liquid adsorption of organic compounds on hematite fe2o3 … · 2017-09-04 · liquid adsorption of...
TRANSCRIPT

The University of Manchester Research
Liquid Adsorption of Organic Compounds on Hematite -Fe2O3 using ReaxFFDOI:10.1021/acs.langmuir.7b02374
Document VersionAccepted author manuscript
Link to publication record in Manchester Research Explorer
Citation for published version (APA):Chia, C. L., Avendano , C., Siperstein, F., & Filip, S. (2017). Liquid Adsorption of Organic Compounds on Hematite-Fe2O3 using ReaxFF. Langmuir, 33(42), 11257-11263. https://doi.org/10.1021/acs.langmuir.7b02374
Published in:Langmuir
Citing this paperPlease note that where the full-text provided on Manchester Research Explorer is the Author Accepted Manuscriptor Proof version this may differ from the final Published version. If citing, it is advised that you check and use thepublisher's definitive version.
General rightsCopyright and moral rights for the publications made accessible in the Research Explorer are retained by theauthors and/or other copyright owners and it is a condition of accessing publications that users recognise andabide by the legal requirements associated with these rights.
Takedown policyIf you believe that this document breaches copyright please refer to the University of Manchester’s TakedownProcedures [http://man.ac.uk/04Y6Bo] or contact [email protected] providingrelevant details, so we can investigate your claim.
Download date:26. Feb. 2020

Liquid Adsorption of Organic Compounds on
Hematite α-Fe2O3α-Fe2O3α-Fe2O3 using ReaxFF
Chung-Lim Chia,† Carlos Avendano,† Flor R. Siperstein,∗,† and Sorin Filip‡
†School Chemical Engineering and Analytical Science, The University of Manchester,
Sackville street, Manchester, M3 9PL, U. K.
‡BP Formulated Products Technology, Research and Innovation, Technology Centre,
Whitchurch Hill, Pangbourne, Berkshire, RG8 7QR, U.K.
E-mail: [email protected]
Abstract
ReaxFF-based molecular dynamics simulations are used in this work to study the
effect of polarity of adsorbed molecules in the liquid phase on the structure and po-
larization of hematite (α-Fe2O3). We compared adsorption of organic molecules with
different polarity on a rigid hematite surface and on a flexible and polarizable surface.
We show that the displacements of surface atoms and surface polarization in a flexible
hematite model is proportional to the adsorbed molecule’s polarity. The increase in
electrostatic interactions resulting from charge transfer in the outermost solid atoms
in a flexible hematite model results in better defined adsorbed layers, but less ordered
than those obtained assuming a rigid solid. These results suggest that care must be
taken when parametrising empirical transferable force fields as the calculated charges
on a solid slab in vacuum may not be representative of a real system, especially when
the solid is in contact with a polar liquid.
1

Introduction
Studies of liquid adsorption on solid surfaces often consider that the solid substrate is in-
ert and its structure and properties unmodified by the presence of the adsorbate. This
approximation, used since the pioneering work of Langmuir,1 remains a common practice
in modern molecular simulation studies for predicting adsorption isotherms and adsorbent
screening.2–12 This has been considered a well-founded assumption when a solid surface or
rigid porous material is put in contact with a low to moderate pressured gas,13–21 but it
has been shown to be inappropriate for modelling some complex porous materials such as
Metal Organic Frameworks (MOFs) where a flexible framework can adopt open or closed
configurations22–24 or where linkers can rotate,25 changing the porous network environment.
This assumption has also been proved to be unsuitable for the study of polymers of intrinsic
microporosity (PIM) designed to have a rigid and contorted backbone which swells upon
adsorption of carbon dioxide at high pressures.26 Nevertheless, non-porous solids are often
considered inert, regardless of the density and polarity of the liquid.27–29 In this work we
study the effect that organic molecules of different polarity have on the structure of a crystal
surface, as well as the effect on the adsorbed phase properties that results from neglect-
ing changes in the solid structure. We focus our attention to adsorption of small organic
molecules such as ethanol (C2H5OH), toluene (C6H5-CH3), and iso-octane (C8H18), which
are commonly used as solvents in the chemical industry, on hematite (α-Fe2O3) which is often
formed on the surface of iron-containing alloys30,31 that are extensively used for construction
of equipment and mechanical structures.
Molecular simulations provides a systematic and simple approach to test how different
approximations used to describe the solid affect the properties of the adsorbed liquid as it is
possible to switch on and off different interactions to assess their relevance in the observed
properties. One of the challenges in using molecular simulations to study the solid-liquid
interface, however, is the availability of suitable force fields (FFs) to describe simultaneously
the properties of both liquid and solid phases. A large effort has been devoted to the devel-
2

opment of classical force fields for metal oxides such as ClayFF32,33 and CHARMM water
contact angle,34 which have been shown to be suitable for the description of non-reactive sur-
faces. A different approach involves using ab initio methods, but they are computationally
expensive making them unsuitable to study large systems. Therefore, QM related adsorption
studies35,36 focus on single molecule adsorption or gas phase systems. In this context, the
reactive force field (ReaxFF) approach has become a powerful methodology to incorporate
chemical reactivity in classical molecular simulations.37,38 ReaxFF offers a less computation-
ally intensive alternative to quantum-based molecular dynamics (MD) simulations, and has
some advantages over classical force fields, such as the ability to model reactive systems.38
It is well known that water can dissociate on the hematite surface, thus ReaxFF is an inter-
esting approach to study the structure and properties of polar fluids on hematite, because
even when no dissociation is expected for the selected molecules, distortion in bond lengths
may be observed which would not be captured by classical force fields.
ReaxFF has been used to study several metal oxide/water interfaces.39,40 Little attention,
however, has been given to the interface between iron oxide and organic solvent molecules,
despite the industrial relevance of these systems. The key questions, which are the aim of this
present work, are to assess the effect that surface distortions can have on the properties of the
adsorbed liquid, as well as the ability of a solvent to affect the solid structure and properties.
In this work, we study the adsorption of selected organic molecules with different polarity:
ethanol, toluene and iso-octane using ReaxFF molecular dynamics (MD) simulations. We
show that the polarity of the liquid has a strong influence on the structure of the iron oxide
surface model, modifying the positions of the outermost surface atoms and their partial
charges, which can lead to larger electrostatic interactions and stronger adsorption of the
liquid, even in the absence of chemical reactions. This increase in the solid-liquid interactions
results in better defined adsorbed layers, but more disorganised than when an inert solid is
considered.
3

Methodology
Hematite (α-Fe2O3) is the most stable iron oxide at ambient conditions, and its (0001)
surface is the most stable according to DFT calculations.41 Nevertheless, this surface is
difficult to prepare experimentally, and at least three terminations have been proposed in the
literature.42 In this work we focus exclusively on the iron terminated α-Fe2O3 (0001), taking
the initial crystallographic positions of the atoms from reference 43 and the corresponding
ReaxFF parameters from the literature.40,44
ReaxFF is a bond order-based force field. The bond order is defined as a continuous
function of the interatomic distance and takes into account the σ, π and π-π bond type
contributions. Unlike classical force fields, ReaxFF includes potential energy terms that are
dependent on the bond order parameter (coordination number) and its general form is given
by:
Esys = Ebond + Elp + Eover + Eunder (1)
+Eangle + Etor + EvdW + Ecoul,
where Esys is the total energy of the system, and the bond order dependents include the Ebond
energy contribution from the formation of chemical bonds, the over coordination penalty en-
ergy Eover, the under coordination energy Eunder, the lone pair energy Elp, and angle Eangle
and torsion Etor energies. Non-bonded van der Waals EvdW and Coulombic Ecoul interac-
tions are also included. Detailed description of ReaxFF method can be found in references [
37,45]. The point charges on the atoms are allowed to vary during the MD simulation and
are calculated based on the geometry of all the atoms using the Electronegativity Equal-
ization method (EEM) developed by Mortier et al.,46 which is a methodology that is less
computationally expensive than the commonly used Charge Equilibration (Qeq) method.47
The cut off radii for both bonded and non-bonded interactions are set to 10.0 A.
4

Two different ReaxFF parametrizations were used in the present study: the FeC-FF pa-
rameterization by Zou et al.44 and the FeOH-FF parametrization developed by Aryanpour
et al.40 The FeC-FF was developed to describe the surface energy and equation of state for
iron carbides (Fe5C2 and Fe3C), and was also used to model the adsorption and desorption of
hydrocarbons on iron and cementite surfaces, but unfortunately does not contain parameters
for Fe-O interactions. The Fe-O interaction parameters were taken from the FeOH-FF which
was developed to describe the properties of iron oxides with different oxidation states, as
well as their interaction with water. This force field shows good agreement with ab initio cal-
culations and experimental data for heats of formation and lattice parameters for hematite,
goethite, lepidocrocite, akaganeite, wustite and magnetite. A combination of the two force
fields was necessary to describe the interactions of organic molecules on iron oxide. Details
of the parameters used for the simulations are in presented in the Supporting Information
(SI).
The structure of bulk hematite obtained using MD-NV T simulations (constant number
of atoms, volume and temperature) with the ReaxFF parameters provided in the SI, at 298 K
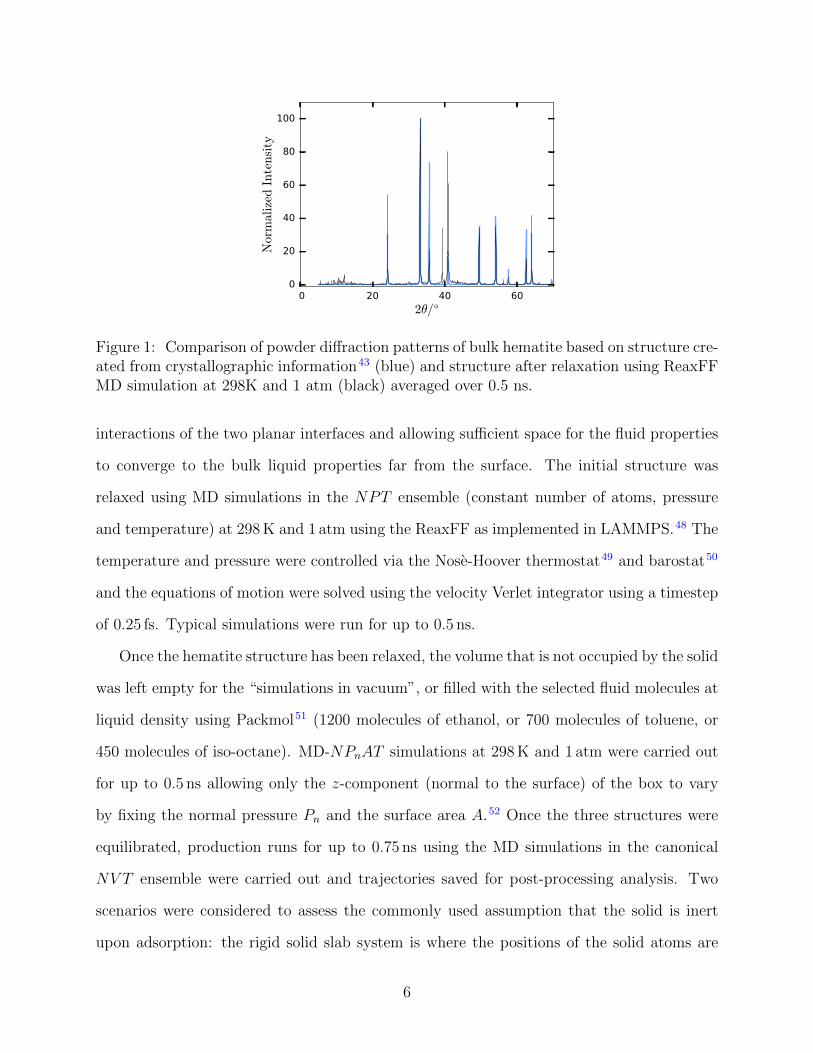
and 1 atm is in good agreement with experimental crystallographic data43 as shown in Figure
1. The powder diffraction patterns showed that locations of the major peaks are retained,
with noticeable differences in intensity for the peaks at 2θ = 24◦, 35◦, 39◦ and 41◦. Some
noise is also observed in the diffraction pattern of the relaxed structure. Further validations
of ReaxFF force field with the bulk hematite equation of state, heat of formation and lattice
parameters, as well as the properties of the fluid molecules considered in this work can be
found in the SI.
Adsorption studies were carried oun in a simulation box of dimensions 41.1 A× 35.6 A×
200.0 A containing 4×4×3 unit cells of hematite, constructed with the initial configuration of
the atoms taken from experimental crystallographic information.43 This creates a hematite
slab with the (0001) crystallographic plane perpendicular to the z direction. The large
dimension of the box in the z direction was necessary to model the surface without having self-
5
0 20 40 60
2θ/°
0
20
40
60
80
100
Nor
mal
ized
Inte
nsi
ty
Figure 1: Comparison of powder diffraction patterns of bulk hematite based on structure cre-ated from crystallographic information43 (blue) and structure after relaxation using ReaxFFMD simulation at 298K and 1 atm (black) averaged over 0.5 ns.
interactions of the two planar interfaces and allowing sufficient space for the fluid properties
to converge to the bulk liquid properties far from the surface. The initial structure was
relaxed using MD simulations in the NPT ensemble (constant number of atoms, pressure
and temperature) at 298 K and 1 atm using the ReaxFF as implemented in LAMMPS.48 The
temperature and pressure were controlled via the Nose-Hoover thermostat49 and barostat50
and the equations of motion were solved using the velocity Verlet integrator using a timestep
of 0.25 fs. Typical simulations were run for up to 0.5 ns.
Once the hematite structure has been relaxed, the volume that is not occupied by the solid
was left empty for the “simulations in vacuum”, or filled with the selected fluid molecules at
liquid density using Packmol51 (1200 molecules of ethanol, or 700 molecules of toluene, or
450 molecules of iso-octane). MD-NPnAT simulations at 298 K and 1 atm were carried out
for up to 0.5 ns allowing only the z-component (normal to the surface) of the box to vary
by fixing the normal pressure Pn and the surface area A.52 Once the three structures were
equilibrated, production runs for up to 0.75 ns using the MD simulations in the canonical
NV T ensemble were carried out and trajectories saved for post-processing analysis. Two
scenarios were considered to assess the commonly used assumption that the solid is inert
upon adsorption: the rigid solid slab system is where the positions of the solid atoms are
6

fixed in the coordinates obtained after the relaxation in vacuum, and the flexible solid slab
system where all the atoms in the solid were allowed to move during the simulation.
Different metrics were used to analyse the structure of the adsorbed liquids. The density
profile along z-axis was computed for all molecules using position of the centres of mass of
each molecule in the liquid phase. The orientational order parameters profiles P1(z) and
P2(z) along the z-direction were used to determine the order of the adsorbed molecules in
different layers. These parameters are based on first and second order Legendre polynomials
given by
P1(z) =1
Nf (z)
Nf (z)∑i=1
ui · z, (2)
and
P2(z) =1
Nf (z)
Nf (z)∑i=1
[3
2(ui · z)2 − 1
2
], (3)
where Nf (z) is the local number of liquid molecules at position z, z is the unit vector normal
to the solid slab, and ui is the unit vector describing the orientation of molecule i as defined
in Figure 2. We used the carbon-oxygen (C-O) bond to define ethanol’s orientation, a vector
perpendicular to the aromatic ring to define toluene’s orientation, and the vector connecting
the two carbons with branching points for iso-octane. Note that P1 provides information
with respect to both orientation and direction of the molecule and takes the limiting values
of 1, -1, and 0 corresponding to parallel, antiparallel, or perpendicular orientations of ui with
respect to the surface’s normal vector z, respectively.The parameter P2 provides information
only of the alignment of the molecular vectors irrespective of the direction of the molecular
axis and takes the limiting values of 1, 0, and -0.5 for full alignment, isotropic order, and
perpendicular alignment of ui with respect to z, respectively.
7

Figure 2: Definition of molecular unit vectors u used for the analysis of the orientationalorder. Molecules correspond to (a) ethanol, (b) toluene, and (c) iso-octane.
Results and Discussion
The structure of the adsorbed fluids studied (ethanol, toluene and iso-octane) on the rigid
hematite slab is shown in Figure 3. Individual simulation snapshots for the flexible solid slab
are qualitatively very similar to the images shown in Figure 3 and have not been included.
The simulation snapshots clearly show three different regions in the fluid: the contact layer,
the second layer and the “bulk” fluid. Two dimensional (2D) probability density maps for
these regions obtained as ensemble averages using the molecules’ centre of mass are also
shown in Figure 3. As one could expect, in all systems, adsorption is more localised in the
first layer, and it is evident that as the fluid polarity increases, adsorption becomes more
localized.
The formation of layers is confirmed in Figure 4 where the density profiles for all fluids in
the rigid and flexible slab systems are shown. The layers are better defined, and slightly more
intense for ethanol and toluene in the flexible slab systems, but they are shifted to slightly
larger distances. This shift is a consequence of the expansion of hematite in the presence
of ethanol and toluene. Only the outermost surface atoms of the solid are responsible for
this expansion, as the ethanol density profiles are practically indistinguishable for studies
carried out in systems with different slab thicknesses: a thin slab made of 3 unit cells in
the z−direction, and a thick slab with 5 unit cells in the z−direction. The density profiles
in systems with thin slabs shown in Figure 4 consider the origin as the slab’s center, but
8

the origin for the results of the thick slab is the slab’s center minus the length of a unit
cell in the z−direction, to enable the comparison with the thin slabs. Integral adsorption
properties can be calculated from the density profiles, including the position of the Gibbs
equimolecular surface and the integral amount adsorbed. These properties are included in
the SI for completeness.
The orientational order parameters P1 and P2 for all fluids in the rigid and flexible systems
are shown in Figure 5. The first layer of adsorbed ethanol has P1 ∼ 0 and P2 ∼ −0.5,
indicating that the C-O bond in ethanol is parallel to the surface as shown in Figure 4(a)’s
snapshot. However, P1 ∼ 0.5 and P2 ∼ −0 in the second layer, suggesting that although
a clear layer is formed, there is no significant orientational order of the molecules. It is
important to note that even though the layering is better defined for the flexible slab, the
ordering in the first layer is slightly higher in the rigid slab as shown by the higher values of
P2 in Figure 5.
The results for the orientational order parameter P1 for toluene are not particularly mean-
ingful due to the molecule’s planar symmetry, but P2 clearly shows that toluene molecules
adsorb flat on to the surface, with an average tilted angle of approximately 35◦ with respect
to the normal direction of the slab. Again, the layering peaks for toluene are better defined
in the case of flexible solid, but the first layer has higher order in the rigid slab system.
Iso-octane does not show any particular ordering on the surface, and the density profiles
are practically identical in both type of simulations, suggesting that the polarity of the
molecules indeed have an effect of the structure of hematite near the surface.
The structure of the adsorbed layers is expected to be sensitive to the approximations
made when modelling the solid surface. It is known that the structure of the adsorbent can
induce order in the structure of the liquid layer: a smooth surface can significantly favor
ordering in the adsorbed liquid,53 while rough surfaces are also known to reduce the order
of the adsorbed layers.54–57 Therefore, intuitively, one can expect that a rigid solid would
favor the formation of well defined ordered layers while the vibrational motion of surface
9

Figure 3: Snapshots and 2D probability density maps of the system consisting a rigidhematite slab in contact with (a) ethanol, (b) toluene, and (c) iso-octane. In the snapshots,the first adsorbed layer is coloured blue, second layer is coloured green and the rest arecoloured pink. The density maps show the degree of mobility in the corresponding layers.
10

0 10 20 30
r/Å
0
1
2
3
ρ/ρ
0
(a)
0 10 20 30
r/Å
0
1
2
3
ρ/ρ
0
(b)
0 10 20 30
r/Å
0
1
2
3
ρ/ρ
0
(c)
Figure 4: Density profiles along the z-axis normalized by the bulk liquid density ρ0 andvisualisations of a typical molecule’s orientation for (a) ethanol (b) toluene, and (c) iso-octane. Results obtained for the flexible slab are shown in red, while the results for the rigidslab are in black. The simulation results for ethanol on the thick solid slab is represented bya dotted line, where the origin is taken as the center of the slab minus the length of one unitcell taken from the relaxation of the hematite slab in vacuum. Vertical dashed lines indicatethe position of the outermost atoms in the fixed solid structure.
11

0 10 20 30
r/Å
-1
0
1
P1
(a)
0 10 20 30
r/Å
-1
0
1
P2
0 10 20 30
r/Å
-1
0
1
P1
(b)
0 10 20 30
r/Å
-1
0
1
P2
0 10 20 30
r/Å
-1
0
1
P1
(c)
0 10 20 30
r/Å
-1
0
1
P2
Figure 5: Order parameter P1 and P2 along the z-axis perpendicular to the surface for (a)ethanol, (b) toluene, and (c) iso-octane. Results obtained for the flexible slab are shown inred, while the results the rigid slab are in black. Vertical dashed lines indicate the positionof the outermost atoms in the fixed solid structure.
atoms would dampen the ordering of the adsorbed layer. We observed better defined layers
but with lower orientational order in the flexible hematite slab simulations than in the rigid
ones, especially for more polar fluids. This suggests that adsorption is stronger in the flexible
system resulting in better defined adsorbed layers, but that bond vibrations of the outermost
atoms may affect the orientational order of the adsorbed layers.
Results from the ReaxFF simulations in the rigid and flexible solid models show significant
differences in the bond lenghts of the surface atoms. Figure 6 shows the bond lengths of the
outermost layer of the flexible hematite slab in contact with different fluids. Note that the
Fe-O bond lengths in the rigid systems are the same as the ones reported for the flexible
system in vacuum. Despite the similarity of the distributions, it is clear that the Fe-O bond
length distribution in the presence of ethanol is non-symetrical and slightly wider than for
the other fluids, suggesting that surface bond vibrations could affect the order of ethanol
molecules on the contact layer, leading to lower values for P2. It should be noted that in
12

addition of the hematite slab expansion of approximately 0.35 A when exposed to ethanol,
the C-O bond in ethanol also increases in the first adsorbed layer by 0.14 A. The smaller
expansions of the hematite slab in the presence of toluene and iso-octane are consistent with
the similar ordering of the adsorbed molecules for the rigid and flexible slab simulations in
these systems.
1.0 1.5 2.0 2.5 3.0
Fe-O Distance / Å
0.00
0.05
0.10
0.15
Pro
bab
ility
Figure 6: Probability distribution function of Fe-O bond length in the outermost layer of theflexible solid in contact with ethanol (solid line), toluene (dotted line), iso-octane (dashedline), and vacuum (red).
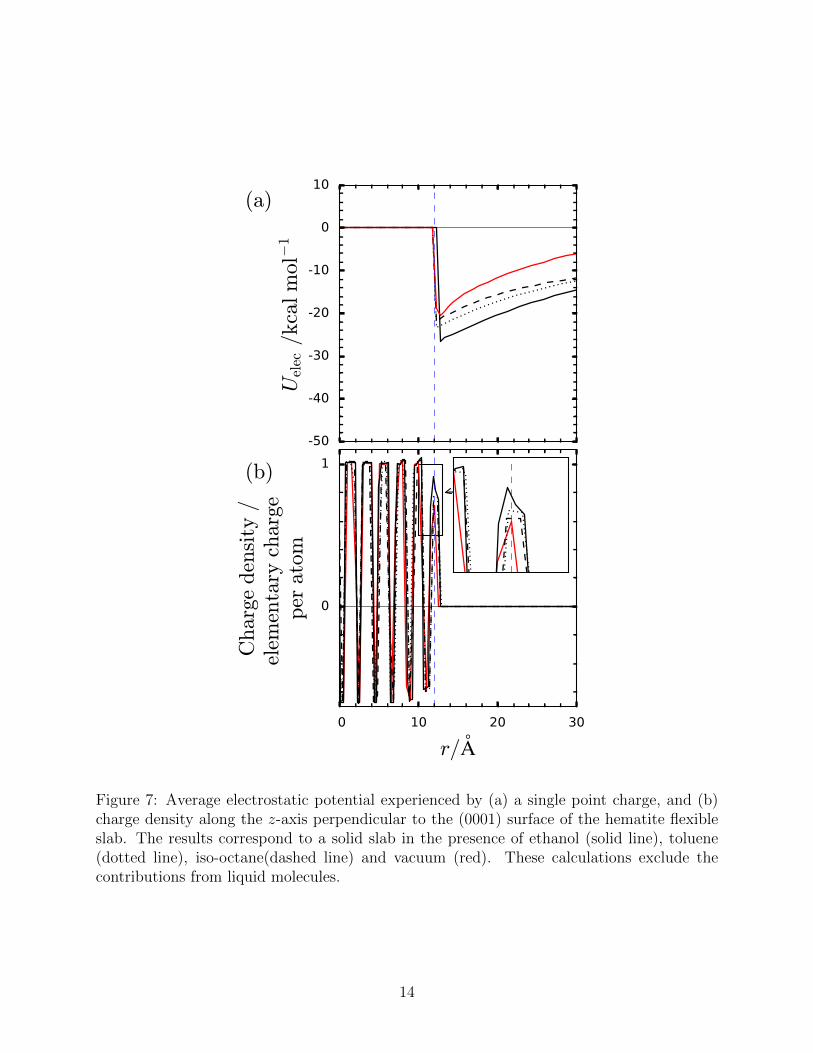
The stronger attraction of liquid molecules to the surface is a result of the increase in
non-bonded electrostatic interactions. Figure 7(a) shows that the electrostatic potential
calculated for a single point charge at a distance r from the surface increases with increasing
the fluid molecule’s polarity. The data shown represents an average potential over the x− y
plane, considering 200 configurations taken over 0.75 ns. Only the charges of the hematite
atoms were taken into account for this calculations and the contributions from all the liquid
molecules were ignored. This suggests that polar molecules are able to polarise surface atoms
in the solid slab, generating a stronger electrostatic potential and increasing the charges on
the solid surface shown in Figure 7(b). No significant difference is observed when thick slabs
are used (see SI).
13
r/Å-50
-40
-30
-20
-10
0
10
Uel
ec/k
calm
ol−
1
(a)
0 10 20 30
r/Å
0
1
Char
geden
sity/
elem
enta
rych
arge
per
atom
(b)
Figure 7: Average electrostatic potential experienced by (a) a single point charge, and (b)charge density along the z-axis perpendicular to the (0001) surface of the hematite flexibleslab. The results correspond to a solid slab in the presence of ethanol (solid line), toluene(dotted line), iso-octane(dashed line) and vacuum (red). These calculations exclude thecontributions from liquid molecules.
14

The polarization of the solid slab in ReaxFF simulations is observed as a result of the
charge transfer at the interface, which is possible because the charges in all atoms are calcu-
lated at every time step of the simulation. A similar charge transfer phenomenon has been
observed in a DFT study where the adsorption of single benzene on hematite causes the
reduction in electronic band gap due to shift in conduction band of the Fe atom that can
lead to electron transfer58 . Although there is no mention of structural distortion in the
study reported in reference 58, it can be seen from visual inspection that the interacting
iron atom protrude slightly from the surface. This induced structural change in hematite
has not been quantified using ab initio methods to the best of our knowledge.
Despite the observed charge transfer, we did not observed any dissociation of ethanol on
hematite at the conditions studied, in contrast to what has been observed of water adsorption
on hematite using first principle calculations.36 The Fe(solid)-O(H2O) and O(solid)-H(H2O)
distances observed prior to water dissociation using first principle calculations are signif-
icantly smaller than the Fe(solid)-O(ethanol) and O(solid)-H(ethanol) distances obtain in
this work with ReaxFF, preventing the path for a transition state and molecule dissociation.
In this work, we did not considered single molecule adsorption, therefore it is not possible to
indicate if the observations are a result of a collective phenomena and fluid-fluid interactions,
or that ethanol does not dissociate on hematite.
Conclusions
In this work we show that significant differences are observed in the structure of adsorbed
molecules at liquid densities when departing from the approximation of an inert solid surface.
The changes arise from the polarisation of the outermost surface atoms in the solid slab, and
they are proportional to the polarity of the adsorbed molecules, as the difference in the
first layer’s density increases with the polarity of the adsorbed molecules, i.e. ethanol >
toluene > iso-octane. This suggest that care should be taken when parameterising empirical
15

transferable force fields as the calculated charges on a solid slab in vacuum may not be
representative of a real system, especially when the solid is in contact with highly polar
liquids.
The degree of order in the adsorbed layers is larger in the rigid slab model, despite having
a lower density than in the flexible model, probably due to the solid vibrations.
Acknowledgements
The authors would like to acknowledge the funding and technical support from BP through
the BP International Centre for Advanced Materials (BP-ICAM) which made this research
possible. CLC also acknowledges the support from the University of Manchester Alumni
Scholarship. The authors would like to acknowledge the assistance given by IT Services and
the use of the Computational Shared Facility at The University of Manchester.
References
(1) Langmuir, I. The adsorption of gases on plane surfaces of glass, mica and platinum. J.
Am. Chem. Soc. 1918, 40, 1361–1403.
(2) Yazaydin, A. O.; Snurr, R. Q.; Park, T.-H.; Koh, K.; Liu, J.; LeVan, M. D.; Benin, A. I.;
Jakubczak, P.; Lanuza, M.; Galloway, D. B.; Low, J. J.; Willis, R. R. Screening of Metal-
Organic Frameworks for Carbon Dioxide Capture from Flue Gas Using a Combined
Experimental and Modeling Approach. J. Am. Chem. Soc. 2009, 131, 18198.
(3) Lin, L.-C.; Berger, A. H.; Martin, R. L.; Kim, J.; Swisher, J. A.; Jariwala, K.;
Rycroft, C. H.; Bhown, A. S.; Deem, M. W.; Haranczyk, M.; Smit, B. In silico screening
of carbon-capture materials. Nat. Mater. 2012, 11, 633–641.
(4) Wu, D.; Yang, Q.; Zhong, C.; Liu, D.; Huang, H.; Zhang, W.; Maurin, G. Revealing
16

the Structure-Property Relationships of Metal-Organic Frameworks for CO2 Capture
from Flue Gas. Langmuir 2012, 28, 12094–12099.
(5) Yu, K.; McDaniel, J. G.; Schmidt, J. R. An efficient multi-scale lattice model approach
to screening nano-porous adsorbents. J. Chem. Phys. 2012, 137, 244102.
(6) Krishna, R.; van Baten, J. M. In silico screening of metal-organic frameworks in sepa-
ration applications. Phys. Chem. Chem. Phys. 2011, 13, 10593–10616.
(7) Sun, W.; Lin, L.-C.; Peng, X.; Smit, B. Computational screening of porous metal-
organic frameworks and zeolites for the removal of SO2 and NOx from flue gases. AIChE
J 2014, 60, 2314–2323.
(8) Basdogan, Y.; Sezginel, K. B.; Keskin, S. Identifying Highly Selective Metal Organic
Frameworks for CH4/H2 Separations Using Computational Tools. Ind. Eng. Chem. Res.
2015, 54, 8479–8491.
(9) Li, S.; Chung, Y. G.; Snurr, R. Q. High-Throughput Screening of Metal-Organic Frame-
works for CO2 Capture in the Presence of Water. Langmuir 2016, 32, 10368–10376.
(10) Sumer, Z.; Keskin, S. Ranking of MOF Adsorbents for CO2 Separations: A Molecular
Simulation Study. Ind. Eng. Chem. Res. 2016, 55, 10404–10419.
(11) Bahamon, D.; Vega, L. F. Systematic evaluation of materials for post-combustion CO2
capture in a Temperature Swing Adsorption process. Chem. Eng. J. 2016, 284, 438–
447.
(12) Moghadam, P. Z.; Fairen-Jimenez, D.; Snurr, R. Q. Efficient identification of hydropho-
bic MOFs: application in the capture of toxic industrial chemicals. J. Mater. Chem. A
2016, 4, 529–536.
(13) McCallum, C.; Bandosz, T.; McGrother, S.; Muller, E.; Gubbins, K. A molecular model
17

for adsorption of water on activated carbon: Comparison of simulation and experiment.
Langmuir 1999, 15, 533–544.
(14) Brennan, J.; Bandosz, T.; Thomson, K.; Gubbins, K. Water in porous carbons. Colloid
Surf. A-Physicochem. Eng. Asp. 2001, 187, 539–568.
(15) Muller, E.; Rull, L.; Vega, L.; Gubbins, K. Adsorption of water on activated carbons:
A molecular simulation study. J. Phys. Chem. 1996, 100, 1189–1196.
(16) Karra, J. R.; Walton, K. S. Molecular Simulations and Experimental Studies of CO2,
CO, and N2 Adsorption in Metal-Organic Frameworks. J. Phys. Chem. C 2010, 114,
15735–15740.
(17) Atci, E.; Erucar, I.; Keskin, S. Adsorption and Transport of CH4, CO2, H2 Mixtures
in a Bio-MOF Material from Molecular Simulations. J. Phys. Chem. C 2011, 115,
6833–6840.
(18) Liu, B.; Sun, C.; Chen, G. Molecular simulation studies of separation of CH4/H2 mix-
ture in metal-organic frameworks with interpenetration and mixed-ligand. Chem. Eng.
Sci. 2011, 66, 3012–3019.
(19) Guo, H.-C.; Shi, F.; Ma, Z.-F.; Liu, X.-Q. Molecular Simulation for Adsorption and
Separation of CH4/H2 in Zeolitic Imidazolate Frameworks. J. Phys. Chem. C 2010,
114, 12158–12165.
(20) Al-Jadir, T. M.; Siperstein, F. R. Monte Carlo Simulation of Adsorption of Polar and
Nonpolar Gases in (FP)YEu Metal-Organic Framework. J. Chem. Eng. Data 2016, 61,
4209–4214.
(21) Al-Janabi, N.; Fan, X.; Siperstein, F. R. Assessment of MOF’s Quality: Quantifying
Defect Content in Crystalline Porous Materials. J. Phys. Chem. Lett. 2016, 7, 1490–
1494.
18

(22) Schneemann, A.; Bon, V.; Schwedler, I.; Senkovska, I.; Kaskel, S.; Fischer, R. A. Flex-
ible metal-organic frameworks. Chem. Soc. Rev. 2014, 43, 6062–6096.
(23) Li, J.-R.; Sculley, J.; Zhou, H.-C. MetalOrganic Frameworks for Separations. Chem.
Rev. 2012, 112, 869–932.
(24) Siderius, D. W.; Mahynski, N. A.; Shen, V. K. Relationship between pore-size distri-
bution and flexibility of adsorbent materials: statistical mechanics and future material
characterization techniques. Adsorption 2017, 23, 593–602.
(25) Hobday, C. L.; Marshall, R. J.; Murphie, C. F.; Sotelo, J.; Richards, T.; Allan, D. R.;
Dren, T.; Coudert, F.-X.; Forgan, R. S.; Morrison, C. A.; Moggach, S. A.; Bennett, T. D.
A Computational and Experimental Approach Linking Disorder, High-Pressure Behav-
ior, and Mechanical Properties in UiO Frameworks. Angew. Chem. Int. Edit. 2016, 55,
2401–2405.
(26) Gonciaruk, A.; Althumayri, K.; Harrison, W. J.; Budd, P. M.; Siperstein, F. R. PIM-
1/graphene composite: A combined experimental and molecular simulation study. Mi-
cropor. Mesopor. Mat. 2015, 209, 126 – 134.
(27) Argyris, D.; Ho, T.; Cole, D. R.; Striolo, A. Molecular Dynamics Studies of Interfacial
Water at the Alumina Surface. J. Phys. Chem. C 2011, 115, 2038–2046.
(28) Doig, M.; Camp, P. J. The structures of hexadecylamine films adsorbed on iron-oxide
surfaces in dodecane and hexadecane. Phys. Chem. Chem. Phys. 2015, 17, 5248–5255.
(29) Strange, N.; Fernndez-Caoto, D.; Larese, J. Z. Thermodynamic and Modeling Study of
n-Octane, n-Nonane, and n-Decane Films on MgO(100). J. Phys. Chem. C 2016, 120,
18631–18641.
(30) Dwivedi, D.; Lepkova, K.; Becker, T. Carbon steel corrosion: a review of key surface
properties and characterization methods. RSC Adv. 2017, 7, 4580–4610.
19

(31) Olsson, C.-O.; Landolt, D. Passive films on stainless steelschemistry, structure and
growth. Electrochim. Acta. 2003, 48, 1093 – 1104.
(32) Cygan, R. T.; Liang, J.-J.; Kalinichev, A. G. Molecular models of hydroxide, oxyhy-
droxide, and clay phases and the development of a general force field. J. Phys. Chem.
B 2004, 108, 1255–1266.
(33) Shahsavari, R.; Pellenq, R. J. M.; Ulm, F.-J. Empirical force fields for complex hydrated
calcio-silicate layered materials. Phys. Chem. Chem. Phys. 2011, 13, 1002–1011.
(34) Cruz-Chu, E.; Aksimentiev, A.; Schulten, K. Water-silica force field for simulating
nanodevices. J. Phys. Chem. B 2006, 110, 21497–21508.
(35) Huang, W.; Ranke, W.; Schloegl, R. Reduction of an alpha-Fe(2)O(3)(0001) film using
atomic hydrogen. J. Phys. Chem. C 2007, 111, 2198–2204.
(36) Nguyen, M.-T.; Seriani, N.; Gebauer, R. Water adsorption and dissociation on -
Fe2O3(0001): PBE+U calculations. J. Chem. Phys. 2013, 138, 194709.
(37) van Duin, A. C. T.; Dasgupta, S.; Lorant, F.; Goddard, W. A. ReaxFF: A Reactive
Force Field for Hydrocarbons. J. Phys. Chem. A 2001, 105, 9396–9409.
(38) Senftle, T. P.; Hong, S.; Islam, M. M.; Kylasa, S. B.; Zheng, Y.; Shin, Y. K.; Junker-
meier, C.; Engel-Herbert, R.; Janik, M. J.; Aktulga, H. M.; Verstraelen, T.; Grama, A.;
van Duin, A. C. T. The ReaxFF reactive force-field: development, applications and
future directions. NPJ Comput. Mater. 2016, 2, 15011.
(39) Huang, L.; Gubbins, K. E.; Li, L.; Lu, X. Water on Titanium Dioxide Surface: A
Revisiting by Reactive Molecular Dynamics Simulations. Langmuir 2014, 30, 14832–
14840.
(40) Aryanpour, M.; van Duin, A. C.; Kubicki, J. D. Development of a Reactive Force Field
for Iron-Oxyhydroxide Systems. J. Phys. Chem. A 2010, 114, 6298–6307.
20

(41) Guo, H.; Barnard, A. S. Thermodynamic modelling of nanomorphologies of hematite
and goethite. J. Mater. Chem. 2011, 21, 11566–11577.
(42) Parkinson, G. S. Iron oxide surfaces. Surf. Sci. Rep 2016, 71, 272 – 365.
(43) Finger, L. W.; Hazen, R. M. Crystal structure and isothermal compression of Fe2O3,
Cr2O3, and V2O3 to 50 kbars. J. Appl. Phys. 1980, 51, 5362–5367.
(44) Zou, C.; Van Duin, A. Investigation of Complex Iron Surface Catalytic Chemistry Using
the ReaxFF Reactive Force Field Method. JOM 2012, 64, 1426–1437.
(45) Chenoweth, K.; van Duin, A. C. T.; Goddard, W. A., III ReaxFF reactive force field
for molecular dynamics simulations of hydrocarbon oxidation. J. Phys. Chem. A 2008,
112, 1040–1053.
(46) Mortier, W. J.; Ghosh, S. K.; Shankar, S. Electronegativity-equalization method for the
calculation of atomic charges in molecules. J. Am. Chem. Soc. 1986, 108, 4315–4320.
(47) Rappe, A. K.; Goddard III, W. A. Charge equilibration for molecular dynamics simu-
lations. J. Phys. Chem. 1991, 95, 3358–3363.
(48) Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput.
Phys. 1995, 117, 1 – 19.
(49) Hoover, W. G. Canonical dynamics - equilibrium phase-space distributions. Phys. Rev.
A 1985, 31, 1695–1697.
(50) Hoover, W. G. Constant-pressure equations of motion. Phys. Rev. A 1986, 34, 2499–
2500.
(51) Martinez, L.; Andrade, R.; Birgin, E. G.; Martnez, J. M. PACKMOL: A package for
building initial configurations for molecular dynamics simulations. J. Comput. Chem.
2009, 30, 2157–2164.
21

(52) Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular
dynamics method. J. Appl. Phys. 1981, 52, 7182–7190.
(53) Cracknell, R. F.; Nicholson, D.; Tennison, S. R.; Bromhead, J. Adsorption and selec-
tivity of carbon dioxide with methane and nitrogen in slit-shaped carbonaceous micro-
pores: Simulation and experiment. Adsorption 1996, 2, 193–203.
(54) Gordillo, M. C.; Mart, J. Effect of Surface Roughness on the Static and Dynamic
Properties of Water Adsorbed on Graphene. J. Phys. Chem. B 2010, 114, 4583–4589.
(55) Coasne, B.; Pellenq, R. J.-M. Grand canonical Monte Carlo simulation of argon ad-
sorption at the surface of silica nanopores: Effect of pore size, pore morphology, and
surface roughness. J. Chem. Phys. 2004, 120, 2913–2922.
(56) Coasne, B.; Hung, F.; Pellenq, R.; Siperstein, F.; Gubbins, K. Adsorption of sample
gases in MCM-41 materials: The role of surface roughness. Langmuir 2006, 22, 194–
202.
(57) Coasne, B.; Hung, F.; Siperstein, F.; Gubbins, K. Molecular simulation of gas adsorp-
tion in realistic models of silica nanopores. Ann. Chim.-Sci. Mat. 2005, 30, 375–383.
(58) Dzade, N.; Roldan, A.; de Leeuw, N. A Density Functional Theory Study of the Ad-
sorption of Benzene on Hematite (α - Fe2O3) Surfaces. Minerals 2014, 4, 89.
22