limited service laboratory checklist · college of american pathologists revised: 09/27/2007...
TRANSCRIPT

Revised: 09/27/2007
COMMISSION ON LABORATORY ACCREDITATION
Laboratory Accreditation Program
LIMITED SERVICE LABORATORY CHECKLIST
Disclaimer and Copyright Notice The College of American Pathologists (CAP) Checklists are posted on the CAP's Web site for information only. If you are enrolled in the CAP's Laboratory Accreditation Program and are preparing for an inspection, you must use the Checklists that were mailed in your application or reapplication packet, not those posted on the Web site. The Checklists undergo regular revision and Checklists may be revised after you receive your packet. If a Checklist has been updated since receiving your packet, you will be inspected based upon the Checklists that were mailed. If you have any questions about the use of Checklists in the inspection process, please e-mail the CAP ([email protected]), or call (800) 323-4040, ext. 6065. All Checklists are ©2007. College of American Pathologists. All rights reserved.

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 2 of 178
LIMITED SERVICE LABORATORY
OUTLINE
SUMMARY OF CHANGES ...............................................................................................................................................4 INSPECTION TECHNIQUES – KEY POINTS..................................................................................................................7 INTRODUCTION................................................................................................................................................................9
DEFINITION...................................................................................................................................................................9 PURPOSE AND USE .....................................................................................................................................................9 ATTESTATION............................................................................................................................................................10
LABORATORY SAFETY.................................................................................................................................................11 PROFICIENCY TESTING ................................................................................................................................................11 QUALITY MANAGEMENT AND QUALITY CONTROL.............................................................................................16
GENERAL ISSUES ......................................................................................................................................................17 PROCEDURE MANUAL.............................................................................................................................................20 INSTRUMENTS AND EQUIPMENT..........................................................................................................................23
Glassware and Pipettes .............................................................................................................................................24 Automatic Pipettes - Fixed Volume Adjustable and/or Micropipettes .....................................................................25 Thermometers ...........................................................................................................................................................27 Temperature-Dependent Equipment .........................................................................................................................27 Centrifuges................................................................................................................................................................28 Analytic Balances .....................................................................................................................................................29 Multiple Analysis Automated Instruments and Systems ..........................................................................................31 Colorimeters and Spectrophotometers ......................................................................................................................32
Instrument Maintenance ................................................................................................................................................33 REAGENTS ..................................................................................................................................................................35 RESULTS REPORTING...............................................................................................................................................39 QUALITY CONTROL AND CALIBRATION – WAIVED TESTS ...........................................................................43
HEMATOLOGY AND COAGULATION SECTION.......................................................................................................44 SPECIMEN COLLECTION AND HANDLING..........................................................................................................44 SPECIMEN COLLECTION AND HANDLING - COAGULATION..........................................................................48 AUTOMATED AND SEMI-AUTOMATED HEMATOLOGY CBC SYSTEMS.......................................................52
CBC Instrument Calibration .....................................................................................................................................52 Fresh Whole Blood ...................................................................................................................................................53 Commercial Calibrators ............................................................................................................................................56 CBC INSTRUMENT QUALITY CONTROL .........................................................................................................57
Stabilized Controls ...............................................................................................................................................58 Moving Averages.................................................................................................................................................60 Retained Patient Specimens .................................................................................................................................62
ERROR DETECTION AND VERIFICATION .......................................................................................................63 GENERAL INSTRUMENT ISSUES.......................................................................................................................67
Automated Differential Counters .........................................................................................................................68 MANUAL HEMATOCRIT (MICROHEMATOCRIT, PACKED CELL VOLUME) ............................................71 MANUAL (HEMOCYTOMETER) WBC, RBC, AND PLT COUNTS (BLOOD) ................................................72 MANUAL BLOOD FILM EXAMINATION (DIFFERENTIAL COUNT) ............................................................74 AUTOMATED RETICULOCYTES........................................................................................................................77 MANUAL RETICULOCYTES................................................................................................................................80
ABNORMAL HEMOGLOBIN DETECTION .............................................................................................................81 BLOOD COAGULATION STUDIES ..........................................................................................................................82
Interinstrument Comparisons (Coagulation).............................................................................................................84 RESULTS REPORTING - COAGULATION ..............................................................................................................85
Photo-Optical Coagulation Systems .........................................................................................................................91 Electromechanical Coagulation Systems ..................................................................................................................92 D-Dimer Studies .......................................................................................................................................................93

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 3 of 178
Coagulation Tests Based on Direct Measurement of Analytes .................................................................................96 AUTOMATED/GENERAL CHEMISTRY SECTION ...................................................................................................100
CALIBRATION AND STANDARDS........................................................................................................................100 CONTROLS ................................................................................................................................................................110
Blood Gas Specimens .............................................................................................................................................115 BLOOD GAS INSTRUMENTS..................................................................................................................................117 IMMUNOASSAYS.....................................................................................................................................................119 THERAPEUTIC DRUG MONITORING ...................................................................................................................120 DRUG OF ABUSE SCREENING ..............................................................................................................................121
Legal Drug of Abuse Specimen Collection ............................................................................................................122 Screening for Drugs of Abuse.................................................................................................................................123
URINALYSIS AND CLINICAL MICROSCOPY SECTION.........................................................................................123 SPECIMEN COLLECTION AND HANDLING........................................................................................................123 INSTRUMENTS AND EQUIPMENT........................................................................................................................125 PROCEDURES AND TEST SYSTEMS ....................................................................................................................126 URINALYSIS - MANUAL MICROSCOPY..............................................................................................................127
Dipstick Readers .....................................................................................................................................................130 BODY FLUIDS................................................................................................................................................................131
BODY FLUID CELL COUNTING – MANUAL .......................................................................................................131 BODY FLUID CELL COUNTING – INSTRUMENTS.............................................................................................132 BODY FLUID NUCLEATED CELL DIFFERENTIALS ..........................................................................................134 SEMEN ANALYSIS...................................................................................................................................................138
Requisitions, Specimen Receipt and Results Reporting .........................................................................................138 Sperm Motility........................................................................................................................................................141 Stained Smear - Sperm Differential ........................................................................................................................144
AUTOMATED SEMEN ANALYSIS INSTRUMENTS ............................................................................................147 Calibration and Quality Control..............................................................................................................................147
MICROBIOLOGY SECTION .........................................................................................................................................152 SPECIMEN COLLECTION AND HANDLING........................................................................................................152 REPORTING OF RESULTS ......................................................................................................................................155 MEDIA........................................................................................................................................................................158 DIRECT SPECIMEN EXAMINATION.....................................................................................................................160 STAINS .......................................................................................................................................................................162 BIOSAFETY ...............................................................................................................................................................164
IMMUNOLOGY/IMMUNOHEMATOLOGY SECTION..............................................................................................167 SYPHILIS SEROLOGY .............................................................................................................................................173
PHYSICAL FACILITIES ................................................................................................................................................174

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 4 of 178
SUMMARY OF CHANGES LIMITED SERVICE LABORATORY Checklist
9/27/2007 Edition
The following questions have been added, revised, or deleted in this edition of the checklist, or in the two editions immediately previous to this one. If this checklist was created for a reapplication, on-site inspection or self-evaluation it has been customized based on the laboratory's activity menu. The listing below is comprehensive; therefore some of the questions included may not appear in the customized checklist. Such questions are not applicable to the testing performed by the laboratory. Note: For revised checklist questions, a comparison of the previous and current text may be found on the CAP website. Click on Laboratory Accreditation, Checklists, and then click the column marked Changes for the particular checklist of interest. NEW Checklist Questions Question Effective Date LSV.00050 09/27/2007 LSV.43671 09/27/2007 LSV.43802 09/27/2007 LSV.43804 09/27/2007 LSV.43807 09/27/2007 LSV.43809 09/27/2007 LSV.43833 09/27/2007 LSV.43861 09/27/2007 LSV.43862 09/27/2007 LSV.43863 09/27/2007 LSV.43914 09/27/2007 LSV.43962 09/27/2007 LSV.44010 09/27/2007 LSV.44058 09/27/2007 LSV.44106 09/27/2007 LSV.44154 09/27/2007 LSV.44202 09/27/2007 LSV.44250 09/27/2007 LSV.44298 09/27/2007 LSV.44346 09/27/2007 LSV.44394 09/27/2007 LSV.01527 10/31/2006 LSV.01555 10/31/2006 LSV.36815 10/31/2006 LSV.37064 10/31/2006 LSV.37068 10/31/2006 LSV.37072 10/31/2006

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 5 of 178
LSV.37076 10/31/2006 LSV.38709 10/31/2006 LSV.38710 10/31/2006 LSV.38939 10/31/2006 LSV.39038 10/31/2006 LSV.39137 10/31/2006 LSV.39236 10/31/2006 LSV.39335 10/31/2006 LSV.39434 10/31/2006 LSV.39533 10/31/2006 LSV.39632 10/31/2006 LSV.39731 10/31/2006 LSV.39830 10/31/2006 LSV.39929 10/31/2006 LSV.40028 10/31/2006 LSV.00450 04/06/2006 LSV.00475 04/06/2006 LSV.00900 04/06/2006 LSV.37520 04/06/2006 LSV.46565 04/06/2006 LSV.46570 04/06/2006 LSV.46575 04/06/2006 REVISED Checklist Questions Question Effective Date LSV.00350 09/27/2007 LSV.36940 09/27/2007 LSV.38540 09/27/2007 LSV.40690 09/27/2007 LSV.41170 09/27/2007 LSV.43250 09/27/2007 LSV.43842 09/27/2007 LSV.44690 09/27/2007 LSV.45676 09/27/2007 LSV.45810 09/27/2007 LSV.46450 09/27/2007 LSV.37060 10/31/2006 LSV.40660 10/31/2006 LSV.41370 10/31/2006 LSV.45900 10/31/2006 LSV.01600 04/06/2006 LSV.38708 04/06/2006 LSV.40610 04/06/2006 LSV.43848 04/06/2006

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 6 of 178
LSV.46601 04/06/2006 DELETED Checklist Questions Question Effective Date LSV.00260 09/27/2007 LSV.40930 09/27/2007 LSV.41960 09/27/2007 LSV.41990 09/27/2007 LSV.42130 09/27/2007 LSV.42210 09/27/2007 LSV.43490 09/27/2007 LSV.43510 09/27/2007 LSV.43520 09/27/2007 LSV.46210 09/27/2007 LSV.46250 09/27/2007 LSV.36700 10/31/2006 LSV.36800 10/31/2006 LSV.37191 10/31/2006 LSV.38677 10/31/2006 LSV.43530 10/31/2006 LSV.01200 04/06/2006 LSV.01300 04/06/2006 LSV.01400 04/06/2006 LSV.38000 04/06/2006 LSV.45225 04/06/2006

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 7 of 178
The checklists used in connection with the inspection of laboratories by the Commission on Laboratory Accreditation (“CLA”) of the College of American Pathologists have been created by the College and are copyrighted works of the College. The College has authorized copying and use of the checklists by College inspectors in conducting laboratory inspections for the CLA and by laboratories that are preparing for such inspections. Except as permitted by section 107 of the Copyright Act, 17 U.S.C. sec. 107, any other use of the checklists constitutes infringement of the College’s copyrights in the checklists. The College will take appropriate legal action to protect these copyrights.
CONTINUING EDUCATION INFORMATION Beginning January 2008, you may earn continuing education credits (CME/CE) by completing an online Inspection Preparation activity that includes review of this checklist. Prior to reviewing the checklist, log on to the CAP Web site at <www.cap.org <http://www.cap.org>>, click the Education Programs tab, then select Laboratory Accreditation Program (LAP) Education Activities, and Inspection Preparation for complete instructions and enrollment information.
****************************************************************
INSPECTION TECHNIQUES – KEY POINTS
**************************************************************** I. READ – OBSERVE – ASK – the three methods of eliciting information during the inspection process. These three methods may be used throughout the day in no particular order. Plan the inspection in a way that allows adequate time for all three components. READ = Review of Records and Documents Document review verifies that procedures and manuals are complete, current, available to staff, accurate and reviewed, and describe good laboratory practice. Make notes of any questions you may have, or processes you would like to observe as you read the documentation. OBSERVE – ASK = Direct Observation and Asking Questions Observing and asking questions accomplish the following:
1. Verifies that the actual practice matches the written policy or procedure 2. Ensures that the laboratory processes are appropriate for the testing performed 3. Ensures that outcomes for any problem areas, such as PT failures and issues/problems
identified through the quality management process, have been adequately investigated and resolved
4. Ensures that previously cited deficiencies have been corrected Use the following techniques: Observe laboratory practices – look at what the laboratory is actually doing. Compare the
written policy/procedure to what you actually observe in the laboratory to ensure the written

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 8 of 178
policy/procedure accurately reflects laboratory practice. Note if practice deviates from the documented policies/procedures.
Ask open ended, probing questions – these are starting points that will allow you to obtain large
amounts of information, and help you clarify your understanding of the documentation you’ve seen and observations you’ve made. This eliminates the need to ask every single checklist question, as the dialogue between you and the laboratory may address multiple checklist questions.
Ask open-ended questions that start with phrases such as “show me how…” or “tell me about
…” or “what would you do if…”. By asking questions that are open-ended, or by posing a hypothetical problem, you will avoid “cookbook” answers. For example, ask “Could you show me the specimen transport policy and show me how you ensure optimum specimen quality?” This will help you to determine how well the technical staff is trained, whether or not they are adhering to the lab’s procedures and policies, and give you a feel for the general level of performance of the laboratory.
Ask follow-up questions for clarification. Generally, it is best not to ask the checklist questions verbatim. For example, instead of asking the checklist question “Is there documentation of corrective action when control results exceed defined tolerance limits?” ask, “What would you do if the SD or CV doubles one month?” A follow-up probing question could be, “What would you do if you could not identify an obvious cause for the change in SD or CV?”
II. Evaluate Selected Specimens and Tests in Detail For the Laboratory General Checklist: Follow a specimen through the laboratory. By following a specimen from collection to test result, you can cover multiple checklist questions in the Laboratory General checklist: questions on the specimen collection manual; phlebotomy; verbal orders; identification of patients and specimens; accessioning; and result reporting, including appropriate reference ranges, retention of test records, maintaining confidentiality of patient data, and proper handling of critical results and revisions to reports.
For the individual laboratory sections: Consult the laboratory’s activity menu and focus on tests that potentially have the greatest impact on patient care. Examples of such tests include HIV antibodies, hepatitis B surface antigen, urine drugs of abuse, quantitative beta-hCG, cultures of blood or CSF, acid-fast cultures, prothrombin time and INR reporting, and compatibility testing and unexpected antibody detection. Other potentially high-impact tests may be identified by looking at very high or low volume tests in the particular laboratory, or problems identified by reviewing the Variant Proficiency Testing Performance Report. To evaluate preanalytic and postanalytic issues: Choose a representative specimen and “follow" the specimen through the laboratory or section of the laboratory, reviewing appropriate records in the preanalytic and postanalytic categories. To evaluate analytic processes: Choose 2 or 3 analytes and perform a comprehensive review of records, including procedure manuals, quality control and proficiency testing records, instrument maintenance records and method performance validations for the last 2 years, selecting timeframes at

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 9 of 178
the beginning, mid-point, and end of this timeframe. Compare instrument print-outs to patient reports and proficiency testing results to ensure accurate data entry. If problems are identified, choose additional tests or months to review. III. Verify that proficiency testing problem have been resolved: From the inspector’s packet, review the Variant PT Performance Report that identifies, by analyte, all of the PT scores below 100%. Correlate any PT problems to QC or maintenance records from the same time period. Be thorough when reviewing these representative records, selecting data from the beginning, middle and end of the period since the last on-site inspection. IV. Review correction of previous deficiencies: Review the list of deficiencies from the previous on-site inspection provided in the inspector’s packet. Ensure that they have been appropriately addressed.
*****************************************************************************
INTRODUCTION
***************************************************************************** --------------------------------------------------------- DEFINITION --------------------------------------------------------- Medical or clinical laboratories whose scope of offered services is limited to generally less than 10-20 commonly performed laboratory tests or procedures (irrespective of work-load volumes). Some special function laboratories fit the description of limited service laboratories. A limited service laboratory (e.g., satellite lab, stat lab, outpatient clinic, emergency service, etc.) is one that typically performs limited chemistry tests (e.g., bilirubin, creatinine, cholesterol, glucose) and/or limited hematology tests (e.g., hemoglobin, hematocrit, WBC count, blood film morphology) and/or urinalysis and/or limited immunology/serology (e.g., pregnancy tests, rheumatoid factor, infectious mononucleosis) and/or limited bacteriology (direct smears and plating) and/or limited immunohematology (e.g., ABO grouping, Rh typing). -------------------------------------------------------- PURPOSE AND USE -------------------------------------------------------- It is applicable to laboratories described above, and is intended to relieve the laboratory inspector of the burden of completing multiple checklists during on-site visits to such facilities. If a site qualifies as a "limited service laboratory," and is a free-standing entity with its own director and CLIA-88 number, the Laboratory General and Team Leader checklists must also be used with this

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 10 of 178
Checklist. In other words, the Limited Service Laboratory Checklist cannot be used alone in that setting. On the other hand, if the limited service laboratory is administratively and medically part of a central laboratory at the same site, then one copy of each of the Laboratory General and Team Leader checklists should suffice for both the central laboratory sections and the limited service laboratory. In such cases, the limited service laboratory is viewed as a multifunctional section of the central laboratory, and must share the same federal CLIA-88 number. This checklist CANNOT be used alone if the limited laboratory service includes anatomic pathology and/or cytopathology, flow cytometry, molecular pathology, histocompatibility, cytogenetics, or point of care testing. Section-specific checklists must be used for each of those disciplines. Further, if the limited service laboratory performs testing in areas covered in the section-specific checklists that are not specifically represented herein (e.g., blood storage, coagulation factor assays, chromatography, electrophoresis, microbiology cultures/sensitivities, maternal alpha-fetoprotein testing), use of this checklist is insufficient for accreditation. This checklist may be used in conjunction with up to 2 section-specific checklists. Laboratories that require more than 2 section-specific checklists in addition to this checklist (with the exception of the Point of Care Testing checklist) must use the section-specific checklists for all testing disciplines and may NOT use this checklist for accreditation. This special checklist utilizes questions/items already in existing section-specific checklists (e.g., hematology, chemistry, microbiology, etc.). Only slight editorial changes have been made for the sake of maintaining context. If a limited service laboratory has a scope of service in a particular discipline that exceeds those addressed in this checklist, a section-specific checklist may be required. The laboratory and inspector must be familiar with the scope of those section-specific checklists. In summary, this checklist does not represent any compromise of CAP Standards for accreditation or difference with respect to CLIA-88 regulatory requirements. If a section of this checklist covers an area this is not pertinent to the laboratory's scope of activity, mark all questions in that section "N/A." Certain requirements in this checklist are now different for waived tests, versus nonwaived tests. Please refer to the checklist sections on Quality Management; Reagents; Quality Control and Calibration—Waived Tests; and Dipstick Readers. The current list of tests waived under CLIA-88 may be found at http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfClia/analyteswaived.cfm. -------------------------------------------------------- ATTESTATION -------------------------------------------------------- This Limited Service Laboratory Checklist is not valid for a College of American Pathologists inspection unless it is used for the purpose intended. If there is any doubt, the regular discipline-specific checklists must be used. In order to ensure that both the laboratory director and inspector understand the limitations of this checklist, their signatures on the Inspector's Summation

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 11 of 178
Report validate that they are in mutual agreement that the laboratory concerned meets the criteria for use of the Limited Service Laboratory Checklist.
*****************************************************************************
LABORATORY SAFETY
***************************************************************************** The inspector should review relevant questions from the Safety section of the Laboratory General checklist, to assure that the limited service laboratory is in compliance. Please elaborate upon the location and the details of each deficiency in the Inspector's Summation Report.
*****************************************************************************
PROFICIENCY TESTING
***************************************************************************** Definitions: Proficiency testing (PT) is defined as determination of laboratory testing performance by means of interlaboratory comparisons, in which a PT program periodically sends multiple specimens to members of a group of laboratories for analysis and/or identification; the program then compares each laboratory’s results with those of other laboratories in the group and/or with an assigned value…(adapted from Clinical Laboratory Standards Institute Harmonized Terminology Database; available at http://www.nccls.org/). Alternative assessment is defined as determination of laboratory testing performance by means other than PT--for example, split-sample testing, testing by a different method, etc. College of American Pathologists (CAP) accredited laboratories must participate in the CAP Surveys, or a CAP-approved alternative proficiency testing program. This must include attempted enrollment in Surveys with graded analytes matching those for which the laboratory performs patient testing. Enrollment in CAP Surveys containing ungraded analytes is strongly encouraged. **NEW** 09/27/2007 LSV.00050 Phase I N/A YES NO Does the laboratory’s current CAP Activity Menu accurately reflect the testing performed? NOTE: An accurate Activity Menu is required to properly assess a laboratory’s compliance with proficiency testing requirements. The accuracy of the Activity Menu can be assessed by inquiry of responsible individuals, and by examination of the laboratory’s test requisition(s), computer order

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 12 of 178
screens, procedure manuals, or patient reports. All tests performed by the laboratory should be listed on the Activity Menu, and visa versa. If tests are identified that are not included on the laboratory’s test menu, the inspector should contact the CAP (800-323-4040) for instructions. Please note that unusual or esoteric tests performed in the laboratory section may not be specifically listed on the laboratory's activity menu but may be identified on the activity menu as a miscellaneous code. Further information may be found with the laboratory's instrumentation list. Some activities are also included on the Master Activity Menu using more generic groupings or panels instead of listing the individual tests. The Master Activity Menu represents only those analytes that are directly measured. Calculations are not included. COMMENTARY: N/A REFERENCE: Department of Health and Human Services, Centers for Medicare and Medicaid Services. Clinical laboratory improvement amendments of 1988; final rule. Fed Register. 2004(Oct 1): 985 [42CFR493.51]. LSV.00100 Phase II N/A YES NO Does the laboratory participate in the appropriate required CAP Surveys or another proficiency testing (PT) program accepted by CAP for the patient testing performed? NOTE: The list of analytes for which CAP requires proficiency testing is available on the CAP website [http://www.cap.org/] or by phoning 800-323-4040 (or 847-832-7000), option 1. A laboratory’s participation in proficiency testing must include all analytes on this list for which it performs patient testing. Participation in proficiency testing may be through CAP Surveys or another proficiency testing provider accepted by CAP. Laboratories will not be penalized if they are unable to participate in an oversubscribed program. If unable to participate, however, the laboratory must implement an alternative assessment procedure for the affected analytes. For regulated analytes, if the CAP and CAP-accepted PT programs are oversubscribed, CMS requires the laboratory to attempt to enroll in another CMS-approved PT program. COMMENTARY: N/A REFERENCES: 1) Department of Health and Human Services, Centers for Medicare and Medicaid Services. Clinical laboratory improvement amendments of 1988; final rule. Fed Register. 1992(Feb 28):7146 [42CFR493.801]; 2) Tholen DW. Reference values and participant means as targets in proficiency testing. Arch Pathol Lab Med. 1993;117:885-889; 3) Borsotti M. External quality assessment scheme in Tuscany, Italy. Ann 1st Super Sanita. 1995;31:175-186; 4) Westgard JO, et al.

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 13 of 178
Laboratory precision performance. State of the art versus operating specifications that assure the analytical quality required by clinical laboratory improvement amendments proficiency testing. Arch Pathol Lab Med. 1996;120:621-625; 5) NCCLS. Continuous Quality Improvement: Integrating Five Key Quality System Components; Approved Guideline—Second Edition. NCCLS document GP22-A2 (ISBN 1-56238-552-6). NCCLS, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 19087-1898 USA, 2004; 6) Ross JW, et al. The accuracy of laboratory measurements in clinical chemistry. A study of 11 routine chemistry analytes in the College of American Pathologists chemistry survey with fresh frozen serum, definitive methods, and reference materials. Arch Pathol Lab Med. 1998;122:587-608; 7) College of American Pathologists, Commission on Laboratory Accreditation. Standards for laboratory accreditation; standard III. Northfield, IL: CAP, 1998; 8) Dale JC, Hamrick HJ. Neonatal bilirubin testing practices. Reports from 312 laboratories enrolled in the College of American Pathologists Excel proficiency testing program. Arch Pathol Lab Med. 2000;124:1425-1428; 9) Plebani M, et al. External quality assessment for serum proteins: state of the art. Clin Chem. 2001;47(suppl):A35; 10) Panteghini M, et al. External quality assessment scheme for biochemical markers of cardiac damage. Clin Chem. 2001;47(suppl):A195; 11) Wilson JF, et al. Primary standardization of assays for anticonvulsant drugs: comparison of accuracy and precision. Clin Chem. 2002;48:1963-1969; 12) Taylor A, et al. Comparison of procedures for evaluating laboratory performance in external quality assessment schemes for lead in blood and aluminum in serum demonstrates the need for common quality specifications. Clin Chem. 2002;48:2000-2007. LSV.00200 Phase II N/A YES NO Does the laboratory integrate all proficiency testing samples within the routine workload, and are those samples analyzed by personnel who routinely test patient samples, using the same primary method systems as for patient samples? NOTE: Replicate analysis of Surveys samples is acceptable only if patient specimens are routinely analyzed in the same manner. With respect to morphologic examinations (identification of cell types and microorganisms; review of electrophoretic patterns, etc.), group review and consensus identifications are permitted only for unknown samples that would ordinarily be reviewed by more than one person in an actual patient sample. If the laboratory uses multiple methods for an analyte, Surveys samples should be analyzed by the primary method. The educational purposes of proficiency testing are best served by a rotation that allows all technologists to be involved in the proficiency testing program. Proficiency testing records must be retained and can be an important part of the competency and continuing education documentation in the personnel files of the individuals. When external proficiency testing materials are not available, the semi-annual alternative performance assessment process should also be integrated within the routine workload, if practical. COMMENTARY: N/A

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 14 of 178
REFERENCES: 1) Department of Health and Human Services, Centers for Medicare and Medicaid Services. Clinical laboratory improvement amendments of 1988; final rule. Fed Register. 1992(Feb 28):7146 [42CFR493.801(b)]; 2) Shahangian S, et al. Toward optimal PT use. Med Lab Observ. 2000;32(4):32-43; 3) Parsons PJ. Evaluation of blood lead proficiency testing: comparison of open and blind paradigms. Clin Chem. 2001;47:322-330. **REVISED** 09/27/2007 LSV.00350 Phase II N/A YES NO Is there ongoing evaluation of PT and alternative assessment results, with prompt corrective action taken for unacceptable results? NOTE: Compliance with this item can be examined by selecting a sample of PT evaluation results and alternative assessment records. Special attention should be devoted to unacceptable results. Compliance requires that all of the following are true:
1. There is documented evidence of ongoing review of all PT reports and alternative assessment results by the laboratory director or the director’s designee. Reviews should be completed within one month of the date reports and results become available to the laboratory.
2. All “unacceptable” PT results and alternative assessment test result have been investigated.
3. Corrective action has been initiated for all unacceptable PT and alternative assessment results. Corrective action is appropriate to the nature and magnitude of the problem; it might consist of staff education, instrument recalibration, change in procedures, institution of new clerical checks, discontinuation of patient testing for the analyte or discipline in question, or other appropriate measures.
4. Primary records related to PT and alternative assessment testing are retained for two years (unless a longer retention period is required elsewhere in this checklist for specific analytes or disciplines). These include all instrument tapes, work cards, computer printouts, evaluation reports, evidence of review, and documentation of follow-up/corrective action.
COMMENTARY: N/A REFERENCES: 1) Ehrmeyer SS, et al. Use of alternative rules (other than the 1-2s) for evaluating interlaboratory performance data. Clin Chem. 1988:34:250-256; 2) Klee GG, Forsman RW. A user's classification of problems identified by proficiency testing surveys. Arch Pathol Lab Med. 1988;112:371-373; 3) Department of Health and Human Services, Centers for Medicare and Medicaid Services. Clinical laboratory improvement amendments of 1988; final rule. Fed Register. 1992(Feb 28):7173 [42CFR493.1407(e)(4)(iv)]; 4) Steindel SJ, et al. Reasons for proficiency testing failures in

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 15 of 178
clinical chemistry and blood gas analysis. A College of American Pathologists Q-Probes study in 655 laboratories. Arch Pathol Lab Med. 1996;120:1094-1101; 5) Clinical and Laboratory Standards Institute (CLSI). Using Proficiency Testing to Improve the Clinical Laboratory; Approved Guideline—Second Edition. CLSI document GP27-A2 (ISBN 1-56238-632-8). Clinical and Laboratory Standards Institute, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 19087-1898 USA, 2007; 6) Shahangian S, et. al. Toward optimal PT use. Med Lab Observ. 2000;32(4):32-43; 7) Zaki Z, et al. Self-improvement by participant interpretation of proficiency testing data from events with 2 to 5 samples. Clin Chem. 2000;46:A70. LSV.00425 Phase II N/A YES NO For tests for which CAP does not require PT, does the laboratory at least semiannually 1) participate in external PT, or 2) exercise an alternative performance assessment system for determining the reliability of analytic testing? NOTE: Appropriate alternative performance assessment procedures may include: split sample analysis with reference or other laboratories, split samples with an established in-house method, assayed material, regional pools, clinical validation by chart review, or other suitable and documented means. It is the responsibility of the laboratory director to define such alternative performance assessment procedures, as applicable, in accordance with good clinical and scientific laboratory practice. Participation in ungraded/educational proficiency testing programs also satisfies this checklist question. Semiannual alternative assessment must be performed on tests for which PT is not available. The list of analytes for which CAP requires proficiency testing is available on the CAP website [http://www.cap.org/] or by phoning 800-323-4040 (or 847-832-7000), option 1. COMMENTARY: N/A REFERENCES: 1) Department of Health and Human Services, Centers for Medicare and Medicaid Services. Clinical laboratory improvement amendments of 1988; final rule. Fed Register. 1992(Feb 28):7184 [42CFR493.1709]; 2) Shahangian S, et al. A system to monitor a portion of the total testing process in medical clinics and laboratories. Feasibility of a split-specimen design. Arch Pathol Lab Med. 1998;122:503-511; 3) Shahangian S, Cohn RD. Variability of laboratory test results. Am J Clin Pathol. 2000;113:521-527; 4) NCCLS. Assessment of laboratory tests when proficiency testing is not available; approved guideline. NCCLS document GP29-A. [ISBN 1-56238-479-1]. NCCLS, 940 West Valley Road, Suite 1400, Wayne, PA 19087-1898 USA, 2002; 5) Marks V. False-positive immunoassay results: a multicenter survey of erroneous immunoassay results from assays of 74 analytes in 10 donors from 66 laboratories in seven countries. Clin Chem. 2002;48:2008-2016.

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 16 of 178
**NEW** 04/06/2006 LSV.00450 Phase II N/A YES NO Is there a policy that prohibits interlaboratory communication about proficiency testing samples until after the deadline for submission of data to the proficiency testing provider? COMMENTARY: N/A REFERENCES: 1) Department of Health and Human Services, Centers for Medicare and Medicaid Services. Clinical laboratory improvement amendments of 1988; final rule. Fed Register. 1992(Feb 28):7146 [42CFR493.801(b)(3)]; 2) Bierig JR. Comparing PT results can put a lab’s CLIA license on the line. Northfield, IL: College of American Pathologists CAP Today. 2002;16(2):84-87. **NEW** 04/06/2006 LSV.00475 Phase II N/A YES NO Is there a policy that prohibits referral of proficiency testing specimens to another laboratory? NOTE: Under CLIA-88 regulations, there is a strict prohibition against referring proficiency testing specimens to another laboratory. In other words, the laboratory may not refer a proficiency testing specimen to a laboratory with a different CLIA number (even if the second laboratory is in the same health care system). COMMENTARY: N/A REFERENCE: Department of Health and Human Services, Centers for Medicare & Medicaid Services. Clinical laboratory improvement amendments of 1988; final rule. Fed Register. 1992(Feb 28): [42CFR493.801(b)(4)].
*****************************************************************************
QUALITY MANAGEMENT AND QUALITY CONTROL
*****************************************************************************

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 17 of 178
-------------------------------------------------------- GENERAL ISSUES -------------------------------------------------------- LSV.00500 Phase II N/A YES NO Does the limited service laboratory have a written quality management/quality control (QM/QC) program? NOTE: The QM/QC program in the limited service laboratory must be clearly defined and documented. The program must ensure quality throughout the preanalytic, analytic, and post-analytic (reporting) phases of testing, including patient identification and preparation; specimen collection, identification, preservation, transportation, and processing; and accurate, timely result reporting. The program must be capable of detecting problems in the laboratory’s systems, and identifying opportunities for system improvement. The laboratory must be able to develop plans of corrective/preventive action based on data from its QM system. All QM questions in the Laboratory General Checklist pertain to the limited service laboratory. COMMENTARY: N/A REFERENCE: Department of Health and Human Services, Centers for Medicare and Medicaid Services. Clinical laboratory improvement amendments of 1988; final rule. Fed Register. 1992(Feb 28):7176 [42CFR493.1445(e)(5)]. LSV.00600 Phase II N/A YES NO Is there a documented procedure describing methods for patient identification, patient preparation, specimen collection and labeling, specimen preservation, and conditions for transportation, and storage before testing, consistent with good laboratory practice? COMMENTARY: N/A LSV.00700 Phase II N/A YES NO Is there evidence of ongoing evaluation of records of controls, instrument maintenance and function, temperature, etc, for all procedures as required?

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 18 of 178
COMMENTARY: N/A **NEW** 04/06/2006 LSV.00900 Phase II N/A YES NO Are quality control data reviewed and assessed at least monthly by the laboratory director or designee? COMMENTARY: N/A LSV.01100 Phase II N/A YES NO Is there a documented system in operation to detect and correct significant clerical and analytical errors, and unusual laboratory results, in a timely manner? NOTE: The laboratory must have a documented system in operation to detect and correct significant clerical and analytical errors, and unusual laboratory results. One common method is review of results by a qualified person (technologist, supervisor, pathologist) before release from the laboratory, but there is no requirement for supervisory review of all reported data. The selective use of delta checks also may be useful in detecting clerical errors in consecutive samples from the same patient/client. In computerized laboratories, there should be automatic "traps" for improbable results. The system for detecting clerical errors, significant analytical errors, and unusual laboratory results must provide for timely correction of errors, i.e., before results become available for clinical decision making. For suspected errors detected by the end user after reporting, corrections must be promptly made if such errors are confirmed by the laboratory. Each procedure must include a listing of common situations that may cause analytically inaccurate results, together with a defined protocol for dealing with such analytic errors or interferences. This may require alternate testing methods; in some situations, it may not be possible to report results for some or all of the tests requested. The intent of this requirement is NOT to require verification of all results outside the reference (normal) range. COMMENTARY: N/A

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 19 of 178
REFERENCE: Houwen B, Duffin D. Delta checks for random error detection in hematology tests. Lab Med. 1989;20:410-417. LSV.01500 Phase II N/A YES NO In the absence of on-site supervisors, are the results of tests performed by personnel reviewed by the laboratory director or general supervisor within 24 hours? NOTE: The CAP does NOT require supervisory review of all test results. This question addresses only that situation defined under CLIA-88 for "high complexity testing" performed by trained high school graduates qualified under 42CFR493.1489(b)(5) when a qualified general supervisor is not present. COMMENTARY: N/A REFERENCE: Department of Health and Human Services, Centers for Medicare and Medicaid Services. Clinical laboratory improvement amendments of 1988; final rule. Fed Register. 1992(Feb 28):7182 [42CFR493.1463(a)(3) and 42CFR493.1463(c)]: 7183 [42CFR493.1489(b)(1) and 42CFR493.1489(b)(5)]. **NEW** 10/31/2006 LSV.01527 Phase II N/A YES NO If the laboratory uses more than one instrument to test for a given analyte, are the instruments checked against each other at least twice a year for correlation of patient/client results? NOTE: This question applies to quantitative tests performed on the same or different instrument makes/models. This comparison must include all instruments. The use of fresh human samples (whole blood, serum, plasma, urine, etc.), rather than stabilized commercial controls, is important to directly address the issue of whether a patient/client sample yields the same results on all of the laboratory's instruments. Statistical agreement of commercial control materials across instruments does not guarantee comparability of patient/client specimen results because of potential matrix effects. In cases when pre-analytical stability of patient/client specimens is a limiting factor, alternative protocols based on QC or reference materials may be necessary but the materials used should be validated to have the same response as fresh human samples for the instruments/methods involved. This checklist requirement applies only to instruments/methods accredited under a single CAP number. COMMENTARY:

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 20 of 178
N/A REFERENCES: 1) Department of Health and Human Services, Centers for Medicare and Medicaid Services. Medicare, Medicaid and CLIA programs; CLIA fee collection; correction and final rule. Fed Register. 2003(Jan 24):5236 [42CFR493.1281(a)]; 2) Podczasy JJ, et al. Clinical evaluation of the Accu-Chek Advantage blood glucose monitoring system. Lab Med. 1997;28:462-466; 3) Ross JW, et al. The accuracy of laboratory measurements in clinical chemistry: a study of eleven analytes in the College of American Pathologists Chemistry Survey with fresh frozen serum, definitive methods and reference methods. Arch Pathol Lab Med. 1998;122:587-608. **NEW** 10/31/2006 LSV.01555 Phase II N/A YES NO For tests that are waived under CLIA-88, does the laboratory follow manufacturer instructions? COMMENTARY: N/A -------------------------------------------------------- PROCEDURE MANUAL -------------------------------------------------------- The procedure manual should be used by personnel at the workbench and should include: test principle, clinical significance, specimen type, required reagents, test calibration, quality control, procedural steps, calculations, reference intervals, and interpretation of results. The manual should address relevant pre-analytic and post-analytic considerations, as well as the analytic activities of the laboratory. The specific style and format of procedure manuals are at the discretion of the laboratory director. The inspection team should review the procedure manual in detail to understand the laboratory's standard operating procedures, ensure that all significant information and instructions are included, and that actual practice matches the contents of the procedure manuals. **REVISED** 04/06/2006 LSV.01600 Phase II N/A YES NO Is a complete procedure manual available at the workbench or in the work area?
NOTE 1: The use of inserts provided by manufacturers is not acceptable in place of a procedure manual. However, such inserts may be used as part of a procedure description, if

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 21 of 178
the insert accurately and precisely describes the procedure as performed in the laboratory. Any variation from this printed or electronic procedure must be detailed in the procedure manual. In all cases, appropriate reviews must occur.
NOTE 2: A manufacturer's procedure manual for an instrument/reagent system may be acceptable as a component of the overall departmental procedures. Any modification to or deviation from the procedure manual must be clearly documented. NOTE 3: Card files or similar systems that summarize key information are acceptable for use as quick reference at the workbench provided that:
a. A complete manual is available for reference b. The card file or similar system corresponds to the complete manual and is subject to
document control
NOTE 4: Electronic (computerized) manuals are fully acceptable. There is no requirement for paper copies to be available for the routine operation of the laboratory, so long as the electronic versions are readily available to all personnel. However, procedures must be available to laboratory personnel when the electronic versions are inaccessible (e.g., during laboratory information system or network downtime); thus, the laboratory must maintain either paper copies or electronic copies on CD or other media that can be accessed via designated computers. All procedures, in either electronic or paper form, must be readily available for review by the inspector at the time of the CAP inspection. Electronic versions of procedures must be subjected to proper document control (i.e., only authorized persons may make changes, changes are dated/signed (manual or electronic), and there is documentation of annual review). Documentation of review of electronic procedures may be accomplished by including statements such as “reviewed by [name of reviewer] on [date of review]” in the electronic record. Alternatively, paper review sheets may be used to document review of electronic procedures. Documentation of review by a secure electronic signature is NOT required.
COMMENTARY: N/A REFERENCES: 1) Department of Health and Human Services, Centers for Medicare and Medicaid Services. Clinical laboratory improvement amendments of 1988; final rule. Fed Register. 1992(Feb 28):7164 [42CFR493.1211]; 2) van Leeuwen AM. 6 Steps to building an efficiency tool. Advance/Laboratory. 1999:8(6):88-91; 3) Borkowski A, et al. Intranet-based quality improvement documentation at the Veterans Affairs Maryland health care system. Mod. Pathol. 2001;14:1-5; 4) Clinical and Laboratory Standards Institute (CLSI). Laboratory Documents: Development and Control; Approved Guideline—Fifth Edition. CLSI document GP2-A5 (ISBN 1-56238-600-X). Clinical and Laboratory Standards Institute, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 19087-1898 USA, 2006.

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 22 of 178
LSV.01800 Phase II N/A YES NO Is there documentation of at least annual review of all procedures by the current laboratory director or designee? NOTE: The director must ensure that the collection of policies and technical protocols is complete, current, and has been thoroughly reviewed by a knowledgeable person. Technical approaches must be scientifically valid and clinically relevant. To minimize the burden on the laboratory and reviewer(s), it is suggested that a schedule be developed whereby roughly 1/12 of all procedures are reviewed monthly. Paper/electronic signature review must be at the level of each procedure, or as multiple signatures on a listing of named procedures. A single signature on a Title Page or Index of all procedures is not sufficient documentation that each procedure has been carefully reviewed. Signature or initials on each page of a procedure is not required. COMMENTARY: N/A REFERENCES: 1) Department of Health and Human Services, Centers for Medicare and Medicaid Services. Clinical laboratory improvement amendments of 1988; final rule. Fed Register. 1992(Feb 28):7173 [42CFR493.1407(e)(13)]; 2) Borkowski A, et al. Intranet-based quality improvement documentation at the Veterans Affairs Maryland health care system. Mod. Pathol. 2001;14:1-5. LSV.01850 Phase II N/A YES NO Does the director (or a designee who meets CAP director qualifications) review and approve all new policies and procedures, as well as substantial changes to existing documents, before implementation? NOTE: Current practice must match the policy and procedure documents. COMMENTARY: N/A REFERENCE: Department of Health and Human Services, Centers for Medicare and Medicaid Services. Clinical laboratory improvement amendments of 1988; final rule. Fed Register. 1992(Feb 28):7164 [42CFR493.1211(f)].

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 23 of 178
LSV.01900 Phase II N/A YES NO Does the laboratory have a system documenting that all personnel are knowledgeable about the contents of procedure manuals (including changes) relevant to the scope of their testing activities? NOTE: The form of this system is at the discretion of the laboratory director. Annual procedure sign-off by testing personnel is not specifically required. COMMENTARY: N/A LSV.02000 Phase II N/A YES NO If there is a change in directorship, does the new director ensure (over a reasonable period of time) that laboratory procedures are well-documented and undergo at least annual review? COMMENTARY: N/A REFERENCE: Department of Health and Human Services, Centers for Medicare and Medicaid Services. Clinical laboratory improvement amendments of 1988; final rule. Fed Register. 1992(Feb 28):7164 [42CFR493.1211(e)]. LSV.02100 Phase II N/A YES NO When a procedure is discontinued, is a paper or electronic copy maintained for at least 2 years, recording initial date of use, and retirement date? COMMENTARY: N/A REFERENCE: Department of Health and Human Services, Centers for Medicare and Medicaid Services. Clinical laboratory improvement amendments of 1988; final rule. Fed Register. 1992(Feb 28):7164 [42CFR493.1211(g)]. -------------------------------------------------------- INSTRUMENTS AND EQUIPMENT --------------------------------------------------------

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 24 of 178
................................................................. Glassware and Pipettes ................................................................. LSV.02200 Phase II N/A YES NO Are glass volumetric flasks of certified accuracy (Class A, National Institute of Standards and Technology (NIST) standard or equivalent) or if non-certified volumetric glassware is used, are all items checked for accuracy of calibration before initial use? COMMENTARY: N/A LSV.02500 Phase II N/A YES NO Are glass volumetric pipettes of certified accuracy (Class A); or are they checked by gravimetric, colorimetric, or some other verification procedure before initial use? NOTE: The following Table shows the American Society for Testing and Materials' calibration (accuracy) specifications for Class A volumetric pipettes:
Nominal Capacity (mL) Variation (± mL)
0.5 - 2 0.006
3 - 7 0.01
8 - 10 0.02
15 - 30 0.03
40 - 50 0.05
100 0.08 Reconstitution of lyophilized calibrators, controls, or proficiency testing materials, or any other tasks requiring accurate volumetric measurement, must be performed only with measuring devices of Class A accuracy, or those for which accuracy has been defined and deemed acceptable for the intended use. COMMENTARY: N/A

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 25 of 178
REFERENCES: 1) Curtis RH. Performance verification of manual action pipets. Part I. Am Clin Lab. 1994;12(7):8-9; 2) Curtis RH. Performance verification of manual action pipets. Part II. Am Clin Lab. 1994;12(9):16-17; 3) Perrier S, et al. Micro-pipette calibration using a ratiometric photometer-reagent system as compared to the gravimetric method. Clin Chem. 1995;41:S183; 4) American Society for Testing and Materials. Standard specification for glass volumetric (transfer) pipets, designation E 969-95. Philadelphia, PA: ASTM, 1995; 5) Johnson B. Calibration to dye for: Artel's new pipette calibration system. Scientist. 1999;13(12):14; 6) Connors M, Curtis R. Pipetting error: a real problem with a simple solution. Parts I and II. Am Lab News. 1999;31(13):20-22; 7) Skeen GA, Ashwood ER. Using spectrophotometry to evaluate volumetric devices. Lab Med. 2000;31:478-479. LSV.02600 Phase I N/A YES NO Is the use of less precise measuring devices such as serological plastic pipettes and graduated cylinders limited to situations where the accuracy and precision of calibrated glass pipettes are not required? NOTE: In contrast with the more stringent accuracy requirements of glass pipettes, ASTM requirements for plastic pipettes are +/- 3% of the stated volume. The procedure manual should specify when the use of non-class A measuring devices is permissible. COMMENTARY: N/A REFERENCE: American Society for Testing and Materials. Standard specification for serological pipets, disposable plastic, designation E 934-88, In 1993 Annual Book of ASTM Standards, section 14 (general methods and instrumentation). Philadelphia, PA: ASTM, 1993:14.02:485-486. ................................................................ Automatic Pipettes - Fixed Volume Adjustable and/or Micropipettes ................................................................. Automatic pipettes and diluting devices of all types must be checked for accuracy and reproducibility before being placed in service and periodically thereafter. LSV.02700 Phase II N/A YES NO Is there a documented procedure defining how pipettes are checked for accuracy of calibration (gravimetric, colorimetric, volumetric or other verification procedure) before being placed in service initially, and results documented?

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 26 of 178
NOTE: Automatic pipettes (fixed volume adjustable and/or micropipettes) must be checked for calibration accuracy either by volumetric, colorimetric, gravimetric, or other means before being placed in service initially, and results documented. COMMENTARY: N/A REFERENCES: 1) Curtis RH. Performance verification of manual action pipets. Part I. Am Clin Lab. 1994;12(7):8-9; 2) Curtis RH. Performance verification of manual action pipets. Part II. Am Clin Lab. 1994;12(9):16-17; 3) Perrier S, et al. Micro-pipette calibration using a ratiometric photometer-reagent system as compared to the gravimetric method. Clin Chem. 1995;41:S183; 4) Bray W. Software for the gravimetric calibration testing of pipets. Am Clin Lab. Oct 1995 (available on the internet at http://www.labtronics.com/pt_art.htm); 5) Kroll MH, et al (eds). Laboratory instrument evaluation, verification & maintenance manual, 5th edition. Northfield, IL: College of American Pathologists, 1999:126-127; 6) Johnson B. Calibration to dye for: Artel's new pipette calibration system. Scientist. 1999;13(12):14; 7) Connors M, Curtis R. Pipetting error: a real problem with a simple solution. Parts I and II. Am Lab News. 1999;31(13):20-22; 8) Skeen GA, Ashwood ER. Using spectrophotometry to evaluate volumetric devices. Lab Med. 2000;31:478-479. LSV.02800 Phase II N/A YES NO Are automatic pipettes used for quantitative dispensing checked for accuracy and reproducibility (gravimetric, colorimetric or other verification procedure) at specified, periodic intervals, and are the results recorded? NOTE: For analytic instruments with integral automatic pipettors, the accuracy and precision of the pipetting system should be checked periodically, unless that is not practical for the end-user laboratory. Manufacturers' recommendations should be followed. COMMENTARY: N/A REFERENCES: 1) Curtis RH. Performance verification of manual action pipets. Part I. Am Clin Lab. 1994;12(7):8-9; 2) Curtis RH. Performance verification of manual action pipets. Part II. Am Clin Lab. 1994;12(9):16-17; 3) Perrier S, et al. Micro-pipette calibration using a ratiometric photometer-reagent system as compared to the gravimetric method. Clin Chem. 1995;41:S183; 4) Bray W. Software for the gravimetric calibration testing of pipets. Am Clin Lab. Oct 1995 (available on the internet at http://www.labtronics.com/pt_art.htm); 5) Kroll MH, et al (eds). Laboratory instrument evaluation, verification & maintenance manual, 5th edition. Northfield, IL: College of American Pathologists, 1999:126-127; 6) Johnson B. Calibration to dye for: Artel's new pipette calibration system. Scientist. 1999;13(12):14; 7) Connors M, Curtis R. Pipetting error: a real problem with a simple solution. Parts I and II. Am Lab News. 1999;31(13):20-22; 8) Skeen GA, Ashwood ER. Using spectrophotometry to evaluate volumetric devices. Lab Med. 2000;31:478-479.

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 27 of 178
................................................................. Thermometers ................................................................. LSV.02900 Phase II N/A YES NO Is an appropriate thermometric standard device of known accuracy (NIST-certified or guaranteed by manufacturer to meet NIST Standards) available? NOTE: Thermometers should be present on all temperature-controlled instruments and environments and checked daily. Thermometric standard devices should be recalibrated or recertified prior to the date of expiration of the guarantee of calibration. COMMENTARY: N/A LSV.03000 Phase II N/A YES NO Are all non-certified thermometers in use checked against an appropriate thermometric standard device before initial use? COMMENTARY: N/A ................................................................. Temperature-Dependent Equipment ................................................................. LSV.35350 Phase II N/A YES NO Are temperatures checked and recorded appropriately for the following types of equipment?
1. Water baths 2. Dry baths (heating blocks) 3. Instrument components (water baths, dialyzers, heating baths) 4. Incubators and ovens (where temperature control is necessary for a procedure) 5. Refrigerators and freezers

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 28 of 178
NOTE: Temperature-dependent equipment containing reagents and patient specimens must be monitored daily, as equipment failures could affect accuracy of patient test results. Items such as water baths and heat blocks used for procedures need only be checked on days of patient testing. COMMENTARY: N/A LSV.35400 Phase II N/A YES NO Have acceptable ranges been defined for all temperature-dependent equipment? COMMENTARY: N/A LSV.35430 Phase II N/A YES NO Is there evidence of corrective action taken if acceptable temperature ranges for refrigerators and/or freezers are exceeded, including evaluation of contents for adverse effects? NOTE: If acceptable temperature ranges for refrigerators and/or freezers are exceeded, reagents, controls, calibrators, etc. must be evaluated for possible adverse effects, with documentation of results. COMMENTARY: N/A ................................................................. Centrifuges ................................................................. LSV.35490 Phase II N/A YES NO Are all centrifuges used in the laboratory clean and properly maintained? COMMENTARY: N/A

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 29 of 178
LSV.35520 Phase II N/A YES NO Is there a documented protocol and schedule for maintenance of all centrifuges (i.e., cleaning, changing brushes, etc.)? COMMENTARY: N/A LSV.35550 Phase II N/A YES NO Are the operating speeds of all centrifuges checked periodically as needed for the intended use and is this done in a safe manner? NOTE: Periodic verification of centrifuge operating speeds is not required if the centrifuge is used only for phase-separation in extraction procedures. For centrifuges having a safety mechanism preventing the opening of the lid while in operation, the checks of rpm should be performed only by an authorized service representative of the manufacturer or an appropriately trained clinical engineer. COMMENTARY: N/A ................................................................. Analytic Balances ................................................................. LSV.35610 Phase I N/A YES NO Are balances cleaned, serviced and checked periodically only by qualified service personnel (i.e., service contract or as needed)? COMMENTARY: N/A LSV.35640 Phase I N/A YES NO Are analytic balances mounted such that vibrations do not interfere with readings? COMMENTARY:

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 30 of 178
N/A LSV.35670 Phase II N/A YES NO Are standard weights of the appropriate ANSI/ASTM Class available and used for checking accuracy? NOTE: The verification of accuracy of the analytical balance must be performed on a regular schedule to ensure accurate creation of analytical calibrators and/or weighed-in controls from standard materials, as well as when gravimetrically checking the accuracy of pipettes. There are three general types of balances in use. First, many contemporary balance designs use force transducers of various designs to provide mass readings. These balances typically have built-in certified calibration weights that are utilized automatically each time of use. The second type of balance employs a force transducer design that uses external weights for calibration each time the balance is used. Typically a single mass at the maximum weighing range, in conjunction with a zero point for the pan, is used for calibration of a force transducer balance design. The third type of balance, an older design, is a mechanical balance beam with internal moveable or external calibration weights. This design may have an electronic read-out. In all cases, verification of accuracy over the weighing range with external calibrated masses is required on a periodic schedule appropriate to the use of the balance. Balances must be checked at least every 6 months if used for weighing out materials to make up standard solutions for method calibration. For other purposes, annual verification may be adequate. Accuracy must be verified when a new balance is installed and whenever a balance is moved. External validation of accuracy requires the appropriate class of ASTM specification weights. ASTM Class 1 weights are appropriate for calibrating high precision analytical balances (0.01 to 0.1 mg limit of precision). ASTM Class 2 weights are appropriate for calibrating precision top-loading balances (0.001 to 0.01 g precision).w ASTM Class 3 weights are appropriate for calibrating moderate precision balances, (0.01 to 0.1 g precision). Periodic external validation of accuracy is required to ensure that internal weights have not deteriorated from adsorption of surface film or corrosion; and to ensure that electronics remain correctly calibrated. COMMENTARY: N/A REFERENCES: 1) American Society for Testing and Materials. Standard specification for laboratory weights and precision mass standards, designation E 617-97 (Reapproved 2003). ASTM International, West Conshohocken, PA 19428-2959 (www.astm.org; 2) American Society for Testing and Materials. E 898-88 (Reapproved 2000). Standard method of testing top-loading, direct reading laboratory scales and balances. ASTM International, West Conshohocken, PA 19428-2959 (www.astm.org).

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 31 of 178
LSV.35685 Phase II N/A YES NO Are results of periodic accuracy checks recorded? NOTE: Mass readings should be recorded in a log book. The deviations in log book readings should be no more than the precision required in the applications for which the balance is used. Acceptable ranges for readings should be specified. COMMENTARY: N/A LSV.35700 Phase II N/A YES NO Are weights well-maintained (clean, in a covered container, not corroded) and are appropriate lifting or handling devices available? NOTE: Weights must be well-maintained (covered when not in use, not corroded) and only be handled by devices that will not allow residual contaminants to remain on the masses. Certified masses will only meet their specifications if maintained in pristine condition. COMMENTARY: N/A ................................................................ Multiple Analysis Automated Instruments and Systems ................................................................. A variety of instruments and equipment are used to support the performance of analytical procedures. All instruments and equipment should be properly operated, maintained, serviced, and monitored to ensure that malfunctions of these instruments and equipment do not adversely affect the analytical results. The inspection team should review the procedures for instrument/equipment operations, maintenance, and monitoring records to ensure that these devices are properly used. The procedures and schedules for instrument maintenance must be as thorough and as frequent as specified by the manufacturer. LSV.35900 Phase II N/A YES NO Are there documented standard procedures for setup, operation, and shutdown of instruments?

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 32 of 178
NOTE: These procedures must be available to the operator. COMMENTARY: N/A ................................................................. Colorimeters and Spectrophotometers ................................................................. LSV.36560 Phase II N/A YES NO Are absorbance and linearity checked periodically with filters or standard solutions, if required by the instrument manufacturer? COMMENTARY: N/A LSV.36600 Phase II N/A YES NO Are filters (filter photometers) clean, free of scratches and in good condition (not deteriorated)? COMMENTARY: N/A LSV.36620 Phase II N/A YES NO Is spectrophotometer wavelength calibration checked regularly with appropriate solutions, filters or emission line source lamps (if required by the manufacturer), and are the results documented? COMMENTARY: N/A LSV.36640 Phase II N/A YES NO Is stray light checked periodically with extinction filters or appropriate solutions, if required by the manufacturer?

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 33 of 178
COMMENTARY: N/A LSV.36660 Phase II N/A YES NO For procedures using calibration curves, are all the curves rerun regularly and/or verified after servicing or recalibration of instruments? COMMENTARY: N/A -------------------------------------------------------- Instrument Maintenance -------------------------------------------------------- Questions in this section apply to ALL instruments in the limited service laboratory. The procedures and schedules for instrument maintenance must be as thorough and as frequent as specified by the manufacturer. LSV.36680 Phase II N/A YES NO Is there a schedule and system AVAILABLE AT THE INSTRUMENT for the regular checking of the critical operating characteristics for all instruments in use? NOTE: This must include, but is not limited to electronic, mechanical, and operational checks. The procedure and schedule must be as thorough and as frequent as specified by the manufacturer. Function checks should be designed to check the critical operating characteristics to detect drift, instability, or malfunction, before the problem is allowed to affect test results. All servicing and repairs should be documented. COMMENTARY: N/A LSV.36720 Phase II N/A YES NO Are instrument function checks documented BY THE TECHNICAL OPERATOR and readily available to detect trends or malfunctions?

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 34 of 178
COMMENTARY: N/A LSV.36740 Phase II N/A YES NO Are tolerance limits for acceptable function documented for specific instruments wherever appropriate? COMMENTARY: N/A LSV.36760 Phase II N/A YES NO Are instructions provided for minor troubleshooting and repairs of instruments (such as manufacturer's service manual)? COMMENTARY: N/A LSV.36780 Phase II N/A YES NO Are records maintained AT OR NEAR each instrument to document all repairs and service procedures? NOTE: A complete record of date when placed in service, serial number, and all repairs and routine service procedures must be maintained for all instruments at or near each instrument. Effective utilization of instruments by the technical staff depends upon the prompt availability of maintenance, repair, and service documentation (copies are acceptable). Laboratory personnel are responsible for the reliability and proper function of their instruments and must have access to this information. Off-site storage, such as with centralized medical maintenance or computer files, is not precluded if the inspector is satisfied that the records can be promptly retrieved. COMMENTARY: N/A

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 35 of 178
-------------------------------------------------------- REAGENTS -------------------------------------------------------- **NEW** 10/31/2006 LSV.36815 Phase II N/A YES NO For all waived tests, does the laboratory follow manufacturer instructions for handling and storing reagents? COMMENTARY: N/A The remaining checklist questions in the Reagents section do not apply to waived tests. The verification of reagent performance is required and must be documented. Any of several methods may be appropriate, such as direct analysis with reference materials, parallel testing of old vs. new reagents, or checking against routine controls. The intent of the questions is for new reagents to be checked by an appropriate method and the results recorded before being placed in service. Where individually packaged reagents/kits are used, there should be criteria established for monitoring reagent quality and stability, based on volume of usage and storage requirements. Processing of periodic "wet controls" to validate reagent quality and operator technique is a typical component of such a system. LSV.36820 Phase II N/A YES NO Are reagents and solutions properly labeled, as applicable and appropriate, with the following elements?
1. Content and quantity, concentration or titer 2. Storage requirements 3. Date prepared or reconstituted by laboratory 4. Expiration date
NOTE: The above elements may be recorded in a log (paper or electronic), rather than on the containers themselves, providing that all containers are identified so as to be traceable to the appropriate data in the log. While useful for inventory management, labeling with "date received" is not routinely required. There is no requirement to routinely label individual containers with "date opened"; however, a new expiration date must be recorded if opening the container changes the expiration date, storage requirement, etc. The inspector will describe specific issues of non-compliance in the Inspector's Summation Report.

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 36 of 178
COMMENTARY: N/A REFERENCES: 1) Department of Health and Human Services, Centers for Medicare and Medicaid Services. Clinical laboratory improvement amendments of 1988; final rule. Fed Register. 1992(Feb 28):7164 [42CFR493.1205(d)]; 2) Gonzales Y, Kampa IS. The effect of various storage environments on reagent strips. Lab Med. 1997;28:135-137; 3) Clinical and Laboratory Standards Institute (CLSI). Laboratory Documents: Development and Control; Approved Guideline—Fifth Edition. CLSI document GP2-A5 (ISBN 1-56238-600-X). Clinical and Laboratory Standards Institute, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 19087-1898 USA, 2006. LSV.36830 Phase II N/A YES NO Are all reagents stored as recommended by the manufacturer? NOTE: Reagents must be stored as recommended by the manufacturer to prevent environmentally-induced alterations that could affect test performance. If ambient storage temperature is indicated, there must be documentation that the defined ambient temperature is maintained and corrective action taken when tolerance limits are exceeded. COMMENTARY: N/A REFERENCE: Gonzales Y, Kampa IS. The effect of various storage environments on reagent strips. Lab Med. 1997;28:135-137. LSV.36840 Phase II N/A YES NO Are all reagents used within their indicated expiration date? NOTE: The laboratory must assign an expiration date to any reagents that do not have a manufacturer-provided expiration date. The assigned expiration date should be based on known stability, frequency of use, storage conditions, and risk of deterioration. COMMENTARY: N/A REFERENCE: Department of Health and Human Services, Centers for Medicare and Medicaid Services. Clinical laboratory improvement amendments of 1988; final rule. Fed Register. 1992(Feb 28):7164 [42CFR493.1205(e)(1)].

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 37 of 178
LSV.36850 Phase I N/A YES NO Are common interferences evaluated for all analytes measured with each reagent system, or is credible information available? NOTE: Common interferences should be documented for each analyte measured with each reagent system. Documentation from information in the literature is acceptable as long as the information is specific for the reagent system in use in the laboratory. COMMENTARY: N/A REFERENCES: 1) Letellier G, Dejarlais F. Analytic interference of drugs in clinical chemistry. 1: Study of twenty drugs on seven different instruments. Clin Biochem. 1985;18:345-351; 2) Clinical and Laboratory Standards Institute (CLSI). Interference Testing in Clinical Chemistry; Approved Guideline—Second Edition. CLSI document EP7-A2 (ISBN 1-56238-584-4). Clinical and Laboratory Standards Institute, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 19087-1898 USA, 2005; 3) Kroll MH, Elin RJ. Interference with clinical laboratory analyses. Clin Chem. 1994;40:1996-2005; 4) Kazmierczak SC, Catrou PG. Analytical interference. More than just a laboratory problem. Am J Clin Pathol. 2000;113:9-11; 5) Tang Z, et al. Effects of drugs on glucose measurements with handheld glucose meters and a portable glucose analyzer. Am J Clin Pathol. 2000;113:75-80; 6) Tang Z, et al. Effects of pH on glucose measurements with handheld glucose meters and a portable glucose analyzer for point-of-care testing. Arch Pathol Lab Med. 2000;124:577-582; 7) Young DS. Effects of drugs on clinical laboratory tests, 5th ed. Washington, DC: American Association for Clinical Chemistry, 2000. LSV.36860 Phase II N/A YES NO If there are multiple components of a reagent kit, does the laboratory use components of reagent kits only within the kit lot unless otherwise specified by the manufacturer? COMMENTARY: N/A LSV.36900 Phase II N/A YES NO Are new reagent lots and/or shipments validated before or concurrent with use for patient testing?

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 38 of 178
NOTE: New reagent lots and/or shipments must be tested in parallel with old lots before or concurrently with being placed in service to ensure that the calibration of the new lot of reagent has maintained consistent results for patient specimens. Good clinical laboratory science includes patient-based comparisons when possible, since it is patient specimens that are tested. For quantitative tests, reagent validation is most reliably performed by assaying the same patient specimens with both the old and new lots to ensure consistent results. For qualitative tests, minimum cross-checking includes retesting at least one known positive and one known negative patient sample from the old reagent lot against the new reagent lot, ensuring that the same results are obtained with the new lot. A weakly positive sample should also be used in systems where patient results are reported in that fashion. Some method manufacturers provide reference materials or QC products specifically intended to validate successful calibration of their methods; these should be used when available. Such materials have method-specific, and, where appropriate, reagent-lot-specific, target values. Thus, these materials should be used only with the intended methods. Proficiency testing materials with peer group established mean values are acceptable for validation of new reagent lots. Third party general purpose reference materials may be suitable for validation of calibration following reagent lot changes if the material is documented in the package insert or by the method manufacturer to be commutable with patient specimens for the method. A commutable reference material is one that gives the same numeric result as would a patient specimen containing the same quantity of analyte in the analytic method under discussion; e.g., matrix effects are absent. Commutability between a reference material and patient specimens can be demonstrated using the protocol in NCCLS EP14-A2. QC material may be an acceptable alternative for validating calibration following reagent lot changes. However, the laboratory should be aware that QC material may be affected by matrix interference between different reagent lots. Thus, even if QC results show no change following a reagent lot change, a calibration inconsistency for patient specimens could exist nonetheless, masked by matrix interference affecting the QC material (a “false negative” validation). The use of patient samples to confirm the absence of matrix interference may be helpful. It is acceptable to use QC material alone to check a new shipment of a reagent lot currently in use, as there should be no change in potential matrix interactions between QC material and different shipments of the same lot number of reagent. COMMENTARY: N/A REFERENCE: Clinical and Laboratory Standards Institute. Evaluation of Matrix Effects; Approved Guideline—Second Edition. CLSI document EP14-A2 (ISBN 1-56238-561-5). Clinical and Laboratory Standards Institute, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 190871898, USA, 2005.

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 39 of 178
-------------------------------------------------------- RESULTS REPORTING -------------------------------------------------------- LSV.36920 Phase II N/A YES NO Are reference intervals (normal ranges) established or verified by the laboratory for the population being tested? NOTE: Age- and sex-specific reference intervals (normal values) must be verified or established by the laboratory. For example, a reference interval can be validated by testing samples from 20 healthy representative individuals; if no more than 2 results fall outside the proposed reference interval, that interval can be considered validated for the population studied (refer to CLSI guideline C28, referenced below). If a formal reference interval study is not possible or practical, then the laboratory should carefully evaluate the use of published data for its own reference ranges, and retain documentation of this evaluation. COMMENTARY: N/A REFERENCES: 1) Fulwood R, et al. Hematological and nutritional biochemistry reference data for persons 6 months-74 years of age: United States, 1976-80. Hyattsville, MD: Dept Health and Human Services pub no (PHS) 83-1682; 2) Department of Health and Human Services, Centers for Medicare and Medicaid Services. Medicare, Medicaid and CLIA programs; CLIA fee collection; correction and final rule. Fed Register. 1992(Jan 19):5231 [42CFR493.1213(b)(2)(i)(F)]; 3) Hicks JM, et al. Vitamin B12 and folate. Pediatric reference ranges. Arch Pathol Lab Med. 1993;117:704-706; 4) Knight JA. Laboratory issues regarding geriatric patients. Lab Med. 1997;28:458-461; 5) Slovacek KJ, et al. Use of age-specific normal ranges for serum prostate-specific antigen. Arch Pathol Lab Med. 1998;122:330-332; 6) Soldin S, Hicks J (eds). Pediatric reference ranges, 3rd edition. Washington, DC: American Association of Clinical Chemistry Press, 1999; 7) NCCLS. How to Define and Determine Reference Intervals in the Clinical Laboratory; Approved Guideline—Second Edition. NCCLS document C28-A2 (ISBN 1-56238-406-6). NCCLS, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 19087-1898, USA 2000; 8) Katzmann JA, et al. Serum reference intervals and diagnostic ranges for free κ and free λ immunoglobulin light chains: relative sensitivity for detection of monoclonal light chains. Clin Chem. 2002;48:1437-1444; 9) Horn PS, Pesce AJ. Effect of ethnicity on reference intervals. Clin Chem. 2002;48:1802-1804; 10) Lahti A, et al Impact of subgroup prevalence on partitioning of Gaussian-distributed reference values. Clin Chem. 2002;48:1987-1999.

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 40 of 178
**REVISED** 09/27/2007 LSV.36940 Phase II N/A YES NO Are all patient/client results reported with reference (normal) intervals or interpretations as appropriate? NOTE: The laboratory must report reference (normal) intervals or interpretations with patient/client results, where such exist. This is important to allow proper interpretation of patient/client data. In addition, the use of high and low flags (generally available with a computerized laboratory information system) is recommended. For urine testing for drugs of abuse, the cut-off value for positive results should be listed, either in the report or in a separate chart/memorandum that is available to clinicians. Under some circumstances it may be appropriate to distribute lists or tables of reference intervals to all users and sites where reports are received. This system is usually fraught with difficulties, but if in place and rigidly controlled, it is acceptable. The results of commercial quality control plasmas that may be used in coagulation assays are internal data for quality assurance purposes, and must NOT be externally reported; if reported with patient results, they may be confused as normal values. COMMENTARY: N/A REFERENCE: Department of Health and Human Services, Centers for Medicare and Medicaid Services. Clinical laboratory improvement amendments of 1988; final rule. Fed Register. 1992(Feb 28):7162 [42CFR493.1109(d)]. LSV.37020 Phase II N/A YES NO Are documented criteria established for immediate notification of a physician or other clinical personnel responsible for patient care when the results of certain tests exceed critical limits important for prompt patient management decisions? NOTE: Criteria for immediate notification of a physician or other clinical personnel responsible for patient care must be established for critical tests (glucose, potassium, calcium, etc.). These criteria may be indicated either in the procedure manual or in a separate policy manual. The bench technologists must be familiar with critical limits for procedures that they perform. COMMENTARY: N/A

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 41 of 178
REFERENCES: 1) Kost GJ. Critical limits for urgent clinician notification at US medical centers. JAMA. 1990;263:704-707; 2) Department of Health and Human Services, Centers for Medicare and Medicaid Services. Clinical laboratory improvement amendments of 1988; final rule. Fed Register. 1992(Feb 28):[42CFR493.1109(f)]; 3) Steindel SJ, Heard NV. Critical values: data analysis and critique. Q-Probes 92-04. Northfield, IL: College of American Pathologists, 1992; 4) Kost GJ. Using critical limits to improve patient outcome. Med Lab Observ. 1993;25(3):22-27; 5) Tate KE, Gardner RM. Computers, quality, and the clinical laboratory: a look at critical values. Proc Annu Symp Comput Appl Med Care. 1993;193-197; 6) Kaufman HW, Collins C. Notifying clients of life-threatening results. Med Lab Observ. 1994;26(8):44-45; 7) Lum G. Evaluation of a laboratory critical limit (alert value) policy for hypercalcemia. Arch Pathol Lab Med. 1996;120:633-636; 8) Emancipator K. Critical values. ASCP practice parameter. Am J Clin Pathol. 1997:108:247-253; 9) Howanitz PJ, Cembrowski GS. Postanalytical quality improvement. A College of American Pathologists Q-Probes study of elevated calcium results in 525 institutions. Arch Pathol Lab Med. 2000;124:504-510; 10) Dalton-Beninato K. Critical value notifications are never welcome news. Lab Med. 2000;31:319-323; 11) Howanitz PJ, et al. Laboratory critical values policies and procedures. A College of American Pathologists Q-probes study in 623 institutions. Arch Pathol Lab Med. 2002;126:663-669; 12) Hortin GL, et al. Intraoperative parathyroid hormone testing. Survey of testing program characteristics. Arch Pathol Lab Med. 2002;126:1045-1049. LSV.37040 Phase II N/A YES NO Is there documentation of prompt notification of the physician (or other clinical personnel responsible for patient care) of results of all critical results? NOTE: Records must be maintained indicating the prompt notification of the appropriate clinical individual after observing results in the critical range. These records should include: date, time, responsible laboratory individual, person notified and test results. In addition, the laboratory should document any failure of attempts to notify the appropriate person of critical results, and document the action taken to prevent recurrence of this problem. COMMENTARY: N/A REFERENCES: 1) Kost GJ. Critical limits for urgent clinician notification at US medical centers. JAMA. 1990;263:704-707; 2) Department of Health and Human Services, Centers for Medicare and Medicaid Services. Clinical laboratory improvement amendments of 1988; final rule. Fed Register. 1992(Feb 28):[42CFR493.1109(f)]; 3) Steindel SJ, Heard NV. Critical values: data analysis and critique. Q-Probes 92-04. Northfield, IL: College of American Pathologists, 1992; 4) Kost GJ. Using critical limits to improve patient outcome. Med Lab Observ. 1993;25(3):22-27; 5) Dalton-Beninato K. Critical value notifications are never welcome news. Lab Med. 2000;31:319-323; 6) Howanitz PJ, et al. Laboratory critical values policies and procedures. A College of American Pathologists Q-probes study in 623 institutions. Arch Pathol Lab Med. 2002;126:663-669.

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 42 of 178
LSV.37050 Phase II N/A YES NO Are routine and STAT results available within a reasonable time? NOTE: A reasonable time for routine daily service, assuming receipt or collection of specimen in the morning is 4 to 8 hours. Emergency or STAT results that do not require additional verification procedures should be reported within 1 hour after specimen receipt in the laboratory. COMMENTARY: N/A REFERENCES: 1) Howanitz PJ, et al. Timeliness of urinalysis. A College of American Pathologists Q-Probes study of 346 small hospitals. Arch Pathol Lab Med. 1997;121:667-672 ; 2) Steindel SJ, Novis DA. Using outlier events to monitor test turnaround time. A College of American Pathologists Q-Probes study in 496 laboratories. Arch Pathol Lab Med. 1999;123:607-614; 3) Manor PG. Turnaround times in the laboratory: a review of the literature. Clin Lab Sci. 1999;12(2):85-89. **REVISED** 10/31/2006 LSV.37060 Phase II N/A YES NO Are there documented protocols for the reporting of toxicology results? NOTE: In addition to the requirements found in the Laboratory General Checklist, the following information must be included in toxicology reports:
1. If appropriate, substances or classes of substances analyzed as part of the toxicology test
2. Specimen type 3. Report status for positive results (i.e., unconfirmed, confirmed or pending confirmation) 4. For immunoassays, the assay cut-off concentration for each drug* 5. If the report includes unconfirmed screening results, a statement that such results are to
be used only for medical (i.e., treatment) purposes. (Unconfirmed screening results must not be used for non-medical purposes, e.g., employment testing, legal testing.)
*The cut-off concentrations may either be included in the report or in a separate chart/memorandum available to clinicians. COMMENTARY: N/A

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 43 of 178
-------------------------------------------------------- QUALITY CONTROL AND CALIBRATION – WAIVED TESTS -------------------------------------------------------- **NEW** 10/31/2006 LSV.37064 Phase II N/A YES NO Do testing personnel follow manufacturer’s instructions for quality control, calibration, calibration verification, and related functions? NOTE: In addition to the above checklist item, the following three checklist questions apply to quality control for waived tests. COMMENTARY: N/A **NEW** 10/31/2006 LSV.37068 Phase II N/A YES NO Are control results documented for quantitative and qualitative tests, as applicable? NOTE: To detect problems and evaluate trends, testing personnel or supervisory staff must review quality control data on days when controls are run. The laboratory director or designee must review QC data at least monthly. Because of the many variables across laboratories, the CAP makes no specific recommendations on the frequency of any additional review of QC data. Internal control results need not be documented for instruments using such controls, if (and only if) an unacceptable instrument control automatically locks the instrument and prevents release of patient results. COMMENTARY: N/A **NEW** 10/31/2006 LSV.37072 Phase II N/A YES NO Is there evidence of corrective action when control results exceed defined acceptability limits?

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 44 of 178
COMMENTARY: N/A **NEW** 10/31/2006 LSV.37076 Phase II N/A YES NO Are the results of controls verified for acceptability before reporting results? COMMENTARY: N/A ======================================================================== NOTE: The remaining questions in this checklist on CONTROLS, CALIBRATION, CALIBRATION VERIFICATION, AMR VALIDATION and INTERINSTRUMENT COMPARISONS do not apply to waived tests. Also, the section “RESULTS REPORTING-COAGULATION” is not applicable to waived tests. ========================================================================
*****************************************************************************
HEMATOLOGY AND COAGULATION SECTION
***************************************************************************** -------------------------------------------------------- SPECIMEN COLLECTION AND HANDLING -------------------------------------------------------- LSV.37080 Phase II N/A YES NO Are all blood specimens collected in anticoagulant for hematology testing mixed thoroughly immediately before analysis? NOTE: There must be documentation that specimen mixing by rotary mixer, rocker, automated sampler, or manual inversions is sufficient to ensure reproducibility of CBC results. Some rocking platforms may be adequate to maintain even cellular distribution of previously well-mixed specimens, but are incapable of fully mixing a settled specimen. For instruments with automated samplers, the laboratory must ensure that the automated mixing time is sufficient to homogeneously disperse the cells in a settled specimen.

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 45 of 178
COMMENTARY: N/A REFERENCES: 1) NCCLS. Procedures and Devices for the Collection of Diagnostic Capillary Blood Specimens; Approved Standard—Fifth Edition. NCCLS document H4-A5 (ISBN 1-56238-538-0). NCCLS, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 19087-1898 USA, 2004; 2) NCCLS. Procedures for the Collection of Diagnostic Blood Specimens by Venipuncture; Approved Standard—Fifth Edition. NCCLS document H3-A5 (ISBN 1-56238-515-1). NCCLS, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 19087-1898 USA, 2003. LSV.37100 Phase II N/A YES NO Are samples for complete blood counts and blood film morphology collected in potassium EDTA? NOTE: Blood specimens for routine hematology tests (e.g., CBC, leukocyte differential) must be collected in potassium EDTA to minimize changes in cell characteristics. Oxalate can cause unsuitable morphologic changes such as cytoplasmic vacuoles, cytoplasmic crystals, and irregular nuclear lobulation. Heparin can cause cellular clumping (especially of platelets), pseudoleukocytosis with pseudothrombocytopenia in some particle counters, and troublesome blue background in Wright-stained blood films. Citrate may be useful in some cases of platelet agglutination due to EDTA, but those CBC data will require adjustment for the effects of dilution. COMMENTARY: N/A REFERENCES: 1) Cohle SD, et al. Effects of storage of blood on stability of hematologic parameters. Am J Clin Pathol. 1981;76:67-79; 2) Savage RA. Pseudoleukocytosis due to EDTA-induced platelet clumping. Am J Clin Pathol. 1984;82:132-133; 3) Rabinovitch A. Anticoagulants, platelets and instrument problems. Am J Clin Pathol. 1984;82:132; 4) NCCLS. Procedures for the Collection of Diagnostic Blood Specimens by Venipuncture; Approved Standard—Fifth Edition. NCCLS document H3-A5 (ISBN 1-56238-515-1). NCCLS, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 19087-1898 USA, 2003; 5) NCCLS. Procedures and Devices for the Collection of Diagnostic Capillary Blood Specimens; Approved Standard—Fifth Edition. NCCLS document H4-A5 (ISBN 1-56238-538-0). NCCLS, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 19087-1898 USA, 2004; 6) NCCLS. Tubes and Additives for Venous Blood Specimen Collection; Approved Standard—Fifth Edition. NCCLS document H1-A5 (ISBN 1-56238-519-4). NCCLS, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 19087-1898 USA, 2003; 7) Broden PN. Anticoagulant and tube effect on selected blood cell parameters using Sysmex NE-series instruments. Sysmex J Intl. 1992;2:112-119; 8) Brunson D, et al. Comparing hematology anticoagulants: K2EDTA vs K3EDTA. Lab Hematol. 1995;1:112-119; 9) Boos MS, et al. Temperature- and storage-dependent changes in hematologic variable and peripheral blood morphology. Am J Clin Pathol. 1998;110:537; 10) Wood BL, et al.

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 46 of 178
Refrigerated storage improves the stability of the complete blood cell count and automated differential. Am J Clin Pathol. 1999;112:687-695. LSV.37110 Phase II N/A YES NO Are there documented criteria for the rejection of unacceptable specimens and the special handling of sub-optimal specimens? NOTE: This question does not imply that all "unsuitable" specimens are discarded or not analyzed. If, for example, a CBC is ordered and there is visible hemolysis, the hemoglobin concentration may still be valid, but other parameters are not. There must be a mechanism to notify clinical personnel responsible for patient care, and to note the condition of the sample on the report if the analytically valid incomplete test results are desired by the ordering physician. The laboratory should record that a dialogue was held with the physician, when such occurs. COMMENTARY: N/A REFERENCES: 1) Department of Health and Human Services, Centers for Medicare and Medicaid Services. Clinical laboratory improvement amendments of 1988; final rule. Fed Register. 2003(Jan 24):7183 [42CFR493. 1249(a) and (b)]; 2) Boos MS, et al. Temperature- and storage-dependent changes in hematologic variable and peripheral blood morphology. Am J Clin Pathol. 1998;110:537. LSV.37120 Phase II N/A YES NO Are samples collected in capillary tubes for microhematocrits or capillary/dilution systems obtained in duplicate whenever possible and adequately labeled with patient identification information throughout the analytic sequence? NOTE: This applies to capillary tubes used for microhematocrit determinations as well as capillary/dilution systems used in proximity to the patient. Microspecimen containers such as those used for capillary blood CBC parameter determinations need not be collected in duplicate. Because of the risk of injury, the use of plain glass capillary tubes is strongly discouraged. COMMENTARY: N/A LSV.37140 Phase II N/A YES NO Are CBC specimens checked for clots (visual, applicator sticks, or automated analyzer histogram inspection/flags) before reporting results?

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 47 of 178
NOTE: This may be done visually or with applicator sticks before testing. Additionally, microclots will often present themselves histographically on automated and semi-automated particle counters or by flagging, and the laboratory must become familiar with such patterns. Finally, platelet clumps or fibrin may be microscopically detected if a blood film is prepared on the same sample. COMMENTARY: N/A LSV.37160 Phase II N/A YES NO Are CBC specimens checked for significant in vitro hemolysis and possible interfering lipemia before reporting results? NOTE: Specimens for complete blood counts must be checked for in vitro hemolysis that may falsely lower the erythrocyte count and the hematocrit, as well as falsely increase the platelet concentration from erythrocyte stroma. Visibly red plasma in a tube of EDTA-anticoagulated settled or centrifuged blood should trigger an investigation of in vivo hemolysis (in which case the CBC data are valid) versus in vitro hemolysis (in which case some or all of the CBC data are not valid and should not be reported). Lipemia may adversely affect the hemoglobin concentration and the leukocyte count. This does not imply that every CBC specimen must be subjected to centrifugation with visual inspection of the plasma supernatant, particularly if this would significantly impair the laboratory's turnaround time. An acceptable alternative for high volume laboratories with automated instrumentation is to examine the numeric data for anomalous results (especially indices), as well as particle histogram inspection. COMMENTARY: N/A REFERENCE: Cantero M, et al. Interference from lipemia in cell count by hematology. Clin Chem. 1996;42:987-988. LSV.37190 Phase I N/A YES NO Has the laboratory clearly defined sample storage conditions and stability for all hematology parameters? NOTE: The laboratory should define sample storage conditions and stability for all hematology parameters, as time- and temperature-dependent alterations can occur, creating spurious results. COMMENTARY: N/A

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 48 of 178
REFERENCES: 1) Boos MS, et al. Temperature- and storage-dependent changes in hematologic variable and peripheral blood morphology. Am J Clin Pathol. 1998;110:537; 2) Gulati GL, et al. Changes in automated complete blood cell count and differential leukocyte count results induced by storage of blood at room temperature. Arch Pathol Lab Med. 2002;126:336-342. ----------------------------------------------------------------- SPECIMEN COLLECTION AND HANDLING - COAGULATION ----------------------------------------------------------------- LSV.37192 Phase I N/A YES NO Is there a documented policy regarding clearing (flushing) of the volume of intravenous lines before drawing samples for hemostasis testing? NOTE: Collection of blood for coagulation testing through intravenous lines that have been previously flushed with heparin should be avoided, if possible. If the blood must be drawn through an indwelling catheter, possible heparin contamination and specimen dilution should be considered. When obtaining specimens from indwelling lines that may be contain heparin, the line should be flushed with 5 mL of saline, and the first 5 mL of blood or 6-times the line volume (dead space volume of the catheter) be drawn off and discarded before the coagulation tube is filled. COMMENTARY: N/A REFERENCES: 1) Lew JKL, et al. Intra-arterial blood sampling for clotting studies. Effects of heparin contamination. Anesthesia. 1991;46:719-721; 2) Konopad E, et al. Comparison of PT and aPTT values drawn by venipuncture and arterial line using three discard volumes. Am J Crit Care. 1992;3:94-101; 3) Konopad E, et al. Comparison of PT and aPTT values drawn by venipuncture and arterial line using three discard volumes. Am J Crit Care. 1992;3:94-101; 4) Laxson CJ, Titler MG. Drawing coagulation studies from arterial lines; an integrative literature review. Am J Critical Care. 1994; 1:16-24; 5) Adcock DM, et al. Are discard tubes necessary in coagulation studies? Lab Med. 1997;28:530-533; 6) Brigden ML, et al. Prothrombin time determination. The lack of need for a discard tube and 24-hour stability. Lab Med. 1997;108:422-426; 7) NCCLS. Collection, Transport, and Processing of Blood Specimens for Testing Plasma-Based Coagulation Assays; Approved Guideline—Fourth Edition. NCCLS document H21-A4 (ISBN 1-56238-521-6). NCCLS, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 19087-1898 USA, 2003; 8) NCCLS. Procedures for the Collection of Diagnostic Blood Specimens by Venipuncture; Approved Standard—Fifth Edition. NCCLS document H3-A5 (ISBN 1-56238-515-1). NCCLS, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 19087-1898 USA, 2003.

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 49 of 178
LSV.37193 Phase I N/A YES NO Are all coagulation specimens collected into 3.2% buffered sodium citrate? NOTE: Sodium citrate is effective as an anticoagulant due to its mild calcium-chelating properties. Of the 2 commercially available forms of citrate, 3.2% buffered sodium citrate (109 mmol/L of the dihydrate form of trisodium citrate Na3C6H5O7·2H2O) is the recommended anticoagulant for coagulation testing. The citrate concentration in 3.8% sodium citrate is higher and its use may result in falsely lengthened clotting times with calcium-dependent coagulation tests (i.e., PT and aPTT) with slightly underfilled samples and with samples with high hematocrits. If the laboratory does not adhere to recommendations for use of 3.2% buffered sodium citrate, it must have data on file to demonstrate that the alternative citrate concentration produces accurate and precise coagulation results. Coagulation testing cannot be performed in samples collected in EDTA due to the more potent calcium chelation. Heparinized tubes are not appropriate due to the inhibitory effect of heparin on multiple coagulation proteins. COMMENTARY: N/A REFERENCES: 1) Adcock DM, et al. Effect of 3.2% vs 3.8% sodium citrate concentration on routine coagulation testing. Am J Clin Pathol. 1997;107:105-110; 2) Reneke, J et al. Prolonged prothrombin time and activated partial thromboplastin time due to underfilled specimen tubes with 109 mmol/L (3.2%) citrate anticoagulant. Am J Clin Pathol. 1998;109:754-757; 3) NCCLS. Collection, Transport, and Processing of Blood Specimens for Testing Plasma-Based Coagulation Assays; Approved Guideline—Fourth Edition. NCCLS document H21-A4 (ISBN 1-56238-521-6). NCCLS, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 19087-1898 USA, 2003. LSV.37194 Phase I N/A YES NO Are there documented guidelines for rejection of under- or overfilled collection tubes? NOTE: The recommended proportion of blood to the sodium citrate anticoagulant volume is 9:1. Inadequate filling of the collection device will decrease this ratio, and may lead to inaccurate results for calcium-dependent clotting tests, such as the PT and aPTT. Conversely over-filling of the collection device will increase this ratio and may lead to clotted samples or inaccurate results. COMMENTARY: N/A REFERENCES: 1) Peterson P, Gottfried EL. The effects of inaccurate blood sample volume on prothrombin time (PT) and activated partial thromboplastin time. Thromb Haemost. 1982;47:101-103; 2) Adcock DM, et al. Effect of 3.2% vs 3.8% sodium citrate concentration on routine coagulation testing. Am J Clin Pathol. 1997;107:105-110; 3) Reneke J, et al. Prolonged prothrombin time and

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 50 of 178
activated partial thromboplastin time due to underfilled specimen tubes with 109 mmol/L (3.2%) citrate anticoagulant. Am J Clin Pathol. 1998;109:754-757; 4) NCCLS. Collection, Transport, and Processing of Blood Specimens for Testing Plasma-Based Coagulation Assays; Approved Guideline—Fourth Edition. NCCLS document H21-A4 (ISBN 1-56238-521-6). NCCLS, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 19087-1898 USA, 2003. LSV.37195 Phase I N/A YES NO Are there documented guidelines for detection and special handling of specimens with elevated hematocrits? NOTE: A hematocrit value >55% may lead to spurious coagulation results. The citrate anticoagulant distributes only in the plasma and not into the blood cells. For this reason, plasma citrate concentration will be increased if the patient’s hematocrit is greater than 55%, leading to prolonged PT and aPTT results, as well as erroneous results for other calcium-dependent clotting tests such as clottable protein C/protein S. Accordingly, there should be a documented procedure for detection and special handling of polycythemic specimens. If possible, a new phlebotomy should be performed, using a reduced volume of sodium citrate, adjusted for the elevated hematocrit. Conversely, there are no current data to support a recommendation for adjusting the citrate concentration in the presence of severe anemia (hematocrit <20%). COMMENTARY: N/A REFERENCES: 1) NCCLS. Collection, Transport, and Processing of Blood Specimens for Testing Plasma-Based Coagulation Assays; Approved Guideline—Fourth Edition. NCCLS document H21-A4 (ISBN 1-56238-521-6). NCCLS, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 19087-1898 USA, 2003; 2) Siegel JE, et al. Effect (or lack of it) of severe anemia on PT and APTT results. Am J Clin Pathol. 1998; 110:106-110; 3) Siegel JE, et al. Monitoring heparin therapy. APTT results from partial- vs full-draw tubes. Am J Clin Pathol. 1998;110:184-187. LSV.37196 Phase II N/A YES NO Are coagulation specimens checked for clots (visual, applicator sticks, or by analysis of testing results) before testing or reporting results? NOTE: Coagulation specimens must be checked for clots of any size. Clotted specimens will have extremely low levels of fibrinogen and variably decreased levels of other coagulation proteins, so that results of the PT, aPTT, fibrinogen and other coagulation assays will be inaccurate or unobtainable. Checking for clots may be done with applicator sticks or by visual inspection of centrifuged plasma for small clots. This may also be performed by analysis of results (waveform analysis or delta checks). Additionally, when a clot is not detected during PT and aPTT testing and, where the fibrinogen level is <25 mg/dl, it should be suspected that the sample is actually serum. This may be important when

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 51 of 178
coagulation specimens are received as centrifuged, frozen “plasma”. Centrifuged plasma and serum cannot be distinguished by visual inspection alone. There should be a mechanism in place to identify these specimens appropriately and/or to reject the sample as a probable serum sample. Laboratories should be encouraged to work with their clients that perform sample processing to ensure that they practice appropriate specimen handling for coagulation specimens. COMMENTARY: N/A REFERENCES: 1) NCCLS. Collection, Transport, and Processing of Blood Specimens for Testing Plasma-Based Coagulation Assays; Approved Guideline—Fourth Edition. NCCLS document H21-A4 (ISBN 1-56238-521-6). NCCLS, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 19087-1898 USA, 2003; 2) Arkin CF. Collection, handling, storage of coagulation specimens. Advance/Lab. 2002;11(1);33-38. LSV.37197 Phase II N/A YES NO Are coagulation tests promptly performed on fresh plasma, or is the platelet-poor plasma frozen until testing can be performed? NOTE: After blood collection, there is progressive degradation of the labile coagulation factors V and VIII, leading to increasing prolongation of the aPTT and PT. The allowable time interval between specimen collection and sample testing depends on the temperature encountered during transport and storage of the specimen. Optimally, for specimens that are uncentrifuged or centrifuged with plasma remaining on top of the cells in an unopened tube kept at 2-4oC or 18-24oC, the following guidelines should be observed:
1. Prothrombin time – 24 hours 2. aPTT – 4 hours 3. Other coagulation assays (e.g., thrombin time, protein Cfactor V and factor VIII) - 4
hours
If testing cannot be performed within these times, platelet-poor plasma should be removed from the cells and frozen at –20oC for up to 2 weeks or at –70oC for up to 6 months. A frost-free freezer should not be used, because of warming cycles. If a laboratory has established an allowable time interval different than that detailed above, data must be available to verify that coagulation testing is valid in the time interval established. COMMENTARY: N/A REFERENCES: 1) NCCLS. Collection, Transport, and Processing of Blood Specimens for Testing Plasma-Based Coagulation Assays; Approved Guideline—Fourth Edition. NCCLS document H21-A4

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 52 of 178
(ISBN 1-56238-521-6). NCCLS, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 19087-1898 USA, 2003; 2) Adcock DM, et al. The effect of time and temperature variables on routine coagulation tests. Blood Coag Fibrinolysis. 1998;9:463-470; 3) Neofotistos D, et al. Stability of plasma for add-on PT and aPTT tests. Am J Clin Pathol. 1998;109:758-763; 4) Davis KD, et al. Use of different thromboplastin reagents causes greater variability in international normalized ratio results than prolonged room temperature storage of specimens. Arch Pathol Lab Med. 1998;122:972-977. LSV.37198 Phase II N/A YES NO Are platelet functional studies (platelet aggregation or initial platelet function test) performed within an appropriate period after venipuncture? NOTE: Following venipuncture, platelets continue to activate in vitro, so that platelet functionality becomes abnormal after a period of approximately 4 hours. The laboratory should ensure that platelet aggregation studies are completed within 4 hours from the time of phlebotomy, or erroneous results could be obtained. COMMENTARY: N/A REFERENCE: Triplett DA, et al. Platelet function: laboratory evaluation and clinical application. Chicago, IL: American Society for Clinical Pathology, 1978. -------------------------------------------------------- AUTOMATED AND SEMI-AUTOMATED HEMATOLOGY CBC SYSTEMS -------------------------------------------------------- ................................................................. CBC Instrument Calibration ................................................................. Several different methods may be used for calibration and longitudinal process control of the automated Complete Blood Count (CBC) instrument. The laboratory should have a document that describes in detail the procedures for calibration and calibration verification. Calibration techniques include: A) the use of multiple analyzed fresh whole blood specimens, and B) the use of manufactured, stabilized preparations of red cells, white cells (or white cell surrogates) and platelets (or platelet surrogates). Typically, a laboratory uses one of these two approaches as their primary calibration technique, with the other for backup, or for verification of the primary method, or on an emergency basis.

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 53 of 178
When stabilized whole blood or other commercial preparations are used for the periodic recalibration of automated instruments, the target values for the measured parameters must have been assigned by using primary reference procedures. The laboratory may assign such values or the manufacturer may certify that the target values were derived through primary reference procedures. All calibration techniques should include periodic verifications of analyzer hemoglobin measurements against a certified hemoglobin preparation (ICSH/WHO international haemiglobincyanide standard), or material that has been certified by its manufacturer as being derived from the certified international haemiglobincyanide standard using reference procedures. Ordinary commercial control materials are not suitable for instrument calibration. LSV.37200 Phase II N/A YES NO If precalibrated instruments are used, are the manufacturer's calibrations verified with appropriate control materials for the system? NOTE: This question does not apply to CBC instruments that can be calibrated by the laboratory. COMMENTARY: N/A REFERENCE: van Assendelft OW, Buursma A. Reference method for the measurement of hemoglobin. Lab Hematol. 1995;1:154-155. ................................................................. Fresh Whole Blood ................................................................. LSV.37240 Phase II N/A YES NO Is there a document defining the specific procedural steps for the periodic calibration of the analyzer with fresh whole blood specimens? NOTE: The laboratory must have criteria that define when recalibration is necessary, based upon the data from the daily quality control system. COMMENTARY: N/A REFERENCE: NCCLS. Calibration and Quality Control of Automated Hematology Analyzers; Proposed Standard. NCCLS document H38-P [ISBN 1-56238-398-1]. NCCLS, 940 West Valley Road, Wayne, Pennsylvania 19087, U.S.A 1999.

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 54 of 178
LSV.37260 Phase II N/A YES NO Does the initial or primary instrument calibration include duplicate analysis of at least 10 fresh whole blood samples that have been analyzed by reference methods? NOTE: The exact number of whole blood samples and number of replicates may not be as specified above for all instruments. Manufacturers' instructions should be followed. COMMENTARY: N/A REFERENCES: 1) International Committee for Standardization in Haematology' Expert Panel of Haemoglobinometry. Recommendations for reference method for haemoglobinometry in human blood (ICSH standard 1986) and specifications for international haemiglobincyanide reference preparation (3rd edition). Clin Lab Haemat. 1987;9:73-79; 2) Lewis SM, et al. Current concepts in haematology 3: blood count calibration. J Clin Pathol. 1991;144:881-884; 3) NCCLS. Procedure for Determining Packed Cell Volume by the Microhematocrit Method; Approved Standard—Third Edition. NCCLS document H7-A3 (ISBN 1-56238-413-9). NCCLS, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 19087-1898, USA 2000; 4) NCCLS. Reference and Selected Procedures for the Quantitative Determination of Hemoglobin in Blood; Approved Standard—Third Edition. NCCLS document H15-A3 (ISBN 1-56238-425-2). NCCLS, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 19087-1898 USA, 2000; 5) Dickerhoff R, von Ruecker A. Enumeration of platelets by multiparameter flow cytometric flow cytometry using platelet-specific antibodies and fluorescent reference particles. Clin Lab Haematol. 1995;17:163-172; 6) Houwen B, et al. The reference method for hemoglobin measurement revisited. Lab Hematol. 1996;2:86-93. LSV.37280 Phase II N/A YES NO Are criteria established for calibration verification? NOTE: Criteria must be established for calibration verification. Criteria include:
1. At complete changes of reagents (i.e., change in type of reagent from same vendor, or change to a different vendor)
2. When indicated by quality control data 3. After major maintenance or service 4. When recommended by the manufacturer 5. At least every six months
One common method of calibration verification involves processing a commercial calibrator and comparing results to those published by the manufacturer. Linearity studies are not required.

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 55 of 178
COMMENTARY: N/A REFERENCE: Department of Health and Human Services, Centers for Medicare and Medicaid Services. Clinical laboratory improvement amendments of 1988; final rule. Fed Register. 2003(Jan 24):7165 [42CFR493. 1255]. LSV.37300 Phase II N/A YES NO Does the laboratory's procedure for recalibration of CBC instrument parameter(s) require one of the following approaches?
1. Comparison to at least 10 fresh whole blood samples whose values have been determined by duplicate analysis in another instrument known to be accurately calibrated, or
2. Duplicate analysis of at least 10 fresh whole blood specimens by reference methods NOTE: The selection of 10 different blood samples (or alternate protocol recommended by the instrument manufacturer) is needed to accommodate a diversity of matrices, and to have the absolute minimum sample size on which to perform statistical calculations. Such fresh samples must have values within the instrument's operating ranges, and must not generate flags indicative of possible abnormalities. COMMENTARY: N/A REFERENCE: NCCLS. Calibration and Quality Control of Automated Hematology Analyzers; Proposed Standard. NCCLS document H38-P [ISBN 1-56238-398-1]. NCCLS, 940 West Valley Road, Wayne, Pennsylvania 19087, U.S.A 1999. LSV.37330 Phase I N/A YES NO Following whole blood calibration, is there a documented procedure for calibration verification? NOTE: Following whole blood calibration, the laboratory should have a documented procedure for verifying that whole blood calibration has been successful, i.e., that the accuracy of test results is established. COMMENTARY: N/A

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 56 of 178
REFERENCE: NCCLS. Calibration and Quality Control of Automated Hematology Analyzers; Proposed Standard. NCCLS document H38-P [ISBN 1-56238-398-1]. NCCLS, 940 West Valley Road, Wayne, Pennsylvania 19087, U.S.A 1999. ................................................................. Commercial Calibrators ................................................................. Commercially available calibrator materials represent a convenient way to ensure that CBC instruments yield accurate results. Because of differences in technology, such calibrators are typically instrument-specific, and are cleared by the Food and Drug Administration for such use. These calibrators have more rigorous assignment of target values than ordinary commercial QC materials, and the latter must not be used for routine instrument calibration. LSV.37340 Phase II N/A YES NO Is there a document defining the specific procedural steps for the periodic calibration of the analyzer with stabilized materials whose target values have been certified by the manufacturer using primary reference procedures? NOTE: The laboratory must have criteria that define when recalibration is necessary, based upon the data from the daily quality control system. COMMENTARY: N/A REFERENCES: 1) Gilmer PR, Williams LJ. The status of methods of calibration in hematology. Am J Clin Pathol. 1980;74:600-605; 2) Lewis SM, et al. Current concepts in haematology 3: blood count calibration. J Clin Pathol. 1991;144:881-884; 3) NCCLS. Calibration and Quality Control of Automated Hematology Analyzers; Proposed Standard. NCCLS document H38-P [ISBN 1-56238-398-1]. NCCLS, 940 West Valley Road, Wayne, Pennsylvania 19087, U.S.A 1999. LSV.37370 Phase II N/A YES NO Are criteria established for calibration verification? NOTE: Criteria must be established for calibration verification. Criteria include:
1. At complete changes of reagents (i.e., change in type of reagent from same vendor, or change to a different vendor)
2. When indicated by quality control data

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 57 of 178
3. After major maintenance or service 4. When recommended by the manufacturer 5. At least every six months
One common method of calibration verification involves processing a commercial calibrator and comparing results to those published by the manufacturer. Linearity studies are not required. COMMENTARY: N/A REFERENCE: Department of Health and Human Services, Centers for Medicare and Medicaid Services. Clinical laboratory improvement amendments of 1988; final rule. Fed Register. 2003(Jan 24):7165 [42CFR493. 1255]. LSV.37380 Phase II N/A YES NO Does the laboratory's procedure for recalibration of a parameter(s) require analysis of stabilized whole blood or other commercial preparations, the parameters of which have been certified by the manufacturer? COMMENTARY: N/A LSV.37390 Phase I N/A YES NO Following calibration with commercial calibrators, is there a documented procedure for calibration verification? COMMENTARY: N/A ................................................................. CBC INSTRUMENT QUALITY CONTROL ................................................................. Longitudinal process quality control (QC) procedures for individual instruments or inter-instrument comparisons may include:
1. Use of preserved or stabilized whole blood controls 2. "Moving average" monitoring

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 58 of 178
3. Retained patient specimens, or 4. Some combination of the above
For laboratories subject to CLIA-88, at least 2 different controls must be assayed and evaluated every 24 hours. For each QC procedure employed, the laboratory must have appropriate QC ranges. For example, expected recovery ranges for commercial control materials are NOT the same as between-run SD ranges, and are probably too wide for daily QC of a single instrument. The laboratory should calculate its own imprecision statistics for each instrument. ................................................................. Stabilized Controls ................................................................. LSV.37460 Phase II N/A YES NO Are 2 different stabilized control specimens analyzed and results recorded during each 24-hours of analyzer use? NOTE: Stabilized control materials must be at 2 different analytic levels (i.e., "normal" and "high"). Three levels of control is a conceptual carryover from clinical chemistry, and does not apply to hematology particle counting. Dilute, "low-level" (e.g., leukopenic and thrombocytopenic) "oncology" controls are less informative indicators of calibration status, and are neither required nor recommended. For example, a 10% calibration bias will be numerically most apparent in a high-level control, less apparent in a normal-level control, and perhaps inapparent in a low-level control; it would be quite extraordinary for a low-level control to indicate a calibration problem that is not revealed by the other controls. There should be some relationship between the frequency of control runs and the numbers of patient specimens processed. If the frequency of commercial control use is less than 2 per 24 hours, one or more of the additional approaches to QC must be employed to produce a total of at least 2 different data points per 24 hours. COMMENTARY: N/A REFERENCES: 1) Lott JA, et al. Synthetic materials for platelet quality control. Am J Med Technol. 1983;49:43-48; 2) Yacko M, et al. Multiple methods for platelet enumeration. Observation of a newly introduced bias. Am J Clin Pathol. 1987;87:109-112; 3) Department of Health and Human Services, Centers for Medicare and Medicaid Services. Clinical laboratory improvement amendments of 1988; final rule. Fed Register. 2003(Jan 24):7168 42CFR493. 1256(d)]; 4) Dotson MA. Methods to monitor and control systematic error. In: clinical hematology: principles, procedures, correlations, 2nd edition. Stiene-Martin EA, et al, eds. Philadelphia, PA: Lippincott, 1998:579-590; 5) Fink NE, et al. Evaluation and additional recommendations for preparing a whole blood control material. Rev Saude Publica. 1998;32:107-11; 6) Springer W, et al. Evaluation of a new reagent for preserving fresh blood

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 59 of 178
samples and its potential usefulness for internal quality control of multichannel hematology analyzers. Am J Clin Pathol. 1999;111:387-396. LSV.37480 Phase II N/A YES NO If commercially ASSAYED controls are used for CBC instruments, do control values correspond to the methodology and have target values (mean and QC ranges) been verified or established by the laboratory? NOTE: Most commercial controls have expected recovery ranges for each parameter, provided by the manufacturer. The mean of such ranges may not be the exact target value in a given laboratory. Each laboratory should assign its own initial target value, based on initial analysis of the material; this target value should fall within the recovery range supplied by the manufacturer, but need not exactly match the package insert mean. The laboratory should establish specific recovery ranges that accommodate known changes in product attributes, assuming that calibration status has not changed. COMMENTARY: N/A LSV.37500 Phase II N/A YES NO If UNASSAYED controls are used, has a statistically valid target mean, and range been established for each lot by repetitive analysis in runs that include previously tested control materials? COMMENTARY: N/A REFERENCES: 1) Fink NE, et al. Evaluation and additional recommendations for preparing a whole blood control material. Rev Saude Publica. 1998;32:107-111; 2) Springer W, et al. Evaluation of a new reagent for preserving fresh blood samples and its potential usefulness for internal quality control of multichannel hematology analyzers. Am J Clin Pathol. 1999;111:387-396. **NEW** 04/06/2006 LSV.37520 Phase II N/A YES NO Is there documentation of corrective action when control results exceed defined acceptability limits?

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 60 of 178
NOTE: Patient test results obtained in an analytically unacceptable test run or since the last acceptable test run must be re-evaluated to determine if there is a significant clinical difference in patient/client results. Re-evaluation may or may not include re-testing patient samples, depending on the circumstances. Even if patient samples are no longer available, test results can be re-evaluated to search for evidence of an out-of-control condition that might have affected patient results. For example, evaluation could include comparison of patient means for the run in question to historical patient means, and/or review of selected patient results against previous results to see if there are consistent biases (all results higher or lower currently than previously) for the test(s) in question). COMMENTARY: N/A REFERENCE: Department of Health and Human Services, Centers for Medicare and Medicaid Services. Clinical laboratory improvement amendments of 1988; final rule. Fed Register. 2003(Oct 1):1046[42CFR493.1282(b)(2)]. ................................................................. Moving Averages ................................................................. The technique of weighted moving averages (derived from multiple batch analysis of patient samples) is acceptably sensitive to drifts or shifts in analyzer calibration if a supplemental QC routine (stabilized control material or retained patient specimens) is employed. The latter is needed to detect random error and to avoid bias due to masking of drift by characteristics of the subpopulations within each individual batch. Laboratories analyzing fewer than 100 CBC specimens daily (long term average) should not use moving averages as the primary method for process control, as this would not generate sufficient data within a day to be of value. Depending on the particular instrument, there may be "on-board" moving average analyses for RBC indices only. In such cases, additional QC techniques are required for WBC, PLT and WBC differential parameters. However, some laboratories have found the mathematical logic of moving averages, modified average of normals, etc, applicable to other CBC parameters, and some instruments have these capabilities built into their software. Or, such calculations may be performed with an associated computer. LSV.37540 Phase II N/A YES NO Are control limits for moving averages appropriately sensitive?

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 61 of 178
NOTE: There must be documentation of the method used to establish the moving average, the frequency of calculation (batch size), and a definition of the basis for selection of upper and lower limits. Control limits for moving averages must be appropriately sensitive such that significant calibration alterations are always detected. Recalibration is not required for minor calibration variations of no clinical consequence. In other words, there should be a high probability for error detection and a low probability for false rejection. COMMENTARY: N/A REFERENCES: 1) Bull BS, et al. A study of various estimators for the derivation of quality control procedures from patient erythrocyte indices. Am J Clin Pathol. 1974;61:473-481; 2) Talamo TS, et al. Microcomputer assisted hematology quality control using a modified average of normals program. Am J Clin Pathol. 1981; 76:707-712; 3) Bull BS, Korpman RA. Autocalibration of hematology analyzers. J Clin Lab Automation. 1983;3:111-116; 4) Cembrowski GS, Westgard JO. Quality control of multichannel hematology analyzers: evaluation of Bull's algorithm. Am J Clin Pathol. 1985;83:337-345; 5) Bull BS, Hay KL. Are red blood cells indexes international? Arch Pathol Lab Med. 1985;109:604-606; 6) Levy WC, et al. Preserved blood versus patient data for quality control - Bull's algorithm revisited. Am J Clin Pathol. 1986;85:719-721; 7) Levy WC, et al. The incorporation of red blood cell index mean data into quality control programs. Am J Clin Pathol. 1986;86:193-199; 8) Lunetzky ES, Cembrowski GS. Performance characteristics of Bull's multirule algorithm for the quality control of multichannel hematology analyzers. Am J Clin Pathol. 1987;88:634-638; 9) NCCLS. Calibration and Quality Control of Automated Hematology Analyzers; Proposed Standard. NCCLS document H38-P [ISBN 1-56238-398-1]. NCCLS, 940 West Valley Road, Wayne, Pennsylvania 19087, U.S.A 1999. LSV.37600 Phase II N/A YES NO If a "moving averages" system is combined with another control system, (e.g., commercial controls or retained patient specimens) is the process well defined and appropriately sensitive to drift in analyzer calibration? COMMENTARY: N/A REFERENCES: 1) Bull BS, et al. A study of various estimators for the derivation of quality control procedures from patient erythrocyte indices. Am J Clin Pathol. 1974;61:473-481; 2) Talamo TS, et al. Microcomputer assisted hematology quality control using a modified average of normals program. Am J Clin Pathol. 1981; 76:707-712; 3) Bull BS, Korpman RA. Autocalibration of hematology analyzers. J Clin Lab Automation. 1983;3:111-116; 4) Cembrowski GS, Westgard JO. Quality control of multichannel hematology analyzers: evaluation of Bull's algorithm. Am J Clin Pathol. 1985;83:337-345; 5) Bull BS, Hay KL. Are red blood cells indexes international? Arch Pathol Lab Med. 1985;109:604-606; 6) Levy WC, et al. Preserved blood versus patient data for quality control -

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 62 of 178
Bull's algorithm revisited. Am J Clin Pathol. 1986;85:719-721; 7) Levy WC, et al. The incorporation of red blood cell index mean data into quality control programs. Am J Clin Pathol. 1986;86:193-199; 8) Lunetzky ES, Cembrowski GS. Performance characteristics of Bull's multirule algorithm for the quality control of multichannel hematology analyzers. Am J Clin Pathol. 1987;88:634-638; 9) Hackney JR, Cembrowski GS. Evaluation of intralaboratory quality control schemes, In Cavil I. Quality Control. Edinburgh, UK: Churchill Livingstone, 1990; 10) Lewis SM, et al. Current concepts in haematology 3: blood count calibration. J Clin Pathol. 1991;144:881-884. ................................................................. Retained Patient Specimens ................................................................. Use of retained patient specimens alone is inadequate for routine QC, and must be considered as a supplemental procedure, in combination with another QC system. Retained patient specimens, while conveniently available, present some difficulties in mathematically defining "agreement" between CBC results separated in time, as these are not stabilized samples. This is in contrast to commercial control materials that have been treated to reduce time-dependent degradation. LSV.37606 Phase I N/A YES NO When the laboratory uses retained patient samples, are statistically defined limits used to determine agreement of sequential assays of a given sample? NOTE: Allowance should be made for time-dependent alterations in data from such labile samples. COMMENTARY: N/A LSV.37612 Phase I N/A YES NO Is there a defined range of CBC values for which these limits are applicable? NOTE: Because imprecision (standard deviation, coefficient of variation) is dependent upon the hematologic target value, the laboratory should restrict the use of these limits to appropriate ranges of CBC values. COMMENTARY: N/A

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 63 of 178
……………............................................................. ERROR DETECTION AND VERIFICATION ……………............................................................. LSV.37650 Phase I N/A YES NO Are there data that periodically compare all results obtained for patient specimens analyzed in the multiple sampling modes of the CBC analyzer (e.g., "open" and "closed" modes) to ensure that they are in agreement? NOTE: Different modes may involve dilution or a different sample path before analysis. When samples are analyzed in more than one mode, it is important to ensure that all modes function properly. Re-analysis of a previously analyzed sample should be performed in the alternate mode(s), and results should agree with the initial mode within the tolerance limits established for agreement by the hematology laboratory's quality control program, and any recommendations by the instrument manufacturer. COMMENTARY: N/A LSV.37660 Phase II N/A YES NO Is there a documented procedure available and in use for detecting and correcting automated WBC counts for the presence of nucleated red cells or megakaryocytes? NOTE: The effect of nucleated erythrocytes and blood megakaryocytes on the apparent WBC count varies with the system used for analysis. Each laboratory should evaluate its system(s) and develop appropriate detection and correction procedures. This is important to prevent reporting a falsely high WBC concentration. With some automated CBC instruments, nucleated erythrocytes or megakaryocytes may present themselves histographically or cytographically, and this can serve as an indicator for careful stained blood film inspection. The laboratory must establish if its particular instrument(s) includes some or all nucleated non-leukocytes in its apparent WBC "count". COMMENTARY: N/A REFERENCES: 1) Culp NB, Wallace J. Correcting the NE-series WBC count for nucleated RBCs. Sysmex J Intl. 1991;1:62-65; 2) Bridgen ML, Dalal BI. Cell counter-related abnormalities. Lab Med. 1999;30:325-334.

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 64 of 178
LSV.37680 Phase II N/A YES NO Is a documented procedure in use to detect other spurious CBC instrument results that may be clinically significant (e.g., pseudomacrocytosis from rouleaux or agglutinates; pseudoleukocytosis with erroneous hemoglobin, falsely low erythrocyte count and hematocrit; hyperlipemias)? NOTE: Analytic sources of error with automated instruments depend on the type of instrument and reagents used by the laboratory. Common potential errors for the hemogram (without platelets) include pseudomacrocytosis (due to microclots, cold agglutinins, rouleaux, or osmotic matrix effects), pseudoleukocytosis (due to platelet agglutination, giant platelets, unlysed erythrocytes, nucleated erythrocytes, megakaryocytes, red cell inclusions, cryoproteins, circulating mucin), erroneous hemoglobin and indices (due to lipemia or leukocytosis), falsely low red cell concentration and hematocrit (due to in vitro hemolysis or extreme microcytosis), and falsely depressed results for all parameters (due to clots). COMMENTARY: N/A REFERENCES: 1) Gagne CC, et al. Effect of hyperchylomicronemia on the measurement of hemoglobin. Am J Clin Pathol. 1977;68:584-586; 2) Cornbleet J. Spurious results from automated hematology cell counters. Lab Med. 1983;14:509-514; 3) Gloster ES, et al. Spurious platelet counts associated with bacteremia. Am J Hematol. 1985;18:329-332; 4) Savage RA. Analytic inaccuracy resulting from hematology specimen characteristics. Three cases of clinically misleading artifacts affecting white blood cell and platelet counts. Am J Clin Pathol. 1989;92:295-299; 5) Rohr LR, Rivers FM. Spurious automated leukopenia due to in vitro granulocyte aggregation. Am J Clin Pathol. 1990;93:572-574; 6) Robbins SH, et al. Cold-induced granulocyte aggregation. A cause of pseudoleukopenia. Arch Pathol Lab Med. 1991;115:155-157; 7) Gulati GG, et al. Interference by cryoproteins in the blood with automated CBCs. Lab Med. 1995;26:138-142; 8) Cantero M, et al. Interference from lipemia in cell count by hematology. Clin Chem. 1996;42:987-988; 9) Bowen KL. Clinical pathology rounds. Erroneous leukocyte counts and cold agglutinins. Lab Med. 1997;28:247-250; 10) Bridgen ML, Dalal BI. Cell counter-related abnormalities. Lab Med. 1999;30:325-334. LSV.37700 Phase I N/A YES NO Are red cell indices (MCV, MCH, MCHC) monitored routinely to detect random errors? NOTE: Patient sample red cell indices (Wintrobe indices or MCV, MCH, MCHC) should be monitored routinely to detect random errors, instrument malfunction, or spurious results. If semiautomated methods are used, indices should be calculated. On many automated instruments, the MCHC is the most useful parameter to ensure accuracy of the red cell parameters in individual patient samples. Since MCHC varies over a narrow range, an abnormal MCHC will often flag potentially spurious red cell parameters. Truly elevated MCHCs may be seen with spherocytosis, while decreased MCHCs can accompany a low MCV in severe iron deficiency anemia. If such RBC abnormalities are not present on the blood film, one or more of the measured RBC parameters is likely erroneous.

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 65 of 178
Incorrect data may be due to instrument malfunction or to problems with the blood sample itself. Some examples include: spuriously elevated MCVs and MCHCs with cold agglutinins, falsely elevated MCHCs with lipemia and plasma paraproteins, spuriously low MCHCs with leukocytosis and osmotic effects such as hyperglycemia altering MCV. MCV and MCH are fairly constant for each patient, and monitoring these indices in a delta check error detection program may provide rapid patient-based detection of instrument malfunction or specimen misidentification. COMMENTARY: N/A REFERENCES: 1) Cornbleet J. How laboratories use RBC indices. Arch Intern Med. 1983;143:1490; 2) Houwen B. Random errors in haematology tests: a process control approach. Clin Lab Haemat. 1990;12:157-168; 3) Gulati GG, et al. Interference by cryoproteins in the blood with automated CBCs. Lab Med. 1995;26:138-142; 4) Cantero M, et al. Interference from lipemia in cell count by hematology. Clin Chem. 1996;42:987-988; 5) Bridgen ML, Dalal BI. Cell counter-related abnormalities. Lab Med. 1999;30:325-334. LSV.37720 Phase II N/A YES NO Are upper and lower limits of all reportable parameters on the CBC instrument defined so that results that fall outside these limits are verified before reporting? NOTE: The laboratory must initially establish or verify the reportable range for each parameter of its automated or semi-automated CBC instrument. In particular, the laboratory must have data on its instrument's accuracy with thrombocytopenic and leukopenic samples. Platelet concentrations below the established lower limits must be reanalyzed by another method (e.g., manual hemocytometry, or semiquantitative blood film estimates, or fluorescence flow cytometry using specific platelet monoclonal antibodies). Particle (WBC, RBC, PLT) concentrations above the established upper limits must, as clinically needed, be reanalyzed by doing the minimum dilution necessary to bring the counts into the instrument's analytic range. When clinically appropriate, apparent analyte concentrations that are lower or higher than the reportable range may be reported as "less than" the lower limit or "greater than" the higher limit. COMMENTARY: N/A REFERENCES: 1) Department of Health and Human Services, Centers for Medicare and Medicaid Services. Clinical laboratory improvement amendments of 1988; final rule. Fed Register. 2003(Jan 24):7164 [42CFR493.1253]; 2) Hanseler E, et al. Estimation of the lower limits of manual and automated platelet counting. Am J Clin Pathol. 1996;105:782-787; 3) Ault KA. Implementation of the immunological platelet count on a hematology analyzer - the Abbott Cell-Dyn 4000. Lab Hematol. 1997;3:125-128.

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 66 of 178
LSV.37740 Phase II N/A YES NO Is there an adequate system (such as microscopic correlation with the blood film) to prevent reporting of spurious thrombocytopenia when platelet clumps, giant platelets, or platelet satellitism are present? NOTE: When PLT satellitosis (satellitism), significant numbers of giant PLT and/or PLT clumps are suspected/detected by cyto/histographic abnormalities or instrument rejection of a PLT result, the PLT concentration must be independently verified. Correlation with a well-prepared blood film must be made. If PLT are clumped after collection in an EDTA-anticoagulated tube that was well-mixed at the time of collection, this may represent in vitro EDTA-induced changes; PLT should be quantified from blood collected directly into a counting diluent, or by use of a different anticoagulant (e.g., liquid sodium citrate with subsequent adjustment for dilution) or by estimation from a non-anticoagulated blood film. COMMENTARY: N/A REFERENCES: 1) Hyun BH, et al. Platelet satellitosis. Chicago, IL: American Society of Clinical Pathologists Check Sample H-78, 1976; 2) Veenhoven WA, et al. Pseudothrombocytopenia due to agglutinins. Am J Clin Pathol. 1979;72:1005-1008; 3) Gloster ES, et al. Spurious platelet counts associated with bacteremia. Am J Hematol. 1985;18:329-332; 4) Cunningham VL, Brandt JT. Spurious thrombocytopenia due to EDTA-independent cold-reactive agglutinins. Am J Clin Pathol. 1992;97:359-362; 5) Hanseler E, et al. Estimation of the lower limits of manual and automated platelet counting. Am J Clin Pathol. 1996;105:782-787; 6) Bridgen ML, Dalal BU. Cell counter-related abnormalities. Lab Med. 1999;30:325-334; 7) Kunicka JE, et al. Improved platelet counting using two-dimensional laser light scatter. Am J Clin Pathol. 2000;114;114:283-289. LSV.37760 Phase II N/A YES NO If significant numbers of unlysed RBC, giant platelets and/or platelet clumps are suspected/detected, is the WBC concentration rechecked by another method or are blood films examined to prevent reporting spuriously high WBC concentrations? NOTE: When unlysed RBC, PLT satellitosis, significant numbers of giant PLT and/or PLT clumps are suspected/detected by histographic abnormalities or instrument rejection of a the PLT result, the WBC count must be verified manually, by automated counting after collection into a different anticoagulant, by automated counting in a lyse-resistant mode, or by semiquantitative blood film evaluation to prevent reporting spuriously high WBC concentrations. COMMENTARY: N/A

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 67 of 178
REFERENCES: 1) Savage RA. Pseudoleukocytosis due to EDTA-induced platelet clumping. Am J Clin Pathol. 1984;81:317-322; 2) Rabinovitch A. Anticoagulants, platelets, and instrument problems. Am J Clin Pathol. 1984;82:132-133; 3) Cunningham VL, Brandt JT. Spurious thrombocytopenia due to EDTA-independent cold-reactive agglutinins. Am J Clin Pathol. 1992;97:359-362; 4) Dorner K, et al. Improved automated leukocyte counting and differential in newborns achieved by the haematology analyser Cell-Dyn 3500. Clin Lab Haem. 1995;17:23-30; 5) Bridgen ML, Dalal BU. Cell counter-related abnormalities. Lab Med. 1999;30:325-334; 6) Kunicka JE, et al. Improved platelet counting using two-dimensional laser light scatter. Am J Clin Pathol. 2000;114;114:283-289. LSV.37780 Phase II N/A YES NO If significant numbers of microcytic erythrocytes and/or small cell fragments are detected/suspected, is the platelet count determined or verified using an alternate method? NOTE: When a significant number of interfering particles are identified at the upper or lower PLT counting threshold (by inspection of the PLT histogram or instrument flag), the PLT concentration must be determined or verified by an alternate method. Such methods could include alternate instrumentation, hemocytometry, or blood film estimate, depending upon the PLT concentration and the degree of clinical accuracy required. COMMENTARY: N/A REFERENCES: 1) Morton BD, et al. Pappenheimer bodies: an additional cause for a spurious platelet count. Am J Clin Pathol. 1980;74:310-311; 2) Akware AM, et al. Spuriously elevated platelet counts due to microspherocytosis. Am J Clin Pathol. 1982;77:220-221; 3) Gloster ES, et al. Spurious elevated platelet counts associated with bacteremia. Am J Hematol. 1985;18:329-332; 4) Bridgen ML, Dalal BI. Cell counter-related abnormalities. Lab Med. 1999;30:325-334; 5) Li S, Salhany KE. Spurious elevation of automated platelet counts in secondary acute monocytic leukemia associated with tumor lysis syndrome. Arch Pathol Lab Med. 1999;123:1111-1114; 6) Kunicka JE, et al. Improved platelet counting using two-dimensional laser light scatter. Am J Clin Pathol. 2000;114;114:283-289. ------------------------------------------------------------ GENERAL INSTRUMENT ISSUES ------------------------------------------------------------ LSV.37840 Phase II N/A YES NO Is there a regular schedule for routine function checks and maintenance procedures of the cell counting system?

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 68 of 178
COMMENTARY: N/A LSV.37860 Phase II N/A YES NO Are function checks conveniently recorded or plotted to readily detect instrument malfunction? COMMENTARY: N/A LSV.37880 Phase II N/A YES NO Are performance or tolerance limits defined for each instrument, component or procedure in the system? COMMENTARY: N/A LSV.37900 Phase II N/A YES NO If semi-automated methods are used with manual dilutions, are there appropriate documented time limits for assay after dilution? NOTE: Minimum time limits for reading white cells and hemoglobin are necessary to ensure complete lysis of red cells. Maximum time limits for red cell and white cell counting after dilution should be set so lysis of the cells will not spuriously lower the counts. Time limitations established for fresh human blood samples may not apply to preserved commercial control materials. COMMENTARY: N/A ................................................................ Automated Differential Counters ................................................................

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 69 of 178
LSV.37960 Phase II N/A YES NO Is there documentation to indicate that the automated method was carefully evaluated in the laboratory against a previously validated automated method or a manual method before it was placed into routine use? NOTE: The laboratory should have the results of its evaluation studies, either in summary form or actual data, available to the inspector for review. The laboratory is not required to verify the manufacturer's studies on flagging of abnormal cells, although studies of the local patient population are recommended. The references that accompany this checklist question may be consulted for examples of specific evaluation protocols. COMMENTARY: N/A REFERENCES: 1) Rümke CL. The statistically expected variability in differential leukocyte counts. In: Differential leukocyte counting, CAP conference/Aspen. Northfield, IL: CAP, 1977:39-45; 2) Kalish RJ, Becker K. Evaluation of the Coulter S-Plus V three-part differential in a community hospital, including criteria for its use. Am J Clin Pathol. 1986;86:751-755; 3) Ross DW, Bentley SA. Evaluation of an automated hematology system (Technicon H-1). Arch Pathol Lab Med. 1986;110:803-808; 4) Clinical and Laboratory Standards Institute (CLSI). Reference Leukocyte (WBC) Differential Count (Proportional) and Evaluation of Instrumental Methods; Approved Standard—Second Edition. CLSI document H20-A2 (ISBN 1-56238-628-X). Clinical and Laboratory Standards Institute, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 19087-1898 USA, 2007; 5) Miers MK, et al. White blood cell differentials as performed by the Technicon H-1; evaluation and implementation in a tertiary care hospital. Lab Med. 1991;22:99-106; 6) Hallawell R, et al. An evaluation of the Sysmex NE-8000 hematology analyzer. Am J Clin Pathol. 1991;96:594-601; 7) Cornbleet PJ, et al. Evaluation of the Cell-Dyn 3000 differential. Am J Clin Pathol. 1992;98:603-614; 8) NCCLS. Method Comparison and Bias Estimation Using Patient Samples; Approved Guideline—Second Edition. NCCLS document EP9-A2 (ISBN 1-56238-472-4). NCCLS, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 19087-1898 USA, 2002; 9) Krause JR. The automated white blood cell differential. A current perspective. Hematol Oncol Clin North Am. 1994;8:605-16; 10) Goyzueta FG, et al. Automated differential white blood cell counts in the young pediatric population. Lab Med. 1996;27:48-52; 11) Gulati GL, et al. Suspect flags and regional flags on the Coulter-STKS. An assessment. Lab Med. 1999;30:675-680; 12) Grimaldi E, Scopacasa F. Evaluation of the Abbott CELL-DYN 4000 hematology analyzer. Am J Clin Pathol. 2000;113:497-505. LSV.37980 Phase II N/A YES NO Does the quality control procedure define limits of agreement with WBC subclasses from manually counted blood films or commercially available material containing at least two classes of white cells and/or surrogate particles?

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 70 of 178
NOTE: For pattern recognition microscopy systems, QC can be done by periodic processing of prepared control slides and maintenance/analysis of Levey-Jennings charts. For flow-through systems, at least two approaches are reasonable: 1) comparison of instrument differentials on fresh blood samples with a conventional manual differential count, and/or 2) use of commercially available stabilized leukocytes and/or particle surrogate control material. The automated instrument and reference determinations should be treated as replicate manual differentials and evaluated using the ± 2 or 3 S.D. agreement limits of Rümke. For commercial controls, mixed leukocyte subclasses (e.g., "mononuclear" or "large unclassified cells") or "remainder" fractions do not need to be assessed with QC procedures. The commercial material should contain surrogate particles to measure total neutrophils, total granulocytes, total lymphoid cells, monocytes, eosinophils, and basophils, if these subtypes are enumerated by the instrument and reported by the laboratory. If discrete populations of abnormal cells are identified and enumerated by the instrument (e.g., nucleated RBC, blasts), then the QC material must contain surrogate particles to evaluate accuracy. COMMENTARY: N/A REFERENCES: 1) Rümke CL. The statistically expected variability in differential leukocyte counts. In: Differential leukocyte counting, CAP conference/Aspen. Northfield, IL: CAP, 1977:39-45; 2) Kalish RJ, Becker K. Evaluation of the Coulter S-Plus V three-part differential in a community hospital, including criteria for its use. Am J Clin Pathol. 1986;86:751-755; 3) Ross DW, Bentley SA. Evaluation of an automated hematology system (Technicon H-1). Arch Pathol Lab Med. 1986;110:803-808; 4) Clinical and Laboratory Standards Institute (CLSI). Reference Leukocyte (WBC) Differential Count (Proportional) and Evaluation of Instrumental Methods; Approved Standard—Second Edition. CLSI document H20-A2 (ISBN 1-56238-628-X). Clinical and Laboratory Standards Institute, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 19087-1898 USA, 2007; 5) Miers MK, et al. White blood cell differentials as performed by the Technicon H-1; evaluation and implementation in a tertiary care hospital. Lab Med. 1991;22:99-106; 6) Hallawell R, et al. An evaluation of the Sysmex NE-8000 hematology analyzer. Am J Clin Pathol. 1991;96:594-601; 7) Cornbleet PJ, et al. Evaluation of the Cell-dyn 3000 differential. Am J Clin Pathol. 1992;98:603-614; 8) NCCLS. Method Comparison and Bias Estimation Using Patient Samples; Approved Guideline—Second Edition. NCCLS document EP9-A2 (ISBN 1-56238-472-4). NCCLS, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 19087-1898 USA, 2002; 9) Krause JR. The automated white blood cell differential. A current perspective. Hematol Oncol Clin North Am. 1994;8:605-16; 10) Goyzueta FG, et al. Automated differential white blood cell counts in the young pediatric population. Lab Med. 1996;27:48-52; 11) Gulati GL, et al. Suspect flags and regional flags on the Coulter-STKS. An assessment. Lab Med. 1999;30:675-680; 12) Grimaldi E, Scopacasa F. Evaluation of the Abbott CELL-DYN 4000 hematology analyzer. Am J Clin Pathol. 2000;113:497-505.

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 71 of 178
LSV.38020 Phase II N/A YES NO Has the laboratory established criteria for checking and reviewing leukocyte differential counter data, histograms, and/or blood smears for clinically important results flagged by the automated differential counter? NOTE: Clinically important results include pathologic quantities of normal cell types and abnormal cells. Flagging mechanisms include those within the particular instrument, inspection of histographic/cytographic displays, laboratory criteria based on local experience, and awareness of published evaluations. COMMENTARY: N/A REFERENCES: 1) Rümke CL. The statistically expected variability in differential leukocyte counts. In: Differential leukocyte counting, CAP conference/Aspen. Northfield, IL: CAP, 1977:39-45; 2) Payne BA, Pierre RV. Using the three-part differential: part II. Implementation of the system. Lab Med. 1986;17:517-522; 3) Kalish RJ, Becker K. Evaluation of the Coulter S-Plus V three-part differential in a community hospital, including criteria for its use. Am J Clin Pathol. 1986;86:751-755; 4) Ross DW, Bentley SA. Evaluation of an automated hematology system (Technicon H-1). Arch Pathol Lab Med. 1986;110:803-808; 5) Clinical and Laboratory Standards Institute (CLSI). Reference Leukocyte (WBC) Differential Count (Proportional) and Evaluation of Instrumental Methods; Approved Standard—Second Edition. CLSI document H20-A2 (ISBN 1-56238-628-X). Clinical and Laboratory Standards Institute, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 19087-1898 USA, 2007; 6) Miers MK, et al. White blood cell differentials as performed by the Technicon H-1; evaluation and implementation in a tertiary care hospital. Lab Med. 1991;22:99-106; 7) Hallawell R, et al. An evaluation of the Sysmex NE-8000 hematology analyzer. Am J Clin Pathol. 1991;96:594-601; 8) Cornbleet PJ, et al. Evaluation of the Cell-dyn 3000 differential. Am J Clin Pathol. 1992;98:603-614; 9) NCCLS. Method Comparison and Bias Estimation Using Patient Samples; Approved Guideline—Second Edition. NCCLS document EP9-A2 (ISBN 1-56238-472-4). NCCLS, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 19087-1898 USA, 2002; 10) Krause JR. The automated white blood cell differential. A current perspective. Hematol Oncol Clin North Am. 1994;8:605-16; 11) Goyzueta FG, et al. Automated differential white blood cell counts in the young pediatric population. Lab Med. 1996;27:48-52; 12) Gulati GL, et al. Suspect flags and regional flags on the Coulter-STKS. An assessment. Lab Med. 1999;30:675-680. ................................................................. MANUAL HEMATOCRIT (MICROHEMATOCRIT, PACKED CELL VOLUME) ................................................................. LSV.38240 Phase II N/A YES NO Is the speed of the microhematocrit centrifuge checked at specified intervals?

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 72 of 178
NOTE: Relative centrifugal field (RCF) must be sufficient to achieve maximum packing of cells. The centrifuge must be capable of sustaining a relative centrifugal field (rcf) of 10,000 to 15,000 at the periphery for 5 minutes. COMMENTARY: N/A REFERENCE: NCCLS. Procedure for Determining Packed Cell Volume by the Microhematocrit Method; Approved Standard—Third Edition. NCCLS document H7-A3 (ISBN 1-56238-413-9). NCCLS, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 19087-1898, USA 2000. LSV.38260 Phase II N/A YES NO If a mechanical timer is used, is its accuracy checked at specified intervals? NOTE: Not applicable to electronic timers. COMMENTARY: N/A LSV.38280 Phase II N/A YES NO Is the constant packing time (minimum spin to reach maximum packing of cells) established before initial use and reassessed when there has been a change in either the speed or time? COMMENTARY: N/A REFERENCE: NCCLS. Procedure for Determining Packed Cell Volume by the Microhematocrit Method; Approved Standard—Third Edition. NCCLS document H7-A3 (ISBN 1-56238-413-9). NCCLS, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 19087-1898, USA 2000. ................................................................. MANUAL (HEMOCYTOMETER) WBC, RBC, AND PLT COUNTS (BLOOD) ................................................................. Hemocytometry RBC counts are not recommended because of the level of imprecision and inability to verify results against a stained blood film.

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 73 of 178
LSV.38380 Phase II N/A YES NO If WBC or PLT counts are performed manually by pipette dilution and chamber count, is each sample counted in duplicate, plating both chambers of a standard hemocytometer? COMMENTARY: N/A LSV.38400 Phase II N/A YES NO When there is leukopenia or thrombocytopenia, does the manual hemocytometer procedure require a technique to offset the increased error associated with counting smaller numbers of cells in the hemocytometer? NOTE: The laboratory protocol must specify an increased number of cells counted (e.g., increased number of hemocytometer squares enumerated, or a lesser specimen dilution) when there is leukopenia or thrombocytopenia, in order to avoid increasing the imprecision of particle counting, which is governed by binomial distributions and Poisson statistics. COMMENTARY: N/A REFERENCES: 1) Barnett RN. Clinical laboratory statistics, 2nd ed. Boston, MA: Little, Brown, 1979:30-33, 101-103; 2) Miale JB. Laboratory medicine hematology, 6th ed. St Louis, MO: CV Mosby, 1982:373-374; 3) Savage RA, Rabinovitch AL. Number of squares on manual platelet counts: when is enough enough? Northfield, IL: College of American Pathologists Summing Up. 1986;xv(4):5-63; 4) Savage RA. Evaluate your practice for platelet counts. Northfield, IL: College of American Pathologists Summing Up, Fall 1987. LSV.38420 Phase II N/A YES NO Is a system documented for assuring that dilution fluids and reagents are free of contaminants that may spuriously change the true cell counts? NOTE: Suggested checks include pH, osmolality and background counts. COMMENTARY: N/A

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 74 of 178
LSV.38440 Phase I N/A YES NO Is at least one cell count control specimen analyzed, or a procedural control employed for each 8 hours of patient testing? NOTE: For leukocytes and platelets, this requirement can be met with assayed liquid control material, a previously assayed patient sample, or comparison with a visual blood film concentration estimate. Visual estimates are not appropriate for erythrocyte hemocytometry. COMMENTARY: N/A REFERENCE: Department of Health and Human Services, Centers for Medicare and Medicaid Services. Clinical laboratory improvement amendments of 1988; final rule. Fed Register. 2003(Jan 24):7168 [42CFR493. 1256(d)]. LSV.38460 Phase II N/A YES NO For hemocytometry platelets, is the manual count correlated with a platelet estimate from a properly prepared blood film? COMMENTARY: N/A REFERENCES: 1) Abbey AP, Belliveau RR. Enumeration of platelets. Am J Clin Pathol. 1978;69:55-56; 2) Nosanchuk JS, et al. The analytic basis for the use of platelet estimates from peripheral blood smears. Laboratory and clinical applications. Am J Clin Pathol. 1978;69:383-387; 3) Mogadam L. Application of the Miller's disc for the estimation and quality control of the platelet count. Lab Med. 1980;11:131-132. ................................................................. MANUAL BLOOD FILM EXAMINATION (DIFFERENTIAL COUNT) ................................................................. LSV.38500 Phase II N/A YES NO Is the quality of blood films satisfactory (properly stained, free of precipitate, good cell distribution)? COMMENTARY:

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 75 of 178
N/A REFERENCES: 1) Wenk RE. Comparison of five methods for preparing blood smears. Am J Med Technol. 1976;42:71-78; 2) College of American Pathologists. Differential leukocyte counting. CAP conference/aspen. Northfield, IL: CAP, 1977; 3) Stiene-Martihn EA. Causes for poor leukocyte distribution in manual spreader-slide blood films. Am J Med Technol. 1980;46:624-632; 4) Lewis SM. Blood film evaluations as a quality control activity. Clin Lab Haematol. 1990;12:119-127; 5) Turgeon ML. Clinical hematology, theories and procedures, 2nd ed. Boston, MA: Little, Brown, 1993;16-25; 6) Dacie JV, Lewis SM. Practical hematology, 8th ed. New York, NY: Churchill Livingstone, 1995;83-89. LSV.38520 Phase II N/A YES NO Are slides uniquely identified? NOTE: Slides or coverslips must be uniquely identified by element(s) such as specimen or accession number, or patient name and/or number. COMMENTARY: N/A **REVISED** 09/27/2007 LSV.38540 Phase II N/A YES NO Does the hematology laboratory have a documented system to ensure consistency of morphologic observations among all personnel performing blood cell microscopy? NOTE: Suggested methods to accomplish this include:
1. Circulation of blood films with defined leukocyte differential distributions and specific qualitative abnormalities of each class of cells (WBC, RBC, PLT), and/or
2. Multi-headed microscopy, and/or 3. Use of blood or marrow photomicrographs with referee and consensus identifications
(e.g., former CAP surveys photomicrographs) The procedure manual should include definitions of semiquantitative measurements such as 1+, 2+, 3+, etc. In the case of comparative blood film WBC differentials, the method of Rümke is recommended to define statistical agreement between observers (Rümke CL. The statistically expected variability in differential leukocyte counts. In: Differential Leukocyte Counting, CAP Conference/Aspen. Northfield, IL: CAP, 1977:39-45.

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 76 of 178
COMMENTARY: N/A REFERENCES: 1) Rümke CL. The statistically expected variability in differential leukocyte counts, In: Differential leukocyte counting, CAP conference/Aspen. Northfield, IL: CAP, 1977:39-45; 2) Wood B, et al. Teaching the clinical interpretation of peripheral blood smears to second-year medical school class using the peripheral blood-tutor computer program. Am J Clin Pathol. 1998;109:514-520; 3) College of American Pathologists. Surveys hematology glossary. Northfield, IL: CAP, 1999:1-26; 4) Brigden ML, Dalal BI. Morphologic abnormalities, pseudosyndromes, and spurious test results. Lab Med. 1999;30:397-405; 5) Haun DE, et al. A better way to assess WBC differential counting skills. Lab Med. 2000;31:329-333. LSV.38560 Phase II N/A YES NO Are blood films retained for at least one week for possible review and/or reference? NOTE: It may be desirable to retain outpatient films for a longer period and significantly abnormal films indefinitely for teaching purposes. COMMENTARY: N/A LSV.38580 Phase II N/A YES NO Do the laboratory staff fully assess, and accurately report, RBC and PLT morphology as part of the performance of a WBC differential and/or blood film review? NOTE: The laboratory must have a system to ensure that technical personnel have fully assessed all morphologic findings in each patient film. Each laboratory director should, in consultation with the medical staff, determine which morphologic findings are reportable. For example, minor degrees of anisocytosis and poikilocytosis without specific types of RBC abnormalities may be considered within the normal spectrum and not reportable to the chart. For RBC abnormalities that are reported, the laboratory must define a qualitative or semiquantitative grading system. When defined abnormalities (e.g., spherocytes, target cells, fragments, etc.) are present, non-specific listings of "anisocytosis" and/or "poikilocytosis" may not provide additional clinically useful information. COMMENTARY: N/A

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 77 of 178
REFERENCES: 1) Napoli V, et al. A semiquantitative estimate method for reporting abnormal RBC morphology. Lab Med. 1980;11:111-116; 2) Krause JR. Red-cell abnormalities in the blood smear: disease correlations. Lab Mgmt. 1985;23(10):29-35; 3) Bell A, Lofsness KG. A photo essay on red cell morphology. J Med Tech. 1986;3:85-93; 4) Lewis SM. Blood film evaluations as a quality control activity. Clin Lab Haematol. 1990;12:119-127. LSV.38620 Phase II N/A YES NO Are there documented criteria with specified findings for blood films that must be reviewed by the pathologist, supervisor or other technologist qualified in hematomorphology, and is there evidence of such review? COMMENTARY: N/A REFERENCES: 1) Peterson P, et al. Physician review of the peripheral blood smear: when and why. An opinion. Lab Hematol. 2001;7:175-179; 2) Gulati GL, et al. Criteria for blood smear review. Lab Med. 2002;33:374-377. LSV.38640 Phase I N/A YES NO Is there a file of unusual slides for teaching/reference purposes? COMMENTARY: N/A ------------------------------------------------------------ AUTOMATED RETICULOCYTES ------------------------------------------------------------ LSV.38642 Phase I N/A YES NO Is there documentation to indicate that the automated method was carefully evaluated in the laboratory against previous manual or automated reticulocyte methods before use for patient reporting? COMMENTARY: N/A

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 78 of 178
REFERENCES: 1) Davis BH, et al. Flow cytometric reticulocyte analysis: multiinstitutional laboratory correlation study. Am J Clin Pathol. 1994;102:468-477; 2) Buttarello M, et al. Laboratory evaluation of the Miles H-3 automated reticulocyte counter. A comparative study with manual reference method and Sysmex R-1000. Arch Pathol Lab Med. 1995;119:1141-1148; 3) Buttarello M, et al. Reticulocyte quantification by Coulter MAXM VCS (volume, conductivity, light scatter) technology. Clin Chem. 1996;42:1930-1937; 4) Cunningham MT, et al. Effect of normalization on the intermethod variability of reticulocyte counting. Lab Hematol. 1996;2:94-98; 5) Davis BH, et al. Immature reticulocyte fraction (IRF) and reticulocyte counts. Comparison of Cell Dyn 4000, Sysmex R-3000, thiazole orange flow cytometry, and manual counts. Lab Hematol. 1996;2:144-150; 6) NCCLS. Methods for Reticulocyte Counting (Automated Blood Cell Counters, Flow Cytometry, and Supravital Dyes); Approved Guideline—Second Edition. NCCLS document H44-A2 (ISBN 1-56238-527-5). NCCLS, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 19087-1898 USA, 2004; 7) Lacombe F, et al. Automated reticulocyte counting and immature reticulocyte fraction measurement. Am J Clin Pathol. 1999;112:677-686; 8) Buttarello M, et al. Flow cytometric reticulocyte counting. Parallel evaluation of five fully automated analyzers: an NCCLS-ICSH approach. Am J Clin Pathol. 2001;115:100-111; 9) Riley RS, et al. Reticulocyte enumeration: past & present. Lab Med. 2001;10:599-608; 10) Buttarello M, et al. Five fully automated methods for performing immature reticulocyte fraction. Comparison in diagnosis of bone marrow aplasia. Am J Clin Pathol. 2002;117:871-879. LSV.38644 Phase I N/A YES NO Is there a procedure in place to determine the strength and stability of the detection dye used for flow-through instrument quantification of reticulocytes? NOTE: The laboratory must have a procedure to determine the strength and stability of the detection dye for reticulocytes. Automated reticulocyte enumeration depends upon fluorescent RNA (or DNA-RNA)-binding dyes such as acridine orange, pyronine Y, propidium iodine, thioflavin T, auramine O, etc., as the detection agent. Accurate determination of the RNA (or DNA-RNA) binding depends on the stoichiometric properties of each dye. The dye should be at saturating concentration, and the concentration of the dye in its buffer should be noted on the reagent container. Determination of the saturating concentration of the dye must be performed by correlative study of automated results with a manual method on a series of samples of low, normal, and high reticulocyte concentrations, with preference given to detection of low reticulocyte counts. These procedures are not required of commercial kits cleared by the Food and Drug Administration (FDA) and used according to manufacturer's instructions. COMMENTARY: N/A REFERENCES: 1) Crissman HA, Steinkamp JA. Cytochemical techniques for multivariate analysis of DNA and other cellular constituents, In: Melamed MR, Lindmo T, Mendelsohn ML, eds. Flow cytometry and sorting, 2nd ed. New York, NY: Wiley-Liss, 1990:227-248; 2) Darzynkiewicz Z, Kaperscinski J. Acridine orange: a versatile probe of nucleic acids and other cell constituents, In:

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 79 of 178
Melamed MR, Lindmo T, Mendelsohn ML, eds. Flow cytometry and sorting, 2nd ed. New York, NY: Wiley-Liss, 1990:291-314; 3) Riley RS, Ross W. Reticulocyte enumeration, In: Riley RS, Makin EJ, Ross W, eds. Clinical applications of flow cytometry. New York, NY: Igaku-shoin, 1993:582-611. LSV.38646 Phase I N/A YES NO Is there a quality control program in place to determine the reproducibility (precision) of reticulocyte quantification? NOTE: This may be accomplished by running 2 levels of control every 24 hours. Preferably, QC material should include both normal and abnormal values. COMMENTARY: N/A REFERENCES: 1) Savage RA, et al. Analytic inaccuracy and imprecision in reticulocyte counting: a preliminary report from the College of American Pathologists reticulocyte project. Blood Cells. 1981;11:97-112; 2) Peebles DA, et al. Analysis of manual reticulocyte counting. Am J Clin Pathol. 1981;76:713-7171; 3) Hackney JR, et al. Automated reticulocyte counting by image analysis and flow cytometry. Lab Med. 1989;20:551-555; 4) Tsuda I, Tatsumi N. Reticulocytes in human preserved blood as control material for automated reticulocyte counters. Am J Clin Pathol. 1990;93:109-110; 5) Hohenwallner W, et al. The reticulocytes; automatic counting, indication and interpretation. Sysmex J Intl. 1992;2:120-135; 6) Batjer JD, et al. Predicting bone marrow transplant engraftment by automated flow cytometric reticulocyte analysis. Lab Med. 1994;25:22-26; 7) Davis BH, et al. Flow cytometric reticulocyte analysis. Multi-institutional interlaboratory correlation study. Am J Clin Pathol. 1994;102:468-477; 8) Cunningham MT, et al. Effect of normalization on the intermethod variability of reticulocyte counting. Lab Hematol. 1996;2:94-98; 9) Koepke JA. Update on reticulocyte counting. Lab Med. 1999;30:339-343; 10) NCCLS. Methods for Reticulocyte Counting (Automated Blood Cell Counters, Flow Cytometry, and Supravital Dyes); Approved Guideline—Second Edition. NCCLS document H44-A2 (ISBN 1-56238-527-5). NCCLS, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 19087-1898 USA, 2004. LSV.38648 Phase I N/A YES NO Are there documented criteria for identifying samples that may give spurious reticulocyte results by the automated method? NOTE: Since all DNA- and RNA-containing cells will stain with DNA-RNA fluorescent dyes, a procedure must be in place to identify when the instrument cannot discriminate such stained particles from true reticulocytes. Potential interferences include Howell-Jolly bodies, nucleated erythrocytes, Heinz bodies, basophilic stippling of red cells, macrothrombocytes, megakaryocyte fragments, platelet clumps, and malaria or other intracellular organisms. Erythrocyte agglutination also may give spuriously high results, as may very high leukocytosis or thrombocytosis. Interfering particles may

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 80 of 178
vary, depending on instrumentation, dye, and reaction conditions. Based upon initial evaluation of the instrument by the laboratory, criteria must be developed to detect samples with potentially erroneous results. This may be accomplished through flagging algorithms incorporated in the instrument and by examination of a blood film from every sample to ensure absence of relevant interferences. COMMENTARY: N/A REFERENCES: 1) Jacobberger HW, et al. Flow cytometric analysis of blood cells stained with the cyanine dye Dioc1[3]: reticulocyte quantification. Cytometry. 1984;5:589-600; 2) Davis BH, et al. Utility of flow cytometric reticulocyte quantification as a predictor of engraftment in autologous bone marrow transplantation. Am J Hematol. 1989;32:81-87; 3) Davis BH, Bigelow NC. Flow cytometric quantification using thiazole orange provides clinically useful reticulocyte maturity index. Arch Pathol Lab Med. 1989;113:684-689; 4) Hackney JR, et al. Automated reticulocyte counting by image analysis and flow cytometry. Lab Med. 1989;20:551-555; 5) Coulet M, Bezou MJ. Utilization of the automated reticulocyte counter Sysmex R-1000. Sysmex J. 1990;13:393-406; 6) Wells DA, et al. Effect of iron status on reticulocyte mean channel fluorescence. Am J Clin Pathol. 1992;97:130-134; 7) Riley RS, Ross W. Reticulocyte enumeration, In: Riley RS, Makin EJ, Ross W, eds. Clinical applications of flow cytometry. New York, NY: Igaku-shoin, 1993:582-611; 8) Batjer JD, et al. Predicting bone marrow transplant engraftment by automated flow cytometric reticulocyte analysis. Lab Med. 1994;25:22-26; 9) Lofsness KG, et al. Evaluation of automated reticulocyte counts and their reliability in the presence of Howell-Jolly bodies. Am J Clin Pathol. 1994;101:85-90. ----------------------------------------------------------- MANUAL RETICULOCYTES ----------------------------------------------------------- LSV.38650 Phase II N/A YES NO Examine a blood film stained for reticulocytes. Is it satisfactory? COMMENTARY: N/A REFERENCE: Fannon M, et al. Effect of staining and storage times on reticulocyte counts. Lab Med. 1982;13:431-433. LSV.38652 Phase II N/A YES NO Is the reported reticulocyte concentration based on a minimum sample size of 1,000 RBC?

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 81 of 178
NOTE: The reticulocyte concentration must be based on a total erythrocyte count of at least 1000 RBC, as reticulocytes are statistically rare events in comparison with the number of mature erythrocytes. The greatest cause of analytical error in manual reticulocyte counting is interobserver error, or at least variation in what to call a reticulocyte (especially the so-called Type IV cells of Heilmeyer). The second most common contribution to analytical error is the size of the sample counted. If one counts 100 cells, even with a Miller disk, the chance of error will be greater than if one counts 1000 RBC, as defined by Poisson statistics. The Miller disk is a counting aid to create a standard area. When using a Miller disc, a minimum of 112 cells must be counted in the small square, as this is equivalent to 1008 cells in the large square. However, it is imperative that the reticulocytes themselves are counted in the large square that contains at least 1000 cells. COMMENTARY: N/A REFERENCES: 1) Greenberg ER, Beck R. The effects of sample size on reticulocyte counting and stool examination. The binomial and Poisson distributions in laboratory medicine. Arch Pathol Lab Med. 1984;108:396-398; 2) Savage RA, et al. Analytic inaccuracy and imprecision in reticulocyte counting: a preliminary report from the College of American Pathologists reticulocyte project. Blood Cells. 1985;11:97-112; 3) NCCLS. Methods for Reticulocyte Counting (Automated Blood Cell Counters, Flow Cytometry, and Supravital Dyes); Approved Guideline—Second Edition. NCCLS document H44-A2 (ISBN 1-56238-527-5). NCCLS, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 19087-1898 USA, 2004; 4) Koepke JA. Update on reticulocyte counting. Lab Med. 1999;30:339-343. ---------------------------------------------------------------- ABNORMAL HEMOGLOBIN DETECTION ----------------------------------------------------------------- LSV.38658 Phase II N/A YES NO Are all samples that appear to have Hb S in the primary screening (by whatever method) further examined to confirm the presence of Hb S by solubility testing or other acceptable methods? NOTE: If the laboratory screens for sickling hemoglobins by solubility testing, a second test must be performed to confirm the presence of a sickling hemoglobin. If the laboratory does no other testing for abnormal hemoglobins, a comment must be attached to the report of positive solubility results, recommending that additional confirmatory testing be performed. For primary screening by electrophoresis or other separation methods, all samples with hemoglobins migrating in the "S" positions or peak must be tested for solubility or by other acceptable confirmatory testing for sickling hemoglobin(s). Known sickling and non-sickling controls must both be included with each run of patient specimens tested.

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 82 of 178
COMMENTARY: N/A ----------------------------------------------------------- BLOOD COAGULATION STUDIES ----------------------------------------------------------- Laboratories serving acute care hospitals should be able to perform a sufficient variety of coagulation tests to evaluate common coagulation disorders. This may not apply to independent laboratories, and if so, questions should be marked "N/A." LSV.38665 Phase II N/A YES NO Are tests for common coagulation problems available? NOTE: Laboratories serving acute care hospitals must be able to perform a sufficient variety of coagulation tests to evaluate common coagulation disorders. Such tests typically include platelet concentration, prothrombin time, activated partial thromboplastin time, fibrinogen assay, fibrin(ogen) degradation products or D-dimer, and platelet function tests such as bleeding time or a whole blood in vitro platelet function test. COMMENTARY: N/A REFERENCES: 1) NCCLS. One-stage prothrombin time (PT) test and activated partial thromboplastin time (aPTT) test; approved guideline H47-A. Wayne, PA: NCCLS, 1996; 2) Badve S, Burns ER. D-dimer measurements unhelpful for ruling in DIC. Lab Med. 2000;31:383-386; 3) Staikos L, et al. Improved anticoagulation management using point of care activated partial thromboplastin time. Clin Chem. 2000;46:A3; 4) Lopez E, et al. Comparison of a POCT for prothrombin time measurement with conventional laboratory testing in patients with oral anticoagulant therapy. Clin Chem. 2000;46:A14; 5) Mammen EF, et al. PFA-100 system: a new method for assessment of platelet dysfunction. Sem Thromb Hemost. 1998;24:195-202; 6) Clinical and Laboratory Standards Institute (CLSI). Performance of the Bleeding Time Test; Approved Guideline—Second Edition. CLSI document H45-A2 (ISBN 1-56238-571-2). Clinical and Laboratory Standards Institute, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 19087-1898 USA, 2005; 7) Taylor FB, et al. Towards definition, clinical and laboratory criteria, and a scoring system for disseminated intravascular coagulation. Thromb Haemos 2001;86:1327-1330.

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 83 of 178
LSV.38670 Phase II N/A YES NO Are tests for defining or monitoring disseminated intravascular coagulation (DIC) problems available, if applicable to the patient population served? COMMENTARY: N/A REFERENCES: 1) Bovill EG. Laboratory diagnosis of disseminated intravascular coagulation. Sem Hematol. 1994;31(2;suppl)35-29; 2) NCCLS. Procedure for the Determination of Fibrinogen in Plasma; Approved Guideline—Second Edition. NCCLS document H30-A2 (ISBN 1-56238-439-2). NCCLS, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 19087-1898 USA, 2001; 3) Yu M, et al. Screening tests of disseminated intravascular coagulation: guidelines for rapid and specific laboratory diagnosis. Crit Care Med. 2000;28:1777-1780. LSV.38675 Phase I N/A YES NO Is there a system to periodically measure the actual platelet concentration of the usual "platelet-poor" plasma used for many coagulation tests? NOTE: Platelet-poor plasma is particularly important in patients with the lupus anticoagulant, and in samples to be frozen for later testing. Platelet-poor plasma should have a residual platelet concentration of less than 10 X 109/L. This is important because platelet membranes form a procoagulant surface that can accelerate coagulation and spuriously shorten clotting times. It is particularly important in patients with the lupus anticoagulant; due to the high content of lipid in the platelet plasma membrane, increased platelets in samples with the lupus anticoagulant can cause the antiphospholipid antibody to bind to the platelet membrane, thus effectively removing it from plasma. In this circumstance, the presence of lupus anticoagulant may not be detected during diagnostic testing. Samples to be frozen should be "platelet-poor” because plasma contaminated with significant numbers of platelets may yield different analytic results after thawing, due to lysis of platelets. COMMENTARY: N/A REFERENCES: 1) Koepke JA, et al. Pre-instrumental variables in coagulation testing. Am J Clin Pathol. 1975;64:591-596; 2) Lupus Anticoagulant Working Party. Guidelines on testing for the lupus anticoagulant. J Clin Pathol. 1991;44:885-889; 3) Middleton AL, Oakley E. Activated partial thromboplastin time (aPTT): Review of Methods. Chicago, IL: American Society of Clinical Pathologists Check Sample PTS 91-8, 1991; 4) Brien W, et al. Lupus anticoagulant testing: effect of the platelet count on the activated partial thromboplastin time. Brit J Biomed Sci 1993;50:114-116.

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 84 of 178
LSV.38680 Phase II N/A YES NO Are coagulation tests (e.g., PT, aPTT, fibrinogen, and factor assays) performed at 37°C? COMMENTARY: N/A REFERENCES: 1) National Committee for Clinical Laboratory Standards. One-stage prothrombin time (PT) test and activated partial thromboplastin time (aPTT) test; approved guideline H47-A. Wayne, PA: NCCLS, 1996; 2) NCCLS. Determination of factor coagulant activities; approved guideline H48-A. Wayne, PA: NCCLS, 1997; 3) NCCLS. Procedure for the Determination of Fibrinogen in Plasma; Approved Guideline—Second Edition. NCCLS document H30-A2 (ISBN 1-56238-439-2). NCCLS, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 19087-1898 USA, 2001. ................................................................. Interinstrument Comparisons (Coagulation) ................................................................. The laboratory may use fresh patient or donor specimens analyzed on a primary instrument for daily QC of a secondary instrument. The selection of these materials (rather than commercial controls) is important to directly address the issue of whether a patient sample yields the same results on all of the laboratory's instruments. If the laboratory has only one instrument for patient testing, this section does not apply. LSV.38686 Phase II N/A YES NO If the laboratory has more than one instrument (same or different makes/models) for performing coagulation assays, are they checked against each other at least twice a year for correlation of patient results? NOTE: This question would only apply when the different instruments/reagents are producing the same reportable result. For example, some laboratories may use multiple aPTT reagents with variable sensitivity to the lupus anticoagulant. If these are defined as separate tests, then this question does not apply unless each type of aPTT test is performed on more than one analyzer. The use of fresh blood/plasma samples (rather than commercial controls) is important to directly address the issue of whether a patient sample yields the same results on all of the laboratory's instruments. Statistical agreement of commercial control materials across instruments does not guarantee comparability of patient specimen results.

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 85 of 178
When the same assay is performed on multiple instruments, it is not necessary to directly compare all combinations of instruments. For example, one instrument may be selected as the reference and all other instruments compared against it. This checklist requirement applies only to instruments/methods accredited under a single CAP number. COMMENTARY: N/A REFERENCE: Department of Health and Human Services, Centers for Medicare and Medicaid Services. Medicare, Medicaid and CLIA programs; CLIA fee collection; correction and final rule. Fed Register. 2003(Jan 24):5236 [42CFR493.1281(a)]. LSV.38692 Phase I N/A YES NO Are there defined tolerance limits for results agreement of interinstrument assays? NOTE: The laboratory should clearly define numeric ranges of agreement between multiple coagulation instruments used by the laboratory for a given assay. These ranges must be statistically defined, and not simply qualitative. COMMENTARY: N/A ----------------------------------------------------------------- RESULTS REPORTING - COAGULATION ----------------------------------------------------------------- ABBREVIATIONS: aPTT = activated partial thromboplastin time(s); INR = International Normalized Ratio(s); ISI = International Sensitivity Index; PT = prothrombin time(s) LSV.38696 Phase II N/A YES NO For PT, is there documentation that the ISI is appropriate to the particular PT reagent and instrumentation used? NOTE: The laboratory must demonstrate appropriateness of its ISI, a measurement of the sensitivity with which thromboplastin reagents detect decreased levels of vitamin K-dependent coagulation factors. The ISI used should be appropriate for the particular reagent-instrument combination. Acceptable documentation would include information from the instrument/reagent manufacturer, or

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 86 of 178
use of an ISI calculated from laboratory specimens. The ISI for a particular reagent varies, depending on the instrument used for PT testing. This is especially true for photo-optical vs. electromechanical instruments, but may also vary among different instruments within the same classification. COMMENTARY: N/A REFERENCES: 1) NCCLS. One-stage prothrombin time (PT) test and activated partial thromboplastin time (aPTT) test; approved guideline H47-A. Wayne, PA: NCCLS, 1996; 2) Fairweather RB, et al. College of American Pathologists Conference XXXI on laboratory monitoring of oral anticoagulant therapy. Arch Pathol Lab Med. 1998;122:768-781; 3) Ansell J, et al. Managing oral anticoagulant therapy. Chest 2001;119:22s-38s. LSV.38700 Phase II N/A YES NO Is the calculation of the INR appropriately adjusted for every new lot of PT reagent, changes in types of reagent, or change in instrumentation? NOTE: The calculation of the INR must be appropriately adjusted for every new lot of PT reagent, changes in types of reagent, or changes in instrumentation. The ISI value usually changes with each new lot of PT reagent. The ISI affects the sensitivity with which the PT reagent will be able to detect decreased levels of the vitamin K-dependent coagulation factors. This change in sensitivity will affect the calculation of the INR value. The laboratory should be able to provide documentation that the calculation of the INR is correct and appropriate for the new ISI value. These changes should be implemented wherever the INR is calculated, whether by the coagulation instrument, laboratory information system, or manually. It is critical to calculate and report appropriate INR values. Reporting erroneous INR values may lead to use of excessive or insufficient oral anticoagulant medication, which may result in bleeding or thrombotic complications in patients. COMMENTARY: N/A REFERENCES: 1) NCCLS. One-stage prothrombin time (PT) test and activated partial thromboplastin time (aPTT) test; approved guideline H47-A. Wayne, PA: NCCLS, 1996; 2) Fairweather RB, et al. College of American Pathologists Conference XXXI on laboratory monitoring of oral anticoagulant therapy. Arch Pathol Lab Med. 1998;122:768-781; 3) Ansell J, et al. Managing oral anticoagulant therapy. Chest 2001;119:22s-38s.

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 87 of 178
LSV.38704 Phase II N/A YES NO Is the appropriate geometric mean of the PT reference interval used in the INR calculation? NOTE: The appropriate mean of the PT reference interval must be used in the INR calculation, given by the formula: INR=(PT of patient / PT of geometric mean normal population)ISI The mean normal population value may change when the specimen collection process, instrument, reagent lot, or reagent changes. The geometric and arithmetic means may not be identical and the geometric mean is the preferred value. When the distribution of values is distributed normally, the GM, the arithmetic mean, the median and the mode of the population being studied are identical theoretically. These values diverge from each other, however, as the population distribution becomes more skewed. The geometric mean is a more appropriate estimate of the average value than the arithmetic mean when the population of interest is lognormally distributed because the geometric mean takes skewing into account. The geometric mean (GM) is the antilog of the arithmetic mean of the logarithms of the individual normal PT values of interest. Calculation of the geometric mean is indicated below; this calculation is available in many spreadsheet programs, such as Microsoft Excel. GM = antilog [(log(X1) + log(X2) + log(X3) + . . . log(Xn))/n]. COMMENTARY: N/A REFERENCES: 1) NCCLS. One-stage prothrombin time (PT) test and activated partial thromboplastin time (aPTT) test; approved guideline H47-A. Wayne, PA: NCCLS, 1996; 2) Fairweather RB, et al. College of American Pathologists Conference XXXI on laboratory monitoring of oral anticoagulant therapy. Arch Pathol Lab Med. 1998;122:768-781; 3) Ansell J, et al. Managing oral anticoagulant therapy. Chest 2001;119:22s-38s; 4) Critchfield GC, Bennett ST. The influence of the reference mean prothrombin time on the international normalized ratio. Am J Clin Pathol. 1994 Dec;102(6):806-11. **REVISED** 04/06/2006 LSV.38708 Phase II N/A YES NO Are there checks of patient reports for correct INR calculations, patient values, and reference ranges under the following circumstances?
1. Change in lot or type of PT reagent 2. Change in instrument

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 88 of 178
3. Establishment of new PT reference range 4. Change in INR calculation 5. At defined intervals, in the absence of the above changes
NOTE: It is suggested that the calculations be checked at the following INR values: 2.0 and 3.0. Patient reports should be checked at least once per year, even in the absence of changes to the test system and calculations. This question applies whether the INR is calculated by the coagulation analyzer or by the laboratory information system. COMMENTARY: N/A REFERENCES: 1) NCCLS. One-stage prothrombin time (PT) test and activated partial thromboplastin time (aPTT) test; approved guideline H47-A. Wayne, PA: NCCLS, 1996; 2) Fairweather RB, et al. College of American Pathologists Conference XXXI on laboratory monitoring of oral anticoagulant therapy. Arch Pathol Lab Med. 1998;122:768-781; 3) Ansell J, et al. Managing oral anticoagulant therapy. Chest 2001;119:22s-38s. **NEW** 10/31/2006 LSV.38709 Phase I N/A YES NO Is there documentation that the aPTT-based heparin therapeutic range is established and subsequently validated when appropriate? NOTE: The heparin-responsiveness of aPTT reagents may change from lot to lot and among different reagents used on different instrument platforms. For this reason, it is necessary to establish the heparin therapeutic range for the aPTT assay with each change of coagulation instrument and/or reagent type. The therapeutic range must be validated with each new lot of a given aPTT reagent. COMMENTARY: N/A REFERENCES: 1) Rosborough TK. Comparison of anti-factor Xa heparin activity and activated partial thromboplastin time in 2,773 plasma samples from unfractionated heparin-treated patients. Am J Clin Pathol. 1997;108:662-668; 2) Olson JD, et al. College of American Pathologists conference XXXI on laboratory monitoring of anticoagulant therapy. Laboratory monitoring of unfractionated heparin therapy. Arch Pathol Lab Med. 1998;122:782-798; 3) Smythe MA, et al. Use of the activated partial thromboplastin time for heparin monitoring. Am J Clin Pathol. 2001;115:148-155; 4) Smythe MA, et al. Different heparin lots. Does it matter? Arch Pathol Lab Med. 2001;125:1458-1462; 5) Hirsh J, et al. Guide to anticoagulant therapy. Heparin: a statement for healthcare officials from the American Heart Association. Circulation. 2001 19:2994-3018; 6) Hirsh J, Raschke R. Heparin and

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 89 of 178
low-molecular-weight heparin: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest. 2004 Sep;126(3 Suppl):188S-203S. **NEW** 10/31/2006 LSV.38710 Phase I N/A YES NO Is there documentation that the aPTT-based heparin therapeutic range is established and validated using an appropriate technique? NOTE: The aPTT is commonly used to monitor the anticoagulant effects of unfractionated heparin. The therapeutic range for heparin therapy should be established by using ex vivo plasma samples anticoagulated with 3.2% sodium citrate obtained from patients receiving current therapy with unfractionated heparin. This can be accomplished one of two ways. (1) The aPTT and heparin activity is measured for each sample and the aPTT range is calculated by comparison to heparin activity or (2) the aPTT of patient samples using the new lot or aPTT method is compared to the prior aPTT lot. It is recommended that the first method be used initially to establish the therapeutic range before starting patient testing with a new instrument or new reagent, while the second method can be used for validation of the therapeutic range with subsequent reagent lot changes. It is not best practice to use plasma samples spiked with heparin in vitro to calculate the therapeutic range, as differences in heparin binding proteins in vitro may lead to overestimation of the therapeutic range. COMMENTARY: N/A REFERENCES: 1) Rosborough TK. Comparison of anti-factor Xa heparin activity and activated partial thromboplastin time in 2,773 plasma samples from unfractionated heparin-treated patients. Am J Clin Pathol. 1997;108:662-668; 2) Olson JD, et al. College of American Pathologists conference XXXI on laboratory monitoring of anticoagulant therapy. Laboratory monitoring of unfractionated heparin therapy. Arch Pathol Lab Med. 1998;122:782-798; 3) Smythe MA, et al. Use of the activated partial thromboplastin time for heparin monitoring. Am J Clin Pathol. 2001;115:148-155; 4) Smythe MA, et al. Different heparin lots. Does it matter? Arch Pathol Lab Med. 2001;125:1458-1462; 5) Hirsh J, et al. Guide to anticoagulant therapy. Heparin: a statement for healthcare officials from the American Heart Association. Circulation. 2001 19:2994-3018; 6) Hirsh J, Raschke R, Heparin and low-molecular-weight heparin: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest. 2004 Sep;126(3 Suppl):188S-203S. LSV.38712 Phase I N/A YES NO Are reference intervals for PT and aPTT current for the reagent or lot number, and have they been appropriately determined?

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 90 of 178
NOTE: Because of the variability between different types of PT and aPTT reagents, and even different lots of PT and aPTT reagents, there may be significant changes in the reference interval after a change of the type or lot of reagent. For this reason, the laboratory should establish or verify the reference interval with each change of lot or change in reagent. COMMENTARY: N/A LSV.38716 Phase I N/A YES NO Are recommendations available to clinicians concerning which laboratory tests to use for monitoring heparin, low molecular weight heparin, direct thrombin inhibitors (e.g., lepirudin, bivalirudin, argatroban) and/or oral anticoagulant therapy, and the therapeutic range for the tests? NOTE: For oral anticoagulant agents (e.g., warfarin), the prothrombin time (PT/INR) is recommended, although many other methods are still in use. In addition, more than a dozen methods are in use for monitoring heparin and low molecular weight heparin therapy. For unfractionated heparin the activated partial thromboplastin time (aPTT) and/or activated clotting time are commonly used, but the heparin assay (factor Xa inhibition) may also be employed. For low molecular weight heparin or danaparoid, monitoring is often not necessary, but the heparin assay (Xa inhibition assay) may be used in certain circumstances, as the aPTT is generally insensitive to the effect of these agents. Direct thrombin inhibitors are becoming more widely utilized and these drugs are often monitored using the aPTT. The tests available should be applicable to the anticoagulant drugs available on the pharmacy formulary at the medical institution. The laboratory should work closely with the pharmacy or therapeutics committee to ensure that appropriate assays are available for the drugs in use by physicians, and that information is available on the test values that indicate that the anticoagulant is in a therapeutic range. COMMENTARY: N/A REFERENCES: 1) Rosborough TK. Comparison of anti-factor Xa heparin activity and activated partial thromboplastin time in 2,773 plasma samples from unfractionated heparin-treated patients. Am J Clin Pathol. 1997;108:662-668; 2) Haraldsson HM, et al. Performance of prothrombin-proconvertin time as a monitoring test of oral anticoagulation therapy. Am J Clin Pathol. 1997;108:662-668; 3) Baker BE, et al. Inability of the activated partial thromboplastin time to predict heparin levels: time to reassess guidelines for heparin assays. Arch Intern Med. 1997;157:2475-2479; 4) Brigden, ML et al. INR reporting in Canadian medical laboratories. An update. Am J Clin Pathol. 1998;109:589-594; 5) Leech BF, Carter CJ. Falsely elevated INR results due to the sensitivity of a thromboplastin reagent to heparin. Am J Clin Pathol. 1998;109:764-768; 6) Fairweather RB, et al. College of American Pathologists conference XXXI on laboratory monitoring of oral anticoagulant therapy. Arch Pathol Lab Med. 1998;122:768-781; 7) Olson JD, et al. College of American Pathologists conference XXXI

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 91 of 178
on laboratory monitoring of oral anticoagulant therapy. Laboratory monitoring of unfractionated heparin therapy. Arch Pathol Lab Med. 1998;122:782-798; 8) Lousberg TR, et al. Evaluation of excessive anticoagulation in a group model health maintenance organization. Arch Intern Med. 1998;158:528-534; 9) Davis KD, et al. Use of different thromboplastin reagents causes greater variability in international normalized ratio results than prolonged room temperature storage of specimens. Arch Pathol Lab Med. 1998;122:972-977; 10) Koerner SD, Fuller RE. Comparison of a portable capillary whole blood coagulation monitor and standard laboratory methods for determining international normalized ratio. Mil Med. 1998;163:820-825; 11) Laposata M, et al. College of American Pathologists conference XXXI on laboratory monitoring of low-molecular-weight heparin, danaparoid, hirudin and related compounds, and argatroban. Arch Pathol Lab Med. 1998;122:799-807; 12) Smythe MA, et al. Use of the activated partial thromboplastin time for heparin monitoring. Am J Clin Pathol. 2001;115:148-155; 13) Smythe MA, et al. Different heparin lots. Does it matter? Arch Pathol Lab Med. 2001;125:1458-1462; 14) Ansell J, et al. Managing oral anticoagulant therapy. Chest. 2001;119:22s-38s; 15) Hirsh J, et al. Heparin and low-molecular-weight heparin: Mechanisms of action, pharmacokinetics, dosing, monitoring, efficacy and safety. Chest. 2001; 119:64s-94s; 16) Hirsh J, et al. Guide to anticoagulant therapy. Heparin: a statement for healthcare officials from the American Heart Association. Circulation. 2001 19:2994-3018. ................................................................. Photo-Optical Coagulation Systems ................................................................. LSV.38720 Phase II N/A YES NO Is the photo-optical coagulation testing system (for PT, aPTT, etc.) checked with 2 different levels of control material during each 8 hours of patient testing, and each time there is a change in reagents? COMMENTARY: N/A LSV.38740 Phase II N/A YES NO Are tolerance limits defined for each instrument, component or procedure in the system? COMMENTARY: N/A

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 92 of 178
LSV.38760 Phase I N/A YES NO Are guidelines established for determining when alternative procedures should be performed (e.g., lipemia, hyperbilirubinemia, turbidity, etc.)? NOTE: Very long clotting times may not be reproducible on an automated coagulation instrument. Criteria should be established by each laboratory for performance of the PT or aPTT by an alternate technique (e.g., manual method) when the readable range of the instrument is exceeded. In addition, criteria should be provided for performance of alternate procedures in the presence of significant hyperbilirubinemia or lipemia, paradoxically short aPTTs and nonduplicating aPTTs. COMMENTARY: N/A ................................................................. Electromechanical Coagulation Systems ................................................................. LSV.38800 Phase II N/A YES NO Is the electromechanical coagulation system (for PT, aPTT, fibrinogen, and other coagulation assays) checked with 2 different levels of control material during each 8 hours of patient testing, and each time there is a change in reagents? COMMENTARY: N/A LSV.38820 Phase II N/A YES NO Are tolerance limits defined for each instrument, component or procedure in the system? COMMENTARY: N/A LSV.38840 Phase II N/A YES NO If the electromechanical system has reusable probes to detect a clot, are documented guidelines for cleaning the probes available?
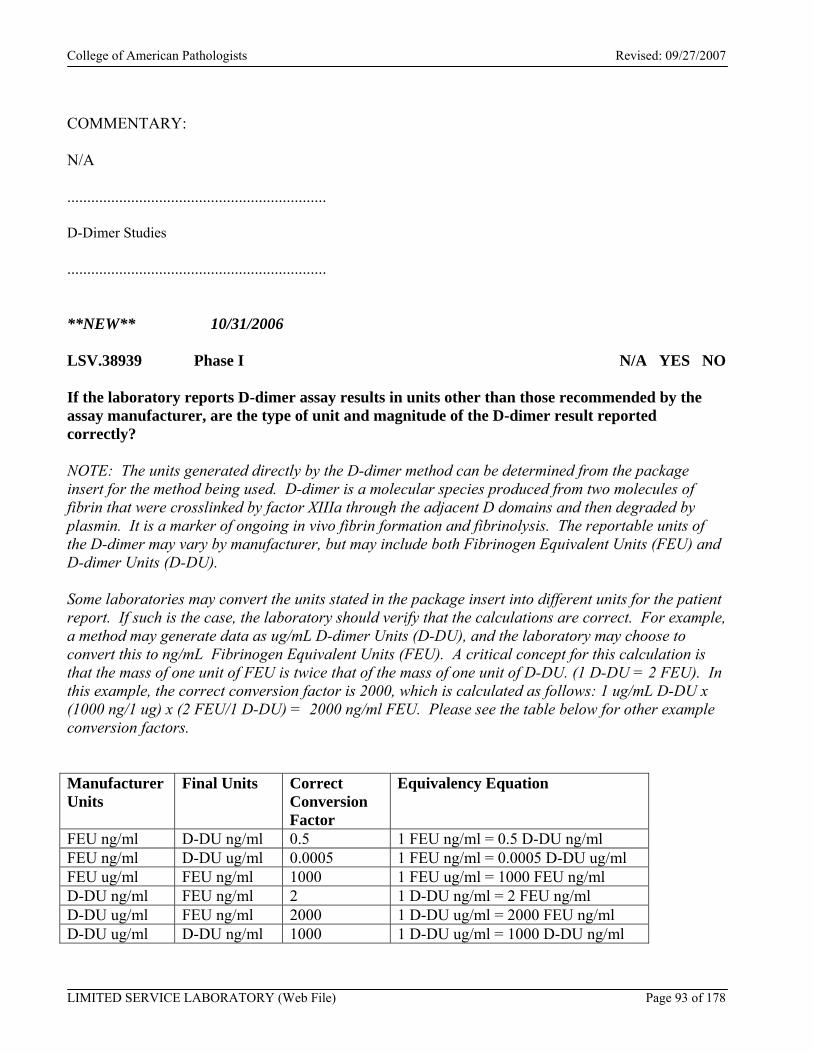
College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 93 of 178
COMMENTARY: N/A ................................................................. D-Dimer Studies ................................................................. **NEW** 10/31/2006 LSV.38939 Phase I N/A YES NO If the laboratory reports D-dimer assay results in units other than those recommended by the assay manufacturer, are the type of unit and magnitude of the D-dimer result reported correctly? NOTE: The units generated directly by the D-dimer method can be determined from the package insert for the method being used. D-dimer is a molecular species produced from two molecules of fibrin that were crosslinked by factor XIIIa through the adjacent D domains and then degraded by plasmin. It is a marker of ongoing in vivo fibrin formation and fibrinolysis. The reportable units of the D-dimer may vary by manufacturer, but may include both Fibrinogen Equivalent Units (FEU) and D-dimer Units (D-DU). Some laboratories may convert the units stated in the package insert into different units for the patient report. If such is the case, the laboratory should verify that the calculations are correct. For example, a method may generate data as ug/mL D-dimer Units (D-DU), and the laboratory may choose to convert this to ng/mL Fibrinogen Equivalent Units (FEU). A critical concept for this calculation is that the mass of one unit of FEU is twice that of the mass of one unit of D-DU. (1 D-DU = 2 FEU). In this example, the correct conversion factor is 2000, which is calculated as follows: 1 ug/mL D-DU x (1000 ng/1 ug) x (2 FEU/1 D-DU) = 2000 ng/ml FEU. Please see the table below for other example conversion factors. Manufacturer Units
Final Units Correct Conversion Factor
Equivalency Equation
FEU ng/ml D-DU ng/ml 0.5 1 FEU ng/ml = 0.5 D-DU ng/ml FEU ng/ml D-DU ug/ml 0.0005 1 FEU ng/ml = 0.0005 D-DU ug/ml FEU ug/ml FEU ng/ml 1000 1 FEU ug/ml = 1000 FEU ng/ml D-DU ng/ml FEU ng/ml 2 1 D-DU ng/ml = 2 FEU ng/ml D-DU ug/ml FEU ng/ml 2000 1 D-DU ug/ml = 2000 FEU ng/ml D-DU ug/ml D-DU ng/ml 1000 1 D-DU ug/ml = 1000 D-DU ng/ml

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 94 of 178
COMMENTARY: N/A REFERENCES: 1) Dempfle CE. D-dimer assays: The current status and new assay technologies. Thromb Res. 2005 Aug 30; [Epub ahead of print]; 2) Olson J, Cunningham M, Brandt J, et al. Use of the D-Dimer for Exclusion of VTE: Difficulties Uncovered through the Proficiency Testing Program of the College of American Pathologists (CAP). J Thromb Hemostasis, Abstract, August 2005; 3) Spannagl M, Haverkate F, Reinauer H, Meijer P. The performance of quantitative D-dimer assays in laboratory routine. Blood Coagul Fibrinolysis. 2005 Sep;16(6):439-43; 4) Gardiner C, Pennaneac’h C, Walford C, et al. An evaluation of rapid D-dimer assays for the exclusion of deep vein thrombosis. Br J Haematol. 2005 Mar;128(6):842-8; 5) Perrier A. Review: several factors are associated with the performance of D-dimer assays for detecting deep venous thrombosis. ACP J Club. 2004 Nov-Dec;141(3):76. **NEW** 10/31/2006 LSV.39038 Phase I N/A YES NO If calculations (including conversion of units) are used to obtain D-dimer results, does the laboratory document periodic checks to ensure that the calculations are correct? NOTE: The D-dimer may be a calculated value. In some institutions, the D-dimer result may calculated by the coagulation analyzer, while in others it may be calculated by the laboratory information system. The accuracy of calculations should be periodically verified. Calculations should be checked for all instruments or methodologies are used to generate reportable D-dimer values. It is suggested that the calculations be checked at both a normal and elevated D-dimer value. Calculations should be checked at least once per year, whenever the instrument or manufacturer of D-dimer reagent is changed, whenever a new D-dimer reference range is established, or if a component of the calculation is changed. COMMENTARY: N/A REFERENCES: 1) Dempfle CE. D-dimer assays: The current status and new assay technologies. Thromb Res. 2005 Aug 30; [Epub ahead of print]; 2) Olson J, Cunningham M, Brandt J, et al. Use of the D-Dimer for Exclusion of VTE: Difficulties Uncovered through the Proficiency Testing Program of the College of American Pathologists (CAP). J Thromb Hemostasis, Abstract, August 2005; 3) Spannagl M, Haverkate F, Reinauer H, Meijer P. The performance of quantitative D-dimer assays in laboratory routine. Blood Coagul Fibrinolysis. 2005 Sep;16(6):439-43; 4) Gardiner C, Pennaneac’h C, Walford C, et al. An evaluation of rapid D-dimer assays for the exclusion of deep vein thrombosis. Br J Haematol. 2005 Mar;128(6):842-8; 5) Perrier A. Review: several factors are associated with the

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 95 of 178
performance of D-dimer assays for detecting deep venous thrombosis. ACP J Club. 2004 Nov-Dec;141(3):76. **NEW** 10/31/2006 LSV.39137 Phase I N/A YES NO If a D-dimer method is used in the evaluation of venous thromboembolism, has the method been validated for this purpose? NOTE: If a D-dimer method is used in the evaluation of deep vein thrombosis and/or pulmonary embolism, the cut-off value for exclusion of these conditions should be validated, either in the peer-reviewed published literature, by the laboratory, or, for FDA-approved methods only, by the manufacturer of the method. The validation study(ies) should establish whether the cut-off is applicable to all patients, or only those with certain pre-test probabilities of having venous thromboembolism. If the laboratory modifies the manufacturer’s recommendations for an FDA-approved method, then the laboratory should perform the appropriate validation studies. Typically, manual latex agglutination D-dimer or FDP (fibrin degradation products) assays are not adequately sensitive to be used for evaluation of deep vein thrombosis and/or pulmonary embolism. COMMENTARY: N/A REFERENCES: 1) Olson J, Cunningham M, Brandt J, et al. Use of the D-Dimer for Exclusion of VTE: Difficulties Uncovered through the Proficiency Testing Program of the College of American Pathologists (CAP). J Thromb Hemostasis, Abstract, August 2005; 2) Spannagl M, Haverkate F, Reinauer H, Meijer P. The performance of quantitative D-dimer assays in laboratory routine. Blood Coagul Fibrinolysis. 2005 Sep;16(6):439-43; 3) Goodacre S, Sampson FC, Sutton AJ, et al. Variation in the diagnostic performance of D-dimer for suspected deep vein thrombosis. QJM. 2005 Jul;98(7):513-27. Epub 2005 Jun 13; 4) Gardiner C, Pennaneac’h C, Walford C, et al. An evaluation of rapid D-dimer assays for the exclusion of deep vein thrombosis. Br J Haematol. 2005 Mar;128(6):842-8; 5) Diamond S, Goldweber R, Katz S. Use of D-dimer to aid in excluding deep venous thrombosis in ambulatory patients. Am J Surg. 2005 Jan;189(1):23-6; 6) Wolf SJ, McCubbin TR, Feldhaus KM, et al. Prospective validation of Wells Criteria in the evaluation of patients with suspected pulmonary embolism. Ann Emerg Med. 2004 Nov;44(5):503-10; 7) Gould MK. Review: of the various D-dimer assays, negative ELISA results are most useful for excluding a diagnosis of deep venous thrombosis or pulmonary embolism. ACP J Club. 2004 Nov-Dec;141(3):77; 8) Stein PD, Hull RD, Patel KC, et al. D-dimer for the exclusion of acute venous thrombosis and pulmonary embolism: a systematic review. Ann Intern Med. 2004 Apr 20;140(8):589-602.

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 96 of 178
**NEW** 10/31/2006 LSV.39236 Phase I N/A YES NO If a D-dimer test is used for exclusion of deep vein thrombosis and/or pulmonary embolism, does the laboratory report the cutoff value for exclusion of venous thromboembolism, as well as the reference range? NOTE: The cut-off can be indicated within the patient report, or by a written memorandum to clinicians. The former is preferable. COMMENTARY: N/A **NEW** 10/31/2006 LSV.39335 Phase I N/A YES NO If a D-dimer test is not used for exclusion of deep vein thrombosis and/or pulmonary embolism, does the laboratory inform clinicians that the test should not be used to exclude deep vein thrombosis or pulmonary embolism? NOTE: This disclaimer may be included in the laboratory report, or in a written memorandum to clinicians. The former is preferable. COMMENTARY: N/A ................................................................. Coagulation Tests Based on Direct Measurement of Analytes ................................................................. This section applies to tests that quantitate an analyte (for example, fibrinogen, D-dimer, protein C, factor assays, heparin anti-Xa inhibition) by directly measuring the concentration or activity of the analyte using an immunologic or other method. The section includes requirements for calibration, calibration verification and analytic measurement range validation, and quality control. Additional discussion of the concepts of calibration, calibration verification and analytic measurement range validation can be found in the Introduction to the Calibration and Standards section of the Chemistry and Toxicology checklist.

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 97 of 178
This section does NOT apply to clot-based hemostasis assays, such as PT, aPTT, thrombin time, dilute Russell’s Viper Venom test, hexagonal phase phospholipid neutralization, platelet neutralization, clottable protein C assays, clottable protein S assays, or platelet function assays. **NEW** 10/31/2006 LSV.39434 Phase II N/A YES NO Are calibration procedures for each method adequate, and are the calibration results documented? COMMENTARY: N/A REFERENCES: 1) Department of Health and Human Services, Centers for Medicare & Medicaid Services. Clinical laboratory improvement amendments of 1988; final rule. Fed Register. 1992(Feb 28):7165 [42CFR493.1217]; 2) Department of Health and Human Services, Centers for Medicare & Medicaid Services. Medicare, Medicaid and CLIA Programs; Laboratory Requirements Relating to Quality Systems and Certain Personnel Qualifications; final rule. Fed Register. 2003(Jan 24):3707 [42CFR493.1255]; 3) Kroll MH, Emancipator K. A theoretical evaluation of linearity. Clin Chem. 1993;39:405-413; 4) NCCLS. Evaluation of matrix effects; proposed guideline EP14-P. Wayne, PA: NCCLS, 1998; 5) Miller WG. “Quality control.” Professional Practice in Clinical Chemistry: A Companion Text, ed DR Dufour. Washington, DC: AACC Press, 1999:12-1 to 12-22; 6) Kroll MH, et al. Evaluation of the extent of nonlinearity in reportable range studies. Arch Pathol Lab Med. 2000;124:1331-1338. **NEW** 10/31/2006 LSV.39533 Phase II N/A YES NO Are all calibration materials properly labeled as to content and calibration values? NOTE: Complete values need not necessarily be recorded directly on each vial of calibrator material, so long as there is a clear indication where specific values may be found for each analyte tested and each analyzer used by the laboratory. COMMENTARY: N/A

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 98 of 178
**NEW** 10/31/2006 LSV.39632 Phase II N/A YES NO Do calibration materials include dates placed in service and expiration dates? NOTE: The dates may be recorded in a log (paper or electronic), rather than on the containers themselves, providing that all containers are identified so as to be traceable to the appropriate data in the log. COMMENTARY: N/A **NEW** 10/31/2006 LSV.39731 Phase II N/A YES NO Are criteria established for frequency of recalibration or calibration verification, and the acceptability of results? NOTE: Criteria typically include:
1. at changes of reagent lots for chemically or physically active or critical components, unless the laboratory can demonstrate that the use of different lots does not affect the accuracy of patient/client test results and the range used to report patient/client test data
2. QC fails to meet established criteria 3. after major maintenance or service 4. when recommended by the manufacturer 5. At least every 6 months
Materials that may be used for calibration verification include, but are not limited to:
1. calibrators used to calibrate the analytical measurement system 2. materials provided by the analytical measurement system vendor for the purpose of
calibration verification 3. previously tested unaltered patient/client specimens 4. primary or secondary standards or reference materials with matrix characteristics and
target values appropriate for the method COMMENTARY: N/A

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 99 of 178
REFERENCES: 1) Department of Health and Human Services, Centers for Medicare and Medicaid Services. Clinical laboratory improvement amendments of 1988; final rule. Fed Register. 2003(Jan 24):3707[42CFR493.1255(b)(3)]; 2) Miller WG. “Quality control.” Professional Practice in Clinical Chemistry: A Companion Text, ed DR Dufour. Washington, DC: AACC Press, 1999:12-1 to 12-22. **NEW** 10/31/2006 LSV.39830 Phase II N/A YES NO Is the method system recalibrated when calibration verification fails to meet the established criteria of the laboratory? COMMENTARY: N/A **NEW** 10/31/2006 LSV.39929 Phase II N/A YES NO Has an acceptable range been established or verified for each lot of control material? NOTE: For unassayed controls, the laboratory must establish an acceptable range by repetitive analysis in runs that include previously tested control material. For assayed controls, the laboratory must verify recovery ranges supplied by the manufacturer. COMMENTARY: N/A REFERENCES: 1) NCCLS. Evaluation of precision performance of clinical chemistry devices; approved guideline EP5-A. Wayne, PA: NCCLS, 1998; 2) NCCLS. User demonstration of performance for precision and accuracy; approved guideline EP15-A. Wayne, PA: NCCLS, 2002; 3) Ross JW, Lawson NS. Analytic goals, concentration relationships, and the state of the art of clinical laboratory precision. Arch Pathol Lab Med 1995;119:495-513; 4) Steindel SJ, Tetrault G. Quality control practices for calcium, cholesterol, digoxin, and hemoglobin. A College of American Pathologists Q-Probes study in 505 hospital laboratories. Arch Pathol Lab Med 1998;122:401-408; 5) NCCLS. Quality Management for Unit-Use Testing; Approved Guideline. NCCLS document EP18-A (ISBN 1-56238-481-3). NCCLS, 940 West Valley Road, Suite 1400, Wayne, PA 19087-1898 USA, 2002.

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 100 of 178
*****************************************************************************
AUTOMATED/GENERAL CHEMISTRY SECTION
***************************************************************************** -------------------------------------------------------- CALIBRATION AND STANDARDS -------------------------------------------------------- **NEW** 10/31/2006 LSV.40028 Phase II N/A YES NO For waived tests, do testing personnel follow manufacturer instructions for calibration, calibration verification, and related functions? COMMENTARY: N/A ======================================================================== NOTE: The remaining questions in this checklist on CALIBRATION, CALIBRATION VERIFICATION, ANALYTIC MEASUREMENT RANGE (AMR), CLINICALLY REPORTABLE RANGE(CRR) and INTERINSTRUMENT COMPARISONS do not apply to waived tests. ======================================================================== The following requirements for calibration, calibration verification and analytic measurement range validation apply only to analyses that provide truly quantitative measurements expressed in mass units per unit volume (e.g. gm/L or mg/ml) OR in units traceable to a reference preparation or standard that is calibrated in mass units per unit volume. If these criteria are not met, the measurement is NOT quantitative and this section is not applicable. This introduction discusses the processes of calibration, calibration verification, and analytical measurement range validation (AMR). CALIBRATION is the set of operations that establish, under specified conditions, the relationship between reagent system/instrument response and the corresponding concentration/activity values of an analyte. Calibration procedures are typically specified by a method manufacturer, but may also be established by the laboratory. The term “calibration” has the same meaning in this checklist as in the U.S. CLIA-88 regulations.

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 101 of 178
However, the term “calibration verification,” as used in this checklist, carries a more restrictive meaning than in CLIA-88. As defined in the January, 2003 revision of CLIA-88, “calibration verification” refers to 2 distinct processes: 1) verification of correct method calibration and 2) validation of the reportable range. This checklist restricts the use of the term “calibration verification” to the first process. The checklist uses a different term, “analytical measurement range (AMR) validation,” to refer to the second process. All of these processes—calibration, calibration verification, and AMR validation--are required to ensure the continued accuracy of a test method. These concepts are further elaborated below. In this checklist, CALIBRATION VERIFICATION denotes the process of confirming that the current calibration settings remain valid for a method. If calibration verification confirms that the current calibration settings are valid, it is not necessary to perform a complete calibration or recalibration of the method. Calibration verification can be accomplished in several ways. If the method manufacturer provides a calibration validation or verification process, it should be followed. Other techniques include (1) assay of the current method calibration materials as unknown specimens, and determination that the correct target values are recovered, and (2) assay of matrix-appropriate materials with target values that are specific for the method. Each laboratory must define limits for accepting or rejecting tests of calibration verification. Materials for calibration verification must have a matrix appropriate for the clinical specimens assayed by that method, and target values appropriate for the measurement system. Materials may include, but are not limited to:
5. Calibrators used to calibrate the analytical measurement system 6. Materials provided by the analytical measurement system vendor for the purpose of
calibration verification 7. Previously tested unaltered patient/client specimens 8. Primary or secondary standards or reference materials with matrix characteristics and
target values appropriate for the method, 9. Proficiency testing material or proficiency testing validated material with matrix
characteristics and target values appropriate for the method In general, routine control materials are not suitable for calibration verification, except in situations where the material is specifically designated by the method manufacturer as suitable for verification of the method's calibration process. The ANALYTICAL MEASUREMENT RANGE (AMR) is the range of analyte values that a method can directly measure on the specimen without any dilution, concentration, or other pretreatment not part of the usual assay process. AMR VALIDATION is the process of confirming that the assay system will correctly recover the concentration or activity of the analyte over the AMR. The materials used for validation must be known to have matrix characteristics appropriate for the method. The matrix of the sample (i.e., the environment in which the sample is suspended or dissolved) may influence the measurement of the analyte. In many cases, the method manufacturer will recommend suitable materials. The test specimens must have analyte values which, at a minimum, are near the low,

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 102 of 178
midpoint, and high values of the AMR. Specimen target values can be established by comparison with peer group values for reference materials, by assignment of reference or comparative method values, and by dilution or admixture ratios of one or more specimens with known values. Each laboratory must define limits for accepting or rejecting validation tests of the AMR. Materials for AMR validation should have a matrix appropriate for the clinical specimens assayed by that method, and target values appropriate for the measurement system. Materials may include, but are not limited to:
1. Linearity material of appropriate matrix, e.g., CAP Survey-based or other suitable linearity verification material
2. Proficiency testing survey material or proficiency testing survey-validated material 3. Previously tested patient/client specimens, unaltered 4. Previously tested patient/client specimens, altered by admixture with other specimens,
dilution, spiking in known amounts of an analyte, or other technique 5. Primary or secondary standards or reference materials with matrix characteristics and
target values appropriate for the method 6. Calibrators used to calibrate the analytic measurement system 7. Control materials, if they adequately span the AMR
RECALIBRATION / CALIBRATION VERIFICATION and AMR VALIDATION INTERVALS: Recalibration or calibration verification, and AMR validation, must be performed at least once every 6 months, as specified under CLIA-88 regulations at 42CFR493.1255(b)(3). Successful calibration verification certifies that the calibration is still valid; unsuccessful calibration verification requires remedial action, which usually includes recalibration and AMR revalidation. The performance of recalibration or a calibration verification procedure resets the calendar to a new maximum 6-month interval before the next required reassessment. Methods that are recalibrated more frequently than every 6 months do not require a separate calibration verification procedure. In addition to the every 6 month requirement, laboratories must perform recalibration or calibration verification and AMR validation at changes in major system components, and at changes of lots of chemically or physically active reagents unless the laboratory can demonstrate that changing reagent lot numbers does not affect the range used to report patient/client test results. The laboratory director should determine what constitutes a major system component change or a change in reagents that would require calibration verification and revalidation of the AMR. Manufacturers’ instructions should be followed. The laboratory should establish other criteria, as appropriate, for recalibration/calibration verification. These include but are not limited to failure of quality control to meet established criteria, and major maintenance or service to the instrument. LSV.40130 Phase II N/A YES NO Are calibration procedures for each method adequate, and are the calibration results documented?

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 103 of 178
COMMENTARY: N/A REFERENCES: 1) Department of Health and Human Services, Centers for Medicare and Medicaid Services. Clinical laboratory improvement amendments of 1988; final rule. Fed Register. 2003(Jan 24):3707 [42CFR493.1255]; 2) Department of Health and Human Services, Centers for Medicare & Medicaid Services. Medicare, Medicaid and CLIA Programs; Laboratory Requirements Relating to Quality Systems and Certain Personnel Qualifications; Final Rule. Fed Register. 2003(Jan 24):3707 [42CFR493.1255]; 3) Kroll MH, Emancipator K. A theoretical evaluation of linearity. Clin Chem. 1993;39:405-413; 4) Clinical and Laboratory Standards Institute. Evaluation of Matrix Effects; Approved Guideline—Second Edition. CLSI document EP14-A2 (ISBN 1-56238-561-5). Clinical and Laboratory Standards Institute, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 190871898, USA, 2005; 5) Miller WG. Quality control, In Professional practice in clinical chemistry: a companion text, ed DR Dufour. Washington, DC: AACC Press, 1999:12-1 to 12-22; 6) Kroll MH, et al. Evaluation of the extent of nonlinearity in reportable range studies. Arch Pathol Lab Med. 2000;124:1331-1338. LSV.40210 Phase II N/A YES NO Are high quality materials with method- and matrix-appropriate target values used for calibration and calibration verification whenever possible? NOTE: Calibration materials establish the relationship between method/instrument response and the corresponding concentration/activities of an analyte. They have defined analyte target values and appropriate matrix characteristics for the clinical specimens and specific assay method. Many instrument systems require calibration materials with system-specific target values to produce accurate results for clinical specimens. COMMENTARY: N/A REFERENCE: Clinical and Laboratory Standards Institute. Evaluation of Matrix Effects; Approved Guideline—Second Edition. CLSI document EP14-A2 (ISBN 1-56238-561-5). Clinical and Laboratory Standards Institute, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 190871898, USA, 2005. LSV.40290 Phase II N/A YES NO Are all calibration materials properly labeled as to content and calibration values?

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 104 of 178
NOTE: Complete values need not necessarily be recorded directly on each vial of calibrator material, so long as there is a clear indication where specific values may be found for each analyte tested and each analyzer used by the laboratory. COMMENTARY: N/A LSV.40370 Phase II N/A YES NO Do calibration materials include dates placed in service and expiration dates? NOTE: The dates may be recorded in a log (paper or electronic), rather than on the containers themselves, providing that all containers are identified so as to be traceable to the appropriate data in the log. COMMENTARY: N/A LSV.40530 Phase II N/A YES NO Are criteria established for frequency of recalibration or calibration verification, and the acceptability of results? NOTE: Criteria typically include:
1. At changes of reagent lots for chemically or physically active or critical components, unless the laboratory can demonstrate that the use of different lots does not affect the accuracy of patient/client test results and the range used to report patient/client test data
2. QC fails to meet established criteria 3. After major maintenance or service 4. When recommended by the manufacturer 5. At least every 6 months
COMMENTARY: N/A REFERENCES: 1) Department of Health and Human Services, Centers for Medicare and Medicaid Services. Clinical laboratory improvement amendments of 1988; final rule. Fed Register. 2003(Jan 24):3707[42CFR493.1255(b)(3)]; 2) Miller WG. Quality control, In Professional practice in clinical chemistry: a companion text, ed DR Dufour. Washington, DC: AACC Press, 1999:12-1 to 12-22.

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 105 of 178
LSV.40550 Phase II N/A YES NO Is the method system recalibrated when calibration verification fails to meet the established criteria of the laboratory? COMMENTARY: N/A **REVISED** 04/06/2006 LSV.40610 Phase II N/A YES NO Is validation of the analytical measurement range (AMR) performed with matrix-appropriate materials which include the low, mid and high range of the AMR, and is the process documented? NOTE: The AMR is the range of analyte values that a method can directly measure on the specimen without any dilution, concentration, or other pretreatment that is not part of the usual assay process. Validation of the AMR is the process of confirming that the assay system will correctly recover the concentration or activity of the analyte over the AMR. The materials used for validation must be known to have matrix characteristics appropriate for the method. The test specimens must have analyte values that as a minimum are near the low, midpoint, and high values of the AMR. Guidelines for analyte levels near the low and high range of the AMR should be determined by the laboratory director. Factors to consider are the expected analytic imprecision near the limits, the clinical impact of errors near the limits, and the availability of test specimens near the limits. It may be difficult to obtain specimens with values near the limits for some analytes (e.g., T uptake, free thyroxine, prolactin, FSH). In such cases, reasonable procedures should be adopted based on available specimen materials. The method manufacturer’s instructions for validating the AMR should be followed, when available. Specimen target values can be established by comparison with peer group values for reference materials, by assignment of reference or comparison method values, and by dilution ratios of one or more specimens with known values. Each laboratory must define limits for accepting or rejecting validation tests of the AMR. The AMR must be revalidated at least every 6 months, and following changes in major system components or lots of analytically critical reagents (unless the laboratory can demonstrate that changing reagent lot numbers does not affect the range used to report patient test results, and control values are not adversely affected). If the materials used for calibration or for calibration verification include low, midpoint, and high values that are near the stated AMR, then separate AMR validation is not required.

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 106 of 178
Materials that may be used for AMR validation include, but are not limited to:
1. Linearity material of appropriate matrix, e.g., CAP Survey-based or other suitable linearity verification material
2. Proficiency testing survey material or proficiency testing survey-validated material 3. Previously tested patient/client specimens, unaltered 4. Previously tested patient/client specimens, altered by admixture with other specimens,
dilution, spiking in known amounts of an analyte, or other technique 5. Primary or secondary standards or reference materials with matrix characteristics and
target values appropriate for the method 6. Calibrators used to calibrate the analytic measurement system 7. Control materials, if they adequately span the AMR
COMMENTARY: N/A REFERENCE: Department of Health and Human Services, Centers for Medicare and Medicaid Services. Clinical laboratory improvement amendments of 1988; final rule. Fed Register. 2003(Jan 24):3707 [42CFR493.1255]. LSV.40650 Phase II N/A YES NO Are criteria established for validating the analytical measurement range, and is compliance documented? NOTE: If the materials used for calibration or for calibration verification include low, midpoint, and high values that are near the stated AMR, and if calibration verification data are within the laboratory's acceptance criteria, the AMR has been validated; no additional procedures are required. If the calibration and/or calibration verification materials do not span the full AMR, or the laboratory extends the AMR beyond the manufacturer's stated range, the AMR must be validated by assaying materials reasonably near the lowest and highest values of the AMR. Some instruments have integral automatic dilution systems when a result exceeds the AMR. Validation of the AMR refers to the inherent measurement range of the method, not to the range extended by an automatic dilution process. A separate validation of the automatic dilution process should be performed. COMMENTARY: N/A

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 107 of 178
LSV.40653 Phase II N/A YES NO Are results falling outside the AMR limits reviewed and reassayed if necessary before reporting? NOTE: The AMR is the range of analyte values that a method can directly measure on the specimen without any dilution, concentration, or other pretreatment not part of the usual assay process. Apparent analyte values that are lower or higher than the AMR do not routinely require repeat analysis if the result is reported as less than the lower limit, or greater than the upper limit, respectively, and the laboratory has evidence that the low result is not due to sampling/dilution errors, immunologic "hook effects," etc. In special cases, the procedure should state if an analyte cannot be diluted or concentrated, or if there are limitations to the amount of dilution or concentration that can be successfully used. The CLINICALLY REPORTABLE RANGE (CRR) is the range of analyte values that a method can report as a quantitative result, allowing for specimen dilution, concentration or other pretreatment used to extend the direct AMR. For example, if it is desired to report a result that exceeds the AMR, the specimen is commonly diluted to bring the analyte into that range, the diluted specimen is reassayed, and the final result calculated using the dilution factor. The establishment of the CRR is a medical judgment made by the laboratory director, and is based in part on the assay technology. The CRR is typically established at the time of initial method validation, and it does not need to be re-evaluated unless the methodology changes for the analyte. The method manufacturer frequently will specify the AMR and procedures to use for dilution or concentration of specimens with values outside the AMR. The lower limit of the CRR is typically the lower limit of the AMR of the method, as verified during method validation and stated in the procedure manual. Values lower than this limit will be reported as less than the limit. The upper limit of the CRR is typically not specified unless there is a measurement limitation of a dilution protocol for an analyte. It is acceptable to dilute until a value in the AMR is achieved. The diluent should be specified for each analyte that can be successfully diluted to bring its quantity into the AMR. An example of a CRR with both low and high limits is given for hCG as follows: Assume that the AMR is 3-1,000 mIU/mL. A laboratory director establishes that, for proper patient/client care, the CRR is 5-1,000,000 mIU/mL. The lower CRR is based on medical judgment that lower values are not diagnostic for pregnancy, while values above the upper CRR are not useful for diagnostic or prognostic purposes. Patient/client specimens with analytical measured results of <3, 3, or 4 mIU/mL are reported as "<5 mIU/mL"; specimens with analytic results >1,000 are diluted and rerun to obtain quantitative values up to 1,000,000 mIU/mL; specimens with analytic results >1,000,000 mIU/mL are reported as ">1,000,000 mIU/mL". An example of a CRR with only a low limit is given for aspartate aminotransferase (AST) as follows: assume the AMR is 4-900 IU/L. A laboratory director establishes that numeric values <4 are not clinically useful, while values >900 are clinically useful. Patient/client specimens with measured results of <4 are reported as "<4 IU/L"; specimens with analytic results >900 are diluted and

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 108 of 178
reassayed until a quantitative result is obtained. In this case, the upper CRR is not specified because specimens are diluted until a quantitative value is obtained. COMMENTARY: N/A REFERENCE: Department of Health and Human Services, Centers for Medicare and Medicaid Services. Clinical laboratory improvement amendments of 1988; final rule. Fed Register. 1992(Feb 28):7164 [42CFR493.1213]. LSV.40656 Phase II N/A YES NO Are dilution protocols and diluents (or concentration protocols) specified for all methods for which the CRR exceeds the AMR? NOTE: When a test result exceeds the AMR, the laboratory may dilute or concentrate the specimen to adjust the analyte content to be within the AMR, then repeat the assay to obtain a quantitative result. The procedure manual must include the protocol for dilution or concentration, any diluents or other components used in the process, and the calculation of the final reportable result. If there is a limit to the amount of dilution or concentration that is appropriate for an analyte, the procedure must state the limit, and specify how to report results that exceed the CRR. COMMENTARY: N/A **REVISED** 10/31/2006 LSV.40660 Phase I N/A YES NO For qualitative tests that use a cut-off value to distinguish positive from negative, is the cut-off value established initially, and verified every 6 months thereafter? NOTE: This checklist question applies only to certain tests that report qualitative results based on a quantitative measurement using a threshold (cut-off value) to discriminate between a positive and negative clinical interpretation (primarily, urine drugs of abuse). The cut-off value that distinguishes a positive from a negative result should be established when the test is initially placed in service, and verified every six months thereafter. If the value of a calibrator or calibration verification material is near that of the cut-off, then the process of calibration or calibration verification satisfies this checklist question. Verification of the cut-off should also be performed at changes of lots of analytically critical reagents (unless the laboratory director has determined that such changes do not affect the cut-off); after

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 109 of 178
replacement of major instrument components; after major service to the instrument; and failure of quality control to meet established criteria. Appropriate materials for establishment and verification of the cut-off are identical to those recommended for calibration verification (listed in the introduction to the Calibration and Standards section of the Chemistry and Toxicology checklist). Note that QC materials are acceptable if the material is specifically designated by the method manufacturer as suitable for verification of the method's calibration process. COMMENTARY: N/A LSV.40670 Phase II N/A YES NO If the laboratory uses more than one instrument to test for a given analyte, are the instruments checked against each other at least twice a year for correlation of patient/client results? NOTE: This question applies to quantitative tests performed on the same or different instrument makes/models. This comparison must include all instruments. The use of fresh human samples (whole blood, serum, plasma, urine, etc.), rather than stabilized commercial controls, is important to directly address the issue of whether a patient/client sample yields the same results on all of the laboratory's instruments. Statistical agreement of commercial control materials across instruments does not guarantee comparability of patient/client specimen results because of potential matrix effects. In cases when pre-analytical stability of patient/client specimens is a limiting factor, alternative protocols based on QC or reference materials may be necessary but the materials used should be validated to have the same response as fresh human samples for the instruments/methods involved. This checklist requirement applies only to instruments/methods accredited under a single CAP number. COMMENTARY: N/A REFERENCES: 1) Department of Health and Human Services, Centers for Medicare and Medicaid Services. Medicare, Medicaid and CLIA programs; CLIA fee collection; correction and final rule. Fed Register. 1993(Jan 19):5236 [42CFR493.1709(a)]; 2) Podczasy JJ, et al. Clinical evaluation of the Accu-Chek Advantage blood glucose monitoring system. Lab Med. 1997;28:462-466; 3) Ross JW, et al. The accuracy of laboratory measurements in clinical chemistry: a study of eleven analytes in the College of American Pathologists Chemistry Survey with fresh frozen serum, definitive methods and reference methods. Arch Pathol Lab Med. 1998;122:587-608.

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 110 of 178
-------------------------------------------------------- CONTROLS -------------------------------------------------------- Controls are used to ensure that a test system is performing correctly. Traditionally, controls are samples that act as surrogates for patient/client specimens, periodically processed like a patient/client sample to monitor the ongoing performance of the entire analytic process. Under certain circumstances, other types of controls (electronic, procedural, built-in) may be used. (Details are in the checklist requirements in this section, below.) **REVISED** 09/27/2007 LSV.40690 Phase II N/A YES NO Are controls run daily for quantitative and qualitative tests? Controls should verify assay performance at relevant decision points. The selection of these points may be based on clinical criteria, or, for certain tests (e.g., urine drugs of abuse), administrative criteria. NOTE 1: Except for tests meeting the criteria in Note 2, below, daily external controls must be run as follows: a. For quantitative tests, 2 controls at 2 different concentrations must be run daily unless a different requirement is specifically required by this checklist (e.g, for blood gas testing, coagulation tests). b. For qualitative tests, a positive and negative control must be run daily. Control testing is not necessary on days when patient testing is not performed. Note 2: Daily controls may be limited to electronic/procedural/internal controls for tests meeting the following criteria:
1. For quantitative tests, the test system includes 2 levels of electronic/procedural/internal controls that are run daily
2. For qualitative tests, the test system includes an electronic/procedural/internal control run daily
3. The system is FDA-cleared or approved 4. The system is not classified as highly complex under CLIA-88 5. The laboratory has performed and documented studies to validate the adequacy of
limiting daily QC to the internal controls. To initially validate quantitative tests using built-in liquid controls, both external and the internal controls should be run daily for at least 20 days. If results are not concordant (based on manufacturer precision data), the laboratory director should determine the course of action (e.g., perform further studies, continue to run daily external controls, etc.). For devices using other types of

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 111 of 178
internal controls (e.g., electronic or process controls, etc.), the validation protocol should be defined by the laboratory director.
6. External controls are run for each new lot number or shipment of test materials.* Regarding the positive external control for qualitative tests, best practice is to run a weak positive control, to maximize detection of problems with the test system.
7. External controls are run at a frequency not less than that recommended by the test manufacturer.
* Repetition of the initial validation study is not required when running external controls with new lots/shipments of thest materials. COMMENTARY: N/A REFERENCES: 1) Department of Health and Human Services, Centers for Medicare and Medicaid Services. Medicare, Medicaid and CLIA programs; CLIA fee collection; correction and final rule. Fed Register. 1993(Jan 19):5232 [42CFR493.1218(b)(1, 2)]; 2) Steindel SJ, Tetrault G. Quality control practices for calcium, cholesterol, digoxin, and hemoglobin. A College of American Pathologists Q-Probes study in 505 hospital laboratories. Arch Pathol Lab Med. 1998;122:401-408; 3) Voss EM, et al. Determining acceptability of blood glucose meters. Statistical methods for determining error. Lab Med. 1996;27:601-606; 4) Clinical and Laboratory Standards Institute (CLSI). Statistical Quality Control for Quantitative Measurement Procedures: Principles and Definitions; Approved Guideline—Third Edition. CLSI document C24-A3 (ISBN 1-56238-613-1). Clinical and Laboratory Standards Institute, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 19087-1898 USA, 2006; 5) Ye JJ, et al. Performance evaluation and planning for patient-based quality control procedures. Am J Clin Pathol. 2000;113:240-248. LSV.40850 Phase II N/A YES NO For quantitative tests, has a valid acceptable range been established or verified for each lot of control material? NOTE: For unassayed controls, the laboratory must establish a valid acceptable range by repetitive analysis in runs that include previously tested control material. For assayed controls, the laboratory must verify the acceptability ranges supplied by the manufacturer. COMMENTARY: N/A REFERENCES: 1) NCCLS. Evaluation of Precision Performance of Quantitative Measurement Methods; Approved Guideline—Second Edition. NCCLS document EP5-A2 (ISBN 1-56238-542-9). NCCLS, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 19087- 1898 USA, 2004; 2)

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 112 of 178
Clinical and Laboratory Standards Institute (CLSI). User Verification of Performance for Precision and Trueness; Approved Guideline—Second Edition. CLSI document EP15-A2 (ISBN 1-56238-574-7). Clinical and Laboratory Standards Institute, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 19087-1898 USA, 2005; 3) Ross JW, Lawson NS. Analytic goals, concentration relationships, and the state of the art of clinical laboratory precision. Arch Pathol Lab Med 1995;119:495-513; 4) Steindel SJ, Tetrault G. Quality control practices for calcium, cholesterol, digoxin, and hemoglobin. A College of American Pathologists Q-Probes study in 505 hospital laboratories. Arch Pathol Lab Med 1998;122:401-408; 5) NCCLS. Quality Management for Unit-Use Testing; Approved Guideline. NCCLS document EP18-A (ISBN 1-56238-481-3). NCCLS, 940 West Valley Road, Suite 1400, Wayne, PA 19087-1898 USA, 2002. LSV.40950 Phase II N/A YES NO If a calibrator obtained from an outside supplier is used as a control, is it a different lot number from that used to calibrate the method? NOTE: In general, calibrators should not be used as QC materials. However, this practice may be necessary for some methods when a separate control product is not available. In such cases, the calibrator used as a control must, whenever possible, be from a different lot number than that used to calibrate the method. COMMENTARY: N/A REFERENCE: Department of Health and Human Services, Centers for Medicare and Medicaid Services. Clinical laboratory improvement amendments of 1988; final rule. Fed Register. 2003(Jan 24):3708 [42CFR493.1256(d)(9)]. LSV.40970 Phase II N/A YES NO If the laboratory performs test procedures for which calibration and control materials are not commercially available, have guidelines been established to verify the accuracy of patient test results? COMMENTARY: N/A LSV.41010 Phase II N/A YES NO Are quality control data organized and presented so they can be evaluated daily by the technical staff to detect problems, trends, etc.?

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 113 of 178
NOTE: Results of controls must be recorded or plotted to readily detect a malfunction in the instrument or in the analytic system. These control records must be readily available to the person performing the test. COMMENTARY: N/A REFERENCE: Clinical and Laboratory Standards Institute (CLSI). Statistical Quality Control for Quantitative Measurement Procedures: Principles and Definitions; Approved Guideline—Third Edition. CLSI document C24-A3 (ISBN 1-56238-613-1). Clinical and Laboratory Standards Institute, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 19087-1898 USA, 2006. LSV.41130 Phase II N/A YES NO For numeric QC data, are quality control statistics (e.g., SD and CV) calculated at specified intervals to define and monitor analytic imprecision? NOTE: The laboratory must evaluate the imprecision statistics (e.g., SD and CV) at specified intervals to confirm that the test system is performing within acceptable limits. For whole blood methods, where stabilized whole blood or other suitable material is not available for QC, such statistics may be generated from previous patient/client samples using the SD of duplicate pairs. This checklist requirement does not apply to external controls run only to validate new lots/shipments of test materials. However the laboratory should have defined acceptable limits for such controls (either from the manufacturer, or developed by the laboratory). COMMENTARY: N/A REFERENCES: 1) Mukherjee KL. Introductory mathematics for the clinical laboratory. Chicago: American Society of Clinical Pathology, 1979:81-94; 2) Barnett RN. Clinical laboratory statistics, 2nd ed. Boston, MA: Little, Brown, 1979; 3) Weisbrodt IM. Statistics for the clinical laboratory. Philadelphia. PA: JB Lippincott, 1985; 4) Matthews DF, Farewell VT. Understanding and using medical statistics. New York, NY: Karger, 1988; 5) Department of Health and Human Services, Centers for Medicare and Medicaid Services. Clinical laboratory improvement amendments of 1988; final rule. Fed Register. 1992(Feb 28):7146 [42CFR493.1218(d)]; 6) Ross JW, Lawson NS. Analytic goals, concentrations relationships, and the state of the art for clinical laboratory precision. Arch Pathol Lab Med. 1995;119:495-513; 7) NCCLS. Laboratory Statistics—Standard Deviation; A Report. NCCLS document EP13-R (ISBN 1-56238-277-2). NCCLS, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 19087, 1995; 8) Clinical and Laboratory Standards Institute (CLSI). Statistical Quality Control for Quantitative Measurement Procedures: Principles and Definitions; Approved Guideline—Third Edition. CLSI document C24-A3 (ISBN 1-56238-613-1). Clinical and

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 114 of 178
Laboratory Standards Institute, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 19087-1898 USA, 2006; 9) Brooks ZC, et al. Critical systematic error supports used of varied QC rules in routine chemistry. Clin Chem. 2000;46:A70. **REVISED** 09/27/2007 LSV.41170 Phase II N/A YES NO Is there documentation of corrective action when control results exceed defined acceptability limits? NOTE: The QC program must provide written guidelines defining the acceptability limits for the control materials for each method. This definition could be as simple as a control value of 3 SD from the mean of the control data (e.g. 13s), or it could involve a more complex set of criteria for accepting or rejecting the run. It is recognized that practice varies between laboratories and depends to some extent on the performance characteristics of the analyte and analyzer in question. The important point is that the laboratory must have written criteria for accepting or rejecting the run. Patient/client test results obtained in an analytically unacceptable test run or since the last acceptable test run must be re- evaluated to determine if there is a significant clinical difference in patient/client results. Re-evaluation may or may not include re-testing patient samples, depending on the circumstances. Even if patient samples are no longer available, test results can be re-evaluated to search for evidence of an out-of-control condition that might have affected patient results. COMMENTARY: N/A LSV.41250 Phase II N/A YES NO Are control specimens tested in the same manner and by the same personnel as patient/client samples? NOTE: It is implicit in quality control (QC) that control specimens are tested in the same manner as patient/client specimens. Moreover, QC specimens must be analyzed by personnel who routinely perform patient/client testing - this does not imply that each operator must perform QC daily, so long as each instrument and/or test system has QC performed at required frequencies, and all analysts participate in QC on a regular basis. To the extent possible, all steps of the testing process must be controlled, recognizing that pre-analytic and post-analytic variables may differ from those encountered with patients/clients. COMMENTARY:

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 115 of 178
N/A REFERENCES: 1) Department of Health and Human Services, Centers for Medicare and Medicaid Services. Clinical laboratory improvement amendments of 1988; final rule. Fed Register. 1992(Feb 28):7166 [42CFR493.1218(c)]; 2) ibid, 2003(Jan 24):3708[42CFR493.1256(d)(7-8). LSV.41330 Phase II N/A YES NO Are the results of controls verified for acceptability before reporting results? NOTE: Control results must be reviewed before reporting patient/client results. It is implicit in quality control that patient/client test results will not be reported when controls do not yield acceptable results. Controls must be run prior to reporting patient results after a change of analytically critical reagents, major preventive maintenance, or change of a critical instrument component. COMMENTARY: N/A REFERENCES: 1) Department of Health and Human Services, Centers for Medicare and Medicaid Services. Clinical laboratory improvement amendments of 1988; final rule. Fed Register. 1992(Feb 28):7166 [42CFR493.1218(e)]; 2) Department of Health and Human Services, Centers for Medicare and Medicaid Services. Clinical laboratory improvement amendments of 1988; final rule. Fed Register. 2003(Jan 24):3708 [42CFR493.1256(d)(6)]. ................................................................. Blood Gas Specimens ................................................................. LSV.41350 Phase II N/A YES NO Are personnel performing arterial punctures knowledgeable about the more significant complications of this procedure compared with venipuncture? COMMENTARY: N/A REFERENCE: NCCLS. Procedures for the Collection of Arterial Blood Specimens; Approved Standard—Fourth Edition. NCCLS document H11-A4 (ISBN 1-56238-545-3). NCCLS, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 19087-1898 USA, 2004.

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 116 of 178
**REVISED** 10/31/2006 LSV.41370 Phase II N/A YES NO For radial artery sampling, is a test for collateral circulation performed and documented before arterial puncture, as applicable? NOTE: The various technologies available have been evaluated in the published literature. Consensus should be established between the laboratory and involved clinicians to define in which patients and under what circumstances such a test is medically useful in averting potential patient injury. The site from where the sample was obtained should be documented. COMMENTARY: N/A REFERENCES: 1) Vaghadia H, et al. Evaluation of a postocclusive circulatory hyperaemia (PORCH) test for the assessment of ulnar collateral circulation. Can J Anaesth. 1988;35:591-598; 2) Cheng EY, et al. Evaluation of the palmar circulation by pulse oximetry. J Clin Monit. 1989;5:1-3; 3) Levinsohn DG, et al. The Allen's test: analysis of four methods. J Hand Surg. 1991;16:279-282; 4) Fuhrman TM, et al. Evaluation of collateral circulation of the hand. J Clin Monit. 1992;8:28-32; 5) Furhman TM, et al. Evaluation of digital blood pressure, plethysmography, and the modified Allen's test as a means of evaluating the collateral circulation to the hand. Anaesthesia. 1992;47:959-961; 6) Fuhrman TM, McSweeney E. Noninvasive evaluation of the collateral circulation to the hand. Acad Emerg Med. 1995;2:195-199; 7) O'Mara K, Sullivan B. A simple bedside test to identify ulnar collateral flow. Ann Intern Med. 1995;123:637; 8) Starnes SL, et al. Noninvasive evaluation of hand circulation before radial artery harvest for coronary artery bypass grafting. J Thorac Cardiovasc Surg. 1999;117:261-266; 9) Cable DG, et al. The Allen test. Ann Thorac Surg. 1999;67:876-877. LSV.41390 Phase II N/A YES NO Is there a system to prevent ambient air contamination of blood gas samples before analysis? COMMENTARY: N/A REFERENCES: 1) Ishikawa S, et al. The effects of air bubbles and time delay on blood gas analysis. Ann Allergy. 1974;33:72-77; 2) Mueller RG, et al. Bubbles in samples for blood gas determinations. Am J Clin Pathol. 1976;65:242-249; 3) Madiedo G, et al. Air bubbles and temperature effect on blood gas analysis. J Clin Pathol. 1980;33:864-867; 4) Biswas CK, et al. Blood gas analysis: effect of air bubbles in syringe and delay in estimation. Brit Med J. 1982;284:923-927; 5) McKane MH, et al. Sending blood gas specimens through pressurized transport tube systems exaggerates the error in

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 117 of 178
oxygen tension measurements created by the presence of air bubbles. Anesth Analg. 1995;81:179-182; 6) Astles JR, et al. Pneumatic transport exacerbates interference of room air contamination in blood gas samples. Arch Pathol Lab Med. 1996;120:642-647. -------------------------------------------------------- BLOOD GAS INSTRUMENTS -------------------------------------------------------- If the institution has blood gas testing sites that are medically and/or administratively separate from the main laboratory, and the blood gas testing medical director is different from the central laboratory medical director, then the Chemistry and Toxicology Checklist must be completed. If blood gas analyses are performed in a central laboratory with other automated chemistry assays, and/or testing occurs physically separate from the main laboratory, but the medical director is the same as the main laboratory, then complete this section. LSV.41410 Phase II N/A YES NO Are there documented procedures for operation, calibration and function checks of all blood gas instruments? COMMENTARY: N/A LSV.41490 Phase II N/A YES NO Are the materials used for calibration of the pH, CO2, and O2 sensors either in conformance with the instrument manufacturer's specifications or traceable to NIST Standard Reference Materials? COMMENTARY: N/A LSV.41570 Phase II N/A YES NO Is the calibration rechecked periodically using barometric pressure if appropriate? NOTE: Instruments used continually must be recalibrated periodically. Instruments used infrequently must be recalibrated each time of use. Some instruments are self-calibrating; however, there must be some defined procedure for verifying the reliability of this process. For all instruments, calibration

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 118 of 178
and calibration verification must be performed according to manufacturer's specifications with at least the frequency recommended by the manufacturer. COMMENTARY: N/A REFERENCE: Department of Health and Human Services, Centers for Medicare and Medicaid Services. Clinical laboratory improvement amendments of 1988; final rule. Fed Register. 2003(Jan 24):3709 [42CFR493.1267(a)]. LSV.41650 Phase II N/A YES NO Is a minimum of 1 quality control specimen for pH, pCO2 and pO2 (tonometered sample or liquid control material) analyzed at least every 8 hours of operation when patient specimens are tested? COMMENTARY: N/A REFERENCES: 1) Department of Health and Human Services, Centers for Medicare and Medicaid Services. Medicare, Medicaid and CLIA programs; CLIA fee collection; correction and final rule. Fed Register. 1993(Jan 19) [42CFR493.1245(b)]; 2) Clinical and Laboratory Standards Institute (CLSI). Statistical Quality Control for Quantitative Measurement Procedures: Principles and Definitions; Approved Guideline—Third Edition. CLSI document C24-A3 (ISBN 1-56238-613-1). Clinical and Laboratory Standards Institute, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 19087-1898 USA, 2006. LSV.41730 Phase II N/A YES NO Do the control materials for pH, pCO2 and pO2 represent both high and low values on each day of patient testing? COMMENTARY: N/A REFERENCES: 1) Department of Health and Human Services, Centers for Medicare and Medicaid Services. Medicare, Medicaid and CLIA programs; CLIA fee collection; correction and final rule. Fed Register. 1993(Jan 19) [42CFR493.1245(b)]; 2) Clinical and Laboratory Standards Institute (CLSI). Statistical Quality Control for Quantitative Measurement Procedures: Principles and Definitions; Approved Guideline—Third Edition. CLSI document C24-A3 (ISBN 1-56238-613-1). Clinical and Laboratory Standards Institute, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 19087-1898

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 119 of 178
USA, 2006; 3) Ng VL, et al. The rise and fall of i-STAT point-of-care blood gas testing in an acute care hospital. Am J Clin Pathol. 2000;114:128-138. LSV.41810 Phase II N/A YES NO Is at least one sample of control material for pH, pCO2 and pO2 included each time patient/client specimens are tested, except for automated instruments that internally calibrate at least once every 30 minutes of use? COMMENTARY: N/A REFERENCES: 1) Department of Health and Human Services, Centers for Medicare and Medicaid Services. Fed Register. 2003(Jan 24): 3709 [42CFR493.1267(c)]; 2) Clinical and Laboratory Standards Institute (CLSI). Statistical Quality Control for Quantitative Measurement Procedures: Principles and Definitions; Approved Guideline—Third Edition. CLSI document C24-A3 (ISBN 1-56238-613-1). Clinical and Laboratory Standards Institute, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 19087-1898 USA, 2006. --------------------------------------------------------- IMMUNOASSAYS --------------------------------------------------------- LSV.41816 Phase II N/A YES NO If immunoassay reagents are NOT used according to instrument and/or kit manufacturer's instructions, are there validation data to support that modifications produce reliable results consistent with the original assay claims of the kit manufacturer? NOTE: For modifications such as dilution enrichments, there must be validation data to support that the modification is equivalent to kit manufacturers' original assay claims. It is not required that the modified assay show equivalent reaction characteristics (i.e., assay signal response) but the laboratory must validate the original assay's claims of sensitivity and specificity for the detection of the target analyte. If all assays follow manufacturer's instructions, mark this question "N/A." COMMENTARY: N/A

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 120 of 178
LSV.41822 Phase II N/A YES NO Has the laboratory evaluated its automatic pipetting systems for carryover? NOTE: The laboratory must have procedures in place for evaluating whether carryover effects are present. This question applies to both stand-alone pipette systems and to sample pipettes integrated with analytic instruments. In practice, carryover is a problem only for analytes with a wide clinical range of analyte concentration, such that a minute degree of carry-over could have significant clinical implications. Examples include immunoassays such as β-HCG, certain enzymes (e.g., CK), and certain drugs of abuse (e.g., benzoylecgonine [cocaine metabolite], which may be present in high concentrations). The laboratory should select representative examples of such analytes for carryover studies. Evaluation for carryover is not required for automatic pipettes that use disposable tips. One suggested method to study carryover is to run known high patient samples, followed by known low samples to see if the results of the low-level material are affected. If carryover is detected, the laboratory must determine the analyte concentration above which subsequent samples may be affected, and define this value in the procedure. Results of each analytical run must be reviewed to ensure that no results exceed this level. If results that exceed the defined level are detected, then the appropriate course of action must be defined (repeat analysis of subsequent samples, for example). Carryover studies must be performed, as applicable, as part of the initial evaluation of an instrument. (The laboratory may use the data from carryover studies performed by instrument manufacturers, as appropriate.) It is recommended that carryover studies be repeated periodically thereafter, particularly after major maintenance or repair of the pipetting assembly of the instrument. COMMENTARY: N/A REFERENCE: NCCLS. Preliminary evaluation of quantitative clinical laboratory methods – second edition; approved guideline EP10-A2. Wayne, PA: NCCLS, 2002. --------------------------------------------------------- THERAPEUTIC DRUG MONITORING --------------------------------------------------------- LSV.41828 Phase I N/A YES NO Where such information is important, has the laboratory provided information to clinical personnel of the optimal specimen collection time in relation to drug dosing?

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 121 of 178
COMMENTARY: N/A REFERENCES: 1) Nicholson PW, et al. Ideal sampling time for drug assays. Br J Clin Pharm. 1980;9:467-470; 2) NCCLS. Development of requisition forms for therapeutic drug monitoring and/or overdose toxicology; approved guideline T/DM1-A. Wayne, PA: NCCLS, 1991; 3) Howanitz PJ, Steindel SJ. Digoxin therapeutic drug monitoring practices. A College of American Pathologists Q-Probes study of 666 institutions and 18679 toxic levels. Arch Pathol Lab Med. 1993;117:684-690; 4) Schoenenberge RA, et al. Appropriateness of antiepileptic drug level monitoring. JAMA. 1995;274:1622-1626; 5) Williamson KM, et al. Digoxin toxicity: an evaluation in current clinical practice. Arch Intern Med. 1998;158:2444-2499. LSV.41834 Phase II N/A YES NO Where applicable, are TDM results reported in relation to patient dosing and/or timing information? NOTE: The intent is to have a mechanism whereby the clinician can easily and accurately link TDM results from the laboratory to the dosage and time of drug administration. Ideally, the test results, dose and administration time would be reported in juxtaposition on the patient chart. This may be the responsibility of the laboratory, or an integrating function of reported laboratory analytic data with clinical information from other sources. COMMENTARY: N/A REFERENCES: 1) Elin RJ. Computer-assisted therapeutic drug monitoring. Clin Lab Med. 1987;7:485-492; 2) Howanitz PJ, Steindel SJ. Digoxin therapeutic drug monitoring practices. A College of American Pathologists Q-Probes study of 666 institutions and 18679 toxic levels. Arch Pathol Lab Med. 1993;117:684-690; 3) Schoenenberge RA, et al. Appropriateness of antiepileptic drug level monitoring. JAMA. 1995;274:1622-1626; 4) Williamson KM, et al. Digoxin toxicity: an evaluation in current clinical practice. Arch Intern Med. 1998;158:2444-2499; 5) Steele BW, et al. An evaluation of analytic goals for assays of drugs. A College of American Pathologists therapeutic drug monitoring survey study. Arch Pathol Lab Med. 2001;125:729-735. --------------------------------------------------- DRUG OF ABUSE SCREENING ---------------------------------------------------

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 122 of 178
…………………………………………………….. Legal Drug of Abuse Specimen Collection …………………………………………………….. The following requirements are applicable for the collection of legal specimens only. Drug of abuse testing for medical treatment purposes does not require the use of external chain of custody documents or restricted-access storage areas. If legal specimen collection is not performed, mark all questions in this section not applicable. LSV.41840 Phase II N/A YES NO Does the specimen receiving/accessioning procedure require documentation of type of specimen, verification of specimen identity, completeness of external chain-of-custody documents, and integrity (tamper-evident) of the transmittal or shipping container? COMMENTARY: N/A LSV.41870 Phase II N/A YES NO Does the laboratory properly complete appropriate sections of external chain-of-custody documents? COMMENTARY: N/A LSV.41900 Phase II N/A YES NO Are the original specimens always maintained in the original containers, and in a limited-access secured area, when not in the possession of an authorized individual? NOTE: The original specimens must always be maintained either in the direct custody of an authorized individual, or be in a locked secured area accessible only to authorized individuals. This locked and limited-access area may be a refrigerator, freezer, or storage room within the laboratory. COMMENTARY: N/A

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 123 of 178
LSV.41930 Phase II N/A YES NO Is access to the limited-access area restricted to authorized laboratory personnel? COMMENTARY: N/A ………………………………………………. Screening for Drugs of Abuse ………………………………………………. This section is intended for laboratories performing drug of abuse screening for medical purposes only. If legal testing is performed, or if confirmation of positive screen results is performed, then the Chemistry and Toxicology Checklist must be used. Legal testing typically includes specimens whose results will be used to determine if individuals will: 1) be referred back to the criminal justice system; 2) lose their job; 3) lose professional credentials, etc. Specimens that may have legal implications include DOA testing on mothers and babies, when positive findings may result in infant custody issues, testing for drug rehabilitation programs to determine if a participant is continuing to use drugs while undergoing therapy, and testing referred by law enforcement agencies (such as motor vehicle accidents in Emergency Services Departments). Laboratories that receive these types of specimens should be aware of the potential legal implications of such testing, and should discuss with the referring physicians the necessity of providing testing on specimens that meet “forensic” standards of testing. If DOA screening is performed on these types of specimens, the Chemistry and Toxicology Checklist must be used.
*****************************************************************************
URINALYSIS AND CLINICAL MICROSCOPY SECTION
***************************************************************************** ----------------------------------------------------------- SPECIMEN COLLECTION AND HANDLING ----------------------------------------------------------- LSV.42050 Phase II N/A YES NO Are instructions provided for patients for proper collection of clean voided urine specimens (i.e., in nursing procedure manual or in specimen collection area)?

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 124 of 178
NOTE: Proper collection of urine specimens is important to avoid contamination, or deterioration of constituents. Instructions must be available to nurses or aides collecting specimens in the hospital and posted in the ambulatory patient collection area to instruct patients on proper technique; graphic instructions are often quite useful. COMMENTARY: N/A REFERENCE: NCCLS. Routine urinalysis and collection, transportation, and preservation of urine specimens – second edition; approved guideline GP16-A2. Wayne, PA: NCCLS, 2001. LSV.42290 Phase II N/A YES NO Are urine specimens examined within 1-2 hours of collection? NOTE: If testing is unavoidably delayed (night collection, etc.), provisions must be made for appropriate preservation of specimens to maintain integrity of cells and formed elements. Refrigeration of urine may be acceptable since it inhibits bacterial growth, but it does not prevent the lytic effects of low specific gravity or alkaline pH. Urine crystal formation may be induced by refrigeration. Preparations that contain boric acid/sorbitol or release formaldehyde may be effective preservatives for some, but not all, urine tests. There should be a method of indicating whether preservative has been added to the sample, and the laboratory should have specified any preanalytical errors attributable to such preservatives. COMMENTARY: N/A REFERENCES: 1) Weinstein MP. Clinical evaluation of a urine transport kit with lyophilized preservative for culture urinalysis and sediment microscopy. Diag Microbiol Infect Dis. 1985;3:501-508; 2) Haber MH. Quality assurance in urinalysis. Clinics in Lab Med. 1998;8:432-436; 3) NCCLS. Routine urinalysis and collection, transportation, and preservation of urine specimens – second edition; approved guideline GP16-A2. Wayne, PA: NCCLS, 2001; 4) Howanitz PJ, et al. Timeliness of urinalysis. A College of American Pathologists Q-Probes study of 346 small hospitals. Arch Pathol Lab Med. 1997;121:667-672; 5) Rafael J, et al. Is a preservative required for routine urinalysis? Am J Clin Pathol. 1997;108:344; 6) Semeniuk H, et al. Evaluation of the leukocyte esterase and nitrite urine dipstick screening tests for detection of bacteriuria in women with suspected uncomplicated urinary tract infections. J Clin Microbiol. 1999;37:3051-3052. LSV.42370 Phase II N/A YES NO Is there a documented policy defining the method for urine preservation (refrigeration or specified preservative) within the laboratory for all tests when analysis is to be delayed?

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 125 of 178
COMMENTARY: N/A REFERENCES: 1) Weinstein MP. Clinical evaluation of a urine transport kit with lyophilized preservative for culture, urinalysis and sediment microscopy. Diag Microbiol Infect Dis. 1985;3:501-508; 2) Haber MH. Quality assurance in urinalysis. Clinics in Lab Med. 1988;8:432-436; 3) NCCLS. Routine urinalysis and collection, transportation, and preservation of urine specimens – second edition; approved guideline GP16-A2. Wayne, PA: NCCLS, 2001; 4) Howanitz PJ, et al. Timeliness of urinalysis. A College of American Pathologists Q-Probes study of 346 small hospitals. Arch Pathol Lab Med. 1997;121:667-672; 5) Rafael J, et al. Is a preservative required for routine urinalysis? Am J Clin Pathol. 1997;108:344. ----------------------------------------------------------- INSTRUMENTS AND EQUIPMENT ----------------------------------------------------------- LSV.42450 Phase II N/A YES NO Are refractometers with specific gravity capability checked periodically with appropriate solutions of known specific gravity and/or refractive index? NOTE: Distilled water (sp. gr. = 1.0000) and 5% NaCl (sp. gr. = 1.0225) can verify total solids (T.S.) meter calibration. For specific gravity determination. by dipstick, manufacturers' recommendations must be followed. COMMENTARY: N/A REFERENCE: Haber MH. Quality assurance in urinalysis. Clinics in Lab Med. 1988;8:432-436. LSV.42490 Phase II N/A YES NO Is there a documented routine maintenance and function verification schedule for all instruments and equipment in the urinalysis and clinical microscopy section(s) of the laboratory? NOTE: The effective utilization of instruments by the technical staff depends upon the prompt availability of maintenance, repair, and service documentation (copies are acceptable). Laboratory personnel are responsible for the reliability and proper function of their instruments and must have access to this information. Off-site storage, such as with centralized medical maintenance or computer files, is not precluded if the inspector is satisfied that the records can be promptly retrieved.

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 126 of 178
COMMENTARY: N/A ----------------------------------------------------------- PROCEDURES AND TEST SYSTEMS ----------------------------------------------------------- The elements of a macroscopic urinalysis vary according to the patient population served by a laboratory and the needs of clinicians. A complete routine urinalysis should include at least the following: glucose, protein, blood/hemoglobin, leukocyte esterase and nitrite. Other analytes (e.g., color, clarity, turbidity, specific gravity, bilirubin, ketones, pH and urobilinogen) are optional for CAP accreditation, but their utility should be reviewed with the medical staff served by the laboratory. There are few occasions when the color, clarity and odor of urine are of clinical significance. LSV.42610 Phase I N/A YES NO Does the routine dipstick urinalysis (whether read manually or by electronic reader) include, as clinically applicable?
1. Glucose 2. Protein 3. Blood or hemoglobin 4. Nitrite 5. Leukocyte esterase
NOTE: The Inspector must provide specific details of any deficiencies in Part B (Deficiency Summary) of the Inspector's Summation Report. COMMENTARY: N/A REFERENCES: 1) Semeniuk H, et al. Evaluation of the leukocyte esterase and nitrite urine dipstick screening tests for detection of bacteriuria in women with suspected uncomplicated urinary tract infections. J Clin Microbiol. 1999;37:3051-3052; 2) De Buys Roessingh AS, et al. Dipstick measurements of urine specific gravity are unreliable. Arch Dis Child. 2001;85:155-157; 3) Voinescu GC, et al. The relationship between urine osmolality and specific gravity. Am J Med Sci. 2002;323:39-42.

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 127 of 178
LSV.43010 Phase II N/A YES NO Is there a documented policy indicating when pediatric specimens should be tested for reducing substances other than glucose? NOTE: Such a policy should be based on consultation with the pediatric clinical staff. The policy should include instructions for dealing with those urines tested and found to be negative with glucose-specific tapes or strips. There is no requirement for routine performance of reducing substance testing in adult urines. COMMENTARY: N/A ----------------------------------------------------------- URINALYSIS - MANUAL MICROSCOPY ----------------------------------------------------------- LSV.43090 Phase I N/A YES NO Are manual microscopic examinations of urine sediment performed as part of complete urinalysis testing, or are there specific, documented criteria defining the circumstances under which the microscopic examination may be omitted? NOTE: There is evidence that in random urinalysis screening (hospital admissions, insurance physicals), urines that are yellow and clear and have negative chemical reactions have a markedly low yield on microscopic examination. Optimal service may entail protocols defining when microscopic examination of urine sediment should or should not be done. COMMENTARY: N/A REFERENCES: 1) Wenz B, Lampasso JA. Eliminating unnecessary urine microscopy. Results and performance characteristics of an algorithm based on chemical reagent strip testing. Am J Clin Pathol. 1989;92:78-81; 2) Schumann GB, Friedman SK. Comparing slide systems for microscopic urinalysis. Lab Med. 1996;27:270-277; 3) Hooper DW. Detecting GD and preeclampsia: effectiveness of routine urine screening for glucose and protein. J Reprod Med. 1996;41:885-888; 4) Jou WW, Powers RD. Utility of dipstick analysis as a guide to management of adults with suspected infection or hematuria. South Med J. 1998;91:266-269; 5) van Nostrand JD, et al. Poor predictive ability of urinalysis and microscopic examination to detect urinary tract infection. Am J Clin Pathol. 2000;113:709-713; 6) Ringsrud KM. Cells in the urine sediment. Lab Med. 2001;32:153-155; 7) Roggeman S, Zaman Z. Safely reducing manual urine microscopy analyses by combining urine flow cytometer and strip results. Am J Clin Pathol. 2001;116:872-878.

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 128 of 178
LSV.43130 Phase II N/A YES NO For post-vasectomy checks for reproductive sterility, is a concentrating technique employed on seminal fluid? NOTE: For post-vasectomy checks for reproductive sterility, a concentrating technique must be employed on the seminal fluid sample. Without such an approach, the presence of both motile and non-motile sperm may not be detected. COMMENTARY: N/A REFERENCES: 1) Jones CD, Cornbleet PJ. Wright-Giemsa cytology of body fluids. Techniques for optimal cytocentrifuge slide preparation. Lab Med. 1997;28:713-716; 2) Jaffe TM, et al. Sperm pellet analysis: a technique to detect the presence of sperm in men considered to have azoospermia by routine semen analysis. J Urol. 1998;159:1548-1550. LSV.43170 Phase II N/A YES NO Are reference materials (atlases, charts or photomicrographs) available to assist in the microscopic identification of sediment constituents? COMMENTARY: N/A REFERENCES: 1) Jao W, et al. An atlas of urinary sediment. Chicago, IL: Abbott Laboratories, 1980; 2) Haber MH. Urinary sediment: a textbook atlas. Chicago, IL: American Society of Clinical Pathology, 198; 3) Graff L. A handbook of routine urinalysis. Philadelphia, PA: JB Lippincott, 1983; 4) Bradley M. Urine crystals - identification and significance. Lab Med. 1982;13:348-353; 5) Zaharopoulos P, Wong JY. Matrix crystals in urine sediments. Lab Med. 1988;19:429-431; 6) Brunzel NA. Fundamentals of urine and body fluid analysis. Philadelphia, PA: WB Saunders, 1994; 7) King C. Comparison of methods for detecting indinavir crystals in urine. Am J Clin Pathol. 1998;110:540; 8) College of American Pathologists. Surveys hematology glossary. Northfield, IL: CAP, 1999:27-36; 9) Hortin GL, et al. Detection of indinavir crystals in urine. Dependence on method of analysis. Arch Pathol Lab Med. 2000;124:246-250; 10) Ringsrud KM. Cells in the urine sediment. Lab Med. 2001;32:153-155; 11) Ringsrud KM. Casts in the urine sediment. Lab Med. 2001;32:191-193.

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 129 of 178
**REVISED** 09/27/2007 LSV.43250 Phase II N/A YES NO Does the urinalysis section of the laboratory have a defined, documented system to ensure consistency of morphologic observations among all personnel performing urine sediment microscopy? NOTE: Suggested methods to accomplish this include:
1. Circulation of preserved urine sediments with defined abnormalities involving leukocytes, erythrocytes, casts, bacteria, yeast, etc.
2. Multi-headed microscopy 3. Use of urine sediment photomicrographs with referee and consensus identifications
(e.g., former CAP surveys clinical microscopy photomicrographs) The procedure manual should include definitions of semiquantitative measurements such as 1+, 2+, 3+, etc. COMMENTARY: N/A REFERENCES: 1) College of American Pathologists. Surveys hematology glossary. Northfield, IL: CAP, 1999:27-36; 2) Astion ML, et al. A web-based system for assessing competency is microscopic urinalysis. Clin Chem. 2000;46:A36; 3) Ringsrud KM. Cells in the urine sediment. Lab Med. 2001;32:153-155; 4) Ringsrud KM. Casts in the urine sediment. Lab Med. 2001;32:191-193; 5) Kim A, et al. Web-based competency assessment system for microscopic urinalysis. Clin Chem. 2002;48:1608-1611. LSV.43330 Phase II N/A YES NO Is there a documented procedure for correlation of microscopic sediment findings (such as casts, RBC, or WBC) with macroscopic results (presence of protein, positive occult blood, positive leukocyte esterase, etc.)? COMMENTARY: N/A REFERENCE: van Nostrand JD, et al. Poor predictive ability of urinalysis and microscopic examination to detect urinary tract infection. Am J Clin Pathol. 2000;113:709-713.

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 130 of 178
................................................................. Dipstick Readers ................................................................. LSV.43570 Phase I N/A YES NO Before placing a new dipstick reader into service, has the laboratory performed comparisons between the new instrument and a manual method or another automated method? NOTE: When changing readers (same or different manufacturer), a new comparative study should be performed between the new instrument(s) and either a manual method or another automated method.. The laboratory should have the results of its evaluation studies, either in summary form or actual data, available to the Inspector for review. COMMENTARY: N/A REFERENCES: 1) Ng RH, et al. Multicenter evaluation of a urine analyzer. Lab Med. 1993;24:783-789; 2) Dias VC, et al. Evaluation of the CLINITEK ATLAS for routine macroscopic urinalysis. Clin Biochem. 1996;29:217-223; 3) Penders J, et al. Quantitative evaluation of urinalysis test strips. Clin Chem. 2002;48:2236-2241. LSV.43650 Phase I N/A YES NO Are there criteria for identifying urine samples that may give erroneous results by the dipstick reader and thus require evaluation by alternate means (visual examination or other confirmatory method)? NOTE: Criteria should be given for identifying urine samples that may give erroneous results by the dipstick reader, and thus require confirmation by other means, such as visual examination. Intensely colored urine samples may result in false positive dipstick reactions with automated reflectance readers. However, the anomalous color will be apparent when visual evaluation is performed. COMMENTARY: N/A REFERENCE: De Buys Roessingh AS, et al. Dipstick measurements of urine specific gravity are unreliable. Arch Dis Child. 2001;85:155-157.

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 131 of 178
*****************************************************************************
BODY FLUIDS
***************************************************************************** ----------------------------------------------------------- BODY FLUID CELL COUNTING – MANUAL ----------------------------------------------------------- LSV.43662 Phase II N/A YES NO Are certified pipettes or commercial dilution systems used when diluting body fluid samples? COMMENTARY: N/A LSV.43668 Phase II N/A YES NO Is the diluting fluid checked periodically for non-specimen background particulates and changed when indicated? NOTE: Checking can be done by examining samples of these fluids under the microscope at defined intervals. If commercial microdilution systems are used, each lot must be examined visually for uniformity of filling and clarity. If diluting fluids are prepared by the laboratory, they must be prepared aseptically; refrigeration is recommended to prevent contamination with microorganisms. COMMENTARY: N/A **NEW** 09/27/2007 LSV.43671 Phase I N/A YES NO Are the lines in the counting or motility chambers bright, and are the chambers clean and free of scratches? NOTE: The inspector should examine several chambers. COMMENTARY:

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 132 of 178
N/A LSV.43674 Phase II N/A YES NO For samples counted using a standard hemocytometer, is each body fluid sample counted in duplicate? NOTE: Limits of agreement between replicate counts must be defined. COMMENTARY: N/A LSV.43680 Phase II N/A YES NO Does the laboratory indicate (as part of the report) that results may be inaccurate if the fluid specimen is partially clotted or has cell clumps or debris on the counting chamber? COMMENTARY: N/A LSV.43692 Phase I N/A YES NO Is there an additional procedure beyond unstained bright-field microscopic visualization of cells on the hemocytometer to ensure the accurate distinction of erythrocytes from other cell types? NOTE: Suggested techniques include acid rinsing of the fluid sample to lyse erythrocytes after initially counting all cells, the addition of a stain such as methylene blue to improve recognition of non-erythrocytes, or phase microscopy. COMMENTARY: N/A ----------------------------------------------------------- BODY FLUID CELL COUNTING – INSTRUMENTS -----------------------------------------------------------

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 133 of 178
LSV.43698 Phase II N/A YES NO Are instrument background counts performed on the diluent fluid and lysing agent to check for contamination that might affect cell counts? NOTE: This can be done by processing these fluids on the instrument used for cell counting and checking for the presence of significant background in the diluting fluids and lysing agents. COMMENTARY: N/A LSV.43704 Phase I N/A YES NO Has the laboratory established a sensitivity limit for each class of particle counted? NOTE: The sensitivity limit is readily defined by performing serial dilutions of cells and graphically plotting expected recovery against observed count. COMMENTARY: N/A REFERENCES: 1) International Committee for Standardization in Haematology (ICSH). Protocol for evaluation of automated blood cell counters. Clin Lab Haemat. 1984;6:69-84; 2) Heaney LG, et al. Electronic cell counting to measure total cell numbers in bronchoalveolar lavage fluid. Eur Respir J. 1994;7:1527-1531; 3) Dietz LJ, et al. Volumetric capillary cytometry: a new method for absolute cell enumeration. Cytometry. 1996:23:177-186; 4) Salinas M, et al. Comparison of manual and automated cell counts in EDTA preserved synovial fluids. Ann Rheum Dis. 1997;56:622-626; 5) Subira D, et al. Flow cytometric analysis of cerebrospinal fluid samples and its usefulness in routine clinical practice. Am J Clin Pathol. 2002;117:952-958. LSV.43710 Phase II N/A YES NO Has the laboratory defined lower limits for counting body fluid cells (erythrocytes, nucleated cells) below which the use of automated or semi-automated cell counters is not reliable? NOTE: The laboratory must have an appropriate protocol which limits the use of automated or semiautomated instruments for cell counting in the very low concentration ranges often seen with body fluids. The lower limit selected must reflect the particular instrument's background count and sensitivity. COMMENTARY:

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 134 of 178
N/A REFERENCES: 1) International Committee for Standardization in Haematology (ICSH). Protocol for evaluation of automated blood cell counters. Clin Lab Haemat. 1984;6:69-84; 2) Subira D, et al. Flow cytometric analysis of cerebrospinal fluid samples and its usefulness in routine clinical practice. Am J Clin Pathol. 2002;117:952-958. LSV.43716 Phase I N/A YES NO Does the laboratory have a procedure to detect clumps of cells or debris that may give spurious cell counts? NOTE: The procedure should include performing macroscopic assessment of body fluid samples processed on cell counting instruments. Instrument generated flags and findings on microscopic examination that suggest the presence of debris are important observations and may require the performance of a wet mount. Marked clumping or clots precludes reporting an automated count. The laboratory report should note the limited accuracy of cell counts in these situations, and include a description of the specimen problem. COMMENTARY: N/A LSV.43722 Phase II N/A YES NO Are 2 different stabilized control specimens analyzed and results recorded at least daily? NOTE: If the same instrument is used for cell counting of peripheral blood specimens, there is no need to have separate control runs for body fluid cell counting. COMMENTARY: N/A ----------------------------------------------------------- BODY FLUID NUCLEATED CELL DIFFERENTIALS ----------------------------------------------------------- LSV.43734 Phase I N/A YES NO Is the method for differentiating body fluid cells appropriate for the intended clinical use?

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 135 of 178
NOTE: The laboratory should use stained cytocentrifuge preparations to facilitate quantitative differentials and complete classification of nucleated cell types in body fluids, as opposed to performing differentials of unstained hemocytometer preparations. Differentials based on supravitally-stained hemocytometer preparations, wedge smears and drop preparations are considered suboptimal; their use should be limited to clinical circumstances requiring differentiation of polymorphonuclear from mononuclear cells (e.g., bacterial meningitis). Further sub-classification of nucleated cells, particularly the detection of malignant cells, should be performed using slide preparation methods that provide optimal cell recovery and morphologic detail, such as cytocentrifugation. Cytocentrifuge preparations provide excellent morphologic detail, deliver a high yield of cells even when the concentration is low, and have a high rate of detection for malignant cells. In cases of leukemia or lymphoma, Romanowsky-stained cytospin slides show excellent morphologic correlation with blood and bone marrow smears. If the laboratory uses an alternate slide preparation method or stain for sub-classification of body fluid mononuclear cells and/or detection of malignant cells, it must demonstrate from literature or in-house studies that this technique is equivalent in cell yield/recovery and morphologic detail to Romanowsky-stained cytocentrifuge preparations. COMMENTARY: N/A REFERENCES: 1) Mengel M. The use of the cytocentrifuge in the diagnosis of meningitis. Am J Clin Pathol. 1985;84:212-216; 2) Ricevuti G, et al. Meningeal leukemia diagnosed by cytocentrifuge study of cerebrospinal fluid. Arch Neurol. 1986;43:466-470; 3) Davey DD, et al. Millipore filter vs cytocentrifuge for detection of childhood central nervous system leukemia. Arch Pathol Lab Med. 1986;110:705-708; 4) Clare N, Rone R. Detection of malignancy in body fluids. Lab Med. 1986;17:147-150; 5) Odom LF, et al. Significance of blasts in low-cell cerebrospinal specimens from children with acute lymphoblastic leukemia. Cancer. 1990;66:1748-1754; 6) Craver RD, Carson TH. Hematopoietic elements in cerebrospinal fluid in children. Am J Clin Pathol. 1991;95:532-535; 7) Rippin KP, et al. Clinical evaluation of the slide centrifuge (cytospin) gram's stained smear for the detection of bacteriuria and comparison with the Filtracheck-UTI and UTIscreen. Am J Clin Pathol. 1995;103:316-319; 8) Jones CD, Cornbleet PJ. Wright-giemsa cytology of body fluids. Techniques for optimal cytocentrifuge slide preparation. Lab Med. 1997;28:713-716; 9) Kleine TO, Lehmitz R. Evaluation of cytodiagnosis of cerebrospinal fluid (CSF) cells. Clin Chem. 2000;46:A137. LSV.43740 Phase II N/A YES NO Is the quality of body fluid smears satisfactory (uniform cell distribution, appropriate dilution so cells are not crowded, properly stained, adequate cell yield, ready recognition of cell types that are reported)? COMMENTARY: N/A

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 136 of 178
REFERENCE: Jones CD, Cornbleet PJ. Wright-giemsa cytology of body fluids. Techniques for optimal cytocentrifuge slide preparation. Lab Med. 1997;28:713-716. LSV.43746 Phase II N/A YES NO Does the laboratory have a documented system to ensure consistency of morphologic observations among all personnel performing body fluid cell differentials? NOTE: The section of the laboratory performing body fluid cell differentials must have a documented system that ensures that all personnel report microscopic morphologic data on patient samples in a similar fashion. For serial samples from the same patient, the laboratory must be able to document that all of its staff are consistent with respect to morphologic classification. Suggested methods to accomplish this include:
1. Circulation of body fluid smears with defined nucleated cell differential distributions, and/or
2. Multi-headed microscopy, and/or 3. Use of body fluid photomicrographs with referee and consensus identifications (e.g.,
former CAP Surveys photomicrographs) COMMENTARY: N/A REFERENCES: 1) College of American Pathologists. Surveys hematology glossary. Northfield, IL: CAP, 1999:37-51; 2) Eisner J, et al. Body fluid tutor: an image-based computer program to teach the laboratory analysis of body fluids. Am J Clin Pathol. 2000;114:299. LSV.43752 Phase II N/A YES NO Are slides with suspected malignant cells reviewed by a pathologist or other qualified physician before final results reporting? COMMENTARY: N/A LSV.43758 Phase I N/A YES NO If a body fluid specimen is examined in more than one area of the laboratory, is there a mechanism to compare the data and interpretations from these different areas, particularly when a diagnosis of malignancy is rendered?

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 137 of 178
COMMENTARY: N/A REFERENCES: 1) Clare N, Rone R. Detection of malignancy in body fluids. Lab Med. 1986;17:147-150; 2) Walts AE, Strigle S. Toward optimal use of the cytology laboratory: quality improvement and cerebrospinal fluid specimens. Diagn Cytopathol. 1995;13:357-361. LSV.43764 Phase I N/A YES NO Is there a file of unusual slides and/or an atlas of body fluid cytomorphology readily available to the technologist evaluating the slides, to assist in the identification of cell types? COMMENTARY: N/A REFERENCES: 1) Kolmer HW. Atlas of cerebrospinal fluid cells, 2nd ed. New York, NY: Springer-Verlag, 1977; 2) Dieppe PA, et al. Synovial fluid crystals. Quart J Med. 1979;192:533-553; 3) Glasser L. Body fluids II. Reading the signs in synovia. Diag Med. 1980;3(6):35-50; 4) Glasser L. Body fluids III. Tapping the wealth of information in CSF. Diag Med. 1981;4:23-33; 5) Greening SE, et al. Differential diagnosis in effusion cytology. J Med Tech. 1984;1:885-895; 6) Strasinger SK. Urinalysis and body fluids. A self-instructional text. Philadelphia: FA Davis, 1985:134-186; 7) Hyun BH, Salazar GH. Cerebrospinal fluid cells in leukemias, lymphomas, and myeloma. Lab Med. 1985;16: 667-670; 8) Kjeldsberg CR, Knight JA. Body fluids, 3rd ed. Chicago, IL: American Society of Clinical Pathology, 1993. LSV.43770 Phase I N/A YES NO Are slides retained for future reference? NOTE: All body fluid smears should be retained for at least one week for possible review/reference. Significantly abnormal smears (e.g., those demonstrating microorganisms, cytologically suspicious or overtly malignant cells, etc.) should be retained for longer periods, subject to space availability. COMMENTARY: N/A

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 138 of 178
----------------------------------------------------------- SEMEN ANALYSIS ----------------------------------------------------------- The preceding items in the Body Fluid Cell Counting and Body Fluid Nucleated Cell Differentials are generally applicable to semen analysis. Additional items of importance to this specialized area are identified in this section. LSV.43773 Phase II N/A YES NO For azoospermic specimens, as well as post-vasectomy checks for reproductive sterility, is a concentrating technique employed on seminal fluid? NOTE: Without a concentration technique, the presence of both motile and non-motile sperm may not be detected. COMMENTARY: N/A REFERENCES: 1) Jones CD, Cornbleet PJ. Wright-Giemsa cytology of body fluids. Techniques for optimal cytocentrifuge slide preparation. Lab Med. 1997;28:713-716; 2) Jaffe TM, et al. Sperm pellet analysis: a technique to detect the presence of sperm in men considered to have azoospermia by routine semen analysis. J Urol. 1998;159:1548-1550. NOTE TO INSPECTOR: If the laboratory performs only post-vasectomy checks, mark the remaining semen analysis questions N/A. ................................................................. Requisitions, Specimen Receipt and Results Reporting ........................…..................................... LSV.43776 Phase II N/A YES NO Are there specific patient instructions for collection and prompt delivery of a semen sample to the laboratory? NOTE: This should be written in simple terms in a language readily understood by the patient. Elements should include the need to abstain from ejaculation for 2-7 days before collection of the specimen, avoidance of lubricants and other contamination, completeness of collection, use of the supplied container, maintenance of sample temperature, and prompt delivery.

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 139 of 178
COMMENTARY: N/A LSV.43782 Phase II N/A YES NO Are semen specimens accompanied by the following collection information, and are records maintained on the following?
1. Method of collection 2. Type of specimen container 3. Days of abstinence 4. Collection or transport problems (e.g., incomplete specimen, exposure to
temperature extremes) 5. Abnormalities of liquefaction 6. Time of specimen receipt and analysis
NOTE: The Inspector must provide specific details of any deficiencies in Part B (Deficiency Summary) of the Inspector's Summation Report. COMMENTARY: N/A REFERENCE: World Health Organization laboratory manual for the examination of human semen and sperm-cervical mucus interaction, fourth edition. New York: Cambridge University Press, 1999. LSV.43788 Phase II N/A YES NO Are all semen specimens given sufficient time for liquefaction before testing? COMMENTARY: N/A REFERENCE: World Health Organization laboratory manual for the examination of human semen and sperm-cervical mucus interaction, fourth edition. New York, NY: Cambridge University Press, 1999. LSV.43794 Phase II N/A YES NO Are semen specimens mixed thoroughly before testing?

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 140 of 178
COMMENTARY: N/A REFERENCE: World Health Organization laboratory manual for the examination of human semen and sperm-cervical mucus interaction, fourth edition. New York, NY: Cambridge University Press, 1999. LSV.43800 Phase II N/A YES NO Are all characteristics of the semen specimens noted and reported (e.g., gelatinous clumps, viscosity, contaminants, erythrocytes)? COMMENTARY: N/A REFERENCES: 1) Haugen TB, Grotmol T. pH of human semen. Int J. Androl. 1998;21:105-108; 2) World Health Organization laboratory manual for the examination of human semen and sperm-cervical mucus interaction, fourth edition. New York, NY: Cambridge University Press, 1999. **NEW** 09/27/2007 LSV.43802 Phase I N/A YES NO Is there an additional procedure besides unstained bright-field microscopy to ensure the accurate distinction of leukocytes from other round cells (e.g., Wright's or PAP stain, leukocyte alkaline phosphatase, myeloperoxidase)? COMMENTARY: N/A REFERENCES: 1) World Health Organization laboratory manual for the examination of human semen and sperm-cervical mucus interaction, 4th edition. New York: Cambridge University Press, 1999; 2) Fishel TJ, et al. Increased polymorphonuclear granulocytes in seminal plasma in relation to sperm morphology. Hum Reprod. 1997;12:2418-2421; 3) Zimmermann BS, et al. Relationship of bacteriological characteristics to semen indices and its influence on fertilization and pregnancy rates after IVF. Acta Obstet Gynecol Scand. 1997;76:964-968; 4) Trum JW, et al. Value of detecting leukocytospermia in the diagnosis of genital tract infection in subfertile men. Fertil Steril. 1998;70:315-319.

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 141 of 178
................................................................ Sperm Motility ................................................................ **NEW** 09/27/2007 LSV.43804 Phase I N/A YES NO Does a procedure exist to verify the sperm motility method used (e.g., video tapes/digital images of specimens with known percent motility and/or specific motion quality) and is it exercised at least semi-annually? COMMENTARY: N/A REFERENCES: 1) Mortimer D. Practical laboratory andrology. New York, NY: Oxford University Press, 1994; 2) Yeung CH, et al. A technique for standardization and quality control of subjective sperm motility assessments in semen analysis. Fertil Steril. 1997;67:1156-1158. **NEW** 09/27/2007 LSV.43807 Phase II N/A YES NO Are manual measures of percent sperm motility quantified in a standardized manner? NOTE: The laboratory must have a documented method for determining and reporting sperm motility in their procedure manual that describes how sperm are assessed and counted (percent motility) and is based on a reference method, such as the current World Health Organization (WHO) Standards. COMMENTARY: N/A REFERENCES: 1) World Health Organization laboratory manual for the examination of human semen and sperm-cervical mucus interaction, 4th edition. New York, NY: Cambridge University Press, 1999; 2) Yeung CH, et al. A technique for standardization and quality control of subjective sperm motility assessments in semen analysis. Fertil Steril. 1997;67:1156-1158.

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 142 of 178
**NEW** 09/27/2007 LSV.43809 Phase II N/A YES NO Is forward progression of sperm evaluated? NOTE: This includes training and assessing the competency of personnel to perform and document the forward progression. COMMENTARY: N/A REFERENCES: 1) World Health Organization laboratory manual for the examination of human semen and sperm-cervical mucus interaction, 4th edition. New York, NY: Cambridge University Press, 1999; 2) Vulcano GJ, et al. A lineal equation for the classification of progressive and hyperactive spermatozoa. Math Biosci. 1998;149:77-93. LSV.43812 Phase II N/A YES NO Is sperm motility percent and progression routinely evaluated within one hour of collection? NOTE: Exceptions must be documented on the final report. COMMENTARY: N/A REFERENCE: World Health Organization laboratory manual for the examination of human semen and sperm-cervical mucus interaction, fourth edition. New York, NY:Cambridge University Press, 1999. LSV.43818 Phase II N/A YES NO Has the laboratory established a standard specimen temperature range for semen analysis assessment, and are deviations from this temperature noted on the report? NOTE: The laboratory must establish a standard specimen temperature range for semen evaluation assessment, and deviations from this temperature must be noted on the report, since specimen motility is temperature-dependent. Specimens may be maintained at 37ºC by use of an incubator and heated microscopic stage. Alternatively, a defined room temperature range is acceptable. COMMENTARY:

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 143 of 178
N/A REFERENCE: World Health Organization laboratory manual for the examination of human semen and sperm-cervical mucus interaction, fourth edition. New York, NY: Cambridge University Press, 1999. LSV.43824 Phase II N/A YES NO Does the laboratory have an appropriate procedure for evaluating a sufficient number of separate and randomly chosen microscopic fields and motile sperm cells? COMMENTARY: N/A REFERENCE: World Health Organization laboratory manual for the examination of human semen and sperm-cervical mucus interaction, fourth edition. New York, NY: Cambridge University Press, 1999. **NEW** 09/27/2007 LSV.43833 Phase I N/A YES NO Does the laboratory perform viability testing on specimens with low percent motility (e.g., less than 30%)? NOTE: Non-motile sperm may represent forms that were originally non-viable in the ejaculate, or previously motile forms that have subsequently lost motility. Thus, viability assessment is useful in making the distinction, and is commonly performed with a dye-exclusion method such as eosin-nigrosin. If viability testing is not performed on samples with decreased motility, the laboratory should include a comment on the patient report that the decreased motility may be due to either non-viable or non-motile sperm. COMMENTARY: N/A REFERENCES: 1) World Health Organization laboratory manual for the examination of human semen and sperm-cervical mucus interaction: 4th edition. New York, NY: Cambridge University Press, 1999; 2) Gunalp S, et al. A study of semen parameters with emphasis on sperm morphology in a fertile population: an attempt to develop clinical thresholds. Hum Repro. 2001;16:110-114.

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 144 of 178
................................................................ Stained Smear - Sperm Differential ................................................................ **REVISED** 09/27/2007 LSV.43842 Phase II N/A YES NO Is the sperm morphology classification method used indicated on the report? NOTE: Different classification systems have different reference intervals for normality. To improve the consistency and usefulness of reporting, CAP recommends the use of the most current edition of the WHO Standards and the Kruger classification system, and discontinuing the use of older classification systems. COMMENTARY: N/A REFERENCES: 1) Kruger, T.F., et al. Sperm morphology features as a prognostic factor in vitro fertilization. Fertility and Sterility 46:1118-1123, 1986; 2) World Health Organization laboratory manual for the examination of human semen and sperm-cervical mucus interaction, fourth edition. New York, NY: Cambridge University Press, 1999; 3) Gunalp S, et al. A study of semen parameters with emphasis on sperm morphology in a fertile population: an attempt to develop clinical thresholds. Hum Repro. 2001;16:110-114. **REVISED** 04/06/2006 LSV.43848 Phase II N/A YES NO Does the laboratory have a documented system to ensure consistency of morphologic observations among all personnel performing microscopic morphologic classification of sperm and other cells? NOTE: Suggested elements of such a system may include:
1. Circulation of stained semen smears with defined specific qualitative abnormalities of sperm
2. Multi-headed microscopy 3. Use of current published references
COMMENTARY:

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 145 of 178
N/A REFERENCES: 1) Souter VL, et al. Laboratory techniques for semen analysis; a Scottish survey. Health Bull (Edinb). 1997;55:140-149; 2) Baker DJ, Witmyer J. Semen analysis training tool. Chicago, IL: American Society of Clinical Pathologists Press, 1998; 3) World Health Organization laboratory manual for the examination of human semen and semen-cervical mucus interaction, fourth edition. New York, NY: Cambridge University Press, 1999; 4) Kruger T, Frenken D. Atlas of Human Sperm Morphology Evaluation; Taylor & Frances, 2004; 5) Glassy E. CAP Color Atlas of Hematology, 1998. LSV.43854 Phase I N/A YES NO Is an individual with expertise in sperm morphology (the pathologist, laboratory director, supervisor, or other technologist) available for consultation, when needed? COMMENTARY: N/A REFERENCE: Guidelines for human embryology and andrology laboratories. Fertil Steril. 1992;58 (suppl 1): section ii, a. LSV.43860 Phase I N/A YES NO Is there a file of unusual slides, or current reference of sperm morphology, available for training and reference? COMMENTARY: N/A REFERENCES: 1) World Health Organization laboratory manual for the examination of human semen and semen-cervical mucus interaction, 4th edition. New York, NY: Cambridge University Press, 1999; 2) Kruger T, Frenken, D. Atlas of Human Sperm Morphology Evaluation, Taylor & Frances, 2004. **NEW** 09/27/2007 LSV.43861 Phase II N/A YES NO Are stains used to facilitate classification of cell types (as opposed to performing differentials of unstained preparations)? COMMENTARY:

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 146 of 178
N/A REFERENCE: Coetzee K, et al. Predictive value of normal sperm morphology: a structured literature review. Hum Reprod Update. 1998;4:73-82. **NEW** 09/27/2007 LSV.43862 Phase II N/A YES NO Are all stains checked for contamination and reactivity each day of use? COMMENTARY: N/A REFERENCES: 1) Department of Health and Human Services, Centers for Medicare and Medicaid Services. Clinical laboratory improvement amendments of 1988; final rule. Fed Register. 2003(Jan 24):7166 [42CFR493.1256(e)(2)]; 2) Mortimer D. Practical laboratory andrology. New York, NY: Oxford University Press, 1994. **NEW** 09/27/2007 LSV.43863 Phase II N/A YES NO Are slides adequately identified? NOTE: Slide identification must include a unique specimen or accession number; patient name and/or number; date. COMMENTARY: N/A REFERENCE: Guidelines for human embryology and andrology laboratories. Fertil Steril. 1992;58 (suppl 1): IV, D. LSV.43866 Phase II N/A YES NO Are the stains used (Wright’s, Papanicolaou, eosin-nigrosin, etc.) of sufficient quality to demonstrate the cellular characteristics for which they are designed? NOTE: The selection of stains used for semen analysis is at the discretion of the director.

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 147 of 178
COMMENTARY: N/A ----------------------------------------------------------------- AUTOMATED SEMEN ANALYSIS INSTRUMENTS ----------------------------------------------------------------- Various systems are in use and some checklist items may not apply to every system. The checklist items are intended to check factors common to automated systems. Inspectors should use individual judgment in applying the questions to the particular type of system being used. **NEW** 09/27/2007 LSV.43914 Phase I N/A YES NO Where applicable, are the optical and imaging components used those recommended by the manufacturer or a validated substitution? COMMENTARY: N/A ................................................................. Calibration and Quality Control ................................................................. Several different methods may be used for calibration and quality control in the automated analysis of semen characteristics. "Calibration" techniques include use of:
1. Multiple analyzed sperm specimens 2. Stabilized preparations of sperm cells (e.g., fixed or preserved) 3. Sperm surrogates (e.g., latex particles) 4. Videotaped sperm specimens
NOTE: If stabilized control materials are used, they should represent different analytic levels (e.g., normal and high). Similarly, retained patient specimens should be of differing counts and/or motility, as applicable.

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 148 of 178
**NEW** 09/27/2007 LSV.43962 Phase II N/A YES NO Is calibration verified with materials appropriate to the reportable range of the instrument, and is verification documented? NOTE: The quality control procedure for the automated instrument must include calibration and evaluation using defined limits of agreement with manually counted semen smears or stored digital images, as appropriate for the particular system. Automated semen analysis laboratories must periodically establish that their analysis equipment is functioning correctly and there is a protocol to determine if the analysis is in control. COMMENTARY: N/A REFERENCE: Guidelines for human embryology and andrology laboratories. Fertil Steril. 1992;58(suppl 1):I, B, 6. **NEW** 09/27/2007 LSV.44010 Phase II N/A YES NO Does the laboratory perform and document calibration and quality control methods for the analyzer during each day of use, using an appropriate number of controls with varying ranges of values? COMMENTARY: N/A REFERENCE: Guidelines for human embryology and andrology laboratories. Fertil Steril. 1992;58(suppl 1):I, B, 6. **NEW** 09/27/2007 LSV.44058 Phase II N/A YES NO Does the laboratory have a procedure for recalibration of instrument parameter(s) when problems are encountered? COMMENTARY:

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 149 of 178
N/A REFERENCE: World Health Organization. Laboratory manual for the examination of human semen and sperm-cervical mucus interaction, 4th edition. New York, NY: Cambridge University Press, 1999. **NEW** 09/27/2007 LSV.44106 Phase II N/A YES NO Has the material used for calibration been validated using primary reference procedures (e.g., manual counts)? COMMENTARY: N/A REFERENCES: 1) World Health Organization. Laboratory manual for the examination of human semen and sperm-cervical mucus interaction, 4th edition. New York, NY: Cambridge University Press, 1999; 2) Krause W. [Value of computer-assisted sperm analysis (CASA). reproducibility--online documentation--prognostic value]. [Article in German]. Fortschr Med. 1996;114:470-473; 3) Tsuji T, et al. Automated sperm concentration analysis with a new flow cytometry-based device, S_FCM. Am J Clin Pathol. 2002;117:401-408. **NEW** 09/27/2007 LSV.44154 Phase II N/A YES NO If a manual method is used as the system control for automated or semi-automated sperm counts, is its accuracy verified and documented at intervals appropriate for laboratory volume? COMMENTARY: N/A REFERENCES: 1) Mortimer D. Practical laboratory andrology. New York, NY: Oxford University Press. 1994; 2) Lenzi A. Computer-aided semen analysis (CASA) 10 years later: a test-bed for the European scientific andrological community. Int J Androl. 1997;20:1-2; 3) Mahmoud AM, et al. Performance of the sperm quality analyser in predicting the outcome of assisted reproduction. Int J Androl. 1998;21:41-46; 4) Tsuji T, et al. Automated sperm concentration analysis with a new flow cytometry-based device, S_FCM. Am J Clin Pathol. 2002;117:401-408.

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 150 of 178
**NEW** 09/27/2007 LSV.44202 Phase II N/A YES NO Are tolerance limits established for the value of each quality control sample? COMMENTARY: N/A **NEW** 09/27/2007 LSV.44250 Phase II N/A YES NO If sperm motility and/or count are assessed by automated analysis, is a procedure established to determine that the concentration of the specimen is within the range appropriate for automated counting? COMMENTARY: N/A REFERENCES: 1) Vantman DD, et al. Computer-assisted semen analysis: evaluation of method and assessment of the influence of sperm concentration on linear velocity determination. Fertil Steril. 1988;49:510-515; 2) Yeung CH, et al. A technique for standardization and quality control of subjective sperm motility assessments in semen analysis. Fertil Steril. 1997;67:1156-1158; 3) Sidhu RS, et al. accuracy of computer-assisted semen analysis in prefreeze and post-thaw specimens with high and low sperm counts and motility. Urology. 1998;51:306-312; 4) Tsuji T, et al. Automated sperm concentration analysis with a new flow cytometry-based device, S_FCM. Am J Clin Pathol. 2002;117:401-408. **NEW** 09/27/2007 LSV.44298 Phase I N/A YES NO Has the laboratory established reference intervals (normal values) for the motion variables measured? COMMENTARY: N/A REFERENCE: Schieferstein G, et al. Sperm motility index. Arch Androl. 1998;40:43-48.

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 151 of 178
**NEW** 09/27/2007 LSV.44346 Phase II N/A YES NO Are upper and lower limits of all reportable parameters on instruments defined so that results that fall outside these limits are verified before reporting? NOTE: Results that fall outside of these limits may be verified by repeating the test, using an alternative method or diluting/concentrating the specimen, as appropriate. COMMENTARY: N/A REFERENCE: Mortimer D. Practical laboratory andrology. New York, NY: Oxford University Press. 1994. **NEW** 09/27/2007 LSV.44394 Phase II N/A YES NO Are criteria established for method calibration verification? NOTE: Criteria must be established for method calibration verification. Criteria for determining the need for calibration verification typically include:
1. At complete changes of reagents, unless the laboratory can demonstrate that changing reagent lots does not affect either the range used to report patient test results or the control values
2. When indicated by quality control data 3. After major maintenance or service 4. When recommended by the manufacturer 5. At least every six months
COMMENTARY: N/A REFERENCE: Department of Health and Human Services, Centers for Medicare and Medicaid Services. Clinical laboratory improvement amendments of 1988; final rule. Fed Register. 2003(Jan 24):7165 [42 CFR 493.1255].

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 152 of 178
*****************************************************************************
MICROBIOLOGY SECTION
***************************************************************************** Microbiology activities for this Checklist are LIMITED to specimen collection, direct microscopy for organisms (e.g., Gram stain for bacteria, potassium hydroxide for fungi, pinworm preparation) and initial culture plating or similar activity before transport to a microbiology laboratory. If the limited service laboratory performs any cultures (includes reporting “No growth”) or antimicrobial susceptibility testing, this Checklist is NOT sufficient, and Checklist 4 (Microbiology) must be employed. -------------------------------------------------------- SPECIMEN COLLECTION AND HANDLING -------------------------------------------------------- Culture specimens are often collected by nurses or others outside the laboratory. An important aspect of quality control is the provision of adequate instructions to ensure proper collection and handling of specimens before they are received by the laboratory. LSV.44450 Phase II N/A YES NO Are there criteria for establishing specimen acceptability? NOTE: This could include important issues such as absence of gross external contamination, adequate specimen type/quantity, suitable preservation, prevention of dried swabs, and correct use of transport media when required. COMMENTARY: N/A LSV.44530 Phase I N/A YES NO Are unacceptable sputum samples not cultured (or cultured only by special request) and is the health care provider or submitting laboratory notified so another specimen can be collected without delay, if clinically indicated? NOTE: It is suggested that the laboratory notify an appropriate caregiver about an inadequate specimen even in an outpatient setting, such as a reference laboratory. Notification can be by phone or computer report. The laboratory may implement written agreements with particular providers or submitting laboratories defining protocols for handling sputum samples.

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 153 of 178
COMMENTARY: N/A REFERENCES: 1) Bartlett RC. Medical microbiology: quality cost and clinical relevance. New York, NY: Wiley, 1974:24-31; 2) Carroll KC. Laboratory Diagnosis of Lower Respiratory Tract Infection: Controversy and Conundrums. J Clin Microbiol. 2002;3115-3120. LSV.44610 Phase II N/A YES NO Are sterile techniques for drawing and handling of blood cultures defined, made available to individuals responsible for specimen collection, and practiced? NOTE: It is recommended that blood culture statistics, including number of contaminated cultures, be maintained and reviewed regularly by the laboratory director. The laboratory should establish a threshold for an acceptable rate of contamination. Tracking the contamination rate and providing feedback to phlebotomists or other persons drawing cultures has been shown to reduce contamination rates. Other measures to monitor include types of skin disinfection, volume of blood drawn, number of culture sets drawn, number of single cultures and line draws. COMMENTARY: N/A REFERENCES: 1) Bates DW, et al. Contaminant blood cultures and resource utilization: the true consequences of false-positive results. JAMA. 1991;265:365-369; 2) Schifman RB, Pindur A. The effect of skin disinfection materials on reducing blood culture contamination. Am J Clin Pathol. 1991;99:536-538; 3) Strand CL, et al. Effect of iodophor versus iodine skin preparation on blood culture contamination rate. JAMA. 1993;269:1004-1006; 4) Spitalnic SJ, et al. The significance of changing needles when inoculating blood cultures; a meta-analysis. Clin Infect Dis. 1995;21:1103-1106; 5) Gibb AP, et al. Reduction in blood culture contamination rate by feedback to phlebotomists. Arch Pathol Lab Med. 1997;121:50-507; 6) Schifman RB, et al. Blood culture contamination. A College of American Pathologists Q-probes study involving 640 institutions and 497 134 specimens from adult patients. Arch Pathol Lab Med. 1998;122:216-221; 7) DesJardin JA, et al. Clinical utility of blood cultures drawn from indwelling central venous catheters in hospitalized patients with cancer. Ann Int Med. 1999;131:641-647; 8) Ernst DJ. Controlling blood culture contamination rates. Med Lab Observ. 2000;32(5):36-47; 9) Ruge DG, et al. Reduction of blood culture contamination rates by establishment of policy for central intravenous catheters. Lab Med. 2002;33:797-800.

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 154 of 178
**REVISED** 09/27/2007 LSV.44690 Phase I N/A YES NO Are recommendations for the appropriate volume of blood per culture available to those collecting the specimens? NOTE: In adults, at least 20 mL of blood per culture set (2 bottles) should be collected for culture. Larger volumes of blood increase the yield of true positive cultures. The laboratory should periodically monitor collected blood volumes and provide feedback to clinical staff. Automated blood culture systems approved or cleared by the FDA may use slightly smaller volumes per culture set and are acceptable. COMMENTARY: N/A REFERENCES: 1) Kellogg JA, et al. Justification and implementation of a policy requiring two blood cultures when one is ordered. Lab Med. 1994;25:323-330; 2) Li J, et al. Effects of volume and periodicity on blood cultures. J Clin Microbiol. 1994;32:2829-2831; 3) Washington J, Ilstrup. Blood cultures: issues and controversies. Rev Infect Dis. 1986;8:792-802; 4) Schiffman RB, et al. Blood culture quality improvement. A College of American Pathologists Q-probes study involving 909 institutions and 289 572 blood culture sets. Arch Pathol Lab Med. 1996;120:999-1002; 5) Arendup M, et al. Diagnosing bacteremia at a Danish hospital using one early large blood volume for culture. Scand J Infect Dis. 1996;28:609-614; 6) Wilson ML. Clinically relevant, cost-effective clinical microbiology. Strategies to decrease unnecessary testing. Am J Clin Pathol. 1997;107:154-165; 7) Goonewardene S. Evaluating the efficiency of the workup of multiple positive blood cultures for individual patients in a university hospital setting. Am J Clin Pathol. 1998;110:542; 8) Novis DA, et al. Solitary blood cultures. A College of American Pathologists Q-Probes study of 132778 blood culture sets in 333 small hospitals. Arch Pathol Lab Med. 2001;125:1290-1294. LSV.44710 Phase II N/A YES NO Are all specimens recorded in an accession book, day book, worksheet, computer or other comparable document? COMMENTARY: N/A

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 155 of 178
LSV.44740 Phase I N/A YES NO Do requests for isolation include source of specimen and, when appropriate, type of infection and/or organism expected? COMMENTARY: N/A LSV.44770 Phase II N/A YES NO Are there documented instructions for microbiology specimen collection and handling that include all of the following?
1. Method for proper collection of culture specimens from different sources 2. Need for prompt delivery of specimens to ensure minimum delay and prompt
processing (CSF, wound cultures, anaerobes) 3. Use of transport media when necessary 4. Method for preservation of specimens if processing is delayed (e.g., refrigeration of
urines) 5. Procedures for safe handling of specimens (tightly sealed containers, no external
spillage) 6. Proper labeling of culture specimens
NOTE: The Inspector must provide specific details of any deficiencies in Part B (Deficiency Summary) of the Inspector's Summation Report. COMMENTARY: N/A REFERENCE: Miller JM, et al. Specimen collection, transport, and storage. In: Murrary PR, et al, ed. Manual of Clinical Microbiology, 8th ed. Washington, DC: ASM Press; 2003. -------------------------------------------------------- REPORTING OF RESULTS -------------------------------------------------------- LSV.45180 Phase I N/A YES NO When indicated, are preliminary reports promptly generated? COMMENTARY:

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 156 of 178
N/A LSV.45200 Phase II N/A YES NO Are documented criteria established for immediate notification of a physician or other clinical personnel responsible for patient care when results of certain tests exceed critical limits important for prompt patient management decisions? NOTE: These criteria, referred to as “critical results,” are best developed by the laboratory in consultation with user clinicians. These may be indicated in the procedure manual and/or in a separate manual or policy. The bench technologists must be familiar with the critical results for the procedures that they perform. COMMENTARY: N/A REFERENCES: 1) Department of Health and Human Services, Centers for Medicare and Medicaid Services. Clinical laboratory improvement amendments of 1988; final rule. Fed Register. 2003(Jan 24):[42CFR493. 1251(b)(13); 493.1291(g)]; 2) Steindel SJ, Heard NV. Critical values: data analysis and critique. Q-Probes 92-04. Northfield, IL: College of American Pathologists, 1992; 3) Kost GJ. Using critical limits to improve patient outcome. Med Lab Observ. 1993;25(3):22-27; 4) Tate KE, Gardner RM. Computers, quality, and the clinical laboratory: a look at critical values. Proc Annu Symp Comput Appl Med Care. 1993;193-197; 5) Kaufman HW, Collins C. Notifying clients of life-threatening results. Med Lab Observ. 1994;26(8):44-45; 6) Lum G. Evaluation of a laboratory critical limit (alert value) policy for hypercalcemia. Arch Pathol Lab Med. 1996;120:633-636; 7) Emancipator K. Critical values. ASCP practice parameter. Am J Clin Pathol. 1997:108:247-253; 8) Dalton-Beninato K. Critical value notifications are never welcome news. Lab Med. 2000;31:319-323; 9) Howanitz PJ, et al. Laboratory critical values policies and procedures. A College of American Pathologists Q-probes study in 623 institutions. Arch Pathol Lab Med. 20002;126:663-669; 10) Kost GJ. Critical limits for urgent clinician notification at US medical centers. JAMA. 1990;263:704-707; 11) Department of Health and Human Services, Centers for Medicare and Medicaid Services. Clinical laboratory improvement amendments of 1988; final rule. Fed Register. 2003(Jan 24): [42CFR493. 1251(b)(13); 493.1291(g)]; 12) Steindel SJ, Heard NV. Critical values: data analysis and critique. Q-probes 92-04. Northfield, IL: College of American Pathologists, 1992; 13) Kost GJ. Using critical limits to improve patient outcome. Med Lab Observ. 1993;25(3):22-27; 14) Dalton-Beninato K. Critical value notifications are never welcome news. Lab Med. 2000;31:319-323. The following questions apply to laboratories that may not perform cultures, but do perform routine screening procedures for bacterial meningitis and Group B streptococcus (GBS). Inspectors should evaluate if there are sufficient procedures for collecting, processing, and reporting of clinical specimens submitted for bacterial meningitis screens and GBS.

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 157 of 178
LSV.45260 Phase II N/A YES NO If antigen-detection methods are used, are back-up cultures performed on both positive and negative CSF specimens? NOTE: Total dependence on a bacterial antigen test for the diagnosis of bacterial meningitis does NOT meet accreditational requirements. The sensitivity and, especially in urine, the specificity of such tests are not adequate. In addition, meningitis may be caused by bacteria not covered by the antigen tests. Thus, culture is essential for evaluation of bacterial meningitis, and must be performed on the patient specimen - if not performed by the present laboratory, the Inspector must seek evidence that culture has been performed elsewhere. COMMENTARY: N/A REFERENCES: 1) Forward KR. Prospective evaluation of bacterial antigen detection in cerebral spinal fluid in the diagnosis of bacterial meningitis in a predominantly adult hospital. Diagn Micro Infect Dis. 1988;11:61-63; 2) Maxson S, et al. Clinical usefulness of cerebrospinal fluid bacterial antigen studies. J Pediat. 1994; 125:235-238; 3) Finlay FO, et al. Latex agglutination testing in bacterial meningitis. Arch Dis Child. 1995;73:160-161; 4) Rathore MH, et al. Latex particle agglutination tests on the cerebrospinal fluid. A reappraisal. J Florida Med Assoc. 1995;82:21-23; 5) Kiska DL, et al. Quality assurance study of bacterial antigen testing of cerebrospinal fluid. J Clin Micro. 1995;33:1141-1144; 6) Perkins MD, et al. Rapid bacterial antigen detection is not clinically useful. J Clin Micro. 1995;33:1486-1491. LSV.45295 Phase I N/A YES NO Are group B streptococcus screens from pregnant women collected and cultured in accordance with the current guidelines? NOTE: In 2002, the CDC released revised guidelines for the prevention of perinatal Group B streptococcal disease. The new guidelines recommend universal prenatal screening for vaginal and rectal Group B streptococcal (GBS) colonization of all pregnant women at 35-37 weeks gestation. Procedures for collecting and processing clinical specimens for GBS culture and performing susceptibility testing to clindamycin and erythromycin are also included in the guidelines COMMENTARY: N/A REFERENCES: 1) Center for Disease Control and Prevention, 2002. Prevention of perinatal Group B streptococcal disease. MMWR 51(RR-11);1-22; 2) Baron, EJ. Laboratory support for prevention of perinatal Group B streptococcal disease: Commentary on the guidelines on screening for Group B streptococci during pregnancy. Clin Micro Newsletter. Vol 25, No 9, May, 2003; 3) Schrag, SJ, et al.

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 158 of 178
A population-based comparison of strategies to prevent early-onset Group B streptococcal disease in neonates. N Engl J Med 2002;347:233-9. -------------------------------------------------------- MEDIA -------------------------------------------------------- The laboratory has the responsibility for ensuring that all media used, whether purchased or prepared by the laboratory, are sterile, able to support growth appropriately and are appropriately reactive biochemically. This will ordinarily require that the laboratory maintain a stock of reference organisms and test the media before or concurrent with use. Explicit documentation of such testing is essential. For prepared, purchased media the laboratory must have explicit documentation that each lot of purchased medium has been tested for sterility, ability to support growth of appropriate organisms and biochemical reactivity at the time of preparation or concurrent with use in the laboratory. The recipient laboratory must have a copy of National Committee for Clinical Laboratory Standards (NCCLS) Document Number M-22-A3 (Quality Assurance for Commercially Prepared Microbiological Culture Media) as a reference source. The manufacturer or preparer must document to the user that their quality control activities meet the NCCLS guidelines, or are otherwise equivalent. The laboratory director may wish to have a signed contractual arrangement with his/her selected manufacturer to cover all expected quality control and documentation. For each lot the preparer will certify that quality control performance was acceptable and maintain a record of test results and the lot numbers for ALL media for at least two years. The user laboratory may record that fact in place of the more detailed documentation of media performance. The user must visually examine each shipment for breakage, contamination, appearance, or evidence of freezing or overheating. Transportation of media/reagents under unfavorable environmental conditions may adversely affect product performance. The user laboratory must continue to test each lot of media except those listed as being exempt from such testing in the tables in M22-A3, using quality control methods employed for media manufactured in-house (which are also listed in the M22 tables). In addition, each shipment of a commercial identification system must be tested for appropriate performance. If more than one lot number is received per shipment, each lot number must be tested. The user must control media for critical reactions that are not tested by the preparer, even though the media may be exempted from testing for other purposes, as specified in M-22-A3 tables. The director is responsible for the quality and performance of media and must document all media failures and the resultant corrective action taken. LSV.45330 Phase II N/A YES NO Does the laboratory have documentation that its media supplier carries out the quality assurance guidelines enumerated in NCCLS Document M22-A3?

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 159 of 178
NOTE: The laboratory has the responsibility for ensuring that all media used, whether purchased or prepared by the laboratory, are sterile, able to support growth appropriately and are appropriately reactive biochemically. This will ordinarily require that the laboratory maintain a stock of reference organisms and test the media before or concurrent with use. Explicit documentation of such testing is essential. For prepared, purchased media, the laboratory must have explicit documentation that each lot of purchased medium has been tested for sterility, ability to support growth of appropriate organisms and biochemical reactivity at the time of preparation or concurrent with use in the laboratory. The recipient laboratory must have a copy of the NCCLS document number M22-A3 (Quality assurance for commercially prepared microbiological culture media) as a reference source. The manufacturer or preparer must document to the user that their quality control activities meet the NCCLS guidelines, or are otherwise equivalent. The laboratory director may wish to have a signed contractual arrangement with his/her selected manufacturer to cover all expected quality control and documentation thereof. For each lot, the preparer will certify that quality control performance was acceptable, and maintain a record of test results and the lot numbers for all media for at least 2 years. The user laboratory may record that fact in place of the more detailed documentation of media performance. The user must visually examine each shipment for breakage, contamination, appearance, or evidence of freezing or overheating. Transportation of media/reagents under unfavorable environmental conditions may adversely affect product performance. The user laboratory must continue to test each lot of media except those listed as being exempt from such testing in the tables in M22-A3, using quality control methods that are used for media manufactured in-house. In addition, each shipment or lot, if more than one lot number is received per shipment of a commercial identification system must be tested for appropriate performance. The user must control media for critical reactions that are not tested by the preparer, even though the media may be exempted from testing for other purposes, as specified in M22-A3 tables. The director is responsible for the quality and performance of media, and must document all media failures and the resultant corrective action taken. COMMENTARY: N/A REFERENCE: NCCLS. Quality assurance for commercially prepared microbiological culture media - third edition; approved standard M22-A3. Wayne, PA: NCCLS, 2004. LSV.45360 Phase I N/A YES NO Does the laboratory have documentation that each shipment of purchased media is examined for breakage, contamination, appearance, and evidence of freezing or overheating? COMMENTARY:

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 160 of 178
N/A LSV.45390 Phase I N/A YES NO Does the laboratory have documentation that an appropriate sample of each purchased medium that is not listed in M22-A3 as exempt from testing is checked for each of the following?
1. Ability to support growth (where applicable) by means of stock cultures or by parallel testing with previous batches
2. Biochemical reactivity, where appropriate NOTE: The Inspector must provide specific details of any deficiencies in Part B (Deficiency Summary) of the Inspector's Summation Report. COMMENTARY: N/A REFERENCE: NCCLS. Quality assurance for commercially prepared microbiological culture media - third edition; approved standard M22-A3. Wayne, PA: NCCLS, 2004. LSV.45490 Phase II N/A YES NO Are all media in visibly satisfactory condition (with expiration date, plates smooth, adequately hydrated, uncontaminated, appropriate color and thickness, tubed media not dried or loose from sides)? COMMENTARY: N/A -------------------------------------------------------- DIRECT SPECIMEN EXAMINATION -------------------------------------------------------- LSV.45570 Phase II N/A YES NO Are reference organisms used to check stains, reagents and susceptibility test methods? COMMENTARY:

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 161 of 178
N/A REFERENCE: Jones RN, et al. Method preferences and test accuracy of antimicrobial susceptibility testing. Updates from the College of American Pathologists microbiology surveys program (2000). Arch Pathol Lab Med. 2001;125:1285-1289. LSV.45650 Phase II N/A YES NO Are control specimens tested in the same manner and by the same personnel as patient samples? NOTE: It is implicit in quality control (QC) that control specimens are tested in the same manner as patient specimens. Moreover, QC specimens must be analyzed by personnel who routinely perform patient testing. This does not imply that each operator must perform QC daily, so long as each instrument and/or test system has QC performed at required frequencies, and all analysts participate in QC on a regular basis. To the extent possible, all steps of the testing process must be controlled, recognizing that preanalytic and postanalytic variables may differ from those encountered with patients. COMMENTARY: N/A REFERENCE: Department of Health and Human Services, Centers for Medicare and Medicaid Services. Clinical laboratory improvement amendments of 1988; final rule. Fed Register. 2003(Jan 24):7166 [42CFR493. 1256(d)(8)]. 1256(f)]. **REVISED** 09/27/2007 LSV.45676 Phase II N/A YES NO For direct antigen tests on patient specimens that DO include internal controls, is a positive and negative external control tested and documented with each new kit lot number or separate shipments of a given lot number? NOTE: Manufacturers' recommendations must be followed. For direct antigen tests on patient specimens that include an internal control, a positive and negative external control (organism or antigen extract) must be tested and documented with each new kit lot number or separate shipments of a given lot number. Internal quality control tests can be used for subsequent testing, based upon manufacturers' recommendations. For panels or batteries, controls must be employed for each antigen sought. For tests classified as "high complexity" under CLIA-88, or that include an extraction phase, the system must be checked each day of use with a positive organism. COMMENTARY:

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 162 of 178
N/A LSV.45702 Phase II N/A YES NO For direct antigen tests on patient specimens that do NOT include internal controls, is a positive and negative control tested and documented each day of patient testing? NOTE: For panels or batteries, controls must be employed for each antigen sought in patient specimens. For each test system that requires an antigen extraction phase, the system must be checked with an appropriate positive control that will detect problems in the extraction process. COMMENTARY: N/A REFERENCE: Department of Health and Human Services, Centers for Medicare and Medicaid Services. Fed Register. 2003(Jan 24):3708 [42CFR493.1256(d)]. LSV.45730 Phase II N/A YES NO Are the results of controls reviewed for acceptability before reporting patient results? COMMENTARY: N/A REFERENCE: Department of Health and Human Services, Centers for Medicare and Medicaid Services. Clinical laboratory improvement amendments of 1988; final rule. Fed Register. 2003(Jan 24):7166 [42CFR493. 1256(f)]. --------------------------------------------------- STAINS --------------------------------------------------- **REVISED** 09/27/2007 LSV.45810 Phase II N/A YES NO Is the gram staining procedure checked and recorded for each new batch of stains and at least weekly against known gram-positive and gram-negative control organisms?

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 163 of 178
NOTE: Personnel who perform gram stains infrequently should run a gram-positive and gram-negative control each day of testing. COMMENTARY: N/A REFERENCES: 1) August, Hindler, Huber, Sewell. Quality control and quality assurance practices. In: Clinical microbiology, Cumitech 3A. Washington, DC: American Society for Microbiology, 1990; 2) Department of Health and Human services, Centers for Medicare and Medicaid Services. Clinical laboratory improvement amendments of 1988, final rule. Fed Register. 2003(Jan 24):3708 [42CFR493.1261(a)(2)]. LSV.45890 Phase II N/A YES NO Are all non-immunofluorescent, non-immunologic-based stains (other than Gram stains) checked with a positive control and negative control for intended reactivity each day of use, and for each new batch, lot number and shipment? COMMENTARY: N/A **REVISED** 10/31/2006 LSV.45900 Phase II N/A YES NO Does the microbiology section of the laboratory have a defined, documented system to ensure consistency of microscopic observations/interpretations among all personnel performing gram and other organism stains? NOTE: Suggested methods to accomplish this include:
1. Circulation of organisms with defined staining characteristics, and/or 2. Multi-headed microscopy, and/or 3. Use of photomicrographs with referee and participant identifications (e.g., former CAP
microbiology surveys or other photomicrographs from teaching collections) COMMENTARY: N/A REFERENCE: Flournoy DJ. Interpreting the sputum gram stain report. Lab Med. 1998;29:763-768.

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 164 of 178
LSV.45910 Phase II N/A YES NO Are fluorescent stains checked for positive and negative reactivity each time of use? COMMENTARY: N/A REFERENCE: Department of Health and Human Services, Centers for Medicare and Medicaid Services. Clinical laboratory improvement amendments of 1988; final rule. Fed Register. 2003(Jan 24):7146 [42CFR493. 1256(e)(3); 493.1273(a)]. --------------------------------------------------- BIOSAFETY --------------------------------------------------- Items in this section apply to ALL areas of the microbiology laboratory. Additional items for specific subsections (bacteriology, mycobacteriology, mycology, parasitology, and virology) are found under the Laboratory Safety subsections for each of those areas. If any of these safety deficiencies apply only to specific subsections of the laboratory, the Inspector must so specify in Part B of the Inspector's Summation Report. LSV.45911 Phase I N/A YES NO Does the microbiology laboratory have policies and procedures for the recognition of isolates that may be used as agents of bioterrorism? NOTE: Microorganisms likely to be utilized as biological weapons include Bacillus anthracis (anthrax), Brucella species (brucellosis), Clostridium botulium (botulism), Francisella tularensis (tularemia), Yersinia pestis (plague) and variola major (smallpox). As part of an institution-wide plan to prepare and respond to a bioterrorism event, the microbiology laboratory should have policies and procedures for the recognition of isolates that may be used as agents of bioterrorism. COMMENTARY: N/A REFERENCES: 1) Snyder JW. Role of the hospital-based microbiology laboratory in preparation and response to a bioterrorism event. J Clin Microbiol. January, 2003; 2) Gilchrist MJR. Laboratory Safety, Management, and Diagram of Biological Agents Associated with Bioterrorism; 3) Robinson-

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 165 of 178
Dunn B. The microbiology laboratory’s role in response to bioterrorism. Arch Pathol Lab Med. March 2002; 126; 4) Morse SA. Bioterrorism: Laboratory Security. Lab Med. June 2001. LSV.45912 Phase I N/A YES NO Does the laboratory participate in the institution’s bioterrorism response plan? COMMENTARY: N/A REFERENCES: 1) Snyder JW. Role of the hospital-based microbiology laboratory in preparation and response to a bioterrorism event. J Clin Microbiol. January, 2003; 2) Gilchrist MJR. Laboratory Safety, Management, and Diagram of Biological Agents Associated with Bioterrorism; 3) Robinson-Dunn B. The microbiology laboratory’s role in response to bioterrorism. Arch Pathol Lab Med. March 2002; 126; 4) Morse SA. Bioterrorism: Laboratory Security. Lab Med. June 2001. LSV.45915 Phase II N/A YES NO Are there documented policies for handling spills of contaminated materials? COMMENTARY: N/A REFERENCE: Department of Health and Human Services. Biosafety in Microbiological and Biomedical Laboratories, 4th ed. Washington, DC: Superintendent of Documents, Document 017-040-00523-7, May 1993. LSV.45920 Phase II N/A YES NO Is there documentation of daily decontamination of bench tops? COMMENTARY: N/A LSV.45925 Phase II N/A YES NO Are there documented policies and procedures for the safe handling and processing of specimens?

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 166 of 178
NOTE: Suggested topics to be considered in the policies and procedures for the safe handling and processing of specimens include the need for tight sealing of containers, avoiding spills of hazardous materials, requirements for wearing gloves, the need for respirator protection, availability and use of vaccinations, and the potential hazards of sniffing plates. COMMENTARY: N/A REFERENCES: 1) Jamison R, et al. Laboratory Safety in Clinical Microbiology, Cumitech 29, July 1996, ASM Press; Washington DC; 2) Fleming DO, Hunt DL. Biological Safety, Principles and Practices, 3rd ed. ASM Press; Washington DC. LSV.45930 Phase II N/A YES NO Have policies and procedures been developed to minimize the occupational risk of exposure to infectious agents handled in the Microbiology laboratory, in accordance with current recommendations regarding the biosafety levels for working with different organisms? NOTE: The four biosafety levels (B.S.L.) for working with infectious agents are described in the CDC-NIH guidelines (Biosafety in Microbiological and Biomedical Laboratories. U.S. Dept. of Health and Human Services, third edition, 1993). Each B.S.L. consists of combinations of equipment, procedures and techniques, and laboratory design that are appropriate for the type of laboratory and infectious agent handled. The laboratory director is responsible for the maintenance of appropriate precautions in the laboratory to minimize the risk of laboratory personnel infection, based on the types of organisms tested and the nature of the studies performed. COMMENTARY: N/A REFERENCES: 1) Biosafety in microbiological and biomedical laboratories, fourth edition. Washington, DC: U.S. Dept Health and Human Services, 1999; 2) Richmond J. "Arthropod Borne Diseases", Anthology of Biosafety VI: American Biological Safety Association, Mundelein, IL April 2003. LSV.45940 Phase II N/A YES NO Are engineering and work practice controls appropriate to the Biosafety level of the laboratory defined and implemented? NOTE: Each increasing B.S.L. number (BSL-1 to BSL-4) implies increased occupational risk from exposure to an agent or performance of a procedure, and therefore is associated with more stringent control and containment practices.

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 167 of 178
COMMENTARY: N/A REFERENCES: 1) Biosafety in microbiological and biomedical laboratories, fourth edition. Washington, DC: U.S. Dept Health and Human Services, 1999; 2) Richmond J. BSL-4 Laboratories. Anthology of Biosafety V: American Biological Safety Association, Mundelein, IL January 2002; 3) Richmond J. Biosafety Level 3. Anthology of Biosafety VII: American Biological Safety Association, Mundelein, IL December 2003. LSV.46003 Phase II N/A YES NO Is a biological safety cabinet available and properly used for handling specimens or organisms considered highly contagious by airborne routes? COMMENTARY: N/A REFERENCE: Department of Health and Human Services. Biosafety in microbiological and biomedical laboratories, 4th edition. Washington, DC: Superintendent of Documents, Document 017-040-00523-7, May 1993. LSV.46066 Phase II N/A YES NO Is the biologic safety cabinet certified at least annually to ensure that filters are functioning properly and that airflow rates meet specifications? COMMENTARY: N/A
*****************************************************************************
IMMUNOLOGY/IMMUNOHEMATOLOGY SECTION
***************************************************************************** If the limited service laboratory performs any immunohematology (blood bank) testing beyond blood group typing (ABO and Rh), antibody screens and direct antiglobulin testing (DAT), this Checklist is not sufficient.

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 168 of 178
LSV.46130 Phase II N/A YES NO Are new reagent lots and/or shipments checked against old reagent lots or with suitable reference material before or concurrently with being placed in service? NOTE: New reagent lots and/or shipments must be tested in parallel with old lots before or concurrently with being placed in service to ensure that the calibration of the new lot of reagent has maintained consistent results for patient specimens. Good clinical laboratory science includes patient-based comparisons when possible, since it is patient specimens that are tested. For quantitative tests, reagent validation is most reliably performed by assaying the same patient specimens with both the old and new lots to ensure consistent results. For qualitative tests, minimum cross-checking includes retesting at least one known positive and one known negative patient sample from the old reagent lot against the new reagent lot, ensuring that the same results are obtained with the new lot. A weakly positive sample should also be used in systems where patient results are reported in that fashion. Some method manufacturers provide reference materials or QC products specifically intended to validate successful calibration of their methods; these should be used when available. Such materials have method-specific, and, where appropriate, reagent-lot-specific, target values. Thus, these materials should be used only with the intended methods. Proficiency testing materials with peer group established mean values are acceptable for validation of new reagent lots. Third party general-purpose reference materials may be suitable for validation of calibration following reagent lot changes if the material is documented in the package insert or by the method manufacturer to be commutable with patient specimens for the method. A commutable reference material is one that gives the same numeric result, as would a patient specimen containing the same quantity of analyte in the analytic method under discussion; e.g., matrix effects are absent. Commutability between a reference material and patient specimens can be demonstrated using the protocol in NCCLS EP14-A2. QC material may be an acceptable alternative for validating calibration following reagent lot changes. However, the laboratory should be aware that QC material may be affected by matrix interference between different reagent lots. Thus, even if QC results show no change following a reagent lot change, a calibration inconsistency for patient specimens could exist nonetheless, masked by matrix interference affecting the QC material (a “false negative” validation). The use of patient samples to confirm the absence of matrix interference may be helpful. COMMENTARY: N/A REFERENCE: Clinical and Laboratory Standards Institute. Evaluation of Matrix Effects; Approved Guideline—Second Edition. CLSI document EP14-A2 (ISBN 1-56238-561-5). Clinical and Laboratory Standards Institute, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 190871898, USA, 2005.

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 169 of 178
LSV.46370 Phase II N/A YES NO Are package inserts for the typing sera in use available and are typing sera used according to the manufacturers' directions, or if alternative procedures are used, have they been evaluated to justify the changes? NOTE: Testing methods used for ABO, Rh and antibody screening that are different from the manufacturer's instructions, are acceptable provided they are not prohibited by the manufacturer, have been demonstrated to be satisfactory, or have been approved by the U.S. Centers for Biologics Evaluation and Research (CBER). COMMENTARY: N/A REFERENCE: Food and Drug Administration. Guide to inspections of blood banks, Sep 1994. **REVISED** 09/27/2007 LSV.46450 Phase II N/A YES NO Do records document acceptable reactivity and specificity of typing sera and reagent cells on each day of use, including a check against known positive and negative cells or antisera, or are manufacturer’s directions for daily quality control followed? NOTE: Unless manufacturer instructions state otherwise, the following apply:
• Each cell used for antibody detection must be checked each day of use for reactivity of at least one antigen using antisera of 1+ or greater avidity.
• Typing reagents such as anti-D, anti-K, anti-Fy(a), etc., must be checked each day of use.
• Anti-IgG reactivity of antiglobulin reagents may be checked during antibody screening and
crossmatching.
• Typing sera and reagent cells must be checked for reactivity and specificity on each day of use, including a check against known positive and negative cells or antisera.
This checklist requirement can be satisfied by testing one vial of each reagent lot each day of testing. COMMENTARY: N/A

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 170 of 178
REFERENCE: Department of Health and Human Services, Centers for Medicare and Medicaid Services. Clinical laboratory improvement amendments of 1988; final rule. Fed Register. 1992(Feb 28):7171 [42 CFR 493.1269]. LSV.46530 Phase II N/A YES NO Are criteria for agglutination and/or hemolysis defined? NOTE: Criteria must be defined in the procedure manual to provide uniformity of interpretation of positive and negative agglutination and hemolysis results. COMMENTARY: N/A LSV.46540 Phase II N/A YES NO Are observations of all test results recorded properly at the time done? NOTE: Test results must be recorded at the time done in order to reduce the risk of transcription errors from delayed recording. COMMENTARY: N/A LSV.46560 Phase II N/A YES NO Are appropriate control(s) used for anti-D testing? NOTE: High protein anti-D reagents require concurrent testing of an inert preparation of the manufacturer's diluent. Monoclonal anti-D reagents do not ordinarily require a separate reagent control. Incorrect assignment of a D-positive phenotype can be ruled out by observing negative reactions in any tube containing red cells and patient serum, or, alternatively, patient cells suspended in 5% bovine albumin. COMMENTARY: N/A

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 171 of 178
**NEW** 04/06/2006 LSV.46565 Phase II N/A YES NO Are ABO, Rh, and antibody screen test results compared against the same tests performed previously to detect discrepancies and identify patients requiring specially selected units? NOTE: Comparison of records of previous ABO and Rh typing are an essential step in compatibility testing. Available laboratory records for each patient must be routinely searched whenever compatibility testing is performed. If no record of the patient’s blood type is available from previous determination(s), the transfusion service should be aware that there is an increased probability of an incorrect blood type assignment and, consequently, of a hemolytic transfusion reaction. COMMENTARY: N/A **NEW** 04/06/2006 LSV.46570 Phase II N/A YES NO Are records available that document investigation and reconciliation of all cases in which ABO and Rh typing results were not in accord with the patient's historical record? NOTE: Available laboratory records for each patient must be routinely searched whenever compatibility testing is performed. Quality management records must include an investigation of all cases in which the ABO or Rh typing was not in accordance with the patient's laboratory historical record. COMMENTARY: N/A **NEW** 04/06/2006 LSV.46575 Phase I N/A YES NO When a direct antiglobulin test is ordered by a patient's physician, does the test system allow detection of RBC-bound complement as well as IgG? NOTE: This ensures detection of patients with paroxysmal cold hemoglobinuria, cold hemagglutinin disease (CHAD), warm autoimmune hemolytic anemia, and drug-induced hemolytic anemia. For the purpose of diagnosing hemolytic disease of the newborn, use of anti-C3’d is not required.

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 172 of 178
Patients with cold hemagglutinin disease, anemia related to some immune complex-mediated hemolytic states (e.g., drug-induced immune complex hemolytic anemia), and up to 15% of patients with warm antibody autoimmune hemolytic anemia may have a positive direct antiglobulin test from complement coating of their red cells, without detectable IgG on the red cells. COMMENTARY: N/A REFERENCES: 1) Sokol RJ, et al. Autoimmune haemolysis: an 18-year study of 865 cases referred to a regional transfusion centre. Brit Med J. 1981;282:2023-2027; 2) Packman CH, Leddy JP, Cryopathic hemolytic syndromes. In: Beutler E, et al, eds. William's Hematology, 5th ed. New York: McGraw-Hill, 1995:685-691; 3) Vengelen-Tyler V, ed. American Association of Blood Banks Technical Manual, 13th ed. Bethesda, MD: AABB Press, 1999:259-262. LSV.46580 Phase II N/A YES NO When performing an antiglobulin test with anti-IgG or polyspecific antiglobulin reagents, are IgG-coated red blood cells used in all negative antiglobulin tests? NOTE: IgG-coated red blood cells must be used to confirm all negative antiglobulin tests when the antiglobulin reagent used for testing has anti-IgG reactivity. Tests found negative by tube methodology must be checked with the addition of appropriate check cells. If a licensed system is used that does not require the use of IgG- coated cells, an appropriate quality control system must be followed, as recommended by the manufacturer. COMMENTARY: N/A LSV.46583 Phase I N/A YES NO When performing an antiglobulin test with anti-C3 antiglobulin reagents, are C3-coated red blood cells used in all negative antiglobulin tests? NOTE: Complement-coated red blood cells should be used to confirm all negative antiglobulin tests when the antiglobulin reagent used for testing has anti-C3 reactivity. Tests found negative by tube methodology should be checked with the addition of appropriate check cells. If a licensed system is used that does not require the use of C3-coated cells, an appropriate quality control system must be followed, as recommended by the manufacturer. COMMENTARY:

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 173 of 178
N/A ----------------------------------------------------------- SYPHILIS SEROLOGY ----------------------------------------------------------- LSV.46587 Phase II N/A YES NO If antigen is delivered by needles, is the volume of delivery checked under each of the following circumstances?
1. Each time a new needle is used 2. When control patterns cannot be reproduced 3. When the antigen drop does not fall cleanly from the tip
NOTE: The Centers for Medicare and Medicaid Services (CMS) has adopted the following Centers for Disease Control (CDC) recommendations for checking needles used for the RPR and syphilis-related cardiolipin-based tests (e.g., toluidine red unheated serum test [TRUST]). RPR needles must be checked under each of the following circumstances:
1. Each time a new needle is used 2. When control patterns cannot be reproduced 3. When a drop of antigen does not drop cleanly from the tip of the needle
COMMENTARY: N/A LSV.46594 Phase II N/A YES NO Is a negative control, plus positive serum controls of known titer or controls of graded reactivity run each day of patient testing? NOTE: A negative control plus positive serum controls of known titer must be run each day of patient testing. If the laboratory reports graded patient results, then graded controls must be run. COMMENTARY: N/A REFERENCE: Department of Health and Human Services, Centers for Medicare and Medicaid Services. Clinical laboratory improvement amendments of 1988; final rule. Fed Register. 2003(Jan 24):3708 [42 CFR 493.1256(d)(3)].

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 174 of 178
**REVISED** 04/06/2006 LSV.46601 Phase II N/A YES NO Are new reagent lots of antigen for VDRL, RPR, TRUST (toluidine red unheated serum test), and USR (unheated serum reagin) tests checked in parallel with reference reagents to verify that they are of standard reactivity? NOTE: New reagent lots of antigen for VDRL, RPR, TRUST, and USR tests must be checked in parallel with reference reagents to verify that they are of standard reactivity. Because the ability of a reagent to detect specimens with low grade reactivity is necessary for the diagnosis of primary syphilis, serum samples of graded reactivity, including those with weak reactivity, must be used. Reactive serum diluted with nonreactive serum to produce various degrees of reactivity may also be used. Roughness of an antigen can best be detected using fresh serum samples obtained from persons without syphilis. Prior to testing patient samples with a new reagent lot, parallel testing must be performed by using specimens of graded reactivity, including minimum/weakly reactive samples (for example, a reactive, a weakly reactive and a negative specimen). If one set of parallel tests shows borderline results, a second set of parallel tests should be performed. COMMENTARY: N/A REFERENCE: Kennedy EJ, et al. Quality Control. In, SA Larsen et al (eds). A manual of tests for syphilis, 9th ed. Washington, DC: American Public Health Association, 1998; chap 4.
*****************************************************************************
PHYSICAL FACILITIES
***************************************************************************** Sufficient space and utilities need to be provided for the overall workload of the laboratory, and to meet all safety requirements. LSV.46610 Phase I N/A YES NO Is there adequate space for administrative functions? COMMENTARY: N/A

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 175 of 178
LSV.46690 Phase I N/A YES NO Is there adequate space for clerical work? COMMENTARY: N/A LSV.46770 Phase I N/A YES NO Is there adequate space for technical work (bench space)? COMMENTARY: N/A REFERENCES: 1) https://www.irnoise.com/absa/respubs.html; 2) Richmond J. "Perspectives on Laboratory Design", Anthology of Biosafety I: American Biological Safety Association, Mundelein, IL January 1999; 3) Richmond J. "Facility Design Considerations", Anthology of Biosafety II: American Biological Safety Association, Mundelein, IL April 2000; 4) Richmond J. "Application of Principles", Anthology of Biosafety III: American Biological Safety Association, Mundelein, IL May 2000; 5) Richmond J. "Issues In Public Health", Anthology of Biosafety IV: American Biological Safety Association, Mundelein, IL September 2001. LSV.46850 Phase I N/A YES NO Is there adequate space for instruments and equipment? COMMENTARY: N/A LSV.46930 Phase I N/A YES NO Is there adequate space for shelf storage? COMMENTARY: N/A

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 176 of 178
LSV.47010 Phase I N/A YES NO Is there adequate refrigerator/freezer storage? COMMENTARY: N/A LSV.47090 Phase I N/A YES NO Is there adequate incubator space? COMMENTARY: N/A LSV.47170 Phase I N/A YES NO Is the space available efficiently utilized? COMMENTARY: N/A LSV.47250 Phase II N/A YES NO Is sufficient space available so that there is no compromise of the quality of work, (including quality control activities) or safety of personnel? COMMENTARY: N/A LSV.47330 Phase I N/A YES NO Are floors and benches clean, free of clutter and well-maintained? COMMENTARY: N/A

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 177 of 178
LSV.47650 Phase I N/A YES NO Are water taps, sinks and drains adequate for the workload of the laboratory? COMMENTARY: N/A LSV.47800 Phase I N/A YES NO Are electrical outlets adequate for the workload of the laboratory? COMMENTARY: N/A LSV.47900 Phase I N/A YES NO Is ventilation adequate for the workload and types of procedures performed in the laboratory? COMMENTARY: N/A LSV.48000 Phase I N/A YES NO Is lighting adequate? NOTE: Direct sunlight should be avoided because of its extreme variability and the need for low light levels necessary to observe various computer consoles, etc. Lighting control should be sectionalized so general levels of illumination can be controlled in areas of the room if desired. COMMENTARY: N/A LSV.48100 Phase I N/A YES NO Is temperature/humidity control adequate to support the types of procedures and workload of the laboratory? COMMENTARY:

College of American Pathologists Revised: 09/27/2007
LIMITED SERVICE LABORATORY (Web File) Page 178 of 178
N/A LSV.48300 Phase I N/A YES NO Are telephones conveniently located, and are calls easily transferred? COMMENTARY: N/A LSV.48350 Phase II N/A YES NO Is there evidence of actions to correct undesirable environmental conditions and/or inadequate utilities noted at the last on-site inspection? COMMENTARY: N/A