levy cystic fibrosis - national jewish health
TRANSCRIPT

Propert
y of P
resen
ter
Not for
Rep
roduc
tion

Hara Levy, M.D., M.M.Sc.Associate Professor of Pediatrics
Division Head, Department of PediatricsPulmonary Medicine
National Jewish Hospital for Kids
February 5th-8th Keystone ConferenceKeystone, CO
Cystic Fibrosis-The Future is Bright
Propert
y of P
resen
ter
Not for
Rep
roduc
tion

• No financial interests to disclose
Disclosure
Propert
y of P
resen
ter
Not for
Rep
roduc
tion

1. Review current clinical guidelines for the diagnosis and treatment of patients with CF.
2. Evaluate current and emerging therapies and pharmacodynamics and their impact on patients with CF.
Learning Objectives
Propert
y of P
resen
ter
Not for
Rep
roduc
tion

Cystic Fibrosis: A Historical Timeline
Cystic fibrosis (CF) of the pancreas was described by Andersen.
The sweat defect was discovered by diSant'Agnese and colleagues when they noticed that many of the infants presenting with heat prostration during the “great summer heat wave” in New York City had CF.
CF was identified as an autosomal recessive disease.
The fundamental physiologic defects were clearly established by Knowles and colleagues and Quinton as the failure of cAMP regulation of chloride transport.
The genetic defect for CF was located on chromosome 7.
The gene encoding the CF transmembrane conductance regulator (CFTR) was identified by positional cloning.
CFTR was established to be a cAMP‐regulated chloride channel by complementation studies.
1938
1953
1983
1965
1985
19901989
Propert
y of P
resen
ter
Not for
Rep
roduc
tion

1950
1989‐2019 Prop
erty o
f Pres
enter
Not for
Rep
roduc
tion

Cystic Fibrosis: Median Survival Age, 1940-2010
pancreatic enzymes/nutritional support
antistaphylococcal antibiotics
lung Transplant
antipseudomonal antibiotics
DNase
Inhaled tobramycin
airway clearance
0
5
10
15
20
25
30
35
40
1930
1940
1950
1960
1970
1980
1990
2000
2010
Year
Projected Ag
e of Survival (years)
1st pathologicdescription
CF geneidentified
Discoveryof high saltin sweat
Sweat chloridetest developed
1st successfulpregnancy
CF proteinidentified
HT saline
azithro
aztrm
CFTR‐targeting2018:
‐Median predicted survival ~48 y‐53% of patients are ≥ 18 y
Slide courtesy of CFF
Propert
y of P
resen
ter
Not for
Rep
roduc
tion

Clinical GeneticsHigh
> 60
31-59
< 30
Low
Sw
eat C
hlor
ide
(mm
ol/L
)
CFTR-RDCRMS
PIMutation Class I, II, III
PSMutation Class IV, V
Clinical spectrum increasing disease severity
Classical CF
Levy et al., Clinical Genetics, 2016Prop
erty o
f Pres
enter
Not for
Rep
roduc
tion
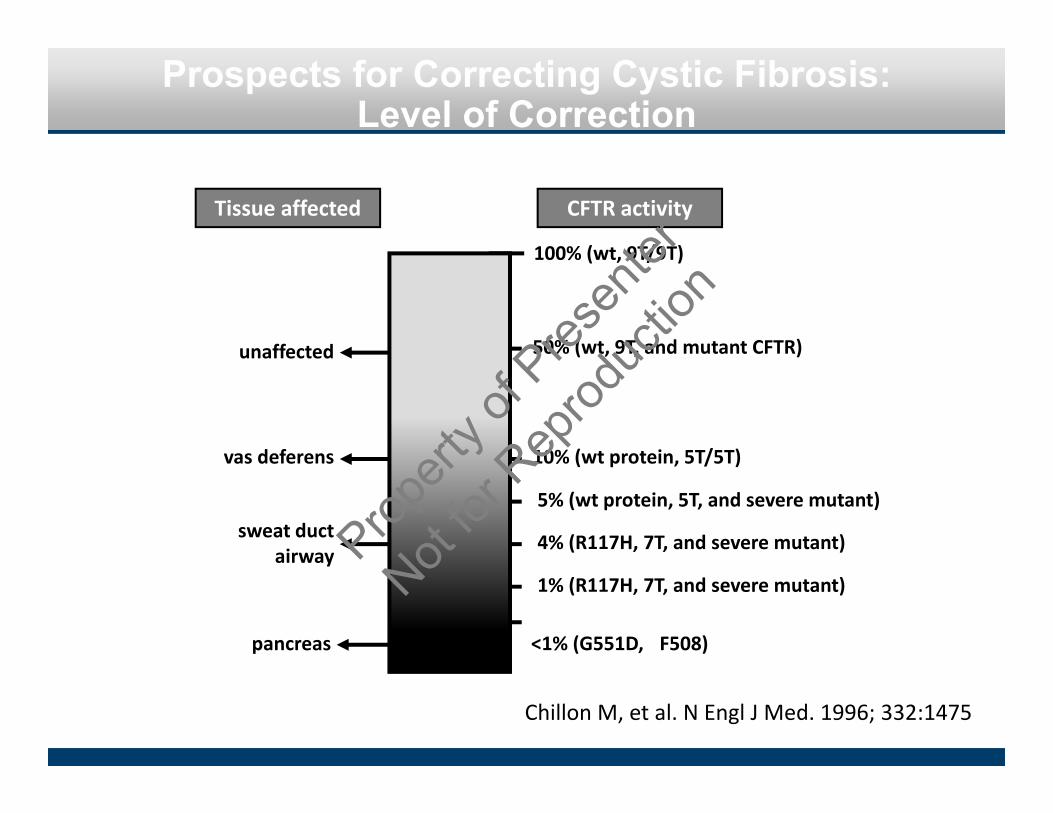
Prospects for Correcting Cystic Fibrosis: Level of Correction
Chillon M, et al. N Engl J Med. 1996; 332:1475
unaffected
100% (wt, 9T/9T)
50% (wt, 9T, and mutant CFTR)
10% (wt protein, 5T/5T)
5% (wt protein, 5T, and severe mutant)
4% (R117H, 7T, and severe mutant)
1% (R117H, 7T, and severe mutant)
<1% (G551D, �F508)
vas deferens
sweat ductairway
pancreas
Tissue affected CFTR activity
Propert
y of P
resen
ter
Not for
Rep
roduc
tion
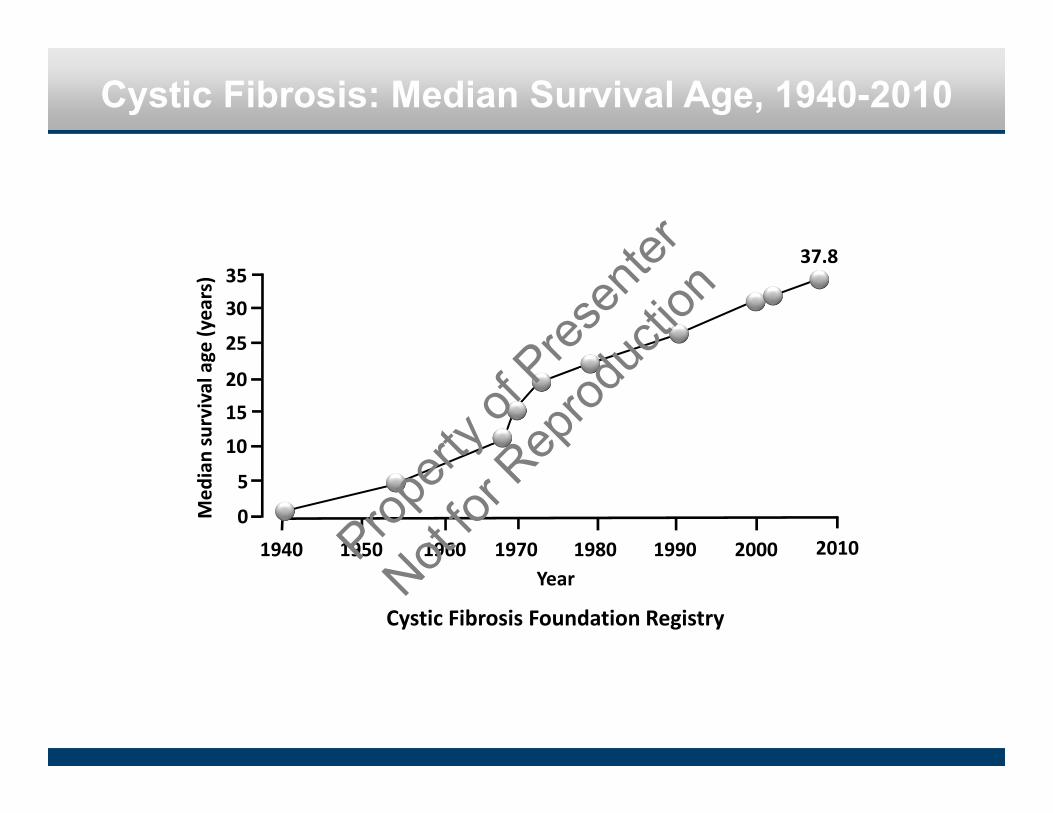
Cystic Fibrosis: Median Survival Age, 1940-2010
0
5
10
1520
25
30
1940 1950 1960 1970 1980 1990 2000
Med
ian survival age (years)
Year
3537.8
Cystic Fibrosis Foundation Registry
2010Propert
y of P
resen
ter
Not for
Rep
roduc
tion

Cystic Fibrosis: Epidemiology
Population
Caucasian (US)
Caucasian (Great Britain)
Hispanic
African American
Native American
Asian (US, England)
Israel
Southern Europe
Epidemiologic
1 in 1,900‐3,700
1 in 2,400‐3,000
1 in 8,000‐9,000
1 in 15,300
1 in 40,000
1 in 10,000
1 in 5,000
1 in 2,000‐4,000
Newborn Screening
1 in 3,400‐3,800
1 in 2,200‐3,200
‐‐
‐‐
‐‐
‐‐
‐‐
‐‐
Propert
y of P
resen
ter
Not for
Rep
roduc
tion

Classes of Cystic Fibrosis-Causing Mutations
Propert
y of P
resen
ter
Not for
Rep
roduc
tion

Cystic Fibrosis: Clinical Phenotype Associated with CFTR Mutations
Milder lung disease
Pancreatic sufficiency
Abnormal sweat chloride
Mild lung disease
Pancreatic sufficiency
Equivocal sweat chloride
R117H (5T)
3849 + 10kB C‐to‐T
2789 + 5 G‐to‐A
R334W
G85E
G91R
R347P
R347H
R347L
R117H (7T)
3849 + 10kB C‐to‐T
G551S
D1152H
Severe lung disease
Pancreatic insufficiency
Abnormal sweat chloride
DF508
G542X
G551D
W1282X
N1303K
R553X
3120 + 1G‐to‐T
1078 del T
R75XProp
erty o
f Pres
enter
Not for
Rep
roduc
tion

Cystic Fibrosis
Organs affected by Cystic Fibrosis
SINUSES: sinusitis (infection)
LUNGS: thick, sticky mucus buildup, bacterial infection and widened airways
SKIN: sweat glands produce salty sweat
LIVER: blocked biliary ducts
PANCREAS: blocked pancreatic ducts
INTESTINES: cannot fully absorb nutrients
REPRODUCTIVE ORGANS: (male and female) complications
Normal airway (cross section)
Airway wall
Airway lined with a thin layer of mucus
Airway with CF (cross section)
Thick, sticky mucus blocks airway
Widened airway
Blood in mucusBacterial
infectionProp
erty o
f Pres
enter
Not for
Rep
roduc
tion
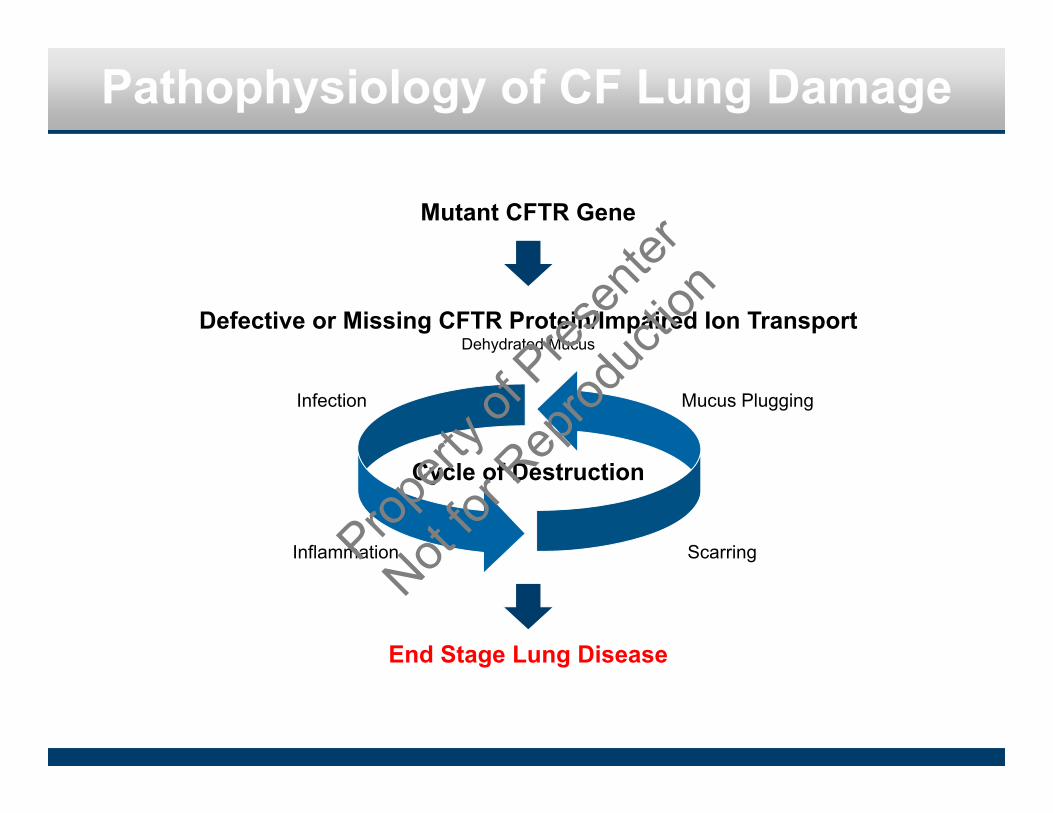
Pathophysiology of CF Lung Damage
Mutant CFTR Gene
Defective or Missing CFTR Protein/Impaired Ion TransportDehydrated Mucus
End Stage Lung Disease
Cycle of Destruction
Infection
Inflammation
Mucus Plugging
ScarringPropert
y of P
resen
ter
Not for
Rep
roduc
tion

WORK IN PROGRESS
Propert
y of P
resen
ter
Not for
Rep
roduc
tion

Wide Spectrum of Disease Severity in CF
Kerem et al NEJM 1990.
Lung function versus age in ∆F508 homozygotesFE
V1(%
pre
dict
ed)
Age (in years)
150
100
50
00 10 20 30 40
Propert
y of P
resen
ter
Not for
Rep
roduc
tion

Pathogenesis of Lung Disease in Cystic Fibrosis
Davis PB, et al. J Respir Crit Care Med. 1996;154:1229
Defective CF gene
Defective/deficient CFTR
Bronchial obstruction
Infection
Bronchiectasis
Inflammation
Abnormal airway surface milieu
Propert
y of P
resen
ter
Not for
Rep
roduc
tion

Treatment of Cystic Fibrosis Lung Disease
Defective CF gene
Defective/deficient CFTR
Bronchial obstruction
Infection
Bronchiectasis
Inflammation
Abnormal airway surface milieu
Decrease mucus viscosityAugment clearance
Chest physiotherapyMucolytics (rhDNase)Hypertonic saline
Decrease bacterial load AntibioticsMacrolides
Reduce host responseCorticosteroids
Ibuprofen
Replace damaged lungs Transplantation
Current focus of modulator and
potentiator therapies
Propert
y of P
resen
ter
Not for
Rep
roduc
tion
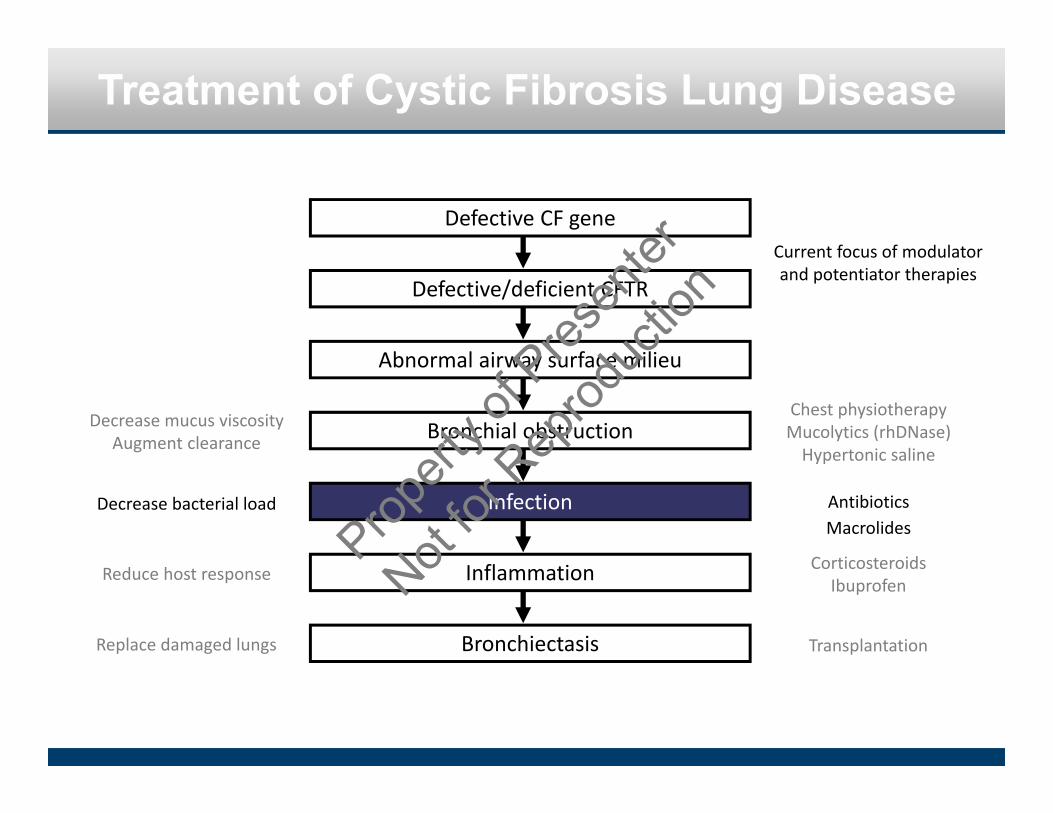
Treatment of Cystic Fibrosis Lung Disease
Defective CF gene
Defective/deficient CFTR
Bronchial obstruction
Infection
Bronchiectasis
Inflammation
Abnormal airway surface milieu
Decrease mucus viscosityAugment clearance
Chest physiotherapyMucolytics (rhDNase)Hypertonic saline
Decrease bacterial load AntibioticsMacrolides
Reduce host response CorticosteroidsIbuprofen
Replace damaged lungs Transplantation
Current focus of modulator and potentiator therapies
Propert
y of P
resen
ter
Not for
Rep
roduc
tion
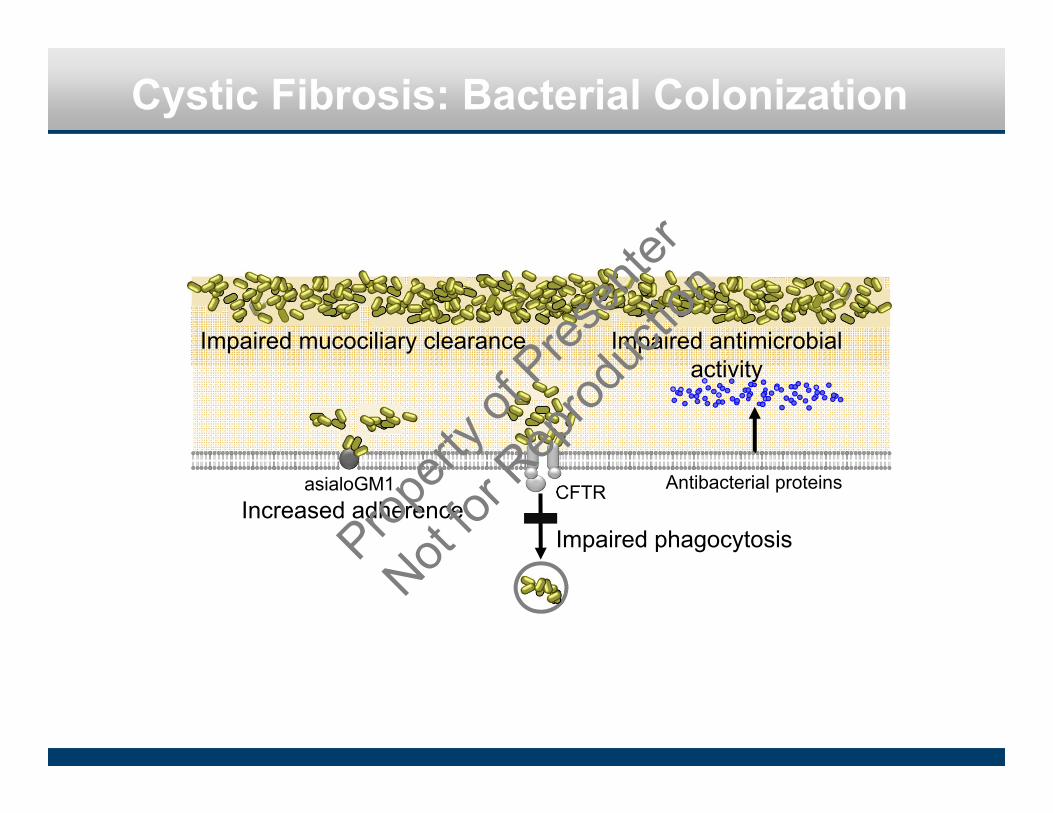
Cystic Fibrosis: Bacterial Colonization
CFTR Antibacterial proteins
Impaired phagocytosisIncreased adherence
Impaired antimicrobial activity
asialoGM1
Impaired mucociliary clearance
Propert
y of P
resen
ter
Not for
Rep
roduc
tion
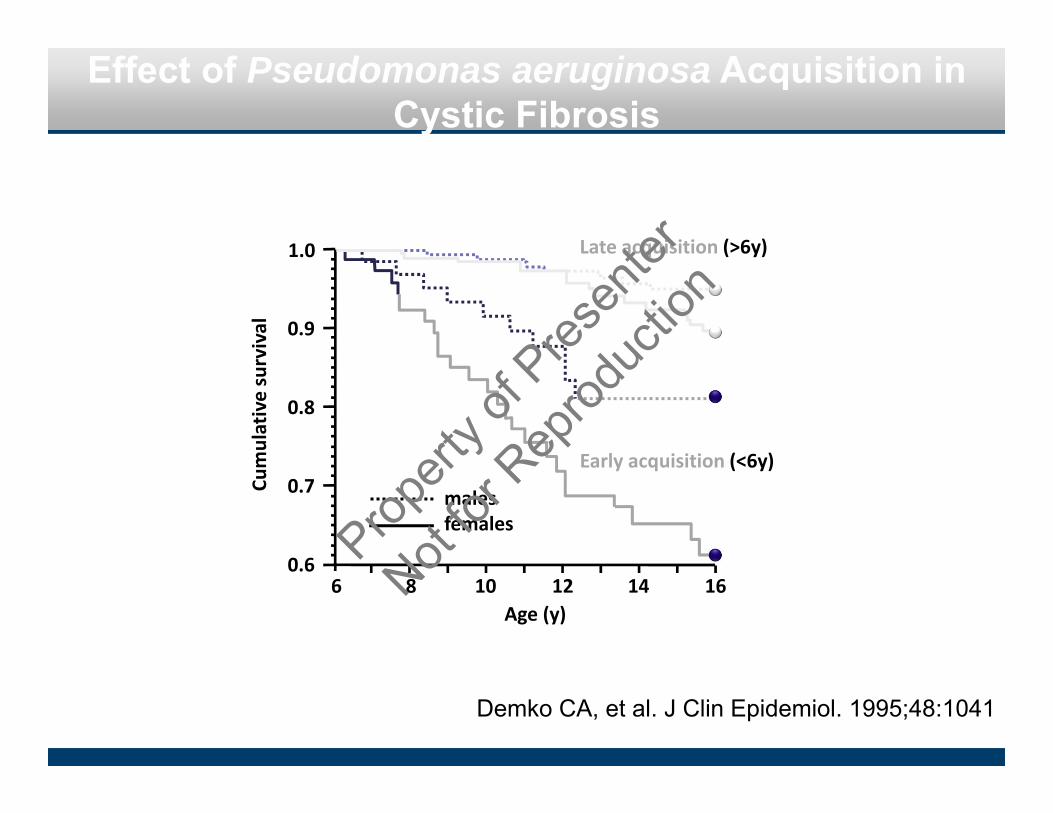
Effect of Pseudomonas aeruginosa Acquisition in Cystic Fibrosis
Demko CA, et al. J Clin Epidemiol. 1995;48:1041
0.9
0.8
0.7
0.6
Cumulative survival
1.0 Late acquisition (>6y)
Early acquisition (<6y)
malesfemales
6 8 10 12 14 16Age (y)
Propert
y of P
resen
ter
Not for
Rep
roduc
tion

Cystic Fibrosis: Organisms Isolated from the Lower Respiratory Tract
100
80
60
40
20
00‐1 2‐5 6‐10 11‐17 18‐24 25‐34 35‐44 >45
P. aeruginosa
S. aureus
H. influenzae
B. cepacia
Age (y)
Percen
tage positive
Data compiled from Cystic Fibrosis Foundation Patient RegistryProp
erty o
f Pres
enter
Not for
Rep
roduc
tion

Treatment of Cystic Fibrosis Lung Disease
Defective CF gene
Defective/deficient CFTR
Bronchial obstruction
Infection
Bronchiectasis
Inflammation
Abnormal airway surface milieu
Decrease mucus viscosityAugment clearance
Chest physiotherapyMucolytics (rhDNase)Hypertonic saline
Decrease bacterial load AntibioticsMacrolides
Reduce host response CorticosteroidsIbuprofen
Replace damaged lungs Transplantation
Propert
y of P
resen
ter
Not for
Rep
roduc
tion

Cystic Fibrosis: Airway Inflammation
respiratory epithelium
IL-8
pmn
Normal Cystic fibrosis
NE TNF-
mac
IL-1O2-
Propert
y of P
resen
ter
Not for
Rep
roduc
tion

Anti-Inflammatory Agents in Cystic Fibrosis: Corticosteroids
A four year, randomized double‐blind, placebo‐controlled trial that compared the efficacy of two doses (1 mg/kg/d and 2 mg/kg/d) of alternate‐day prednisone therapy with placebo in children with CF.
Eigen H, et al. J Pediatr. 1995;126:515.Lai HC, et al. N Engl J Med. 2000;342:851.
�FVC
(% predicted
for a
ge
‐6‐5‐4‐3‐2‐101234
6 12 18 24 30 36 42 48 mos
1 mg/kg 2 mg/kg
placebo
p = 0.0001
high‐dose arm stopped
Propert
y of P
resen
ter
Not for
Rep
roduc
tion
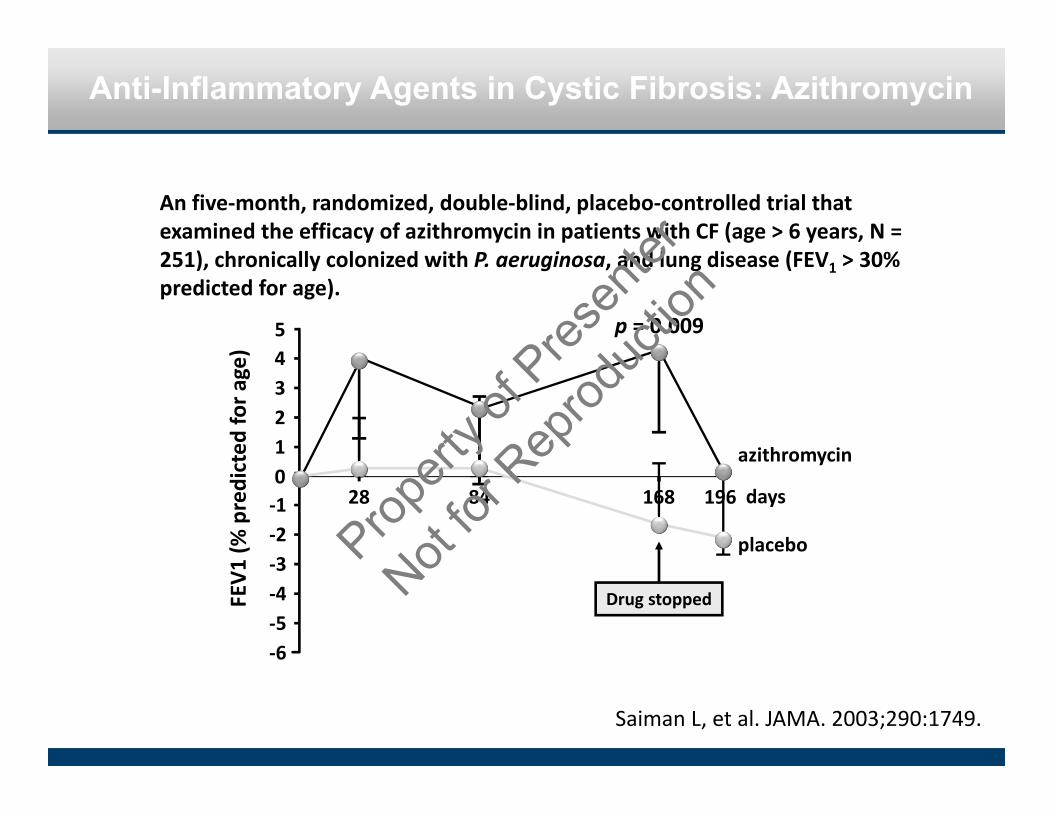
Anti-Inflammatory Agents in Cystic Fibrosis: Azithromycin
An five‐month, randomized, double‐blind, placebo‐controlled trial that examined the efficacy of azithromycin in patients with CF (age > 6 years, N = 251), chronically colonized with P. aeruginosa, and lung disease (FEV1 > 30% predicted for age).
Saiman L, et al. JAMA. 2003;290:1749.
�FEV
1 (%
predicted
for a
ge)
‐6‐5‐4‐3‐2‐101234
28 84 168 196 days
azithromycin
placebo
5 p = 0.009
Drug stopped
Propert
y of P
resen
ter
Not for
Rep
roduc
tion

Treatment of Cystic Fibrosis Lung Disease
Defective CF gene
Defective/deficient CFTR
Bronchial obstruction
Infection
Bronchiectasis
Inflammation
Abnormal airway surface milieu
Decrease mucus viscosityAugment clearance
Mucolytics (rhDNase)Chest physiotherapy
Decrease bacterial load AntibioticsMacrolides
Reduce host response CorticosteroidsIbuprofen
Replace damaged lungs Transplantation
Block Na+ uptakeIncrease Cl‐ efflux
AmilorideUTP/ATP
Hypertonic saline
Propert
y of P
resen
ter
Not for
Rep
roduc
tion

Cystic Fibrosis: Alternative Therapies to Effect Bioelectric Properties of the Respiratory Epithelium
ClCa
CFTR
ENaC
Cl‐
Cl‐Na+ Na+Cl‐
CF Altering other channels
AmilorideUTP/ATPHypertonic
saline
Propert
y of P
resen
ter
Not for
Rep
roduc
tion
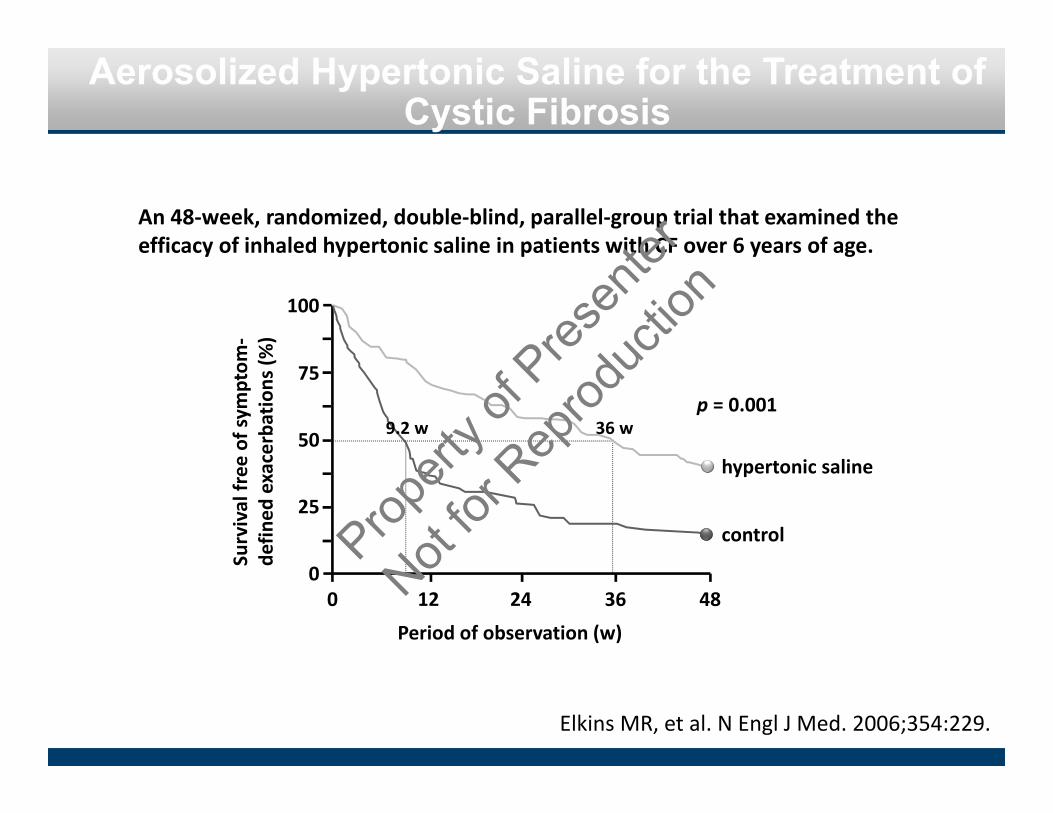
Aerosolized Hypertonic Saline for the Treatment of Cystic Fibrosis
Elkins MR, et al. N Engl J Med. 2006;354:229.
0
25
50
75
100
0 12 24 36 48
Period of observation (w)
Survival free
of sym
ptom
‐de
fined
exacerbations (%
)
hypertonic saline
control
9.2 w 36 w
An 48‐week, randomized, double‐blind, parallel‐group trial that examined the efficacy of inhaled hypertonic saline in patients with CF over 6 years of age.
p = 0.001
Propert
y of P
resen
ter
Not for
Rep
roduc
tion
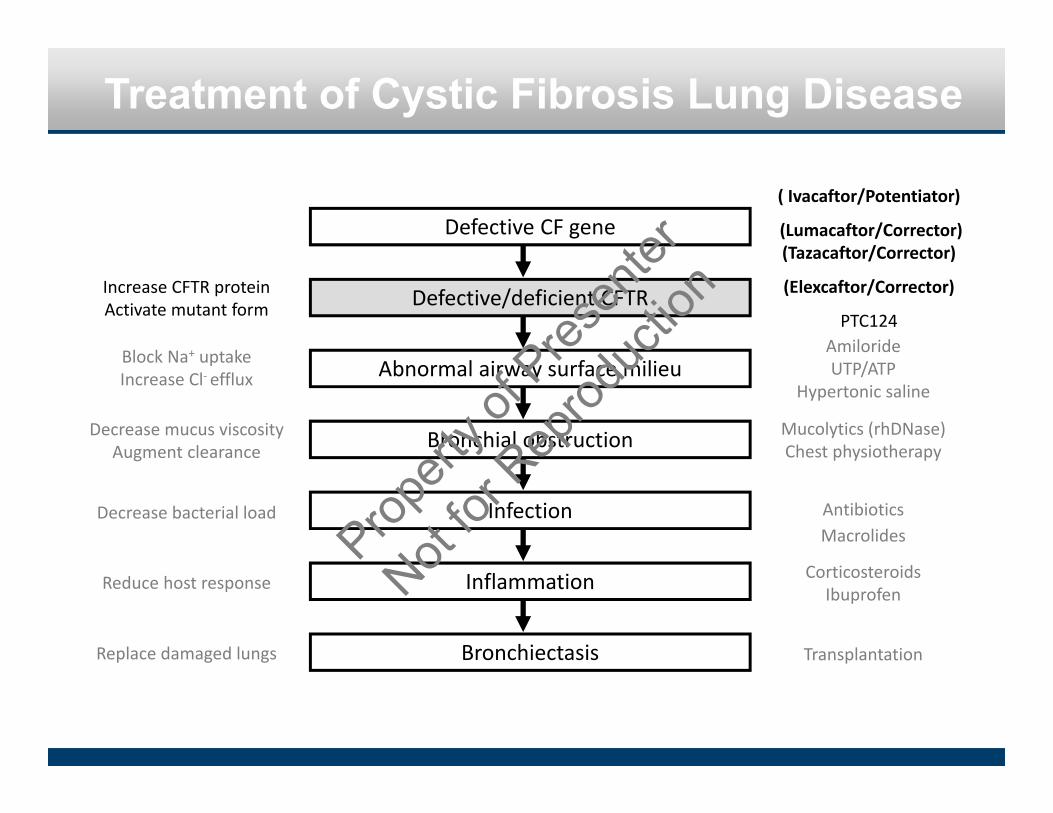
Treatment of Cystic Fibrosis Lung Disease
Defective CF gene
Defective/deficient CFTR
Bronchial obstruction
Infection
Bronchiectasis
Inflammation
Abnormal airway surface milieu
Decrease mucus viscosityAugment clearance
Mucolytics (rhDNase)Chest physiotherapy
Decrease bacterial load AntibioticsMacrolides
Reduce host response CorticosteroidsIbuprofen
Replace damaged lungs Transplantation
Block Na+ uptakeIncrease Cl‐ efflux
( Ivacaftor/Potentiator)
(Lumacaftor/Corrector) (Tazacaftor/Corrector)
(Elexcaftor/Corrector)
PTC124
Increase CFTR proteinActivate mutant form
AmilorideUTP/ATP
Hypertonic saline
Propert
y of P
resen
ter
Not for
Rep
roduc
tion

Golgi
ER
Endosome
Nucleus
Cell membrane
apical trafficking
G551D CFTR
Golgi
ER
Endosome
Nucleus
Cell membrane
Proteasome
degradation
F508 CFTR
Low temperature
Glycerol
Cystic Fibrosis: Correcting CFTR Dysfunction
Zeitlin P. N Engl J Med. 2004;351:606
VX809translation
transcription
post‐translational folding
VX770
VX 770=Ivacaftor=PotentiatorVX 809=Lumacaftor =Corrector
Propert
y of P
resen
ter
Not for
Rep
roduc
tion

25
[Sweat chloride] (m
mol/L)
‐10
‐8
‐6
‐4
‐2
050 100 200
Cystic Fibrosis: Potentiating delF508 CFTR Dysfunction
A two‐week, randomized double‐blind, crossover trial that compared the effect of regular treatment with Lumacaftor with placebo in CF patients with delF508 mutation.
Propert
y of P
resen
ter
Not for
Rep
roduc
tion

Gentamicin-Induced Correction of CFTR Function in Patients with Cystic Fibrosis and CFTR Stop Mutations
Wilschanski M. N Engl J Med. 2003; 349:1433.
0
‐2
‐4
‐6
‐8
0 0.3 0.6 0.9 1.2
Gentamicin concentration (%)
Respon
se of n
asal PD to
chloride‐
free
isop
roterin
ol (m
V)
p = 0.03pre‐treatment
post‐treatment
Propert
y of P
resen
ter
Not for
Rep
roduc
tion

Analysis* Statistic Treatment Difference for TRIKAFTA (N=55) vs Tezacaftor/Ivacaftor# (N=52)
Primary
Absolute change in ppFEV1 from baseline at Week 4 (percentage points) Treatment difference (95% CI) P value 10.0 (7.4, 12.6) P<0.0001
Key Secondary
Absolute change in Sweat Chloride from baseline at Week 4 (mmol/L) Treatment difference (95% CI) P value -45.1 (-50.1, -40.1) P<0.0001
Absolute change in CFQ-R respiratory domain score from baseline at Week 4 (points)
Treatment difference (95% CI) P value 17.4 (11.8, 23.0) P<0.0001
ppFEV1: percent predicted forced expiratory volume in 1 second; CI: confidence interval; CFQ-R: Cystic Fibrosis Questionnaire-Revised. * Baseline for primary and key secondary endpoints is defined as the end of the 4-week tezacaftor/ivacaftor run-in period.# Regimen of tezacaftor 100 mg qd/ivacaftor 150 mg q12hr. USOU TRUJAFTA_21_October 2019
Triple combination therapy (elexacaftor/tezacaftor/ivacaftor; ivacaftor)
For patients (12 yrs and older) with one or two F508del mutations
Propert
y of P
resen
ter
Not for
Rep
roduc
tion

MODULATOR THERAPIES
Propert
y of P
resen
ter
Not for
Rep
roduc
tion

CFF Goals in CFTR Modulator Development
• Enable multiple CFTR modulator options - Patient Choice- Mitigate risk-
• Expand access to rare mutations leveraging in vitro models
• Lessons from highly effective modulator therapy –PROMISE study (TDN)Simplify study (TDN) Real World Research (CFF and NIH)
Propert
y of P
resen
ter
Not for
Rep
roduc
tion

Optimizing Treatment in the Era of Modulators
• Two significant variables‒ Level of CFTR activity provided by modulator ‒ Status of underlying lung disease
• Personalized medicine goes beyond genotype
‒ If there is more than one modulator, which one provides best benefit? ‒ What other therapies will be needed if modulators are started at birth? ‒ What therapies can be stopped if modulators are started later in life?
Propert
y of P
resen
ter
Not for
Rep
roduc
tion

Treatment of Cystic Fibrosis Lung Disease
Defective CF gene
Defective/deficient CFTR
Bronchial obstruction
Infection
Bronchiectasis
Inflammation
Abnormal airway surface milieu
Decrease mucus viscosityAugment clearance
Mucolytics (rhDNase)Chest physiotherapyHypertonic saline
Decrease bacterial load AntibioticsMacrolides
Reduce host response CorticosteroidsIbuprofen
Replace damaged lungs Transplantation
Block Na+ uptakeIncrease Cl‐ efflux
AmilorideUTP/ATP
VX809VX770PTC124
Increase CFTR proteinActivate mutant form
Gene therapyProvide normal gene
Propert
y of P
resen
ter
Not for
Rep
roduc
tion

Pretreatment In vivo transfection In vitro transfectionPretreatment In vivo transfection In vitro transfection
Administration of an Adenovirus Containing the Human CFTR cDNA to the Respiratory Tract of Individuals with Cystic Fibrosis
Crystal RG, et al. Nat Genet. 1994;8:42.Prop
erty o
f Pres
enter
Not for
Rep
roduc
tion

Prospects for Gene Therapy of Cystic Fibrosis: Submucosal Gland
http://www.medicine.mcgill.ca/dynhist/histoimages
epithelium (+)epithelium (+)
submucosal glands (++)submucosal glands (++)
duct (++++)duct (++++)
Sites of CFTR expression in the human airwaySites of CFTR expression in the human airway
Engelhardt JF, et al. J Clin Invest. 1994;93:737.Prop
erty o
f Pres
enter
Not for
Rep
roduc
tion

Prospects for Gene Therapy in CF: Obstacles
• Respiratory epithelial cells versus submucosalglands
• Target receptors?
• Difficulty to bypass physical and functional barriers in the airway
• Immunologic consequences
• Relevant outcome measures
Propert
y of P
resen
ter
Not for
Rep
roduc
tion

What is Our Near-Term Goal?
• 100% of people with CF have access to CFTR-based therapies
– Small molecules– Gene therapy approaches– mRNA therapy– Gene editing/stem cell therapy
• 100% of people with CF have access to the symptomatic therapies that they need but when? Right now one size fits all…
– Antimicrobials– Anti-inflammatories – Mucolytics– Nutritionals
Propert
y of P
resen
ter
Not for
Rep
roduc
tion
Relying on traditional
inputs is like flying blind
• Signs & symptoms
• Clinical exam
• Imaging
• Pulmonary function testingProp
erty o
f Pres
enter
Not for
Rep
roduc
tion
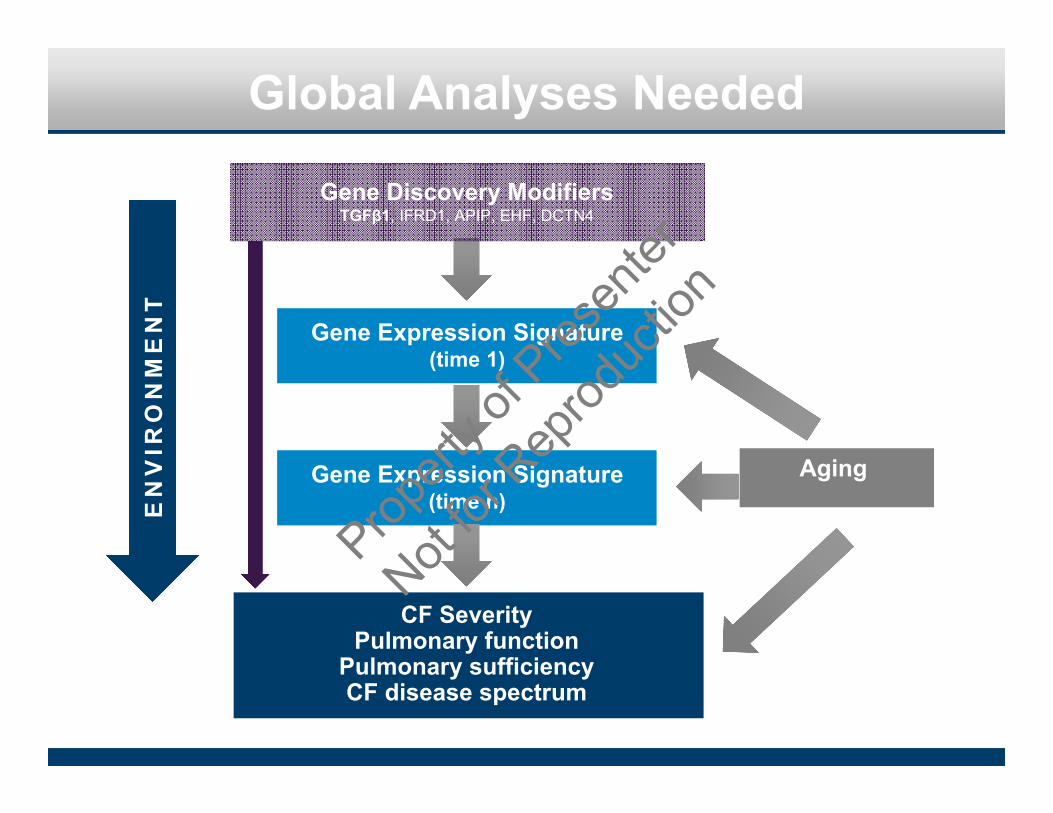
Global Analyses Needed
Aging
EN
VIR
ON
ME
NT
Gene Discovery ModifiersTGFβ1, IFRD1, APIP, EHF, DCTN4
Gene Expression Signature(time 1)
Gene Expression Signature(time n)
CF SeverityPulmonary function
Pulmonary sufficiencyCF disease spectrum
Propert
y of P
resen
ter
Not for
Rep
roduc
tion

Generation of Genomic and EpigenomicSignature
Next-generation sequencingMicroarray analysis
RNA seqCHIP seq
PERSONALIZED MEDICINE
AnalysisTechnical and
statistical validationData base LIMS
support
EpidemiologyDescriptive and
demographic analysis of cohort
Translational Molecular Biology: Cystic Fibrosis
Propert
y of P
resen
ter
Not for
Rep
roduc
tion

Approximately one-third of CF subjects are not responding well to treatment after early diagnosis through newborn screening. This observation confirms our previous observations with another cohort (Lai et al Pediatr 2009; 123:714-722).
These nonresponders are also evident in national data and are suspected to have detrimental genetic modifiers that can be detected by WGS (Corvol, Levy et al Nat Commun. 2015; 6:8382).
Although data from the CF Foundation (CFF) indicate that most children (<18 years) with CF have normal lung function, 20% of CF deaths occur before 20 years of age. This apparent discrepancy may result in part from the wide spectrum of clinical heterogeneity in CF.
Early life events such as bacterial infection, airway inflammation, and structural changes have long-term consequences in patients with CF, as do diet, exercise, treatment regimen, and infections, adding further complexity to disease progression and therapeutic outcome.
This complexity underscores the need for more precise predictors of infection, inflammation, and the structural and functional impacts thereof in CF.
UNKNOWN UNKNOWNS
Propert
y of P
resen
ter
Not for
Rep
roduc
tion

Unique Molecular Signatures Correlate with Disease Severity
PI:HCn=1043
PS:HCn=870
n=224
n=819
n=51
Unionn=1094
A B
PS-HC PI- PI+
-2 +2Fold‐Change
HC CFPS+C
Levy, et al. Physiologic Genomics , Jan 2019
Propert
y of P
resen
ter
Not for
Rep
roduc
tion
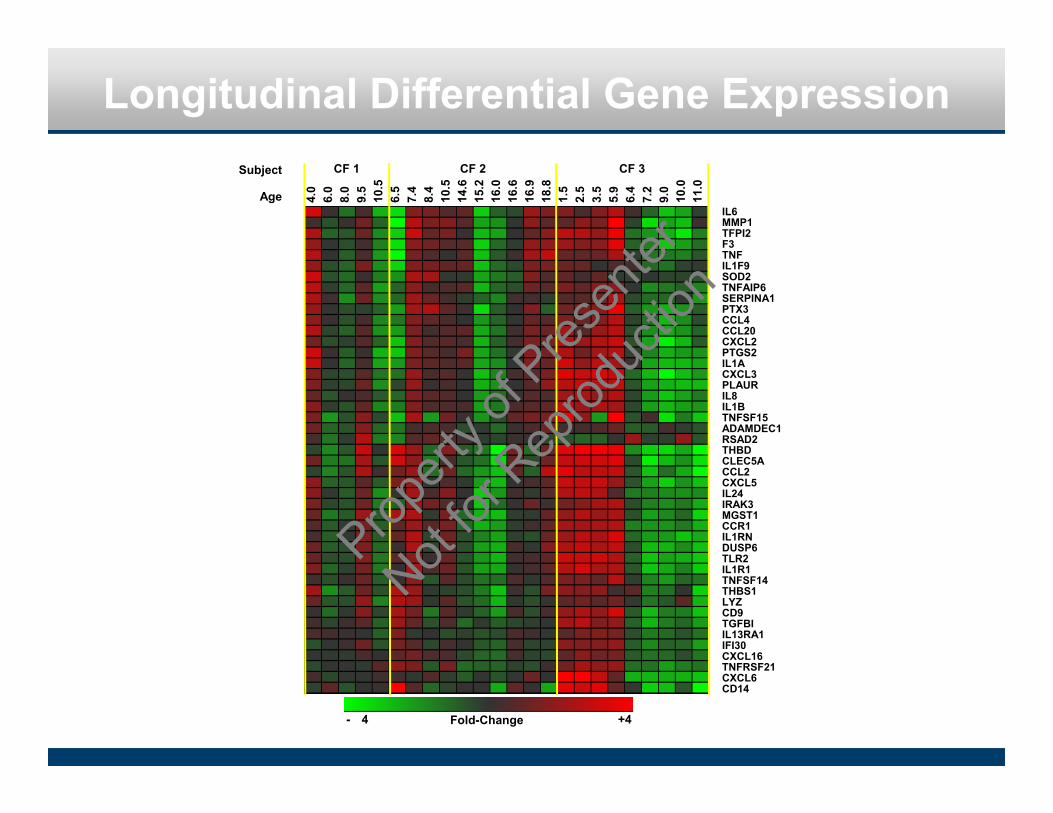
4.0
6.0
8.0
9.5
10.5
6.5
7.4
8.4
10.5
14.6
15.2
16.0
16.6
16.9
18.8
1.5
2.5
3.5
5.9
6.4
7.2
9.0
10.0
11.0
Longitudinal Differential Gene Expression
Fold-Change- 4 +4
IL6MMP1TFPI2F3TNFIL1F9SOD2TNFAIP6SERPINA1PTX3CCL4CCL20CXCL2PTGS2IL1ACXCL3PLAURIL8IL1BTNFSF15ADAMDEC1RSAD2THBDCLEC5ACCL2CXCL5IL24IRAK3MGST1CCR1IL1RNDUSP6TLR2IL1R1TNFSF14THBS1LYZCD9TGFBIIL13RA1IFI30CXCL16TNFRSF21CXCL6CD14
Subject
Age
CF 1 CF 2 CF 3
Propert
y of P
resen
ter
Not for
Rep
roduc
tion

Microarrays have the potential
to predict CF course and therapeutic
response
• Airway clearance• Antibiotics• Hypertonic saline• Mucolytics• Potentiators and
correctors
Propert
y of P
resen
ter
Not for
Rep
roduc
tion

Future DirectionsComparative analyses with RNA-seq to analyze spliced isoforms and non coding RNAs
Application in other cohorts such as COPD looking at CFTR in chronic bronchitis
Comparison of signature in probands of differing CFTR mutation status, parents and to determine impact of CFTR on expression array; can parents and siblings inform relevant genes
Investigate epigenomic underpinnings of expression signature such as miRNA specific profile impacting expression and lncRNA’s
Single cell sequencing comparing CF PBMCs, CF sputum macrophages and epithelial cells over course of infection (Chan Zuckerberg Grant)
1
2
3
4
5Prop
erty o
f Pres
enter
Not for
Rep
roduc
tion

Acknowledgments
Levy Laboratory, National Jewish Health• Xi Zhang, M.D., Ph.D.• Stephanie Jump, B.A.
Collaborators Northwestern Feinberg School of Medicine• Susanna McColley, M.D.• Scott Budinger, M.D.• Alexadra Mischarin, Ph.D.
Collaborators National Jewish Health and Univ Colorado
Russ Bowler, M.D., Ph.D.Camille Moore, Ph.D. Pam Zeitlin, M.D., Ph.D.Katie Hierst, M.D., Ph.D.Eszter Vladar, Ph.D.
Medical College of Wisconsin and University of Wisconsin, Madison Collaborators• Martin Hessner, Ph.D., McGee Center,
Children’s Hospital of Wisconsin
• Shuang Jia M.S., McGee Center, Children’s Hospital of Wisconsin
• Philip Farrell M.D., Ph.D. UW School of Medicine and Public Health
• HuiChuan Lai, Ph.D. UW School of Nutritional Sciences
• Pippa Simpson, Ph.D, Quantitative Health Services Center, Children’s Hospital of Wisconisn
• Amy Pan Quantitative Health Services Center, Children’s Hospital of Wisconsin
Funding Sources• NIH-NHLBI DP2 HL074202
• NIH-NHLBI R21 HL102523-01
• National Jewish Heath
Propert
y of P
resen
ter
Not for
Rep
roduc
tion

Methodology: GeneChip® Scanning and Microarray Analysis
After an overnight hybridization with the labeled mRNA the chips are scanned. A laser scans each DNA spot on the microarray.
A spot with a large amount of labeled mRNA will have a high intensity of light, and a spot with little or no labeled mRNA with have a low light intensity.
The intensity information is sent to a computer for data analysis.
1
2
Propert
y of P
resen
ter
Not for
Rep
roduc
tion
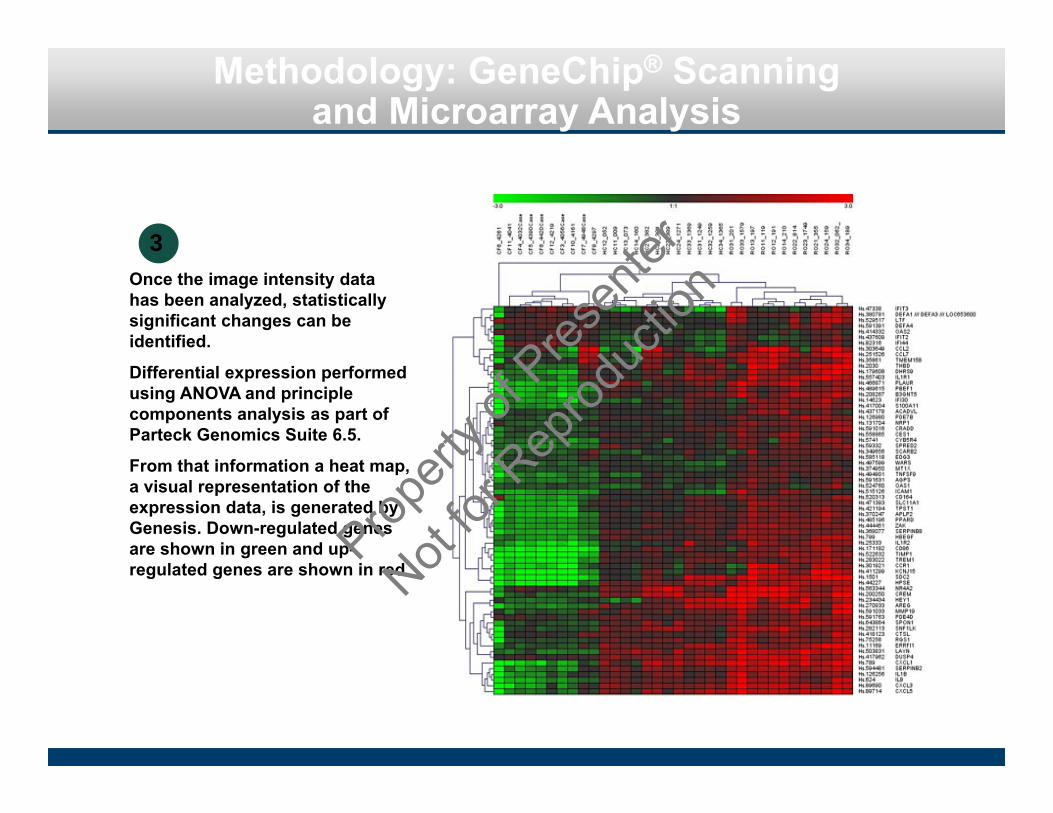
Methodology: GeneChip® Scanning and Microarray Analysis
Once the image intensity data has been analyzed, statistically significant changes can be identified.
Differential expression performed using ANOVA and principle components analysis as part of Parteck Genomics Suite 6.5.
From that information a heat map, a visual representation of the expression data, is generated by Genesis. Down-regulated genes are shown in green and up-regulated genes are shown in red.
3
Propert
y of P
resen
ter
Not for
Rep
roduc
tion