lecture 5 prenatal genetic counseling
TRANSCRIPT

IDENTIFY COMMON CONGENITAL ANOMALIES IN INFANTS AND CHILDREN
• PRENATAL GENETIC EVALUATION AND COUNSELING
----------------------------------------------


GENETIC SCREENING
• SCREENING MAY BE USED IN POPULATION AT RISK FOR A PARTICULAR GENETIC DISORDER
• ONLY APPROPRIATE WHEN THE NATURAL HISTORY OF THE DISEASE IS UNDERSTOOD
• THE SCREEING TEST ARE VALID AND RELIABLE– SENSITIVITY– SPECIFICITY– FALSE-POSITIVE AND FALSE NEGATIVE
RATES ARE ACCEPTABLE– EFFECTIVE THERAPHY IS AVAILABLE– JUSTIFY ITS COST

TYPE OF SCREENING
• HETEROZYGOTE SCREENING
• PRESYMPTOMATIC SCREENING
• PRENATAL DIAGNOSIS
• NEWBORN SCREENING

Heterozygote secreening
• Screening a subceptible population– Ashkenazic jewsTay-Sachs– High frequenzy of heterozygotes in
blackthalasemia
• Screening for person who a carrier for a specific disorder to make informed reproductive choices.
• Consanguineus mating

Presymptomatic genetic screening
• In family history of dominantly inherited disorder– Hutington’s ds– Breast cancer– Adult polycystic kidney disease
• Identifying a definite carrier of the genetic disorder

PRENATAL DIAGNOSIS
• Steele and Breg– Amniotic fluid cells could be cultured– Fetal karyotip can be demonstrated– Pregnant women , on the basis of age– Woman under 35 yrs old secreening for:
• Cystic fibrosis,• DMD• Other commond genetic disorder
• Only 2% prenatal diagnosis are terminated• Because of the fetus has a genetic defect

Indication for prenatal diagnosis
• Maternal age over 35• Previous child with chromosome abnornality• Structural chromosomal abnormality in one
parent• Family history of a neural tube defect• Family history of genetic defect• Fetus is at risk of an identifiable defect• X-linked disorder

TERMINATION OF PREGNANCY
• LEGAL TERMINATION: WHEN THE FETUS IS FOUND TO BE SERIOUSLY ABNORMAL– CHROMOSOMAL DEFECTS– ANATOMICAL ABNORMALITIES
• HOTLY DEBATED AS ABORTION


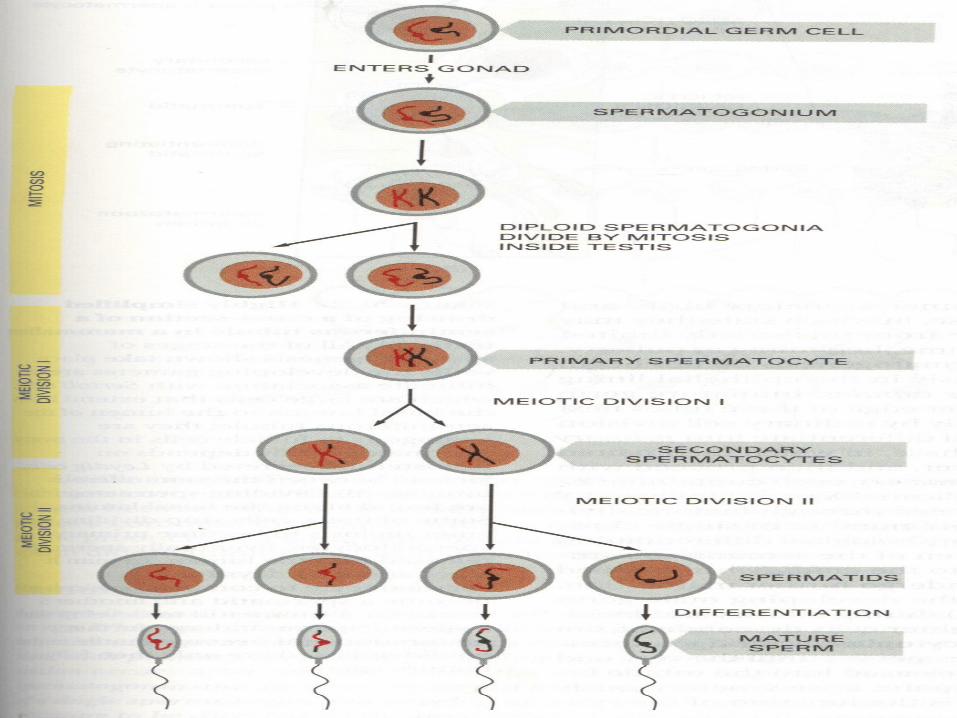
GAMETOGENESIS
• Conversion of germ cells into male and female gametes
• OOGENESIS and SPERMATOGENESIS
• Male gametspermatozoa: 22+Y
• Female gametovum:22+X
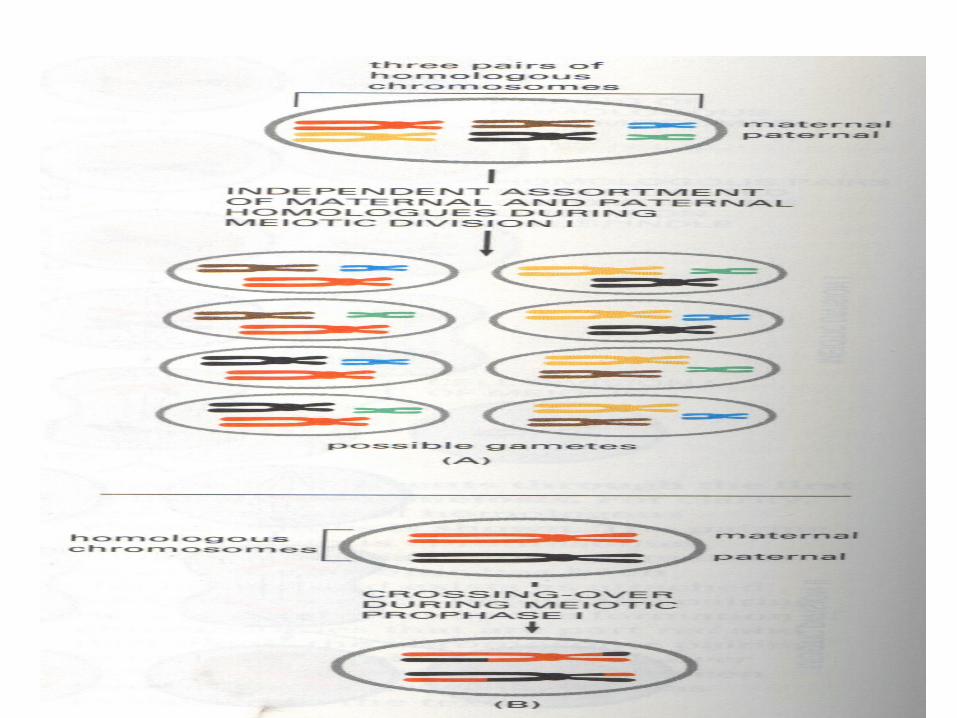
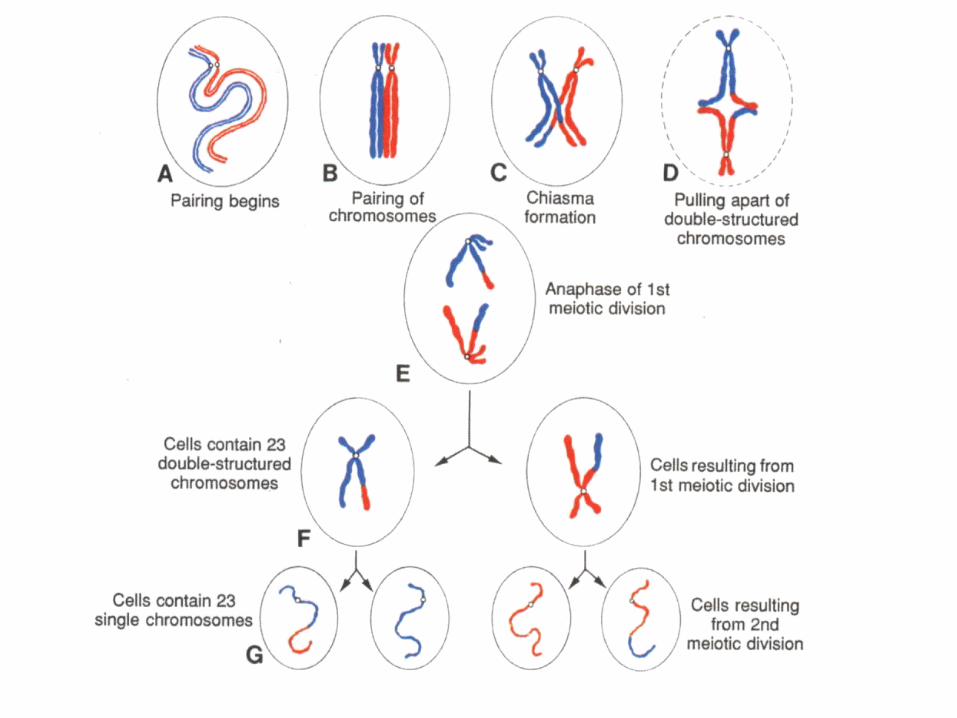
• Mitosis, meiosis I , meiosis II and cytodifferentiation
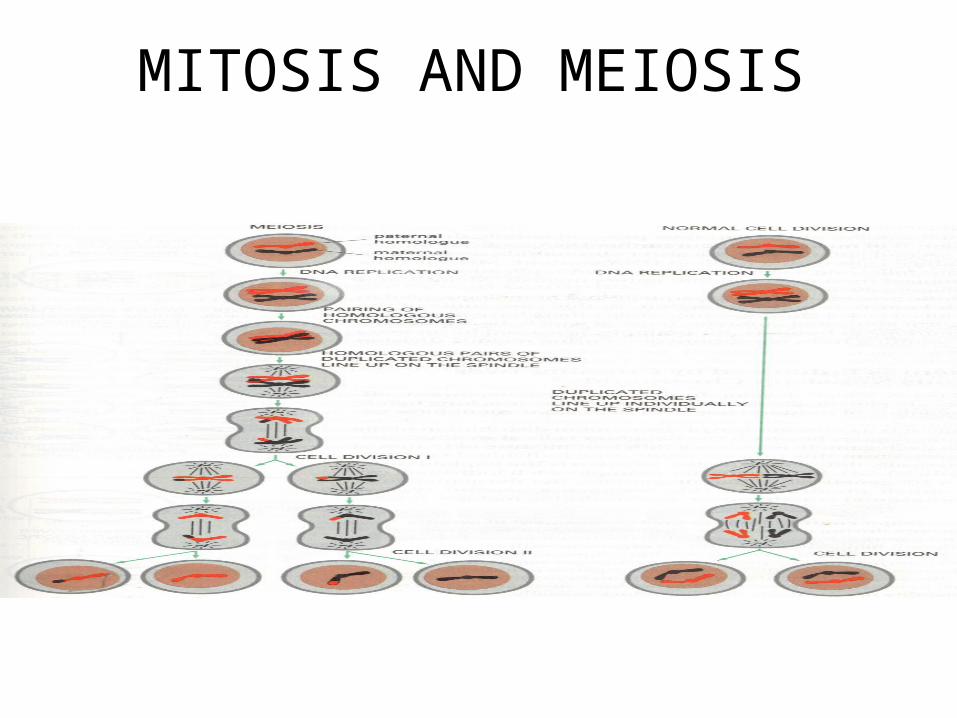
MITOSIS AND MEIOSIS

Morphological changes
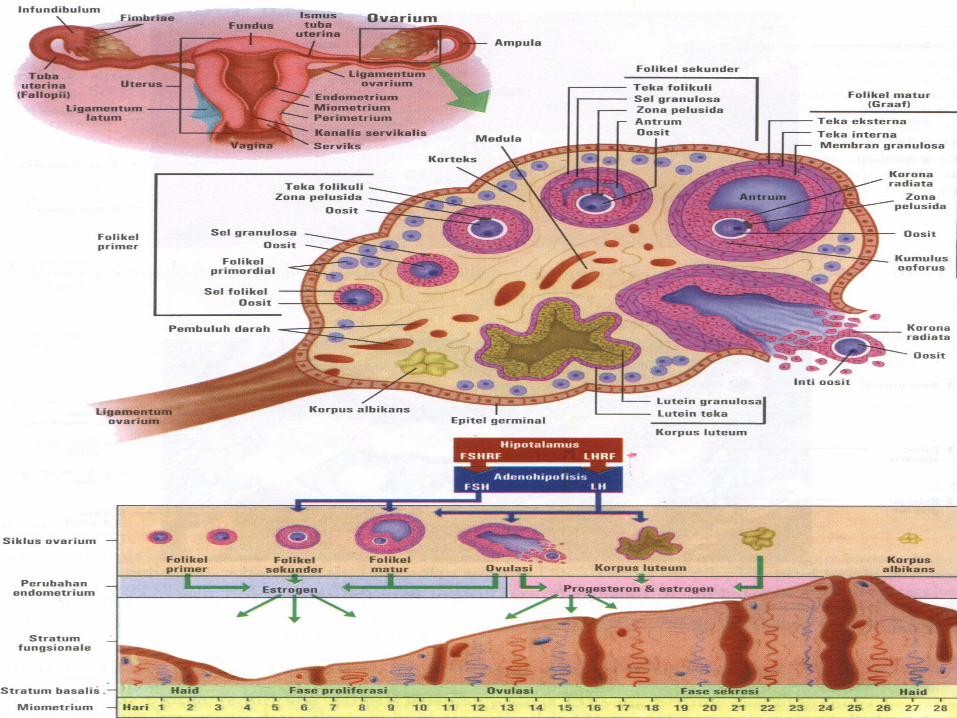
• Oogenesis– Maturation of oocytes begins before birth:
primordial germ celloogoniaprimary oocytes+epithelial cellprimordial follicle and rest in prophase of first meiotic division because of oocyte maturation inhibitor
– Maturation of oocytes continues at puberty• Spermatogenesis
– Maturation of sperm begins at puberty,spermatogonia are transform into spermatozoa

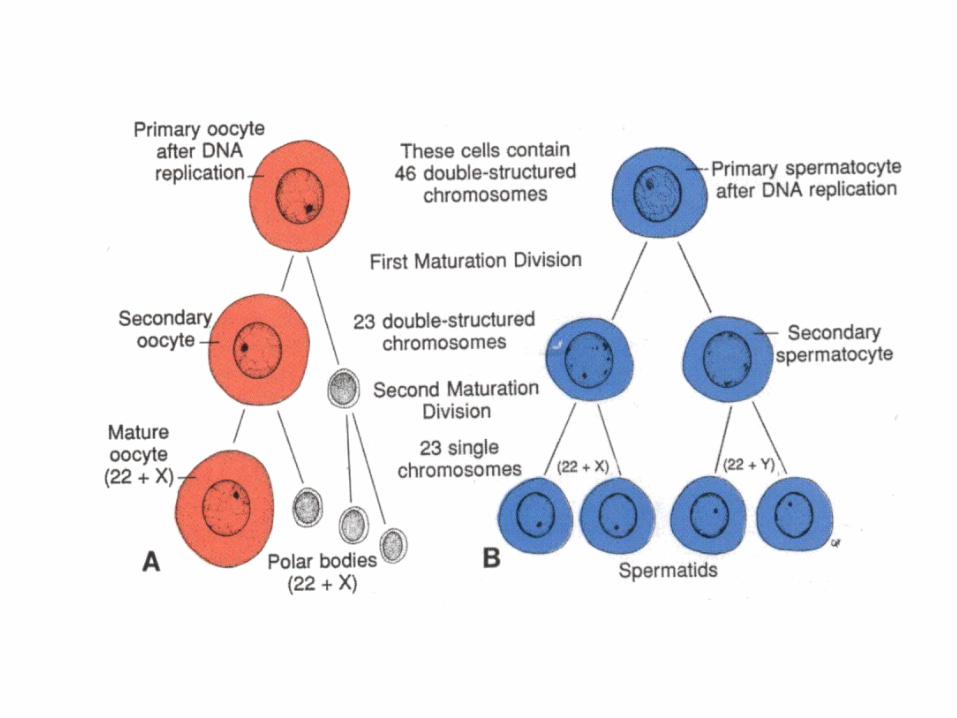
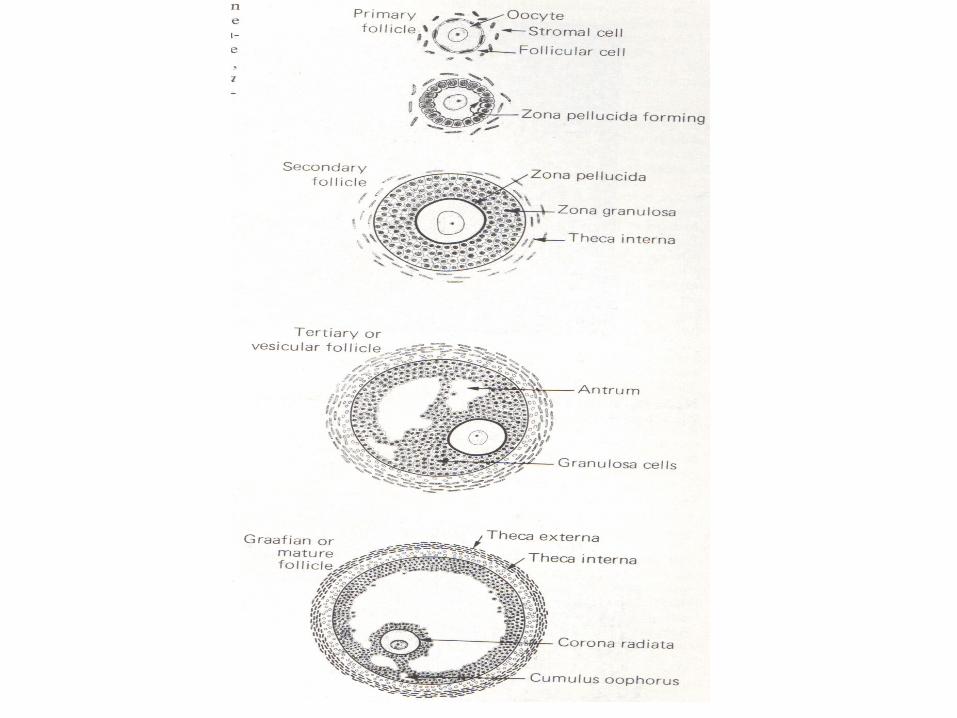
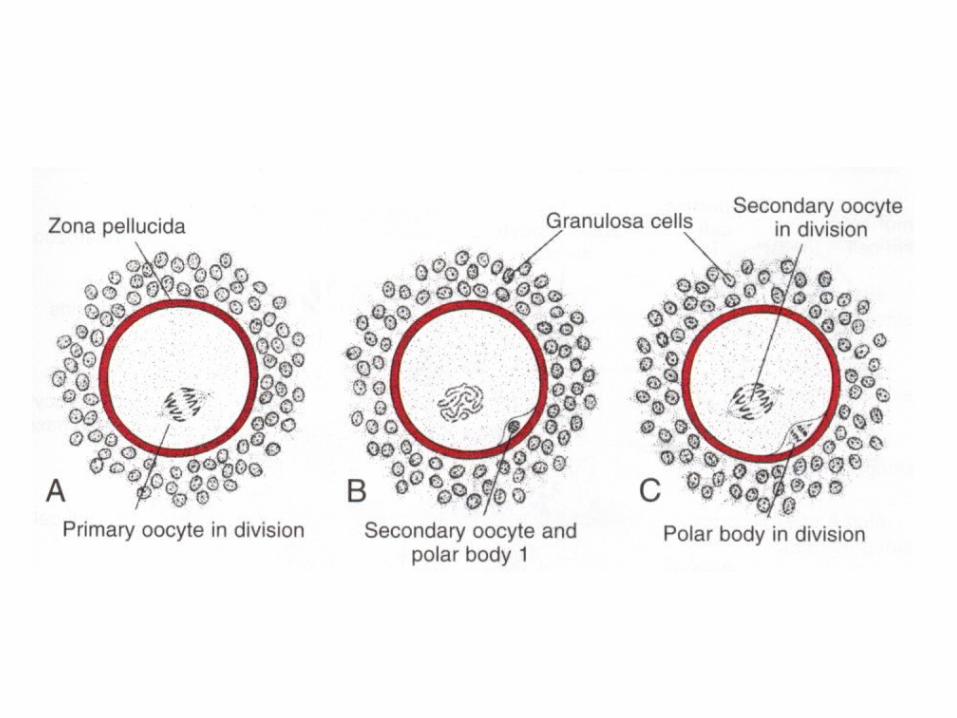
OOGENESIS
• OogoniaOosit IOosit IImature• Folikel primordialFolikel IFolikel
IIFolikel de Graaf• Oosit I enter prophase of the first meiotic
division• This may last 40 or more years and finishes
only when the cell begins its final maturation• Maturation of oocytes contunies at puberty

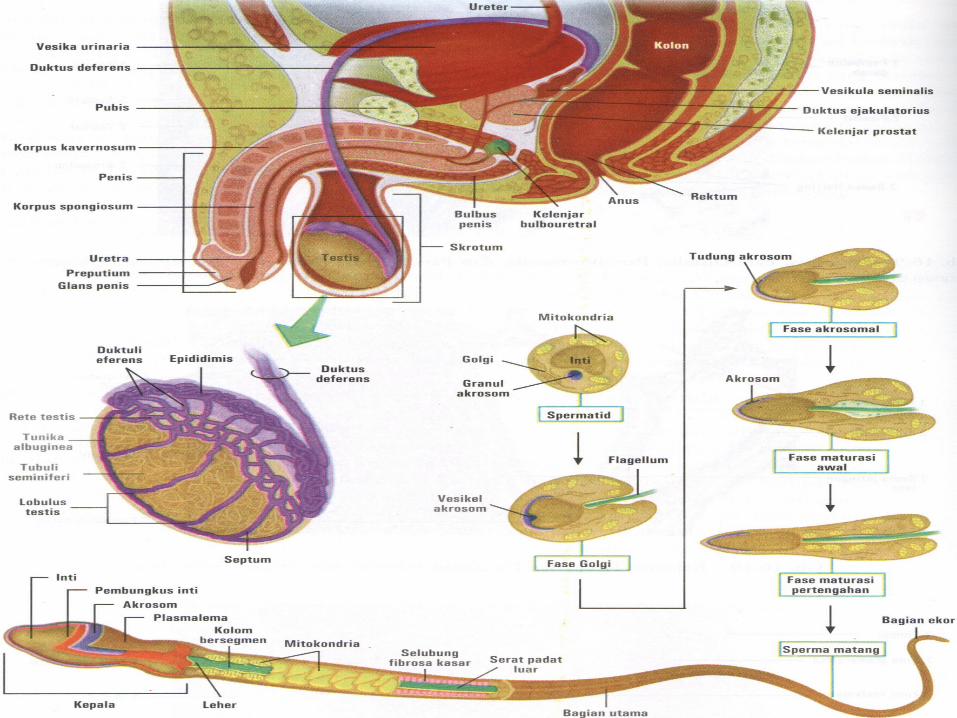

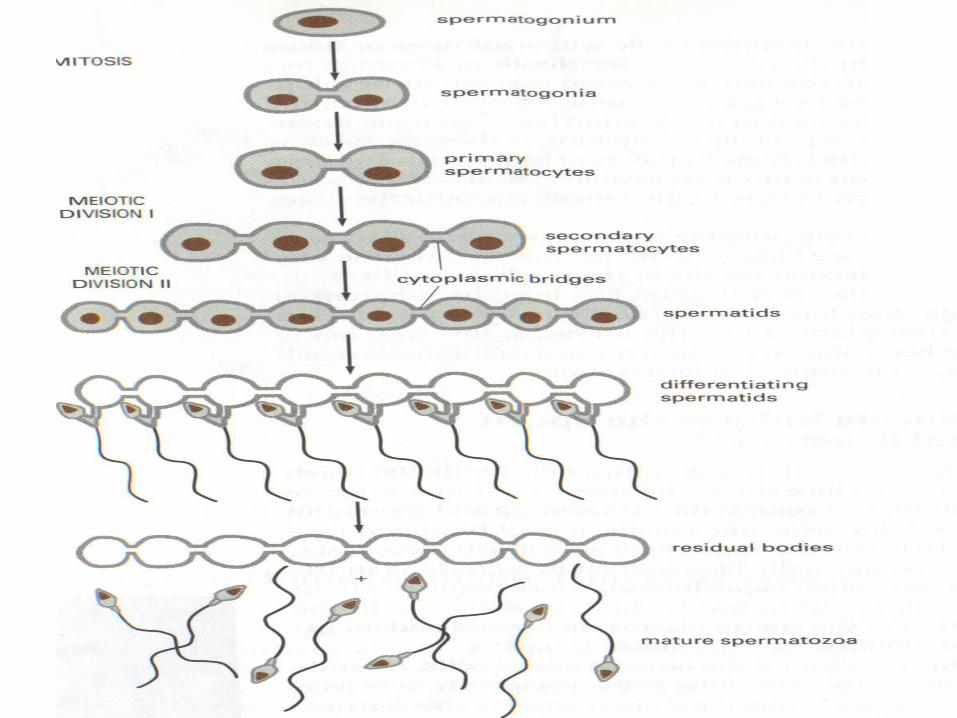
SPERMATOGENESIS
• Maturation of sperm begins at puberty• Spermatogonia transformed into
spermatozoa• Spermiogenesis:
spermatedspermatozoa– Formation of the acrosome– Condensation of the nucleus– Formation of neck, middle piece, tail– Shedding of most of the cytiplasm

CLINICAL CORRELATION
• Birth defect and spontaneous abortions– Chromosomal abnormalities– Mutations
• Non disjunction
• Mosaicism
• Translocation
• Microdeletion/deletion
• Fragile sites
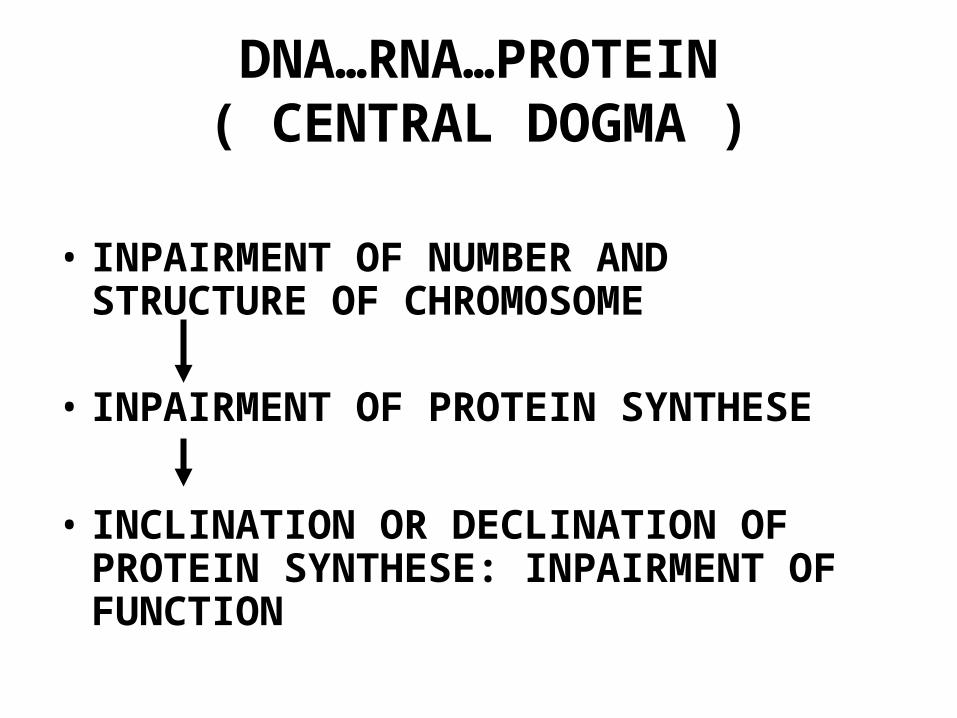
DNA…RNA…PROTEIN( CENTRAL DOGMA )
• INPAIRMENT OF NUMBER AND STRUCTURE OF CHROMOSOME
• INPAIRMENT OF PROTEIN SYNTHESE
• INCLINATION OR DECLINATION OF PROTEIN SYNTHESE: INPAIRMENT OF FUNCTION
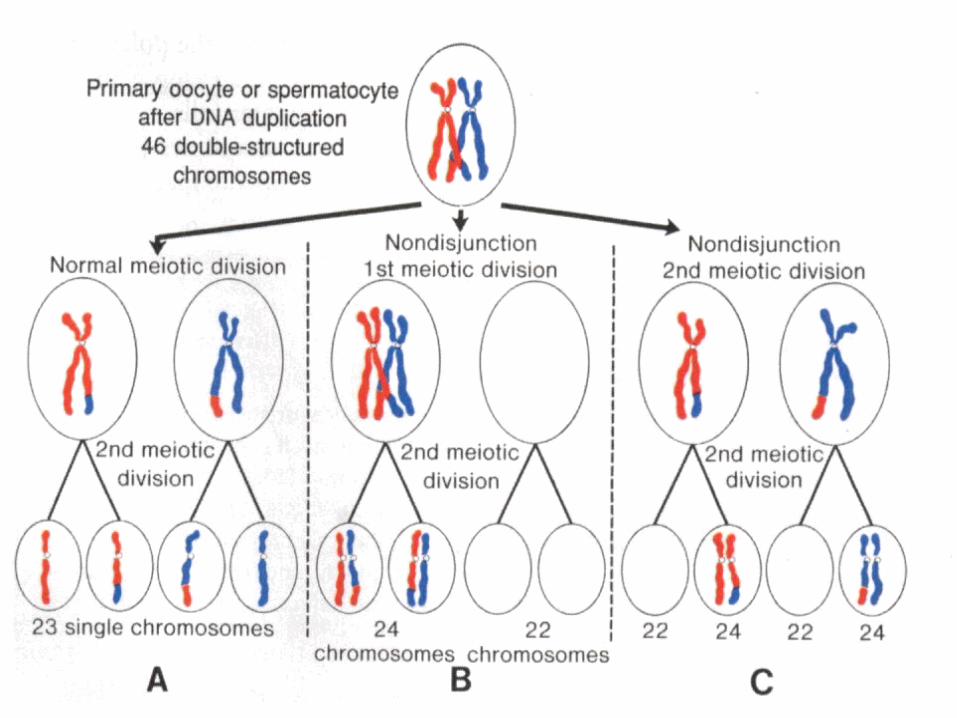
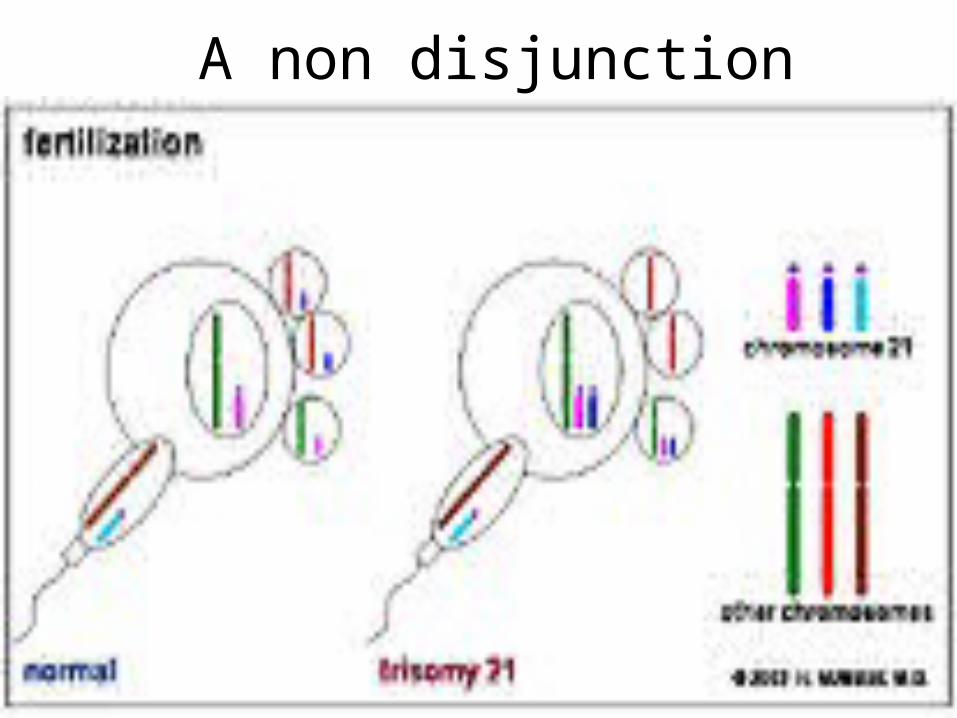
A non disjunction
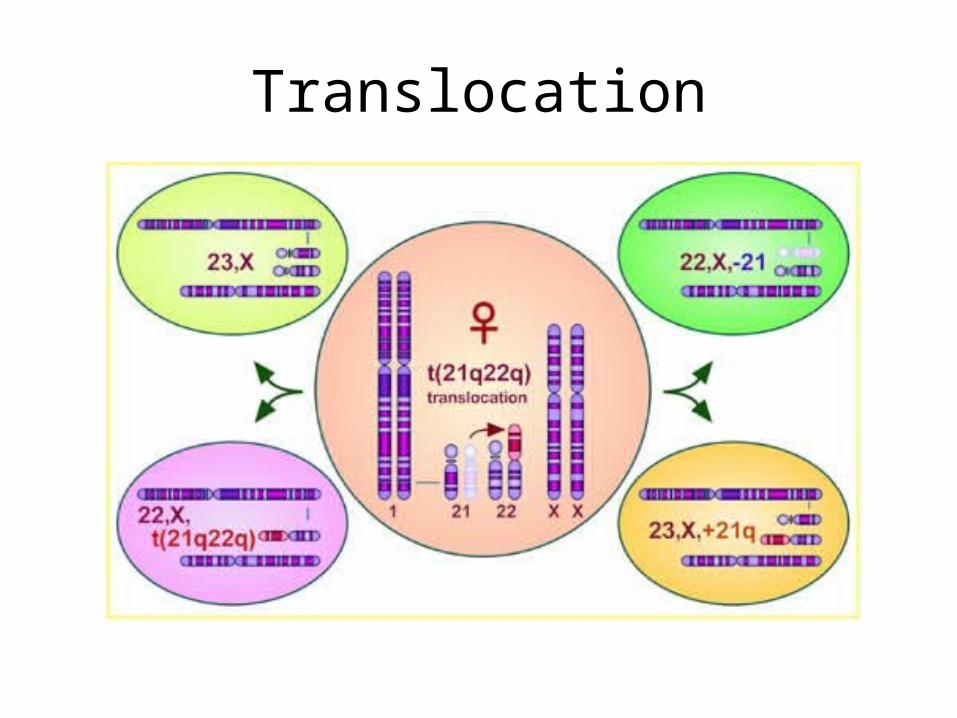
Translocation
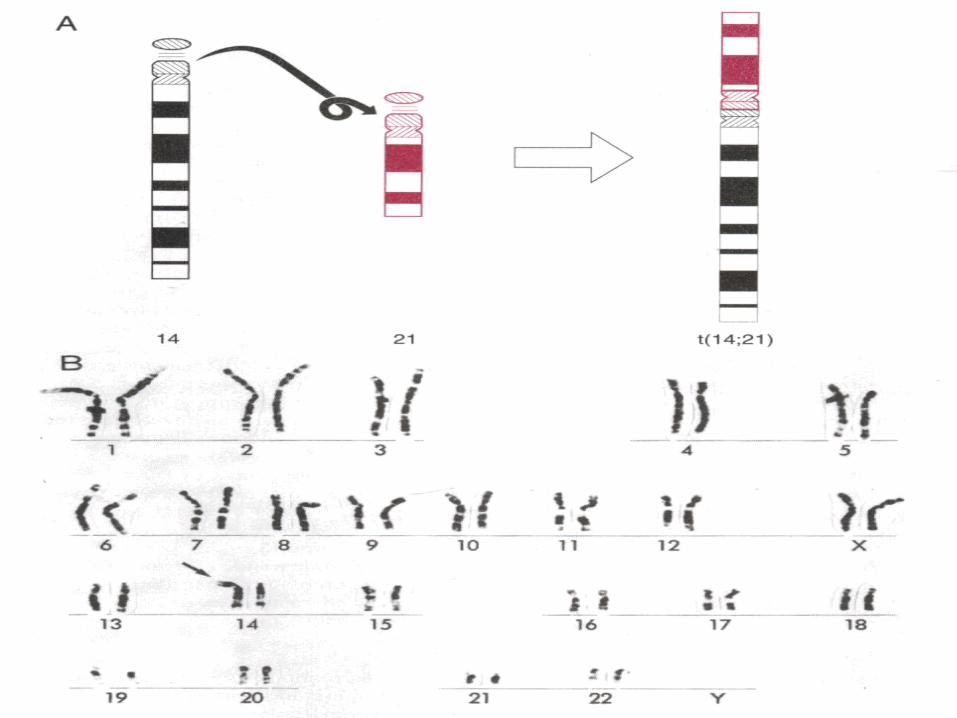
•
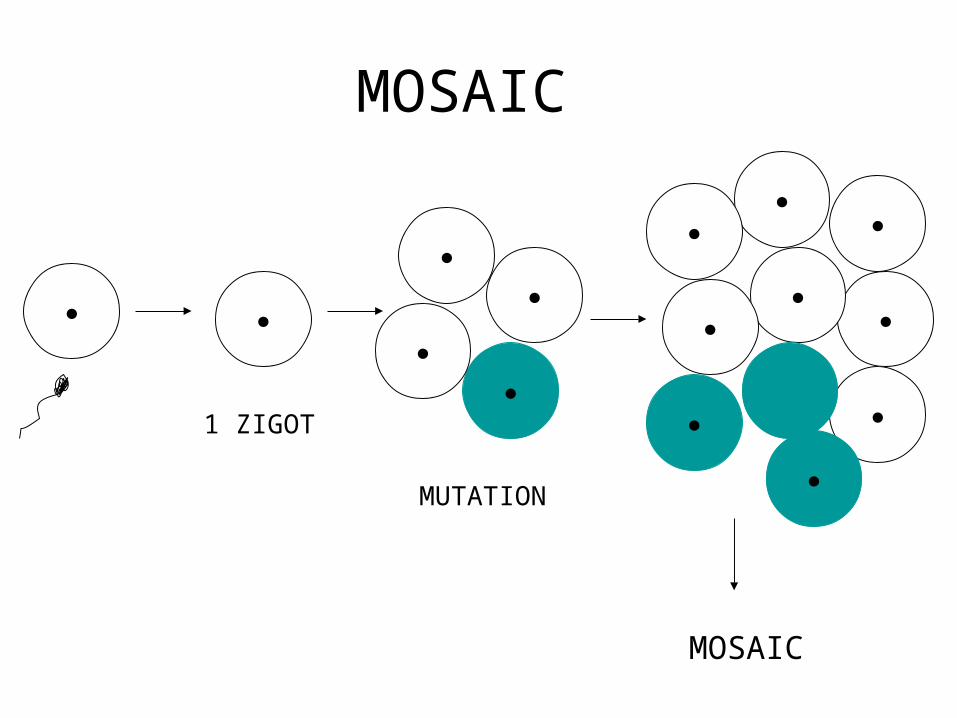
MOSAIC
● ●
●
●
●
●
●
●
●●
●
●
●
●
●
●
MOSAIC
MUTATION
1 ZIGOT
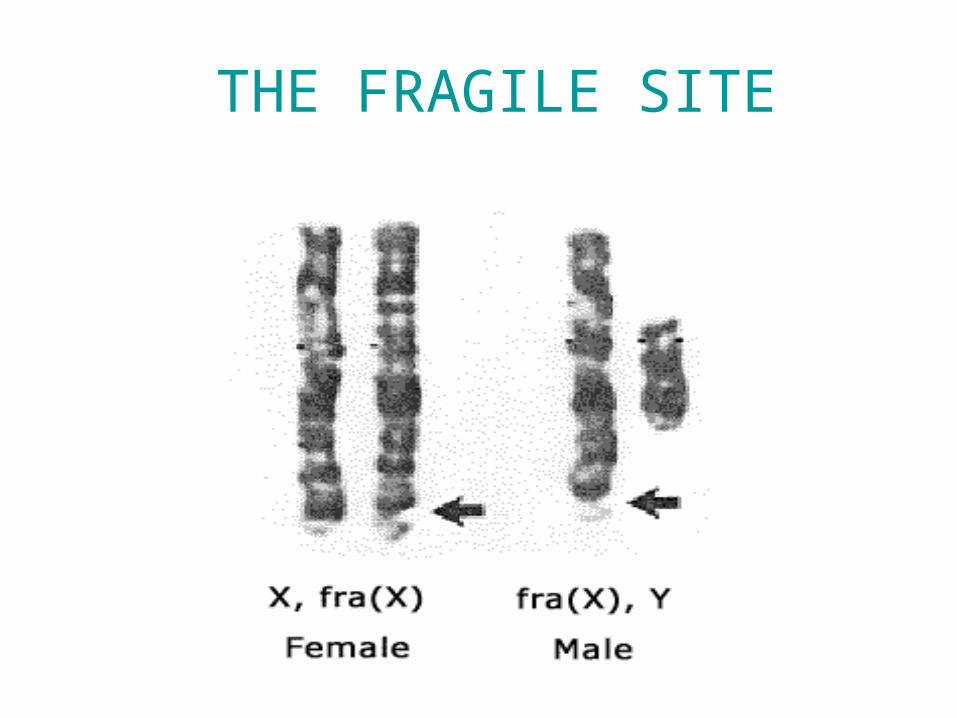
THE FRAGILE SITE

Down syndrome
• Mitotic nondisjunction• Unbalance translocation between 21
and 13, 14 or 15• Mosaicism• Trisomy 21• Genotip: 46,XY. 46,XX• Phenotype:
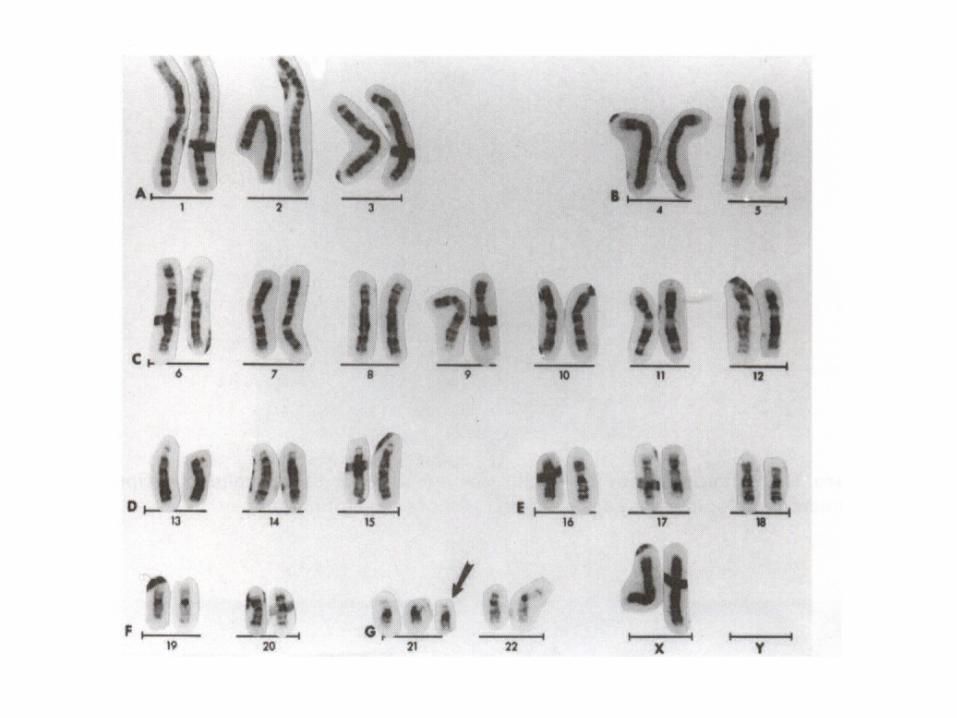
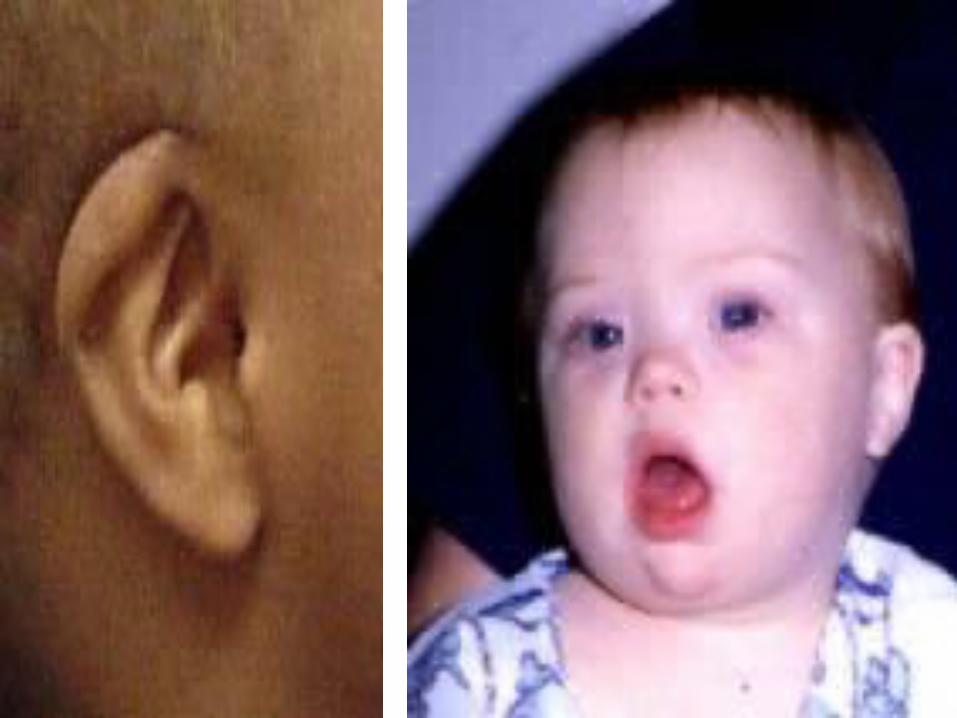
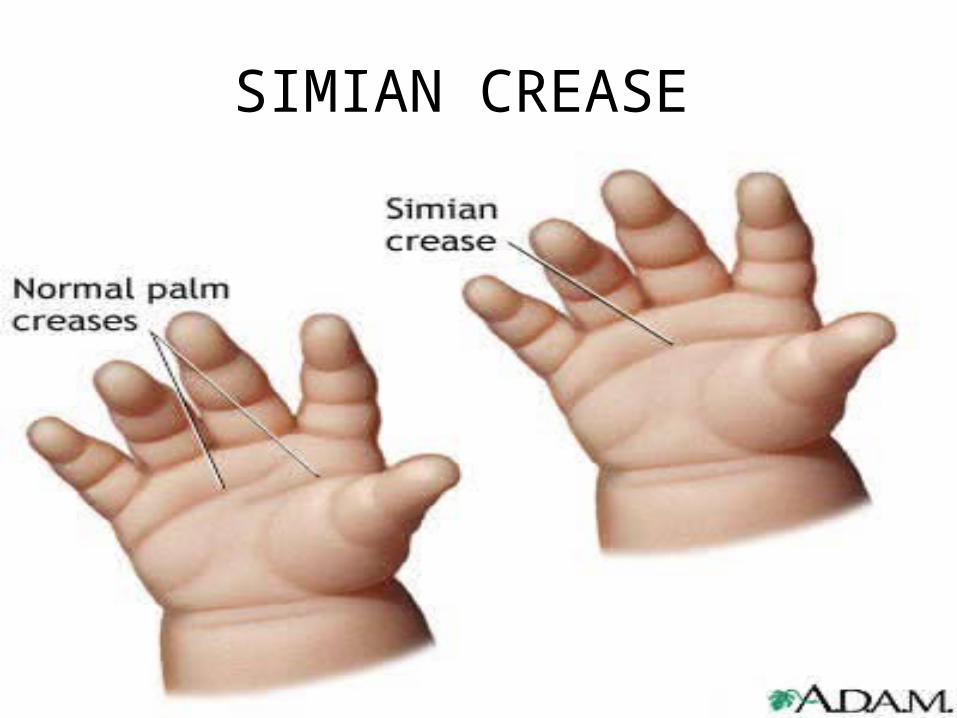
SIMIAN CREASE
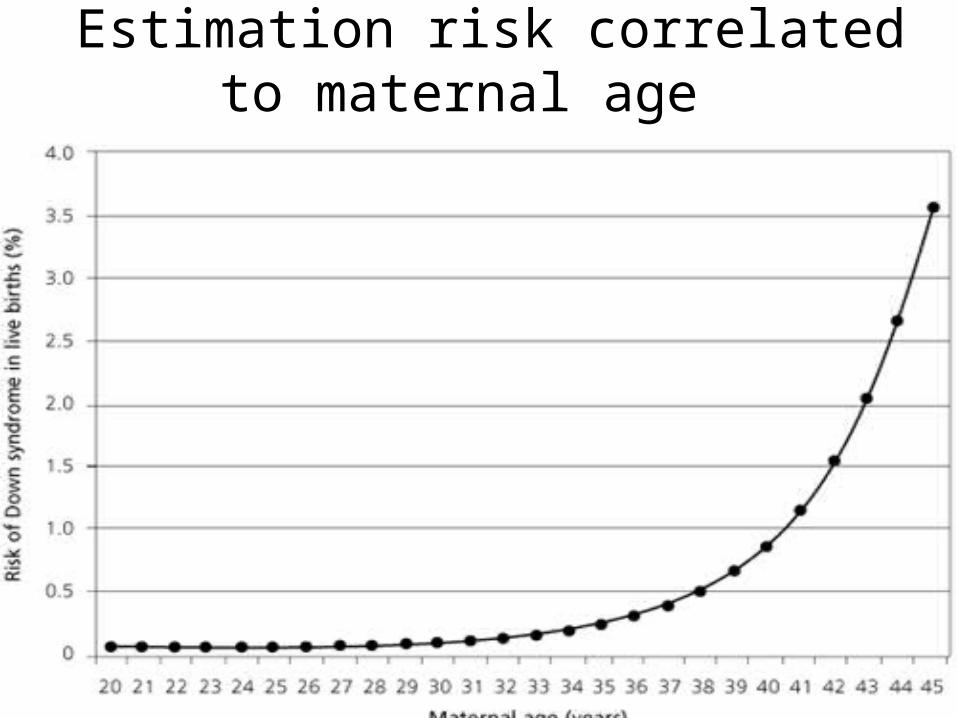
Estimation risk correlated to maternal age

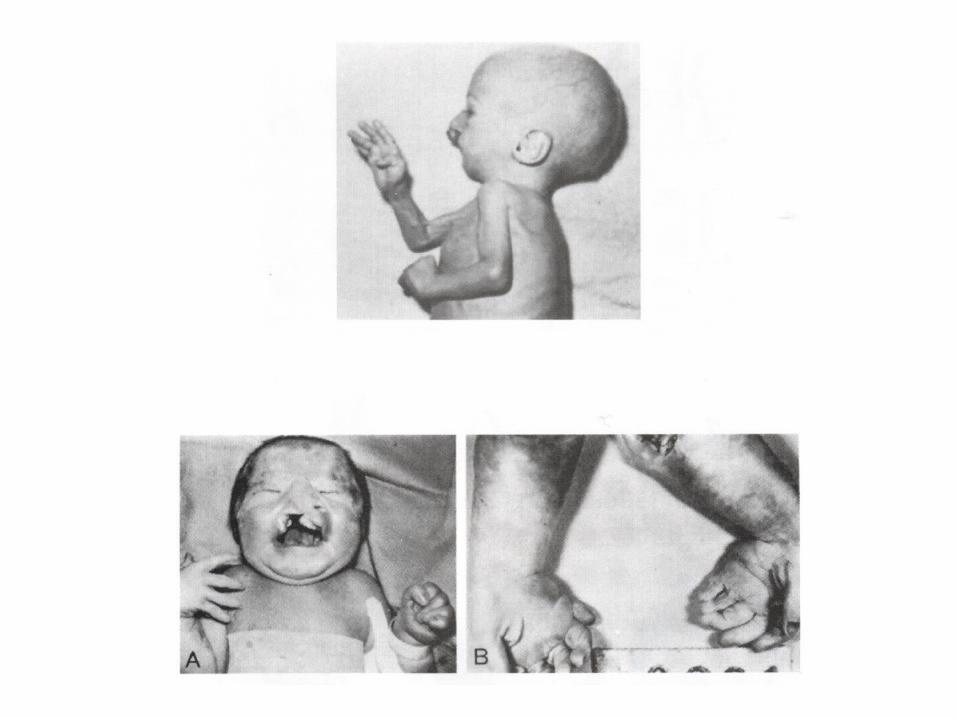
Edward syndrome
• nondisjunction:
• Trisomy 18
• Genotype: 46,XY . 46.XX
• The incidence: 1:5000 newborns and ussually die by age 2 months
• Phenotype:

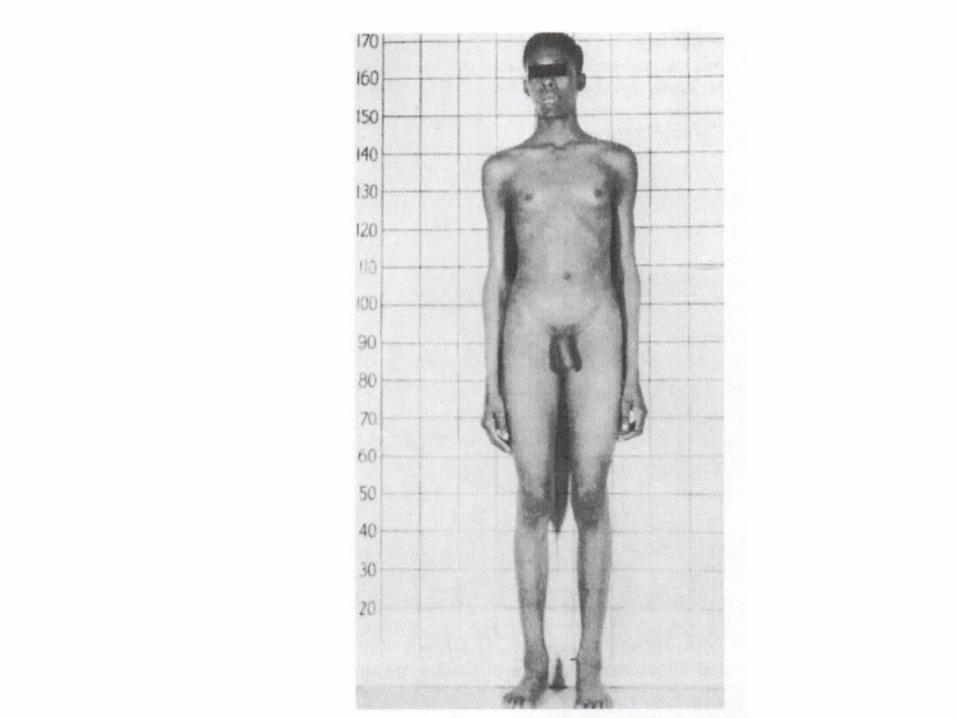
Klinefelter syndrome
• Nondisjunction of XX homologous
• 47,XXY / 48, XXXy
• Phenotype:

Turner syndrome
• Nondisjunction in the male gamete
• Monosomy for X
• Structural abnormality of X
• Genotype: 45,X
• Phenotype:
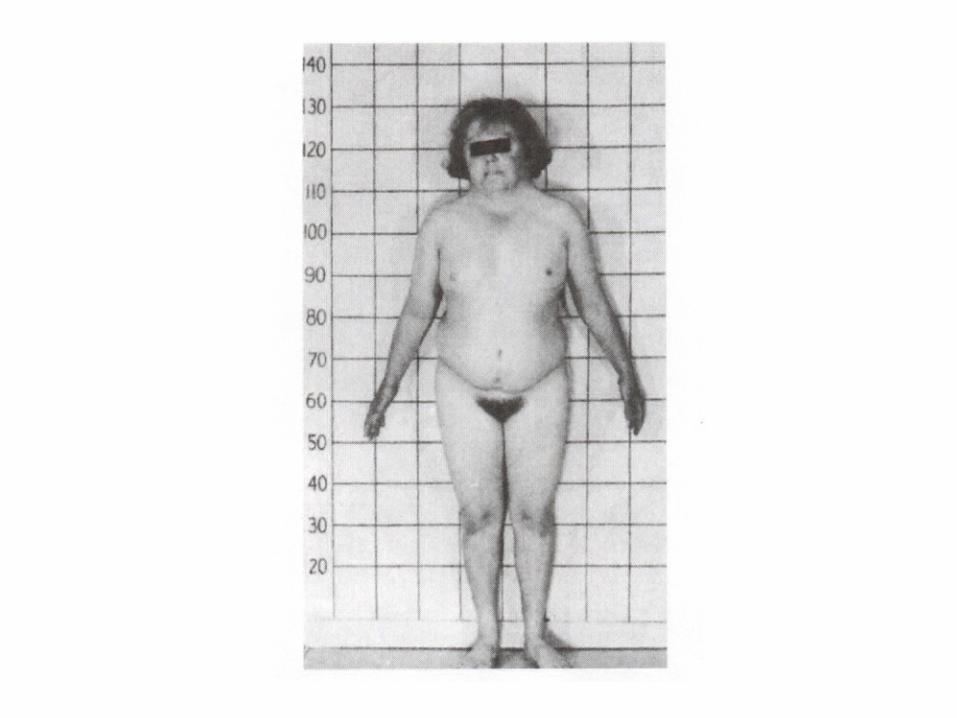

Deletion
• Micro deletio– Genomic imprinting
• Large deletion– Cri-du-chat symdrom:
Partial deletion of short arm chr 5
or chromosomal lost

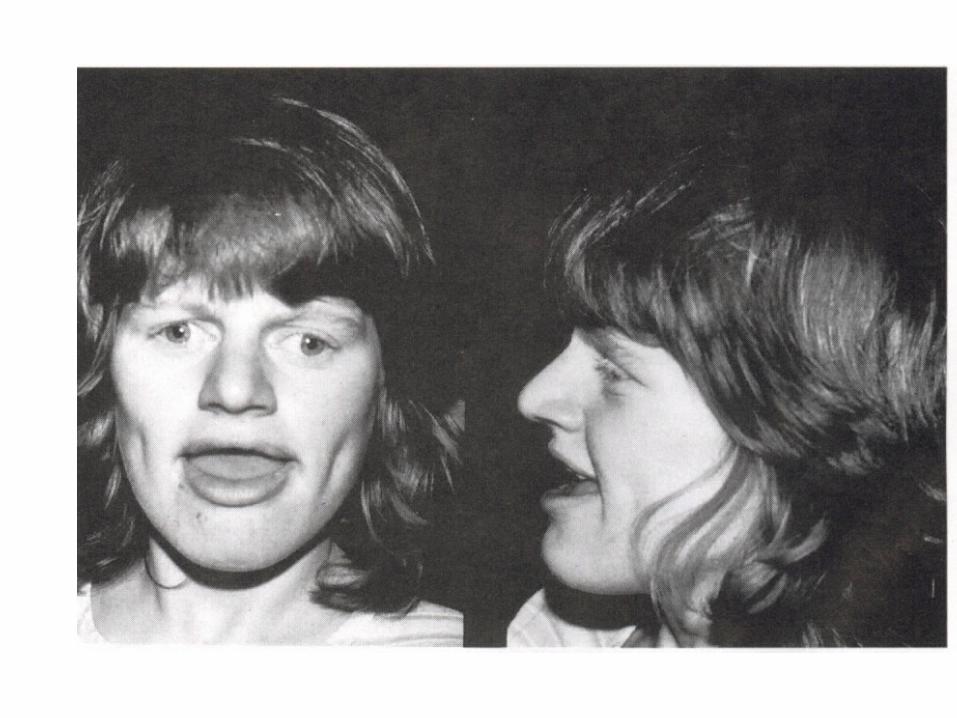
Genomic imprinting
• Exhibit differential expression depending on whether the genetic material is inherited from ( mother or father )
• Angelman syndrome: partial deletion, inherting the deletion on the maternal chromosome 15 ( 15q11-15q13 )
• Fradel willi syndrome:partial deletion, inherting the deletion on the faternal chromosome 15 ( 15q11-15q13 )
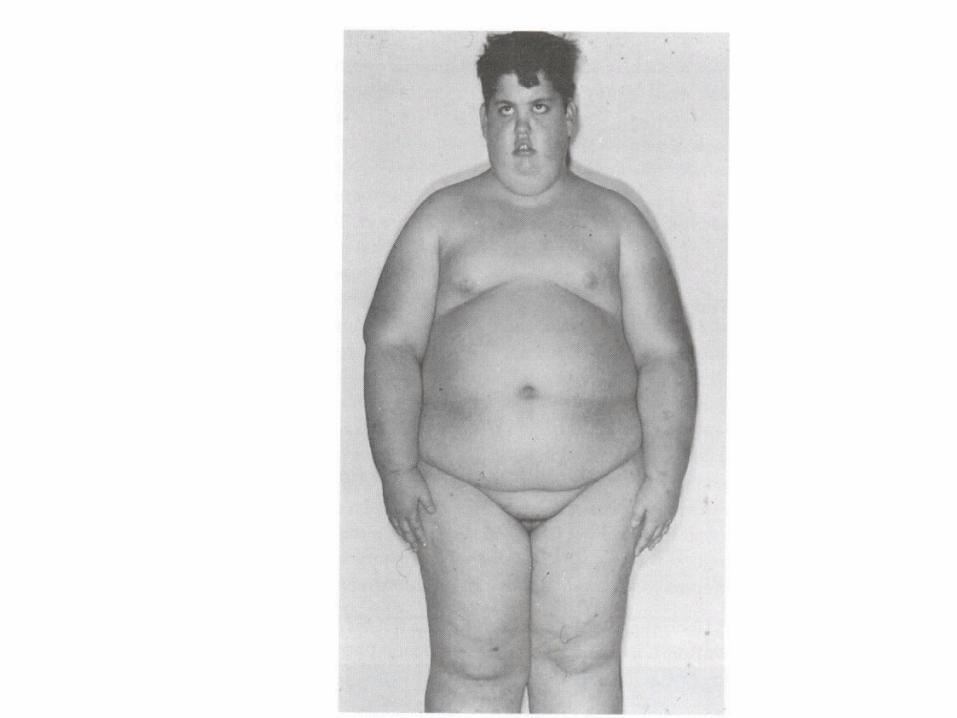

Fragile sites
• A region of chromosome that demonstrate a propensity to separate naturally or under certain manipulation
• Consist of CGG repeat
• The fragile site: long arm of X ( Xq27 )
• Phenotype : fragile X syndrome
• Males are more affected
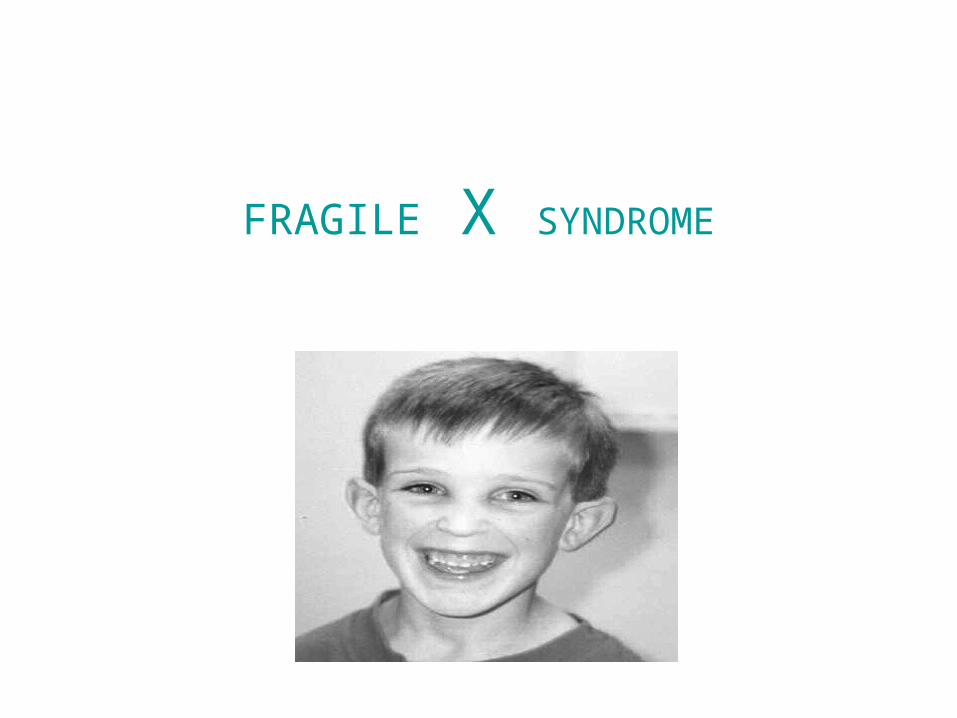
FRAGILE X SYNDROME
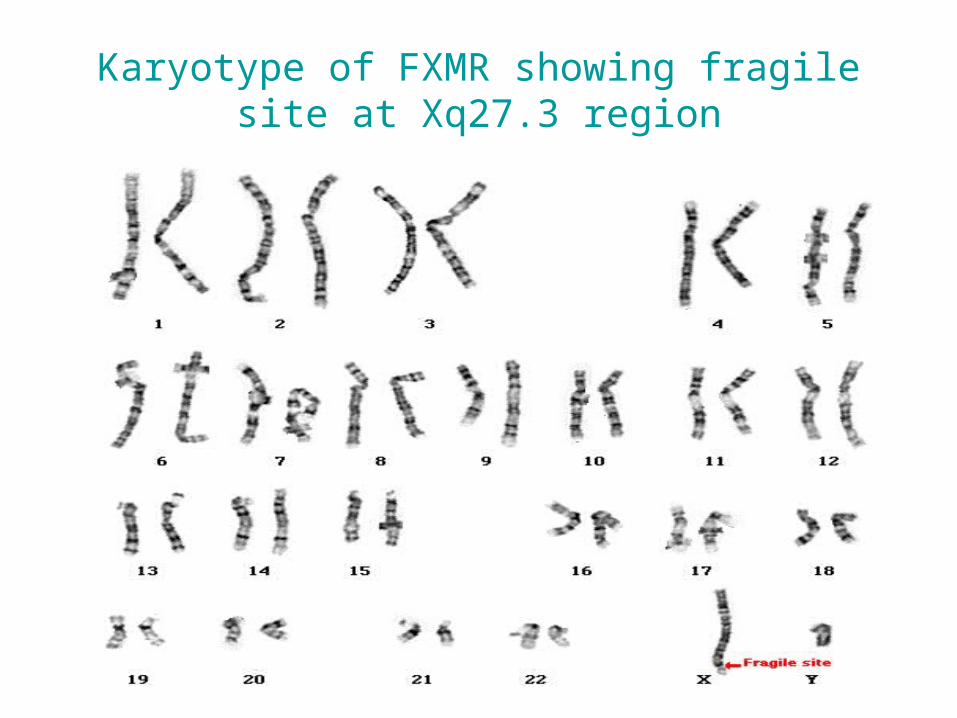
Karyotype of FXMR showing fragile site at Xq27.3 region

COLOUR BLINDNES