medical device single audit program - samed home · medical device single audit program list of...
TRANSCRIPT

MEDICAL DEVICE SINGLE AUDIT PROGRAM
Medical Device Regulatory Conference 2.-3.12.2015

DR. PETER DRECHSLER

IMDRF International Medical Device Regulatory Forum

MISSION
IMDRF/MC/N1FINAL:2014
“The mission of the IMDRF [International Medical Device
Regulators Forum] is to strategically accelerate international
medical device regulatory convergence to promote an efficient
and effective regulatory model for medical devices that is
responsive to emerging challenges in the sector while
protecting and maximizing public health and safety.”

IMDRF born February 2011 as a forum to discuss future directions in medical device regulatory harmonization.
Voluntary group of medical device regulators from around the world who have come together to build on the strong foundational work of the Global Harmonization Task Force on Medical Devices (GHTF)
Accelerate international medical device regulatory harmonization and convergence.

1992 - 2012
Medical device regulatory authorities & industry
Steering committee and 5 Study Groups covering subjects:
Device Classifications; Labeling
Post Market Surveillance, Adverse Event Reporting, Reports Exchange, GMDN
Risk Management, Process Validation
Auditor Training, Regulatory Auditing Guidelines
Clinical Evaluations, Clinical Evidence

IMDRF Management Committee (MC) regulators: Australia, Brazil, Canada, China, the European Union, Japan, Russia, and the USA
Observers: APEC LSIF (Asia Pacific Economic Cooperation Life Science Innovation Forum)
WHO - World Health Organization
Affiliate Organizations: Pan American Health Organization Asian Harmonization Working Party
Working Groups: Standards; MDSAP; Submissions; UDI; NCAR; Software

IMDRF Working group items:
National Competent Authorities Report Exchange Program
Roadmap for Implementation of Unique Device Identification system
Medical Device Single Audit Program
List of Recognized Standards
Regulated Product Submission
Software as a Medical Device

THE PROGRAM

Recognize Auditing Organizations (AO)
Single Audit by AO would: benefit patient health and patient access
leverage regulatory resources
minimize medical device manufacturing disruptions due to multiple regulatory audits
provide global benefit both on short term goals and longer term goals by IMDRF regulators

STATEMENT OF COOPERATION (NOV 2012)
Australia Therapeutics Goods Administration (TGA)
Brazil Agência Nacional de Vigilância Sanitária
(ANVISA)
Canada Health Canada/Santé Canada
United States of America Food and Drug Administration (FDA)

COOPERATION
IMDRF Initial Recognition, Surveillance, and Re-Recognition Criteria for MDSAP
Recognized Auditing Organizations
Standardized Recognized AO Auditor Competency and Competency Maintenance Requirements
Standardized Regulatory Authority Assessor Competency and Competency Maintenance Requirements
Standardized Audit and Assessment Models Auditing of a Manufacturer by an MDSAP Recognized AO
Assessment of MDSAP Recognized AO’s by participating Regulatory Authorities

COOPERATION
Audit duration calculations
MDSAP audit model for consistency
MDSAP report
Standardized Rating System for Manufacturer Audit Findings
Standardized Rating System for Recognized Auditing Organization Assessment Findings
Extremely stringent impartiality requirements

THE PILOT

MDSAP PILOT
Pilot started in January 2014 (for 3 years, to Dec 2016)
CB’s from participating member states can apply to AO’s CMDCAS recognized registrars
Office audit and witnessed audit required Conducted by regulators
September 2014 AO’s started conducting audits
Operational program slated for 2017

PARTICIPATING RAS As of June 2015
USA: U.S. Food and Drug Administration (FDA)
Canada: Health Canada/Santé Canada
Brazil: Agência Nacional de Vigilância Sanitária (ANVISA)
Australia: Therapeutics Goods Administration (TGA)
Japan: Ministry of Health, Labor and Welfare (MHLW) and Pharmaceuticals and Medical Devices Agency (PMDA)

OFFICIAL OBSERVERS As of June 2015
World Health Organization (WHO) Prequalification of In Vitro Diagnostics Program
European Union

AUTHORIZED AO See Mid-Pilot-Report
.
BSI Group America Inc. http://www.bsigroup.com/en-US/
TÜV SÜD America Inc. http://www.tuv-sud-america.com/
SAI Global Cert. Services PTY Ltd. http://www.qmi.com
LNE G-MED http://www.gmed.fr
TÜV USA Inc. http://www.tuv-nord.com/us
Intertek Testing Services NA Inc. http://www.intertek.com
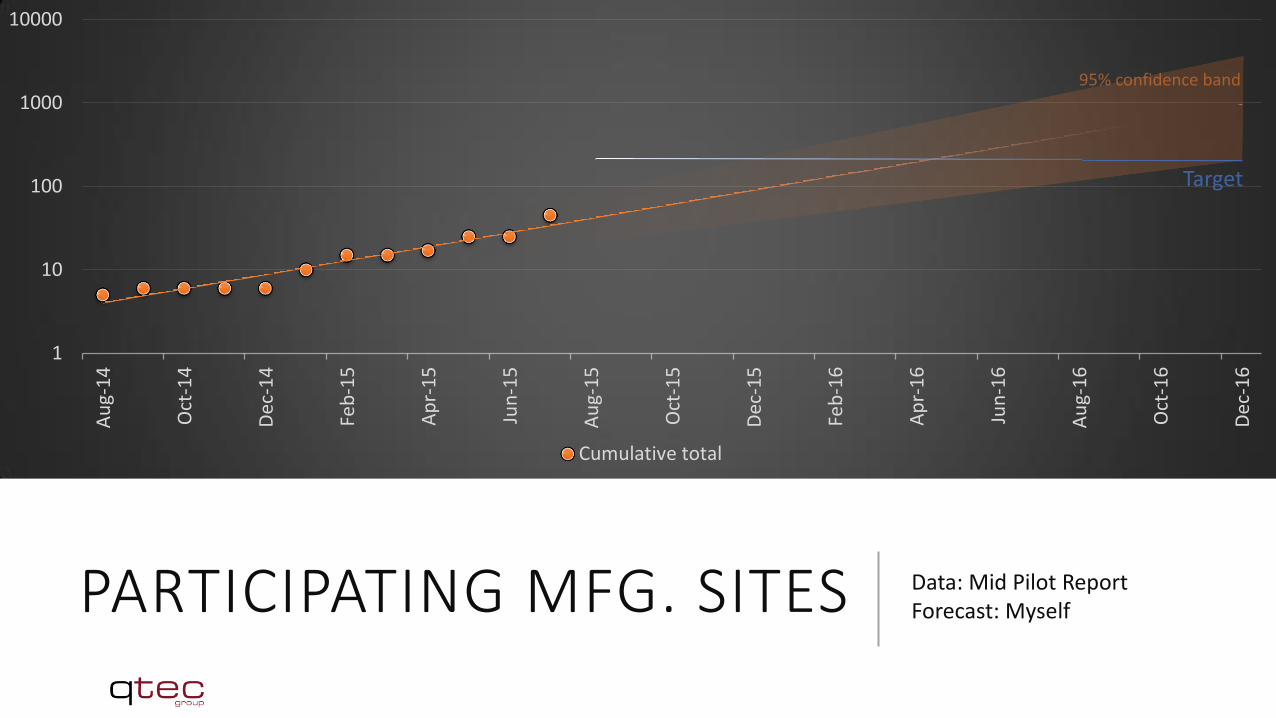
PARTICIPATING MFG. SITES Data: Mid Pilot Report Forecast: Myself
1
10
100
1000
10000
Au
g-1
4
Oct
-14
Dec
-14
Feb
-15
Ap
r-1
5
Jun
-15
Au
g-1
5
Oct
-15
Dec
-15
Feb
-16
Ap
r-1
6
Jun
-16
Au
g-1
6
Oct
-16
Dec
-16
Cumulative total

KIMBERLY TRAUTMAN, FDA Associate Director, International Affairs
Office of the Center Director
Center for Devices and Radiological Health
Personal communication, 2015-10-22: “…MDSAP is definitely going full implementation in January 2017, so manufacturers should take advantage of the ‘learning phase’ while they can…”

BENEFITS

BENEFITS FOR MANUFACTURERS
No additional requirements for manufacturers
Single audit optimizes time and resources
Routine audits are scheduled/planned with manufacturer
Expected to improve predictability
Expected to add additional Regulatory Authorities
Some RA’s will use Pilot audit outcomes as alternatives to own inspections to process applications for market authorization

PROCESS - INTEGRATED OR STAND ALONE, MORE COMPLEX? MDSAP can be integrated into existing audit cycle
CE requirements are not built into MDSAP audit tasks and not considered part of MDSAP audit time, must added
Multi-site manufacturers do not get a certificate unless all relevant sites are audited under MDSAP
No such thing as an MDSAP pre-assessment in traditional sense (all reports go to all RAs, all NC’s count)

MDSAP Pilot routine audits are announced, scheduled by the Auditing Organization with the manufacturer, with a pre-established duration.
The FDA will review MDSAP Pilot audit reports with a level of scrutiny commensurate to the significance of audit findings, taking into account the review and follow-up performed by the Auditing Organization.
Firms have one month to provide their full response to critical nonconformities (grade 4 and 5) to the Auditing Organization (as opposed to 15 working days following a FDA inspection).
Certification documents issued by the Auditing Organization state compliance with applicable US regulations, which may provide a marketing advantage.
FDA will utilize other forms of Advisory Notice, where necessary instead of FDA Warning Letters for MDSAP Audits during the Pilot. Warning Letters will only be considered when the MDSAP audit conclusion reveals an imminent/unreasonable risk to public health.

Health Canada will operate the current Canadian Medical Devices Conformity Assessment System (CMDCAS) and MDSAP in parallel during the three year pilot.
Health Canada will accept either an MDSAP certificate or a CMDCAS certificate for the purpose of obtaining a new (or maintaining an existing) Class II, III, or IV medical device license, pursuant to section 32 of the Canadian Medical Devices Regulations.
Upon the successful conclusion of the pilot, Health Canada intends to implement MDSAP as the mechanism to achieve regulatory compliance for quality management system requirements in Canada.

The Brazilian National Health Surveillance Agency – ANVISA will utilize the outcomes of the program, including the reports, to constitute an important input on ANVISA’s pre-market and post-market assessment procedures, providing, when applicable, key information that are expected to support regulatory technical evaluation on these issues.
Due to recent regulatory changes (RDC 15:2014), ANVISA may use MDSAP Pilot audits in lieu of a premarket inspection by ANVISA to grant ANVISA’s GMP Certificate to manufacturers intending to put medical devices of class III or IV on the Brazilian market. Undergoing an MDSAP Pilot audit may accelerate ANVISA ́s GMP certification process, which is a pre-requisite to the marketing authorization.
ANVISA can also use MDSAP Pilot audits to renew ANVISA’s GMP Certificate bi-annually, as an alternative to an ANVISA comprehensive inspection.

For manufacturers intending to put medical devices of class II, III or IV on the Japanese market, an MDSAP Pilot audit report might be utilized for a desk review instead of a premarket inspection performed by PMDA or registered certification bodies in Japan. An MDSAP Pilot audit report might also be utilized in this manner for periodical post market inspections.
Undergoing an MDSAP Pilot audit may accelerate the Marketing Authorization with fewer burdens as well as reduce some burden for a post market phase.

The TGA will accept MDSAP certificates as evidence of compliance with ISO13485:2003 where the Standard has been used to demonstrate partial compliance with the requirements of an Australian Conformity Assessment Procedure. It is expected that Australian Sponsors may be required to submit to the TGA, additional technical documentation to demonstrate compliance with the requirements of the Essential Principles of Safety and Performance and the manufacturer’s chosen Conformity Assessment Procedure.
The TGA will take into account MDSAP audit reports when deciding whether to issue or maintain a TGA Conformity Assessment Certificate. Under some circumstances a manufacturer may avoid routine TGA inspections.

FURTHER READING

RESOURCES Medical Device Single Audit Program
http://www.fda.gov/MedicalDevices/InternationalPrograms/MDSAPPilot/
http://www.imdrf.org/workitems/wi-mdsap.asp

INFORMATION
Pilot Program Announcement (brief description) - link
Program Announcement (including benefits) - link
MDSAP FAQs - link
Eligible Auditing Organizations – link
MDSAP Audit Procedures & Forms - link

NGEYABONGA – ENKOSI KAKHULU NGIYABONGA – I NKOMU – NDI A LIVHUHA
THANK YOU – NDIYABULELA – KE ITUMETSE KE A LEBOGA – BAIE DANKIE – DANKE SCHÖN

CONTACT DETAILS
Dr. Peter Drechsler ▪ Member of the Executive Board
qtec group ▪ Humboldtstraße 30/32 ▪ 70771 Leinfelden-Echterdingen ▪ Germany
Mobile: +49 173/2462720 ▪ Phone: +49 711/469273-40
Email: [email protected]
Internet: www.qtec-group.com ▪ DoEasy: www.DoEasy.de
Helping you to concentrate on your core competencies: the product and the market.