letterss copies/cv1257.pdfamyotrophic lateral sclerosis (als) is a fatal neurodegenerative condition...
TRANSCRIPT

402 VOLUME 10 | NUMBER 4 | APRIL 2004 NATURE MEDICINE
L E T T E R S
1The Graham Watts Laboratory, Sobell Department of Motor Neuroscience and Movement Disorders, Institute of Neurology, The National Hospital for Neurology andNeurosurgery, University College London, Queen Square, London WC1N 3BG, UK. 2Autonomic Neuroscience Institute, Royal Free and University College MedicalSchool, Rowland Hill Street, London NW2 3PF, UK. Correspondence should be addressed to L.G. ([email protected]).
Published online 21 March 2004; doi:10.1038/nm1021
Amyotrophic lateral sclerosis (ALS) is a fatalneurodegenerative condition in which motoneurons of thespinal cord and motor cortex die, resulting in progressiveparalysis1,2. This condition has no cure3 and results ineventual death, usually within 1–5 years of diagnosis1,2.Although the specific etiology of ALS is unknown, 20% offamilial cases of the disease carry mutations in the geneencoding Cu/Zn superoxide dismutase-1 (SOD1)4. Transgenicmice overexpressing human mutant SOD1 have a phenotypeand pathology that are very similar to that seen in human ALSpatients5,6. Here we show that treatment with arimoclomol, acoinducer of heat shock proteins (HSPs), significantly delaysdisease progression in mice expressing a SOD1 mutant inwhich glycine is substituted with alanine at position 93(SOD1G93A). Arimoclomol-treated SOD1G93A mice showmarked improvement in hind limb muscle function andmotoneuron survival in the later stages of the disease, resultingin a 22% increase in lifespan. Pharmacological activation ofthe heat shock response may therefore be a successfultherapeutic approach to treating ALS, and possibly otherneurodegenerative diseases.
Arimoclomol is an analog of bimoclomol, a hydroxylamine derivativethat acts as a coinducer of HSP expression7. These small-moleculecompounds interact with and amplify the heat shock response,which is an established, powerful cytoprotective mechanism8, atleast under acute conditions. In chronic disease, the heat shockresponse seems to be insufficient to cope with prolonged exposureto a stressful environment. HSPs are upregulated in human ALSpatient tissue9 and in mutant SOD1G93A mice10. In motoneurons,the toxicity of mutant SOD-1 protein may reduce the availability ofHSPs, thereby disrupting their normal chaperone11 and antiapop-totic12 functions and reducing their cytoprotective effects. Strategiesaimed at increasing levels of HSPs may therefore be successful inprotecting motoneurons from cell death in ALS. Indeed, we havepreviously shown that increasing HSP levels by treatment with anHSP coinducer rescues motoneurons from death in a nerve injurymodel of motoneuron degeneration13.
Here we examined whether treatment with arimoclomol could pre-vent the progressive loss of motoneurons and muscle function thatoccurs in SOD1G93A mice. SOD1G93A mice of both sexes were treateddaily with arimoclomol (10 mg/kg intraperitoneally) from 35 d ofage14. In untreated SOD1G93A mice, disease onset occured around 70 dof age, as determined by a reduction in body weight. At 120 d of age,mice were prepared for live, in vivo electrophysiological assessment ofhind limb muscle function to assess disease progression. The number offunctional motor units in each extensor digitorum longus (EDL) mus-cle was determined. Examples of motor unit recordings are shown inFigure 1a–c, and the results are summarized in Figure 1d. Wild-typemice had 28 ± 0.6 motor units in their EDL muscle (n = 11; Fig. 1a),whereas only 8.3 ± 0.7 motor units survived in SOD1G93A mice at 120 dof age (n = 10; Fig. 1b). In arimoclomol-treated SOD1G93A mice, therewas a significant increase in motor unit survival (14.3 ± 0.6 motorunits; n = 10; P ≤ 0.05; Fig. 1c).
EDL is a fast muscle that fatigues quickly when repeatedly stimu-lated, producing a characteristic fatigue pattern from which a ‘fatigueindex’ can be calculated15 (Fig. 1e). Examples of such fatigue traces areshown in Figure 1f–h. Normal EDL in wild-type mice is very fatigable(Fig. 1f). As the disease progressed in SOD1G93A mice, the fatigue char-acteristics of EDL muscle changed substantially and the muscle becamefatigue resistant (Fig. 1g), a phenomenon that was reflected in thefatigue index. Normal EDL muscle had a fatigue index of 0.848 ± 0.028(n = 10), which was reduced to only 0.255 ± 0.04 (n = 10) in SOD1G93A
mice at 120 d of age. Treatment with arimoclomol prevented thischange in the fatigability of EDL muscle. The muscle remained largelyfatigable (Fig. 1h), with a significantly improved fatigue index of 0.416± 0.07 (n = 10; P ≤ 0.05; Fig. 1e). Arimoclomol also prevented thechanges in other contractile characteristics of EDL muscle that usuallyoccur during disease progression in SOD1G93A mice (SupplementaryFig. 1 and Supplementary Note online).
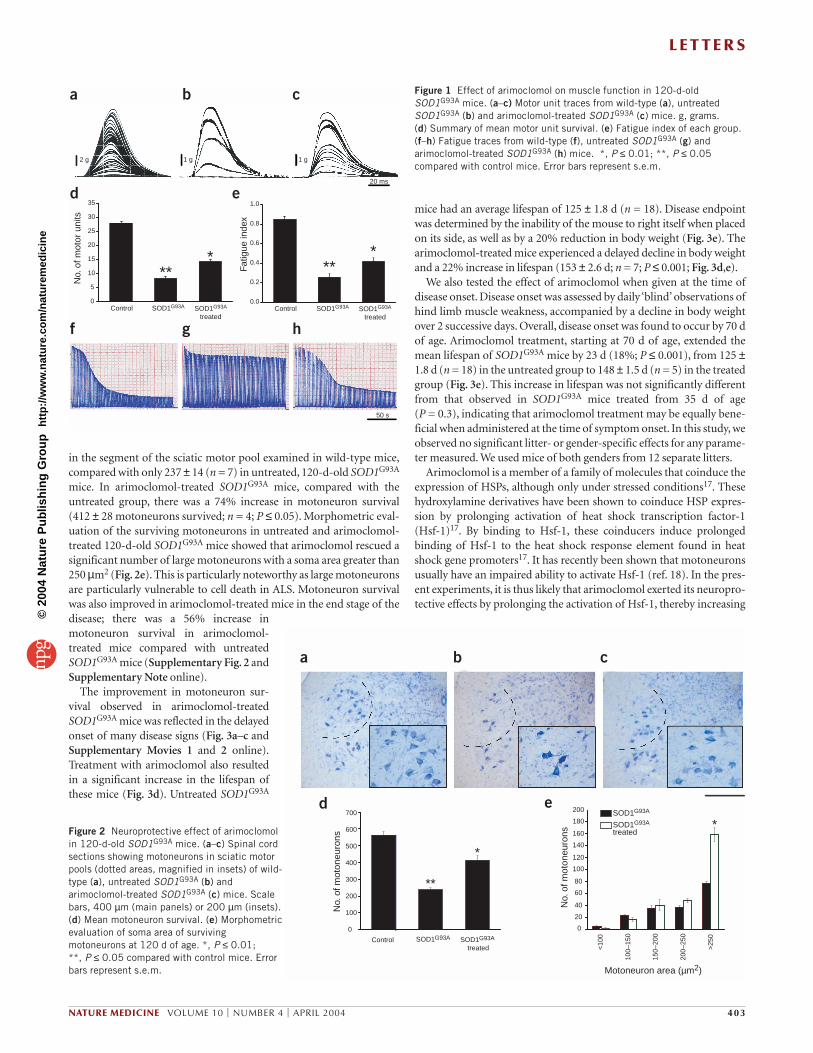
After the physiological experiments were completed, motoneuronsurvival was assessed morphologically by counting the number ofmotoneurons in the sciatic motor pool in the ventral horns from cross-sections of lumbar spinal cord16 (Fig. 2a–c). Treatment with arimoclo-mol significantly increased motoneuron survival in 120-d-oldSOD1G93A mice (Fig. 2d). There were 593 ± 15.8 (n = 13) motoneurons
Treatment with arimoclomol, a coinducer of heat shockproteins, delays disease progression in ALS miceDairin Kieran1, Bernadett Kalmar1, James R T Dick1, Joanna Riddoch-Contreras1, Geoffrey Burnstock2 & Linda Greensmith1
©20
04 N
atur
e P
ublis
hing
Gro
up
http
://w
ww
.nat
ure.
com
/nat
urem
edic
ine
L E T T E R S
NATURE MEDICINE VOLUME 10 | NUMBER 4 | APRIL 2004 403
in the segment of the sciatic motor pool examined in wild-type mice,compared with only 237 ± 14 (n = 7) in untreated, 120-d-old SOD1G93A
mice. In arimoclomol-treated SOD1G93A mice, compared with theuntreated group, there was a 74% increase in motoneuron survival (412 ± 28 motoneurons survived; n = 4; P ≤ 0.05). Morphometric eval-uation of the surviving motoneurons in untreated and arimoclomol-treated 120-d-old SOD1G93A mice showed that arimoclomol rescued asignificant number of large motoneurons with a soma area greater than250 µm2 (Fig. 2e). This is particularly noteworthy as large motoneuronsare particularly vulnerable to cell death in ALS. Motoneuron survivalwas also improved in arimoclomol-treated mice in the end stage of thedisease; there was a 56% increase inmotoneuron survival in arimoclomol-treated mice compared with untreatedSOD1G93A mice (Supplementary Fig. 2 andSupplementary Note online).
The improvement in motoneuron sur-vival observed in arimoclomol-treatedSOD1G93A mice was reflected in the delayedonset of many disease signs (Fig. 3a–c andSupplementary Movies 1 and 2 online).Treatment with arimoclomol also resultedin a significant increase in the lifespan ofthese mice (Fig. 3d). Untreated SOD1G93A
mice had an average lifespan of 125 ± 1.8 d (n = 18). Disease endpointwas determined by the inability of the mouse to right itself when placedon its side, as well as by a 20% reduction in body weight (Fig. 3e). Thearimoclomol-treated mice experienced a delayed decline in body weightand a 22% increase in lifespan (153 ± 2.6 d; n = 7; P ≤ 0.001; Fig. 3d,e).
We also tested the effect of arimoclomol when given at the time ofdisease onset. Disease onset was assessed by daily ‘blind’ observations ofhind limb muscle weakness, accompanied by a decline in body weightover 2 successive days. Overall, disease onset was found to occur by 70 dof age. Arimoclomol treatment, starting at 70 d of age, extended themean lifespan of SOD1G93A mice by 23 d (18%; P ≤ 0.001), from 125 ±1.8 d (n = 18) in the untreated group to 148 ± 1.5 d (n = 5) in the treatedgroup (Fig. 3e). This increase in lifespan was not significantly differentfrom that observed in SOD1G93A mice treated from 35 d of age (P = 0.3), indicating that arimoclomol treatment may be equally bene-ficial when administered at the time of symptom onset. In this study, weobserved no significant litter- or gender-specific effects for any parame-ter measured. We used mice of both genders from 12 separate litters.
Arimoclomol is a member of a family of molecules that coinduce theexpression of HSPs, although only under stressed conditions17. Thesehydroxylamine derivatives have been shown to coinduce HSP expres-sion by prolonging activation of heat shock transcription factor-1 (Hsf-1)17. By binding to Hsf-1, these coinducers induce prolongedbinding of Hsf-1 to the heat shock response element found in heatshock gene promoters17. It has recently been shown that motoneuronsusually have an impaired ability to activate Hsf-1 (ref. 18). In the pres-ent experiments, it is thus likely that arimoclomol exerted its neuropro-tective effects by prolonging the activation of Hsf-1, thereby increasing
50 s
2 g 1 g 1 g
20 ms
Control SOD1G93A0
5
10
15
20
25
30
35
No.
of m
otor
uni
ts
SOD1G93A
treatedControl SOD1G93A SOD1G93A
treated
***
0.0
0.2
0.4
0.6
0.8
1.0
Fatig
ue in
dex
***
a b c
d e
f g h
Figure 1 Effect of arimoclomol on muscle function in 120-d-oldSOD1G93A mice. (a–c) Motor unit traces from wild-type (a), untreatedSOD1G93A (b) and arimoclomol-treated SOD1G93A (c) mice. g, grams. (d) Summary of mean motor unit survival. (e) Fatigue index of each group.(f–h) Fatigue traces from wild-type (f), untreated SOD1G93A (g) andarimoclomol-treated SOD1G93A (h) mice. *, P ≤ 0.01; **, P ≤ 0.05compared with control mice. Error bars represent s.e.m.
<10
0
100–
150
150–
200
200–
250
>25
0
20
40
60
80
100
120
140
160
180
200
No.
of m
oton
euro
ns
Motoneuron area (µm2)
SOD1G93A
SOD1G93A
treated
0
Control SOD1G93A SOD1G93A treated
0
100
200
300
400
500
600
700
No.
of m
oton
euro
ns
**
*
*
a b c
d e
Figure 2 Neuroprotective effect of arimoclomolin 120-d-old SOD1G93A mice. (a–c) Spinal cordsections showing motoneurons in sciatic motorpools (dotted areas, magnified in insets) of wild-type (a), untreated SOD1G93A (b) andarimoclomol-treated SOD1G93A (c) mice. Scalebars, 400 µm (main panels) or 200 µm (insets).(d) Mean motoneuron survival. (e) Morphometricevaluation of soma area of survivingmotoneurons at 120 d of age. *, P ≤ 0.01; **, P ≤ 0.05 compared with control mice. Errorbars represent s.e.m.
©20
04 N
atur
e P
ublis
hing
Gro
up
http
://w
ww
.nat
ure.
com
/nat
urem
edic
ine

L E T T E R S
404 VOLUME 10 | NUMBER 4 | APRIL 2004 NATURE MEDICINE
levels of HSPs in motoneurons. We therefore examined the activationof Hsf-1, as well as the expression of HSPs, in the spinal cords of mice.Hsf-1 was present in the spinal cords of wild-type mice, as well as inuntreated and arimoclomol-treated SOD1G93A mice. However, the Hsf-1 band is shifted in western blot analyses of arimoclomol-treatedSOD1G93A mice. This shift is a characteristic feature of stress-induced
Hsf-1 activation17,19,20 (Fig. 4a), as Hsf-1 becomes phosphorylatedwhen it is activated under stress conditions. This inducibly phosphory-lated form of Hsf-1 is detectable at a higher molecular weight in west-ern blots than the constitutively phosphorylated form, as evidencedhere by the characteristic band shift. Immunostaining for human SOD-1 confirmed that the mutant human protein was present in
11 12 13 14 15 16 17 18 19 20 21 22 23 240
20
40
60
80
100
120
Age (weeks)
% s
urvi
val
SOD1G93A
SOD1G93A (day 35)
SOD1G93A (day 70)
10 11 12 13 14 15 16 17 18 19 20 21 22 23 2414
15
16
17
18
19
20
21
22
Bod
y w
eigh
t (g)
Age (weeks)
SOD1G93A
SOD1G93A (day 35)
a b c
d eFigure 3 Disease signs and lifespan in arimoclomol-treated and untreated SOD1G93A mice. (a,b) UntreatedSOD1G93A mouse (125 d of age) shows significant signsof hind limb muscle wasting, has no toe-spreading reflexand has marked kyphosis (a), and is unable to right itself(b). This mouse was judged to have reached the diseaseendpoint. (c) Arimoclomol-treated, age-matchedSOD1G93A littermate shows definite toe-spreading reflexand has no signs of hind limb muscle wasting orkyphosis, and is able to perform a righting reflex testwith no delay. (d) Lifespan of untreated SOD1G93A miceand those treated with arimoclomol from 35 or 70 d ofage. (e) Decline in body weight during diseaseprogression. Error bars represent s.e.m.
HSP27 HSP-70 HSP-900
102030405060708090
100110120
Pix
el d
ensi
ty (
% o
f wild
type
)
SOD1G93A SOD1G93A treated Untreated Treated
HSP-27
HSP-70
HSP-90
Wild-type SOD1G93A SOD1G93A
treated
Wild-type SOD1G93A SOD1G93A
treated
-Pi-Pc
-Pd
a
b
c
d
e
f
Figure 4 HSP-70 and Hsf-1 expression in arimoclomol-treated and untreated SOD1G93A mice. (a) Western blot analysis of Hsf-1 expression in spinal cordtissue from wild-type, untreated SOD1G93A and arimoclomol-treated SOD1G93A mice. The Hsf-1 monomer is present between 65–75 kDa, but is shifted by8–10 kDa in the arimoclomol-treated SOD1G93A mice. Pi, inducible phosphorylated Hsf-1; Pc, constitutively phosphorylated Hsf-1; Pd, dephosphorylatedHsf-1. (b) Spinal cord sections from untreated and arimoclomol-treated SOD1G93A mice, immunostained for human SOD-1. (c,d) Spinal cord sections fromuntreated (c) and arimoclomol-treated (d) SOD1G93A mice, immunostained for HSP-70. (e) Representative western blots for HSP-27, HSP-70 and HSP-90.(f) Quantitative densitometric analysis of western blots in e. Scale bars, 60 µm.
©20
04 N
atur
e P
ublis
hing
Gro
up
http
://w
ww
.nat
ure.
com
/nat
urem
edic
ine

L E T T E R S
NATURE MEDICINE VOLUME 10 | NUMBER 4 | APRIL 2004 405
motoneurons of untreated and arimoclomol-treated SOD1G93A mice(Fig. 4b), whereas no immunoreactivity was observed in spinal cords ofwild-type mice. Immunostaining also revealed that at 120 d of age,expression of HSP-70 was increased in the lumbar spinal cords of bothuntreated (Fig. 4c) and treated (Fig. 4d) mutant SOD1G93A mice,although there was a clear increase in the intensity of HSP-70immunoreactivity in motoneurons from arimoclomol-treatedSOD1G93A mice. This was verified by quantitative western blot analysis(Fig. 4e,f). The western blots also revealed a slight increase in HSP-27expression in spinal cords of SOD1G93A mice, as previously reported10.Arimoclomol treatment did not increase the expression of HSP-27 anyfurther, but there was a clear increase in HSP-70 and HSP-90 in spinalcords of treated SOD1G93A mice (Fig. 4e,f).
Our results show that treatment with arimoclomol significantlydelays disease progression in SOD1G93A mice, even when administeredafter symptom onset. Arimoclomol prolonged the activation of Hsf-1,resulting in an increase in HSP-70 and HSP-90 expression in SOD1G93A
mice. Thus, agents such as arimoclomal that prolong Hsf-1 activationmay be of therapeutic value in the treatment of ALS, and possibly otherneurodegenerative disorders.
METHODSGenetic background and breeding protocol of SOD1G93A mice. All experimentsdescribed in this study were carried out under license from the UK Home Office,and were approved by the Ethical Review Panel of the Institute of Neurology.Transgenic mice carrying a human SOD1 gene with a G93A mutation(TgN[SOD1-G93A]1Gur) were purchased from The Jackson Laboratory. Thecolony was maintained by breeding male heterozygous carriers to femaleB6SJLF1 hybrids. Each experimental group contained arimoclomol-treated anduntreated littermates; results from transgenic mice were compared to those fromtheir wild-type littermates. Arimoclomol was obtained from Biorex R&D.
Assessment of muscle function and number of motor units. Isometric contrac-tions were elicited by stimulating the EDL motor nerve using square-wave pulsesof 0.02 ms duration and supramaximal intensity via silver-wire electrodes.Contractions were elicited by trains of stimuli at frequencies of 20, 40 and 80 Hz.The half-relaxation time values were measured with the aid of a computer andappropriate software. We assessed the number of motor units in the EDL mus-cles by stimulating the motor nerve with stimuli of increasing intensity, resultingin stepwise increments in twitch tension because of successive recruitment ofmotor axons.
Fatigue test. EDL muscles were stimulated at 40 Hz for 250 ms every second, andcontractions were recorded with a pen recorder (Lectromed Multitrace 2). Thedecrease in tension after 3 min of stimulation was measured, and the fatigueindex was calculated as (initial tetanic tension – tetanic tension after 3 min stim-ulation) ÷ initial tetanic tension.
Motoneuron survival. After transcardiac perfusion with 4% paraformaldehyde,the lumbar region of the spinal cord was removed and 20-µm transverse sec-tions were cut and stained with gallocyanin (a Nissl stain). Gallocyanin-stainedmotoneurons located within the sciatic motor pool, in which a nucleolus wasclearly visible, were counted in each ventral horn on every third section betweenthe L2 and L5 levels of the spinal cord. This method avoids the possibility ofcounting the same cell twice.
Immunocytochemistry. Sections of spinal cord were immunostained with anti-bodies to human SOD-1 (1:500; Sigma) or HSP-70 (1:100; Stressgen), using apreviously described method13.
Western blots. Snap-frozen spinal cords were homogenized with proteaseinhibitors and loaded onto an 8% SDS-PAGE. Blots were incubated with anti-bodies to Hsf-1 (Lab Vision), or to HSP-27, HSP-70 or HSP-90 (all fromStressgen), and visualized by chemiluminescence.
Statistical analysis. Statistical significance was assessed between groups using aMann-Whitney U test. Significance was set at P ≤ 0.05.
Note: Supplementary information is available on the Nature Medicine website.
ACKNOWLEDGMENTSWe thank Biorex R&D (Hungary) for the gift of arimoclomal. L.G. is the GrahamWatts Senior Research Fellow funded by the Brain Research Trust. D.K. is in receiptof a Brain Research Trust Prize Studentship.
COMPETING INTERESTS STATEMENTThe authors declare competing financial interests (see the Nature Medicine websitefor details).
Received 24 November 2003; accepted 2 March 2004Published online at http://www.nature.com/naturemedicine/
1. Rowland, L.P. & Shneider, N.A. Amyotrophic lateral sclerosis. N. Engl. J. Med. 344,1688–1700 (2001).
2. Shaw, P.J. Motor neurone disease. Br. Med. J. 318, 1118–1121 (1999).3. Miller, R.G., Mitchell, J.D., Lyon, M. & Moore, D.H. Riluzole for amyotrophic lateral
sclerosis (ALS)/motor neuron disease (MND). Cochrane Database Syst. Rev.CD001447 (2002).
4. Rosen, D.R. et al. Mutations in Cu/Zn superoxide dismutase gene are associated withfamilial amyotrophic lateral sclerosis. Nature 362, 59–62 (1993).
5. Gurney, M.E. et al. Motor neuron degeneration in mice that express a human Cu,Znsuperoxide dismutase mutation. Science 264, 1772–1775 (1994).
6. Wong, P.C. et al. An adverse property of a familial ALS-linked SOD1 mutation causesmotor neuron disease characterized by vacuolar degeneration of mitochondria.Neuron 14, 1105–1116 (1995).
7. Vigh, L. et al. Bimoclomol: a nontoxic, hydroxylamine derivative with stress protein-inducing activity and cytoprotective effects. Nat. Med. 3, 1150–1154 (1997).
8. Lindquist, S. The heat-shock response. Annu. Rev. Biochem. 55, 1151–1191(1986).
9. Garofalo, O. et al. Ubiquitin and heat shock protein expression in amyotrophic lateralsclerosis. Neuropathol. Appl. Neurobiol. 17, 39–45 (1991).
10. Vleminckx, V. et al. Upregulation of HSP27 in a transgenic model of ALS. J. Neuropathol. Exp. Neurol. 61, 968–974 (2002).
11. Shinder, G.A., Lacourse, M.C., Minotti, S. & Durham, H.D. Mutant Cu/Zn- superoxidedismutase proteins have altered solubility and interact with heat shock/stress proteinsin models of amyotrophic lateral sclerosis. J. Biol. Chem. 276, 12791–12796(2001).
12. Okado-Matsumoto, A. & Fridovich, I. Amyotrophic lateral sclerosis: a proposed mech-anism. Proc. Natl. Acad. Sci. USA 99, 9010–9014 (2002).
13. Kalmar, B. et al. Upregulation of heat shock proteins rescues motoneurones from axo-tomy-induced cell death in neonatal rats. Exp. Neurol. 176, 87–97 (2002).
14. Zhu, S. et al. Minocycline inhibits cytochrome c release and delays progression ofamyotrophic lateral sclerosis in mice. Nature 417, 74–78 (2002).
15. Dick, J., Greensmith, L. & Vrbova, G. Blocking of NMDA receptors during a criticalstage of development reduces the effects of nerve injury at birth on muscles andmotoneurones. Neuromuscul. Disord. 5, 371–382 (1995).
16. White, C.M., Greensmith, L. & Vrbova, G. Repeated stimuli for axonal growth causesmotoneuron death in adult rats: the effect of botulinum toxin followed by partial den-ervation. Neuroscience 95, 1101–1109 (2000).
17. Hargitai, J. et al. Bimoclomol, a heat shock protein co-inducer, acts by the prolongedactivation of heat shock factor-1. Biochem. Biophys. Res. Commun. 307, 689–695(2003).
18. Batulan, Z. et al. High threshold for induction of the stress response in motor neuronsis associated with failure to activate HSF1. J. Neurosci. 23, 5789–5798 (2003).
19. Sarge, K.D., Murphy, S.P. & Morimoto, R.I. Activation of heat shock gene transcrip-tion by heat shock factor 1 involves oligomerization, acquisition of DNA-bindingactivity, and nuclear localization and can occur in the absence of stress. Mol. CellBiol. 13, 1392–1407 (1993).
20. Morrison, A.J., Rush, S.J. & Brown, I.R. Heat shock transcription factors and theHSP70 induction response in brain and kidney of the hyperthermic rat during post-natal development. J. Neurochem. 75, 363–372 (2000).
©20
04 N
atur
e P
ublis
hing
Gro
up
http
://w
ww
.nat
ure.
com
/nat
urem
edic
ine