-time
points during healing, and appeared to show a faster healing rate for this biomaterial compared to
the standard of care, SSD. However, the inconsistency of healing in the SSD treated wounds
contributed to a loss of statistical significance in several outcome measures. Additional studies
using keratin biomaterials that more specifically monitor burn wound progression, preferably in
a non-spontaneously healing burn injury model, are warranted.
Keywords: keratin biomaterial, deep partial thickness burns, hydrogel, crude keratose, alpha
keratose, gamma keratose, collagen, silver sulfadiazine, re-epithelialization, granulation tissue.

______________________________________________________________________________
149
1. Introduction:
Burns are one of the most catastrophic injuries to treat. Often, partial thickness burns
convert to full thickness burns due to inadequate tissue perfusion and increased production of
inflammatory cytokines leading to protein denaturation and necrosis [1]. The phenomenon of
burn injury progression occurs when events downstream of the original damage cause further
tissue loss. As a result, wounds increase in surface area and deepen, progressing from second
degree to third degree burns and increasing overall total body surface area (TBSA) burned,
thereby increasing risk to the patient, healing time, and treatment expense. In 1953, Jackson
described a model of burn injury in terms of three zones of tissue damage: 1) the inner zone of
coagulation, which is characterized by necrotic tissue, 2) an intermediate zone of stasis, which is
characterized by damaged cells that may live or die depending on treatment and other factors,
and 3) an outer zone of hyperemia where cells are stressed but will likely survive if the wound
does not get infected and standard treatment is given [2].
Superficial and partial thickness burns heal from the remaining epithelial structures in the
dermis [3], so the zone of stasis represents an opportunity for treatment as these cells are capable
of survival if an appropriate therapy can be developed that increases their survival, yet limited
clinical research has been directed in this area. In vivo studies however, have shown that
treatments like cerium nitrate, curcumin, activated protein C (APC), recombinant tissue type
plasminogen (r-TPA), TAK-044 (nonselective endothelin receptor antagonist), simvastatin, and
beraprost sodium (prostaglandin I2 analogue) were able to control burn injury progression in rats
by preventing tissue necrosis in the zone of stasis [4-11]. In general these studies have made use
of drug compounds that require intravenous or enteric administration, thus complicating their

______________________________________________________________________________
150
clinical development. Topical application of an efficacious therapeutic agent represents a
simplified, local treatment modality.
Previous studies (Chapter 2) in our lab have shown that crude keratose, which is a
heterogeneous mixture composed of alpha keratose, gamma keratose and keratin associated
proteins (KAPs) fractions increased tissue sparing and enhanced wound healing in both mice
chemical and swine thermal burn studies. Another study in an in vitro thermal injury model using
primary mouse dermal fibroblasts showed that the gamma keratose fraction was able to maintain
increased cell viability compared to the other treatments post thermal stress (Chapter 2). Finally,
gamma keratose was shown to salvage the cells by down regulating many genes involved in cell
death pathways (Chapter 3). Mass spectrometry analysis identified the major constituents of the
gamma fraction as keratins 81, 83, 85, 86, 31, 33A, 33B, 34 with lower than expected molecular
weights, suggesting that these were degraded alpha keratin proteins (Chapter 2). As such, they
represent peptides with biological function different than that of their parent molecules. In the
present study, we hypothesized that the peptides found in the gamma keratose fraction would
provide a therapeutic benefit when used in a topical keratin biomaterial hydrogel formulation.
This postulate was tested by comparing a “crude” keratose hydrogel with a re-formulated
hydrogel, modified keratose hydrogel or MKH, that contained a reduced amount of gamma
keratose, in a swine burn injury model. The amount of gamma keratose was reduced to reflect
the concentration at which gamma keratose was effective in the heat stress cell culture model. A
collagen hydrogel, Coloplast®, and the clinical standard of care, silver sulfadiazine cream (SSD)
were used as controls.

______________________________________________________________________________
151
2. Methods:
2.1. Keratin Protein Extraction: Keratin protein was extracted from human hair using the
oxidative protocol specified by de Guzman et al. [12]. The extracted keratin is referred
to as crude keratose.
2.2. Separation of Alpha and Gamma Keratose Fractions: Crude keratose was further
separated into alpha and gamma keratose fractions by isoelectric precipitation.
Concentrated hydrochloric acid was added dropwise to the crude keratose solution with
stirring until a pH 4.2 was reached. At this pH, the alpha keratose precipitates leaving
the acid-soluble gamma fraction in solution. The insoluble alpha keratose was separated
by fixed angle centrifugation at 1500 rpm (252 RCF) for 15min at 4ºC. After
neutralizing the gamma-containing supernatant to pH 8.4, it was dialyzed against
endotoxin free water using a 5kDa spiral wound cartridge (Millipore, Billerica, MA) for
5 volume washes. Finally, the solution was concentrated to minimal volume, pH
adjusted to 7.4, lyophilized, and sterilized by gamma irradiation at 1 MRad prior to use.
The precipitated alpha keratose was re-dissolved in 0.1M sodium hydroxide (NaOH)
solution and dialyzed against endotoxin free water using a 5kDa spiral wound cartridge
(Millipore) for 5 volume washes. Finally, the solution was concentrated to minimal
volume, pH adjusted to 7.4, lyophilized, and sterilized by gamma irradiation at 1 MRad
prior to use.
2.3. Hydrogel Preparation: The MKH was prepared using a proprietary mixture of alpha
and gamma keratose powder and reconstituted with phosphate buffered saline (PBS) to
form a hydrogel. Crude keratose powder was similarly reconstituted with PBS to form a
15% w/v hydrogel. The gels were made under sterile conditions, centrifuged to remove

______________________________________________________________________________
152
air bubbles and incubated in warm room (37ºC) overnight on a shaker. Gels were loaded
into syringes for easy application onto the burn wounds. Collagen hydrogel
(Woun’Dress® Collagen Hydrogel, Coloplast) and SSD (Watson Pharmaceuticals) were
obtained from commercial sources and used as received.
2.4. Swine burn study: The swine study was approved by the Wake Forest University
Animal Care and Use Committee (ACUC). Fourteen female, Yorkshire swine (Baux-
Mountain, Winston-Salem, NC) weighing 20-25 kg at the time of surgery were used in
this study. The animals were housed in individual pens upon their arrival and allowed to
become acclimated for at least 7 days.
2.5. General Anesthesia and Monitoring: The animals were washed and shaved one day
prior to the surgery, at which time they were pretreated with a transdermal fentanyl patch
(50mcg/h) for pain management. While on study, the animals were fasted each night
prior to the administration of anesthesia. Anesthesia was induced intramuscularly with
ketamine (10mg/kg) and dexmedetomidine (0.05mg/kg). Isoflurane was used to maintain
the animal in an anesthetic state during the surgery and during dressing changes. The
previously applied fentanyl patch was removed and a new patch (75mcg/h) was applied
during surgery, which was replaced every 3 days. Blood pressure, heart rate and body
temperature were monitored during surgery for any complications.
2.6. Burn Wound Creation: Wounds were created using a pressure controlled burn device
(described in Chapter 4). This spring loaded device was made of insulative Delrin®.
Cylindrical brass blocks (360 grade), measuring 3 cm in diameter and 5 cm in height,
were used to create the burn wounds. The blocks were heated in a boiling solution of
polyethylene glycol (PEG; Mn 400; Sigma-Aldrich) and deionized (DI) water. A

______________________________________________________________________________
153
stainless steel post protruded from the top of each block to allow for easy insertion into
the burn device holder. A total of three identical brass blocks were rotated for use during
the burn surgery to assure temperature equilibration and minimize the amount of time
needed to maintain the animal under general anesthesia. As described previously in
Chapter 4, twelve burns were created on each swine, six on either side of the mid dorsal
line between shoulder and the hip. For each time point, twenty four wounds (on two
swine) were randomly assigned to four treatment groups and treatments were
administered 60 minutes after creating burns. Treatments included Coloplast (Collagen
Hydrogel), Crude Keratose, MKH, and SSD. Each treatment had six replicates.
Following the treatments, the wounds were covered with saline soaked non-adhesive
gauze (Telfa, Tyco Healthcare, Mansfield, MA), followed by an occlusive adhesive
dressing (IobanTM 2, 3M, St.Paul, MN), a fabric stocking, a protective plastic shield,
and a nylon jacket to keep the dressings in place and prevent the animal from rubbing
directly on the wounds. Atipamezole (0.05mg/kg) was administered intramuscularly to
reverse the effect of the dexmedetomidine after surgery. Two animals were euthanized at
1, 3, 6, 9, 12, 15 and 30 days after the surgery and tissues harvested for histological
analysis after euthanasia on those respective days. Dressing changes were performed
every three days.
2.7. Digital wound capture: Digital pictures were captured on 0, 1, 3, 6, 9, 12, 15, 21 and 30
days post-surgery during the dressing changes, immediately after cleaning and debriding
the wounds (Nikon-D90, Nikon Inc., Melville, NY). All the images were taken with the
camera placed exactly at the same distance and at the same angle (90º, perpendicular) to
the wound. A ruler and grey scale were located within each photograph for later

______________________________________________________________________________
154
processing of the image. Wound area was measured using ImageJ software (National
Institutes of Health, Bethesda, MD). These data were represented as mean ± standard
deviation.
2.8. Histology: The entire wound was excised from the animal at the designated time point
and half of the tissue was fixed in 10% neutral buffered formalin for 2 days. After
fixation, a tissue cross section closest to the center of the wound was cut and processed
in 70% isopropyl alcohol (IPA), 80% IPA, 95% IPA, 100% IPA and xylene (Leica
ASP300S Tissue processor, Buffalo Grove, IL) overnight. The tissue was embedded in
paraffin, cut in 5 µm sections using a microtome (Leica CM 1850, Leica Microsystems
Inc., Buffalo Grove, IL), and stained with hemotoxylin and eosin (H&E). Representative
sections of each wound were evaluated histomorphometrically by a blinded reviewer.
Burn depth measurement: Digital images of stained tissue sections (1.5cm in length)
were captured (Axio Imager M1 Microscope, Carl Zeiss USA, Thornwood, NY). The
thickness of the normal skin section from the epidermis down to the dermis-fat junction
and the residual dermis in the burned region were measured. An average of three
measurements was taken, one at the burned-normal dermis junction, one in the center of
the burned region, and another on the opposite end of the burned region. Burn depth was
determined by calculating the difference of the thickness of the residual dermis in the
burned region from the thickness of the normal skin section.
Re-epithelialization measurement: The burned region without epithelial cell coverage
(defined as at least one layer of cells staining dark pink) was measured from the stained
tissue sections. The percent re-epithelialization was determined by subtracting the burned
region from the measured radius of the wound (as determined from the digital image for

______________________________________________________________________________
155
the same wound at that time point). Re-epithelialization data was analyzed in two phases:
Early time points that included day 3 and 6, and later time points that included day 9, 12
and 15. This was done because the re-epithelialization curves demonstrated two phase
healing behavior, suggesting that wound healing rates were different between these two
time ranges and that distinct phases of healing (tissue salvage and cell survival followed
by matrix deposition and tissue remodeling) were present.
Granulation tissue thickness measurement: The thickness of the new granulation tissue
(characterized by connective tissue and blood vessels stained pink with H&E) was
determined by taking an average of three measurements obtained throughout the length of
the stained tissue sections as previously described.
3. Data Collection and Statistical Analysis: For visual wound assessment digital images,
wound size values for each treatment group were averaged (e.g. n=36 on day 3, n=30 on day
6, n=24 on day 9, etc.) and reported as mean ± standard deviation (SD). For burn depth, re-
epithelialization and granulation tissue assessment, values for each treatment group from six
replicates were averaged and represented as mean ± standard deviation (SD). Two-way
ANOVA and Bonferroni analyses were performed using Graph pad Prism at 95% confidence
intervals for all the data analyses.
4. Results:
4.1. Visual wound assessment: The eschar (dead skin) typically sloughs off completely
within 9-12 days, so re-epithelialization was observed most distinctly in the digital
images of the wounds from 9 to 30 days. To this end, image analysis data showed a
significant difference between MKH and SSD on days 9 (p<0.05), 12 (p<0.001) and

______________________________________________________________________________
156
15(p<0.05), and crude keratose and SSD on days 12 (p<0.001) and 15 (p<0.05). Also, a
significant difference (p<0.01) was noted between Coloplast and SSD on days 9 and 12.
(Figure 1)
4.2. Histology:
Percent burn depth: On day 3, the average burn depth was minimal in Coloplast
treatment group (26.6%) compared to the other treatment groups (MKH - 30.9%, Crude
KOS – 34.51%, SSD – 34.98%). By day 9, vertical progression was evident in all the
treatments. Crude keratose treatment (5.56%) had a minimal increase in burn depth
compared to the other treatments (Coloplast - 22.5%, MKH - 20.01%, SSD - 9.81%)
(Figure 2 & Table 1). There was no statistically significant difference in burn depth
between the treatments.
Re-epithelialization: Burns treated with crude keratose and MKH demonstrated an
upward trend in re-epithelialization at early time points (days 3and 6) and later time
points (days 9, 12 and 15) (Figure 3&4). The rate of re-epithelialization was relatively
higher in the MKH group (4.1 +/- 0.96) compared to other treatments at early time points
(Coloplast - 3.4 +/- 1.1, crude keratose - 3.4 +/- 1.2, SSD - 1.3 +/- 1.7; Table 2). This
observation corresponded with previous findings that SSD can impede re-
epithelialization [13, 14]. In addition, healing was generally inconsistent across the
replicates in the SSD treatment group as indicated by the higher standard deviations.
At later time points (days 9, 12 and 15), the rate of re-epithelialization was
relatively higher in the MKH treatment group (7.6 +/- 1.2) compared to other treatments
(Coloplast - 6.9 +/- 1.1, crude keratose - 7.3 +/- 1.4, SSD- 5.9 +/- 1.2; Table 3), although
not as markedly as at the early time points. Healing was generally more consistent across

______________________________________________________________________________
157
replicates as indicated by the relatively smaller standard deviations compared to early
stages of healing. When the data was extrapolated to determine a predicted value for the
number of days taken to complete wound closure, crude and MKH treatments required 19
days, whereas both Coloplast and SSD treatments required 24 days (Table 4).
Thickness of granulation tissue: Compared to the other treatments, SSD had a larger
difference (1.115cm) in the thickness of granulation tissue from days 9 to 15 and crude
keratose had the least difference (0.746cm). Differences in all the treatment groups were
not statistically significant (Figure 5).
5. Discussion:
Due to tissue necrosis and burn wound progression, deep partial thickness burns can
convert to full thickness burns, thereby increasing risk to the patient, length of hospital stay, and
the costs associated with treatment. At the early stages after burn injury, the processes of healing
and wound progression are in opposition to each other, and most often progression prevails due
to a lack of specific treatments that can address tissue loss at the level of cellular pathways.
Currently there are no approved treatments that can control burn injury progression by regulating
death signals within the cell. The standard of care is generally focused on avoiding infection
while allowing the burn to take its natural course, followed by excision of dead tissue after some
short time period to facilitate healing from the healthy periphery of the burn. However, the
standard of care, SSD, did not perform as well as the other treatments in this study. One of the
more striking findings was the variability in healing observed in this treatment group. Despite its
beneficial antimicrobial properties [15, 16], the use of SSD as a standard of care has been
questioned due to its reported interference with re-epithelialization [13, 14, 17, 18]. Tian et al.

______________________________________________________________________________
158
suggested that the use of silver nano-particles as a more effective alternative to SSD [17].
Crystalline silver has also been shown to have beneficial wound healing properties [19], but to
our knowledge no comparative testing of these different forms of silver in the same burn model
have been published.
Assessment of digital images of wound areas did show that MKH had a significantly
greater effect on minimizing wound size at days 9, 12, and 15 compared to SSD, but not so
compared to crude keratose and Coloplast. Even though significant difference between MKH
and SSD was observed in the visual wound assessment, histological analyses have not shown
any significant difference at the time points 9, 12 and 15. This might be due to the parameters
considered when measuring a wound visually from digital images. Visually a wound is
considered to be re-epithelialized if it is covered with a thin pink or white opalescent non glossy
covering, but there is a chance that such a coating is present even in the absence of neo-
epidermis [20]. Additionally there is a higher chance for the fragile neo-epidermis to be
accidentally removed during the processing and sectioning of the tissue samples. In the present
study, percent re-epithelialization as determined using stained tissue sections showed that crude
keratose and MKH treated wounds were able to re-epithelialize faster compared to SSD,
especially at early time points where the rate for MKH was 220% higher than SSD. However,
these data were not statistically significant, largely due to the high standard deviation in the SSD
treatment group (the percent relative standard deviation was 131% for SSD versus 23% for
MKH; the R squared for the least squares linear regression of these data was 0.03 for SSD versus
0.54 for MKH). Later stages of re-epithelialization appeared to be similar in all treatment groups.
As expected, the injury model did demonstrate burn injury progression in all treatment
groups, but it was the crude keratose that showed the smallest increase in burn depth compared

______________________________________________________________________________
159
to the other treatments. This finding may indirectly support our hypothesis as the crude keratose
formulation contained the highest level of gamma keratose, which may be the reason that wound
progression was relatively lower. However, this outcome measure also failed to demonstrate
statistical significance.
Granulation tissue depth, a measure of later stages of healing, was highest in the SSD
treatment group and minimal in the crude keratose treatment. However, this may have been due
to the fact that this treatment group experienced the most wound progression, and consequently
had the most extensive tissue loss. Higher amounts of granulation tissue may have been the
normal response to tissue loss and not necessarily indicative of better healing. Regardless, the
differences in this outcome measure were again found to not be statistically significant when
comparing the different treatments.
In order to better understand the apparent lack of statistical power in our experimental
design, power calculations were performed based on the re-epithelialization data. This showed
that two more replicates would be required to show a significant (p<0.05) difference in re-
epithelialization rate between MKH and SSD, and more than 100 replicates would be required to
show a significant difference between MKH and crude keratose or Coloplast. These numbers
point toward improvement in arguably the most important outcome in burn treatment, re-
epithelialization, but they do not necessarily support the conclusion that the amount of gamma
keratose was optimal, and/or that the route of administration was ideal in this study. As was
shown in Chapter 2, the gamma keratose fraction contains large peptide molecules, and delivery
of these compounds through the skin and especially through a thick layer of eschar, would be
expected to be difficult and inefficient. Interestingly, Nunez et al. have shown that soluble
keratose has vasodilation properties when administered intravenously or topically [21].

______________________________________________________________________________
160
Considering that the zone of stasis is characterized by reduced blood circulation due to the
clogging of blood vessels after burn injury [2], perhaps part of the mechanism of tissue sparing
may be due to keratin’s ability to dilate blood vessels in the tissue surrounding the burn.
Moreover, a different route of keratin administration such as intravenous injection may show
even greater efficacy, as has been the case with other compounds tested in burn models.
However, as discussed previously, local delivery is often preferred so a higher dose of gamma
keratose, such as was demonstrated using crude keratose, may represent a viable solution.
Regardless, a better understanding of the mechanism of action, optimal dose, and efficient
administration of the most efficacious compounds will require more research.
6. Conclusion:
In this study, we tried to understand if a particular formulation of keratin hydrogel
containing a reduced concentration of gamma keratose, MKH, could have a beneficial effect on
burn wound healing. We compared MKH to the crude keratose that was previously shown to
promote burn wound healing (Chapter 2) and that contained a fraction of protein, gamma
keratose, which rescued thermally stressed cells (Chapter 2 and 3). The amount of gamma
keratose was reduced in MKH to reflect the concentration that demonstrated efficacy in a cell
culture model of thermal stress. A statistically significant difference in wound size was observed,
but this did not translate to the histological measurements. The rate of early re-epithelialization
appeared faster in the MKH treatment group, particularly at early time points where it was
expected to have its greatest effect, but statistical significance was lost due to variance in the
SSD treatment group. Regardless, the outcome measures in this study were not able to
differentiate MKH (i.e. low gamma keratose concentration) from crude keratose (high keratose

______________________________________________________________________________
161
concentration). More research to investigate the mechanism of keratins in burn wound healing,
and more specifically in burn injury progression, are needed.
Conflict of interest statement
Mark Van Dyke, Ph.D. holds stock and is an officer in the company, KeraNetics LLC, which has
provided partial funding for this research. Wake Forest Health Sciences has a potential financial
interest in KeraNetics, LLC through licensing agreements.

______________________________________________________________________________
162
References:
[1] Singh V, Devgan L, Bhat S, Milner SM. The pathogenesis of burn wound conversion. Ann
Plast Surg. 2007;59:109-15.
[2] Jackson DM. [The diagnosis of the depth of burning]. Br J Surg. 1953;40:588-96.
[3] Wasiak J, Cleland H, Campbell F. Dressings for superficial and partial thickness burns.
Cochrane Database Syst Rev. 2008:CD002106.
[4] Goldman RD, Grin B, Mendez MG, Kuczmarski ER. Intermediate filaments: versatile
building blocks of cell structure. Curr Opin Cell Biol. 2008;20:28-34.
[5] Rogers MA, Langbein L, Praetzel-Wunder S, Winter H, Schweizer J. Human hair keratin-
associated proteins (KAPs). Int Rev Cytol. 2006;251:209-63.
[6] Popescu C, Hocker H. Hair--the most sophisticated biological composite material. Chem Soc
Rev. 2007;36:1282-91.
[7] Carney DH, Redin W, McCroskey L. Role of high-affinity thrombin receptors in postclotting
cellular effects of thrombin. Semin Thromb Hemost. 1992;18:91-103.
[8] Freedberg IM, Tomic-Canic M, Komine M, Blumenberg M. Keratins and the keratinocyte
activation cycle. J Invest Dermatol. 2001;116:633-40.
[9] Norfleet AM, Bergmann JS, Carney DH. Thrombin peptide, TP508, stimulates angiogenic
responses in animal models of dermal wound healing, in chick chorioallantoic membranes, and
in cultured human aortic and microvascular endothelial cells. Gen Pharmacol. 2000;35:249-54.
[10] Freyberg S, Song YH, Muehlberg F, Alt E. Thrombin peptide (TP508) promotes adipose
tissue-derived stem cell proliferation via PI3 kinase/Akt pathway. J Vasc Res. 2009;46:98-102.
[11] Olszewska-Pazdrak B, Hart-Vantassell A, Carney DH. Thrombin peptide TP508 stimulates
rapid nitric oxide production in human endothelial cells. J Vasc Res. 2010;47:203-13.

______________________________________________________________________________
163
[12] de Guzman RC, Merrill MR, Richter JR, Hamzi RI, Greengauz-Roberts OK, Van Dyke ME.
Mechanical and biological properties of keratose biomaterials. Biomaterials. 2011;32:8205-17.
[13] Hussain S, Ferguson C. Best evidence topic report. Silver sulphadiazine cream in burns.
Emerg Med J. 2006;23:929-32.
[14] Lansdown AB. A review of the use of silver in wound care: facts and fallacies. Br J Nurs.
2004;13:S6-19.
[15] Atiyeh BS, Costagliola M, Hayek SN, Dibo SA. Effect of silver on burn wound infection
control and healing: review of the literature. Burns. 2007;33:139-48.
[16] Modak SM, Fox CL, Jr. Binding of silver sulfadiazine to the cellular components of
Pseudomonas aeruginosa. Biochem Pharmacol. 1973;22:2391-404.
[17] Tian J, Wong KK, Ho CM, Lok CN, Yu WY, Che CM, et al. Topical delivery of silver
nanoparticles promotes wound healing. ChemMedChem. 2007;2:129-36.
[18] Demling RH, Leslie DeSanti MD. The rate of re-epithelialization across meshed skin grafts
is increased with exposure to silver. Burns. 2002;28:264-6.
[19] Wright JB, Lam K, Buret AG, Olson ME, Burrell RE. Early healing events in a porcine
model of contaminated wounds: effects of nanocrystalline silver on matrix metalloproteinases,
cell apoptosis, and healing. Wound Repair Regen. 2002;10:141-51.
[20] Singer AJ, Hirth D, McClain SA, Clark RA. Lack of agreement between gross visual and
histological assessment of burn reepithelialization in a porcine burn model. J Burn Care Res.
2012;33:286-90.
[21] Nunez F, Trach S, Burnett L, Handa R, Van Dyke M, Callahan M, et al. Vasoactive
properties of keratin-derived compounds. Microcirculation. 2011;18:663-9.

______________________________________________________________________________
164
Figures and Tables:
Figure 1: Visual wound assessment
MKH has a significant difference compared to silver sulfadiazine (SSD) at days 9 (p<0.05), 12
(p<0.001) and 15(p<0.05). Crude keratose has a significant difference compared to SSD on days
12 (p<0.001) and 15 (p<0.05) and coloplast has a significant difference to SSD days 9 (p<0.01)
and 12 (p<0.01).
0 10 20 30 400.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4 Crude Keratose
MKH
Coloplast
Silver Sulfadiazine
Days
Rela
tive W
ound S
ize
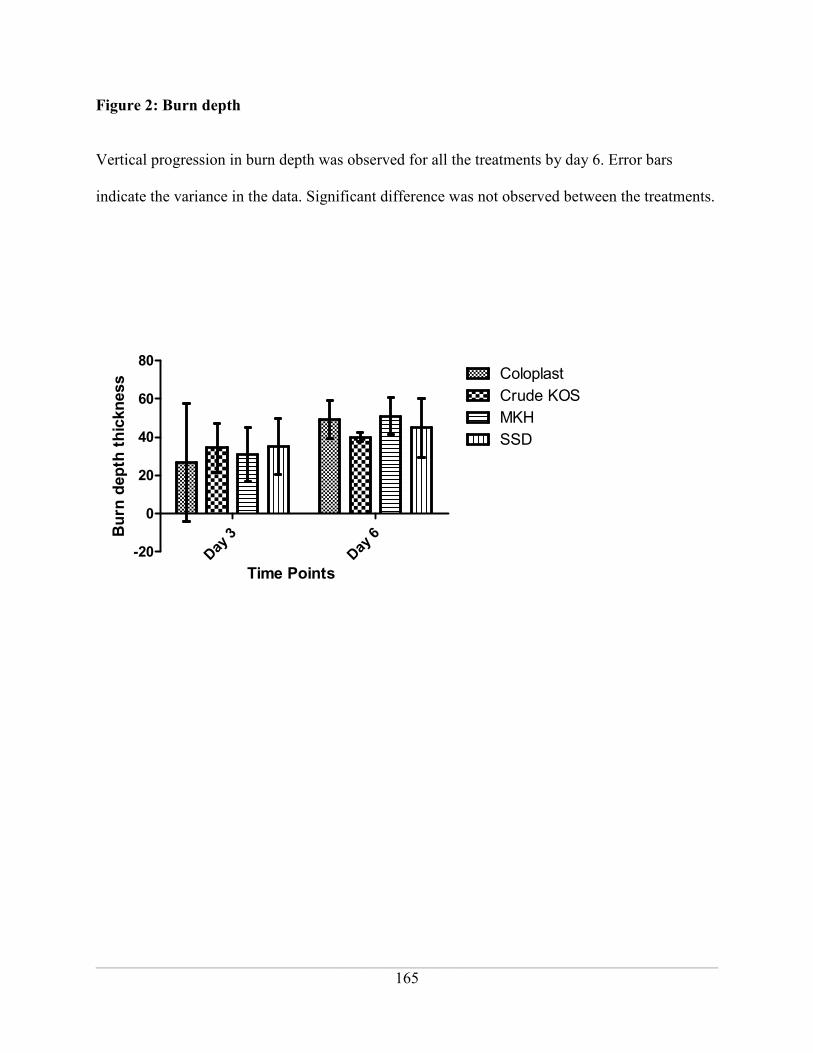
______________________________________________________________________________
165
Figure 2: Burn depth
Vertical progression in burn depth was observed for all the treatments by day 6. Error bars
indicate the variance in the data. Significant difference was not observed between the treatments.
Day
3
Day
6
-20
0
20
40
60
80Coloplast
Crude KOS
MKH
SSD
Time Points
Burn
depth
thic
kness

______________________________________________________________________________
166
Figure 3: Early time points rates of re-epithelialization
Burns treated with keratose (KOS) treatments demonstrated a general upward trend in re-
epithelialization at early stages of treatment, whereas Coloplast and SSD generally trended
downward. There was no statistically significant difference between treatment groups (n=6,
mean +/- standard deviation, p>0.05)
Coloplast
0 3 60
20
40
60
Day
% Re-Epithelialization
Crude KOS
3 60
20
40
60
Day
% Re-Epithelialization
SSD
3 6
-20
0
20
40
60
Day
% Re-Epithelialization
KeraHeal
0 3 60
20
40
60
Day
% Re-Epithelialization
MKH

______________________________________________________________________________
167
Figure 4. Rates of re-epithelialization during later stages of wound healing
Compared to the other treatments, keratose (MKH and CrudeKOS) treatments promote faster re-
epithelialization. There was no statistically significant difference between treatment groups (n=6,
mean +/- standard deviation, p>0.05)
Coloplast
9 12 150
20
40
60
80
100
Day
% Re-Epithelialization
Crude KOS
9 12 150
20
40
60
80
100
Day
% Re-Epithelialization
SSD
9 12 150
20
40
60
80
100
Day
% Re-Epithelialization
KeraHeal
9 12 150
20
40
60
80
100
Day
% Re-Epithelialization
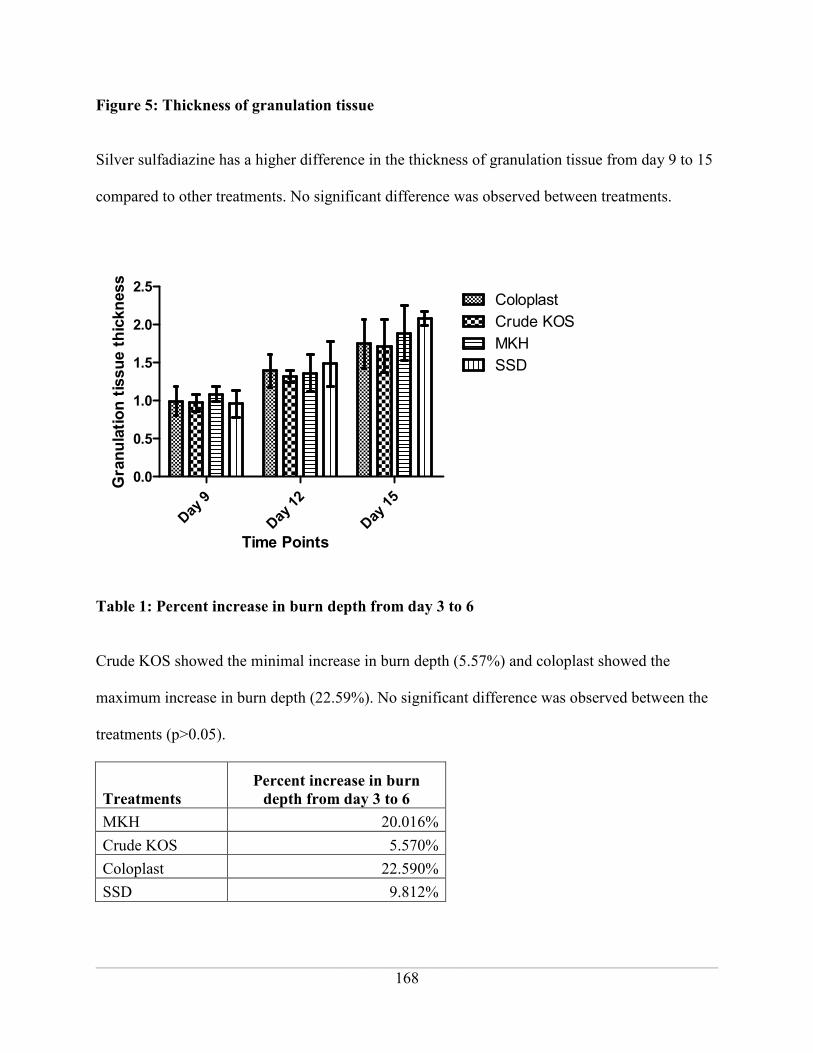
______________________________________________________________________________
168
Figure 5: Thickness of granulation tissue
Silver sulfadiazine has a higher difference in the thickness of granulation tissue from day 9 to 15
compared to other treatments. No significant difference was observed between treatments.
Day
9
Day
12
Day
15
0.0
0.5
1.0
1.5
2.0
2.5Coloplast
Crude KOS
MKH
SSD
Time Points
Gra
nula
tion tis
sue thic
kness
Table 1: Percent increase in burn depth from day 3 to 6
Crude KOS showed the minimal increase in burn depth (5.57%) and coloplast showed the
maximum increase in burn depth (22.59%). No significant difference was observed between the
treatments (p>0.05).
Treatments
Percent increase in burn
depth from day 3 to 6
MKH 20.016%
Crude KOS 5.570%
Coloplast 22.590%
SSD 9.812%

______________________________________________________________________________
169
Table 2. Rates of re-epithelialization at day 3 and day 6.
Compared to silver sulfadiazine, MKH has the highest difference in the rate of re-
epithelialization in the early stages of wound healing (days 3 and 6).
Treatment
Rate (% Re-
Epithelialization/Day) R Squared % Difference vs. SSD
Coloplast 3.4 +/- 1.1 0.40 +160%
Crude KOS 3.4 +/- 1.2 0.35 +160%
MKH 4.1 +/- 0.96 0.54 +220%
SSD 1.3 +/- 1.7 0.03 --
Table 3. Rates of re-epithelialization between days 9 and 15.
Compared to silver sulfadiazine, MKH has the highest difference in the rate of re-
epithelialization in the later stages of wound healing (days 9 to 15).
Treatment
Rate (% Re-
Epithelialization/Day) R Squared % Difference vs. SSD
Coloplast 6.9 +/- 1.1 0.72 17%
Crude KOS 7.3 +/- 1.4 0.64 24%
MKH 7.6 +/- 1.2 0.72 29%
SSD 5.9 +/- 1.2 0.62 --
Table 4. Days to wound closure.
Keratose (Crude KOS and MKH) treatments have a faster wound closure compared to coloplast
and silver sulfadiazine.
Treatment Mean Days to Wound Closure
Coloplast 24.2
Crude KOS 19.5
MKH 19.2
SSD 23.4

______________________________________________________________________________
170
CHAPTER – 6
_____________________________________________________________________________________
DELAYED TREATMENT OF PORCINE DEEP PARTIAL THICKNESS BURNS
SUBJECT TO THERAPEUTIC KERATIN BIOMATERIALS
Authors, Affiliation:
Gaines Ca, Poranki D
a, Van Dyke M
a
aWake Forest School of Medicine, Wake Forest Institute for Regenerative Medicine
Wake Forest School of Medicine, Medical Center Boulevard, Winston Salem, NC 27157
This manuscript was not yet submitted for publication. Deepika Poranki assisted in the design
and execution of the animal study and data analysis. Dr. Mark Van Dyke acted in an advisory
and editorial capacity.

______________________________________________________________________________
171
Abstract
Burn wound healing outcomes rely heavily upon the timely administration of first aid treatments
and can affect the quality of later healing. The previously-reported wound healing capabilities of
keratin biomaterials makes them a viable treatment option as they may be capable of influencing
cell survival in the peri-burn region. In this study, the healing efficacy of deep partial thickness
burns subject to keratin biomaterial treatments 10 hrs after injury was evaluated. While burn
wound digital image examination suggested that keratin biomaterials enhance wound closure
rates at 12 days post-injury, histological evaluation suggested that the standard of care silver
sulfadiazine (SSD) encouraged faster reepithelialization rates for the first 15 days of the 30-day
study. By Day 30, the extent of reepithelialization due to keratin biomaterial treatments was
significantly greater in comparison to SSD (p≤0.001). Measurement and evaluations of
additional endpoints during the second half of the study are needed to determine the true
downstream healing efficacy of keratin biomaterials for the delayed treatment of deep partial
thickness burn wounds.
Keywords: deep partial thickness burns, keratin biomaterials, delayed treatment, swine model,
reepithelialization

______________________________________________________________________________
172
1. Introduction
Burn wound healing outcomes rely heavily upon the timely administration of first aid
treatments. Arguably the most precarious burn wounds to care for, deep partial thickness burns,
are those in which damage extends through the epidermis and well into the reticular dermis.
Unmediated treatment of these wounds is likely to result in burn wound progression, in which
tissue damage moves radially from a centralized region and evolves into a full thickness burn
condition. Within this centralized “zone of coagulation,” damage to blood vessels and dermal
appendages result in an ischemic state that starts within 48 hrs of injury [1-3] and lasts up to 3-7
days post-injury [4, 5]. Fibroblasts, keratinocytes and affiliated dermal cells within the adjacent
“zone of stasis” will succumb to either cell death effectively extending the zone of coagulation
and increasing the size and severity of the wound or revitalization and proliferation based upon
decided treatment modalities. Proper assessment of burn severity dictates the optimal course of
treatment for challenging conditions encountered downstream such as contracture, hypertrophic
scarring and microbial infection that often accompany full thickness burns.
Timely access to first aid burn treatments is often limited by physical proximity to
appropriate medical personnel or facilities specializing in burn care. Burn victims are
responsible for 45,000 hospitalizations in the US, 55% of who were transferred to specialized
“burn units [6].” At the time of injury, the victim may be located hundreds of miles away from
assistance, as often seen in rural communities nationwide or with members of the armed forces
serving overseas needing specialized care. With a total of 128 self-reported burn centers (51
American Burn Association-verified) dispersed throughout the US, Canada and Australia [7],
easy access by the multitudes of severely burned victims may not be easily attainable. While

______________________________________________________________________________
173
Klein [7] reported that approximately 80% of the US lives within 1-2 hrs of ground or air
transport of a verified burn center, many cover large geographic areas in which initial treatment
is transferred to a closer local hospital for initial assessment. As such, actual transfer to a burn
center may result in delays, ultimately leading to longer hospital stays, increased complications
and possible mortality. Sheridan [8] reported that a 5-day delay in transporting pediatric burn
victims with severe burns at >20% total body surface area (TBSA) burned to a designated burn
center resulted in significantly greater incidences of bacterial infection, wound sepsis and longer
wound closure times. MacKenzie [9] found that the in-hospital and one year mortality rates for
patients seen at a trauma center vs. non-trauma center was significantly lower. A study
conducted at the University of Washington Burn Center reported an average transport time post-
injury of 7.2 hrs; initial assessments described wounds with a 17% TBSA [10]. In this study, the
estimated burn size by non-trauma care personnel was overestimated, as TBSA of 15% is the
threshold for fluid resuscitation needed.
According to the United States Army Institute of Surgical Research (USAISR) Burn Center,
hand and facial burns were the most prevalent seen in military personnel, with approximately 73-
76% of combat victims experiencing this type of injury within a 2-year timeframe [11-13]. As
perhaps the most difficult regions of the body to treat, inadequate healing may lead to impaired
functionality and in some cases removal from previous military duties [14]. From 2003 to 2007,
transport time for injured soldiers from Iraq and Afghanistan to the USAISR was up to 4 days
from time of injury to admission to the burn center [12]. While 5.6% of the victims died from
their injuries at the USAISR, the ventilation support provided to over one-third of the victims in-
flight likely attributed to the relatively low casualty rate. These studies suggest that preventative

______________________________________________________________________________
174
measures taken to stabilize burns prior to more extensive treatments are crucial to limiting
negative outcomes.
Spontaneous wound healing largely depends on the following conditions: a) systemic
hydration via fluid resuscitation, b) local hydration of the wound bed to encourage
reepithelialization and c) wound infection control. Microbial control may have the most critical
local effects on progressive wound healing. Stander [15] suggests that microbial control within
the wound bed be maintained if delayed transport to a burn center is over 12 hrs. D’Avignon
[16] suggested the application of silver sulfadiazine (SSD) within 8 hrs of injury in combat-
related burns. Topical application of SSD cream has long been considered the ‘gold standard’
for microbial control [17, 18]. In deep partial thickness burns, sweat glands and hair follicles
may still be present in deeper layers of the dermis. If left untreated, gram-positive bacteria will
migrate from these appendages to colonize the wound within 48 hrs post-injury [19, 20]. By 7-
days post-injury, gram-positive and gram-negative microbes migrate from other regions in the
body to inhabit the burn wound eschar, creating a ripe environment for infection [21-23].
Additionally, researchers suggest that the application of SSD along with gentle debridement is
most effective in containing wound progression if patients cannot be transported for at least 72
hrs [17, 24]. While infection control is important, SSD alone is inadequate to keep the wound on
the path of healing. In fact, some studies have reported that SSD has a deleterious effect on
healing through exhibiting cytotoxic effects that delays wound closure. A study conducted by
Fraser [25] suggested that keratinocytes grown in culture exhibited a significant reduction in cell
number when exposed to SSD for 72 hrs.
An optimal first-line treatment for burns would be to encourage cell proliferation within the
wound bed without causing further infection to the wound. Burns have been characterized as

______________________________________________________________________________
175
having an exaggerated inflammatory phase [19]. Growth factor (i.e. transforming growth factor
beta or TGF-β) infiltration into the wound bed accounts for the initial influx of fibroblasts during
early wound healing, and is often responsible for scarring and contracture [26].
The first line of defense in burn care is the use of topical treatments. From a
physiological standpoint, the primary goals of such treatments are to prevent further tissue loss
and infection, and to promote spontaneous healing. In order for this to occur, revascularization
of the wound must happen within a few days post-injury. One such benefit of topicals is their
convenience and ease of use. This is especially important for victims to be able to treat
themselves at home or on the battlefield. In lieu of SSD, biologic dressings that exhibit some
bioactivity have been proven to enhance wound healing [27-31]. Common themes among these
treatments are non-immunogenicity, biocompatibility, non-toxicity and biodegradability.
A potential wound healing treatment that embodies the aforementioned characteristics, as
well as the added benefit of prompting no spontaneous protolytic activity within the body, are
keratin biomaterials. Keratin proteins derived from human hair have shown promise in a variety
of regenerative medicine applications, and is comprehensively outlined in Rouse et al. [32].
Within cell culture models, keratin hydrogels encouraged cell adhesion and proliferation on par
with collagen hydrogels [33]. Keratin dressings have also been shown to accelerate wound
healing and closure through the stimulation of keratinocyte migration and proliferation along the
wound margin within a porcine partial thickness model [34]. Previous studies conducted by our
lab demonstrate enhanced wound healing rates in a swine contact burn model treated with a
crude extract of keratin hydrogel (Chapter 2). Additionally, keratin hydrogel-treated chemical
burns within a rodent model showed containment of the burn wound (Chapter 2). The present
study was designed to test the healing efficacy of delayed treatment of deep partial thickness

______________________________________________________________________________
176
burns with a crude keratose hydrogel and a modified keratose hydrogel (MKH) within a tightly
controlled swine contact burn model. This delayed treatment scheme was conducted to
compliment a standard treatment paradigm evaluated in our lab (Chapter 5) and more closely
represents cases encountered by first responders in an attempt to provide initial care for burn
wounds and prevent the need for more invasive surgical treatments.
2. Materials & Methods
2.1. Animal study logistics: This study was approved by the Wake Forest Animal Care and
Use Committee. Fourteen female, Yorkshire swine (Baux Mountain, Winston-Salem,
NC) weighing 20-25 kg at the time of surgery were used. The animals were housed in
individual pens and allowed to acclimate to their surroundings for at least 7 days prior to
surgery. Deep partial thickness burns were created on pairs of swine treated and
monitored for 1, 3, 6, 9, 12, 15 or 30 days relative to the date of injury. The order in
which these designated endpoints occurred were randomly selected. One day prior to
surgery, the swine were cleaned and shaved along the dorsum. At this time, a fentanyl
patch (50 mcg/h) was adhered to the shoulder or hip of each animal as an analgesic in
preparation for the upcoming surgery. The animals were fasted the night before the
administration of anesthetization for each procedure conducted throughout the study.
2.2. Burn surgery protocol: Details of the burn surgery protocol used were reported
previously [35]. Briefly, the animals were anesthetized intramuscularly with a
combination of ketamine (10 mg/kg) and dexmedetomidine (0.05 mg/kg), intubated and
sustained on isofluorane during the surgical procedure. Vitals, such as blood pressure
and body temperature, were closely monitored during surgery to ensure animal safety.
Three cm diameter wounds were created using an innovative burn system in which a

______________________________________________________________________________
177
custom-made, pressure-controlled burn device housed a detachable 360 grade brass
block. As the designated heat source, the brass block was immersed in a boiling
negative azeotropic mixture of 80:20 polyethylene glycol (Mn = 400; Sigma-Aldrich, St.
Louis, MO):deionized water to ensure proper heat transfer from the source to the animal.
For each wound, the brass block made contact with the animal’s skin for 20s. Six
wounds were created on each side of the dorsal mid-line, between the shoulder and hip.
A total of 24 wounds were created, 12 per animal (Figure 1). The wounds were allowed
to set for 1 hr prior to being covered with saline-soaked gauze and bandaged as
described previously [35]. For the Day 30 endpoint, black dots were tattooed in the 4
corners surrounding each wound as a means to monitor contracture over time. Post-
surgical care consisted of the intramuscular administration of atipamezole (0.05 mg/kg)
to reverse the sedative effects of the dexmedetomidine and careful monitoring during
recovery.
2.3. Bandage changes:A typical bandage change occurred approximately every 3 days.
Under general anesthesia, all bandaging was removed and the wounds were lightly
debrided with saline-soaked gauze. Digital images of each wound were captured
(Nikon D60, Nikon, Melville, NY); a gray scale (Kodak, Rochester, NY) was included
in the field of view for image optimization along with a ruler to ensure proper scaling
during analysis. After treatment application, the animals were re-bandaged and
monitored during recovery. Saline was administered intravenously every other bandage
change to ensure proper systemic hydration. A new fentanyl patch (75 mcg/h) was
placed on each animal for pain management.

______________________________________________________________________________
178
2.4. Delayed treatment: Test and control treatments were randomly applied to wounds over
the swine pair for each endpoint. Test treatments consisted of Crude and modified
keratose formulations of keratin-based hydrogels, which will be referred to as Crude
KOS and MKH throughout the paper. Keratin proteins were extracted from a
commercial source of human hair via oxidative methods as described in de Guzman
[36]. Crude keratose was further separated into alpha and gamma keratose fractions by
isoelectric precipitation. Concentrated hydrochloric acid was added dropwise to the
crude keratose solution with stirring until a pH 4.2 was reached. At this pH, the alpha
keratose precipitates leaving the acid-soluble gamma fraction in solution. The insoluble
alpha keratose was separated by fixed angle centrifugation at 1500 rpm for 15min at 4ºC.
After neutralizing the gamma-containing supernatant to pH 8.4, it was dialyzed against
endotoxin free water using a 5kDa spiral wound cartridge (Millipore, Billerica, MA) for
5 volume washes. Finally, the solution was concentrated to minimal volume, pH
adjusted to 7.4, lyophilized, and sterilized by gamma irradiation at 1 MRad prior to use.
The precipitated alpha keratose was re-dissolved in 0.1M sodium hydroxide (NaOH)
solution and dialyzed against endotoxin free water using a 5kDa spiral wound cartridge
(Millipore) for 5 volume washes. Finally, the solution was concentrated to minimal
volume, pH adjusted to 7.4, lyophilized, and sterilized by gamma irradiation at 1 MRad
prior to use. Crude KOS and MKH formulations were sterilized via gamma irradiation (2
MRad). Under sterile conditions, powdered formulations were reconstituted in saline
and agitated overnight at 37°C to form 15 weight% hydrogels. Control treatments
consisted of a collagen-based hydrogel wound dressing indicated for the treatment of
superficial and partial thickness burns (Woun’Dres™ Collagen Hydrogel, Coloplast,

______________________________________________________________________________
179
Minneapolis, MN) and 1 wt% silver sulfadiazine (SSD) cream (Watson Pharmaceuticals,
Parsippany, NJ), long considered the standard of care for burn injuries [18]. All topical
treatments were transferred to 10 cc syringes for controlled application, with
approximately 2 cc applied to each wound. Treatment was delayed for approximately 10
hrs post-surgery and administered under general anesthesia. The animals were then
bandaged and allowed to recover as previously stated. All hydrogel treatments were
applied for 15 days, and SSD for 9 days, and was replaced by saline-soaked gauze for
protection during additional healing.
3. Data analysis
3.1. Digital Image Capture: Digital images of the burn wounds at progressive stages of
healing were captured on surgery day and at every subsequent bandage change and end
point. The margin of each wound was traced using Image J software (NIH, Bethesda,
MD); the area of the open wound was calculated. For each series of wounds
corresponding to an endpoint, the average area value was normalized to the surgery day
area value. The data set was analyzed via 2-way ANOVA and Bonferroni post hoc
analyses using GraphPad Prism (GraphPad Software, La Jolla, CA). Additionally, the
series of Day 30 wounds were evaluated for contracture where the circumference of the
“boxed” area around each of the wounds was measured by connecting the dots using
Image J. The data set was analyzed using 2-way ANOVA and Bonferroni post hoc
analyses using GraphPad Prism.
3.2. Histological analysis: At each specified endpoint, a pair of swine was euthanized and
each of the wounds was harvested in full as reported previously [35]. Briefly, the
wounds were cut into quadrants and fixed in 10% neutral buffered formalin for 48 hrs.

______________________________________________________________________________
180
A 2-cm thick slice from the innermost portion of the quadrant of each wound was
sampled and paraffin processed as previously reported. The slice represented the region
of the wound with the greatest degree of damage. The tissue samples were embedded in
paraffin and sectioned into 5 µm thick slices. One tissue section was evaluated per
wound, representing a sample size of 6 for each treatment. Sections stained with
hexatoxylin and eosin (H&E) were used to qualitatively access burn depth and the
relative density of granulation tissue. The extent of reepithelialization was calculated by
subtracting the length of the non-epithelialized burn region of the tissue sample from the
measured radius of the corresponding surgery day digital image of the wound to reveal
the length of the reepithelialized region. The length of the reepithelialized region was
normalized to the surgery day wound radius to determine the % reepithelialization. All
measurements were taken with Image J. Each data set was analyzed via linear
regression up to Day 15. At Day 30, the data was analyzed via 1-way ANOVA and
Bonferroni post hoc analysis. All statistical evaluations were done using GraphPad
Prism.
4. Results
4.1. Digital Image Capture: An approximate 10% increase in measured wound size
for up to 6 days post-injury was observed for all treatments, followed by a steady
decrease to <25% of the original wound size by Day 30 (Figure 2). The fastest rates of
size reduction were seen between Days 9 and 12. The healing patterns among treatments
were not significantly different (p>0.05) over the course of 30 days, except for significant
differences in the extent of healing in wounds treated with the keratin hydrogels vs. SSD
(p<0.001) and Coloplast vs. SSD (p<0.05) at Day 12. In measuring the progressive

______________________________________________________________________________
181
occurrence of wound contracture, the mean distance surrounding each wound expanded
in all treatment groups within the first 6 days of injury (Figure 3). Thereafter, these
“borders” steadily contracted to near baseline with some variation at the later time points.
There was no significant difference in the degree of wound contracture among treatment
groups at any specified time point (p>0.05). On average, MKH and Coloplast exhibited
the least amount of contracture over 30 days of healing, while Crude KOS and SSD
exhibited slightly higher amounts.
4.2. Histological analysis:
Burn Depth: Representative images of the degree of tissue damage incurred as a result
of a deep partial thickness burn wounds at Day 1 post-injury are depicted in Figure 4. In
all cases, the epidermis had been completely destroyed, leaving a gradient of dermal
damage relative to the proximity of the heat source throughout the depth of the tissue.
The upper to mid dermal regions revealed fully coagulated tissue, as defined by the
damaged appendages (e.g., hair follicles, sweat glands) and the absence of distinct
collagen bundles and blood vessels. In the lower dermal regions, next to the adipose
layer of skin, was dermis that incurred a slightly lesser degree of damage than the upper
layers. While distinct collagen bundles were present, they were loosely packed and show
evidence of potentially early coagulation. The depth of burn damage remained relatively
constant over all treatment groups.
Inflammatory Cell Infiltration: At Day 6 post-injury, inflammatory cells infiltrated the
wound margin as evidenced by the purple bands of nuclei depicted in Figure 5. For SSD-
treated wounds, evidence of this band is barely visible and appears to be hidden within

______________________________________________________________________________
182
the adipose layer. The deposition of collagen and new blood vessel formation within
interstitial spaces along and below the lower dermis encouraged cells from fatty layer to
disperse and spread throughout the entire dermal region. Coagulated tissue initially
found toward the top of the dermal layer eventually sloughed off, leaving behind
extensive granulation tissue by Day 12 (Figure 6). Lower densities of granulation tissue
were observed in the Crude KOS and SSD treated wounds relative to the other
treatments.
Re-epithelialization: Minimal amounts of regenerated epithelium were present up to
Day 6 post-injury. Within the first 6 days of treatment, rates of re-epithelialization
among treatment groups showed a general downward trend hovering at 20% or lower
(Figure 7). The MKH treatment showed the greatest stability after Day 3 at
approximately 10% wound closure. Re-epithelialization has progressed to the greatest
degree in wounds treated with SSD as indicated by a sharp initial slope to Day 3 and at
least a 40% greater re-epithelialization rate as compared to that of the keratin treatments
and 28% compared to Coloplast (Table 1). Although healing is generally inconsistent
across replicates as indicated by the large error bars, the tightest control is seen at Day 3
with the SSD treatment. Overall, no statistically significant difference in healing rate was
observed between treatment groups (p>0.05).
Re-epithelialization rates during the later stages of healing indicated an upward trend
among all treatment groups, with the most linear progression demonstrated by MKH
(Figure 8). SSD maintains the fastest re-epithelialization rate from Days 9 to 15, which is
40% greater than that of the MKH and Coloplast treatments and 15% greater than Crude

______________________________________________________________________________
183
KOS (Table 2). Healing was more consistent across replicates in comparison to the
earlier stages of wound healing as evidenced by the smaller error bars, although the
tightest control in this case was demonstrated by the Crude KOS treatment.
Qualitatively, well-organized, multilayered epithelium was present with all treatments at
Day 12 (Figure 9). Distinct rete ridges were seen in the Coloplast and SSD treatments.
In the keratin treatments, the papillary dermis appears to be loosely packed in comparison
to other treatments and may be indicative of mechanical weakness. No statistically
significant difference in healing rate observed between treatment groups was reported
(p>0.05). The degree of re-epithelialization shifts dramatically at Day 30 post-injury as
the hydrogel treatments exhibit significantly greater wound closure than SSD at p≤0.001
(Figure 10).
5. Discussion
Delays in the administration of adequate first aid treatments for burn wounds may have a
deleterious effect on downstream wound healing processes. In our study, keratin biomaterial
treatments were applied to deep partial thickness burns at 10 hrs post-injury in order to evaluate a
more realistic scenario during which potential complications that may arise from delayed burn
care. The 10-hr threshold was chosen based on discussions with burn care clinicians, logistical
constraints for conducting the study within our animal care facility and the desire to inflict
minimal stresses onto the animals during the study.
Quantitative analyses of the burn wound digital images showed significant decreases in
wound size over time in all treatment groups (Figure 2). Wound closure along the burn surface
is a function of the migration and proliferation of keratinocytes originating from healthy dermal
tissue along the wound margin and appendages. The keratin biomaterial treatments showed the

______________________________________________________________________________
184
greatest reduction in wound size at Day 12 post-injury in comparison to SSD. From a
histological standpoint, there was no significant difference in the degree of re-epithelialization
over time between treatment groups (Figure 7). Interestingly, the data exposes a slightly greater
re-epithelialization rate in SSD between Days 9 and 12 in comparison to all other treatment
groups (Table 2). While in direct conflict with recent reports on the stagnated closure of burn
wounds when treated with SSD as mentioned previously, its potential usefulness in healing
cannot be ignored [37, 38]. In the case of the present study, the latter concept was evident at
Days 9-15. To address the apparent discrepancy between the statistically significant wound size
evaluation and non-significant histological evaluations, attention must be brought to the integrity
of the reepithelialized layer [39]. While the hydrogel materials may have encouraged a greater
degree of re-epithelialization at Day 12 as demonstrated by the wound digital images, perhaps
portions of the re-epithelialized layers as defined by migrating keratinocytes were lost during
histological processing [40]. Mechanical stresses incurred during gross tissue preparation or
paraffin block sectioning may be contributing factors. The multilayered appearance of the
emerging epithelium and presence of distinct rete ridges and density of the papillary dermis may
be considered in determining epithelial integrity. The representative H&E images of the
Coloplast- and SSD-treated burns at Day 12 (Figure 9) showed a more robust papillary dermis to
support epithelial growth as compared to the other treatment groups. Interestingly, wound
closure as exhibited by the hydrogel treatments was significantly greater than that of SSD by
Day 30 (Figure 10). This suggests that perhaps the keratin and Coloplast treatments showed the
greatest activity at time points between Days 15 and 30, and that accurate re-epithelialization
rates cannot be determined without more definitive data from this timeframe.

______________________________________________________________________________
185
Beneath the open wound, granulation tissue at various stages of remodeling was present
at Day 12 (Figure 6). An abundance of cells from the fatty layer was dispersed within the
granulation tissue with the Coloplast and MKH treatments. Evidence of densely-packed
collagen deposits with relatively smaller regions of cells from fatty layer dispersed was seen with
the Crude KOS and SSD treatments. The latter condition suggests that these treatments may be
further along in the wound healing process, creating a dermal foundation of greater integrity to
support closure of the open wound.
Up to Day 6 post-injury, the influence of all treatments appears to be minimal. After a
slight increase in the degree of re-epithelialization at Day 3, all treatment groups trended
downward upon reaching Day 6. This was likely due to the formation of wound eschar requiring
cleaning at each bandage change prior to treatment application. Although careful and deliberate
debriding techniques were used, it is possible that growing epithelium at the interface between
the eschar and healthy wound margin was removed as well. Overall, the lack of significant
differences between treatment groups raises the question of how well, or if, penetration of the
topical treatments occur through the eschar to more centralized regions of the burns [41].
Additionally, H&E images indicate relatively consistent initial burn depths across treatments at
Day 1 and a similarly-emerging presence of an inflammatory cell band to begin the processes of
removing damaged tissue and debris from the wounds and collagen deposition.
In contrast to a similar burn study conducted at our lab in which treatments were applied
1 hr post-injury, the treatment of deep partial thickness burns after 10 hrs with keratin
biomaterials for enhanced wound healing prior to Day 15 cannot be validated based on the
outcomes of this study. In the former case, the mean rates of re-epithelialization during the
earlier (up to Day 6) and later (Days 9-15) endpoints were 220% and 29% greater than those for

______________________________________________________________________________
186
SSD, respectively (Chapter 5). The current study demonstrated outcomes in favor of SSD up to
Day 15, with a striking end result of significant wound closure using Crude KOS, MKH and
Coloplast by Day 30. While our surgical protocol was fully qualified to produce consistent deep
partial thickness burns and the wounds were randomly treated as indicated in Figure 1, the
thickness and microstructure of the underlying tissue along the dorsum of the swine may have
also introduced variation that was exposed in our results. Additional endpoints between Days 15
and 30 post-injury needed to provide greater insight into the re-epithelialization activity during
the second half of the study.

______________________________________________________________________________
187
References
[1] Jackson D. The diagnosis of the depth of burning Br J Surg. 1953;40:588.
[2] Hinshaw J. Early changes in the depth of burns. Ann N Y Acad Sci. 1968;150:548-53.
[3] Williams W. Pathophysiology of the burn wound. In: Herndon D, editor. Total Burn Care.
2nd ed. London: Saunders; 2002. p. 514-21.
[4] CC Kao WG. Acute burns. Plast Reconstr Surg. 2000;105:2482-92.
[5] Lanier S, McClain S, Lin F, Singer A, Clark R. Spatiotemporal progression of cell death in
the zone of ischemia surrounding burns. Wound Rep Reg. 2011;19:622-32.
[6] ABA. Burn Incidence and Treatment in the United States: 2011 Fact Sheet. 2011.
[7] Klein M, Kramer C, Nelson J, Rivara F, Gibran N, Concannon T. Geographic Access to Burn
Center Hospitals. JAMA. 2009;302:1774-81.
[8] Sheridan R, Weber J, Prelack K, Petras L, Lydon M, Tompkins R. Early burn center transfer
shortens the length of hospitalization and reduces complications in children with serious burn
injuries. J Burn Care Rehabil. 1999;20:347-50.
[9] MacKenzie E, RIvara F, Jurkovich G, Nathens A, Frey K, Egleston B, et al. A National
Evaluation of the Effect of Trauma-Center Care on Mortality. N ENGL J MED. 2006;354:366-
78.
[10] Klein M, Nathens A, Emerson D, Heimbach D, Gibran N. An Analysis of the Long-
Distance Transport of Burn Patients to a Regional Burn Center. J Burn Care Res. 2007;28:49-55.
[11] Kauvar D, Cancio L, Wolf S, Wade C, Holcomb J. Comparison of Combat and Non-
Combat Burns From Ongoing U.S. Military Operations. Journal of Surgical Research.
2006;132:195-200.

______________________________________________________________________________
188
[12] Hedman T, Renz E, Richard R, Quick C, Dewey W, Barillo D, et al. Incidence and Severity
of Combat Hand Burns After All Army Activity Message. J Trauma. 2008;64:S169-S73.
[13] Baer D, Dubick M, Wenke J, Brown K, McGhee L, Convertino V, et al. Combat Casualty
Care Research At The U.S. Army Insitute of Surgical Research. JR Army Med Corps.
2009;155:327-32.
[14] Zuijlen Pv, Kreis R, Vloemans A, Groenevelt F, Mackie D. The prognostic factors
regarding long-term functional outcome of full-thickness hand burns. Burns. 1999;25:709-14.
[15] Stander M, Wallis L. The Emergency Management and Treatment of Severe Burns. Emerg
Med Int. ;2011:1-5.
[16] D'Avignon L, Chung K, Saffle J, Renz E, Cancio L. Prevention of infections associated with
combat-related burn injuries. J Trauma. 2011;71:S282-S9.
[17] DeSanti L. Pathophysiology and current management of burn injury. Adv Skin Wound
Care. 2005;18:323-32.
[18] Fox C. Silver sulfadiazine, addendum to local therapy. Modern treatment. New York:
Harper and Row; 1967. p. 1259.
[19] Cleland H. The modern management of burns. ADF Health. 2007;8:2-7.
[20] Erol S, Altoparlak U, Akcay M, Celebi F, Parlak M. Changes of microbial flora and wound
colonization in burned patients. Burns. 2004;30:357-61.
[21] Al-Akayleh A. Invasive burn wound infection. Annals of Burns and Fire Disasters. 1999;7.
[22] Altoparlak U, Erol S, Akcay M, Celebi F, Kadanali A. The time-related changes of
antimicrobial resistance patterns and predominant bacterial profiles of burn wounds and body
flora of burned patients. Burns. 2004;30:660-4.

______________________________________________________________________________
189
[23] Macedo Jd, Santos J. Bacterial and fungal colonization of burn wounds. Mem Inst Oswaldo
Cruz. 2005;100:535-9.
[24] WHEPP. Guidelines for the stabilization of burn patients for 72 hours until transfer to a
burn center. Wisconsin2011. p. 1-20.
[25] Fraser J, Cuttle L, Kempf M, Kimble R. Cytotoxicity of topical antimicrobial agents used in
burn wounds in australasia. ANZ J Surg. 2004;74:139-42.
[26] Evers L, Bhavsar D, Mailander P. The biology of burn injury. Experimental Dermatology.
2010;19:777-83.
[27] Sakiel S, Grzybowski J. Clinical application of new bovine collagen membranes as a partial-
thickness burn wound dressing. Polim Med. 1995;25:19-24.
[28] Nunes P, Albuquerque-Junior R, Cavalcante D, Dantas M, Cardoso J, Bezerra M, et al.
Collagen-Based Films Containing Liposome-Loaded Usnic Acid as Dressing for Dermal Burn
Healing. J Biomedicine Biotech. 2011;2011:1-9.
[29] Baoyong L, Jian Z, Denglong C, Min L. Evaluation of a new type of wound dressing made
from recombinant spider silk protein using rat models. Burns. 2010;36:891-6.
[30] Jayakumar R, Prabaharan M, Kumar PS, Nair S, Furuike T, Tamura H. Novel Chitin and
Chitosan Materials in Wound Dressing. Biomedical Engineering: Trends in Materials Science:
InTech; 2011. p. 1-24.
[31] Huang S, Fu X. Naturally derived materials-based cell and drug delivery systems in skin
regeneration. J Controlled Release. 2010;142:149-59.
[32] Rouse J, Dyke MV. A Review of Keratin-Based Biomaterials for Biomedical Applications.
Materials. 2010;3:999-1014.

______________________________________________________________________________
190
[33] Wang X-Q, Kravchuk O, Kimble R. A retrospective review of burn dressings on a porcine
burn model. Burns. 2010;36:680-7.
[34] Pechter P, Gil J, Valdes J, Tomic-Canic M, Pastar I, Stojadinovic O, et al. Keratin dressings
speed epithelialization of deep partial-thickness wounds. Wound Rep Reg. 2012;20:236-42.
[35] Gaines C, Poranki D, Du W, Clark R, Dyke MV. Development of a porcine deep partial
thickness burn model. Burns. 2012.
[36] Guzman Rd, Merrill M, Ritcher J, Hamzi R, Greengauz-Roberts O, Dyke MV. Mechanical
and biological properties of keratose biomaterials. Biomaterials. 2011;32:8205-17.
[37] Kaufman T, Levin M, Hurwitz D. The effect of topical hyperalimentation on wound healing
rate and granulation tissue formation of experimental deep second degree burns in guinea-pigs.
Burns. 1984;10:252-6.
[38] Atiyeh B, Costagliola M, Hayek S, Dibo S. Effect of silver on burn wound infection control
and healing_review of the literature. Burns. 2007;33:139-48.
[39] Santoro M, Gaudino G. Cellular and molecular facets of keratinocyte reepithelization during
wound healing. Exp Cell Res. 2005;304:274-86.
[40] Singer A, Hirth D, McClain S, Clark R. Lack of agreement between gross visual and
histological assessment of burn reepithelialization in a porcine burn model. J Burn Care Res.
2012;33:286-90.
[41] Wang X-Q, Hayes M, Kempf M, Cuttle L, Mill J, Kimble R. The poor penetration of topical
burn agent through burn eschar on a porcine burn model. Burns. 2009;35:901-6.

______________________________________________________________________________
191
Figures and Tables:
Figure 1. Schematic of randomized placement of treatments. The dorsal of a pair of swine is
depicted, with burn wounds color-coded to represent the type of treatment applied. Replicates
are numbered from 1 to 6 to represent a sample size of 6 for each treatment group.
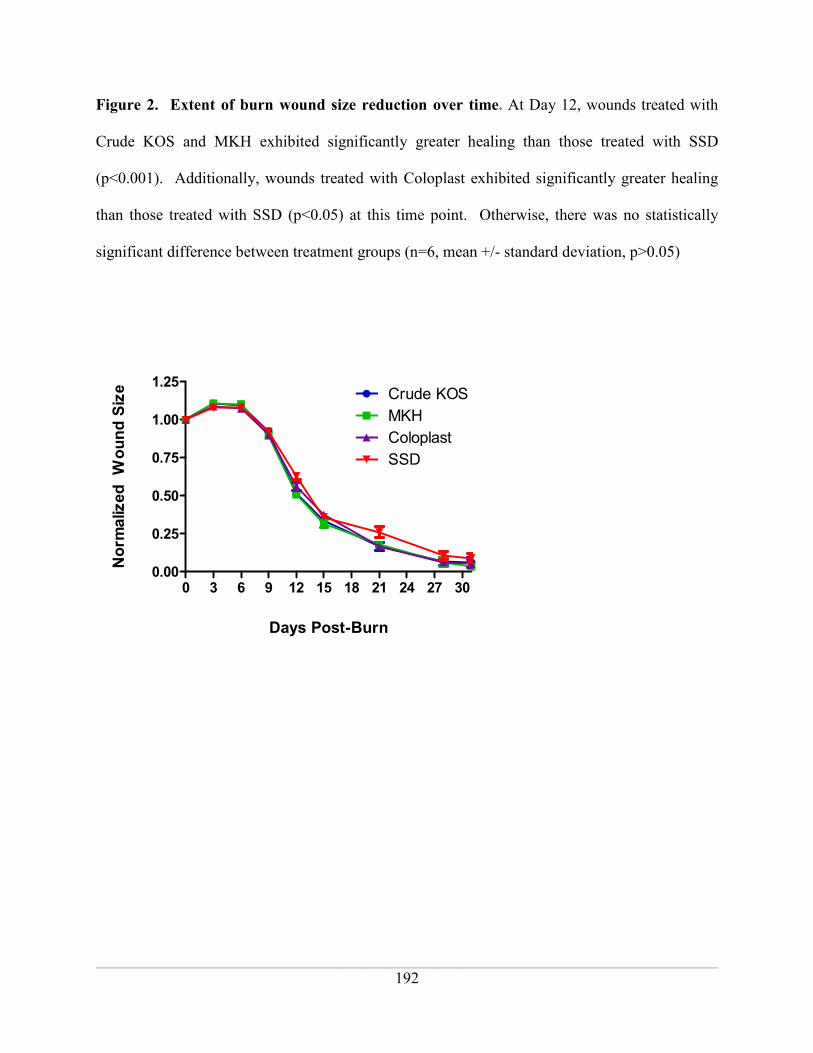
______________________________________________________________________________
192
Figure 2. Extent of burn wound size reduction over time. At Day 12, wounds treated with
Crude KOS and MKH exhibited significantly greater healing than those treated with SSD
(p<0.001). Additionally, wounds treated with Coloplast exhibited significantly greater healing
than those treated with SSD (p<0.05) at this time point. Otherwise, there was no statistically
significant difference between treatment groups (n=6, mean +/- standard deviation, p>0.05)
0 3 6 9 12 15 18 21 24 27 300.00
0.25
0.50
0.75
1.00
1.25Crude KOS
MKH
Coloplast
SSD
Days Post-Burn
Norm
alized W
ound S
ize

______________________________________________________________________________
193
Figure 3. Degree of wound contracture as a function of time. There was no statistically
significant difference between treatment groups (n=6, mean +/- standard deviation, p>0.05). On
average, MKH and Coloplast exhibited the least amount of contracture over 30 days of healing,
while Crude KOS and SSD exhibited slightly higher amounts.
0 3 6 9 12 15 18 21 24 27 30
0.90
0.95
1.00
1.05
1.10
1.15Crude KOS
MKH
Coloplast
SSD
Days Post-Injury
Norm
alized W
ound C
ontr
actu
re

______________________________________________________________________________
194
Figure 4. H&E images of deep partial thickness burn depth at Day 1. Representative
images of burned dermal tissue subject to A) Coloplast, B) Crude KOS, C) MKH D) SSD
treatments at Day 1. Fully coagulated tissue is labeled in the upper to mid dermal region, while
gradients of thermal damage is depicted by arrows within the lower dermal regions. The extent
of dermal damage remains consistent at this end point.
Figure 5. H&E images of deep partial thickness burns at Day 6. Representative images of
burned dermal tissue subject to A) Coloplast, B) Crude KOS, C) MKH D) SSD treatments at
Day 6. Emerging band of inflammatory cells is depicted by arrows within the mid to lower
dermal regions. Cell infiltration in the SSD-treated wounds is barely present in the adipose
region.

______________________________________________________________________________
195
Figure 6. H&E images of deep partial thickness burns at Day 12. Representative images of
granulation tissue subject to A) Coloplast, B) Crude KOS, C) MKH D) SSD treatments at Day
12. Fatty tissue that has been dispersed through the dermal region by the deposition of collagen
via fibroblastic activity is indicated, with scant regions present in the Crude KOS- and SSD-
treated wounds.

______________________________________________________________________________
196
Figure 7. Initial rates of re-epithelialization in deep partial thickness burns. All wounds
treated at 10 hrs post-burn demonstrated a general downward trend in reepithelialization at early
stages of treatment, with the greatest stability observed after Day 3 using MKH. Healing was
generally inconsistent across replicates as indicated by the large error bars, with the greatest
consistency observed at Day 3 using the SSD treatment. There was no statistically significant
difference between treatment groups (n=6, mean +/- standard deviation, p>0.05).
Coloplast
0 3 60
20
40
60
Days Post-Burn
% Reepithelialization
Crude KOS
0 3 60
20
40
60
Days Post-Burn
% Reepithelialization
MKH
0 3 60
20
40
60
Days Post-Burn
% Reepithelialization
SSD
0 3 60
20
40
60
Days Post-Burn
% Reepithelialization

______________________________________________________________________________
197
Figure 8. Rates of re-epithelialization in second degree burns during late stages of healing.
All treatments demonstrated an upward trend in healing. Healing was generally more consistent
across replicates as indicated by the relatively smaller error bars compared to early stages of
healing (see Figure 7). There was no statistically significant difference between treatment
groups (n=6, mean +/- standard deviation, p>0.05).
Coloplast
9 12 150
20
40
60
80
100
Days Post-Burn
% Reepithelialization
Crude KOS
9 12 150
20
40
60
80
100
Days Post-Burn
% Reepithelialization
MKH
9 12 150
20
40
60
80
100
Days Post-Burn
% Reepithelialization
SSD
9 12 150
20
40
60
80
100
Days Post-Burn
% Reepithelialization

______________________________________________________________________________
198
Figure 9. H&E images of deep partial thickness epithelium at Day 12. Representative
images of emerging epithelium subject to A) Coloplast, B) Crude KOS, C) MKH D) SSD
treatments at Day 12. All images show relatively well-organized, multilayered epithelium. Well
defined rete ridges are seen in the Coloplast and SSD treatments. The papillary dermis (as
indicated by arrows) for all treatments except Coloplast and SSD appear to be loosely packed
due to potential mechanical stress or the density of adipose tissue.
Figure 10. Degree of reepithelialization at Day 30 post-burn. Wounds treated with Coloplast,
Crude KOS and MKH KOS exhibited significantly greater healing than those treated with SSD
by Day 30 (n=6, mean +/- standard deviation, p≤0.001).

______________________________________________________________________________
199
Table 1. Initial rates of reepithelialization between Days 0 and 6. SSD exhibited a rate of 1.9
± 1.4 %re-epithelialization/day within the first 6 days of injury, at rates of 28% greater than
Coloplast, 42% greater than Crude KOS and 48% MKH.
Treatment
Rate
(% Reepithelialization/Day) R2 % Difference vs. SSD
Coloplast 2.6 +/- 1.7 0.12 -28%
Crude KOS 2.1 +/- 1.2 0.17 -42%
MKH 1.9 +/- 1.4 0.10 -48%
SSD 3.6 +/- 1.0 0.44 --
Table 2. Rates of reepithelialization between Days 9 and 15. SSD exhibited a rate of 5.7 ±
0.78 %re-epithelialization/day between Days 9 and 15, at rates of 43% greater than Coloplast,
15% greater than Crude KOS and 41% MKH.
Treatment
Rate
(% Reepithelialization/Day) R2 % Difference vs. SSD
Coloplast 3.2 +/- 1.1 0.37 -43%
Crude KOS 4.8 +/- 1.0 0.58 -15%
MKH 3.4 +/- 1.1 0.36 -41%
SSD 5.7 +/- 0.78 0.77 --

______________________________________________________________________________
200
CHAPTER 7
_____________________________________________________________________________________
SUMMARY, CONCLUSIONS AND PROSPECTIVE RESEARCH

______________________________________________________________________________
201
SUMMARY
The overall goal of this dissertation work was to test the wound healing properties of
human hair derived keratin biomaterials. Keratins represent a class of biomaterials that have been
used in a variety of biomedical applications [1-9], but the understanding of how keratin proteins
function in these applications is only at its nascent stages. Keratin biomaterial hydrogels possess
all the physical attributes of a burn wound dressing like providing a moist environment, acting as
a protective barrier and absorbing excess wound fluid. As part of this work, pilot in vivo studies
in mice and swine showed that an oxidized form of keratin biomaterial termed “keratose
hydrogel” was able to promote faster wound healing by preventing an initial increase in wound
area that often accompanies a burn (known a burn wound progression). This keratose hydrogel
was provided as a heterogeneous mixture of proteins representing “alpha” and “gamma”
fractions of keratin. These different fractions have disparate properties including molecular
weights, amino acid content, and solubility characteristics and can be separated by techniques
such as dialysis or isoelectric precipitation. Burn progression is mainly due to tissue necrosis and
therefore, we hypothesized that the interaction of keratin with burned tissue would promote cell
survival and reduce the total body surface area burned. The preceding chapters provide insight
into the differential wound healing activity associated with keratin fractions (alpha and gamma)
and the molecular mechanism by which the oxidized form of keratin biomaterial promotes cell
survival after thermal injury.
The first research aim, as presented in Chapter II, identified the keratose fraction that
displayed differential activity in salvaging thermally stressed cells. An in vitro thermal stress
model similar to Jackson’s burn model was developed in which a large number of cells died from

______________________________________________________________________________
202
the initial heat treatment, additional cells died within hours after heating, and surviving cells re-
established the culture. Using this in vitro thermal stress model, the efficacy of different keratose
preparations (alpha, gamma and crude keratose) in cell survival was tested. Our results showed
that gamma keratose was better able to maintain cell viability compared to crude keratose, alpha
keratose, and fibroblast growth media. As this is the first study to show that this keratose fraction
has particular efficacy for cell salvage after thermal stress, mass spectrometry analysis was
performed to understand the composition of proteins present in the gamma keratose fraction.
Mass spectrometry analysis results suggested that gamma keratose was a degraded protein
product of alpha keratose.
Chapter III provided an in depth analysis of the potential cell survival mechanism(s)
when gamma keratose treatment was given. Using the same thermal stress model (developed in
Chapter II), we attempted to understand whether gamma keratose induces differential expression
of cell death pathway genes as a possible mechanism of its apparent cell salvage activity. RT2
ProfilerTM
PCR array (specific to cell death pathways: apoptosis, necrosis and autophagy) was
used to investigate gene expression in thermally stressed cells treated with gamma keratose
dissolved in the media compared to cells treated with fibroblast growth media only. Our results
showed that gene expression was more significant (i.e. smaller p values) in cells treated with
gamma keratose compared to cells treated with fibroblast growth media at the early time point
post thermal stress. Additionally, we observed that treating the thermally stressed cells once with
gamma keratose was able to maintain cell viability by down regulating the genes involved in
apoptosis, necrosis and autophagy.

______________________________________________________________________________
203
The creation of highly-reproducible burn wounds is essential in the assessment of healing
progression in therapeutic models. Chapter IV explains in detail the development of a porcine
deep partial thickness burn model. This model offered control over variability through two
simple means, a burn device that creates operator-independent pressure, and a heating system
that ensures consistent and uniform heating of the brass blocks. Heating the brass blocks with an
azeotropic solution of polyethyleneglycol (PEG) and water created deeper burns compared to
boiling water only. Using the same in vivo burn model, a modified keratose hydrogel (MKH)
that contained a reduced amount of gamma keratose was tested for its wound healing properties
along with crude keratose, collagen hydrogel and silver sulfadiazine. Results showed that MKH
promoted faster re-epithelialization and wound closure compared to the other treatments. To
demonstrate potential clinical relevance and real-world practicality, a delayed treatment study
was performed with the same treatment groups wherein treatment was delayed 10 hours, thereby
representing transportation and admission to a burn unit. Results showed that MKH was still able
to promote faster re-epithelialization compared to the other treatments, but not with the same
efficacy as more immediate treatment.
LIMITATIONS AND PROSPECTIVE RESEARCH
Further investigation of the composition of the gamma keratose fraction by gel
electrophoresis and mass spectrometry identified degraded alpha keratose proteins with different
molecular weights. As gamma keratose showed differential activity in cell salvage after thermal
stress, further work needs to be done to elucidate whether the differential activity is associated
with a single excised band or multiple alpha keratose peptides. Additionally, the interaction of
gamma keratose with cells needs to be further investigated to better understand the mechanism of

______________________________________________________________________________
204
cell survival (for example, how do(does) the alpha keratose peptide(s) “communicate” with the
cell?). Ultimately, this knowledge will help to develop second generation keratin biomaterials for
wound healing.
Gamma keratose showed an overall down regulation of the genes involved in cell death
pathways (apoptosis, necrosis and autophagy). Autophagy, unlike apoptosis and necrosis
involves cell survival mechanism by maintaining normal cellular function during nutrient
deficient conditions or by removing damaged organelles and aggregated proteins [10, 11].
Therefore, additional molecular studies need to be performed to understand whether autophagy
helps in survival of thermally stressed cells when treated with gamma keratose. Also, in vitro
studies have shown that in stressed conditions like starvation and energy deprivation, cell death
occurs through autophagy when other cell death pathways (like apoptosis) are inhibited [12-14].
Further work needs to be done to understand if the down regulation of apoptotic and necrotic
genes is triggering the activation of the autophagic pathway. In addition, the effect of gamma
keratose on other skin cell types (e.g. endothelial cells, keratinocytes) should be investigated to
gain insight into the total wound healing mechanism(s) of gamma keratose.
In the in vivo studies we performed, there were some limitations that need to be
addressed in future work. Although an in vivo burn model using a burn device and an azeotropic
mixture solution successfully created highly consistent deep partial thickness burns, 3 cm burns
were not critical size wounds that convert to full thickness if proper treatment was not provided.
To study whether keratose treatment is able to prevent the conversion of the zone of stasis region
to necrotic tissue and further limit burn progression, using the contact burn comb model would
have provided more definite information. Moreover, a non-spontaneously healing model could

______________________________________________________________________________
205
have been used in which full wound closure would only happen if autografting is performed. In
adition, the experimental design for the in vivo studies used 24 hours as the earliest time point
for euthanasia, but according to Clark et al. cellular necrosis in the zone of stasis occurs between
one and four hours and apoptosis is responsible for cell death only after 24 hours. Therefore,
including early time points within hours after creation of the burn would provide more insight
into the tissue salvage mechanism of gamma keratose. Another interesting facet of this research
that should be investigated relates to the fact that the zone of stasis is characterized by reduced
blood circulation due to the clogging of blood vessels after burn injury [15]. Nunez et al. have
shown that alpha keratose and KAPs (referred to as crude keratose in this work) had vasodilation
properties when administered intravenously or topically [1]. Future investigations utilizing
keratin as an IV administered fluid may therefore be worth pursuing. In addition, performing
immunohistochemical staining specific to blood vessel may provide more insight into the role of
blood vessel plugging (early time points), or even new blood vessel formation (later time points)
when keratin treatment is given. Like many other compounds, there is likely a limit to the
beneficial dose of gamma keratose. In vitro studies in the lab have shown that very high
concentrations of gamma keratose (i.e. >10mg/mL) can be detrimental for thermally stressed
cells. Determining an optimum ratio of alpha and gamma keratose fractions in pilot studies and
then using that ratio to study burn wound healing may have provided more statistical difference
between treatment groups. Finally, determining the optimum time in which keratose treatment
can promote tissue salvage would be important information for clinical application as most
changes at the cell and molecular level are irreversable.

______________________________________________________________________________
206
CONCLUSIONS
In summary, this dissertation work outlines the importance of keratose biomaterial in the
treatment of burns. In vitro studies demonstrated that the gamma keratose fraction was able to
promote survival of thermally stressed cells by down regulating the genes involved in cell death
pathways. In vivo studies showed that keratose biomaterial was able to promote faster re-
epithelialization and wound closure in deep partial thickness burns. Although further work is
needed to understand the specific mechanism(s) of wound healing in both in vitro and in vivo
studies, particularly those related to burn wound progression, the results so far have indicated the
potential benefit of using keratin biomaterial in burn therapy.

______________________________________________________________________________
207
References
[1] Nunez F, Trach S, Burnett L, Handa R, Van Dyke M, Callahan M, et al. Vasoactive
properties of keratin-derived compounds. Microcirculation. 2011;18:663-9.
[2] Saul JM, Ellenburg MD, de Guzman RC, Van Dyke M. Keratin hydrogels support the
sustained release of bioactive ciprofloxacin. J Biomed Mater Res A. 2011;98:544-53.
[3] Dias GJ, Mahoney P, Swain M, Kelly RJ, Smith RA, Ali MA. Keratin-hydroxyapatite
composites: biocompatibility, osseointegration, and physical properties in an ovine model. J
Biomed Mater Res A. 2010;95:1084-95.
[4] Lu G, Ling K, Zhao P, Xu Z, Deng C, Zheng H, et al. A novel in situ-formed hydrogel
wound dressing by the photocross-linking of a chitosan derivative. Wound Repair Regen.
2010;18:70-9.
[5] Pechter PM, Gil J, Valdes J, Tomic-Canic M, Pastar I, Stojadinovic O, et al. Keratin
dressings speed epithelialization of deep partial-thickness wounds. Wound Repair Regen. 2012.
[6] Richter JR, de Guzman RC, Van Dyke ME. Mechanisms of hepatocyte attachment to keratin
biomaterials. Biomaterials. 2011;32:7555-61.
[7] Shen D, Wang X, Zhang L, Zhao X, Li J, Cheng K, et al. The amelioration of cardiac
dysfunction after myocardial infarction by the injection of keratin biomaterials derived from
human hair. Biomaterials. 2011;32:9290-9.
[8] Sierpinski P, Garrett J, Ma J, Apel P, Klorig D, Smith T, et al. The use of keratin biomaterials
derived from human hair for the promotion of rapid regeneration of peripheral nerves.
Biomaterials. 2008;29:118-28.

______________________________________________________________________________
208
[9] Tachibana A, Nishikawa Y, Nishino M, Kaneko S, Tanabe T, Yamauchi K. Modified keratin
sponge: binding of bone morphogenetic protein-2 and osteoblast differentiation. J Biosci Bioeng.
2006;102:425-9.
[10] Nishida K, Kyoi S, Yamaguchi O, Sadoshima J, Otsu K. The role of autophagy in the heart.
Cell Death Differ. 2009;16:31-8.
[11] Zhang Y, Calderwood SK. Autophagy, protein aggregation and hyperthermia: a mini-
review. Int J Hyperthermia. 2011;27:409-14.
[12] Shimizu S, Kanaseki T, Mizushima N, Mizuta T, Arakawa-Kobayashi S, Thompson CB, et
al. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on
autophagy genes. Nat Cell Biol. 2004;6:1221-8.
[13] Madden DT, Egger L, Bredesen DE. A calpain-like protease inhibits autophagic cell death.
Autophagy. 2007;3:519-22.
[14] Xu Y, Kim SO, Li Y, Han J. Autophagy contributes to caspase-independent macrophage
cell death. J Biol Chem. 2006;281:19179-87.
[15] Jackson DM. [The diagnosis of the depth of burning]. Br J Surg. 1953;40:588-96.

______________________________________________________________________________
209
SCHOLASTIC VITA
DEEPIKA PORANKI_______________________________________________
Wake Forest Institute for Regenerative Medicine
Wake Forest School of Medicine
Medical Center Boulevard
Winston Salem, NC-27157
EDUCATION______________________________________________________
Aug 2012 Ph.D. in Molecular Genetics
Wake Forest University
May 2005 M.S. in Molecular Genetics
Andhra University, INDIA
May 2003 B.S. in Biotechnology
Andhra University, INDIA
PROFESSIONAL EXPERIENCE_____________________________________
Jun 2012 – Aug 2012 Business Development Intern
Aerial Biopharma, Cary, NC
Aug 2007 – Aug 2012 Graduate Research Assistant
Institute for Regenerative Medicine, WFU
Aug 2011 – Apr 2012 Technology Transfer Intern
Office of Technology Asset Management, WFU
May 2011 – Oct 2011 Undergraduate Student Mentor
Institute for Regenerative Medicine, WFU

______________________________________________________________________________
210
Nov 2004 – Mar 2005 Research Intern
Central Tobacco Research Institute, INDIA
Aug 2003- Sep 2004 Graduate Teaching Assistant in Biochemistry
Andhra University, INDIA
HONORS AND AWARDS___________________________________________
Aug 2007 – Aug 2012 Graduate Student Fellowship
Wake Forest University Graduate School
Apr 2012 NC Biotech Center Summer Internship Award
North Carolina Biotech Center
Jan 2012 NABS Research Scholarship
North American Burn Society
Jan 2012 NABS Student Travel Award
North American Burn Society
Jan 2012 Alumni Student Travel Award
Wake Forest University
Nov 2011 Graduate Student Travel Award
Institute for Regenerative Medicine, WFU
Oct 2011 Grand Prize – Poster completion
10th
Annual Charlotte Biotechnology Conference
Jun 2011 AFIRM Fellow’s Career Enrichment Award
Armed Forces Institute for Regenerative Medicine
Jun 2008 Finalist – National Student Scholarship
Paul Foundation, INDIA
May 2005 University Second by GPA – Molecular Genetics
Andhra University, INDIA

______________________________________________________________________________
211
PUBLICATIONS___________________________________________________
Poranki, D *., Gaines, C
*., Du, W., Clark, R.A.F., Van Dyke, M.E. Development of deep partial
thickness burn model. (accepted in Burns Journal)
Poranki, D., Whitener, W., Howse S., Mesen, T., Howse, B., Burnell, J., Greengauz-Roberts,
O., Molnar. J., Van Dyke. M.E. A keratin biomaterial promotes skin regeneration after burns in
vivo and rescue cells after thermal stress in vitro. (manuscript in preparation)
Poranki, D., Van Dyke, M.E. Gamma keratose maintains cell viability in vitro after thermal
stress by regulating the expression of cell death pathway specific genes. (manuscript in
preparation)
Poranki, D., Gaines, C., Van Dyke, M.E. Assesment of deep partial thickness burn treatment
with keratin biomaterial hydrogels in a swine model. (manuscript in preparation)
Gaines, C., Poranki, D., Van Dyke, M.E. Delayed treatment of porcine deep partial thickness
burns subject to therapeutic keratin biomaterials. (manuscript in preparation)
ABSTRACTS – PODIUM PRESENTATIONS__________________________
Poranki ,D., Gaines, C., Van Dyke, M.E., Dec 2011. Development of a pivotal pre-clinical
porcine deep partial thickness burn model and testing of a keratin biomaterial hydrogel product.
Tissue Engineering and Regenerative Medicine North America (TERMIS-NA). Houston, TX
Poranki ,D., Gaines, C., Van Dyke, M.E., Jan 2012. A pre-clinical study testing the
effectiveness of keratin biomaterial hydrogel in porcine deep partial thickness burns. North
American Burn Society. Utah., CO
ABSTRACTS – POSTER PRESENTATIONS__________________________
Poranki, D., Gaines, C., Van Dyke, M.E., Mar 2012. Keratin biomaterial in the treatment of
burns. Wake Forest Institute for Regenerative Medicine Annual Retreat.
Poranki, D., Gaines, C., Van Dyke, M.E., Mar 2012. Keratin biomaterial in the treatment of
burns. Wake Forest University Graduate Student and post-doctoral Research Day.

______________________________________________________________________________
212
Gaines C, Poranki D, Van Dyke ME., Dec 2011. Optimized deep partial thickness swine burn
model for the evaluation of keratin hydrogel treatments. Materials Research Science.
Poranki, D., Gaines, C., Van Dyke, M.E., Nov 2011. Keratin biomaterial helps in the survival of
thermally stressed fibroblasts in vitro and promotes wound healing in vivo. Division of Surgical
Sciences 19th Annual Resident’s and Fellow’s Research Day.
Gaines, C., Poranki, D., Van Dyke, M.E., Nov 2011 Keratin Mediated healing of deep partial
thickness burns. Division of Surgical Sciences 19th Annual Resident’s and Fellow’s Research
Day.
Poranki, D., Gaines, C., Van Dyke, M.E., Nov 2011. Development of a porcine deep partial
(2⁰) thickness burn model. North Carolina Tissue Engineering & Regenerative Medicine
(NCTERM).
Poranki, D., Gaines, C., Van Dyke, M.E., Oct 2011. Keratin Biomaterial for Burn Wound
Healing. 10th Annual Charlotte Biotechnology Conference.
Gaines C, Poranki D, Van Dyke ME., Oct 2011. Optimized Wound Creation in a Deep Partial
Thickness Porcine Burn Model. Biomedical Engineering Society Annual Meeting (BMES).
Poranki, D., Gaines, C., Van Dyke, M.E., Jan 2011. Development of a porcine deep partial (2⁰)
thickness burn model. AFIRM All Hands Meeting.
Poranki, D., Gaines, C., Van Dyke, M.E., Dec 2010. Keratin Biomaterials Augment Burn
Wound Healing. Tissue Engineering & Regenerative Medicine North America (TERMIS-NA).
Poranki, D., Gaines, C., Van Dyke, M.E., Aug 2010. Keratin Biomaterials Promote Cell and
Tissue Survival in Burns. ATACC Meeting.
Poranki, D., Gaines, C., Van Dyke, M.E., Mar 2012. Heat shock response of Keratin
Biomaterial on Primary Mouse Fibroblasts – An in vitro Study. Wake Forest University
Graduate Student and Post Doctoral Research Day.
Poranki, D., Gaines, C., Van Dyke, M.E., Jan 2010 Effect of Keratin Biomaterials on Thermally
Injured Skin cells. AFIRM All Hands Meeting.
Poranki, D., Vinukonda, N., Katakam, A. Data Mining Colorectal Cancer CGH data to
investigate gender disparities. Association of Genetic Technologists Annual Meeting.

______________________________________________________________________________
213
Poranki, D. Are we losing hope for a cure to Down’s syndrome? Annual Meeting of American
College of Clinical Genetics.
Poranki, D., Sarala, K., Aug 2005 An RAPD based genetic diversity study to determine the
genetic similarity among advanced tobacco breeding lines. Central Tobacco Research Institute
Research Day.