mecillinam resistance in e. coli - diva portal1146016/... · · 2017-10-01master degree project...
TRANSCRIPT

Mecillinam Resistance in E. coli – fitness, compensation, and resistance in different environments
Emelie Ekstrand ___________________________________________ Master Degree Project in Infection Biology, 45 credits. Spring 2017 Department of Medical Biochemistry and Microbiology Supervisor: Dan I. Andersson Co-supervisor: Elisabeth Thulin

UPPSALA UNIVERSITY 2017
Emelie Ekstrand 2 (29)
Abstract
The global increase of antibiotic resistant bacteria threatens the modern health care and challenges the therapeutic effects of available antibiotics. The β-lactam mecillinam (Mec) is an exception to this due to a stable clinical resistance prevalence resistance of approximately 3%. It is only used to treat uncomplicated urinary tract infections (UTIs), mainly caused by E. coli.
Mecillinam resistance (MecR) is easily selected for in laboratory settings and linked to >40 genes, including the mrdA gene encoding the Mec target penicillin-binding protein 2. A majority of the known MecR mutations confer a severe fitness cost. Fitness is important for bacteria to survive in the bladder and clinical isolates have been shown to have high fitness. These isolates contain loss-of-function mutations in the cysB gene, which encode a positive regulator of cysteine biosynthesis. In a previous evolution experiment, fitness cost of cysB and mrdA MecR mutations was compensated and the compensatory mutations were identified. Here the compensatory mutations were reconstructed into wildtype (WT) E. coli strain MG1655, and cysB and mrdA backgrounds to study the impact of the mutations on resistance and fitness, using MIC tests and Bioscreen C assays.
Our results show that the mrdA mutants only had partial fitness compensation (significantly lower fitness than WT) for all strains and all strains also increased their MecR. The low fitness is possibly an explanation for the lack of mrdA mutants outside laboratories. Of the clinically relevant cysB mutants the majority lost their resistance when increasing growth rate, some even to levels significantly higher than WT, indicating that ΔcysB mutations are easier to compensate for. One strain (ydjNmx2) however, had a significantly higher growth rate while remaining clinically MecR.

UPPSALA UNIVERSITY 2017
Emelie Ekstrand 3 (29)
Popular Summary
Antibiotics are necessary to treat bacterial infectious diseases but their efficiency is now threatened by globally increasing resistance. Sometimes spontaneous changes occur in the genes of the bacteria, which can give them the ability to resist certain antibiotics. These genetic changes, so-called mutations, often come with the cost of lowering the bacteria’s ability to grow and multiply (referred to as fitness). Resistance to antibiotics may arise or already exist in small populations of bacteria, but might spread to others if beneficial enough.
Few antibiotics that have been in use for long remain effective but mecillinam (Mec) is one of the exceptions. It has been in the market for over 40 years and is still very effective for treatment of uncomplicated urinary tract infections (UTIs). Over 80 % of UTIs are caused by Escherichia coli (E. coli), a bacterium normally living in the intestine of humans. When E. coli are grown in the presence of Mec in laboratory settings it easily acquires mutations making it Mec resistant (MecR), which is unexpected since MecR E. coli rarely is found in the clinic. How MecR arise in E. coli and the cause of the different findings between laboratories and clinics is not yet fully known. This study aims to increase the knowledge in this area and contribute to prolonging the effectiveness of mecillinam as a working antibiotic.
Previous studies have shown that all clinical MecR isolates from UTIs have lost their function of cysB, a gene normally involved in activating the bacterial process of producing the amino acid cysteine that is needed for cell growth. These mutants have a higher fitness compared to other MecR bacteria, such as the mrdA mutation causing changes to Mec’s target. Costly mutations can be compensated for by changes in other parts of the genome and a study identified several of these in mrdA and cysB mutants that were grown for 400 generations without Mec being present.
We moved these compensatory mutations, single or in combinations, into the genome of a standard model (called a wildtype; WT) of E. coli containing the cysB or the mrdA mutation. By reconstructing these strains in the laboratory, we could study how these mutations affected the bacteria with regard to MecR and fitness. The compensatory mutations in the mrdA group did not reach high fitness, but increased MecR. Since all these strains had the fitness-costly mutation in mrdA, the altered target of Mec could explain why they remained resistant to mecillinam and provide a probable explanation of why they are not able to survive clinically. In the cysB group, we saw that all mutants gained higher fitness, some even higher than the WT. Most of them lost the MecR in the process but we found one that gained in fitness and remained MecR.
This study contributes to the understanding of antibiotic resistance by showing the impact fitness has on MecR mutants and their clinical prevalence.

UPPSALA UNIVERSITY 2017
Emelie Ekstrand 4 (29)
Key words
Antibiotic resistance • bacterial genetics • compensatory mutations • cysB • Escherichia coli • mecillinam • mrdA • strain construction
Abbreviations
Amp Ampicillin bp base pair(s) Cam chloramphenicol DMSO dimethyl sulfoxide DNA deoxyribonucleic Acid E. coli Escherichia coli HGT horizontal gene transfer IS insertion sequence Kan kanamycin kb kilo base pair LA Luria-Bertani agar LB Luria-Bertani Mec mecillinam MH Mueller-Hinton MIC minimal inhibitory concentration nt nucleotide(s) PBP penicillin-binding protein PCR polymerase chain reaction Tet tetracycline TBE tris-borate-EDTA us upstream UTI urinary tract infection WSG whole-genome sequencing

UPPSALA UNIVERSITY 2017
Emelie Ekstrand 5 (29)
Introduction
The discovery and introduction of antibiotics revolutionised medicine and they are considered a cornerstone of modern medicine. Alexander Fleming discovered penicillin by chance in the year 1928 [1], although it took until 1941 until Walter Florey and Ernst Boris Chain could introduce the working drug to the market [2]. Penicillin was the first β-lactam discovered, a compound gaining its broad-spectrum antibacterial property from the name-giving β-lactam ring in its chemical structure (highlighted in Figure 2). Between the 1950s and 1970s, also referred to as the “golden age of antibiotics” [3,4], new β-lactams and several new antibiotic groups were discovered and introduced. These miracle medicines changed the society by curing previous potentially lethal infectious diseases and thereby prolonging the average lifespan of the human population [5–8].
From the year 1987, no new classes of broad-spectrum antibiotics have been introduced and few new drugs have reached the market during this “discovery void” [9]. More and more of the antibiotics so frequently used started to fail in treatment as antibiotic resistance spread among bacteria, partly due to the overuse of antibiotics. Despite the need for new effective antibiotics, the emerging resistance, and the need to restrict the use of any new antibacterial drug, made the efforts of antibiotic development an economically risky business.
The increased antibiotic resistance is a global problem and the Centres for Disease Control and Prevention (CDC) and the World Health Organisation (WHO) have announced that tackling this is a priority [10,11]. The large, and sometimes unnecessary use of antibiotics has led to increased amounts being released into the environment, resulting in a high pressure on bacteria to become antibiotic resistant. Bacterial strains become resistant by mutations in the genome or through transfer of resistance plasmids, enabling them to thrive compared to non-resistant bacteria [12–14]. The longer the antibiotic has been on the market and the higher amount that has been prescribed, the more resistant bacteria can be found in clinical settings, indicating a strong correlation between selection pressure and resistance evolution.
Bacteria have a rapid generation time and their dynamic genomes allow for beneficial mutations to spread quickly. Horizontal gene transfer (HGT), including transformation, transduction, and conjugation, enable genetic material to be linearly transferred by exogenous uptake [15–17]. The uptake of external genetic material through the cell membrane(s) and its incorporation in the recipient’s genome is called transformation. Here, the recipient includes the foreign DNA by homologous recombination, requiring similar or identical parts in both genetic structures to occur [18]. In a similar process, transduction, the introduction of genetic material into the cell is mediated by bacteriophages which have packed donor DNA instead of the phage’s [18]. Unlike the two previous HGTs described, conjugation demand cell-to-cell contact. This is mediated by the pili system where the donor bacteria produce a pilus which attaches to a recipient cell and bringing them close together, enabling the copy and sharing of genetic material [18]. HGT does not just occur within the same species but exchange can occur wider, enabling beneficial genes such as antibiotic resistance, to become wide-spread [10,19].
The gain of antibiotic resistance is often linked with fitness cost since energy and resources will be redistributed from growth [12], and it is rare that bacteria gain a high resistance without a loss of fitness [12]. Although, it has been found that in environments with sub-MIC levels of antibiotics selective pressure is maintained while enabling fitness compensations to occur over time [13,20].
To study the mechanisms of antibiotics and the emerging resistance, bacterial model organisms are often used. One of the main models is Escherichia coli (E. coli), known for its large pan-genome and tendency to promiscuously acquire genetic material through HGT [21].

UPPSALA UNIVERSITY 2017
Emelie Ekstrand 6 (29)
This gram-negative, rod-shaped bacterial species is a part of the human microbiota in the larger intestine, and is defined as an opportunistic commensal due to the extra-intestinal infections it can cause. The severity of these infections range from uncomplicated to life-threatening, making this gram-negative bacteria of clinical interest and it has therefore been widely studied for over a century [22].
Figure 1. A schematic figure of E. coli and its cell wall. Figure adapted from Pinho et al. [23]
Gram-negative bacteria are enveloped by a cell membrane, a thin layer of peptidoglycan, a periplasmic space and a non-permeable outer membrane containing specialised channels and lipopolysaccharides (Figure 1) [24–26]. These extra barriers protect the cell by preventing uptake of harmful substances, making it difficult for drugs to enter. An important step in the construction of the cell wall is the cross-linking of peptidoglycans (PGs) through transglycosylation and transpeptidation facilitated by penicillin-binding proteins (PBPs). The resulting polymerisation and cross-linking (via flexible polypeptides) creates a rigid structure surrounding the bacteria [24,25]. Normally, the PBPs bind temporarily to the amino acids D-alanyl-D-alanine (D-ala-D-ala) during transpeptidation before moving on to the next, enable a recycling of the enzyme. This is inhibited by β-lactams, binding to the PBPs instead of D-ala-D-ala due to the similarity of the β-lactam ring and the amino acids. Subsequently, the prevention of cross-linkage of the cell wall cause the cell to lyse as it grows [1,5,27,28].
One type of disease caused by E. coli is urinary tract infections (UTIs), where it is the primary pathogen, found in 81% of all uncomplicated cases that were sampled [29]. UTIs are common in the population, affecting mainly women which have a lifetime incidence of 53-60% [30,31]. It is characterised by singes and frequent urinations due to infection in the bladder and 75% of all cases in Sweden are labelled uncomplicated, meaning the patients have functional anatomically normal urinary tracts [29]. In Sweden, UTI’s are one of the largest reasons for antibiotic out-of-hospital treatment, amounting to 12% of all antibiotics distributed by pharmacies [29,32].

UPPSALA UNIVERSITY 2017
Emelie Ekstrand 7 (29)
Figure 2. The chemical structures of pivmecillinam and its hydrolysis into mecillinam (upper part). In the lower part of the figure are the β-lactam ring is shown next to the core structure of penicillins, a sub-group of β-lactams. The figure is based on information from Sakamoto et al. [33].
One of the first-line drugs for UTIs is the penicillin mecillinam (Mec; Figure 2), an 6β-amidinopenicillanic acid derivate introduced by Lund and Tyrbring in 1972 [34,35], and despite over forty years on the market clinical resistance prevalence remains low, at 3% of all E. coli UTI isolates[32]. The mechanisms for Mec resistance (MecR) are not fully understood and the drug separates itself from the other β-lactams by its unusual chemical structure and low effect on gram-positive bacteria [36,37]. In Sweden, the prodrug pivmecillinam is used, it is hydrolysed in the intestine to its active form, [33] with 50% of the administered dose being excreted via urine [38]. The bactericidal effect of Mec is achieved through binding to a PBP instead of the D-ala-D-ala (Figure 1) like other β-lactams. Mec specifically targets PBP2, which is responsible for the elongation of the rod-shaped cell [39–41], causing the growing bacteria to form large spheres before lysis [27,34–36].
The urine bladder may contain bacteria [42] but due to the flushing out of urine, adhesive properties and high growth rates are demanded for colonisation. The large pan-genome of E. coli includes several virulence factors such as adhesins to attach to the wall of the bladder, and the aerobactin system beneficial for growth in low-iron environments which have been identified in isolates from UTIs [43]. As described earlier, antibiotic resistance often come with a fitness cost, and this is the case also with MecR. Never the less, MecR is easy to acquire in laboratory settings where over forty genes have been linked to it [39,43–49]. Clinical breakpoints have been set by the European Committee on Antimicrobial Susceptibility Testing (EUCAST) at ≤8 mg L-1 for sensitivity and >8 mg L-1 for resistance with an epidemiological cut-off of ≤1 mg L-1 [50]. Mec reaches a high concentration in the bladder and is relatively unchanged throughout the body, causing few adverse effects for the patient while acting with high selective pressure and short adaptation time for the bacteria [51]. The fitness cost of mutations conferring high MecR is thought to be the reason for most MecR mutants not being viable in clinical settings [12].
Despite the distinct contrast between the low prevalence of MecR E. coli in clinical samples and the high frequency of resistance when selecting under laboratory conditions [32,52], few studies have been made within this area. In general, possible differences between in vitro and in vivo conditions are rarely highlighted in published material although they are of high relevance. To better understand the gap, a previous study screened clinical MecR isolates from UTIs for resistance mutations and compared them to laboratory-originated resistant mutants. The clinical isolates all carried mutations inactivating the cysB gene, which encodes a positive regulator of the cysteine regulon (biosynthesis). These ΔcysB mutants had a higher fitness compared to the laboratory isolates which harboured a large variety of mutations with big fitness costs [52].

UPPSALA UNIVERSITY 2017
Emelie Ekstrand 8 (29)
Since a high growth rate is important for the establishment of resistant mutants in the bladder, an evolution experiment was set up where 8 lineages of mutants with high fitness costs (ΔcysB, mrdA, spoT, ppa, aspS and ubiE), to see if the fitness costs could be compensated. These mutants were serially passaged for 200-400 generations without the presence of Mec and sequenced to find growth compensating mutations [unpublished data communicated by M. Knopp and E. Thulin]. Compensatory mutations for spoT, ppa, aspS and ubiE occurred intragenically or in a gene with an opposing function. The ΔcysB was compensated by mutations in the cysteine biosynthesis pathway [53–55]. Unlike the previously mentioned mutants, which lost resistance when acquiring fitness close to the wildtype E. coli, the mrdA mutant only partially compensated the growth rate but maintained MecR [39,45]. The genes in which compensatory mutations occurred in the ΔcysB and mrdA are listed in Table 1 along with their respective gene functions.
Table 1. Genes in whom compensatory mutations were found after evolution experiment of reconstructed MecR ΔcysB and mrdA mutants. The table was based on unpublished data communicated by M. Knopp and E. Thulin.
Gene Function cysB Encodes positive regulator of the cys regulon [52–55] cysP Thiosulphate importer, part of the cysteine biosynthesis [53–55] cysK Cysteine synthetase, part of the cysteine biosynthesis [53–55] dicA Temperature-sensitive TF repressor for genes involved in cell
division [56] nudE Hydrolase breaking down compounds like ADP-ribose and NADH
[57,58] ydjN mut. Cystine/cysteine:cation symporter [54] ydjN dupl. Dupl. containing ydjN, 35-77 kb [54]
mrdA Encodes PBP2 [27] nlpI Lipoprotein linked to cell division and PG dynamics [59] Large dupl. Large dupl. of 300 kb (~200 genes) at position 3366948-3651836
in the E. coli genome [52] pgsA PGP synthase associated with cell membrane [60] sppA del. Del. (3-28 kb) containing part of sppA (protease IV, signal peptide
peptidase) [61] Del., deletion; dupl., duplication; mut, mutation; PG, peptidoglycan; PGP, phosphatidylglycerophosphate; TF, transcription factor
The difference in compensatory mutations may be connected to why some MecR mutants are prevalent in the clinic while others are not.

UPPSALA UNIVERSITY 2017
Emelie Ekstrand 9 (29)
Aim
The aim of this study was to reconstruct and investigate the impact of previously identified compensatory mutations in mecillinam resistant Escherichia coli (E. coli) which would increase the understanding of the differences seen between clinical and laboratory settings. The effect of the mutations on resistance and fitness were examined through their introduction, alone or in combination, into the respective cysB and mrdA mutant backgrounds. The specific objective of the project was to determine the minimal inhibitory concentration (MIC) of mecillinam and fitness (by growth rate measurements) on reconstructed mutants.
Furthermore, to investigate the mechanisms behind very high mecillinam resistance (i.e. mecillinam MICs >256 mg/L) in E. coli, a selection for highly resistant cysB knock out mutants was performed. These mutants were phenotypically tested with MIC assays, and whole-genome sequenced in search for genetic mutations potentially explaining the presence of clinical cysB mutants with a higher MecR, than that caused by only a cysB mutation.
This study could enable a better understanding of mecillinam resistance and the hope is that greater insights of MecR mechanisms may prolong the use of today’s antibiotics and postpone the post antibiotic era.

UPPSALA UNIVERSITY 2017
Emelie Ekstrand 10 (29)
Materials and methods
Bacterial strains and media All bacteria used were derivates of Escherichia coli K12 MG1655 and specific strains used in this study are listed in Table 3 under results; see also supplementary Table S1. Luria-Bertani (LB; Sigma-Aldrich) and Mueller-Hinton (MH; Becton, Dickinson, and Company) were used as liquid medium while LA (LB with 15g L-1 agar) and MH agar were used as solid medium. Plates with minimal media (M9 + 0.4% glucose) were used for phenotypic testing of highly resistant MecR cysB mutants with and without 0.3 mM cysteine (Sigma-Aldrich) supplemented. For counter-selection of cat-sacB-cassette, sucrose agar was used (5%; 875 ml H2O, 10 g Tryptone, 5 g yeast extract, 200 µL 5M NaOH, 15 g agar and 125 mL 40% sucrose). Unless stated otherwise, agar plates were incubated overnight at 37°C. Overnight cultures were incubations of a single colony in 1 mL MH at 37°C, shaking, and strains were saved by freezing the overnight culture in 10% dimethyl sulfoxide (DMSO) at -80°C.
Antibiotics were supplemented to medium when appropriate and in the following concentrations: 100 mg L-1 mecillinam (Mec), 100 mg L-1 ampicillin (Amp), 12.5 mg L-1 chloramphenicol (Cam), 100 mg L-1 kanamycin (Kan), 12.5 mg L-1 tetracycline (Tet). All antibiotics were from Sigma-Aldrich.
PCR and Sanger sequencing For each polymerase chain reaction (PCR) tube with 1 µL PCR template, 25 µL DreamTaq PCR Master Mix (2X) (Thermo Scientific) was used along with 14 µL sterile water (W4502, Sigma-Aldrich), and 5 µL each of forward (FWD) and reverse (REV) 5 µM primers. The standard template for the PCR protocol was, unless stated otherwise, an initial denaturation at 95°C for 5 min, followed by 30 repeats of 30 seconds of 94°C denaturation, 30 sec of appropriate annealing temperature, and 2 min of 72°C elongation. Protocol was finalized by an additional 7 min of 72°C elongation before cooled down to 4°C.
PCR products were screened to ensure correct result using gel electrophoresis with 1% agarose (BioNordika, Stockholm, Sweden) and 1x Tris-borate-EDTA (TBE; 54g Tris, 27.5 g boric acid, 20 mL 0.5 M EDTA). Samples were mixed with DNA Gel Loading Dye (6X) and 4 µL were loaded along with GeneRuler 1kb DNA Ladder as size standard (both from Thermo Scientific). Gel was dyed in Ethidium-bromide solution (BioChemica) for a minimum of 15 min in 0.5 mg L-1 before viewed using an ultraviolet light camera.
To screen for single nucleotide mutations, Sanger sequencing was used. The PCR products of samples of interest were purified using GeneJET PCR Purification Kit (Thermo Scientific) according to their protocol. Of the purified DNA, 15 µL was mixed with 5 µL FWD or REV primer, respectively. The DNA samples were sequenced by Eurofins MWG Operon (Ebersberg, Germany) and all primers used are available in supplementary Table S2.
DNA inserts For each sample, a master mix of 18.5 µL sterile water (W4502, Sigma-Aldrich), along with PCR regents from Thermo Scientific (10 µL 5x Phusion buffer, 5 µL 2mM dNTPs, 5 µL DMSO, 0.3 µL Phusion High-Fidelity DNA polymerase, 0.2 µL MgCl2), and 5 µL each of FWD and REV primers (see Table S2 for primers). One µL of DNA template (one bacterial colony suspended in 50 µL of sterile water) was added to PCR tube containing 49 µL of master mix before proceeding with a Touchdown PCR program according to Table 2.
DNA inserts were purified from the PCR product using GeneJET PCR Purification Kit (Thermo Scientific) according to protocol but with the alternation of using 35 µL of sterile water instead of the instructed 50 µL of elution buffer before the final centrifugation. DNA
UPPSALA UNIVERSITY 2017
Emelie Ekstrand 11 (29)
concentration of the final product was measured using NanoDrop 1000 (Thermo Scientific) before stored at -20°C.
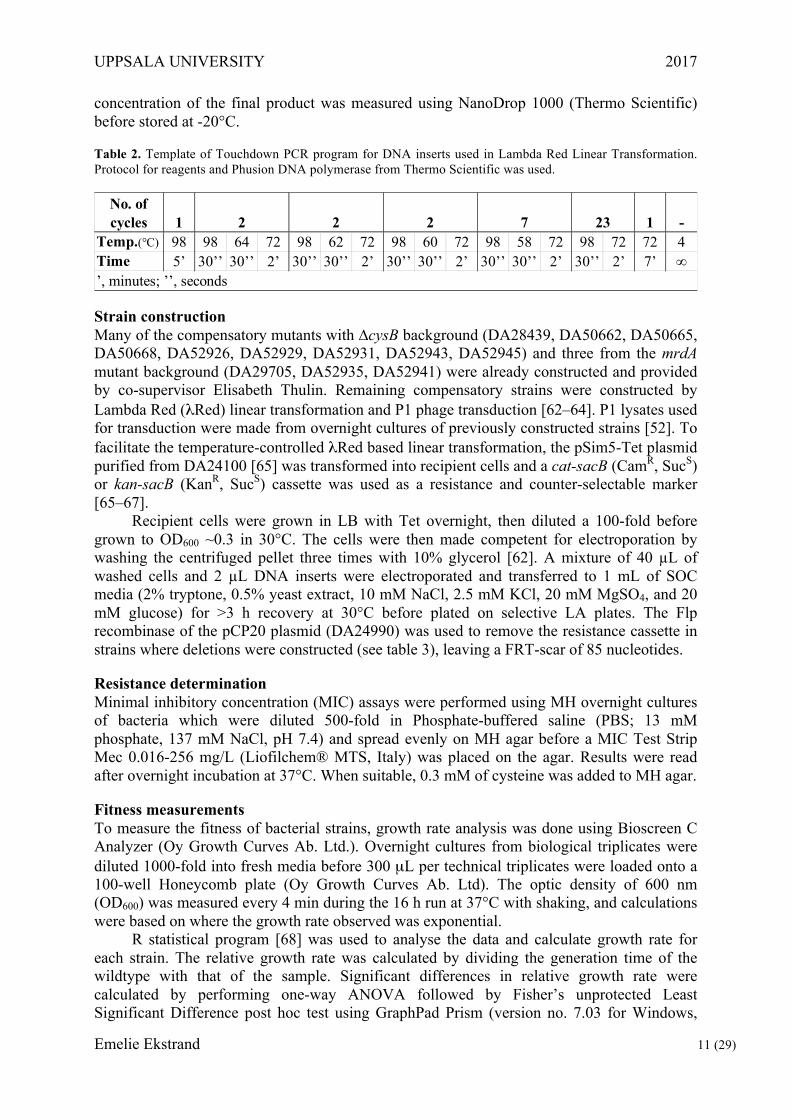
Table 2. Template of Touchdown PCR program for DNA inserts used in Lambda Red Linear Transformation. Protocol for reagents and Phusion DNA polymerase from Thermo Scientific was used.
Temp.(°C) 98 98 64 72 98 62 72 98 60 72 98 58 72 98 72 72 4Time 5’ 30’’ 30’’ 2’ 30’’ 30’’ 2’ 30’’ 30’’ 2’ 30’’ 30’’ 2’ 30’’ 2’ 7’ ∞
2 2 2 7 23
’, minutes; ’’, seconds
1 -No. of cycles 1
Strain construction Many of the compensatory mutants with ∆cysB background (DA28439, DA50662, DA50665, DA50668, DA52926, DA52929, DA52931, DA52943, DA52945) and three from the mrdA mutant background (DA29705, DA52935, DA52941) were already constructed and provided by co-supervisor Elisabeth Thulin. Remaining compensatory strains were constructed by Lambda Red (λRed) linear transformation and P1 phage transduction [62–64]. P1 lysates used for transduction were made from overnight cultures of previously constructed strains [52]. To facilitate the temperature-controlled λRed based linear transformation, the pSim5-Tet plasmid purified from DA24100 [65] was transformed into recipient cells and a cat-sacB (CamR, SucS) or kan-sacB (KanR, SucS) cassette was used as a resistance and counter-selectable marker [65–67].
Recipient cells were grown in LB with Tet overnight, then diluted a 100-fold before grown to OD600 ~0.3 in 30°C. The cells were then made competent for electroporation by washing the centrifuged pellet three times with 10% glycerol [62]. A mixture of 40 µL of washed cells and 2 µL DNA inserts were electroporated and transferred to 1 mL of SOC media (2% tryptone, 0.5% yeast extract, 10 mM NaCl, 2.5 mM KCl, 20 mM MgSO4, and 20 mM glucose) for >3 h recovery at 30°C before plated on selective LA plates. The Flp recombinase of the pCP20 plasmid (DA24990) was used to remove the resistance cassette in strains where deletions were constructed (see table 3), leaving a FRT-scar of 85 nucleotides.
Resistance determination Minimal inhibitory concentration (MIC) assays were performed using MH overnight cultures of bacteria which were diluted 500-fold in Phosphate-buffered saline (PBS; 13 mM phosphate, 137 mM NaCl, pH 7.4) and spread evenly on MH agar before a MIC Test Strip Mec 0.016-256 mg/L (Liofilchem® MTS, Italy) was placed on the agar. Results were read after overnight incubation at 37°C. When suitable, 0.3 mM of cysteine was added to MH agar.
Fitness measurements To measure the fitness of bacterial strains, growth rate analysis was done using Bioscreen C Analyzer (Oy Growth Curves Ab. Ltd.). Overnight cultures from biological triplicates were diluted 1000-fold into fresh media before 300 µL per technical triplicates were loaded onto a 100-well Honeycomb plate (Oy Growth Curves Ab. Ltd). The optic density of 600 nm (OD600) was measured every 4 min during the 16 h run at 37°C with shaking, and calculations were based on where the growth rate observed was exponential.
R statistical program [68] was used to analyse the data and calculate growth rate for each strain. The relative growth rate was calculated by dividing the generation time of the wildtype with that of the sample. Significant differences in relative growth rate were calculated by performing one-way ANOVA followed by Fisher’s unprotected Least Significant Difference post hoc test using GraphPad Prism (version no. 7.03 for Windows,

UPPSALA UNIVERSITY 2017
Emelie Ekstrand 12 (29)
GraphPad Software, La Jolla California USA) between the wildtype E. coli and each of the constructed mutants.
Isolation and whole-genome sequencing of MecR cysB mutants To isolate highly Mec resistant mutants, ten individual overnight cultures of the constructed ∆cysB mutant (DA28439) were diluted ten-fold with PBS of which 100 µL were plated and incubated overnight on MH agar supplemented with100 mg L-1 Mec. Same procedure was repeated but a plating of 200 µL of undiluted culture on MH agar with 200 mg L-1 of Mec. Colonies were re-streaked on freshly made agar of the same Mec concentration, and overnight cultures were started the following day in MH broth. Genetic and phenotypic screening was performed by local sequencing of the cysB gene, MIC assays and growth on minimal media (M9 + 0.4% glucose) agar plates with or without 0.75 mM of cysteine.
Strains were prepared for WGS by MiSeq by purifying overnight cultures using EpiCenter MasterPureTM DNA purification kit (Epicentre) according to cell sample protocol. The final pellet was resuspended in 70 µL of 10 mM Tris-HCl, pH 7.5, to check the quality of sample a gel electrophoresis, 0.7% agarose were run with 500 ng of sample. The DNA concentrations were measured on Thermo Scientific’s Qubit® 2.0 Fluorometer with Qubit® dsDNA BR Assay Kit (Thermo Scientific) according to manufacturer’s protocol. Samples were diluted to a final concentration of 1 mg L-1 before whole-genome sequenced (Illumina). Results were analysed using CLC Genomics Workbench software (version no. 10; CLC bio, Aarhus, Denmark) with a cut-off frequency set to 75%.

UPPSALA UNIVERSITY 2017
Emelie Ekstrand 13 (29)
Results
Reconstructions of compensatory mutations in mrdA and ∆cysB mutants Mutations found in fitness compensated strains carrying a cysB mutation (originating from DA28439) or an mrdA mutation (originating from DA29705) were reconstructed into respective parental strain and the wildtype (DA5438). The mutations were moved using Lambda Red transformation and P1 phage transduction, and the resulting strains were screened for reconstructed mutations using PCR and local sequencing.
Attempts to introduce an nlpI mutation into the mrdA mutant DA29705 were unsuccessful and therefore replaced by the nlpI KO from the Keio strain collection (Keio strain 1324; nlpI knocked out with KanR cassette) [69]. DA54543 was also constructed with the use of a Keio strain (K323; sppA KO, KanR cassette) since the in-lab sppA del could not be inserted with the mrdA and nlpI combination. A complete list of all strains with specified mutations can be found in Table S1. Table 3. Characteristics of constructed compensatory strains; including MIC value and relative growth rate compared to wildtype. All strains are based on the wildtype E. coli MG1655.
Strain Genotype MIC
(mg L-1) Relative
growth rate DA5438 E. coli MG1655, wildtype 0.125 1 DA49341 E. coli MG1655, CamR wildtype 0.125 1 DA28439 Reconstructed phenotypical mut by KO of cysB gene 24 0.59 DA50662 dicA mut into DA28439 32 0.63 DA50665 cysP mut into DA28439 0.25-0.38 1.03 DA50668 nudE mut into DA28439 64 0.63 DA52926 ydjNmx2 mut into DA28439 32-48 1.07 DA52929 cysK mut into DA28439 1-1.5 0.93 DA52931 ydjN dupl. into DA28439 0,25 1.00 DA52943 cysP + cysK mut into DA28439 1.5-2 0.95 DA52945 cysP + ydjNm mut into DA28439 0.25 1.01 DA55120 ydjNm mut into DA28439 24-48 0.67 DA55448 ydjNm mut + ydjN dupl. into DA28439 0,38 1.06 DA29705 Reconstructed mrdA mutant 16 0.29 DA52935 pgsA mut into DA29705 >256 0.53 DA52941 ansA del into DA29705 >256 0.24
DA54156 sppA del into DA29705 >256 0.51
DA54533 Large dupl. into DA29705 >256 0.29
DA54536 pgsA + Large dupl. into DA29705 >256 0.23
DA54538 pgsA mut + sppA del into DA29705 48 0.59 DA54541 sppA del + Large dupl. into DA29705 >256 0.40 DA54543 nlpI KO + sppA KO into DA29705 >256 0.51 DA54545 nlpI KO into DA29705 >256 0.43 DA54547 nlpI KO + Large dupl. into DA29705 24-32 0.66 DA55447 pgsA mut + sppA del + Large dupl. into DA29705 >256 0.70 Del, deletion; dupl, duplication; KO, knock-out; mut, mutation

UPPSALA UNIVERSITY 2017
Emelie Ekstrand 14 (29)
Mecillinam MIC determination of reconstructed compensated strains MIC assays were performed with a minimum of two biological replicates per strain using Mec MIC Test Strips on MH agar, and results can be found in Table 3. The MIC values of the compensatory mutations showed few variants within the strain replicates and within the strain groups (cysB or mrdA). In the clinically relevant cysB mutants, six of the ten strains lost their resistance with the additional mutations while four had MIC values close to or higher than the ∆cysB (DA28439; 24 mg L-1); dicA, ydjNm, ydjNmx2 and (nudE). None of the mrdA reconstructed mutants showed wildtype levels of susceptibility to Mec and all the additional mutations in the mrdA background (DA29705; MIC of 16 mg L-1) increased the resistance to MICs of >256 mg L-1, apart from the nlpI + Large dupl. (LD) at 24-32 mg L-1 and pgsA del + sppA del at 48 mg L-1.
Fitness of MecR compensatory strains Bioscreen analysis was used to assess the fitness (exponential growth rate) of strains with reconstructed compensatory mutations and is visualised in Figures 3 and 4 where the strains were compared with wildtype E. coli. The mutant strains were grown in MH and compared with DA5438 apart from the strains containing Large dupl. or ydjN dupl. which were run in MH with Cam to prevent loss of duplications, and compared with the CamR DA49341 (also originating from DA5438).
Figure 3. The relative growth rate compared to WT (left axis) and MIC (right axis) for cysB mutants. Significances (P value <0.05) were found for 8 of the 11 strains whereof four of the strains had a very strong significance with a P value <0.0001. No significances were found between the WT and ydjN dupl., ydjNmx2+cysP, and cysP respectively.
All compensatory mutations added to the cysB mutant showed an increase of growth rate compared to the parental strain DA28439 (Figure 3), though only the ydjNm + ydjNm dupl. and the ydjNmx2 mutants had a growth rate significantly higher than the WT DA4538, which was set as 1 (P values <0.05 and <0.001 respectively). The growth rates of ydjN dupl.,

UPPSALA UNIVERSITY 2017
Emelie Ekstrand 15 (29)
ydjNmx2 + cysP, and cysP mutants were non-significant compared to the wildtype (P values of 0.87, 0.67 and 0.32 respectively).
The results from the mrdA mutants’ growth rates (Figure 4) showed that all strains had low fitness, when compared to WT (P value <0.0001). The mrdA mutant had a relative growth rate of 0.29 and the additional mutants both increased and decreased the fitness (values written out in Table 3).
Figure 4. The relative growth rate (left Y axis) of the mrdA mutants plotted with the MIC (right Y axis). All mutants, including the single mrdA variant showed significantly low fitness compared with WT with P values <0.0001. The MICs distinguishes from the susceptible WT (0.125 mg L-1) to clinically resistant (>8 mg L-1) for all mutants.
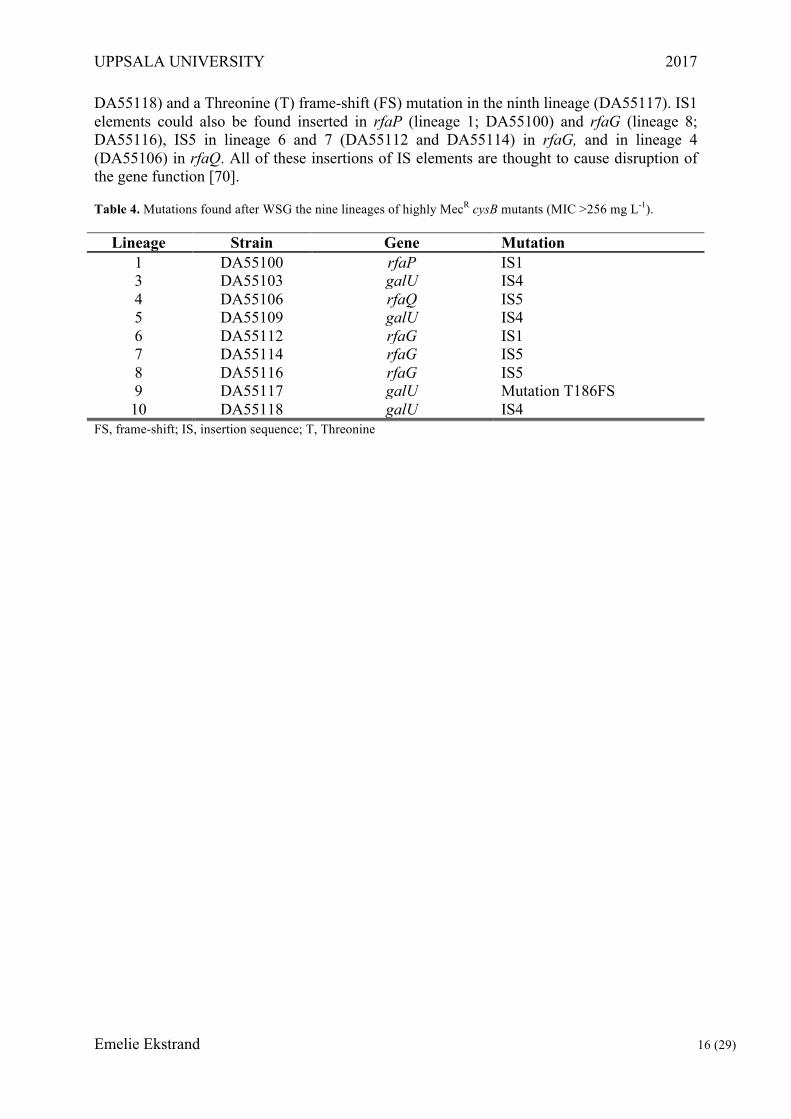
Selection and whole-genome sequencing of highly MecR cysB mutants The selection of MecR mutants was performed to elucidate how cysB mutants can become highly resistant to mecillinam by acquisition of additional mutations, since we have identified clinical isolates that have higher resistance than that conferred by the cysB mutation they carry. Therefore, ten lineages of the constructed cysB mutant DA28439 were plated on MH agar supplemented with Mec at concentrations of 100 or 200 mg L-1 to select for bacteria with higher resistance than the MIC of 24 mg/L that is provided by the cysB deletion. Growth was only observed on the Mec 100 mg L-1 plates and one colony from nine of those initial ten independent cultures, were identified as highly MecR cysB mutants and sent for whole-genome sequencing. These lineages still had the ΔcysB mutation (screened by PCR) and were phenotypically controlled through MICs on MH (>256 mg L-1) and growth on minimal media supplemented with and without cysteine, the latter confirming the cysteine auxotroph phenotype by growth of the strains only when cysteine was supplemented.
Analysis of the WSG data showed mutations, mainly various insertion sequence (IS) elements, in the nonessential genes rfaG, rfaP, rfaQ and galU (Table 4). The galU gene revealed insertions of IS element 4 (IS4) in lineage 3, 5 and 10 (DA55103, DA55109,
UPPSALA UNIVERSITY 2017
Emelie Ekstrand 16 (29)
DA55118) and a Threonine (T) frame-shift (FS) mutation in the ninth lineage (DA55117). IS1 elements could also be found inserted in rfaP (lineage 1; DA55100) and rfaG (lineage 8; DA55116), IS5 in lineage 6 and 7 (DA55112 and DA55114) in rfaG, and in lineage 4 (DA55106) in rfaQ. All of these insertions of IS elements are thought to cause disruption of the gene function [70].
Table 4. Mutations found after WSG the nine lineages of highly MecR cysB mutants (MIC >256 mg L-1).
Lineage Strain Gene Mutation 1 DA55100 rfaP IS1 3 DA55103 galU IS4 4 DA55106 rfaQ IS5 5 DA55109 galU IS4 6 DA55112 rfaG IS1 7 DA55114 rfaG IS5 8 DA55116 rfaG IS5 9 DA55117 galU Mutation T186FS 10 DA55118 galU IS4
FS, frame-shift; IS, insertion sequence; T, Threonine

UPPSALA UNIVERSITY 2017
Emelie Ekstrand 17 (29)
Discussion
Compensatory mutations in MecR ∆cysB and mrdA mutants The reconstruction of previously identified fitness-compensating mutations in cysB and
mrdA mutant backgrounds enabled us to study the individual and combined impact of these mutations. The compensatory mutations arose either in the clinically important cysB or the laboratory relevant mrdA background. By comparisons of MecR and fitness of these mutants, the differences between the clinical and laboratory MecR isolates were investigated [unpublished data communicated by M. Knopp and E. Thulin].
The low prevalence of resistance in the clinical setting may be due to the need of a high growth rate for the bacteria to remain within the bladder [29,32,51]. This, in combination of the high Mec concentration reached during treatments demand a phenotype that is difficult for the bacteria to acquire. For example, compensatory mutations might occur in the bacteria but the resulting phenotype does not meet the demand on either fitness or resistance. Examples of this would be when bacteria with high growth rate fail to gain the required level of MecR, or when sufficient resistance is retained but at the cost of fitness being to low, flushing out the MecR bacteria with the urine [29,51,52].
The latter example is in line with the results gained from the group of mutants with the mrdA background. The MecR of these strains increased drastically compared to the original mrdA mutant, but the compensatory mutants remained significantly low in fitness compared to WT, implying a difficulty to compensate for the high cost of the mrdA mutation. The high fitness cost of an impaired PBP2 function, caused by supplementation of Mec or by mutations in the mrdA gene, has been shown in several studies. Morphological studies of E. coli with mrdA mutations resulting in defective PBP2 have shown that alternations preventing the binding of Mec also greatly impair the protein’s physiological function [36]. Instead of the normal rod shape of E. coli, these mutants grew as round cells with a low growth rate even in the absence of Mec. [27,35].
As seen in Figures 4 and Table 3, the mrdA mutation alone does not give a high MIC but the MIC increases greatly when combined with the compensatory mutations. All but two of the reconstructed mrdA background strains had MIC values of Mec >256 mg L-1. The two exceptions (nlpI + LD and pgsA + sppA del.) still had an increase in resistance, though within three concentration steps on the MIC Test Strip of the parental value (24-32 mg L-1, 48 mg L-1 and 16 mg L-1 respectively). The increase in the MIC of Mec could possibly be explained by the effect of the compensatory mutations on the bacterial cell division and enveloping membranes. Mecillinam has been shown to have a low inhibitory effect on the cell division in low concentrations [27,35,36]. These mutations might stabilise the cellular integrity of the bacteria, allowing cell division to continue despite the inhibitory effect from Mec. Hence, the bacteria may avoid the swelling due to accumulated cell mass within the weakened bacterial envelope, leading to lysis.
The selective pressure for MecR along with environmental factors such as fitness cost appear to greatly impact the selection of clinically viable mutations. The results from the compensatory mutations found in the cysB phenotype showed that most of the reconstructed strains lost their MecR in favour of a higher growth rate (seen in Table 3 and Figure 3), even though exceptions were found. Four of the ten reconstructed compensatory strains increased their resistance (dicA, ydjNm, ydjNmx2 and nudE), though the compensations achieved in growth rates were minor. Since the MIC of the cysB mutation is at 24 mg Mec L-1, the slight increase in the values of dicA, ydjNm and ydjNmx2 were considered within error margins of the MIC test that was used. The nudE mutant showed an unexplainable high MIC, doubling

UPPSALA UNIVERSITY 2017
Emelie Ekstrand 18 (29)
that of the parental strain, and due to inconsistent results during repeats of the MIC assay this result will be excluded from further discussions.
The growth rate of strains with the dicA and nudE mutations were both significantly lower than WT (P values of <0.0001), but a slight increase was observed when compared to the cysB parental strain (P values of 0.22 and 0.21 respectively). The strains containing mutations in the promotor of the ydjN gene, ydjNm and ydjNmx2 both were significantly higher in fitness compared to parental growth rate (P values of 0.032 and <0.0001 respectively). However, the single mutation strain, ydjNm, remained significantly lower in fitness than the WT with a P value of 0.03, while interestingly the same result for ydjNmx2 was significantly higher (P value of 0.0095). Excluding the last mentioned cysB compensatory mutation, the collective results of MIC and Bioscreen C assays are concurrent with the general theory that high antibiotic resistance usually comes with a fitness cost [12].
Since a high fitness is rare in strains with high antibiotic resistance, the ydjNmx2 (DA52926) was particularly interesting. This reconstructed strain contained two mutations located in the promotor of the ydjN gene (C to A 60 nt us and T to A 47 nt us). Bioscreen C assay revealed a relative growth rate of 1.07, which was significantly higher (P value of 0,0095) than the WT. This, along with an MIC higher than the clinical breakpoint of Mec (32-48 mg L-1) see Table 3 and S1, distinguished the mutant. The mutations in the promotor caused an upregulation of the ydjN gene without the need of cysB, enabling the normal function of ydjN as a cystine importer. The regained access to cysteine would result in an increased growth rate. The increased concentration of cysteine in the bacteria appear to be low enough to maintain the MecR but sufficient to significantly increase the growth rate. Due to the fact that the ydjNmx2 mutation was not found alone but only in combination with the cysP mutation in the lineages of compensatory mutations, it needs to be further studied to ensure the results of this phenotype.
Furthermore, all the original nlpI mutations found in the compensating strains [unpublished data communicated by M. Knopp and E. Thulin] led to a loss of function. Therefore should the usage of the nlpI KO from the Keio strain K1324 [69] result in the same phenotype as that of the compensatory strains in the aspect of MIC and growth rate. Still, the nlpI strains constructed during this study should be reconstructed by a clean Knock out of the nlpI gene, rather than the transfer from a Keio strain, to ensure that the same results can be achieved. The constructions of the large duplication in the mrdA mutants should also be repeated using a different antibiotic marker, preferably a kan-sacB cassette instead of the cat-sacB, because of problems occurring during reconstructions[66,67].
Increased resistance in ΔcysB mutants by selection on high levels of Mec When analysing the clinical isolates in a previous study, some of them had mecillinam
MICs that were much higher than what is conferred by the cysB mutations they carried. To investigate the cause of this high MecR, a selection of ∆cysB mutants was carried out on MH agar supplemented with Mec at 100 and 200 mg L-1. Successful selection was achieved at a Mec concentration of 100 mg L-1 while the Mec 200 mg L-1 yielded no growth. The MICs of the isolated were all over 256 mg L-1 when grown on MH agar, somewhat conflicting results due to the lack of isolates on MH + Mec 200 mg L-1.
To identify any changes in the genome compared with the parental cysB mutant of these highly MecR mutants, WSG was performed and analysed. The results of the nine highly MecR cysB mutants showed IS elements disrupting non-essential genes (see Table 4). Most of these genes have been linked to mecillinam resistance in previous studies [71–74]. The IS elements found are thought to cause disruptions of the genes due to their large size, the transposable insertion sequences, ranging from 768 bp for IS1 to 1426 bp for IS4, preventing intended function of the products [70].

UPPSALA UNIVERSITY 2017
Emelie Ekstrand 19 (29)
The mutations found in the highly MecR cysB isolates can all be linked to the LPS which extends out from the outer membrane of E. coli. Disruption of the galU gene in connection to MecR has not been identified previously, though mutations in the galE gene have shown to greatly increase MecR in combination with other mutations (including some in cysB) in Salmonella typhimurium [75,76]. The galU gene encodes for the UTP—glucose-1-phosphate uridylyltransferase, an enzyme that is part of the galactose metabolism for E. coli. It also has a central in the synthesis of the lipopolysaccharide (LPS) core used in the cell envelope. Due to similarities in many of the gene functions found in the closely related E. coli and S. typhimurium the impact of the galE mutation in S. typhimurium may be translational to the findings of galU mutations in E. coli. The products of the rfa genes are; LPS glucosyltransferase I for rfaG, LPS core heptose kinase for rfaP, and LPS core heptosyltransferase III for rfaQ [71–73]. In a study performed in S. typhimurium it was shown that mutations in the rfa operon resulted in high levels of MecR in combination with other mutations [75]. This too might be extrapolated to explain the effects seen by the rfa mutations in E. coli.
However, none of the clinical highly MecR cysB mutant strains (MICs >256 mg/L) carried galU mutations and although several had mutations in rfa genes, similar rfa mutations are also found in strains with lower resistance (MICs about 32 mg/L). Any distinctions between the two resistance groups are yet to be unravelled. In conclusion, the connection between fitness and resistance has been further supported by the general results in this study as compensatory mutations increasing fitness resulted in decreased antibiotic resistance. The cause for the difference between MecR E. coli in clinical versus laboratory environments, appear to be linked to the bacteria’s ability to maintain a high growth rate while acquiring clinical resistance. Yet, the complete mechanisms of MecR are still unknown. More studies need to be made to further uncover the mechanisms behind MecR with the hope that increased knowledge of the causative effects behind the antibiotic resistance will postpone the post-antibiotic era.

UPPSALA UNIVERSITY 2017
Emelie Ekstrand 20 (29)
Acknowledgements
I would like to thank Dan I. Andersson for his guidance and giving me the opportunity to do my master thesis in his lab, and my co-supervisor Elisabeth Thulin for her aid and interest to share her knowledge with me. Oskar Ljungkvist for his endless patience and support, my family, and all the amazing people in the D7:3 corridor for turning this into a great experience.

UPPSALA UNIVERSITY 2017
Emelie Ekstrand 21 (29)
References
1. Fleming A. On the Antibacterial Action of Cultures of a Penicillium, with Special Reference to their Use in the Isolation of B. influenzæ. Br J Exp Pathol. 1929;10: 226–236. Available: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2048009/
2. Ligon BL. Penicillin: its discovery and early development. Semin Pediatr Infect Dis. 2004;15: 52–57. doi:10.1053/j.spid.2004.02.001
3. Aminov RI. A Brief History of the Antibiotic Era: Lessons Learned and Challenges for the Future. Front Microbiol. 2010;1. doi:10.3389/fmicb.2010.00134
4. Bérdy J. Thoughts and facts about antibiotics: Where we are now and where we are heading. J Antibiot (Tokyo). 2012;65: 385–395. doi:10.1038/ja.2012.27
5. Hewitt WL. Penicillin-Historical Impact on Infection Control. Ann N Y Acad Sci. 1967;145: 212–215. doi:10.1111/j.1749-6632.1967.tb50219.x
6. Armstrong GL, Conn LA, Pinner RW. Trends in Infectious Disease Mortality in the United States During the 20th Century. JAMA. 1999;281: 61–66. doi:10.1001/jama.281.1.61
7. Lederberg J. Infectious History. Science. 2000;288: 287–293. Available: http://www.jstor.org.ezproxy.its.uu.se/stable/3075144
8. Debabov D. Antibiotic resistance: Origins, mechanisms, approaches to counter. Appl Biochem Microbiol. 2013;49: 665–671. doi:10.1134/S0003683813080024
9. Silver LL. Challenges of Antibacterial Discovery. Clin Microbiol Rev. 2011;24: 71–109. doi:10.1128/CMR.00030-10
10. About Antimicrobial Resistance | Antibiotic/Antimicrobial Resistance | CDC [Internet]. [cited 30 May 2017]. Available: https://www.cdc.gov/drugresistance/about.html
11. WHO | Antibiotic resistance. In: WHO [Internet]. [cited 7 Jun 2017]. Available: http://www.who.int/mediacentre/factsheets/antibiotic-resistance/en/
12. Hughes D, Andersson DI. Evolutionary consequences of drug resistance: shared principles across diverse targets and organisms. Nat Rev Genet. 2015;16: 459–471. doi:10.1038/nrg3922
13. Gullberg E, Albrecht LM, Karlsson C, Sandegren L, Andersson DI. Selection of a Multidrug Resistance Plasmid by Sublethal Levels of Antibiotics and Heavy Metals. mBio. 2014;5: e01918-14. doi:10.1128/mBio.01918-14
14. Roth JR, Andersson DI, Hughes D. The Origin of Mutants under Selection: Interactions of Mutation, Growth, and Selection. EcoSal Plus. 2011;4. doi:10.1128/ecosalplus.5.6.6
15. de la Cruz F, Davies J. Horizontal gene transfer and the origin of species: lessons from bacteria. Trends Microbiol. 2000;8: 128–133. doi:10.1016/S0966-842X(00)01703-0

UPPSALA UNIVERSITY 2017
Emelie Ekstrand 22 (29)
16. Thomas CM, Nielsen KM. Mechanisms of, and Barriers to, Horizontal Gene Transfer between Bacteria. Nat Rev Microbiol. 2005;3: 711–721. doi:10.1038/nrmicro1234
17. Gyles C, Boerlin P. Horizontally Transferred Genetic Elements and Their Role in Pathogenesis of Bacterial Disease. Vet Pathol. 2014;51: 328–340. doi:10.1177/0300985813511131
18. Ochman H, Lawrence JG, Groisman EA. Lateral gene transfer and the nature of bacterial innovation. Nature. 2000;405: 299–304. doi:10.1038/35012500
19. Walsh C. Antibiotics: actions, origins, resistance [Internet]. Washington, D.C.: ASM Press; 2003. Available: https//catalog.hathitrust.org/Record/004319991
20. Andersson DI, Hughes D. Evolution of antibiotic resistance at non-lethal drug concentrations. Drug Resist Updat. 2012;15: 162–172. doi:10.1016/j.drup.2012.03.005
21. Selander RK, Levin BR. Genetic diversity and structure in Escherichia coli populations. Science. 1980;210: 545–547. doi:10.1126/science.6999623
22. Friedmann HC. Escherich and Escherichia. EcoSal Plus. 2014;6. doi:10.1128/ecosalplus.ESP-0025-2013
23. Pinho MG, Kjos M, Veening J-W. How to get (a)round: mechanisms controlling growth and division of coccoid bacteria. Nat Rev Microbiol. 2013;11: 601–614. doi:10.1038/nrmicro3088
24. Kellenberger E, Ryter A. Cell Wall and Cytoplasmic Membrane of Escherichia coli. J Biophys Biochem Cytol. 1958;4: 323–326. Available: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2224488/
25. Beveridge TJ. Structures of Gram-Negative Cell Walls and Their Derived Membrane Vesicles. J Bacteriol. 1999;181: 4725–4733. Available: http://jb.asm.org/content/181/16/4725
26. Brown L, Wolf JM, Prados-Rosales R, Casadevall A. Through the wall: extracellular vesicles in Gram-positive bacteria, mycobacteria and fungi. Nat Rev Microbiol. 2015;13: 620–630. doi:10.1038/nrmicro3480
27. Spratt BG. Distinct penicillin binding proteins involved in the division, elongation, and shape of Escherichia coli K12. Proc Natl Acad Sci U S A. 1975;72: 2999–3003. Available: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC432906/
28. Tomasz A. The Mechanism of the Irreversible Antimicrobial Effects of Penicillins: How the Beta-Lactam Antibiotics Kill and Lyse Bacteria. Annu Rev Microbiol. 1979;33: 113–137. doi:10.1146/annurev.mi.33.100179.000553
29. Medical Products Agency. Nedre urinvägsinfektion hos kvinnor - Information från Läkemedelsverket. 2007. Report No.: 18.
30. Foxman B, Barlow R, D’Arcy H, Gillespie B, Sobel JD. Urinary Tract Infection: Self-Reported Incidence and Associated Costs. Ann Epidemiol. 2000;10: 509–515. doi:10.1016/S1047-2797(00)00072-7

UPPSALA UNIVERSITY 2017
Emelie Ekstrand 23 (29)
31. Griebling TL. Urologic Diseases in America Project: Trends in Resource Use for Urinary Tract Infections in Women. J Urol. 2005;173: 1281–1287. doi:10.1097/01.ju.0000155596.98780.82
32. Swedres-Svarm. Consumption of antibiotics and occurrence of antibiotic resistance in Sweden. 2014;14027. Available: www.folkhalsomyndigheten.se/publicerat-material/
33. Sakamoto F, Ikeda S, Tsukamoto G. Novel ester of 6-((hexahydro-1H-azepin-1-yl)methyleneamino)penicillanic acid, process for its production, and its use as antibacterial agent [Internet]. EP0070477 A1, 1983. Available: http://www.google.com/patents/EP0070477A1
34. Lund F, Tybring L. 6β-Amidinopenicillanic Acids—a New Group of Antibiotics. Nature. 1972;236: 135–137. doi:10.1038/10.1038/newbio236135a0
35. Tybring L, Melchior NH. Mecillinam (FL 1060), a 6β-Amidinopenicillanic Acid Derivative: Bactericidal Action and Synergy In Vitro. Antimicrob Agents Chemother. 1975;8: 271–276. doi:10.1128/AAC.8.3.271
36. Spratt BG. The mechanism of action of mecillinam. J Antimicrob Chemother. 1977;3: 13–19. doi:10.1093/jac/3.suppl_B.13
37. Ball D, Hill J, Scott R. The Basics of General, Organic, and Biological Chemistry, v. 1.0. Chem Dep Books. 2011; Available: http://engagedscholarship.csuohio.edu/scichem_bks/2
38. FASS Vårdpersonal. Pivmecillinam - Selexid® [Internet]. 2016 [cited 14 May 2017]. Available: https://www.fass.se/LIF/product?userType=0&nplId=19771216000013
39. Tamaki S, Matsuzawa H, Matsuhashi M. Cluster of mrdA and mrdB genes responsible for the rod shape and mecillinam sensitivity of Escherichia coli. J Bacteriol. 1980;141: 52–57. Available: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC293528/
40. Vollmer W, Bertsche U. Murein (peptidoglycan) structure, architecture and biosynthesis in Escherichia coli. Biochim Biophys Acta BBA - Biomembr. 2008;1778: 1714–1734. doi:10.1016/j.bbamem.2007.06.007
41. Typas A, Banzhaf M, Gross CA, Vollmer W. From the regulation of peptidoglycan synthesis to bacterial growth and morphology. Nat Rev Microbiol. 2012;10: 123–136. doi:10.1038/nrmicro2677
42. Wolfe AJ, Brubaker L. “Sterile Urine” and the Presence of Bacteria. Eur Urol. 2015;68: 173–174. doi:10.1016/j.eururo.2015.02.041
43. Johnson JR. Virulence factors in Escherichia coli urinary tract infection. Clin Microbiol Rev. 1991;4: 80–128. doi:10.1128/CMR.4.1.80
44. O’Kelly F, Kavanagh S, Manecksha R, Thornhill J, Fennell JP. Characteristics of gram-negative urinary tract infections caused by extended spectrum beta lactamases: pivmecillinam as a treatment option within South Dublin, Ireland. BMC Infect Dis. 2016;16: 620. doi:10.1186/s12879-016-1797-3

UPPSALA UNIVERSITY 2017
Emelie Ekstrand 24 (29)
45. Adler M, Anjum M, Andersson DI, Sandegren L. Combinations of mutations in envZ, ftsI, mrdA, acrB and acrR can cause high-level carbapenem resistance in Escherichia coli. J Antimicrob Chemother. 2016;71: 1188–1198. doi:10.1093/jac/dkv475
46. Poulsen HO, Johansson A, Granholm S, Kahlmeter G, Sundqvist M. High genetic diversity of nitrofurantoin- or mecillinam-resistant Escherichia coli indicates low propensity for clonal spread. J Antimicrob Chemother. 2013;68: 1974–1977. doi:10.1093/jac/dkt159
47. Nicoloff H, Andersson DI. Indirect resistance to several classes of antibiotics in cocultures with resistant bacteria expressing antibiotic-modifying or -degrading enzymes. J Antimicrob Chemother. 2016;71: 100–110. doi:10.1093/jac/dkv312
48. Iwaya M, Jones CW, Khorana J, Strominger JL. Mapping of the mecillinam-resistant, round morphological mutants of Escherichia coli. J Bacteriol. 1978;133: 196–202. Available: http://jb.asm.org/content/133/1/196
49. Vinella D, D’Ari R, Jaffé A, Bouloc P. Penicillin binding protein 2 is dispensable in Escherichia coli when ppGpp synthesis is induced. EMBO J. 1992;11: 1493–1501. Available: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC556598/
50. EUCAST: EUCAST [Internet]. [cited 28 May 2017]. Available: http://www.eucast.org/
51. Kerrn MB, Frimodt-Møller N, Espersen F. Urinary concentrations and urine ex-vivo effect of mecillinam and sulphamethizole. Clin Microbiol Infect. 2004;10: 54–61. doi:10.1111/j.1469-0691.2004.00737.x
52. Thulin E, Sundqvist M, Andersson DI. Amdinocillin (mecillinam) resistance mutations in clinical isolates and laboratory-selected mutants of Escherichia coli. Antimicrob Agents Chemother. 2015; 1718 –1727. doi:10.1128/AAC.04819-14
53. Kredich NM. Biosynthesis of Cysteine. EcoSal Plus. 2008;3. doi:10.1128/ecosalplus.3.6.1.11
54. Nakatani T, Ohtsu I, Nonaka G, Wiriyathanawudhiwong N, Morigasaki S, Takagi H. Enhancement of thioredoxin/glutaredoxin-mediated L-cysteine synthesis from S-sulfocysteine increases L-cysteine production in Escherichia coli. Microb Cell Factories. 2012;11: 62. doi:10.1186/1475-2859-11-62
55. Ohtsu I, Wiriyathanawudhiwong N, Morigasaki S, Nakatani T, Kadokura H, Takagi H. The l-Cysteine/l-Cystine Shuttle System Provides Reducing Equivalents to the Periplasm in Escherichia coli. J Biol Chem. 2010;285: 17479–17487. doi:10.1074/jbc.M109.081356
56. Béjar S, Bouché JP. A new dispensable genetic locus of the terminus region involved in control of cell division in Escherichia coli. Mol Gen Genet MGG. 1985;201: 146–150.
57. Bessman MJ, Frick DN, O’Handley SF. The MutT Proteins or “Nudix” Hydrolases, a Family of Versatile, Widely Distributed, “Housecleaning” Enzymes. J Biol Chem. 1996;271: 25059–25062. doi:10.1074/jbc.271.41.25059

UPPSALA UNIVERSITY 2017
Emelie Ekstrand 25 (29)
58. O’Handley SF, Frick DN, Dunn CA, Bessman MJ. Orf186 Represents a New Member of the Nudix Hydrolases, Active on Adenosine(5′)triphospho(5′)adenosine, ADP-ribose, and NADH. J Biol Chem. 1998;273: 3192–3197. doi:10.1074/jbc.273.6.3192
59. Ohara M, Wu HC, Sankaran K, Rick PD. Identification and Characterization of a New Lipoprotein, NlpI, in Escherichia coli K-12. J Bacteriol. 1999;181: 4318–4325. Available: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC93934/
60. Gopalakrishnan AS, Chen YC, Temkin M, Dowhan W. Structure and expression of the gene locus encoding the phosphatidylglycerophosphate synthase of Escherichia coli. J Biol Chem. 1986;261: 1329–1338. Available: http://www.jbc.org/content/261/3/1329
61. Ichihara S, Beppu N, Mizushima S. Protease IV, a cytoplasmic membrane protein of Escherichia coli, has signal peptide peptidase activity. J Biol Chem. 1984;259: 9853–9857. Available: http://www.jbc.org/content/259/15/9853
62. Näsvall J, Knöppel A, Andersson DI. Duplication-Insertion Recombineering: a fast and scar-free method for efficient transfer of multiple mutations in bacteria. Nucleic Acids Res. 2017;45: e33–e33. doi:10.1093/nar/gkw1078
63. Datta S, Costantino N, Court DL. A set of recombineering plasmids for gram-negative bacteria. Gene. 2006;379: 109–115. doi:10.1016/j.gene.2006.04.018
64. Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A. 2000;97: 6640–6645. Available: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC18686/
65. Koskiniemi S, Pränting M, Gullberg E, Näsvall J, Andersson DI. Activation of cryptic aminoglycoside resistance in Salmonella enterica. Mol Microbiol. 2011;80: 1464–1478. doi:10.1111/j.1365-2958.2011.07657.x
66. Cherepanov PP, Wackernagel W. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene. 1995;158: 9–14. doi:10.1016/0378-1119(95)00193-A
67. Ried JL, Collmer A. An nptI-sacB-sacR cartridge for constructing directed, unmarked mutations in Gram-negative bacteria by marker exchange-eviction mutagenesis. Gene. 1987;57: 239–246. doi:10.1016/0378-1119(87)90127-2
68. R Core Team. R: A Language and Environment for Statistical Computing [Internet]. Vienna, Austria: R Foundation for Statistical Computing; 2012. Available: https://www.R-project.org
69. Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, et al. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol. 2006;2: 2006.0008. doi:10.1038/msb4100050
70. Griffiths AJ, Miller JH, Suzuki DT, Lewontin RC, Gelbart WM. Bacterial insertion sequences. 2000; Available: https://www.ncbi.nlm.nih.gov/books/NBK21779/
71. Parker CT, Kloser AW, Schnaitman CA, Stein MA, Gottesman S, Gibson BW. Role of the rfaG and rfaP genes in determining the lipopolysaccharide core structure and cell

UPPSALA UNIVERSITY 2017
Emelie Ekstrand 26 (29)
surface properties of Escherichia coli K-12. J Bacteriol. 1992;174: 2525–2538. Available: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC205891/
72. Roncero C, Casadaban MJ. Genetic analysis of the genes involved in synthesis of the lipopolysaccharide core in Escherichia coli K-12: three operons in the rfa locus. J Bacteriol. 1992;174: 3250–3260. Available: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC205993/
73. Genevaux P, Bauda P, DuBow MS, Oudega B. Identification of Tn10 insertions in the rfaG, rfaP, and galU genes involved in lipopolysaccharide core biosynthesis that affect Escherichia coli adhesion. Arch Microbiol. 1999;172: 1–8. doi:10.1007/s002030050732
74. Meyer C, Hoffmann C, Haas R, Schubert S. The role of the galU gene of uropathogenic Escherichia coli in modulating macrophage TNF-α response. Int J Med Microbiol. 2015;305: 893–901. doi:10.1016/j.ijmm.2015.09.004
75. Antón DN. Resistance to mecillinam produced by the co-operative action of mutations affecting iipopolysaccharide, spoT, and cya or crp genes of Salmonella typhimurium. Mol Microbiol. 1995;16: 587–595. doi:10.1111/j.1365-2958.1995.tb02421.x
76. Costa CS, Antón DN. High-level resistance to mecillinam produced by inactivation of soluble lytic transglycosylase in Salmonella enterica serovar Typhimurium. FEMS Microbiol Lett. 2006;256: 311–317. doi:10.1111/j.1574-6968.2006.00133.x

UPPSALA UNIVERSITY 2017
Emelie Ekstrand 27 (29)
Supplemental Materials
Table S1. All strains used in this study. Strain Genotype Description DA5438ⁱ E. coli MG1655 Wildtype (IS1 insertion in fadK) DA24100ⁱ MG1655 /pSIM5-Tet Lambda red system TetR plasmid DA24990ⁱ MG1655 /pCP20 Lambda red system AmpR, CamR FLP plasmid
DA27146ⁱ S. enterica del(CRISPR1-cas1)::spc del(CRISPR2)::cat-sacB-T0 / pSIM5-tet
Serovar Typhimurium strain LT2, used for amplification of cat-sacB cassette
DA35728ⁱ Blue-Chr, Acatsac1: amilCP+cat+sacB Used for selection with blue cat-sacB cassette DA49341ⁱ E. coli MG1655 araB::FRT-cat-FRT Used as CamR wildtype DA28439ⁱ cysB::FRT scar Reconstructed phenotypical mutant of cysB gene DA50662ⁱ cysB::FRT scar, 51 nt (A to G) ds of dicA dicA mutant into cysB KO
DA50665ⁱ cysB::FRT scar, 70 nt (G to A) and 44 nt (T to del) us of cysP cysP mutant into cysB KO
DA50668ⁱ cysB::FRT scar, 50 nt (G to T) us of nudE nudE mutant into cysB KO
DA52926ⁱ cysB::FRT scar, 60 nt (C to A) and 47 nt (T to A) us of ydjN ydjNmx2 mutant into cysB KO
DA52929ⁱ cysB::FRT scar, 73 nt (C to T) us of cysK cysK mutant into cysB KO
DA52931ⁱ cysB::FRT scar, forced dupl. cat-sacB cassette inserted by ydjN
ydjN dupl. mutant with cat-sacB cassette into cysB KO
DA52943ⁱ cysB::FRT scar, 73 nt (C to T) us of cysK, 70 nt (G to A) and 44 nt (T to del) us of cysP cysP and cysK mutants into cysB KO
DA52945ⁱ cysB::FRT scar, 60 nt (C to A) and 47 nt (T to A) us of ydjN, 44 nt (T to del) us of cysP cysP and ydjNm mutants into cysB KO
DA55120ⁱⁱ cysB::FRT scar, 47 nt (T to A) us of ydjN ydjNm mutation into cysB KO DA29705ⁱ mrdA(D389G), ybeD::FRT-scar Reconstructed mrdA mutant DA52935ⁱ mrdA(D389G), ybeD::FRT-scar, L16P in pgsA pgsA mutation into mrdA mutant DA52941ⁱ mrdA(D389G), ybeD::FRT-scar, ansA::FRT-scar ansA del into mrdA mutant DA54156ⁱⁱ mrdA(D389G), ybeD::FRT-scar, sppA::FRT-scar sppA del into mrdA mutant DA54533ⁱⁱ mrdA(D389G), ybeD::FRT-scar, Large dupl. Large dupl. into mrdA mutant
DA54536ⁱⁱ mrdA(D389G), ybeD::FRT-scar, L16P in pgsA, Large dupl.
pgsA mutation and Large dupl. into mrdA mutant
DA54538ⁱⁱ mrdA(D389G), ybeD::FRT-scar, pgsA::FRT-scar, sppA::FRT-scar pgsA del and sppA del into mrdA mutant
DA54541ⁱⁱ mrdA(D389G), ybeD::FRT-scar, sppA::FRT-scar, Large dupl. sppA del and Large dupl. into mrdA mutant
DA54543ⁱⁱ mrdA(D389G),ybeD::FRT-scar, nlpI::FRT-scar, sppA::FRT-scar nlpI* and sppA KO* into mrdA mutant
DA54545ⁱⁱ mrdA(D389G), ybeD::FRT-scar, nlpI::FRT-scar nlpI* into mrdA mutant
DA54547ⁱⁱ mrdA(D389G), ybeD::FRT-scar, nlpI::FRT-scar, Large dupl. nlpI* and Large dupl. into mrdA mutant
K323* sppA::FRT-Kan-FRT Keio strain used for sppA KO in DA54543 K1324* nlpI::FRT-Kan-FRT Keio strain used for nlpI constructions *, Keio origin; ⁱ, lab collection; ⁱⁱ, this work; del, deletion; dupl, duplication; KO, knock-out Large dupl., amplification of 300 kb at position 3366948-3651836 in the E. coli genome

UPPSALA UNIVERSITY 2017
Emelie Ekstrand 28 (29)
Table S2. A complete list of primers used. Primer name Sequence Used for sacB_scr_FWD GCTGTACCTCAAGCGAAAGG screening for the cat-sacB cassette, binds to
sacB cysB_FWD GAAAAAGACGGAAAGGCGA amplification of the cysB gene and screening
of the cysB KO cysB_REV GCGAGGCGGGTAATTAGA
cysB_KO_FWD ATGAAATTACAACAACTTCGCTATATTGTTGAGGTGGTCATGTAGGCTGGAGCTGCTTC amplification of the cat cassette used for KO
of the cysB gene cysB_KO_REV TTATTTTTCCGGCAGTTTTATATCTTTAAACATGACCTCACATATGAATATCCTCCTTAGTTCC
cysK_LiTr_FWD AATTCAGCAATCCGGCAAACCCTGAAATTCACGAAAAGACTGTAGGCTGGAGCTGCTTC amplification of the cat-sacB cassette used
for moving the mutation in the cysK gene cysK_LiTr_REV TCCCGGTATCTGGCGGTGTAGCGCCAGTCAATCGTGCTTCCATATGAATATCCTCCTTAG
cysP_LiTr_FWD AACGCGAATACCAGACTGCGCGTACTCCACCGCCAGCGATTGTAGGCTGGAGCTGCTTC amplification of the cat-sacB cassette used
for moving the mutation in the cysP gene cysP_LiTr_REV TTTCCCGAACCCGAAAACGTCGGGTAACGCGCGTTATACCCATATGAATATCCTCCTTAG
dicA FWD GGAACGGGGTGATAGTGA amplification of the dicA gene and screening of the dicA mut dicA REV GTATAGTCTGGCGCGATG
dicA_LiTr_FWD CAGAAAATTAATGCAGCGCAGGATCTGAAATGGGCTAATGTGTAGGCTGGAGCTGCTTC amplification of the cat-sacB cassette used
for moving the mutation in the dicA gene dicA_LiTr_REV ATCTATCTTGTTAGTTATGACTAACAATAAAGGTGTTTTACATATGAATATCCTCCTTAG
nudE_LiTr_FWD TCTCTGGCGCTGAATGCCGAAAGCAACAACGTCATGATGCTGTAGGCTGGAGCTGCTTC amplification of the cat-sacB cassette used
for moving the mutation in the nudE gene nudE_LiTr_REV CGCTGGCGCATATGATGGATTTGCTGGAAGACCCTGACTTCATATGAATATCCTCCTTAG
ydjN FWD AATGGTGCTGGGTTATTTGA amplification and screening of the ydjN gene, including promotor ydjN REV AAGACTAACGGCATAACGA
ydjN_LiTr_FWD GTCTGAACGCCATTGAAAGTAACTATGTTGGTAAAGTCTCTGTAGGCTGGAGCTGCTTC amplification of the cat-sacB cassette used
for moving the mutation in the ydjN gene ydjN_LiTr_REV AAGAAACGACTTTGGGTGATCGATGTCCGGTAACAAACACCATATGAATATCCTCCTTAG
ydjN_dupl_scr_FWD GTTTTGTATAGGGGCCGTTGamplification and screening of the ydjN gene ydjN_dupl_scr_REV AATAAGATCAGGAGAACGGG
ydjN_dupl_LiTr_FWD GTCAGCGAAAAAAATGTCCAACTTATCAATACATTCCTGGTGTAGGCTGGAGCTGCTTC amplification of the cat-sacB cassette used
for forced duplication of the ydjN gene ydjN_dupl_LiTr_REV TTAGTATTCAACCTGATGCTTAATAAGCCGATGAAACTGGCATATGAATATCCTCCTTAG
mrdA_FWD ATTTTACGCTCCATCATGCCamplification of the mrdA gene mrdA_REV TGTCGCCTGCCTGTAAAG
mrdA_LiTr_FWD AGTTGCCTTTGCTGCTTGGTTTTACCGTTGGGGTGTAGTCTGTAGGCTGGAGCTGCTTC amplification of the Cam cassette used for
KO of the ybeD gene (linked to mrdA) mrdA_LiTr_REV TGATCAGGTGGTTGAAGTGGTACAGCGCCATGCGCCAGGTCATATGAATATCCTCCTTAG
ansA_scr_FWD TCGTCGGCCACATTATTTamplification and screening of the ansA gene ansA_scr_REV TTAGCGACATCCACCGTA
ansA_del_LiTr_FWD ATGCAAAAGAAATCAATTTACGTTGCCTACACGGGCGGGATGTAGGCTGGAGCTGCTTC amplification of the cat-sacB cassette used
for moving the mutation in the ansA gene ansA _del_LiTr_REV TTAATCATCCGGCGTCAGTTCGCCGCGCAGGTTTTGGCTCCATATGAATATCCTCCTTAG
nlpI_K1324_FWD AGTACGTGTGCCTCAAAAamplification and screening of the nlpI gene nlpI_K1324_REV CGTATCCGTCTGAGCATT

UPPSALA UNIVERSITY 2017
Emelie Ekstrand 29 (29)
Large dupl_FWD ACGCAACAGACAAATCA amplification and screening of the Large dupl. Large dupl_REV ATACAGCTAACGTCTGGC
Large_dupl_LiTr_FWD ATTGCTGAACAACGAGCTGACTTACCAACTAATATAAAAGTGTAGGCTGGAGCTGCTTC amplification of the cat-sacB cassette used
for moving the Large dupl. Large_dupl_LiTr_REV ATCAGTAATTTTACATTGTTCATTTTGCCTCCACCAGGACCATATGAATATCCTCCTTAG
pgsA_scr FWD TCTTTTAAACTGGAGCGGGamplification and screening of the pgsA del pgsA_scr REV CGAGGAAATTGCAAAAGTG
pgsA_LiTr_FWD AGAGACATCAATGTTTCAACGACCAGAAGATCTTTTCTGCTGTAGGCTGGAGCTGCTTC amplification of the cat-sacB cassette used
for moving the mutation in the pgsA gene pgsA_LiTr_REV TTTCGGCGTAATTTTCAGCAAACGATCAAAAGTGGTGAAACATATGAATATCCTCCTTAG
sppA_scr_FWD TGATTAACTCCTGTCGTGamplification and screening of the sppA del sppA_scr_REV AGGAAATATGGGGCGAAA
sppA_del_LiTr_FWD ATGCGAACCCTTTGGCGATTTATTGCCGGATTTTTTAAATTGTAGGCTGGAGCTGCTTC amplification of the cat-sacB cassette used
for moving the mutation in the sppA del sppA_del_LiTr_REV TTAACGCATGTTGGCGCAGGTCAGGCAAAACGCATAACGGCATATGAATATCCTCCTTAG