lecture 1, 2, 3 pharmacokinetic modeling and drug …...2020/02/03 · 1 1 pharmacokinetic modeling...
TRANSCRIPT

1
11
Pharmacokinetic Modeling and Drug Design
V. Frecer
Department of Physical Chemistry of Drugs
Faculty of Pharmacy, Comenius University in Bratislava
Elective subject, year 4, SS, 2L/1S
2019-2020
Lecture 1, 2, 3
2
List of lectures:
• Lecture 1,2 - Introduction to pharmacokinetics: Transport and fate ofa drug in organism; Drug design and development; Molecular structure and pharmacokinetic parameters
• Lecture 3 - Physicochemical principles of drug distribution
• Lecture 4 - Pharmacokinetic models of drug disposition
• Lecture 5 - Pharmacokinetic compartment models
• Lecture 6 - Nonlinear pharmacokinetic models
• Lecture 7 - Perfusion pharmacokinetic models
• Lecture 8 - Physiological pharmacokinetic models
• Lecture 9 - Pharmacokinetic models of drug-receptor binding
• Lecture 10 - Methods of prediction of transport properties of compounds
Contents

2
3
Literature
Recommended literature:
• ATKINS, Peter W. – DE PAULA, Julio: Physical Chemistry: Thermodynamics, Structure, and Change, 10th Ed., Oxford University Press, Oxford, UK, 2014.
• BOROUJERDI, Mehdi: Pharmacokinetics and Toxicokinetics, CRC Press, Boca Raton, FL, U.S.A. 2015.
• JAMBHEKAR, Sunil S. - BREEN, Philip J.: Basic Pharmacokinetics, 2nd Ed., Pharmaceutical Press, London, UK, 2012.
• KERNS, Edward H. - DI, Li: Drug-like Properties: Concepts, Structure Design and Methods, Elsevier, Burlington, MA, U.S.A., 2008.
• PATRICK, Graham L.: An Introduction to Medicinal Chemistry, 5th Ed., Oxford University Press, Oxford, UK, 2013.
4
Lecture 1
Introduction to Pharmacokinetics:
Transport and fate of a drug in organism
Drug design and development
Molecular structure and pharmacokinetic parameters
3
5
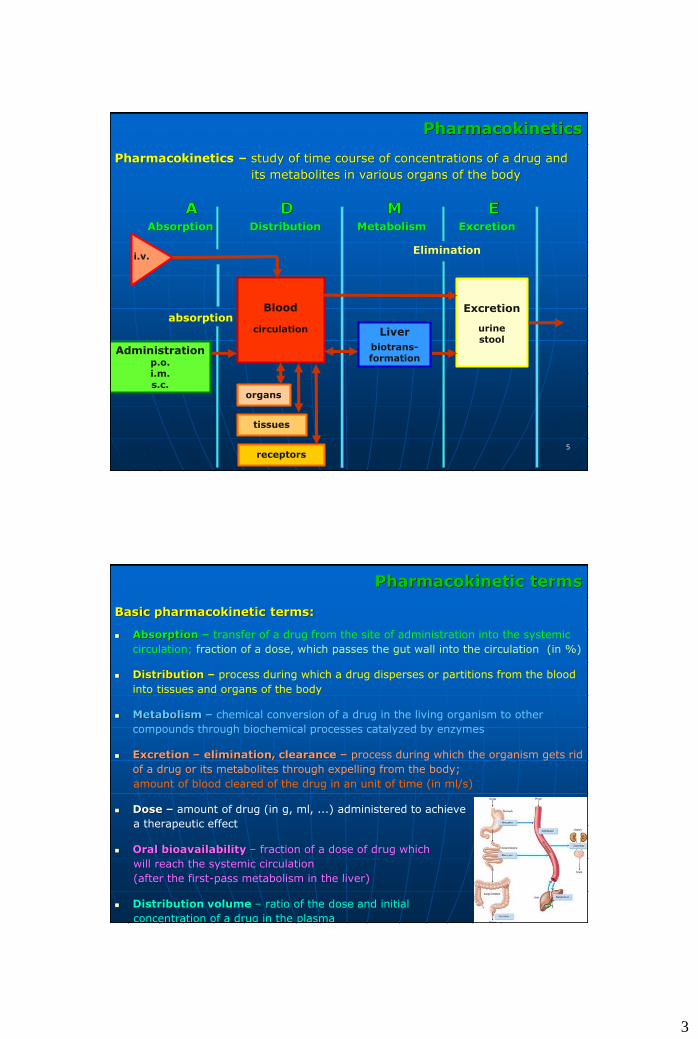
Pharmacokinetics
Pharmacokinetics – study of time course of concentrations of a drug and
its metabolites in various organs of the body
Absorption Distribution Metabolism Excretion
Elimination
Administrationp.o.i.m.s.c.
Blood
circulation Liver
biotrans-formation
Excretion
urinestool
organs
tissues
receptors
i.v.
absorption
6
Pharmacokinetic terms
Basic pharmacokinetic terms:
Absorption – transfer of a drug from the site of administration into the systemic
circulation; fraction of a dose, which passes the gut wall into the circulation (in %)
Distribution – process during which a drug disperses or partitions from the blood
into tissues and organs of the body
Metabolism – chemical conversion of a drug in the living organism to other
compounds through biochemical processes catalyzed by enzymes
Excretion – elimination, clearance – process during which the organism gets rid
of a drug or its metabolites through expelling from the body;
amount of blood cleared of the drug in an unit of time (in ml/s)
Dose – amount of drug (in g, ml, ...) administered to achieve
a therapeutic effect
Oral bioavailability – fraction of a dose of drug which
will reach the systemic circulation
(after the first-pass metabolism in the liver)
Distribution volume – ratio of the dose and initial
concentration of a drug in the plasma

4
7
Transport and fate of a drug in the organism
absorption
metabolism
distribution
binding
Excretion
drug drug
dissolved
bound
metabolite free
drug bound
free
cleared
cleared
GITskin
muscleslungs
body fluidsand tissues
urinestoolbile
breathsaliva, sweat
biologiceffect
Pharmacodyna-mic phase
interaction drug-receptor
at site of action
drug available for biological
action
bioavailability
Pharmacokinetic phase
absorption
distribution
metabolism
excretion
drug binding
drug available for absorption
pharmaceutical availability
Pharmaceutic phase
disintegration of drug form
dissolution of released active
substance
extra vascularadministrationof drug dose
Interaction of a drug with an organism:
8
Transport of drugs across biological membranes
Model of cellular
membrane:
Fluid bilayerof phospholipids
(phosphatidylcholine)with proteins
thickness: 75 – 100 nm
membranechannels - pores
diameter:~4 nm Bates TR, Gibaldi M. Biopharmaceutics. Lea & Febiger, Philadelphia, USA, 1970.
Transport of drugs:
passive diffusion of molecules across membranes (lipophilic compounds, non-dissociated polar molecules, logPo,w, pKa), driving force: concentration gradient
passage of compounds through pores in cellular membrane (soluble electrolytes and ions with dimensions up to the pore diameter, dependent of the charge in the pore opening, d < 4 nm, Mw < 200 Da)
transport facilitated by specific carriers (active transport against conc. and electrochem. grad., facilitated diffusion in direction of conc. and electrochem. g.)
pinocytosis (large molecules engulfed by the membrane)

5
9
Dissociation and distribution of drugs
B[100]
OH- BH+
[1]
Total: [101]
B[100]
BH+ OH-
[1]
Total: [101]
pH = 7,0
weak base pKa =5,0 weak base pKa =5,0
B[100]
OH- BH+
[1]
Total: [101]
B[100]
BH+ OH-
[10000]
Total: [10100]
pH = 7,0 pH = 7,0 pH = 3,0
Dissociation: acid AH + H2O H3O+ + A- base B + H2O BH+ + OH-
pKa=-log[H3O+][A-]/[AH] Henderson-Hasselbach: pKa – pH = log[AH]/[A-]
pKa – pH = log[BH+]/[B]
pKa – pH = 5 – 7 = -2 acid conc. [A-] = 100 and [AH] = 1
base conc. [BH+] = 1 and [B] = 100 (pKb = 14 – pKa = 9)
pKa – pH = 5 – 3 = 2 acid conc. [A-] = 1 and [AH] = 100
base conc. [BH+] = 100 and [B] = 1 (pKb = 14 – pKa = 9)
Diffusion: across membranes only the neutral non-dissociated form, equilibrium in each phase
10
Absorption of drugs
Absorption of drugs from GIT into systemic circulation
Absorption depends on the structure and physicochemical properties of molecules
Lipinski „Rule of five“ - oral bioavailability: Mw < 500 Da, 5 HB prot. don.,
10 HB prot. accep., logPo/w 5 (2245 per oral drugs from WDI, 1997)
Properties, which determine ADME
- molecular structure (composition, topology, 3D-str.)
- molecular mass Mw,
- polar and hydrophobic molecular surface, Ap An,
- number of hydrogen bonds, HBpd and HBpa,
- number of rotatable bonds, Nrot,
- partitioning coefficient octanol/water, logPo/w,
- solubility in water, logSw,
- blood/brain partitioning coef., logPBB,
- permeability of Caco-2 cells,
- binding to serum proteins, logKsp,
- binding to serum albumines, logKhsa,
- number of possible metabolic reactions
Lipinski CR. et al. Adv. Drug Deliv. Rev. 23, 3-25 (1997).
absorption of molecules of a drug in small intestine
molecular structure determines physicochemical properties of compounds

6
11
Significance of ADME properties for drug design
Role of ADME/Tox properties in termination of drug design projects:
ADME
- unfavorable pharmacokinetic profile (39 %)
Toxicity
- toxicity in animals (11 %)
- harmful side effects in man (10 %)
12
Prediction of ADME properties of drugs
Calculation of physicochemical properties determining ADME (W. Jorgensen)
- partitioning coefficient octanol/water logPo/w, QikProp
- water solubility logSw,
- blood/brain distribution coefficient logPBB,
- binding to serum albumins logKhsa,
Physicochemical properties (descriptors) calculated with help of molecular mechanics (MM), quantum chemistry (QM), computer simulations, ... are correlated with experimental quantities (logPo/w, logSw, ...) for large sets of compounds (700 molecules, including 500 drugs)
QikProp, Schrödinger, release 2016-4 , LLC, New York, NY, U.S.A.
2016.
Duffy EM, Jorgensen WL. J. Am. Chem. Soc. 122, 2878-2888 (2000).
Jorgensen WL, Duffy EM. Bioorg. Med. Chem. Lett. 10, 1155-1158
(2000).

7
1313
Stages of drug development
identify disease
isolate proteininvolved in disease
find a molecule effective
against protein
preclinical testing
compound synthesis scale up
drug formulation
human clinical trialsFDA approval
drug in clinical practice
explore molecular basis of disease (biochemistry, pharmacological target
identification, …)
isolate protein, determine crystal structure, protein homology modeling,
binding site determination, …
rational drug design, combinatorial chemistry, virtual screening, QSAR,
lead compound identification, …
development of a new drug: ~15 years, costs: >800 mil. US$
14
selectivity
efficacytoxicity
metabolism
absorptionexcretion
distribution
potentialdrug
Requirements for new drugs
• genetics
• cell and molecular biology
• bioinformatics
• structural biology
• biochemistry
• computer-assisted drug design
• medicinal chemistry
• toxicology
• chemical technology
• clinical pharmacology
• medicine
• ...
Drug discovery – multidisciplinary research

8
151515151515
Role of computations in drug development
Identification and validation of site of action
Genetics, Molecular biology
Bioinformatics
Determination of 3D structure
Crystallographic analysis
NMR spectra, Homology modeling
Medicinal chemistry
Organic synthesis
Combinatorial chemistry
Peptide chemistry
Biological testing
High throughput screening
in vitro, in vivo screening
Molecular design
Molecular modeling
Computational chemistry
Computer graphics
Optimization of hits
QSAR, Toxicology, ADME
Clinical tests
Pharmacology, Pharmacokin.
Finding of active ligands
Screening of databases
16161616
Force field-based simulations
Quantumchemical calculations
QSAR analyses
Homology protein modeling
Molecular diversity
Computer-assisted combinatorial chemistry
In silico screening
Docking of small molecules
Prediction of ligand-receptor binding affinity
Pharmacophore models
ADME properties prediction
Solvent effect calculations
Data mining
Bioinformatics
Chemiformatics
Molecular graphics
…
Computational methods in biomedical research

9
17
Combinatorial (parallel)
synthesis (same chemistry)
• in solid phase
• in solution
103-105 compunds/experiment
Traditional (serial) synthesis
1 researcher =
~50 compounds/year
A + B AB
BmA1 B1
A2 B2
A3 B3
. .
. .
An(acids)
Bm(amines) n x m (amides)
B1 B2
A2
A1
An
HTS
Traditional and combinatorial drug design
181818
Design of virtual combinatorial libraries
3D structure
of protein
pharmaco-
phore
protein
family
no
information
focused diverse
Info
rmati
on
ab
ou
t ta
rget
Design structure
based
Design based
on pharmacophore
Targeted Sets
Initial libraries
(diversity lib.)
Library size
Chemical space: ~1050 - 1080 compounds Arpád Furka, 1981
(existence of Universe since Big bang: ~5.1018 s)
10
191919
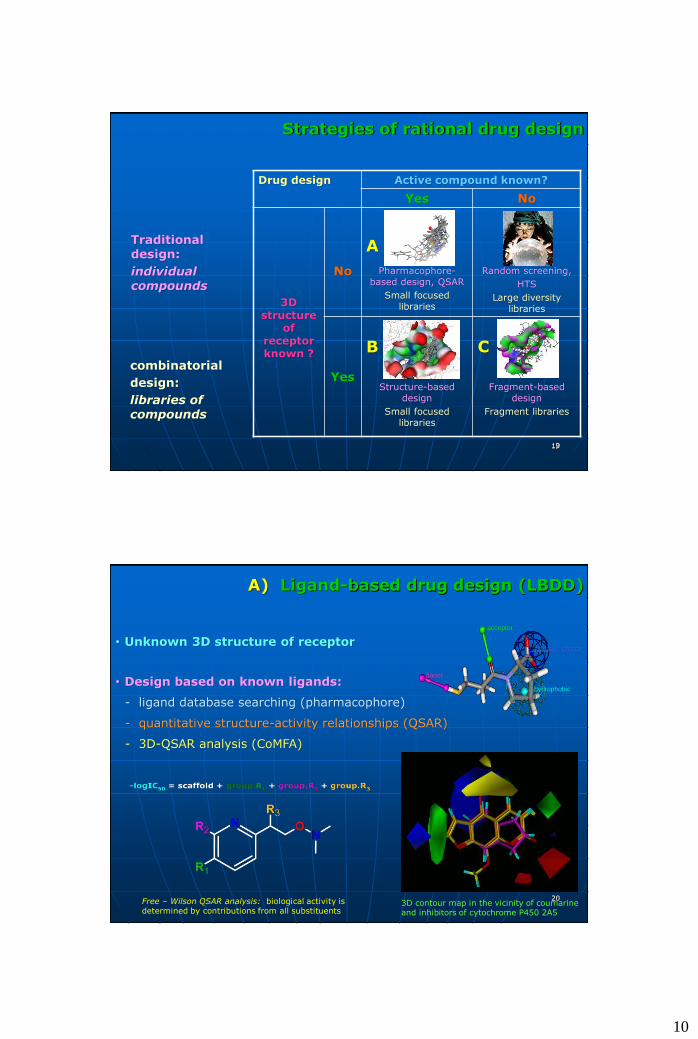
Strategies of rational drug design
1919
Drug design Active compound known?
Yes No
3D structure
of receptor known ?
No
A
Pharmacophore-based design, QSAR
Small focused libraries
Random screening,
HTS
Large diversity libraries
Yes
B
Structure-based design
Small focused libraries
C
Fragment-based design
Fragment libraries
Traditional design:
individual compounds
combinatorial
design:
libraries of compounds
2020
A) Ligand-based drug design (LBDD)
• Unknown 3D structure of receptor
• Design based on known ligands:
- ligand database searching (pharmacophore)
- quantitative structure-activity relationships (QSAR)
- 3D-QSAR analysis (CoMFA)
donor
acceptor
negative charge
hydrophobic
Free – Wilson QSAR analysis: biological activity is determined by contributions from all substituents
3D contour map in the vicinity of coumarine and inhibitors of cytochrome P450 2A5

11
2121
Pharmacophore-based design
Pharmacophore – spatial arrangement of a collection of groups or molecular fragments, which determine its biological activity
Utilization:
- Database searching–• for new chemical structures,
which correspond to pharmacophore
donor
acceptor
charged-neg.
hydrophobic
Pharmacophore of thymidine monophosphate kinase of M. Tuberculosis
Overlap with designed inhibitor
Keita M, et al.: RSC Adv. 4(99), 55853-55866 (2014).
2222
Quantitative structure-activity relationships (QSAR)
• QSAR: chemical, physical, biological properties (activity) – encoded in chemical structure of molecules
- similar molecules ~ similar properties, diverse molecules ~ diverse prop.
- model of biological activity
biological effect = f(molecular structure)
* mechanism of action
* prediction of activities of analogs
Molecular property Descriptor
Lipophilicity p, logPo/w, Rt
Steric properties Mw, Vm, shadow ind., 3D des.
Electronic prop. q, sd, EHOMO, ELUMO, E.A.
Structure, topology HB, W, logZ, ,
Biological effect Descriptor
Binding to receptor structural, electronic, topologic
Oral bioavailability lipophilicity, structural, steric
Metabolic activation structural, electronic

12
2323
• Methods of QSAR:
- L.P. Hammett, R.W. Taft dissociation constants of aromatic acids
pK = pKo - .s
- S.M. Free, J.W. Wilson biol. activity: sum of contributions of substituents
log(1/C) = a.(contrib.R1)+b.(contrib.R2)+...
- C. Hansch, A. Leo biol. activity: lipophilicity, steric, electronic prop.
log(1/C) = a.π2 + b.π + c.Es +d.s + e
Quantitative structure-activity relationships (QSAR)
Procedure:iterative approach to
selection of descriptorsand QSAR model
Methods:genetic algorithms
neural networksmachine learning
etc.
2424
3D Quantitative structure-activity relat. (3D-QSAR)
• QSAR models:
- 2D-QSAR models – descriptors derived from molecular structure
- 3D-QSAR models (CoMFA, CoMSIA, CoMMA,...) descriptors – molecular fields (MEP, hydrophobic interactions, ...) in 3D space around molecule
CoMFA models depend on the quality of overlap of compared molecules:
overlap with pharmacophoreoverlap of atoms
overlap of molecular shapes overlap of molecular fields

13
2525
B) Structure-based design (SBDD)
3D structure of pharmacological target (site of action):
• X-ray structures of protein targets (Protein Data Bank, 149,000 3D str. of macromolecules, Feb. 2019)
• NMR structures of proteins in solution
• homology-based protein models
Design of ligands:
• in situ modificat. of known ligands/substrates
• docking and in silico screen. of new molecules
Influenza A virus N1 neuraminidase–oseltamivir (2HU0) hemagglutinin IAV H18N11 (4MC5)
2626
Structure-based drug design (SBDD)
Interactions of ligand in binding site of a receptor, ligand design:
• complementarity of L and R (steric, molecular fields)
• desolvation of L and R
• entropic effects
A B
C D
www.kubinyi.de
binding site ofreceptor
ligand 1
Estimate of binding affinity of ligand to receptor:
Calculation of Gibbs free energy of LR complex formation
DGo = -RT lnKi
molecularfields
ligand 2
shape of binding site structure of binding site
complementarity of ligand and binding site

14
2727
Structure-based drug design (SBDD)
Factors that contribute to recognition and binding of a ligand:
π-π interactions
hydrophobiccontacts
explicitwater molecules
hydrogen bonds(directional interactions)
steric interactions
flexibility of ligandand receptor
desolvation of ligandand receptor
peptidomimetic inhibitor of plasmepsin II
2828
Protein homology (comparative) modeling
Availability of 3D structures of macromolecules (proteins) is limited
• PDB (160,000 X-ray and NMR atomic resolution structures), 148,000 proteins, only 30000 sequences <30% identity
• similar a.a. sequence similar 3D structure (particularly overall fold) similar function of protein
• protein structures are more conserved than sequences amongst homologous proteins (up to 30% sequence identity)
• homology model building of target protein
- template proteins selection (detection of distant evolutionary relationships)
- target-templates sequences alignment (BLAST, FASTA, PFAM, protein threading RaptorX,…)
- target-templates structural alignment and model building (backbone generation, loop modeling, side chains conform., refinement)(Modeller, Jackal, SCRWL, SwissModel, …
- model assessment (What If, ProCheck, PSQS, Ramachandran plot,…)

15
2929
Protein homology (comparative) modeling
Sali A, Kuriyan J.: Trends Biochem. Sci. 22, M20–M24 (1999)
Quality of homology models depends mainly on:
- selection of template proteins
- alignment and sequence identity totemplates
- resolution of template structures
- method of modeling and refinement
3030
Binding affinity of ligand to receptor
Models of ligand-receptor binding:
- lock and key
- induced fit theory
ligand (L) + receptor (R) ligand-receptor (LR) complex
+
Pharmacodynamic effect of ligand:
kas kf kdis [LR] [L]L + R LR LR* Kd = —— Rel. response = —— = ————
kdis k-f kas [Ro] [L] + Kd
binding affinity pharmacol. dissociation effect depends on dose Leffect constant
[L] – concentration of free ligand
Binding affinity of L: lnKd = DGo/RT property of complex LR

16
3131
Prediction of binding affinity of ligand
Molecular modeling – calculation of binding affinity – modeling of LRaq
pKd = -log10Kd = aDGcom,aq/2,303RT + b
Thermodynamic cycle:
DGcom,g
Lg + Rg LRg gas phase
Gsol(L) Gsol(R) Gsol(LR)
DGcom,aq
Laq + Raq LRaq aqueous solution
Gibbs free energy of formation of LRaq:
DGcom,aq = [Gg(LR) - Gg (L) - Gg (R)] + [Gsol(LR) - Gsol(L) - Gsol(R)]
where:Gg = Etot + thermal corrections of internal energy and entropy
3232
Calculations of ligand-receptor interactions
• Energies of molecules and intermolecular interactions:
- quantum mechanical methods (QM)
- molecular mechanics methods (MM, force fields)
- hybrid QM/MM methods
- scoring functions
• Molecular simulations, statistically averaged quantities:
- MC simulations, MD simulations
- conformational searching
• Solvation:
- explicit models (discrete solvent molecules)
- implicit models (GBSA - Still et al., PB - Honig et al., PCM - Miertuš et al.)
• Models of estimate of DGcom,aq used in drug research:
- MM-PBSA/GBSA (Kollman et al.)
- LIE (Åqvist et al.)
- MM-PCM (Frecer, Miertuš)
- …

17
3333
Methods of computational chemistry
Quantum chemical methods (ab initio, semiempiric, DFT methods)
Schrödinger equation
ĤΨ(r1,…,rn,R1,…,RN) = EΨ(r1,…,rn,R1,…,RN)
Ĥ – Hamilton operator of total e., Ψ – wave function (observable physicalquantities), E – energy of molecule, ri, Ri – radius vectors of electrons and nuclei
I. R. Levine
hyper surface E(r,R)
Science 2007;315:1561-1565.
propanal
343434
Methods of computational chemistry
Molecular mechanics - potential energy of molecule
U(x1,…,x3N) = ½∑vkv(l-lo)2 + ½∑uku(θ-θo)
2 +
+ ½∑tVnt[1+cos(nω-γ)] +
+ ½∑i∑j(qiqj/εrij) +
+ ½∑i∑j[Aij/rij12-Bij/rij
6] +Force field constants: parameterized against experiment and ab initio calculations
+ ½∑i∑j[Cij/rij12-Dij/rij
1 0]
Molecules 2014;19(10):15735
bonding
nonbonding
electrostatic
dispersion-repulsion

18
3535
Methods of computational chemistry
Hybrid quantum mechanical/molecular mechanical methods (QM/MM):
• Binding site – QM region
Schrödinger equation: ĤΨ = EΨ
• Bulk protein – MM region
molecular mechanics: E = bonding + nonbonding terms
• Solvent – MM region
- explicit hydration (MM)
- implicit hydration – classical models (Born, Jano, PCM, PB, GBSA, ....)
• Link between QM and MM regions of protein (dummy atoms, frontier atomic orbitals, ...)
Nobel price for chemistry 2013: QM/MM methodsM. Karplus, A. Warshel, M. Levitt
3636
Polarizable Continuum Model of solvation (PCM)
MEP on molecular surface of imidazole
Solvent – continuous homogeneous polarizable medium (dielectric constant)
Reaction field – elst. interaction with the molecule incavity
Quantum chemical model a:
Gsol,els = <Ψ|Ĥo+VRF|Ψ> - <Ψo|Ĥo|Ψo> - ½ <Ψ|VRF|Ψ>
Classical model b:
Gsol,els = ½ j (qj VRF,j – mj ●ERF,j)
a Miertus S, Scrocco E, Tomasi J.: Chem. Phys. 55(1), 117-129 (1981).
Miertus S, Tomasi J.: Chem. Phys. 65(2), 239-245 (1982).
b Frecer V, Miertus S.: Int. J. Quant. Chem. 42(5), 1449-1469 (1991).
Miertus S, Frecer V.: J. Math. Chem. 10(1), 183-204 (1992).
vdW cavity - aminopropanone

19
3737
Molecular simulations
• Experimental biological activities:
Ki, IC50 – thermodynamic quantities observed on large ensembles of molecules
(V = 1μL, c = 1 μM 6·1011 molecules of enzyme at [T, p])
(macroscopic quantities averaged over a set of most probable configurations of statistical ensemble at thermal equilibrium )
• Prediction of Ki, IC50 through calculations of L-R binding affinity:
lnKi = - DG°bin/RT
averaging of DG°bin over statistical ensemble of LR configurations DG°bin
• Calculation of thermodynamic quantities - molecular simulations:
- Monte Carlo simulations
- molecular dynamics
dihydrofolate reductase in a periodic box of water
3838
Monte Carlo simulations
• MC simulations:
- sampling of configurational space by generating states Ai (random changes of internal coordinates)
- calculation of averaged phys. chem. quantities: A = 1/n·∑i
nAi
- configuration Ai+1 is generated from configuration Ai (Markov chain)
- configuration Ai+1 is accepted to statistical ensemble if: Ei+1 < Ei
if : Ei+1 > Ei random number generator (0<R<1)
Boltzmann probability of state Ai+1 (Metropolis importance sampling)
• R > exp[-(Ei+1-Ei)/kT] Ai+1 is accepted
• R < exp[-(Ei+1-Ei)/kT] Ai+1 is rejected
Tapia L, et al.: Bioinformatics 23(13), i539-i548 (2007).
evolution of population of conformations during
protein folding; Monte Carlo simulation

20
3939
Molecular dynamics
• Molecular dynamics:
- method of statistical mechanics: simulation of evolution of complex systems, calculation of time-averaged physicochemical quantities
- generation of MD trajectories (system configurations) – numerical integration of Newton equations of motion (dt = 1 fs) in a constantly changing force field:
fxi = miaxi = mi(d2xi/dt2) = -∂U(x1,…,x3N)/∂xi
axi(t) = fxi/mi vxi(t+dt) = vxi(t) + axi(t)·dt
xi(t+dt) = xi(t) + vi(t)·dt + ½ ai(t)·dt2
U(x1,…,x3N) – potential energy (FF)
xi = xi(t), vi = vi(t) i= 1, ... N
trajectory
van Eijk M, et al.: J. Biol. Chem. 287, 26666-26677 (2012).
time-course of RMSD of a loop of porcine and human surfactant protein D (collectin)
4040
Molecular dynamics
• Applications of molecular dynamics:
- calculation of thermodynamic quantities of systems - ensemble averaged values
Ā = 1/n·∑i
nA(ti)
- kinetic quantities
- conformational transitions
- refinement of structures
- tracking of dynamic behavior of macromolecules: thermal fluctuations
Software: CHARMM, AMBER, GROMOS, NAMD, GROMACS, MacroModel, Tinker, ...
two snapshots of a protein in different times of an MD simulation
a consequence of thermal fluctuations is that proteins are “breathing”
which can lead to transitions between various conformations

21
4141
Docking
• Virtual (in silico) screening of libraries of compounds:
- 3D structure of macromolecular receptor
- binding site of ligand
- docking: generation of ligand poses in bindingsite of receptor
- scoring functions
- ranking of poses and ligands
- selection of perspective ligands for synthesis and activity testing
Plasmodium falciparum enoyl-acyl carrier protein reductase
active site ofPfENROverlap of generated poses of
selected analogs of triclosan
Frecer V., Megnassan E., Miertus S. Eur. J. Med. Chem. 44(7), 3009-3019 (2009).
4242
Docking
• Scoring functions:
- force field-based (AMBER, CHARMM, MMFF, OPLS, ..)physically correct, suitable for organic molecules
- empirical based on frequency of occurrence of specific types of interactions
- knowledge-based - obtained from analysis of crystal structures of LRcomplexes, prefer frequently occurring interactions and geometries
• Famous scoring functions:
AutoDock, LigScore, ChemScore, GlideScore, PLP, PMF, LUDI, FlexXMMFF(tot, vdW), OPLS(tot, vdW), HINT, ICM, Validate, DrugScore, ...
• Docking to rigid or flexible receptor
Kitchen, D. B., et al.: Nat. Rev. Drug Discov. 3, 935-949 (2004).
Moitessier, N. et al.: Brit. J. Pharmacol. 153, 7-26 (2008).

22
4343
C) Fragment-based drug design (FBDD)
• Ligand is constructed from fragments in the binding site of receptor
- fragment - small molecule (<300 Da), weaker binding in a pocket of the active site, fragments are linked into a larger molecule (500-700 Da)
- positions and orientations of fragments are retained in molecules
Hajduk PJ, M, et al.: J. Am. Chem. Soc. 119(25), 5818-5827 (1997).
Ki = 17 mM Ki = 20 μM
Ki = 15 nM
SAR by NMRdesign of nonpetidic
inhibitors of metalloprotease
stromelysin
fragment 1 fragment 2
linker
www.kubinyi.de
4444
Fragment-based drug design (FBDD)
Hunk A, et al.: Angew. Chemie Int. Ed. 48(45), 8452-8456 (2009).
inhibitor panthotenate synthase of M.
tuberculosisdesigned by FBDD
• FBDD includes 3 steps:
- design of high quality library of fragments
- docking, scoring and ranking of fragments
- augmenting, combination and joining of fragments into final ligand
• Software for FBDD:
- GRID, MCSS, SPROUT, MUSIC, LUDI, SkelGen, Superstar, SEED, FFLD, GANDI, eHiTS, Caveat, HOOK, Recore, Allegrow, Confirm, MED-SuMo, LEGEND, GROWMOL, LigBuilder, SMoG, HOOK, PRO_LIGAND, SPLICE/RACHEL, CLIX, LORE, GEMINI, ...

23
45
Examples of design of bioactive compounds by molecular modeling
46
Kubinyi H., In: Computer Applications in Pharmaceutical Research and Development, Ekins S. (Ed.), John Wiley & Sons, Inc., (2006).
Drugs developed by means of molecular modeling:
Dorzolamide (Trusopt – Merck, 1995)Inhibitor of carbonic anhydrase therapy of glaucoma, occular drops
Imatinib (STI-571, Gleevec, Glivec – Novartis, 2001)
Inhibitor of tyrosine kinase, cancer therapy (chronic myelogenic leukemia)
Nelfinavir (AG1343, Viracept - Aguron Pharm., Pfizer, 1997)
Inhibitor of aspartic protease of HIV-1 virustherapy of HIV-1 infection and AIDS
Examples of successful molecular design of drugs

24
47
Examples of drug design projects
4848
Design of inhibitors of protease of HIV-1 virus
Peptidomimetic inhibitors of aspartic protease of HIV-1 (HIV PR)
Crystal structure of HIV PR with inhibitor XV-638 (1BV9)
HIV PR essential enzymenecessary for viral maturation
selected inhibitors of HIV PRapproved by FDA
HIV-1 virus

25
4949
Drug resistance of HIV-1 virus
mutations that cause drug resistance of HIV-1 to PR inhibitors
Viral resistance to drug occurs under selective pressure:
- known HIV PR mutations
R8K, R8Q, V32I, K45I, M46I/M46L/M46F, I47V, G48V, F53L, A71V, V82A/V82I/V82T, I84V, L89M and their combinations
Constant need to develop new antiviral compounds, which act on wider spectrum of HIV PR mutants
50
Peptidomimetic inhibitors of HIV PR
Three series of C2-symmetric HIV PR inhibitors:
- contain non-cleavable isosteres of peptide bond
(prepared in collaboration with University of Trieste, Prof. F. Benedetti)
dihydroxyethylenediamine core--Phe-Ψ[CHOH-CHOH]-Phe--
hydroxyethylenediamine core--Phe-Ψ[CH2-CHOH]-Phe--
Dihydroxyethylenediamine core--Phe-Ψ[CHOH-CHOH]-Pro--
Lopinavir, Abbott Laboratories

26
5151
Peptidomimetic inhibitors of HIV PR
--Phe-Ψ[CHOH-CHOH]-Phe– inhibitors designed to compensate the V82A, V82I, V82F mutations of HIV PR
10 structures proposed
Frecer V, Miertus S, Tossi A, Romeo D. Drug Des. Disc. 15(4), 211-231 (1998).
Burello E, Bologa C, Frecer V, Miertus S. Mol. Phys. 100(19) 3187-3198 (2002).
5252
hydroxyetylénediamínové jadro --Phe-Ψ[CH2-CHOH]-Phe--
32 štruktúr navrhnutých
Frecer V. Miertus S. Macromol. Chem. Phys. 203(10-11), 1650-1657 (2002).
Frecer V, Jedinak A, Tossi A, Berti F, Benedetti F, Romeo D, Miertus S. Lett. Drug Des. Disc. 2(8), 638-646 (2005).
T17 v aktívnom centre HIV PR
Peptidomimetické inhibítory HIV PR

27
53535353
Dihydroxyethylenediamine core --Phe-Ψ[CHOH-CHOH]-Pro--
~100 structures proposed
Frecer V. Miertus S.: Macromol. Chem. Phys. 203(10-11), 1650-1657 (2002).
Frecer V, Jedinak A, Tossi A, Berti F, Benedetti F, Romeo D, Miertus S.: Lett. Drug Des. Disc. 2(8), 638-646 (2005).
Frecer V, Berti F, Benedetti F, Miertus S.: J. Mol. Graphics Modell. 27(3), 376-378 (2008).
FP23 - best drug candidate
FP23 in catalytic center of HIV PR
favorable ADME properties
Peptidomimetic inhibitors of HIV PR
54
Experimental verification of predicted inhibitory potencies of designed peptidomimetics towards the HIV PR: group of Profs. Benedetti and Berti, University of Trieste, Italy
Confirmed inhibition constants in the low nanomolar conventration range
Synthesis and testing of inhibitors of HIV PR
Frecer V, Jedinák A, Tossi A, Berti F, Benedetti F, Romeo D, Miertus S. Lett. Drug Des. Disc. 2(8), 638-646 (2005).
Berti F, Frecer V, Miertus S.: Curr. Pharm. Des. 20(3), 3398-3411 (2014).

28
5555
drug-likecharacter
of designed inhibitors
Confirmed inhibition constants in low nanomolar concentration range
Frecer V, Berti F, Benedetti F, Miertus S. J. Mol. Graphics Modell. 27(3), 376-378 (2008).
Berti F, Frecer V, Miertus S.: Curr. Pharm. Des. 20(3), 3398-3411 (2014).
Synthesis and testing of inhibitors of HIV PR
Experimental verification of predicted inhibitory potencies of designed peptidomimetics towards the HIV PR: group of Profs. Benedetti and Berti, University of Trieste, Italy
56
Thank you

29
5757
Collaborators:ICS-UNIDO, UCM-Trnava University of TriesteS. Miertus D. RomeoJ. Miertus F. Benedetti
F. BertiInternational Fellows A. Tossi E. Burello (Italy) S. PriclS. DeNardi (Italy) P. BraiucaC. Bologa (USA) A. Jedinák (Slovakia) National University of SingaporeA. Nair (India) J.L. DingE. Megnassan (Ivory Coast) B. HoM. Keita (Ivory Coast) D.H.P. LowM. Kabeláč (Czech Rep.) D. Cerin (Slovenia) University of MilanR. Kothamarti (India) P. SeneciL. Owono (Cameroon) T. Udommaneethanakit (Thailand) Xeptagen, VeniceT. Rungrotmongkol (Thailand) G. FassinaM. Malaisree (Thailand) P. PengoD. Kong (China) L. Beneduce
5858
Plasmodium falciparum – virulentná forma malárie v krajinách tretieho sveta
Rezistentné formy P. falciparum – potrebné nové “dostupné” liečivá zacielené proti novým farmakologickým targetom
Target - enzýmy FAS-II biochemickej syntézy mastných kyselín
enoyl-acyl carrier protein reduktáza esenciálny enzým FAS-II pathway (PfENR)
kryštálová štruktúra PfENR s peptidomimetickým inhibítorom TCL11
Freundlich JS, et al. J. Biol. Chem. 282(35), 25436-25444 (2007).
séria PfENR inhibítorov odvodených od triclosanu, aktívne v nanomolárnych koncentráciách
Príklad 2. SBDD: Kombinatorický dizajn inhibítorov
Anopheles gambiae

30
5959
- predpoveď väzbovej afinity: kotvenie inhibítorov do kryštálovej štruktúry PfENR-TCL11 (IC50 =76 nM)
- QSAR model pre tréningový set: pIC50 = -logIC50 = a.LUDI + btarget specific scoring function LUDI (prispôsobená pre PfENR)
- predpoveď ADME vlastností
- QSAR model aplikovaný na in silico skríning kombinatorickej knižniceanalógov
inhibítory PfENR tréningového setu
pIC50 = -6.3473 + 0.0069·LUDI
n = 16, R2 = 0.83, F-test = 65.7
Enoyl-acyl carrier protein reduktáza - P. falciparum
aktívne centrum PfENR
V. Frecer, E. Megnassan, S. Miertus, Eur. J. Med. Chem. 44(7), 3009-3019 (2009).
6060
Kombinatorická knižnica inhibítorov
60
Peptidomimetický skafold, 3 R-groups
počiatočná diversity library
40 (R1) x 23 (R2) x 11 (R3) =
= knižnica10120 analógov
zacielená knižnica
8 (R1) x 5 (R2) x 3 (R3) =
= knižnica120 analógov
R-groups zacielenej (výslednej) knižnice
Kombinatorická knižnica - 3 substitučné miesta R1, R2, R3
8 R1 5 R2 3 R3

31
61
Navrhnuté inhibítory PfENR
Vybrané štruktúry najlepších kandidátov na inhibítory PfENR
Frecer V, Megnassan E, Miertus S. Eur. J. Med. Chem. 44(7), 3009-3019 (2009).
TCL-40-9-11 preložená s TCL11
Predpovedaná IC50 = 13 nM
62
Ďakujem za pozornosť