kinetic resolution of unnatural and rarely occurring amino
TRANSCRIPT

Reprinted from the Journal of the American Chemical Society, 1989, /l l-, 635'1.Copyright @ 198^9 by the American Chemical Society and reprinted by permission of the copyright owner.
Kinetic Resolution of Unnatural and Rarely Occurring AminoAcids: Enantioselective Hydrolysis of l/-Acyl Amino AcidsCataly zed by Acylase Ir
H. Keith Chenault,2 Jiirgen Dahmer,3 and George M. Whitesides*
Contribution from the Department of Chemistrl', Ilaruard Llniuersitl,,Cambridge, Massachusetts 02138. Receiued October 25, 1988
Abstract: Acylase I (aminoacylase; N-acylamino-acid amidohydrolase, EC 3.5.1.14, from porcine kidney and the fungusAspergi l lus) is a broadly appl icable enzymatic catalyst for the kinetic resolut ion of unnatural and rarely occurring a-aminoacids. I ts enantioselectivi ty for the hydrolysis of N-acyl L-a-amino acids is nearly absolute, yet i t accepts substrates havinga wide range of structure and functional i ty. This paper reports the init ial rates of enzyme-catalyzed hydrolysis of over 50N-acyl amino acids and analogues, the stabi l i t ies of the enzymes in aqueous and aqueous/organic solut ions, and the effectsof dif ferent acyl groups and metal ions on the rates of enzymatic hydrolysis. E,leven a-amino and a-methyl a-amino acidswere resolved on a 2-29-9 scale. Crude L- and o-amino acid products had general ly ' >90% ee. The uti l i ty of resolved aminoacids as chiral synthons was i l lustrated by the preparation of (R)- and (S)-l-butene oxide and the diastereoselective (cis:trans,7-8:1 ) iodolactonization of three 2-amino-4-alkenoic acid derivatives.
Acylase I (aminoacylase; N-acylamino-acid amidohydrolase,EC 3.5.1.14) catalyzes the enantioselective hydrolysis of N-acylL-amino acids (eq 1).+7 Acylase I enzymes isolated from porcine
?o'" fl acyrase | - ?oo- :o'* fl.5rA*, *,An,*"* + ,/-"A*,
(1)
kidney f i*ol and the fungus ,trp,rrgit tu, ,o ioo, are com-mercial ly avai lable, inexpensive, stable in solut ion, and high inspecific activity. Here we describe the use of these enzymes ascatalysts for the kinetic resolution of a-amino acids. We reportthe range of substrates accepted by each enzyme, factors influ-encing the activi t ies and stabi l i t ies of the enzymes, and methodsfor the preparative-scale resolution of representative compounds.Both l- and o-amino acid products were obtained with high(general ly >90Vo) enantiomeric excesses.
Amino acids are important as biomedically active compoundssand as chiral direct ing auxi l iar iese'10 and chiral synthonse'r l-13 in
(l) This research was supported by the National Institutes of Health,Grant GM 30367, and by a grant from Firmenich, SA.
(2) NIH Train ing Grant Trainee, 1984-1985 (Grant 5-T32-GM-07598).(3) Deutsche Forschungsgemeinschaft Postdoctoral Fellow, I 986- I 987.(4) Greenstein, J. P.; Winitz, M. Chemistry of the Amino Acids; Wiley:
New York, l96l ; Vol . 2, pp 1753-1767.(5) Greenstein, J. P. Methods Enzymol.1957, 3,554-570.(6) Birnbaum, S. M.; Levintow, L.; Kingsley, R. B.; Greenstein, J. P. .r.
Biol. Chem. 1952, 194,455-470.(7) (a) Chibata, I.;Tosa, T.; Sato, T.; Mori, T. Methods Enzymol.1976,
44,'146-7 59. (b) Immobilized Enzymes'. Research and Deuelopment: Chi-bata, L, Ed. ;Wi ley: New York, 1978;pp 168-178. (c) Tosa, T. ; Mor i , T. ;Fuse, N.; Chibata, L Biotechnol. Bioeng. 1967,9,603-615.
(8) (a) Barrett, G. C., Ed. Chemistry and Biochemistry of the AminoAcids:Chapman and Hall: London, 1985. (b) Wagner,l.;Musso, H. Angew.Chem., Int. Ed. Engl.1983, 22,816-828. (c) Montreuil, J. In ComprehensiueBiochemistry; Neuberger, A., van Deenen, L. L. M., Eds.; Elsevier: Am-sterdam, The Nether lands, 1982;Vol . l98, Part I I , pp l -188. (d) Pais, M.;Jarreau, F.-X. In Chemistry and Biochemistry of Amino Acids, Peptides, andProteins; Weinstein, B., Ed.; Marcel Dekker: New York, l97l;Vol. l, ppl2'l-157. (e) Amino Acids, Pept., Proteins (Spec. Period. Rep.) 1976, 8,326-327. (0 Davies, J. S. In Chemistry and Biochemistry of Amino Acids,Peptides, and Proteins; Weinstein, B., Ed.; Marcel Dekker: New York, 1977;Vol. 4, pp l-27. (g) Amino Acids, Pept., Proteins (Spec. Period Rep.) 1983,t4,432-502.
(9) Coppola, G. M.; Schuster, H. F. Asymmetric Synthesis'. Constuctionof Chiral Molecules Using Amino Acids: Wiley: New York, 1987.
(10) (a) Evans, D. A. In Asymmetric Synthesis; Morrison, J. D., Ed.;Academic: Or lando, FL, 1984; Vol . 3, pp 2-110. (b) Lutomski , K. A. ;Meyers, A. l. Ibid. pp 213-274. (c) Enders, D. Ibid. pp 275-3a1. (d)Martens, J. Top. Curr. Chem.1984, 125,165-246.
o Refcrencc -1 I . h Ref 'e rence J9. 'Refe rencc 24. d Reference 38." Rcference . l I ' Re ferencc 40 g Ref 'erencc J5. ' Reference 36.ispeci f ic actrv i t \ was deternt ined as descr ibed in Resul ts and Exper i -mental Sect ion. /Costs are fbr enzymes as research biochemicals (1988pr i ce l i s t , S igma Chemica l Co . ) and thus a re upper l im i t s . Approx i -mately 1000 U wi l l catalyze the hydrolysis o i i mol of acyl- t - -aminoacid per day.
organic synthesis. Both o- and L-amino acidsr4 and a-methyl
amino ac ids have impor tan t uses and a re thus des i rab le i n ho -
mochiral form Methods for preparing enantiomerically enriched
amino ac ids such as f c rn ren ta t i on and enzymat i c ca ta l ys i s , l 5
( l l ) (a) t -ubel l . W. D.; Rapoport . H. J. Am" Chem. Soc. 1987, 109,236-239. (b) Kunz, H. ; Lerchen, H. G. Tetrahedron Let t .1987,28,1873-1876; 1985, 26,525'l-5260. (c) t{ernot, S.; Larcheveque, M.; Petit, Y. Synth.Commun.1986 , 16 . 183-190 . (d ) Hag iwara , l l . ; K imura , K . ; Uda , F I . " r .Chem. Soc. Chem. Commun.1986, 860-861. (e) Mzengeza, S. ; Yang, C.M.; Whitney, R. A. J. Am. Chem. Soc. 1987. 109,276-2'7 '7. (0 Schur ig, V. ;Leyrer, U. ; Wistuba,D. J. Org. Chenr. l986, J l , 242-245. (g) Maurer, P.J. ; Knudson, C. G.; Palkowitz, A. D. ; Rapoport , H. . / . Org. Chem.l985, 50.325-332. (h) Knudson, C. G.; Rapoport, H. J. Org. Chem. 1983, 48,2260-2266. (i) Mori, K.: Sasaki, M.; Tamada, S.; Suguro, T.; Masuda, S.Tetrahedron 1979. 3 5. I 601 -l 605.
( l2) Kurokawa, N.; Ohfune, Y. J. Am. Chem. Soc. 1986, 108,6041-6043.(13) Ohfune, Y. ; Kurokawa, N. Tetrahedron Let t .1985,26, 5307-5308.( 1a) By convention. we refer to the configuration at the a-carbon of amino
acids as o or l. The more mmmon, naturally occurring L-amino acids all havethe (S) configuration, except for r--cysteine, which, because of priorit izingsulfur over oxygen, has the (R) configuration.
(15) (a) Kirk-Othmer Encyclopedia af Chemical Technology,3rd ed.;Wi ley: New York, 1978; Vol . 2, pp 376"410. (b) Considine, D. M., Ed.Chemical and Process Technology Encyclopedia; McGraw-Hill: New York,1974; pp 93-106. (c) Aida, K. , Chibata, I . , Nakayama, K. , Takinami, K. ,Yamada, H., Eds. Biotechnology of Amino Acid Produclion; Kodansha:Tokyo, 1986. (d) Hami l ton, B. K. ; Hsiao, H. Y. ;Swann, W. E. ; Anderson,D. M.; Delente, J. J . Trends Biotechnol . l985, J,64-68. (e) Yamada. H. ;Shimizu, S.; Shimada, H.; Tani, Y.; Takahashi, S.l Ohashi, T. Biochimie 1984,62,395-399. (l) \ 'amada, H.; Kumagai, H. Pure Appl. Chem. 1978, 50,ll lT-1127 . (g) Yonaha. K.; Soda, K. ln .Aduances in Biochemical Engi-neering Biotechnology; Fiechter. A., Ed.;Springer-Verlag: Berlin, 1986;Vol.33, pp 95-130. (h) Chibata, I.; Tosa, T.; Sato, T. Methods Enzy'mol. 1976,44 .739-746 .
1989 American Chemical Society
Table I. Pronerties of Acvlase I
porc ine k idney Aspergillus
no . o f subun i t sMW (d imer )op t imum p l lK . ( ch lo roacc t r l -A la ) . m \ ' lZn2+ pe r subun i tKo*.1Zn2+;, Ml(dirr(Co2+), Messent ia l Cys per subuni td ipept idase act ivysp ac t i vy , t U / *gdo l l a rs / 1000 [ , ' J
2a
85 500'7 .0 . -9 .0d6. ( td
l "
1 6 - l 0 e
1O-re
lo,n
yes/
302 . 1
2u73 000D6.0 -8 .5D6 . 3 DJ '
l0-r0/10-1 sIga
no"0 . 3 02 . 3
0002-7863 lS9 l rs l l -6354$01.s0 /0 @

L'nantioselectiDe Hydrolysis of N-Acyl Amino Acids
Chart I. Amino Acids Tested As Their Acyl Derivatives for Substrate Activity with Acylase
coo-I
RANH.*
A m .
I
t 9 8 9 6 3 5 5
R comoound comoound R comoound
CHs
cH3cH2
7ct-t,
(cH3)2cH
(cH3)3c
IA"r,
4cu,
ZVCH2
*cx,%"r,
C"*,I
4"r,
v r 1 2
1
2
3
4
5
z>,\A^cH,
ctcH2
HOCH2
g O l A C H z
lo,,..#cl,
ro lcur) f cH,
xo]:^cH,
,oX"*,
ruc -cx,
H@C^CHz
xcoc:AcHz
€",cooH
o cooHi l l
4H, ,Vct ,H
o
, r *Aar ,
il)cr,
/n-o cH,
Ctrt'""
D"*,G"*,0..^"*,
1r1C14CHz etr'*""
1 7
1 8
1 9
20
2 1
22
Z J
24
25
26
27
28
29
30
31
6
7
I
9
1 0
1 1
1 2
1 ?
1 4
1 5
1 6
34
35
36
37\^cH,
A"*
$cuz
chemica l reso lu t ion, l6 and asymmetr ic synthes is lT-22 u;1 1 . ruu.l i m i t a t i o n s .
\ \ 'e have examined k inet ic reso lu t ions us ing acr lase I as acc inren ient route to unnatura l and uncommon amino ac id : h . r r rnph rgh ( )9Sc r ' r . ) enan t i omer i c excesses . l r ( j r ccn : t c rn c l i i l . ^ : - r - : -
and ch ibat " . , u1.7 '28.2e have s tud ied acr lase I i r . rm F l rcrnc krdner
Char t I I . r r -Me thv l ,Aminc rF o r S u b s t r a t c \ c t i v i t r r r i t h
{ s T h c i r { c r I D c r r r . r t i r c rAc ids Tes ted\ c r l a s e I
l J a ^ n n -r r ? v V V V
?,^ Nr ' , , *
conF U
r l 6 t ra r Grec 'n ' t e r : . . J P . \ \ . : : l z . \ l ( -hemts t r r o , l ' t he . l n t tno . c ids .\ \ r . e r \ g u \ ' , r 1 k . t g b . . \ o i , . p p -
1 - i - ' 6 0 t b ) E g u s a . S . : S i s i d o . \ { . .I : r ; r . s : : \ 8 : , , ' i Chenr 5 , . , c Jpn l9 t l 6 . - {9 .3175-3 . l78 . ( c ) Bo l l i nge r , F .\ \ J \ {ed Chem 1911. la. l ' t3-311 (d) Bajgrowicz, J. A. ; Cossec, B. ;Prgrere. C . Jacqure r . R. ; \ ' ia l lefont , P. Tetrahedron Let t . 1984, 25,i '59- I i9 : . (e ) Terashima. S. ; Achiwa, K. ; Yamada, S. Chem. Pharm. Bul l .1966. i 4. I 138- l 143. ( l ) Yamaguchi , T. ; Nishimura, K. ; Shinbo, T. ; Sugiura,\1. Chem. Let t . 1985, 1549-1552. (g) Viret , J . ; Patzel t , H. ; Col let , A. Tet-rahedron Lett. 1986, 27, 5865-5868.
i l7) (a) Morrison, J. D., Ed. Asymmetric Synthesis, Chiral Catalysis:\cademic: Or lando, FL, 1985; Vol . 5. (b) Knowles, W. S. ,4cc. Chem. 'Res.19 t3 . 16 . 106-112 . ( c ) V ineyard , B . D . ;Know les , W.S . ;Sabacky , M. J . / .r l r t l ( -ata l .1983, 19,159-169. (d) col lman, J, p. ; Hegedus,L.s. pr incipresan,:l .lpplications of Organotransition Metal Chemislry; Universitv Science:\ l r l l \ a l l ey , CA, 1980 ;pp 341-349 . (e ) V igneron , J . p . ;Kagan , H . ;Horeau ,- \ Terrahedron Leu.1968, 5681-5683. ( f ) Corey, E. J. ; McCaul ly , R. J. ;Sachdev. H. S. " / . Am. Chem. Soc.1970.92,2476-2488 (e) Corey, E. J. ;S. ichdev, H. S. ; Gougoutas, J. Z. : Saenger, W. Ib id. 2488-t5} l .
tt.!) (a) Schollkopf, U Top. Curr. Chem. t983, 109, 65-84. (b)Scholl.kopf, U. Pure Appl. Chem. 1983, jJ, 1799-1806. (c) Schollkopf, U.I 'et rahedron 1983,39,2085-2091. (d) Schol lkopf , U. ; Busse, U. ; Lonsky,R . Hinr ichs, R. L iebigs Ann. Chem. 1986, 21 50-2163.
t l9) (a) Seebach, D. ; Mi l ler , D. D. ; Mul ler , S. ; Weber,T. Helu. Chim.l i ' ra 1985, 68,949-952. (b) Seebach, D. ; Aebi , J . D. ; Naef, R. ; Weber, T.Heb. Chim. Acta 1985,68, 144-1559 (c) Ikegami, S.; Hayama, T.; Katsuki,T : ) 'amaguchi, M.; Tetrahedron Lett. 1986, 27,3403-3406.
(20) (a) Evans, D. A.; Weber, A. E. "r. Am. Chem. Soc. 19g6. /0g.6157-6761. (b) Evans, D. A. ; Sjogren, E. B. ; Weber, A. E. ; Conn, R. E.Tetrahedron Lett. 1987, 28,39-42. (c) Seebach, D.; Juaristi, E.; Miller, D.D ; Schickli, C.; Weber, T. Helu. Chim. Acta 1987, 70, 237-26t. (d) Ito, y.;Sa*amura, M.; Hayashi ,T. J. Am. Chem. Soc. 1986, t0S,64} i -e406.
(21) (a) Wi l l iams, R. M,; Sincla i r , P. J. ;Zhai , D. ;Chen, D. J. Am. Chem.Soc 1988, I 10, 1547-155'7. (b) Sincla i r , P. ; Zhai , D. ; Reibenspies. J. :\ \ ' r l l iams, R. M. " / . Am. Chem. Soc. 1986, 108, I103-1104. (c) Oppolzer,\\ ' ; Pederosa, R.; Moretti, R. Tetrahedron Lett. 1986, 27,83t-93a. (d)Schollkopf. U.; Neubauer, H. J.; Hauptrief, M. Angew. Chem., Int. Ed. Engi.1985. 24. 1066-1067.
cH2+l
HOCH2
HOOC^CH2
-s cH.
(\V.*,Ho'r^il
9.r,OH
HoYAl
9"*,
G.,r"t'\ )N-H
39
40
41
42
and (Aspergillus oryzae), respectively. The physical and kineticpropert ies of both enzymes have been studied.3ca0 Propert iesrelevant to their applications in synthesis are summarized in TableI .
Our purpose in this work was to establ ish the breadth of ut i l i tyof acylase I for resolving amino acids and analogues. We havemeasured both init ial rates and enantioselectivi t ies of hydrolysescatalyzed by acylase I. Only isolated resolut ions of unnaturalamino acids using acylase I have been reported previously.al

6356 J . Am. Chem. Soc. , Vo l . I I1 , No. 16, 1989
Results
Potential Substrates. Charts I-III list compounds examinedas potential substrates. These compounds comprise acyl derivativesof simple a-amino acids (l-38), a-methyl a-amino acids (39-46),
B-amino acids (47, 48), an a-aminophosphonic acid (49), and twoa-hydroxy acids (50, 5l), as well as heterocycl ic compounds 52and 53. Acyl groups employed were acetyl (A), chloroacetyl (CA),and methoxyacetyl (MA).42
Many of the amino ac ids (8 , 9 ,12-14.19, 20, 23-29.31, 35,37, 38) were made by the phase-transfer-catalyzed (PTC) al-kylat iona3 of ethyl N-(diphenylmethylene)glycinate (sqg (ChartIV). This procedure worked well and general l l gave free aminoac id in 30-55% y ie ld . Ket imine 54 uas impor tant as the g l1 'c ineenola te equiva lent . In our hands. PTC a lk l la t ion o f the a ld imine.ethy l A- [ (4-ch lorophenv l )methy lene]g l ic inate (55) .a3 led on l \ tohydro lys is o f 55.
Dieth l ' l acetamidomalonate (56) is another usefu l g l lc ineequivalent. Alkylat ion of 56 in sodium ethoxide/ethanol. fol lowedby saponif icat ion and decarboxylat ion, gave acetyl amino acidsl0-A, l l -A,45 16-A, and 36-4 in 40-65Va yields. This procedurewas especially useful for preparing compounds for enzymatic
(22) (a) B,vans, D. A.; Britton, T. C.; Dorow, R. L.; Dellaria, J. F. J. Am.Chem. Soc. 1986, /08,6395-6397. (b) Gennar i , C. ;Colombo, L. ; Berol in i ,G. J. Am. Chem. Soc. 1986, 108,6394-6395. (c) Tr imble, L. A. ; Vederas,J. C. /. Am. Chem. Soc. 1986, 108,6397-6399. (d) Oppolzer, W.; Moretti,R. Heb. Chim. Acta 1986,69,1923-1926. (e) Evans, D. A. ; El lman, J. A. ;Dorow, R. L. Tetrahedron Let t . 1987, 28, l123-1126.
(23) Other enzymat ic k inet ic resolut ions of amino acids or amino acidprecursors: (a) Chen, S.-T. : Wang. K -T. : Wong. C.-H. J Chent. Sr. tc . . ( 'hent .C o m m u n . 1 9 8 6 . l 5 l 4 - 1 5 1 6 . ( b ) L a l o n d e . J . J . ; B e r g b r e i t e r . D E : \ \ ' o n q .C . -H J . Org . Chem,1988 . -5J . 2323-2321 . ( c ) Roper . J \1 . Bauer . D PS y n t h e s i s 1 9 8 3 , l 0 1 l - 1 0 4 3 1 d ; A n a n t h a r a m a i a h . G \ 1 . R o e s k e . R \ \Te t rahedron k t t . l f f l 2 . : J . 33 - r5 -1316 te ) Tu rk . J . : Panse . G T : \ {a rsha l l .G . R . . f . Org . Chem. 1975 . 40 .953-955 t f ) B . lo rk l rng . F . Boure l l e . J .Hjalmarsson. M.; Hul t . K. ; \or in. T . J . Chem. Scrc. . Chem. Commun. 1981 .l 0 4 l - 1 0 4 2 .
(24 ) Rao , K . R . ; B i rnbaum, S , M. ; K ings le l , R . B . ; Greens te in . J .P . J .Bio l . Chem. 1952. 198. 501-524.
(25) Fu. S.-C. J. ; Bi rnbaum, S. M. " / . Am. Chem. Soc. 1953, 75,918-920.(26) Fu, S.-C. J. ; Bi rnbaum, S. M.; Greenstein, J. P. J. Am. Chem. Soc.
1954 . 76 .6054-6058 .(27) Marshal l , R. ; Birnbaum, S. M.;Greenstein, J. P. J. Am. Chem. Soc.
1956, 7 8, 4636-4642.(28) Tosa, T. ; Mor i , T. ; Fuse, N. ; Chibata, l . Enzymologia 1967,32,
I 5 3 - t 6 8 .(29) Tosa, T,; Mori, T.; Chibata, L Enzymologia 1971,40. 49-63.(30) Kordel, W.;Schneider,F. Hoppe-Seyler's Z. Physiol. Chem. 1916.
3 5 7 . t t 0 9 - l l r 5 .(31) Kordel , W.; Schneider, F. Biochim. Biophvs. .4cta 1916. 445.
446-457.(32) Kordel , W.; Schneider, F. Z. I ' ia tur forsch. . C 1971. J. 'C. 337-341 .(33) Kordel , W.; Schneider, F. Z. IVatur forsch. . C 1971. 3)C. 342-344.(34) Frey, J. ; Kordel , W.; Schneider, F. Z . \ 'a tur forsch. . C 1917, 32C,'t
69-'t7 6.(35) Kumpe, E. ; Lof f ler . H.-G.; Schneider. F. Z. i la tur forscfr . , C 1981.
3 6 C , 9 5 r - 9 5 5 .(36) Szajdni , B. ; Kiss, A. ; Boross, L. Acta Biochim. Biophys. Acad. Sci .
Hung. 1980, 1 5, 29-31.(37) Lof f ler . H.-G.;Schneider, F. Bio l . Chem. Hoppe-Seyler 1987,368,
48 r -48 5.(38) Kordel, W.; Schneider, F. Hoppe-Seyler's Z. Physiol. Chem.1975.
356 ,915-920 .(39) Gentzen, L; Loffler, H.-G.; Schneider, F. Z. Naturforscfr., C 1980,
JJC, 544-550.(a0) Gilles, I.; Loffler, H.-G.; Schneider, F. Z. Naturforsch., C 1981, J6C.'t
5t-'7 54.(41) (a) Kel ler , J . W.; Hami l ton, B. J. Tetrahedron Leu. 1986, 27,
1249-1250. (b) Schwyzer, R.; Karlaganis, G. Ann. Chem.1973, 1298-1309.(c) Jansen, A. C. A. ; Weustnik, R. J. M.;Ker l ing, K. E. T. ;Havinga, E. Recl .Trau. Chim. Pays-Bas 1969,88, 819-827. (d) Baldwin, J. E.; Haber, S. B.;Hoskins, C.; Kruse, L.l. J. Org. Chem.1977,42,1239-1241. (e) Morishima,H.; Yoshizawa, J. ; Ushi j ima, R. ; Takeuchi , T. ; Umezawa, H. J. Ant ib iot .1982, .t5, 1500-1506. (0 Scannell, J. P.; Pruess, D. L.; Demny, T. C.; Sello,L. H. ; Wi l l iams, T. ; Stempel , A. J. Ant ib iot .1972, 25,122-127. (g) Sahn,U.; Knobloch, G.; Wagner, F. " I . Ant ib iot .1973, 26,389-390.
(42) Acyl derivatives of amino acids and amino acid analogues l-51 aredesignated by the appropriate number followed by suffixes indicating the acylgroup: A (acetyl), CA (chloroacetyl), and MA (methoxyacetyl).
(43) O'Donnell, M. J.; Wojciechowski, K. J. Chem. Soc., Chem. Commun.1 9 8 4 , 3 1 3 - 3 1 5 .
(44) O'Donnell, M. J.; Polt, R. L. J. Org. Chem. 1982,47,2663-2666.(45) Coul ter , A. W.; Tala lay, P. J. Bio l . Chem.1986,243,3238-3247.
Chenault et al.
0 5 1 0 1 5 2 0 2 5 3 0
t , m inFigure l. Colorimetric determination of the initial rates of hydrolysis of.V-acety lmethionine (O), 1V-chloroacety l -a-cyclopropylg lyc ine (O), N-acet ; - l - r ' i -s-r r -croty ' lg lyc ine ( f ) , and . fv--acety l -a-propargylg lyc ine (O)c a t a l r z e d b r P K A .
0 4 0 8 0 1 2 0 1 6 0
t , h
Figure 2. Stability of soluble acylase I from (a) porcine kidney and from(b) Aspergil lus. Enzymes were incubated in 0.10 M phosphate buffer,pH 7.5, at25 "C, under ambient atmosphere (O) and under nitrogen (O).
Chart I I I . Analogues of a-Amino Acids Tested for SubstrateAc t i v in r . r i t h .Ac l l ase I
NH.+
ftr:* {,coo- ?o.t- oH
)-.coo | "1'ANHo+ ACOoH
47 48 49 50
1 . 0
0 .8
o 0 . 6t -
0 .2
0 .0
= r a
;H 1 6
"tQ 1 .4
t , a
2 A
1 . 01 6 01 2 08 04 0
OHI
,1Afcoox\2
51
ooFrun h\A.oo, tt.A.oo,..
52 53
Chart IV. Glycine Enolate Equivalents
N , C O O E T
54
N _ COOET
55
HNAcI
Erooc^cooEt
56
a-amino acids wereresolution, because the product N-acetylsubstrates of acylase L
Free amino acids were generally acylated with the appropriateacyl chloride in 4 N NaOH.25 Alternatively, acetylat ions wereperformed with acetic anhydride/acetic acid. We found, as haveothers,6'46 that chloroacetylat ions of amino acids under Schot-
(a6) (a) Ronwin, E. J. Org. Chem.1953, 18, 127-132. (b) Ronwin, E.Ib id. t546-1553.

Enantioselectiue Hydrolysis of N-Acyl Amino Acids
ten-Baumann type condit ions with either chloroacetyl chlorideor chloroacetic anhydride suffer from side reactions, low yields(often lV2UVo), and difficult purifications. Ronwin reported thechloroacetylation of amino acids in ethyl acetate,6 but yields weresti l l variable and low. we found that a variety of amino acidsgave satisfactory (50-90vo) yields of chloroacetylated product indry, refluxing acetonitrile. The method worked particurarly wellwith a-methyl a-amino acids and other hydrophobic amino acids,but fai led with hydrophil ic amino acids such as alanine andglu tamic ac id .
Sources and Specific Activity of Acylase I. Although acylaseenzymes are present in various mammalian t issues and mi-croorganisms, commercial preparations of acylase I (or amino-acylase) are isolated from porcine kidney or from Aspergillus.Both enzymes may be purified to a specific activity of around 300U/rg," b.ut commercial preparations vary widely in their specificactivi t ies.aT The results presented in this pup.. were obtainedwith enzymes from Sigma (Table I).
Kinetic Analysis. The activi ty of acylase I with potentialsubstrates was determined by measuring the init ial rate of en-zyme-catalyzed hydrolysis of racemic acyl amino acid (, lO m\Iin 0 .10 M potass ium phosphate buf fer . pH 7.5 . ,10 oC) . , { l iquotsof the hydro ly ,s is react ions \ \ 'e re removed per iod ica l l r . quenched* i t h pe rch lo r i c ac id . and assa red f o r f r ee am ino ac rd b r co ro r i -me t r i c de te rm ina t i on u i t h n i nh ld r i n tF i gu re I ) ao The ra tes o r 'h;-droly'sis were cal ibrated br measuring the colorimetr ic respxrnseof the f ree amino ac ids wi th n inh l 'd r in and uere normar ized tothe ra te o f hydro lys is o f A-acety ' l -or - -meth ion ine (AcMet . , V, =100) . AcMet is commerc ia l ly ava i lab le , inexpens ive, and anexcel lent substrate for both enzymes.6.27
control experiments included incubating potential substratesin the absence of enzyme and incubating the enzymes in theabsence of potential substrate. only 22-cAa2 and 40-cA exhibitedspontaneous hydrolysis under the condit ions of the assay, and so22-\ lA and 40-MA were used for measurements of enzymatichrdro l ls is . PKA ( l mg ml- t ) showed no increase in co lor imetr icresponse af ter l0 h . AA, however , a lways exh ib i ted a smal lbackground increase in absorbance or *false activi ty". The ab-sorbance always increased more sharply ini t ial ly and then reveledof f . D ia lys is o f the enzyme d id not e l iminate the phcnomenon.Therefore, when slowly reacting substrates \r 'ere assa\ed rr i th .{.{ .the enzy 'me was a l lowed to rest in buf ier so lur ion l 'o r t ' h be iorca s s a r i n g , a n d a b l a n k r e a c t i o n c o n t a i n i n g o n l r . { . { r r a , r u n i nparal lel. The dif ferences in absorption bet*een rhe assar reacrionand the con t ro l ue re used t o ca l cu la te t he ra te o f enz rma t i chrdro l rs is . In pract rce. a l l compounds that exh ib i ted act iv i i r u i th. { . { uere h ldro l lzed at ra tes a t least l0 t imes greater than thebackground reaction. but the background reaction does r imit thesens i t iv i ty o f the assay.
stability of Acylase I. PKA and AA were stable as lyophilizedpowders (the form in which they are bought) when stored de-siccated at 4 oc and showed no loss of activity after storage for6 months to 2 years. Soluble PKA was sensitive to autoxidationaeunder ambient atmosphere (first-order half life, 36 h) but lost noactivi ty when kept under an atmosphere of nitrogen (Figure 2).Reducing agents, such as mercaptoethanol or dithiothreitol, arenot helpful in preventing oxidative inactivation of pKA. At lowconcentrations, they, themselves, deactivate pKA, presumably byreducing cystine l inkages.3l
Soluble AA showed no sensitivity toward autoxidation (Figure2).s0 The init ial loss of act ivi ty was reproducible but was not
J . Am. Chem. Soc. , Vo l . 111, No. 16. 1989 6357
t , h t , nFigure l . Stabi l i ty of soluble acylase I in the presence of organic co-solvents ,\crlasc I lrom porcine kidney (a. c) and from Aspergillus (b,d)uerc rncubatcd in ( ) l0 \ ,1 phosphate buf fer , pH 7.5 , conta in ing ethanolt : 1 . b ) r r r I ) \ lSO r c . d t . . i t l - ( oC . unde r n i t r ogen . Concen t ra t i ons o f. ) rB .u r l ue \ ) \ , ' . \ en l \ r i e i - c l ( ) ' , r f t . l 0 ' 7 rO ) . - 10 '7 (a \ . 40q , ( t r ) , and50Vor a l
; r csu l t t r l ' p ro t c t r l r t r c dcg rada t ron o f t hc cnzvme o r o f pa r t i a labsorption oi protcrn ro the inner surfaces of the incubation vessels.I t uas i ndependcn r o l t he concen t ra r i on o f AA and was no t a f -fec ted b1 the presence of so lub le bov ine serum a lbumin or byprecoat ing the vessels wi th denatured prote in .
Both enzymes exhibited moderate stabi l i ty ' in the presence oforganic cosolvents (Figure 3) and were catalyt ical ly act ive in themixed-solvent systems. They retained approximately 50Vo of theiraqueous specif ic act ivi t ies in 307o DMSO.
Subshate Specificity. Chart V summarizes data from the initialrates of hydrolysis of a broad range of substrates catalyzed byPKA and AA. I t is a general guide to our results and those ofothers24'28'3e'51's2 and is useful as a predictive tool. Chart V breaksthe act iv i t ies o f subst ra tes down in to component react iv i t ies o frnd i r idua l funct ionu l groups in each of the three var iab le domainstR . R . . R , ) t r l : t t b : t r a t c . ' . ^ \ s a f i r s t app rox ima t i on , t hese com-ponc l t r c . t e l i \ t l tC : . t r c cun tu la t i r c . Thus . t he reaC t i v i t y O f a l a -n r i l l r n rnc * r rh Pk \ i . . r pp ro r i n ra te l r I o rde r o f magn i t ude l essth . rn that o i . \ - . rccr \ l . i l . rnrnc. bur the r i t re o f h1 'dro lys is o f N-(a-ch lor t rprop i t )Dr I t - t r -c r c loherr lg l r c tne r r l -3 orders o f magni tudeless t han t ha t o f - . \ - ch lo roace t , r l r a l i ne . Rc la t i ve ra tes o f i n i t i a lh l d ro l l s i s ( t l ) o f i nd i v i dua l compounds a re rabu la ted i n t heExpe r imen ta l Sec t i on .
Acylase I was quite specific for the hydrolysis of ,V-acyl a-aminocarboxyl ic acids. Within that constraint, however, pKA and AAaccepted a broad range of structure and functionality in both theamino acid (R,) and acyl (R3) moiet ies of substrates. The twoenzymes were mmplementary in their substrate specificities. Bothenzymes showed an aff ini ty for straight-chain alkyl, alkenyl, oralkynyl amino acids of moderate length (C:-Cs). Cis and transdouble bonds were tolerated equally well . AA showed higherrelative activities with aromatic and B-branched amino acids thanPKA. PKA showed preferential activity with <.r-carboxylate aminoacids and accepted substrates in which the a-hydrogen had beenreplaced by a methyl group (Rz = CHr). AA did not accepta-methy l amino ac ids.
Productive binding and turnover of substrates required theterminal carboxylate group. Acylamines, N-acyl amino acid estersand amides,25 and N-acetyl-a-aminophosphonic acid 49-A werenot substrates ( Z, < 0.005 and 0.05 with PKA and AA, respec-t ively), The fai lure of PKA to hydrolyze 49-A contradicted thereported use of PKA to resolve a-aminophosphonic acids.53
(51) Otvos, L.; Moravcsik, E.; Mddy, Gy. Biochem. Biophys. Res. Com-mun. 1971. 44. 1056-1064.
(52) (a) Sato, T.; Mori, T.;Tosa, T.; Chibata, l. Arch. Biochem. Biophys.1971, 147,788-796. (b) Birnbaum, S.M.; Fu, S.-C. J. ; Greenstein, J. -p. . / .Bio l . Chem. 1953, 203.333-338.
100
i 8 0
6 6 0(g
^ 4 0o\
1 6 01 2 0
1 0 0
> 8 0=o " "(!
-o 4o
1 6 01 2 0
(47) It is important to note that some manufacturers and authors cite unitsof acylase actjyi-ty as micromoles of substrate hydrolyzed per hour instead ofpe r minute. Different manufacturers and authors aiso use different temper-atures, pH, substrates, and concentrations to measure specific activity.Throughout this paper, a unit (U) designates the amount of inzyme requiredto hydrolyze I pmol of substrate per minute under the standaid assay con-ditions outlined in the Experimental Section. N-Acetyl-nl-methionine is thestandard substrate unless another is specified.
(48) Rosen, H. Arch. Biochem. Biophys.1957,67,10-15.(49) Autoxidation probably involves oxidation of an essential cysteine
residue. See; References 3l and 36.(50) AA lacks free thiol groups.3e
0
0 0

6358 J .
Chart V.
Am. Chem. Soc., Vol. 1l l , No. 16, 1989
Reactivities of Substituents in Substrates of Acylase I
Chenault et al.
trr. fao.t fr
R, TA, ,H
R3R2R1 reactivitya
cH3(cH2)Gs, (cH3)2cH , ) " r" , - - - l *o
4 p n ) , - " , c H g 6 c ^ " , ) " ,
OH
A..(cHz)o, , HocHz . \ - -cH2 , c tcH2
Nc(cHr'r , Hooc (cHJag ,c cx.1cHryo,,s^ cH,
0*., zr.,-" d 0,.r.,". lo0.*.'
i.,.,dr':'Q7"'
cH3, czHs
XCHa (X = Cl , Br, CH3O)
XCHaCH? (X = Cl , Br)
H ,o c .Hu d
H.NCHrd
good, > 10 %
acacH3(cH2)o , 4(cHde (, , ' HolcHr;".
ot
,r* Aar,
cH3(cH2)c1c t t1c t ;e
t -nc t l t tH r lef a i r , 1 - 1 0 %
HOI
cH3cH
cn.lcHr;r-, ,r
Hru\zcH'1 / t\ * N
nril lcnr;. o t
(cH3)3c , U'"'"'." ,, ,oV.r.
NHe* O
H.NA* r ,a r r , . , r cTJNHCH, I
C H , e cH,cr rcu lc t r , l e poor , 0 .0 '1 -1 %
aReactivities oi substiruents (Rr, Rr, R3) are expressed relative to the reactivities of the corresponding substituents of the model substrate, N'acerylmerhionine (40 mM racemici Rr = a HjscHrcHr, R! = H, Rr = CHr). Contains data from ref 6, 48, 52, 62, 66, and 14. See Results andExpirimental Section for derails. ,eA only; ieacti"ity *itn pfe i. fii.. 'PKA only; reactivity with AA is fair. dAA only: reactivity with PKA ispoor. 'AA only; no reactivity with PKA. /Data for PKA only. 'PKA only; no reactivity with AA.
Acylase I was specific for the position and chemical nature ofthe amide bond in substrates. N-Acyl B-amino acids 47-A and53 were not substrates. Although 53 was an N-acyl B-amino acid,i t also resembled an N-acyl a-amino acid in that the amidecarbonyl resided 7 to the carboxylate group. Compound 48-Awas hydrolyzed by PKA ( Z, = 0.06) but, apparently, with littleor no enantioselectivi ty. The rate of hydrolysis of 48-A ( init ialconc€ntration, 40 mM) remained constant through 72Vo conversionand showed no curvature around 50Vo conversion. An inflectionaround 50% conversion would have been expected if the enzymaticcatalysis had been appreciably enantioselective. Replacing thehydrogen on the amide nitrogen with an alkyl group destroyedsubstrate activi ty: N-Methyl and N-ethyl N-acyl amino acids(tert iary amides) were not substrates.2s Likewise, replacing theamide nitrogen by oxygen destroyed substrate activity: O-Acetyllactic acid (50-A) and O-acetylmandelic acid (51-A), esteranalogues of acyl amino acids, were not substrates.
(53) Telegdi, J.; Moravcsik, E.; Otvos, L. In Proceedings of the Interna-tional Conference on the Chemistry and Biotechnology of Biologically ActiueNatural Products, /st; Atanasova, 8., Ed.; Bulgarian Academy of Science:Sofia, Bulgaria, 1981; Vol. 3, pp 221-225.
Other compounds not accepted as substrates included acylderivatives of prol ine and aspart ic acid. Fai lure of acylase I tohydrolyze acyl aspartates must result from unfavorable chargeinteractions. Ac1'l derivatives of asparagine and aspartate p-esterswere substrates ( V, - l ) , and 35-MA, a methyl ketone analogueoi acyl aspartate, reacted with PKA at a relat ive rate of 4lVo.Pyroglutamic acid (52) was not hydrolyzed by acylase [.
Effect of Acyl Groups on the Rates of Enzymatic Hydrolysis.PKA and AA accept a range of unsubstituted and co-substitutedacetyl and propionyl moieties as the acyl group in substrates (ChartV).7'24'26'5r'54 Both enzymes hydrolyze formyl, benzoyl, and glycylamino acids, although AA does so at relat ive rates higher thanthose of PKA. PKA but not AA tolerates branching at the aposition of acyl groups. PKA therefore hydrolyzes dipeptides anda-chloroacyl or a-methylacyl amino acids; AA does not.
We examined acetyl, chloroacetyl, and methoxyacetyl deriva-tives for use in preparative resolutions of amino acids. Acetylamino acids are good substrates of acylase 1aa'28 and are readilyprepared. Chloroacetyl amino acids have particularly high en-
(54) Price, V. E.; Gilbert, J. B.; Greenstein, J. P. J. Biol. Chem.1949, 179,| 169-t t7 4.
Enantioselectiue Hydrolysis of N-Acyl Amino Acids
amino acid
2.5
g 2.0(g
g 1.s
t 1 .0
I o.s
0 .0
J. Am. Chem. Soc. , Vo l . I I I , No. 16. t989 6359
70
30
1 5
< at a
v
o
0
0 4 0 B 0 1 2 0 1 6 0
V.1no Co2+)
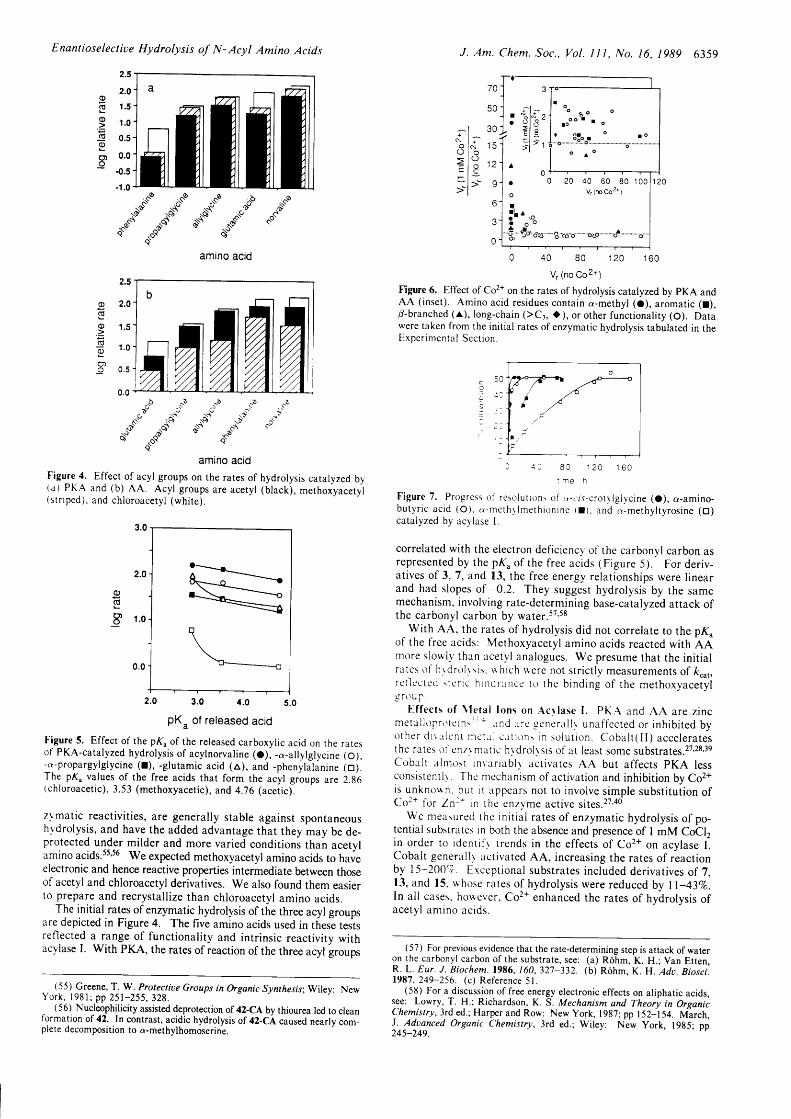
Figure 6. Effect of Co2+ on the rates of hydrolysis catalyzed by pKA andAA ( inset) . Amino acid residues contain a-methyl (O), aromat ic ( r ) ,p-branched (r) , long-chain ( )Cr, i ) , or other funct ional i ty (O). Datawere taken from the initial rates of enzymatic hydrolysis tabulated in theExDer imen ta l Scc t ion .
2.5
2.0oct 1.5
g 1'o(s 0.5(l)
o) 0.0o
-0.5
r lN l +
O l oo l o
EI?3 ls> l
r' /^t
-//''.-b 3 ro +o
"l.o- .-o' .f a.' i
.-"" J tt-.q "--
Jq.t"
.of e*oa
amtno acto
Figure 4. Effect of acyl groups on the rates of hydrolysis catalyzed by(a) PKA and (b) AA. Acyl groups are acety l (b lack), methoiyacety l( s t r i ped ) , and ch lo roace ty l (wh i te ) .
2.0 3.0 4.0 5.0
pKa of released acid
Figure 5. Effect of the pK" of the released carboxylic acid on the ratesof PKA-catalyzed hydrolysis of acylnorval ine (o), -a-al lylglycine (o),-o-propargylglycine ( l) , -glutamic acid (a), and -phenylalanine 1o1.The pK" values of the free acids that form the acyl groups are 2.g6(chloroacetic), 3.53 (methoxyacetic), and 4.76 (aceticl .
zlmatic reactivi t ies, are general ly stable against spontaneoush'drolysis, and have the added advantage that they may be de-protected under milder and more varied condit ionl than acetylamino
""16r.s5's6 we expected methoxyacetyl amino acids to have
electronic and hence reactive properties intermediate between thoseof acetyl and chloroacetyl derivatives. we also found them easierro prepare and recrystallize than chloroacetyl amino acids.
The initial rates of enzymatic hydrolysis of tire three acyl groupsare depicted in Figure 4. The five amino acids used in these testsref lected a range of functional i ty and intr insic reactivi ty witha* lase I. with PKA, the rates of reaction of the three acyl groupt
(55) Greene, T. W . Prote-ctiue Groups in Organic Synthesis; Wiley: NewY o r k , l 9 8 l ; p p 2 5 1 - 2 5 5 , 3 2 8 .
^ (56) Nuc^leophilicity assisted deprotection of 42-cAby thiourea led to clean
lormation of 42. ln contrast, acidic hydrolysis of 42-cL caused neariy com-plete decomposition to a.methylhomoserine.
2.0
od
I 1.0
8 0 1 2 0 1 6 0' i m e . h
Figure 7. Progress of resolurrtrns of tr-r. i . i -crotr lglycine (O), a-amino-butyr ic ac id (O.1. a-mcrhr lmeth ion ine ( l ) . and a-methy l ty ros ine (O)catalyzed by acylase L
correlated with the electron deficiency' of the carbonyl carbon asrepresented by the pK, of the free acids (Figure 5). For deriv-at ives of 3, 7, and 13, the free energy relat ionships were l inearand had slopes of -0.2. They suggest hydrolysis by the samemechanism, involving rate-determining base- catalyzed attack ofthe carbonyl carbon by water.57's8
With AA. the rates of hydrolysis did not correlate to the pK"of the free acids: Methoxyacetyl amino acids reacted with AAmore slowly' than acetvl analogues. we presume that the init ialrates of ' hr drolr sis. rr hich \r 'ere not str ict ly measurements of k""1,r c t l ec t cd : r c r i e h rnd runcc t o rhe b ind ing o f t he me thoxyace ty lg r 0 u f
f - f fec ts o f \ te ta l lons on. {cr lase I . PK.{ and AA are z incme ta l l op r r l t c l n \ " ' . r nd . , r . o . - n . r . l l r una f f ec ted o r i nh ib i t ed byo the r d r r r l cn r n r c ra i c . r r i t r ns i n so lu t i on . coba l t ( l l ) acce le ra testhe ratc-s ol 'cnzr matic hrdrolrsis of at least some substrates.27,28,3ecobal t a lnrosr rnrar iab l l ac t ivates AA but a f fec ts pKA lessconsistenrlr. Thc mechanism of act ivat ion and inhibit ion by co2+is unknorrn . but i r r ippcars not to invo lve s imple subst i tu i ion o fCor+ for ln : * in the cnzyme act ive s i tes .27,ad
We measured the init ial rates of enzymatic hydrolysis of po-tential substratcs in both the absence and presence of l mM Coclrin order to identi l ' r trends in the effects of co2+ on acyrase I lcobalt general lr act ivated AA, increasing the rates of reactionby l5-2007. Erceptional substrates included derivatives of 7,13, and 15. whose rares of hydrolysis were reduced by l l -43Vo.ln al l cases. however, co2+ enhanced the rates of hydrolysis ofacety l amino ac ids.
(57) For previous evidence that the rate-determining step is attack of wateron the carbonyl carbon of the substrate, see: (a) Rohm, K. H.; Van Etten,R. L. Eur. J. Biochem. 1986, t60,327-332. (b) Rohm, K. H. Adu. Biosci.1987,249-256. (c) Reference 51.
(58) For a discussion of free energy electronic effects on aliphatic acids,s99: Lowry, T. H.; Richardson, K. S. Mechanism and Theory in OrganicChemistry,3rd ed.; Harper and Row: New york, l9g7; pp t52:154. M"arch,J..Adua-nced Organic Chemistry,3rd ed.; Wiley: New york, l9g5; pp245-249.
0.0
o 2 0 4 0 6 0 8 0 1 o o l 1 2 0
R\=-.

6360 J. Am. Chem. Soc., Vol. I I I . No. 16. 1989
Table I I . Resolut ions of Amino Acids Using Acylase I
Chenault et al.
racemlcsubstrate (mmol) enzyme Co2+
% yield %t ee
iso lat ion
2-A (200)2-CA (70)3 - A ( 3 1 )7-CA (s2)l 0 - A ( 1 7 )l l - A ( s 7 )ls -cA (8)16-A (24)36-A ( r4)42-CA ( le)43 -CA (15 )44-CA ( ls )44-CA (7)
PKAPKAAAPKAAAPKAPKAAAAAPKAPKAPKAPKA
nonoyesnoyesnoyesyesyesyesyesyesyes
) z
4 l- 12
J J
3 84'742504 1J I
46646 3
EtOH ppt /EtOAc extrion exchion exchEtOAc extr /EtOH pption exchEIOH pp t /E tOAc ex t rEtOAc extr / . ion exchEtOAc ex t r , / i on exchEtOAc ex t r / i on cxchEtOAc ex t r , / i on e xchEtOAc ex t r / i on exchEtOH ppt /EtOAc extrEtOAc extr / ion exch
4040J J
4 lJ J
A A+ +1 -) l
504 55 l
l -
-t0
>99 .5>99 .5>99 .5>99 .5
99>99 .5
9995999 39 r9 5q 1
>99 .5>99.5>99 .5>99 .5
93>99.5
8498
a
a
804748
oThe f ree amino acid was not isolated.
With PKA, the effects of Co2+ were less consistent and oftenmore dramatic than with AA. Cobalt general ly inhibited thereactions of fair-to-good substrates but enhanced the reactivitiesof poor-to-fair substrates (Figure 6). In particular, Co2+ activatedPKA toward acyl derivatives of hydrophobic or sterically hinderedamino ac ids, Aromat ic , long-chain ( )C; ) , /3-branched and a-methyl amino acids al l showed dramatical ly increascd reactivi t iesin the presence of Co2+. \o such t rend ex is tcd for A, { .
Preparative Resolut ions of Amino Acids L sing Acylase I. Ttrdemonst ra te the use of PKA and . , \A for preparar i \e reso lur ionsof unnatura l and uncommon amino ac ids. ue sub. ;ected I I com-pounds represent ing the fu l l range of react iv in u i th ac l lase I (1 .= 0 .005 -169 ) t o enzyma t i c hy ' d ro l ys i s (Tab le I I ) . Reso lu t i onswere performed on 2-29 g of start ing material at pH 7-8. Init ialconcent ra t ions o f racemic subst ra te were 0.1-0.5 M. Whenneeded, I mM CoCl2 was included in reaction mixtures to enhancethe rates of reaction. Progress of the reactions was monitoredby assaying al iquots for free amino acid by colorimetr ic deter-mination with ninhydrin (Figure 7).48 Periodic, manual additionof a few drops of base maintained the pH of reactions at 7-8.
Resolutions of good substrates with PKA ran about 24 h andwerc usually performed under ambient atmosphere. Under thesecondit ions, PKA retained approximately 507o of i ts original ac-t iv i ty . Under anaerob ic condi t ions, however . PKA mainta inedfu l l ac t iv i ty . To protect PKA f rom autox idat ion in reso lur ion:las t ing longer than 24 h, subst ra te so lu t ions \ \ere spargcd r r i thnitrogen before adding PKA and then kept under an armosphereof n i t rogen dur ing the react ions. Resolu t ions us ing .A.A d id notrequi re anaerob ic condi t ions.
Af ter reso lu t ions \ r 'e re complete . acr lase I was denatured andthe products were scparated b1 d i f ferent ia l so lub i l i ty or ion-ex-change chromatographl. \ \ 'e used various protocols, dependingon the so lub i l i t ies o f the products and the s ize o f the react ions(see Exper imenta l Sect ion) . The most genera l , s imple, andhigh-yielding protocol. houever. involved acidifying the productmixture and extracting the acrl o-amino acid into ethyl acetate.The crude acyl o-amino acid. which was contaminated by freecarboxyl ic acid cleaved from the L enantiomer, was hydrolyzedin ref luxing 2 N hydrochloric acid to recover free o-amino acidor washed with ether to obtain reasonably pure N-acyl o-aminoacid. The acidic reaction mixture was applied to a column ofDowex-50 (H*) . E lu t ion o f the co lumn wi th I N aqueous am-monia provided the neutral, salt-free L-amino acid. Anotherprotocol involved precipitating the r--amino acid by adding ethanolto the near-neutral (pH 6) reaction mixture. Filtration removedthe t--amino acid, and acidif icat ion fol lowed by extraction gavecrude l /-acyl o-amino acid.
Other protocols were used as times. Sequential application ofa neutral product solution to columns of Dowex-50 (H*) and thenDowex-l (OH-) fol lowed by elut ion with ammonia and formicacid, respectively, gave L-amino and crude acyl o-amino acids,respectively. In an alternative to ion-exchange chromatography,amino acids were freed from salt and carboxylic acids by dissolvingthe dry, amino acid hydrochloride in methanol, f i l ter ing, and
adding excess propv lene ox ide (u 'arn ing: cancer suspect agent ) .Propylene oxide consumed hydrochloric acid, and the neutralamino acid precipitated from solut ion. In general, neutral(zwitterionic) amino acids were water soluble but not organicsoluble, and N-acyl amino acids were soluble in ethanol, acetone,and ethy l acetate .
Resolutions of a-Methyl a-Amino Acids. To test the practicall imi ts o f reso lu t ions o f very low act iv i ty ' subst ra tes wi th acy laseI . uc u ' cd n rcn rb rane -enc losed acv lase I se t o reso l ve cv -me thy l -r r c l h i ( ) n r n c t . l 2 ) . , - r 1 r L - t h r l p h c n r l a l a n i n c ( 4 3 ) . a n d a - m e t h y l -t \ r o \ i nc ( . 1 { ) f hc l ce hn rquc o l ' n t c r r rb r r rnc -enc losed ca ta l ys i sr \ l E E C ' ) " . r i l , r r i c d t h c u : c o f l a r u c q u a n t r t i c s t l f p r o t e i n i n s m a l lrcaction volun.rc: rrnd thc rcc()\ 'er\ and rcuse oi the soluble enzyme.PKA 1- : g) uas enc losed in d ia l rs is tub ing and used success ive lyto cata lyze the h idro ly 'ses o f 44-CA (2.0 g) , 43-CA (3.9 g) , and42-C.A (4.4 g). Reactions were kept under an atmosphere ofnitrogen. After completion of each resolut ion, the reaction wasterminated by removing the dialysis tubing containing enzymefrom the reaction mixture. Products remaining within the mem-brane were recovered by dialysis, and products from the reactionmix ture and d ia l l ' za tes wcre separated b1, ion-exchange chro-ma tog raph r . , \ f t c r ca tu l r z i ng t h r ce reac t i ons ove r a pe r i od o f36 davs . t he cnz r n t c r c ta i ned 55 ' ' . o f i t s o r i g i na l ac t i v i t y .
( -hcmica l ' , i c ld : . rnd cnant ionrcr ic pur i t ics o f products were fa i rt t r eo t )d I n gcnc r . r l . t hc cnun t i osc lec t i i i t 1 f o r t he hy "d ro l ys i s o fthc t cn. rn t l ( )nrcr \ \ . i \ sood. but unfar orab le Michael is constantslcd to incomplc tc conrcrs ion of the L enant iomer and subsequentenantiomeric contamination of the o isomer. A separate resolut ionof 44 under similar condit ions gave results virtual ly identical withthe f irst.
Enantioselectivity and Absolute Stereoselectivity of Acylase I.The enantiomeric puri t ies and absolute configurations of theproducts of preparative resolut ions catal.vzed by acylase I weredetermined by 'H NMR analys is o f the (R)- (+) -MTPA amidesof the corresponding amino acid methyl esters.60'61 In al l cases,measurements of enantiomeric ercess were performed on crude,unrecrystal l ized products. Thus. the measurements ref lect theintrinsic enantioselectivitl of the resolution process. Methyl estersof amino acids wcre prepared without racemization by the actionof thionyl chloride/methanol or diazomethane/methanol on thefree amino acids.
The rH NMR s ignals o f the (+) -MTPA methoxy groups ofI and o-amino acid derivatives appeared at approximately 3.35and 3.50 ppm. respectively, in agreement with previous stereo-chemical correlat ions.6l With derivatives of cr-methyl a-aminoacids, the cr-methyl signals could also be analyzed. Again, inagreement with previous stereochemical correlat ions,60'61 the
(59) Membrane-enclosed enzymatic catalysis (MEEC): Bednarski, M. D.;Chenaul t , H. K. ; Simon, E. S. ; Whi tesides, G. M. " / . Am. Chem. Soc. 1987,t09 .1283-128s .
(60) Dale, J. A. ; Mosher, H. S. J. Am. Chem. Soc. 1973,95, 512-519.(61) (a) Yamaguchi , S. ln Asymmetr ic . Synthesis; Morr ison, J. D. , Ed. ;
Academic Press: New York, 1983; Vol . l , pp 125-152. (b) Yasahara, F. ;Yamaguchi, S. Tetrahedron Lett. 1980, 2l,2821-2830.

Lnant ioselect iue H1'droly.e i5 of l i -Ac1, l Amino Acids
Scheme I . Conversion of a-Aminobutyr ic Acid to (R)- and(S) -Bu tene Ox ideo
J . . ' 1 n r . ( h e n t . t r . , c , I , t l . l l 1 . . \ ' o . 1 6 , 1 9 8 9 6 3 6 1
asymmet r i c i nduc t i on i n t hc e l ec t roph i l i c c r c l i za t i ons o f 2 - sub -s t i tu ted 4-a lkenoic ac ids, c is se lecr i r i r r io r the cvc l izat ions o f2-amino-4-alkenoic acid derivatives is * el l documented. l2, l 3'66 Thero le o f n i t rogen in s tereodi rect ion is unkno*n,
Discussion
Acy lase I is a usefu l cata lys t for the reso lu t ion o f unnatura lamino acids on a mult igram scale. I ts general stabi l i ty in solut ional lows i t to be used in soluble form, and membrane-enclosure ofthe enzyme allows large quantities of protein to be used to cataly'zethe reactions of even very low activi ty substrates. The practicallaboratory appl icat ion of PKA extends to the resolut ion of sub-strates having ) l%, of the activi ty of AcMet on a l0-100-g scaleand to the resolut ion of substrates having 0.001-lVo activi ty ona l -5-g sca le . AA exh ib i ts a somewhat more l imi ted subst ra tespec i f ic i ty than PKA. The spec i f ic act iv i ty o f AA, which is100-fold lower rhan rhar of PKA. limits the scale of its applicabilityp ropo r t i 0na tc l r .
. . \ l though both [)K..\ and .. \ .A have been used to resolve manyni . l lu ra l r tnd 'ontc unnetur l l amino ac ids, enant iOmer iC pur i t ies ,i l ' :cp , r1qg1l . r i . r l l . hurg t lggn rc-por ted for the (o f ten repeated ly): ' l ! r \ \ ' . . i l l rzc t j nr , , t juc t . \ lanr o i the determinat ions o f enan-l ) , r ; 11g ' ' . l L : : : \ h . : r c j gpgndcd on va lues o f op t i ca l r o ta t i on andh. r rc thcrc l , r rc bccn . lp f \ r \ ) \ tmat ions at best . Here, we have shownth . i t t hc i n t r r n . r . cn . l n l i r ) \ c l cc t t r i t r o f t he reso lu t i on s tep , i t se l f ,r : h igh. ( . \n erccpt ion i : t rur rcso lur ion o f 43. ) Fur thermore, theabsolute sensc of the enantrosclcct ir in rcnrl ins constant, even withunna tu ra l and c1 -me th r l am ino ac ids .
A prob lem more preva lent than nonspec i f ic hydro lys is is theincomplete hydrolysis of the I enantiomer, causing enantiomericcontamination of the o product. Our measurements of initial ratesof hydrolysis did not evaluate the Michaelis constants (K.) ofsubstrates expl ici t ly. Substrates having high K, ()10 mM, asapparently ls-CA,43-CA, and 44-CA do) wil l not afford highlyenantiomerical ly enriched o products without recrystal l izat ion.
Thc s1'ntheses of butene oxide and compounds 57-59 representonlv tuo gcncrel prtrcedurcs b1'which the resolved a-amino centermur bc t r : rn \ f ( ) rnrcd t , i d i f ' fc rcnt funct iona l i ty or used to induce. l \ \ , n . t n tc l r \ . l i ' t . \ \ l r c rc l r lcd s tcrcocenters . In par t iCUlar . Otheri ' , , r - r r : , r1 'g lgg i ; , rph i l re crc l rz . r t ion t ) r or idat ion o f double bonds near: h c . : n r r n ( r g r i r u n : l r r r b c r n d u c e d b r t h e t r - c h i r a l c e n t e r . 5 7
\ \ e bc- i ic . rc rhc u 'c o i acr lase I is a pract ica l method for thep rcpa ra t i on o f ' cnan t i omer i ca l l r pu re , unna tu ra l am ino ac ids o /knon'n ab.solute wn"f iguration. I t is practical even for the prep-aration of research quanti t ies ( l-5 g) of very low activi ty sub-strates. such as the a-methyl a-amino acids (at least the t_ en-antiomer). We note that the resolut ions described here have notbecn specifically optimized and that the optical purities of alreadycnantiomerical ly enriched amino acids are readi ly improved byrecrvstal l izat ion. In general, though, these resolut ions provide,in a .single process. both enantiomers of an amino acid having highcnantiomeric puri t ies. Like any resolut ion, a disadvantage of themethcxl is that the maximum chemical vield of anv one enantiomeris -50 '7 .
Experimental Section\{aterials and \lethods. Organic chemicals were from Aldrich and
Fluka Biochemicals were from Sigma. Crotyl bromide was dist i l ledfrom calcium hrdride. oxalyl chloride was dist i l led and stored undernitrogen. Drisopropylamine and tr iethylamine were dist i l led from cal-cium hrdnde and srored under nitrogen. Methylene chloride and ace-tonitr i le ucrc ire:hly dist i l led from calcium hydride. THF was freshlydist i l led froni sodium/benzophenone. Ethanol was dried over 2-A mo-lecular sic''cs. Rcactions requiring anhydrous conditions were performedin oven- or f lame-dried glassware under an atmosphere of nitrogen.
1[ { z rndrrC \MR spect ra were recorded on Bruker AM-300 andAM-2,s0 instruments with peaks referenced to CHCI3 (tH d 7.26, t3C 6
(66) Ohfune, Y.; Hori, K.; Sakaitani, M. Tetahedron Lett. 1986, 27.6079-608 2
(67) (a) Tamaru, Y. ; Hojo, M.; Higashimura, H. ; Yoshida, Z. J. Am.Chem. Soc, 1988, 1 10.3994-4002. (b) Garner, P.; Park, J. M. "I. Org. Chem.1988, 5J, 2979-2984. (c) Cardil lo, G.; Orena, M.; Sandri, S. J. Org. Chem.1986 , J1 . 7 13 - t l 1 .
JX.*"I
,/,\/ \
!t.- AcNH
|"r.V\coo- Sc,ooH " -$coo-
l ll r i i l i i it lcl c l
gcoor J.**
1, . . .1l r v , v l i v , v+l
o Reac t ion cond i t i ons : ( i ) acy lase l . 40% , " - i e ld o i each p r rduc t r rhco r e t i c a l m a x i m u m i s 5 0 ? ) : ( i i ) I \ H C l . r e f l u r . 9 0 ' 7 : t r r r ) \ . r \ OH C l . 9 l ' 7 : ( i v ) B H . i T H F . 9 0 " , . ( v t K O H . 7 5 ' ; . q i 9 6 ' , e c
Scheme I I . I odo lac ton iza r ion o f o -A l l r l s l r c ine Der i ra r i res
H . R .
R2
7 - A , R 1 = R 2 = H'to-A, R1 = H, R2 = CH311-A, R1 = CH3, R2 = H
t r -methy l s igna l o f L-amino ac ids appeared downf ie ld o f rhat o fthe o enantiomorphs. When possible. optical rotat ions of prMucrs\r'ere compared to those of known compxrunds. In all clrsr-s. rrcr luscI ca ta l yzed t he hyd ro l vs i s o f t he . \ . - ac r l I - am ino r r c rd n l
Enant iomer ic excess measuremr 'n ts \ \ere ca l ibra tcd br . tddrns0 . - i - 5 0q o f t he (+ ) - \ 1TP . . \ de r i r u r i r c , - r i t he co r r c :Fond ,nn I . r " -c e n r i c a m i n o u c i d t o t h e d e r i r a r r r e o i r e s t l l r c d . r m r n o . i c i i . . \d i as te reomcr i c impu r i t r o f 0 .1 ) ' ; cou ld be de tec red .
L'se of Resohed Amino Acids as Chiral Svnthons. To demon-strate the use of resolved unnatural amino acids as chiral synthons,\ \ e p repa red (R ) - and (S ) - l - bu tene ox ide f r om a -am inobu ty r i cac id (Scheme I ) and three ch i ra l lac tones v ia iodo lacton izat ionof subst i tu ted a-a l ly lg lyc ines (Scheme I I ) .
r- and o-Aminobutyric acids (ee )99.5Vo), obtained by reso-lut ion (Table II) , were converted in three chemical steps to (R)-and (S)-l-butene oxide, respectively, in 6l%a overal l yield. Theoptical puri t ies of the epoxides were determined by 'H NMRspectroscopy in the presence of Eu(hfc)3.63 Both had a 96Va ee.
Kinetical ly control led iodolactonizations6a of o-7-A, D-10-CA,and o-11-A, recovered from the resolut ions of the correspondingracemates, gave lactones 58-60 (cis:trans, 7-8: l) . The diaste-reomeric rat ios of the products were measured by tH NMR.Assignment of the configurations of the major and minor dia-stereomers was performed by NOE difference spectroscopy andby correlation of the lH NMR spectral data of both diastereomersto that of known cis- and trans-2,4-disubsti tuted .y-butyro-lactones.6s'66 Although l i t t le precedent exists, in general, foi I ,3
(62) with a-methyl amino acids, the L enantiomer refers to that enan-tiomer which would normally be considered r, if the a-methyl group were infact a hydrogen. L enantiomers of the c-methyl amino acids diiussbd in thispaper ar€ (.S) in absolute configuration.
(63) Kim, M.-J. ; Whi tesides, G. M. J. Am. Chem. Soc. 1988. 110.2959-2964.
(64) Gonzalez, F.(65) Hussain, S. A.
Soc., Perkin Trans. l
I 7 5 - l 8 l .J. Chem.
H
c _ i s : t r a n s = 7 - B : 1
5 7 , R 1 = R 2 = H58, Rl = H, R2 = CH35 9 R 1 = C H 3 , R 2 = H
6362
Table
J. Am. Chem. Soc., Vol. I I I , No. 16, 1989
III . Direct Chloroacetylat ion of Amino Acids in Acetonitr i le
amlno aclo yield,o Vo
alanineval inenorval inea-al ly lg lyc inephenylalaninea-methylphenylalaninea-methyltyrosinea-methyl-DOPAser ineglutamic acida,e-diaminopimel ic acid
'Yie ld is of isolated, recrystal l ized product . bOnl l srarr ing mater ia lwas recovered. 'React ion of ser ine gave a complex product mi \ l .u! -cthat was not character ized.
77.0; CDCI3), acetone-ds ( tH 6 2.04; acetone-d6), or the methyl s ignalof sodium 3-(trimethylsilyl)propanesulfonate ('H 6 0.00, r3C 6 0.0; DrO;.Elemental analyses were performed by Galbraith and Spang Microana-lyt ical Laborator ies. Mass spectra l data were obtained by H.K.C. andDrs. Todd Wi l l iams and Sher i Ogden (Mass Spectrometry Laboratory,Univers i ty of Cal i fornia, Berkeley) .
Enzymes. All of the results in this paper were obtained with acylaseI from Sigma: porcine kidney (grade I) and Aspergil lus. PKA and AAare also available from Biozyme and Amano, respectively. A preliminarysurvey of the activities of these enzymes indicated that both had activityprof i les s imi lar to their Sigma counterparts. The speci f ic act iv i t ies ofboth enzymes were about half of the corresponding Sigma enz,vmes. buttheir costs per unit of activity were 25 and 50%. respectivelr. ol thc Sigmrrenzymes.
Synthesis of Amino Acids and Analogues. Compounds 8. 9. 12. l{. 19.20,23-29,31, 35, 37, and 38 were prepared br ohase-rrunst 'cr r i lkr i r r r , ' lo f 55 .4J '44 Compounds t0 -A . t6 -A . ,na - tO f \ re re F \ rcFdred
' b r r i r . r r . ,
t ion6E of 56 in sodium ethoxide'ethanol . fo l lo*ed br sap'onrt ic . t l l t r0 .1 f l r ld e c a r b o x l l a t i o n . a s H r d r o l r s i s i n r e f l u r i n g : \ H C I i o r I h r o r 1 . 5 \NaOH for 4 h wi th 3GA) gave the f ree amino acids. Compounds l5 andl7 were prepared by modi f ied Strecker react ions6e of cr ,c lopropane-carboxaldehyde and 2-adamantylethanal ,T0 respecr ivelv. 4-Carboxy-2-pyrrol idone (53) was made by reducing . /V-benzyl-4-carboxy-2-pyrrol idoneTr in sodium/ l iquid ammonia. a-Methyl-a-v inylg lyc ine (39)was made from Dl-a-methylglutamic acid by a method analogous to thatfor preparing t--a-vinylglycine.T2 Compounds ll and ll-A,45 32,13 49,74and 50-A75 were prepared by literature methods.
Acylation of Amino Acids (Schotten-Baumann Conditions). The am-ino acid was dissolved in 3 equiv of 4 N NaOH (4 equiv i f acylat ing adicarboxyl ic acid) , and acyl chlor ide ( l . l equiv) was added in f ive por-t ions and wi th v igorous shaking to the chi l led (<0 oC) solut ion over 50min. Acid i f icat ion to pH 1.5 wi th concentrated HCl, exrract ion wi thethyl acetate, drying the organic layer (MgSOa), and rotar)'evafxrrari()ngave the product as an oil. Trituration with petroleum ether caused theN-acyl amino acid to crystal l ize. The crystals were washed wi th etherand recrystall ized from ethyl acetate/hexane.
Acetylation of Amino Acids (Acetic Anhydride/Acetic Acid). To 2 gof amino acid s lurr ied in 25 mL of g lacia l acet ic acid was added i lmolar equiv of acetic anhydride. The mixture was stirred at room re m-perature until it became homogeneous. After removal of the solvent brrotary evaporat ion, the residue was taken up in acetone and f i l teredRotary evaporation of the fi ltrate gave acetyl amino acid. This merhodwas especially useful for N-acetylation of hydroxy amino acids and anr-inophosphonic acid 49.
(68) Preparation of (bromomethyl)cyclopropane: Meek, J. S.; Rowe. -lV, J. Am. Chem. Soc. 1955, 77, 6675-6677. Preparation of (chloro-methyl) furan: Kirner, W. R. J. Am. Chem. Soc. 1928, J0, 1955-1961Divald, S. ; Chun, M. C. l Joul l ie , M. M. J. Org. Chem.1976,41,2835-2846
(69) Gaudry,R. Can. J. Chem., B 1946,24,301-30'1.( 70) Prepared from cyclopropyl- and I -adamantylmethanol, respectiveli .
br Swern oxidation: Omura, K.; Swern, D. Tetrahedron1978,34, l65l-1660.Aldehydes were previously reported: Smith, L. I.; Rogier, E. R. "/. Am. Chenr.Soc. 1951, 73.4047-4049. Do, K. Q.;Thanei , P. ; Caviezel , M.; Schwyzer.R. Helr ' . Chim. Acta 1979. 62.956-964.
(71) Pavtash, P. L. ; Sparrow, E. ; Gathe, J. C. J. Am. Chem. Soc. 1950.7 2 . t 4 t 5 - 1 4 1 6 .
(72) Hanessian. S.; Sahoo, S. P. Tetrahedron Lett. 1984, 25, 1425-1428.(73) Joucla. M.; El Goumzili, M.; Fouchet, B. Tetahedron Lett. 1986. 27,
l 677- I 680.(74) Oleksyszyn, J. ; Tyka, R. ; Mastalerz, P. Synthesis 1978, 479-480.(75) Fi lachione, E. M.; Lengel , J . H. ; F isher, C. H. J. Am. Chem. Soc.
1944.66. 494-496.
Table IV. Relat ive In i t ia lCatalvzed bv Acvlase I
Chenault et al.
Rates of Hydrolysiso of N-Acyl Amino Acids
l)
1 51 l,s86 1i68960
a
bh
compd
porcine kidney Aspergillus
no Co2* I mM Co2+ no Co2* | mM Co2+
AcMet,V-benzoylmethioninet -AI .CA2-A3-A3-(-A3- \ lA4-A5- ( A6-A7 - A7-CA7-MA8.MA9-MAl0-AIO-CAI l -AI I . C A12-MAl3-AI3-CAI3-MAI4-CAIs -CA| 6 -Al 7 - ( ' \I t i - \I t - ( \l l t \ l \1 9 \ l {20 - \ l {2 l - {22-. {22. \ IA23-l\tA24-I/.A25-MA26-MA27-N/.A28-MA29-MA30-A-r(.)-cA30-\t.\3 r - c ' \-12-\ t . \l.l {-1{- {.15 \t {-16_.\.17-( {,1lJ-\t {-19-\ t {'t0-..\4(f - \1.{. i l - ( . \{ t \ lA{2 .CA43.CA44-C'A45-C'A46-CA47-CA48-A4950-A5 l - A52-A
40 415 6
2 8 5 l6 8 1 4 820 431 8 3 048 35
9 .9 9 .73 5 4 140 236 1 8 4
( ' c
.1r 84e8 1231l 50434 . 9 t 2
100r00 860.23
5 3l 2r q nl 2 l1 Al +
1 . i x40
( '56 :t :l r' t :
o hl 69 128l 63 32954l 1 /t / +t 6
) \ i
L .
( ) - 1 \
t ' - l X.lbt 1
262 . 42 .0
l 0 :
5 .4 x l 0 -21 +
0 . 1 496l 9209 l. l ' )
i l 9
{ t , i
: i
- + il o0..1.10 . 25 . 1 x t 0 - 2D
2 . 8 x l 0 - 3b0 .51 .7 x l 0 -3l)
x l0 -2
5 3501039458
l0-3 | .4
-1 1
l 6t f
. J
l l: 9
286 l18l 0262021r 6 92 )h1 9
l . r6 J
J J
224 1
10521
').+
8 523J . L
68
4086
t3'l) z
c
68761 53 5
t r . O
8 l900 .380.244 . 2
0 . l 3
44l 969: 86 lt :h
l 66 . 11 . 27 . 56 . 2
l 0
I J
27
5 I
l 8
7 . 9
0 . 1 8
3 56 .6
t 43 . t
0 . 1 2 r . 0( c
28 36 680 . 7 1 3 2 4 l0 .9 t4 230 . l 7b0.0269 . 3 X 1 0 2 c c2 .2 x I0 - l1 . 3 c cl . 5 x l 0 - 2 c c4 . 7 x 1 0 - r c c
cccc
c
' ln i t ia l rates of hydrolysis of 40 mM racemic substrate, pH 7.5, 40 'C,
are reported as a percentage of the rate of hvdrolysis of N-acetylmethionine:for PKA, 30 + 5 prmol min-r mg-r (no Cot*) , lor AA, 0.30 + 0.02 pmolmin rmg ' ( l mM Cot*) . 6Relat ive rate is (5 x l0-3To. 'Relat ive rate (5x l0-2 ' / r , . dRel l r t ive rate is ( l x l0-r7c.
Direct Chloroacetylation of Amino Acids. Chloroacetyl chloride ( L05equiv) was added to a dry. round-bottom flask containing 20-100 mmolof anr ino acid and enoush acetoni t r i le to make the f inal solut ion 0.5 M
bD
D
0.06h
h

Enantioselectiue Hydrolysis of N-Acyl Amino Acids
1 7
1 00 200 300 400 500 600
I . m t n
Figure 8. color imetr ic response wi th n inhydr in int r ins ic to acylase Iisolated from Aspergil/us (Sigma; l0 mg in 2.00 mL of buffer). Enzyme*as incubated under assay condi t ions, and al iquots (0.20 mL) werequenched and reacted wi th n inhydr in.
in acid chlor ide. The f lask was f i t ted wi th a ref lux condenser and ani t rogen source. The mixture was heated at ref lux under n i t rogen unt i lthe amino acid d issolved completely (general ly , l - r .5 h) . occasional ly ,a smal l amount of the amino acid d id not d issolve. The color less toorange solution was cooled and evaporated under reduced pressure. Theresidue was st i r red in acetone or ethyl acetate for about l5 min to a l loucomp le te so lu t i on o f the acy l am ino ac id . The so lu t i on was f i l t e red andthe f i l t r a te e 'apora ted under reduced p ressure . The res iduc \ \a \ r c -c r v s t a l l i z e d f r o m e t h r I a c e t a t e / h e r a n e ( T a b l e I I I )
E n z y ' m a t i c . { s s a r s . l n i t i a l r a t e s o l e n z r m a r r c h r d r t l l r : r : r T a b l c I \ rw e r e d c t e r m r n e d b r c o m b i n i n g x 0 s m t r l o f r a c e m r c . i c \ i , r n r I n r r . r r r d , r rl na logue . l 0 !L o i { \ KOH r . l 0 gL fo r d i ca rbo ry l r c ac ids ) . and ac r lasc ,I t 0 . 0 1 - l 0 m g o f P K . . \ o r l 0 - 1 0 m g o f . { . { ) r n 0 1 0 \ , 1 p o r a s s i u mp h o s p h a t e b u f f e r ( * i t h o r r + i t h o u r I m \ l C o C l : ) . p H 7 5 . , 1 0 o C . T h eenz \me \ |as added las t to i n i t i a te reac t ion . The f i na l assay vo lume wasI00 mL. The reac t ion p roceeded a r 40 "C , and s i x a l i quo ts (0 .20 mL)*ere per iodical ly removed and quenched in 0.20 mL of l4zo perchlor icacid. Port ions (0.16 mL) of the acid i f ied al iquots were combined wi th0.84 mL of water, 0.50 mL of sodium cyanide/acetate buf fer , and 0.50mL of 31/ . - n inhydr in, heated to 100 oc for l5 min, and quenched wi th5,00 mL of 507o aqueous 2-propanol, according to the method of Rosen.{The absorbances (570 nm) of the solutions were measured after allowinsthem to coo l fo r l 5 -30 m in .
The acid i f ied react ion al iquots were stable when stored at 4 oc andshowed no s igni f icant hydrolysis of acyl amino acid or thc protc in Thcabsolute intensi ty of the color imetr ic response gcnerared br . rc idr l ' icca l i quo ts s to red in the co ld rema ined v i r tua l l l cons ran r l o r l { h . rnd rhcre la t i ve i n tens i t i es o f a l i quo ts co l l ec ted and s ro red tose thc r r cmr lncdconstant for at least 3 days.
Ra tes o f hyd ro l vs i s * ' e re ca l i b ra ted b r reac t ing kno* n . tmounrs(0 .00 -0 .20 smo l ) o f each amino ac id b r the p rocedurc abo 'e ( see Sup-p lemen ta ry Ma te r ia l f o r de ta i l s ) The ac t i v in o f acy lase I i t se l f wasde te rm ined in a s im i la r manner . w i th AcMet (40 mM racemic ) as thesubstrate. The activities of PKA and AA were measured in the absenceand presence of I mM CoCl2, respectively. Relative rates (l/.) of hy-drolysis of substrates were expressed as percentages of the rate observedw i th AcMet .
Control experiments involved incubating the enzymes ( L0 mg of pKAand l0 mg of AA) in the absence of substrates and potent ia l substrates(40 mM) in the absence of enzyme under assay conditions. Aliquots wereperiodically removed and tested for response with ninhydrin by the me-thod above. AA exhibi ted an intr ins ic * fa lse
act iv i ty ' (F igure g) .consequent ly, when s lowly react ing substrates were assayed wi th AA,the enzyme was allowed to rest in buffer solution for 6 h before assaying,and a blank react ion contain ing only AA was run in paral le l . The di f -ferences in absorpt ion between the assay' react ion and the contro l wereused to ca lcu la te the ra te o f enzymat i c hyd ro l ys i s .
. {c id i f ied al iquots f rom assays of pKA and AA wi th AcMer wererncubated at 4 oc and per iodical ly assayed for response wi th n inhydr into determine the stabi l i ty of the acid i f ied al iquots dur ing storage.
compounds 2l -A, which i tsel f reacted wi th n inhvdr in. and 5l -A were,rssared s imi lar ly to other potent ia l substrates exceDt that the rates ofh rd ro l vs i s were de te rm ined by tH NMR. Each ac id i f i ed a l i quo t wasetaporated under reduced pressure, and the residue was taken upin D2o.Peaks for the a-methine protons of the acylated and f ree ui ids *"r .:n:cgrared to measure the amount of hydrolysis. Hydrolysis of s0-A was:ronrrored by assaying al iquots for L- and o- lact ic acids.76
Stabi l i ty 'of Acylase I . Acylase I (1.0 mg of pKA or l0 mg of AA)!Ar) rncubated at 25 oC in 0.10 M potassium phosphate buf fer , pH 7.5,* ; rh Lrr * i thout organic cosolvents. Buf fer for AA also contain"d t -M
1 . 5
t . +
L b
1 . 3
1 . 2
I I 1 , No . 16 , 1989 6363
L
% DL added
4 .0
2 .0
1 . 0
0 .5
0 .0
: ? . J e 3 . a A-r-r--r---Tt
3 . s 0 3 . 3 AP P H
Figure 9. H \ \l R spectra (300 MHz) at 3.3O-3.60 ppm of the methoxyether peaks ot the (R)-(+)-MTPA der ivat ives of (a) methyl r -a-c is-crotylglycinate. (b) methyl oL-a-crs-crotylglycinate, and (c) a | 0.0-4.}Voof b (base- l ine detai l ) .
CoCl2. Final volume was 2.00 mL. Unless the enzyme was to be exposedto atmospher ic oxygen, a l l solut ions were made in bul fer previouslydegassed wi th n i t rogen, and the incubat ing enz\me solut ion was keptunder an atmosphere of n i t rogen. sealcd * i th a se ptum. Al iquots of theenzyme so lu t i ons wcrc pe r iod ica l l r remored r ra . r r i nge and assayed fo rac t i v i t y .
Reso lu t i on o f \m ino {c ids . T . r d r . : r l l cd \ \ e te r \ \ e re added racemicluc r i . r t r i n , r r i e r r i . y t t j e : . , , r rS f r t i r u r r ' . , , 1 ' f \ ) t r \ \ l um hydrox ide to make thef ' t n . r i r ' , r u t t , r n : i l
- i \ i \ , , , u : r , , o r . : i : r ) c O n t a i n e d I m M C O C I 2 a n dt t e r c dc , i \ \ Sc r : . : : . r t . | : c ! c \ . . r : \ F r l . r l c , tncen t ra t i on o f racemiC acy l. 1 l ' r i I t , , . r ! : \ l , \ . r r r r ' t r i \ ' l \ \ r t h g t r . U : u b : l f u t . € S . a d d i t i O n O f 8 . 3 U O f. : r \ . - : : C I n l : l : , , . , , i . : . \ , I - . 1 n l n ( t a C t d t n l t t a t C d t h C f C a C t i O n ( U n i t S b a S e do n t h c l n i l r . l i r . r l c i r l h , r d r o l r s r s o i t h e s u b s t r a t e b e i n g r e s o l v e d a n dmc: l :u red . rs dc :c r rbcd abore l . \ \ ' r t h poor subs t ra tes . re la t i ve l v fewerun i t s o l ' enz rmat i c ac t i v i t l were used . P rog ress o f the reac t ion wasmoni tored by assaying al iquots for f ree amino acid wi th n inhydr in.a8Resolutions proceeded for 24 h or until the release of free amino acidleveled off. Periodic, manual addition of base maintained the pH of thereaction at approximately 7 .5.
The reaction was adjusted to pH 5.0 with concentrated hydrochloricacid, heated to 60 oC with Norit, and fi ltered. The fi ltrate was acidifiedto pH 1.5 wi th concentrated hydrochlor ic acid and extracted wi th ethylacetate. The aqueous layer was applied to a column of Dowex-50 (H+),and the column was rinsed with water until neutral and then eluted withI N aqueous ammonia. Alternatively, the aqueous layer was evaporatedto dryness by rotary evaporation. The residue was taken up in methanol,filtered, and treated with excess propylene oxide (warning: cancer suspectagent) to g ive the l ree L-amino acid as a precip i tate.
The ethvl acetare layer was dried (MgSOa), fi ltered, and evaporatedby rotary evaporar ion. Tr i turat ion of the oi ly residue wi th ether orether/petro leunr ether mixtures gave the acyl o-amino acid as a sol id.Al ternat ivel r , , the crude oi l could by hydrolyzed by ref lux ing for 2 h in2 N HCl.78 The o-amino acid was recovered from the hydrolysis mixtureby cation-exchange chromatography or precipitation from methanol withpropylenc oxide. s imi lar to the recovery of the l -amino acid.
Resolutions of 42-44 were performed as described above except that5 g of PKA in approximarely 40 mL of degassed 0.1 M potassiumphosphatc buf fer , pt{ 7.5, was enclosed in 13.5 x 2.1 cm of d ia lys istubing (Spectropor 2; MW cutof f , 12000-14000). Af ter d ia lvt rc re-moval of the bulfer salts, the enclosed enzyme was placed in a degassed
, -6 t ra t \oll. F. ln Methods of Enzymatic Analysis,3rd ed.; Bergmeyer,
H L-. Ed. \ 'er lag, Chemie: Weinheim, Germany, lggl ;Vol . O, pp SEZ_5SS., b r Ca*ehn . K . Ib id . pp 588-596 .
(77) Because of i ts h igh solubi l i ty , l i th ium hydroxide was used in resolu-tions of which the L-amino acid product was precipitated from neurral aqueoussolut ion by the addi t ion of ethanol . Both l i th ium and porassrum cat ionsfuncli.oned equally well in purifications by ion-exchange chromarography.
(78) compounds 364 and 42-CA were hvdrolvzed br- refiuxins roi + tr in2.5 N NaoH and in ethanol wirh 2 equiv o i th iourea. iespect i "e ly Acid ichydrolysis destroyed both compounds. Hvdrolvses of .\-acrll a-met'hl,r aminoac ids requ i red more than 2 h - io r como le t ion
'1 .1

6364
% DL added
4.0
2 .0
1 . 0
0.5
0.0
rT--r-T--[-T--l [-r-r-T_-r-T--l3 . s 6 3 . 3 9 3 . 5 S 3 . 3 9
PPI4 PPI1
Figure 10. rH NMR spectra (300 MHz) at 3.30-3.60 ppm of themethoxy ether peaks of the (R)-(+)-MTPA der ivat ives of (a) methylD-a-c is-croty lg lyc inate, (b) methyl DL-d-c i . r -croty lg lyc inate. and (c) a *0 .0 -4 .0% o f b (base- l i ne de ta i l )
so lu t i on o f 44 -CA. pH 7 .5 . and s t r r red gen t l r under n i t r t rgen \ i t c ' r t hc 'r e a c t i o n h a d l e v e l e d o i i a t 5 0 ' 7 c o n r e r s i o n . t h e d i a l r s r s r u b r n g w a \ r e -moved and dia l l zed against d isrr l led. degassed rr ater to remo\ e producrsThe same enz)me'*as used subsequenl l r to h ldrolyze 41CA and 42-CA
Determination of the Enantiomeric Purities of Amino Acids. Theenant iomeric pur i t ies of amino acids were determined by tH NMRanalysis of the (+)-MTPA amides5o of the amino acid methyl esters.Methyl esters were prepared without racemization with methanol/thionylchlorideie or diazomethane/methanol. The (+)-MTPA methoxy groupsof t-- and D-amino acid derivatives appeared as narrow quartets ("Ilrr =
1.0-1.5 Hz) at approximately 6 3.35 and 3.50, respect ively. The a-methyl s inglets of a-methyl amino acid der ivat ives (6 I .69 and 1.65 foro- and L-amino acids, respectively) were also distinguishable. Calibrationby the addi t ion of known amounts of the (+)-MTPA der ivat ive of theappropr iate racemic amino acid showed that a 0.257o diastereomericimpurity (99.5Vo ee for the amino acid) could be detected (Figures 9 andl 0 ) .
Conversion of o- and t--a-Aminobutyric Acid to (S)- and (R)-ButeneOxide. Resolved a-aminobutyric acid was converted in three steps toenantiomerically enriched (96Vo ee) butene oxide as described previous-ly. to
Iodolactonization of 7-A, l0-A, and I l-A. Allylglycine derivatives 7-A,l0-A, and ll-A were iodolactonized under kinetic-controlled conditions.trAnalysis by 'H NMR and \iOED spectroscopy indicated that 57, 58, and59 were fo rmed w i th c i s to t rans ra t i os o f 8 :1 . 8 :1 . and 7 :1 , respec t i ve l y .
Acknowledgment. We thank Jaesang Kim for preparing l0-Aand for performing some of the di rect chloroacety lat ions and theresolut ion of 3-A.
Supplementary Material Available: Calibration of the variable
color imetr ic response of amino acids wi th n inhydr in and tablesof spectroscopic and analyt ical data for potent ia l substrates ofacvlase I and compounds 57-59 (2,5 pages). Ordering informationi s s i v e n o n a n \ c u r r e n t n r a s t h e a d p a g e .
r - 9 r L h l c [ ( H . r r r r . . t S / { n r C h e n r . S o c 1 9 5 6 . 7 8 . 3 8 1 - 3 8 4 .r x t ) r C h e n . r u l t . l J K . K r m . \ , l . - J : \ k i 1 a m a . , A . : M i y a z a w a , T . ; S i m o n , E .
S . \ \ h r t e s r d e s . ( i \ 1 J O r p ( ' h e n t 1 9 8 7 . J l . 2 6 0 8 - 2 6 1 l .