medicinal chemistry introduction, part 2
TRANSCRIPT

Medicinal Chemistry Introduction, Part 2
8 Nov 2018 University Tor Vergata
Vincenzo Summa Ph.D.

The lessons of today
Part one: Chemistry strategy applied from HIT ID validated to lead ID and a little bit behind J Part two: Attrition rate of the drug discovery process Toxicity and its role Examples and considerations about toxicity

What you learnt about the Drug Discovery Process…..
Preclinical candidate
Target Identification
validation
Biology
Hit Identification
Biology Med chem
Lead Optimization
Med Chem
Biology DMPK ADME Legal
Early & late Development
Process Chemistry
Toxicology Clinical Legal
Phase Discipline

What you learnt about the Drug Discovery Process…..
1) The HITs are never the best & most active molecules 2) You need to confirm the HITs, by synthesis or by acquisition of cmpds 3) Primary exploratory SAR 4) Identify a lead, the lead is never the Drug 5) Lead optimization phase: long, difficult, multiparameters needs to be taken
in consideration 6) Identification of Preclinical Candidate.... It should have the potential to
become a drug
HIT to Lead - Lead to preclinical candidate:
Each of these points implies different chemistry strategies to obtain the maximum results in the shorter time frame
Today we will talk about point 3 and 4

Structure activity relathionship (SAR): Linear Path
Lead molecules Synthesis of analogs
Test in primary assay
Test in secondary assay
Test in cell based assay
Test in vitro DMPK
Test in vivo
Test in vitro tox
PK & ADME info in precl. species Candidate for in vivo tox- Preclinical candidate

Structure activity relathionship (SAR): Modern Path
Lead molecules
Synthesis of analogs
Test in primary assay
Test in secondary assay Test in cell based assay
Test in vitro DMPK
Test in vivo Test in vitro tox
PK & ADME info in precl. species
Candidate for in vivo tox- Preclinical candidate

Analogs Synthesis .... A big dilemma First: define the strategy guided by the level of knowledge on the program Assumption: HIT validated! Small SAR information available: explore the scaffold to identify minimal active template (small number of analogs, acquisiton of commercial cmpds) Minimal template known: esploration of the SAR of side chains to improve activity against the targets and selectivity ( large set of compunds needed to explore the chemical space around the core) SAR in conjuntions in vitro DMPK and in vitro tox acitivities to identifiy liabilities (a good number of compounds needed) SAR to fix DMPK and tox issues (focus synthesis to answer specific question or hypothesis) Scale up to fully characterize a small set of compounds both in vitro and in vivo (optimization of the synthesis)

Scaffold exploration definition of minimal requirements Hit Lead investigation: Goal identify the minimal active template – define pharmacophore 1 case: HIV integrase program
The HITs
Inactive compounds
Active compound

Scaffold exploration definition of pharmacophore Hit to Lead investigation: Goal identify the minimal active tamplate – define pharmacofore
1 case: HIV integrase program
Inactive
Minimal template
Acid replacements Less active

Quinolone antibacterial Pharmacophore - Exercise
Active Compounds
Inactive Compounds
Minimal template
?
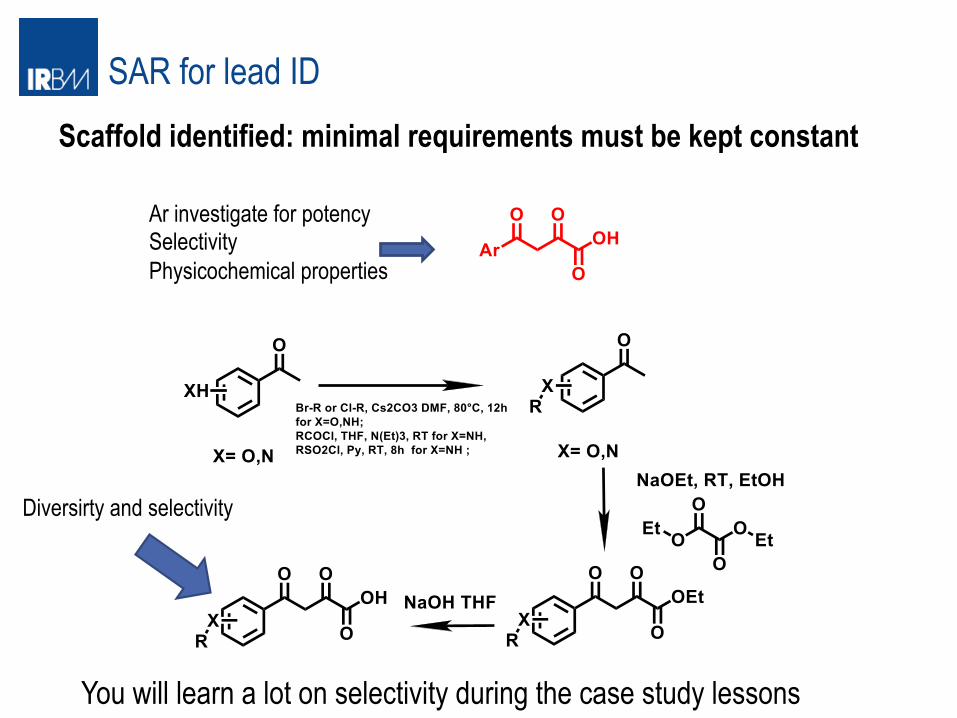
SAR for lead ID Scaffold identified: minimal requirements must be kept constant
Ar investigate for potency Selectivity Physicochemical properties
Diversirty and selectivity
You will learn a lot on selectivity during the case study lessons
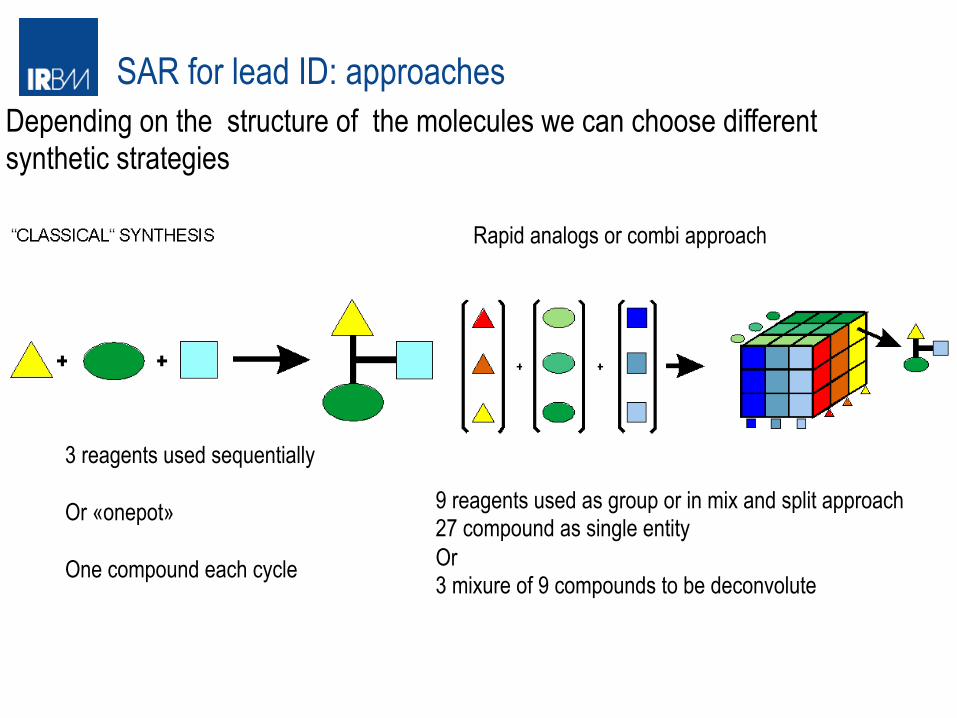
SAR for lead ID: approaches
3 reagents used sequentially Or «onepot» One compound each cycle
Rapid analogs or combi approach
9 reagents used as group or in mix and split approach 27 compound as single entity Or 3 mixure of 9 compounds to be deconvolute
Depending on the structure of the molecules we can choose different synthetic strategies

Solid phase versus solution phase pros and cons
or single

MIX and split approach When do you use this approach? 1) Chemistry must be very efficient and modular (classical application peptide syn) 2) Easy work up of the reaction
Example 3 reagents available X,Y,Z to be combined to form XYZ molecules
At the end we have 3 pools of nine compounds each, ready for testing

YXY
Deconvolution of the mixtures
Active mixture resynthesis of the pool
YXY
Active mixture resynthesis of the single cmpds
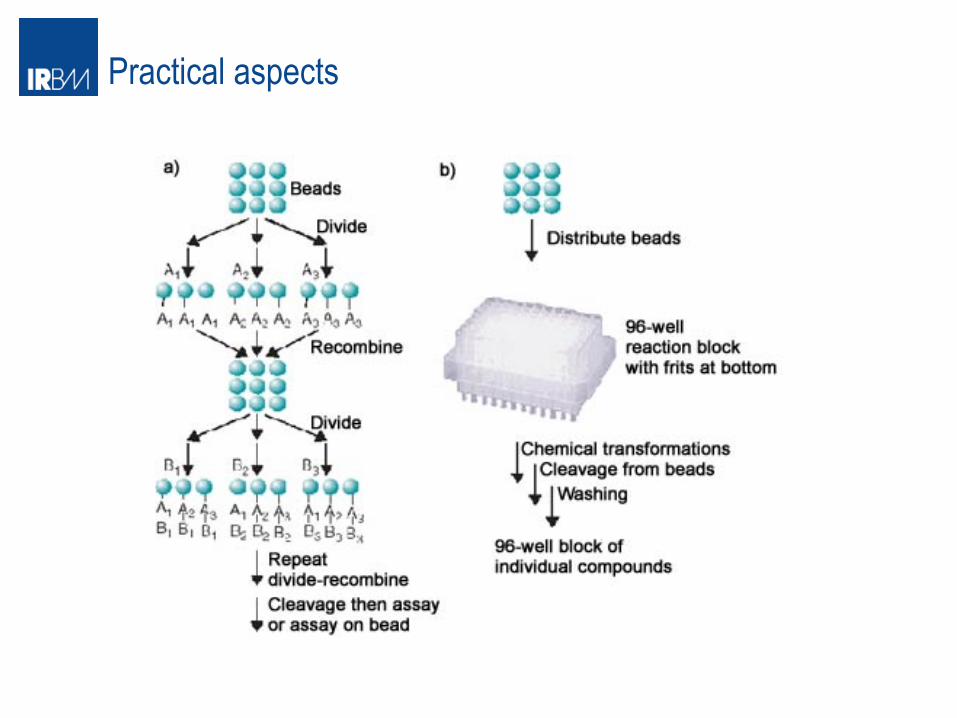
Practical aspects
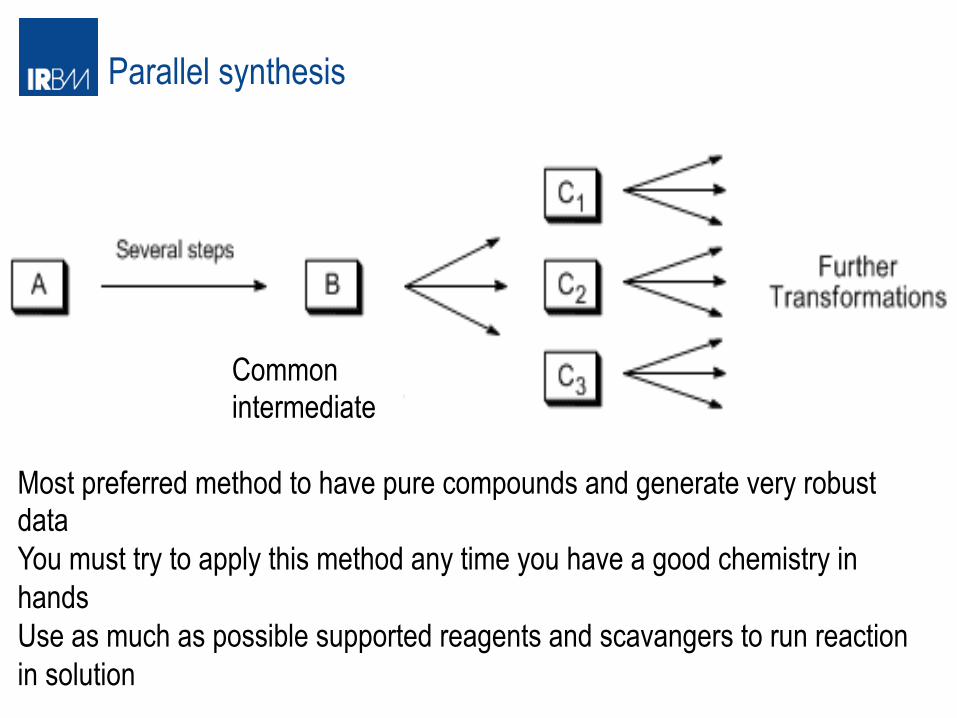
Parallel synthesis
Most preferred method to have pure compounds and generate very robust data You must try to apply this method any time you have a good chemistry in hands Use as much as possible supported reagents and scavangers to run reaction in solution
Common intermediate
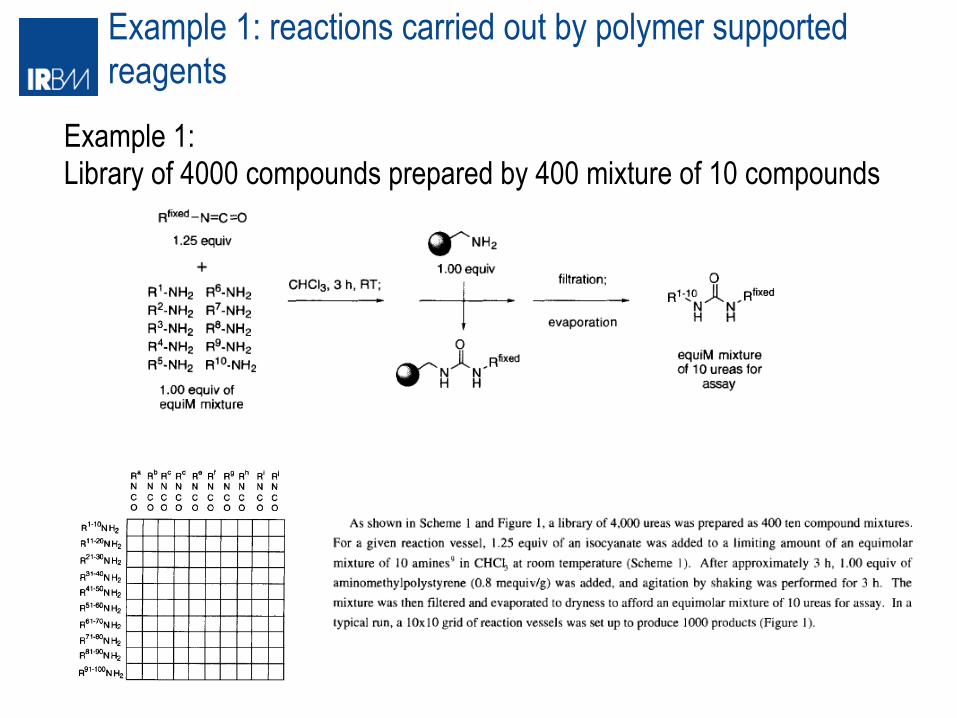
Example 1: reactions carried out by polymer supported reagents
Example 1: Library of 4000 compounds prepared by 400 mixture of 10 compounds

Example 1: Deconvolution of the active pool

Example 1: Comparison standard chemistry vs polymer supported reagent
The biological results are comparable between the two synthetic metodologies With the Polymer supported reagent the synthesis is much more efficient
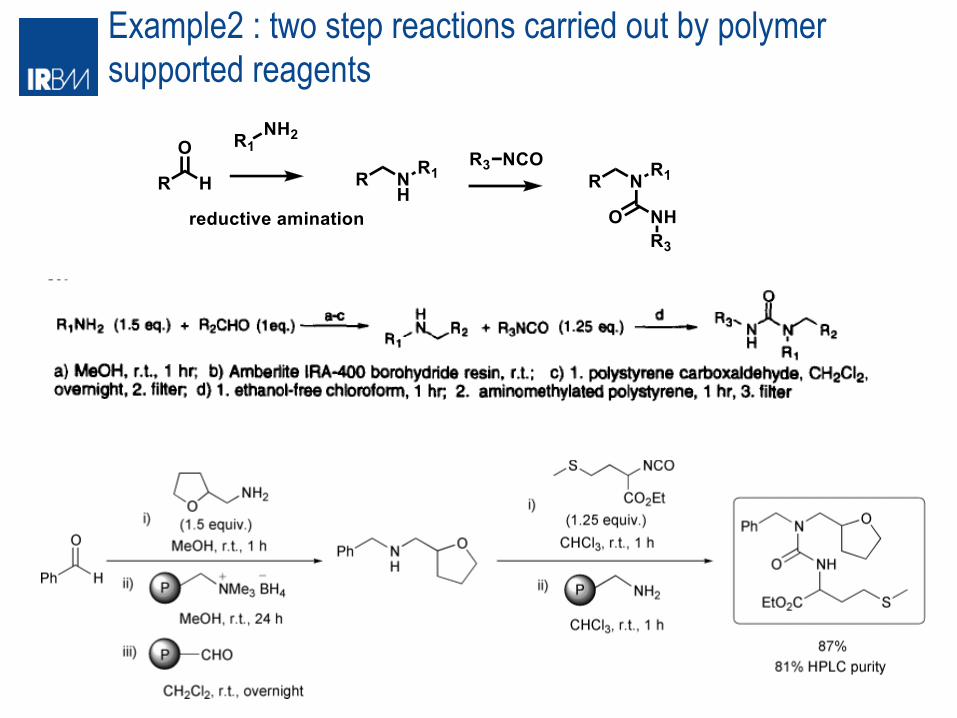
Example2 : two step reactions carried out by polymer supported reagents

Example 3: multistep synthesis carried out by polymer supported reagents synthesis of (±)-oxomaritinidine(16) and (±)- epimaritidine(17)

Consideration about polymer supported reagents
Very convenient for single step and standard reactions Needs to have good number of testing reactions to optimize the procedure per single step Allow to set up a parallel synthesis reducing drasticaly the work up cycle time (dedicated tools are needed) Cost effective methodology Raccomended for screening compounds Not applicable for scale up work or gram scale synthesis

General Drug discovery process
24
TargetID / Val
LeadOpt
EarlyDev
LateDev
LeadID
IRBM capabilities
Collab. Collab.
*Preclinical Candidate
Time & Investment

Efficiency of the drug discovery process
25
Target Identification
Lead Identification
Lead Optimization
Early & late Development

Today are we more effective in discovering new drugs?
The answer is no!!!!!!

...the problems don’t end when the drug is on the market.....

Toxicity and Safety Issues
«all substances are poisons, there is none which is not a poison. The right dose differentiaties a poison and a remedy» Paracelsus (1493-1541) The difference between a potent inhibitor / antagonist/ agonist and a drug: • the most potent compound known is of little use if it is also toxic and
unsafe to administer as a drug • To some extent the same can also be said of excellent inhibitors with
poor pharmacokinetic profiles
Toxicity: • Mechanism based • Non-mechanism based

Mechanism based toxicity
Occurs when the target receptor is involved in additional biological processes other than that desired, or when additional undesirable sequelae can occur due to impacting on the target
Should be predictable
• Question becomes whether there is a likely therapeutic window in humans, or whether the potential side effect can be tollerated
• Investigate carefully the biological logic/ rational for the target
• Make a full use of all biological tools available ( ie human genetics, KO mice, tisssue expresssion of the receptor ligands, antibodies)
• Contemplate careful in vivo exp as soon as the specific compounds become available in the program to evaluate the risks
• Investigate if there is a specie difference for the target

Non Mechanism based toxicity Toxicity is compound (or compound class) specific, NOT due to the biological mechansim involved with the target Most common form of toxicity that Medicinal Chemists have to deal with Important consideration this toxicity can be driven by a metabolite of the parent compound
Strategy to overcome the issue: Define the toxicophore ie indentify the molecualr source of the toxicity within the molecule and the cosequently modify the structure Issue realated to the strategy: can it be done mantaining efficacy a pharmacokinetic profile? Get access to a large set of enzymatic or cellular assays to find off target activities ie Panlabs 1468 diffent assay available to find off target activity https://www.eurofinspanlabs.com/Catalog/AssayCatalog/AssayCatalog.aspx?path=415&leaf=415&track=Add%2f2%2fBrands%2fPanlabs the cost is very high..... Set up in house counterscreening to evaluate a toxicity Structure Activity relathioship in parallel with efficay SAR in early phase of the program

Non mechanism based toxicity examples
Manifestations: General effects mutageniticy and genotoxicity very difficult to rationalize and difficult to solve with rational approach ( with exception of DNA alkylating agent and DNA intercalator)
Cytochrome P450 inhibition: potential for drug drug interations (ie modification od the PK parameters of co-dosed drug)
is it always undesired effect? No not always......
Potential cardiotoxicity due to hERG inhibition

Cytochrome P450 inibition always undesired?
A great number of drugs are metabolized by CYP 450. Inhibition of this detoxifying mechanism can cause severe consequences but:
-We can capitalize on the inhibition of the Cyp 450 to reduce the clearance of drug to enance exposure and half life One example is the use of Ritonavir HIV protease inhibitor not used anymore into the HIV therapy due to the poor activity against the HIV pro mutants and sever side effects.
Ritonavir strong inhibitor of CYP3A4 and 2D6
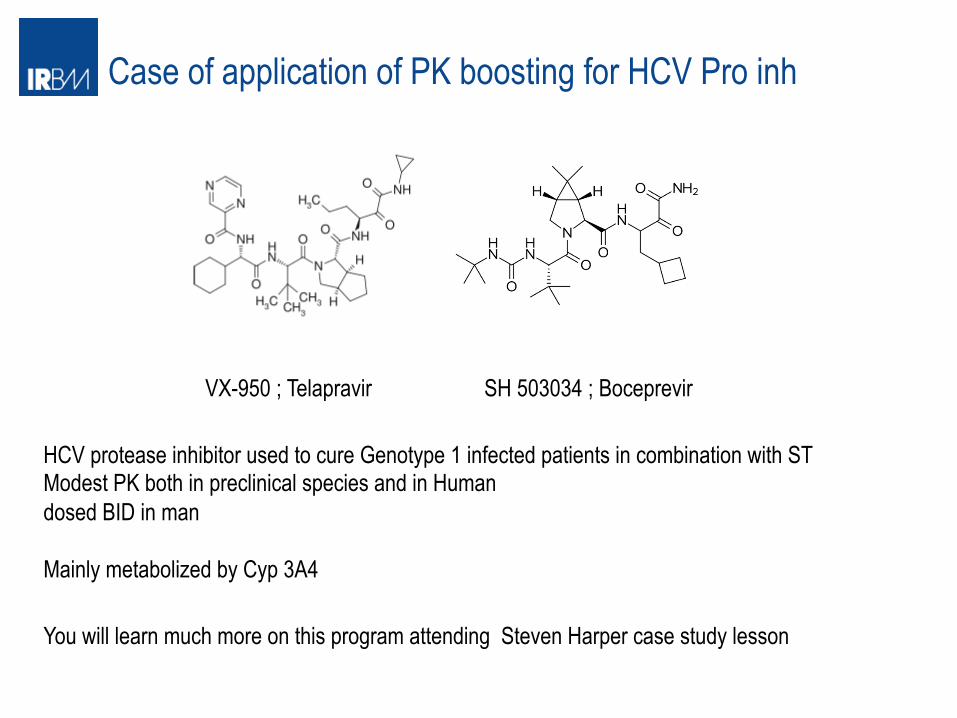
Case of application of PK boosting for HCV Pro inh
VX-950 ; Telapravir SH 503034 ; Boceprevir
HCV protease inhibitor used to cure Genotype 1 infected patients in combination with ST Modest PK both in preclinical species and in Human dosed BID in man Mainly metabolized by Cyp 3A4 You will learn much more on this program attending Steven Harper case study lesson

Combination of Ritonavir as PK boosting

Enance PK profile od HCV Pro inh in presensce of Ritonavir
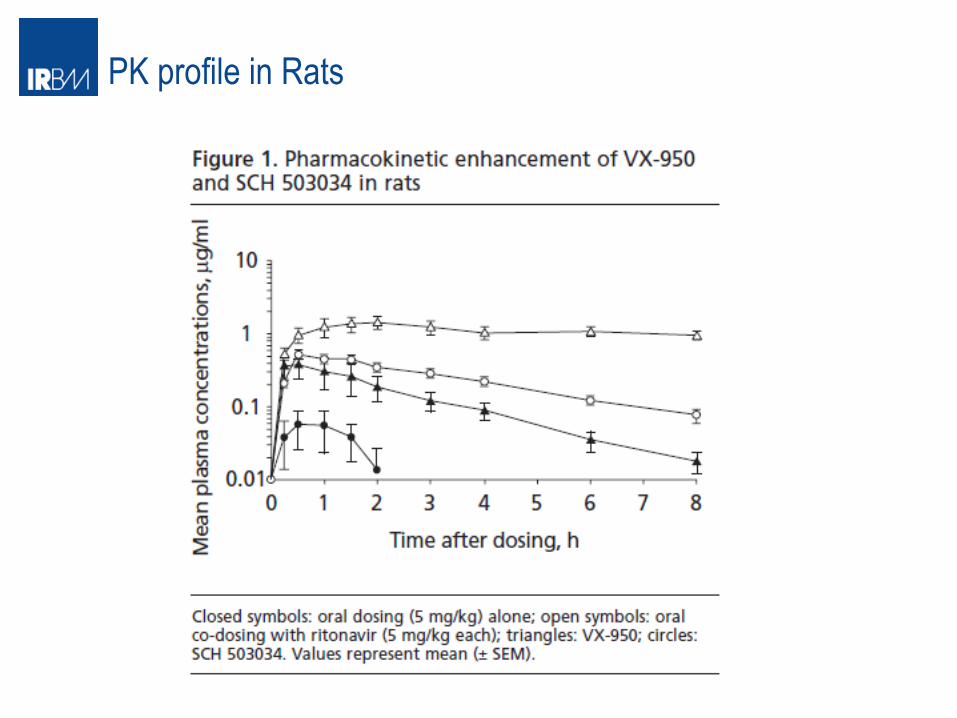
PK profile in Rats

Modeling of Human PK boosted by Ritonavir

Early assessment of cardio safety: why?

Early assessment of cardio safety: how?

Pivotal role of hERG channel in QT polongation

Pivotal role of hERG channel in QT prolongation

Curiosity...hERG is human Ether a go-go related gene

Unsual promiscous target
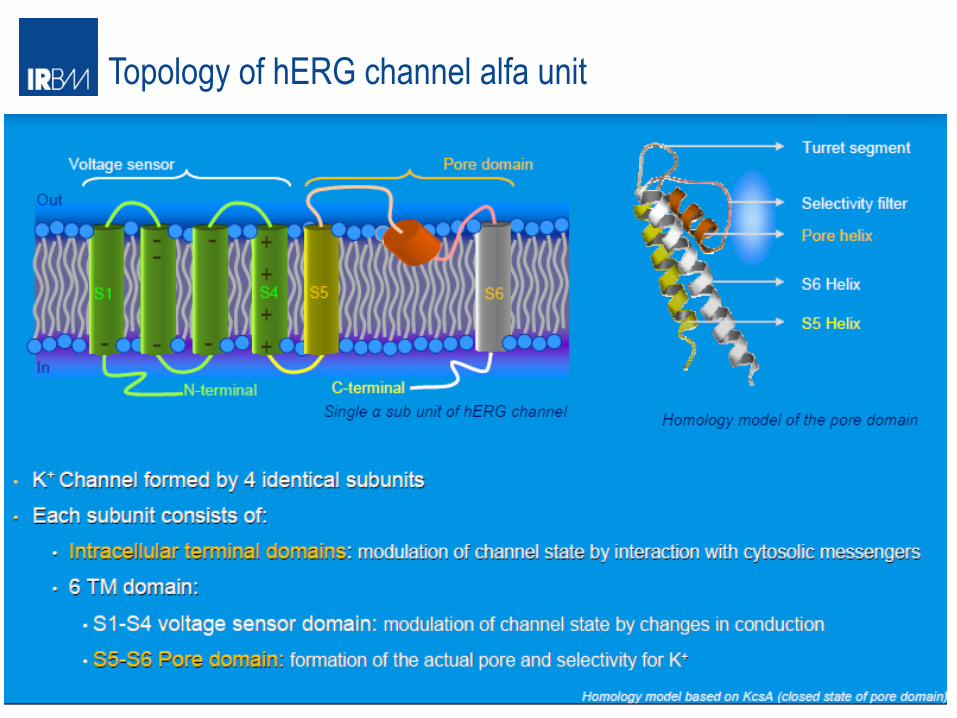
Topology of hERG channel alfa unit
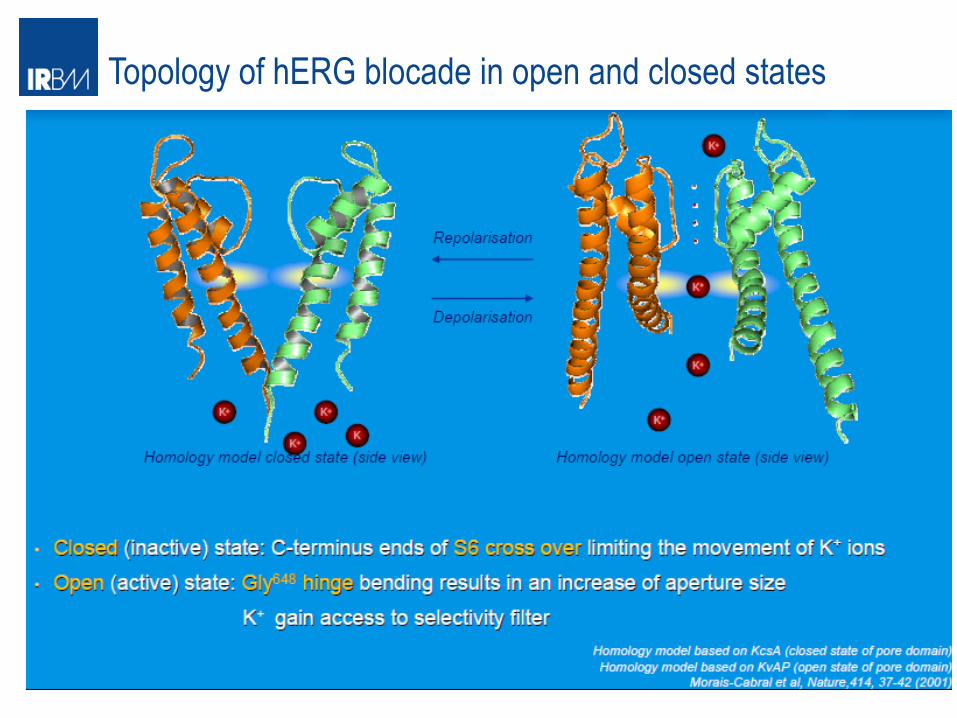
Topology of hERG blocade in open and closed states
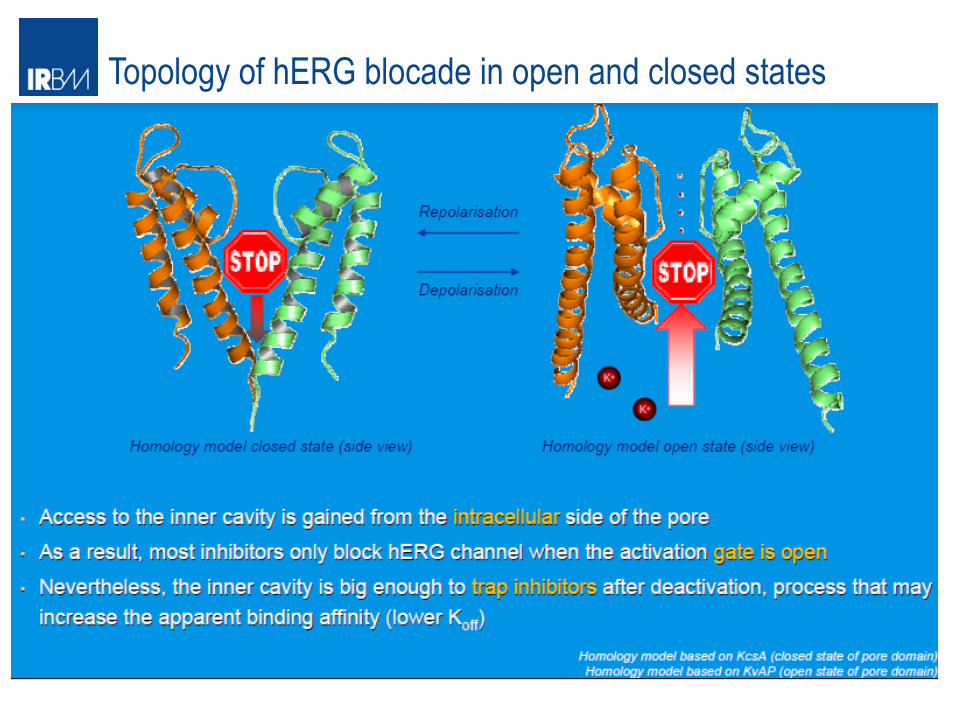
Topology of hERG blocade in open and closed states
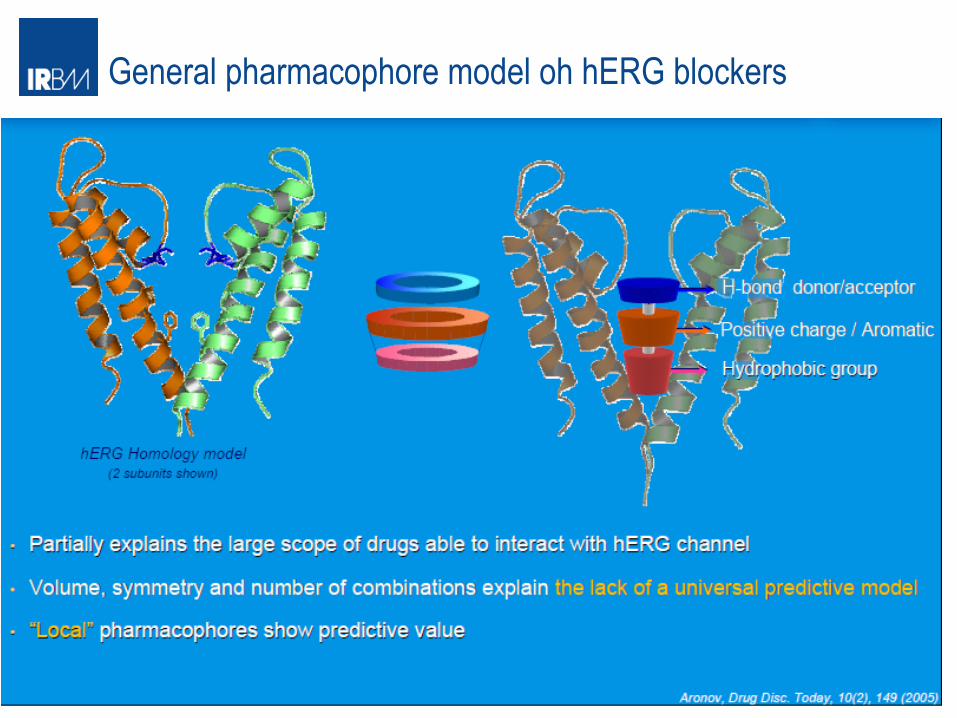
General pharmacophore model oh hERG blockers

Deletion of key interations
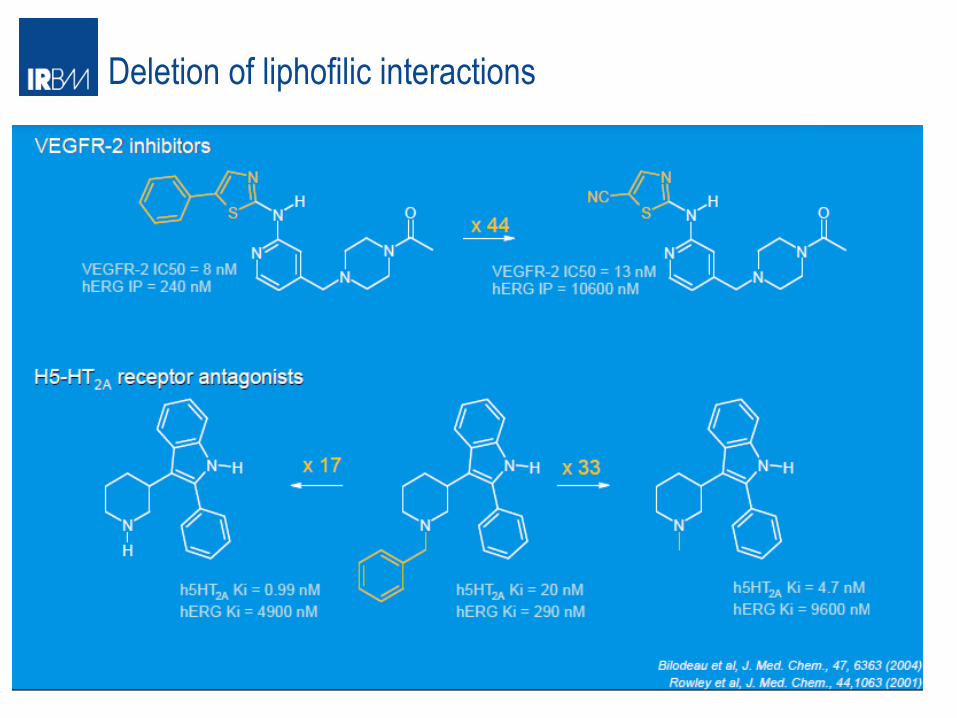
Deletion of liphofilic interactions

Deletion of the basic centre

Deletion of the basic centre

Reduction of hERG activity by modulating the pKA of the amine

Pharmacophore model for basic compounds

Final remarks
