metabolic control analysis under uncertainty: … · engineering, illinois institute of technology,...
TRANSCRIPT

Metabolic Control Analysis under Uncertainty: FrameworkDevelopment and Case Studies
Liqing Wang, _IInancx Birol, and Vassily HatzimanikatisDepartment of Chemical and Biological Engineering, Northwestern University, Evanston, Illinois
ABSTRACT Information about the enzyme kinetics in a metabolic network will enable understanding of the function of thenetwork and quantitative prediction of the network responses to genetic and environmental perturbations. Despite recentadvances in experimental techniques, such information is limited and existing experimental data show extensive variation andthey are based on in vitro experiments. In this article, we present a computational framework based on the well-established(log)linear formalism of metabolic control analysis. The framework employs a Monte Carlo sampling procedure to simulate theuncertainty in the kinetic data and applies statistical tools for the identification of the rate-limiting steps in metabolic networks.We applied the proposed framework to a branched biosynthetic pathway and the yeast glycolysis pathway. Analysis of theresults allowed us to interpret and predict the responses of metabolic networks to genetic and environmental changes, and togain insights on how uncertainty in the kinetic mechanisms and kinetic parameters propagate into the uncertainty in predictingnetwork responses. Some of the practical applications of the proposed approach include the identification of drug targets formetabolic diseases and the guidance for design strategies in metabolic engineering for the purposeful manipulation of themetabolism of industrial organisms.
INTRODUCTION
For more than a century, substantial scientific efforts have
been invested in exploring the cellular metabolism to
understand the properties of its elementary components,
such as enzymes, and distinct subsystems, such as bio-
synthetic pathways. As a result, significant advancements
have been made in this field, which in turn have led to the
appreciation of the importance of studying individual en-
zymes within the context of metabolic networks and their
physiological environment (Bailey, 1991, 1998; Papin et al.,
2003). Metabolic flux analysis (MFA) is a framework that
addresses an important aspect of this problem through the
identification and analysis of the metabolic fluxes, i.e.,
steady-state reaction rates, in metabolic networks (Papout-
sakis, 1984; Vallino and Stephanopoulos, 1993; Varma and
Palsson, 1993a,b). The mass balance equations of metabolic
intermediates and the balance equations of energy and redox
allow the formulation of linear constraints on the chemical
reaction rates around each metabolite. Some of the metabolic
fluxes can be estimated through measurements of the
consumption and production rates of extracellular metabo-
lites, i.e., substrates and products, and through tracer ex-
periments with stable isotopes that allow the estimation of
some key intracellular reactions (Klapa et al., 2003; Sauer
et al., 1997; Schmidt et al., 1999). This experimental
information is used together with the linear constraints to
obtain a quantitative estimation of the metabolic fluxes.
Constraints-based analysis (Price et al., 2003; Varma and
Palsson, 1993a,b) is another MFA approach based also on the
linear constraints on themetabolic reaction rates, and it allows
the investigation of a broad range of properties of metabolic
networks, such as the flux distribution in the metabolic
network, that can support optimal growth rate, physiological
responses of the flux distribution after gene deletion, medium
requirements, and network robustness (Price et al., 2003).
MFA has been widely applied to interpret cellular
physiology as well as to design experiments for redirecting
metabolic fluxes for improved biological performance in
medical and biotechnological applications (Stephanopoulos
and Vallino, 1991; Varma and Palsson, 1993a,b; Yarmush
and Berthiaume, 1997). However, MFA is limited in its
ability to identify how fluxes in the metabolic networks are
reconfigured in response to environmental and genetic
changes since information about the kinetic properties of
individual enzymatic steps in the metabolic networks is not
considered within the analysis.
A variety of conceptual approaches have been developed to
introduce kinetic information into the study of metabolic
networks (Teusink et al., 2000; Vaseghi et al., 1999).
Metabolic control analysis (MCA), initially called metabolic
control theory, was one of the first frameworks developed
for the study of metabolic networks with respect to their
sensitivity to biochemical and environmental variations
(Kacser and Burns, 1973). MCA offers a rigorous theoretical
means for the quantification of the steady-state and dynamic
responses of fluxes and metabolite concentrations induced by
the changes of system parameters such as enzyme activities
(Hatzimanikatis and Bailey, 1997; Kacser and Burns, 1973).
Since its establishment, this conceptual framework has
undergone extensive developments (Fell and Sauro, 1985;
Submitted June 21, 2004, and accepted for publication September 22, 2004.
Address reprint requests to Vassily Hatzimanikatis, E-mail: vassily@
northwestern.edu.
_IInancx Birol’s present address is Dept. of Chemical and Environmental
Engineering, Illinois Institute of Technology, Chicago, IL 60616.
� 2004 by the Biophysical Society
0006-3495/04/12/3750/14 $2.00 doi: 10.1529/biophysj.104.048090
3750 Biophysical Journal Volume 87 December 2004 3750–3763

Hatzimanikatis and Bailey, 1996, 1997; Heinrich and
Rapoport, 1974; Kholodenko and Westerhoff, 1993; Reder,
1988) and attracted significant attention as a powerful tool
in basic biology, biophysics, biotechnology, and medicine
(Berthiaume et al., 2003; Bowden, 1999; Cascante et al.,
2002; Schuster, 1999; Westerhoff and Kell, 1996).
However, a persisting hurdle in MCA is the lack of
comprehensive knowledge of the kinetic properties of the
enzymes in a metabolic network. Although such information
is available for many enzymes (Schomburg et al., 2002) and
biochemical techniques allow measurements of the kinetic
properties of a number of enzymes (Schmidt et al., 1999;
Teusink et al., 2000), such information is generally obtained
from in vitro studies, and inmost of the cases, details about the
in vivo kinetic properties of the enzymes are not accessible. In
addition, even when the intracellular properties are inferred
through sophisticated experiments, these measurements are
subject to variations resulting from differences in experimen-
tal systems and conditions. Furthermore, most experiments
conducted on living organisms to measure their metabolic
properties yield results that are essentially ensemble aver-
ages. Thus, these results are inherently subject to extensive
variations due to the differences between individual cells.
The uncertainty in enzyme kinetic parameters and its
impact on the prediction of the metabolic properties have
attracted considerable attention. A number of approaches
have been proposed that employ parameter space sampling
procedures and statistical analysis tools to study character-
istics of metabolic network under parameter variations
(Almaas et al., 2004; Alves and Savageau, 2000; Petkov
and Maranas, 1997; Pritchard and Kell, 2002; Thomas and
Fell, 1994). In this study, we introduce a computational
framework that enables the statistical characterization of
the kinetic responses of metabolic networks. It integrates
information from biochemistry, genomics, cell physiology,
and MFA, while taking into account the uncertainty
associated with the kinetic information of enzymes in the
network. Based on a previously developed (log)linear MCA
formalism (Hatzimanikatis and Bailey, 1996, 1997; Hatzi-
manikatis et al., 1996), the number of metabolic parameters
required for the analysis are reduced to a minimum, and the
high degree of uncertainty, as well as the partial knowledge
about the kinetic properties of the enzymes in metabolic
pathways, are addressed using a Monte Carlo method that
relies on large-scale computation. Statistical analysis of the
simulation results allows us to identify and characterize
quantitatively the rate-limiting steps in two characteristic
metabolic networks: a branched biosynthetic pathway and the
glycolysis pathway in yeast.
METHODS
Mathematic modeling of metabolic networks
For any metabolic system, the dynamics of the metabolite concentrations can
be expressed by the equation that describes the metabolite mass balances,
dxdt
¼ Nvðx; pe; psÞ; (1)
where x is the metabolite concentration vector, N is the stoichiometric
matrix, v is the metabolic flux vector, pe is the enzyme activity parameter
vector, which includes both kinetic parameters and enzyme concentrations,
and ps is the vector of other system parameters such as temperature and pH
(for nomenclature and dimensions, see Table 1). Reversible fluxes are de-
composed into two separate and opposite irreversible fluxes. The values of
the metabolic fluxes are in general functions of metabolite concentrations,
enzyme kinetic parameters, and other system parameters, such as pH and
temperature. In particular, the forward and backward rate expressions of the
same enzymatic reaction share some of the kinetic parameters.
In manymetabolic networks, the concentration of some of the metabolites
are subject to conservation constraints. Common examples include the com-
pounds involved in energy (ATP, ADP, and AMP) and redox metabolism
(NAD and NADH). The total amount of each group of these compounds,
which are called conserved moieties, remains invariant over the characteristic
response time of themetabolic network, although individual compound levels
may vary (Heinrich et al., 1977; Reich et al., 1976). Consideration of the
conserved moieties requires the introduction of a third parameter vector, pm,which represents the total concentration of the metabolites in each moiety
group. Within each conserved moiety, the conservation constraint requires
that the concentration of some compounds depends on the concentration of
their counterparts. Accordingly, the original set of metabolite concentrations
x is divided into two categories: an independent metabolite concentration
vector, xi, and a dependent metabolite concentration vector, xd. Each element
of the latter belongs to a different conserved moiety.
Conservation relationships among metabolites in a metabolic network
lead to a rank deficiency of the stoichiometric matrix, N, by introducing
linearly dependent rows. The rows that correspond to the mass balances of
the independent metabolites can be extracted from the stoichiometric matrix,
N, and form a new stoichiometric matrix, NR (Heinrich and Schuster, 1996;
Reder, 1988). Consideration of the above constraints, leads to the reduction
of the mass balance equations of the metabolic network (Eq. 1) into the form
dxi
dt¼ NR vðxi; xdðxi; pmÞ; pe; psÞ; (2)
where the set of system parameters, p, consists of the conserved moiety
concentrations pm, the enzyme activity parameters pe, and other parameters
included in ps,
pT ¼ ½pTm...pTe...pTs �: (3)
(Log)linear kinetic formalism of MCA
Within the MCA framework, concentration control coefficients,Cxp; and flux
control coefficients, Cvp; are defined as the fractional change of metabolite
concentrations and metabolic fluxes, respectively, in response to fractional
changes of system parameters (Table 1) (Kacser and Burns, 1973).
Following the established (log)linear model formalism (Hatzimanikatis
et al., 1996; Reder, 1988), we can linearize and scale the system (Eq. 2)
around the steady state, and derive these expressions for the control
coefficients (see Supplementary Material for details):
Cxip ¼�ðNRVEi1NRVEdQiÞ�1½NRVPm
..
.NRVPe
..
.NRVPs�;
(4)
Cvp ¼ ðEi1EdQiÞCxi
p 1 ½Pm...Pe
..
.Ps�: (5)
Here, V is the diagonal matrix whose elements are the steady-state fluxes; Ei
and Ed are the matrices of the elasticities with respect to metabolites, defined
MCA under Uncertainty 3751
Biophysical Journal 87(6) 3750–3763

as the local sensitivities of metabolic fluxes to independent and dependent
metabolite concentrations, respectively; IIm, IIe, and IIs are the matrices of
the elasticities with respect to parameters, i.e., the local sensitivities of
metabolic fluxes to system parameters, pm, pe, and ps, correspondingly(Table 1); andQi is a weight matrix that represents the relative abundance of
dependent metabolites with respect to the abundance of the independent
ones. A second weight matrix, Qm, is also defined, for the relative
abundance of dependent metabolites with respect to the levels of their
corresponding total moieties, which leads to the expression for the matrices
of elasticities with respect to parameters, IIm,
Pm ¼EdQm: (6)
Numerical calculation of control coefficients
Eqs. 4 and 5 suggest that the values of the flux and concentration control
coefficients depend on information from only four levels: system stoichiom-
etry, flux distribution, conservedmoiety compositions, and elasticities. Eqs. 4
and 5 lead to the important observation that the control coefficients do not
depend explicitly on the concentration of the metabolites. The concentration
of the metabolites affects only the values of the elasticities and the local
dynamics of the system.
The stoichiometric matrix, N, can be constructed based on established
biochemical studies and genomic information (Forster et al., 2003; Kanehisa
and Goto, 2000; Krieger et al., 2004), and the reduced stoichiometric matrix,
NR, can be readily deduced from system stoichiometry through the
identification of the conserved moiety groups (Schuster and Hilgetag, 1995).
The values of the net steady-state fluxes in the matrix V can be estimated
based on MFA studies (Varma and Palsson, 1993a,b; Schmidt et al., 1999;
Teusink et al., 2000). Furthermore, each reversible flux is decomposed into
a forward and a backward flux. We can define the equilibrium coefficient, r e
(0,1N), as the ratio of the forward flux rate and the backward reaction rate,
r¼ vfvb: (7)
The equilibrium coefficient is a measure of the value of the reversible steps
relative to the net flux through the reversible enzyme,
r�1¼ vf � vbvb
¼ vnetvb
: (8)
TABLE 1 MCA nomenclature
3752 Wang et al.
Biophysical Journal 87(6) 3750–3763

A r-value close to 1 corresponds to forward and backward fluxes that are
much greater than the net flux and indicates that the enzyme operates near
thermodynamic equilibrium. The values r ¼ 0 and r / N correspond to
irreversible reactions in the backward and forward directions, respectively.
Fluxes in both directions can be calculated from the net flux value and the
equilibrium coefficient.
The matrices of elasticities with respect to metabolites, Ei and Ed, depend
on the local sensitivities of enzymatic reaction rates to metabolite concentra-
tions. For example, for an enzyme that follows irreversible Michaelis-
Menten kinetics,
vi ¼ vmax;i
xjKm;i1xj
; (9)
the elasticity with respect to metabolite, ei,j, can be calculated as
ei;j ¼ @ lnvi@ lnxj
¼ 1
11xj=Km;i
: (10)
Note that ei,j is a function of the scaled substrate concentration xj/Km,i only,
and is bounded between 0 and 1. More generally, it can be easily shown that
values of the elasticities with respect to metabolites for enzymes that follow
other types of common kinetics also lie within well-defined bounds (Segel,
1975) (see Supplementary Material for some examples). For reversible
reactions, as well as for reactions with multiple reactants and products, the
enzyme elasticities with respect to substrates and products are correlated,
since individual elasticities are functions of a common set of metabolite
concentrations. For example, the rate expression of a single-substrate single-
product reaction that follows reversible Michaelis-Menten kinetics can be
written as
vf ¼ vmax;f
a
11a1p; (11)
vb ¼ vmax;b
p
11a1p; (12)
where a and p represent the scaled substrate and product concentrations,
respectively, and the subscripts f and b denote the forward and backward
reactions, respectively. The elasticities of these rate expressions with respect
to substrate and product metabolite can be expressed as
ef;a ¼ 11p
11a1p; (13)
ef;p ¼ �p
11a1p; (14)
eb;a ¼ �a
11a1p; (15)
eb;p ¼ 11a
11a1p: (16)
These relationships suggest that the elasticities of this enzyme kinetic
mechanism are correlated since four elasticities depend on two metabolite
concentrations. Furthermore, the elasticities with respect to the reactant are
in the range of [0,1] (Eqs. 13 and 16) and those with respect to the product
are in the range of [�1,0] (Eqs. 14 and 15) (see Supplementary Material for
the elasticities of enzymes with multiple reactants and products in the
glycolysis pathway case study).
In most of the common rate expressions for enzyme kinetics (Segel,
1975), as in the following case studies, the reaction rate is proportional to
a parameter called maximum reaction rate, vmax, which is the product of the
total enzyme concentration and a catalytic rate constant, commonly called
maximum specific enzyme activity. Under these conditions, the correspond-
ing elasticity with respect to maximum enzyme activity is equal to 1, and
therefore the corresponding elements in the matrix of elasticities with respect
to parameters, IIe, are equal to 1. Some of the most common exceptions
include the enzyme channeling cases (Kholodenko and Westerhoff, 1993),
which will be the subject of future work.
The elements of the weight matrix, Qm, can be estimated based on
knowledge about the relative concentration of the metabolites in the con-
served moieties (see Results and Discussion, below, on how to obtain such
estimates) and the elements of the moiety elasticity matrix, IIm, can be
obtained from Eq. 6. Finally, the elements of the elasticity matrix, IIs,describe the effect of changes in other system parameters and can be
estimated through information about the enzyme kinetics or perturbation
experiments. In the case-studies considered here we do not consider changes
in these parameters, and therefore the elasticity matrix, IIs, is not consideredin this article.
Monte Carlo simulation and stability analysis
The uncertainty in the quantification of metabolic fluxes, enzyme kinetic
properties, and metabolite concentrations, does not allow a precise quanti-
fication of the control coefficients. To address this problem, we have de-
veloped a Monte Carlo sampling methodology that allows us to describe
control coefficients using statistical analysis and representation (Fig. 1).
The method consists of the following steps:
Step 1. We define the stoichiometry based on biochemistry and
genomics knowledge about the system.
Step 2. Steady-state fluxes are estimated based on experimental
measurements and MFA methods.
Step 3. Through stoichiometric analysis, we identify the conserved
moieties and we group metabolites into an independent group and
dependent group.
Step 4. We assume a distribution for the value of the scaled metabolite
concentrations that determine the distributions in the values of the
corresponding elasticities. When details about the rate expressions of
the enzymes are not available, we either use simple random genera-
tion of the elasticity values, or assume ad hoc kinetics. If the system
FIGURE 1 Diagrammatic description of the Monte Carlo simulation
algorithm.
MCA under Uncertainty 3753
Biophysical Journal 87(6) 3750–3763

involves conserved species, we also generate random values for the
relative individual concentrations.
Step 5. For the generated elasticity values and relative values of the
conserved moieties, we test the local stability of the steady state.
Although the values of control coefficients do not depend onmetabolite
concentration levels, for every randomly generated system, i.e., for a set
of elasticities, the metabolite concentration levels will determine the
local stability of the system. Thus, for every system in the population of
the control coefficients, we generate random values for the metabolite
concentration levels within physiological bounds, and we examine the
local stability of the system based on the eigenvalues of the Jacobian
matrix,
J¼NRVðEi1EdQiÞX�1
i ; (17)
where Xi is a diagonal matrix of the corresponding metabolite
concentrations. For feasible values of the control coefficients, all the
eigenvalues of this matrix should have negative real parts for each
sample.
Step 6. If the steady state is not stable, we discard the set of values and
return to Step 4 to generate another random system. If the steady state
is stable, we calculate the corresponding control coefficients, store
the data, and return to Step 4 for the next iteration.
After repeating this process multiple times, we generate a population of
control coefficients. Unstable configurations are rejected and only stable
ones are considered for the statistical analysis of the simulated systems.
Although stable states that could correspond to locally unstable steady states,
such as oscillations, have been observed in some metabolic systems, the
current method has been developed for systems whose observable fluxes
suggest that the system is operating at stable quasisteady states, i.e., the
fluxes remain time-invariant over the observable timescale. In such systems,
the naturally occurring condition appears to be stable and we do not
introduce any bias by rejecting unstable solutions.
Monte Carlo simulations: elasticities
Enzyme elasticities associated with most common kinetic mechanisms are
uniquely determined by the scaled metabolite concentrations, which require
a complete knowledge about in vivo metabolite concentration levels and
enzyme kinetic parameters. In general, such information is not available, and
when available, it is subject to uncertainty due to experimental conditions as
discussed above. Furthermore, in vivo conditions are very different from the
in vitro conditions under which the kinetic parameters have been estimated.
To simulate these uncertainties in the elasticities, we introduced the
following Monte Carlo sampling technique.
Most of the enzymatic reactions initiate with the binding of the substrate
or regulatory metabolite, S, on the active site, A, of the enzyme
A1S�!kon AS
AS�!koff A1S
: (18)
According to this model, the free active site of the enzyme, A, binds
reversibly to metabolite, S, to form the enzyme-substrate complex, AS, with
an on-rate constant, kon, and an off-rate constant, koff. The total active site
concentration is the sum of the concentration of the free active site and the
active site-metabolite complex,
½AT� ¼ ½A�1 ½AS�; (19)
where [�] represents the concentration value of the corresponding species.
The degree of the saturation of the active site can then be defined as
sA ¼ ½AS�½AT� ¼
½S�=Km
½S�=Km11; (20)
where
Km ¼ koffkon
; (21)
and the scaled metabolite concentration can therefore be expressed in terms
of sA as
½S�Km
¼ sA
1�sA
: (22)
Assuming that the population of the active site in each species is evenly
distributed between nonsaturation and full saturation, we uniformly sample
sA by assigning random numbers between 0 and 1. Eq. 22 provides a general
way of sampling scaled metabolite concentrations to generate random
independent samples of scaled metabolite concentrations, which we can then
use to calculate the elasticities with respect to metabolites, and subsequently,
the control coefficients. Note that the distributions of the randomized
elasticities with respect to metabolites will be determined by the particular
enzyme kinetic mechanisms. For example, for enzymes that follow
Michaelis-Menten kinetics, the elasticity with respect to the substrate will
be uniformly distributed between 0 and 1; whereas for enzymes that follow
Hill kinetics, the elasticity with respect to the substrate will be distributed
between 0 and the value of the Hill coefficient, nh, following a bimodal
distribution with peaks at the limiting values (Fig. 2). These observations are
in agreement with the nature of different kinetic mechanisms. Enzymes that
follow Hill kinetics switch between an active form and an inactive form,
depending on the saturation status. Therefore the elasticity falls into two
regimes corresponding to the different enzyme activity states. This type
of information about the enzyme kinetics is captured by the Monte
Carlo sampling and the uncertainty in the enzyme saturation levels, i.e.,
the uncertainty in both enzyme kinetics and metabolite concentrations,
is translated into uncertainty in the control coefficients, which can
be statistically quantified and analyzed. Furthermore, the bounds and
distribution of sA can be refined for systems where detailed knowledge of
the degree of enzyme saturation is available (Fig. 2).
FIGURE 2 Uniform sampling of enzyme saturation degrees and random-
ization of elasticities with respect to metabolites. Histograms of elasticities
with respect to metabolites for Michaelis-Menten kinetics and Hill kinetics
(Hill coefficient ¼ 2) for uniformly sampled enzyme saturation degree, s,
at three scenarios: uniform sampling between 0 and 1 (upper panels)corresponding to the full range of enzyme saturation degrees; uniform
sampling between 0 and 0.5 (middle panel) corresponding to low enzyme
saturation; and uniform sampling between 0.5 and 1 (lower panel)
corresponding to high enzyme saturation.
3754 Wang et al.
Biophysical Journal 87(6) 3750–3763

Monte Carlo simulations: conserved moieties
The calculation of the conserved moiety weight matrices, Qi and Qm, is
illustrated here using the two most prevalent and important conserved
moieties in living cells: the adenylates group (AMP, ADP, and ATP) and the
pyridine nucleotides group (NAD and NADH). Within each moiety, the
individual species will be interconverted to each other through reactions in
the metabolic network, but the total amount of each group always remains
constant (Andersen and von Meyenburg, 1977; Ball and Atkinson, 1975;
Reich, 1974; Reich and Selkov, 1981).
For the adenylates moiety, based on the conservation relationship
½ATP�1 ½ADP�1 ½AMP� ¼ ½AP�; (23)
any of the three cofactors can be selected as a dependent metabolite (e.g.,
AMP) and the other two (ATP and ADP) will be independent metabolites.
From the definition of Qi and Qm, their elements are merely the relative
ratios of concentration of the various species within the conserved moiety
(Table 1):
d ln ½AMP�d ln ½ATP� ¼� ½ATP�
½AMP�; (24)
d ln ½AMP�d ln ½ADP� ¼�½ADP�
½AMP�; (25)
d ln ½AMP�d ln ½AP� ¼ ½AP�
½AMP�: (26)
Uncertainty should be considered again in the analysis since intracellular
metabolite concentrations might be unknown and they usually fluctuate
under different cellular states and environments. In general, each of the
metabolite concentrations could be uniformly sampled from physiological
ranges if experimental measurements exist (Teusink et al., 2000).
For the adenylates pool, in particular, the values of the weight matrix
entries are directly connected with the energetic state of the living cell. All
three adenylates are important regulators of metabolic reactions and, more
generally, many catabolic and anabolic processes are regulated by the energy
status of the cell. An index named energy charge (ec) has been defined to
reflect cellular energy status by quantifying the relative number of high-
energy phosphate bonds in the adenylates moiety (Atkinson, 1968),
ec¼ ½ATP�10:5½ADP�½AP� : (27)
Although the value of energy charge can vary between 0 and 1, living cells
maintain its value within a small range,;0.9 in Escherichia coli and 0.8–0.9
in Saccharomyces cerevisiae (Andersen and vonMeyenburg, 1977; Ball and
Atkinson, 1975). For a given ec value, the steady-state values of intracellularadenylates concentrations have the relations of
½ADP� ¼ 23ec3½AP��2½ATP�; (28)
½AMP� ¼ ð1�23ecÞ½AP�1 ½ATP�: (29)
After substituting these relationships into Eqs. 24–26, we can express the
elements of Qi and Qm as
d ln ½AMP�d ln ½ATP� ¼� ½ATP�=½AP�
1�23ec1 ½ATP�=½AP�; (30)
d ln ½AMP�d ln ½ADP� ¼� 23ec�2½ATP�=½AP�
1�23ec1 ½ATP�=½AP�; (31)
d ln ½AMP�d ln ½AP� ¼ 1
1�23ec1 ½ATP�=½AP�: (32)
These equations suggest that the elements of the adenylates weight matrices
can be calculated based on the values for the ec index and the fraction of
ATP in the adenylates pool. The viable ranges of the ec index has been
experimentally accessed for different organism species and physiological
conditions of the cells (Ball and Atkinson, 1975; Chapman et al., 1971). Eqs.
28 and 29 also suggest that, for all adenylates concentrations to be positive,
the ATP fraction has to be bounded as
23ec�1,½ATP�½AP� ,ec: (33)
Therefore, we uniformly sample ec index values within the physiological
range and, for each ec index, we uniformly sample the relative ATP levels
between the bounds defined by Eq. 33. Thus, the corresponding indices of
the weight matrices are randomized based on these samples (Eqs. 30–32).
Applying similar concepts on the quantification of the pyridine
nucleotides moiety, we start from the linear conservation relationship
½NAD�1 ½NADH� ¼ ½AN�: (34)
If we choose NAD as the dependent metabolite, the corresponding elements
in Qi and Qm are again the relative ratios of moiety concentrations,
d ln ½NAD�d ln ½NADH� ¼�½NADH�
½NAD� ; (35)
d ln ½NAD�d ln ½AN� ¼ ½AN�
½NAD�: (36)
Taking physiological conditions into consideration, the pyridine
nucleotides must be largely oxidized for the glycolytic reactions to proceed.
An index called the catabolic reduction charge (crc) has been defined to
represent the redox status of the cellular condition (Andersen and von
Meyenburg, 1977),
crc¼ ½NADH�½AN� : (37)
After the introduction of this index, Eqs. 35 and 36 can be expressed as
d ln ½NAD�d ln ½NADH� ¼� crc
1� crc; (38)
d ln ½NAD�d ln ½AN� ¼ 1
1� crc: (39)
Therefore, we uniformly sample crc values within the physiological range,
;0.05 in aerobically grown E. coli and 0.001;0.0025 in S. cerevisiae(Andersen and von Meyenburg, 1977; Holzer et al., 1956) and get the
elements of the weight matrices.
Statistical analysis
After feasible configurations of simulation outputs are calculated and
collected, statistical properties of the control coefficients are analyzed. The
primary tool that we use in the studies presented here is that of the
complementary cumulative distribution functions (CCDFs). The comple-
mentary cumulative distribution function, denoted here by F(x), measures
the probability that the random variable X assumes a value greater or equal to
x; that is, F(x)¼ P(X$ x) (Papoulis and Pillai, 2002). For a discrete sample,
like the Monte Carlo simulation used in this study,
FðxÞ ¼ +all
xi$x
pðxiÞ: (40)
The CCDF will provide a measure of the probability that a control
coefficient is greater than a certain value. Further statistical analysis of the
MCA under Uncertainty 3755
Biophysical Journal 87(6) 3750–3763

populations of the control coefficients could also be performed (Pritchard
and Kell, 2002), but it is beyond the scope of this article.
RESULTS AND DISCUSSION
In this section, we will illustrate the application of the MCA
Monte Carlo method using two case studies: a prototypical
module of biosynthetic pathways; and the more complex
yeast glycolytic pathway.
Branched pathway
A primary module of metabolic networks is the branched
pathway which, together with the linear pathway, constitutes
the major building blocks of most metabolic networks. The
splitting ratios of branching fluxes at key nodes essentially
determine the overall distribution of metabolic fluxes in
most biological systems. We chose the branched pathway
presented in Fig. 3 as our first example to illustrate how to
apply our framework on actual pathway models and how to
interpret the simulation results.
Assuming irreversible Michaelis-Menten kinetics for all
five fluxes, we derived an analytical expression for the flux
control coefficients (Table 2). The expressions suggest that
the flux control coefficients depend on the enzyme elasticities
and on the splitting ratios alone. We used the sampling pro-
cedure described above and we generated populations of
control coefficients for different splitting ratios of the flux
through the pathway to study the distribution of the control
coefficient of flux through enzyme 2, v2, with respect to the
activity of enzyme 2, pe2 :Statistical analysis of the simulated results suggests that
the splitting ratio has a critical impact on the control
coefficients distributions. When the flux through enzyme 2
is a small portion of the overall flux, i.e., a splitting ratio
a ¼ v2/(v11v2) ¼ 0.1, in 95% of the cases the control
coefficients of v2 with respect to pe2ðCv2pe2) will be.0.5, with
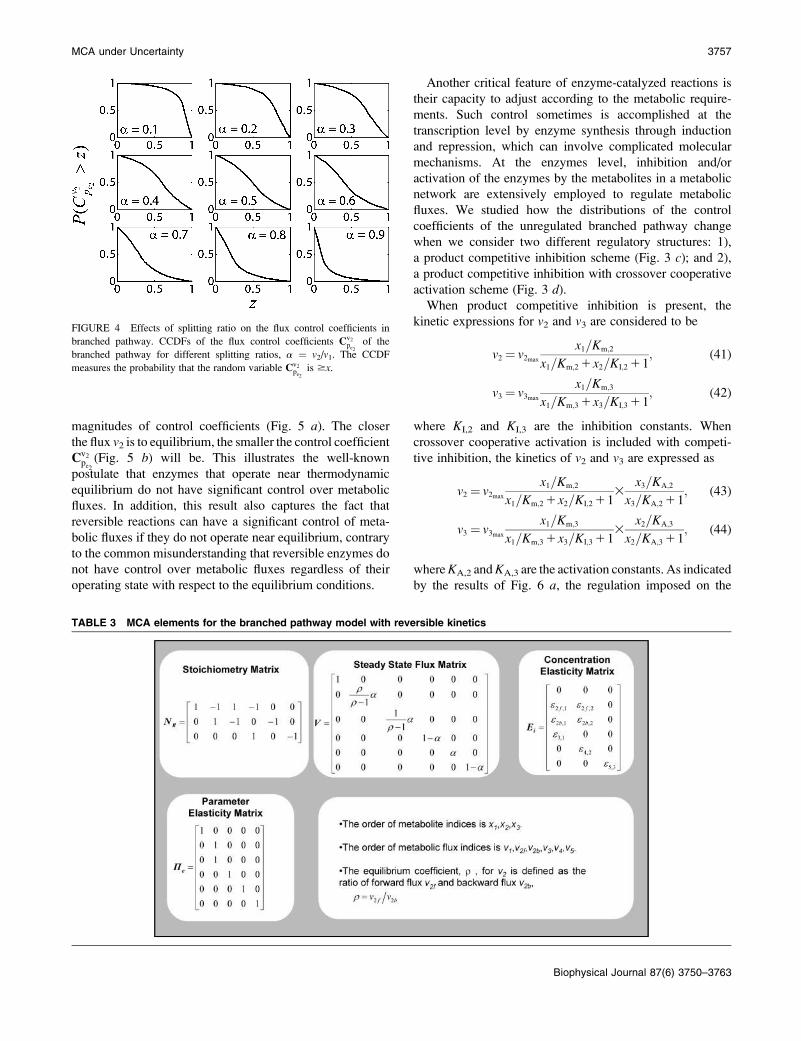
a mean value 0.84 (Fig. 4). As the splitting ratio a increases
from 0.1 to 0.9, the ability to control v2 via manipulation of
pe2 is continuously attenuating, as indicated by decreasing
mean values of Cv2pe2
and the dramatic change in the CCDF
(Fig. 4).
Reversibility also affects the distributions of control
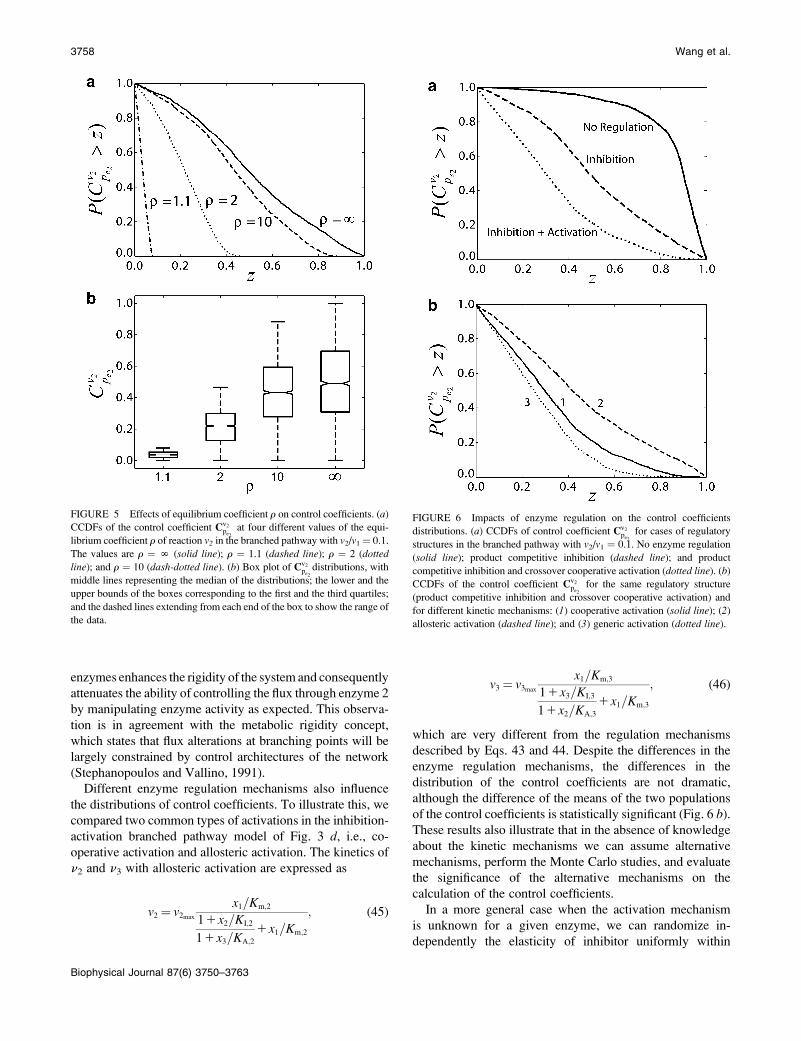
coefficients. If v2 is assumed to be a reversible reaction (Fig.
3 b) (see Table 3 for MCA elements of this model), and we
consider the control of pe2 over the net flux v2, the value ofthe equilibrium coefficient has a profound effect on the
FIGURE 3 Branched pathway models. Four types of branched bio-
synthetic pathways consisting of three metabolites: (a) simple irreversible
branched pathway; (b) branched pathway with a reversible reaction (n2); (c)
branched pathway with feedback inhibition; and (d) branched pathway withfeedback inhibition and crossover activation.
TABLE 2 MCA elements and analytical results for the branched pathway model with irreversible kinetics
3756 Wang et al.
Biophysical Journal 87(6) 3750–3763
magnitudes of control coefficients (Fig. 5 a). The closer
the flux v2 is to equilibrium, the smaller the control coefficient
Cv2pe2(Fig. 5 b) will be. This illustrates the well-known
postulate that enzymes that operate near thermodynamic
equilibrium do not have significant control over metabolic
fluxes. In addition, this result also captures the fact that
reversible reactions can have a significant control of meta-
bolic fluxes if they do not operate near equilibrium, contrary
to the common misunderstanding that reversible enzymes do
not have control over metabolic fluxes regardless of their
operating state with respect to the equilibrium conditions.
Another critical feature of enzyme-catalyzed reactions is
their capacity to adjust according to the metabolic require-
ments. Such control sometimes is accomplished at the
transcription level by enzyme synthesis through induction
and repression, which can involve complicated molecular
mechanisms. At the enzymes level, inhibition and/or
activation of the enzymes by the metabolites in a metabolic
network are extensively employed to regulate metabolic
fluxes. We studied how the distributions of the control
coefficients of the unregulated branched pathway change
when we consider two different regulatory structures: 1),
a product competitive inhibition scheme (Fig. 3 c); and 2),
a product competitive inhibition with crossover cooperative
activation scheme (Fig. 3 d).When product competitive inhibition is present, the
kinetic expressions for v2 and v3 are considered to be
v2 ¼ v2max
x1=Km;2
x1=Km;21x2=KI;211; (41)
v3 ¼ v3max
x1=Km;3
x1=Km;31x3=KI;311; (42)
where KI,2 and KI,3 are the inhibition constants. When
crossover cooperative activation is included with competi-
tive inhibition, the kinetics of v2 and v3 are expressed as
v2 ¼ v2max
x1=Km;2
x1=Km;21x2=KI;2113
x3=KA;2
x3=KA;211; (43)
v3 ¼ v3max
x1=Km;3
x1=Km;31x3=KI;3113
x2=KA;3
x2=KA;311; (44)
whereKA,2 andKA,3 are the activation constants. As indicated
by the results of Fig. 6 a, the regulation imposed on the
TABLE 3 MCA elements for the branched pathway model with reversible kinetics
FIGURE 4 Effects of splitting ratio on the flux control coefficients in
branched pathway. CCDFs of the flux control coefficients Cv2pe2
of the
branched pathway for different splitting ratios, a ¼ v2/v1. The CCDF
measures the probability that the random variable Cv2pe2
is $x.
MCA under Uncertainty 3757
Biophysical Journal 87(6) 3750–3763
enzymes enhances the rigidity of the system and consequently
attenuates the ability of controlling the flux through enzyme 2
by manipulating enzyme activity as expected. This observa-
tion is in agreement with the metabolic rigidity concept,
which states that flux alterations at branching points will be
largely constrained by control architectures of the network
(Stephanopoulos and Vallino, 1991).
Different enzyme regulation mechanisms also influence
the distributions of control coefficients. To illustrate this, we
compared two common types of activations in the inhibition-
activation branched pathway model of Fig. 3 d, i.e., co-operative activation and allosteric activation. The kinetics of
n2 and n3 with allosteric activation are expressed as
v2 ¼ v2max
x1=Km;2
11x2=KI;2
11x3=KA;2
1x1=Km;2
; (45)
v3 ¼ v3max
x1=Km;3
11x3=KI;3
11x2=KA;3
1x1=Km;3
; (46)
which are very different from the regulation mechanisms
described by Eqs. 43 and 44. Despite the differences in the
enzyme regulation mechanisms, the differences in the
distribution of the control coefficients are not dramatic,
although the difference of the means of the two populations
of the control coefficients is statistically significant (Fig. 6 b).These results also illustrate that in the absence of knowledge
about the kinetic mechanisms we can assume alternative
mechanisms, perform the Monte Carlo studies, and evaluate
the significance of the alternative mechanisms on the
calculation of the control coefficients.
In a more general case when the activation mechanism
is unknown for a given enzyme, we can randomize in-
dependently the elasticity of inhibitor uniformly within
FIGURE 5 Effects of equilibrium coefficient r on control coefficients. (a)CCDFs of the control coefficient Cv2
pe2at four different values of the equi-
librium coefficient r of reaction v2 in the branched pathway with v2/v1¼ 0.1.
The values are r ¼ N (solid line); r ¼ 1.1 (dashed line); r ¼ 2 (dottedline); and r ¼ 10 (dash-dotted line). (b) Box plot of Cv2
pe2distributions, with
middle lines representing the median of the distributions; the lower and the
upper bounds of the boxes corresponding to the first and the third quartiles;
and the dashed lines extending from each end of the box to show the range of
the data.
FIGURE 6 Impacts of enzyme regulation on the control coefficients
distributions. (a) CCDFs of control coefficient Cv2pe2
for cases of regulatory
structures in the branched pathway with v2/v1 ¼ 0.1. No enzyme regulation
(solid line); product competitive inhibition (dashed line); and product
competitive inhibition and crossover cooperative activation (dotted line). (b)CCDFs of the control coefficient Cv2
pe2for the same regulatory structure
(product competitive inhibition and crossover cooperative activation) and
for different kinetic mechanisms: (1) cooperative activation (solid line); (2)
allosteric activation (dashed line); and (3) generic activation (dotted line).
3758 Wang et al.
Biophysical Journal 87(6) 3750–3763

[�1,0] and that of activator uniformly within [0,1]. As
shown by curve 3 in Fig. 6 b, the results can still convey
useful information for the control property of the system,
although the exact mechanism of enzyme kinetics and/or
regulation might have a statistically significant impact on
the distribution of the control coefficients. The proposed
computational methodology offers the possibility to compu-
tationally investigate the effects of different enzyme kinetics
by assuming alternative kinetics expressions, or by explicit
randomization of the elasticities, and comparing the distribu-
tions of the control coefficients. Such studies will further
provide suggestions on the importance of knowledge of
enzyme kinetics in deriving conclusions about rate-limiting
enzymes and metabolic system responses to changes in
system parameters.
Glycolysis pathway
The glycolysis pathway is the main pathway of glucose
catabolism in most of the organisms, and is one of the most
complex metabolic pathways. In glycolysis, a molecule of
glucose is degraded to two molecules of pyruvate via a series
of enzyme-catalyzed reactions. ATP and NADH are the
carriers of energy and redox, respectively. Under anaerobic
conditions, pyruvate is further transformed into reduced
products such as ethanol and lactic acid. This fermentation
process is one of the major energy sources for living or-
ganisms under respiration-limited conditions. We studied
the anaerobic metabolism in yeast to illustrate the application
of our framework in such a complex metabolic network.
We based our study on a kinetic model of glycolysis of
nongrowing, anaerobic S. cerevisiae developed by Teusink
et al. (2000) through the combination of in vitro and in vivo
experiments. The model, represented by the pathway shown
in Fig. 7, consists of 19 metabolic fluxes (12 reversible and 7
irreversible fluxes) and 17 intermediates including energy
and redox cofactors. In addition to the primary product of
ethanol, byproducts such as glycerol and succinate are also
considered in the system (see Supplementary Material for
model details).
Considering the mass balance equations around each
metabolite node, a 173 31 stoichiometric matrix N was
constructed, and two conserved moieties were identified in
the system: the pyridine nucleotides ([NADH] 1 [NAD] ¼[AN]) and the adenylates ([ATP] 1 [ADP] 1 [AMP] ¼[AP]). The total concentrations of these moieties, [AN] and
[AP], were accordingly included into the analysis as
additional parameters (described in Methods). NAD and
AMP were selected as the dependent metabolites and the
reduced stoichiometry matrix, NR, was constructed. Based
on the pathway stoichiometry and physiological measure-
ments of product formation rates (Teusink et al., 2000), a
steady-state flux distribution was determined by MFA (see
Supplementary Material for details).
FIGURE 7 Anaerobic glycolytic pathway model of nongrowing yeast,
S. cerevisiae, with glucose as the sole carbon source. Chemical species: Gin,
intracellular glucose; G6P, glucose-6-phosphate; F6P, fructose-6-phos-
phate; FdP, fructose 1,6-diphosphate; GAP, glyceraldehydes-3-phosphate;DHAP, dihydroxy acetone phosphate; BPG, bisphosphoglycerate; 3PG,
3-phosphoglycerate; 2PG, 2-phosphoglycerate; PEP, phosphoenolpyruvate;
PYR, pyruvate; AcAld, acetaldehyde; ETOH, ethanol; ATP, adenosine
triphosphate; ADP, adenosine diphosphate; AMP, adenosine monophos-
phate; NAD and NADH, nicotinamide adenine dinucleotide. Pathway steps
and enzymes (in bold): trans, glucose cross-membrane transport; HK,
hexokinase; PGI, phosphoglucose isomerase; PFK, phosphofructokinase;
ALD, fructose 1,6-diphosphate aldolase; TPI, triose phosphate isomerase;
GAPDH, glyceraldehydes-3-phosphate dehydrogenase; PGK, phosphoglyc-
erate kinase; PGM, phosphoglycerate mutase; ENO, enolase; PYK, pyruvate
kinase; PDC, pyruvate decarboxylase; ADH, alcohol dehydrogenase;
ATPase, net ATP consumption; and AK, adenylate kinase.
MCA under Uncertainty 3759
Biophysical Journal 87(6) 3750–3763

The elasticities with respect to metabolites are calculated
based on the enzyme kinetics mechanism provided by the
model (Teusink et al., 2000) (see Supplementary Material for
details) and the scaled metabolite concentrations generated
by uniformly sampling enzyme saturation degrees. For the
calculation of weight matrix indices, ATP was chosen as the
independent metabolite of the adenylates moiety and its
fraction in the adenylates pool was sampled based on the
uniformly randomized energy charge within its physiolog-
ically measured range, [0.8–0.9] (Ball and Atkinson, 1975),
and Eq. 33. The catabolic reduction charge is randomized
within experimental range, [0.001–0.0025] (Holzer et al.,
1956). After a Monte Carlo simulation, 5495 random
datasets were sampled before the 5000 sample sets, yielding
stable solutions (91% stability), and the distribution of the
flux control coefficients was determined.
In the yeast anaerobic metabolic pathway, the enzyme
alcohol dehydrogenase (ADH) catalyzes the reaction step
leading to ethanol production and the flux through this
enzyme also reflects the overall flux through the glycolysis
pathway since it is the main output flux of the carbon through
glycolysis. We analyzed the distribution of the control
coefficients of the flux through this enzyme with respect to
three key enzymes: phosphofructokinase (PFK), pyruvate
kinase (PYK), and the enzyme facilitating the transport of
glucose across the plasma membrane (transporter). A large
number of enzymes have been identified as hexose trans-
porters. In this study, as in most of the modeling studies, we
lump the function and kinetics of each enzyme into a single
step. PFK and PYK catalyze phosphoryl group transfer reac-
tions during the glucose breakdown and are major points of
regulation in glycolysis (Fig. 7). Similarly, glucose transport
has been shown to exert considerable control on the glycolytic
flux (Reijenga et al., 2001). Hence, a detailed study of the
control coefficients of ADH with respect to these three key
enzymes would be of interest for a better understanding of the
control of the glycolytic fluxes.
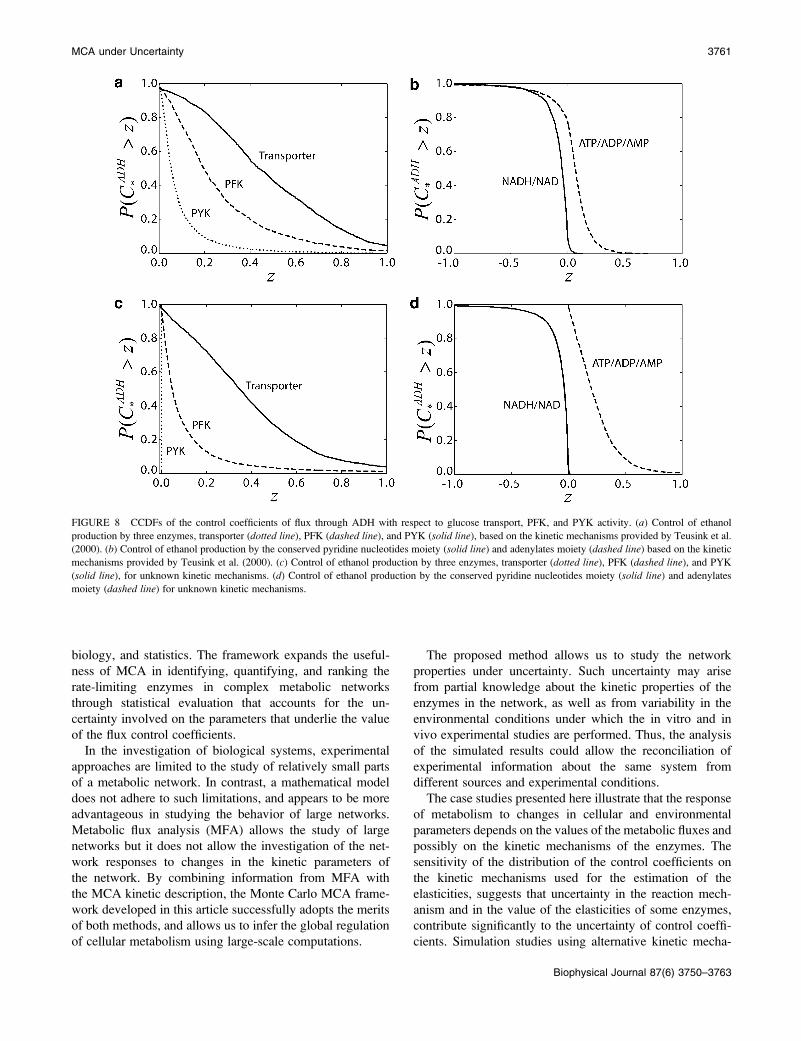
Our result confirmed that glucose uptake exerts the stronger
control on the glycolytic flux, with PFK, and to a lesser extent
PYK, sharing the control (Fig. 8 a). This implies that the
changes in the activity of glucose transporters might lead to
significant changes in anaerobic ethanol production. How-
ever, the mean values of the control coefficients are relatively
low (0.48 for transport, 0.27 for PFK, and 0.09 for PYK),
suggesting that overexpression of any of the enzymes alone
might not lead to significant changes. This is consistent with
experimental studies that have shown that single enzyme
overexpression cannot increase glycolytic flux in yeast
(Schaaff et al., 1989). Furthermore, our results suggest that
simultaneous overexpression of PFK and PYK will also be
ineffective in increasing glycolytic flux, since the probability
of these enzymes, together, to have a flux control coefficient
.0.2, is,0.25. These conclusions are also in agreement with
the previously reported experiments on simultaneous over-
expression of PFK and PYK (Schaaff et al., 1989). Although
these experimental studies have been performed under
conditions different from those used to quantify our Monte
Carlo model, the statistical nature of our framework allows us
to draw some broader conclusions since it could account for
the uncertainties introduced due to the relative differences
between the two experimental systems.
In addition, we studied the control over ethanol production,
and glycolytic flux, exerted by the conserved moieties. As
indicated by Fig. 8 b, increasing the total level of pyridine
nucleotides will likely lead to a decrease of ethanol pro-
duction, whereas increasing adenylates concentration is more
likely to result in an increase of ethanol production. However,
the negative values for some of the control coefficients of the
ethanol productionwith respect to the total level of adenylates
(Fig. 8 b) suggest that there exist some configurations of
elasticities that could lead to a decrease in ethanol production
when the total level of adenylates is increasing. Although
there is no experimental evidence of such coupling between
glycolytic flux and conserved moieties, it illustrates the
potential control of the levels of the conserved moieties on
metabolic fluxes as previously demonstrated for simpler
model systems (Kholodenko et al., 1994).
For many biological organisms under investigation,
systematic and detailed descriptions of enzyme kinetic
mechanisms are absent. This is a typical challenge when
genome-scale metabolic networks are considered (Forster
et al., 2003). To examine the possibility of applying our
framework to such systems, we assumed no information of
enzyme kinetic expressions of the glycolysis model and
randomized all elasticities with respect to metabolites by
uniform sampling between [0,1] for substrates and activator
metabolites, and [�1,0] for products and inhibitor metabo-
lites. In this scenario, we observed similar patterns of control
coefficients distributions (Fig. 8, c and d) as before (Fig. 8,
a and b). However, the distribution of the glycolytic flux
control coefficients with respect to PYK and PFK appear to
be sensitive to the knowledge about the details of enzyme
kinetics, whereas the distribution of the control coefficient
with respect to glucose transport remains almost the same.
These observations suggest that accurate estimation of the
control coefficients of glycolytic flux, with respect to PYK
and PFK, will require knowledge of the kinetic mechanism
of some of the enzymes in the pathway, while glucose
transport is, indeed, one of the most important determin-
ing steps of the glycolytic flux. A more detailed analysis
of the effects of the knowledge of the kinetic mechanisms of
the individual enzymes could help us identify which are the
enzymes for which additional knowledge about their kinetics
is required.
CONCLUDING REMARKS
The statistical and computational MCA framework presented
here combines principles and methods from biochemistry,
mathematical modeling, systems engineering, computational
3760 Wang et al.
Biophysical Journal 87(6) 3750–3763
biology, and statistics. The framework expands the useful-
ness of MCA in identifying, quantifying, and ranking the
rate-limiting enzymes in complex metabolic networks
through statistical evaluation that accounts for the un-
certainty involved on the parameters that underlie the value
of the flux control coefficients.
In the investigation of biological systems, experimental
approaches are limited to the study of relatively small parts
of a metabolic network. In contrast, a mathematical model
does not adhere to such limitations, and appears to be more
advantageous in studying the behavior of large networks.
Metabolic flux analysis (MFA) allows the study of large
networks but it does not allow the investigation of the net-
work responses to changes in the kinetic parameters of
the network. By combining information from MFA with
the MCA kinetic description, the Monte Carlo MCA frame-
work developed in this article successfully adopts the merits
of both methods, and allows us to infer the global regulation
of cellular metabolism using large-scale computations.
The proposed method allows us to study the network
properties under uncertainty. Such uncertainty may arise
from partial knowledge about the kinetic properties of the
enzymes in the network, as well as from variability in the
environmental conditions under which the in vitro and in
vivo experimental studies are performed. Thus, the analysis
of the simulated results could allow the reconciliation of
experimental information about the same system from
different sources and experimental conditions.
The case studies presented here illustrate that the response
of metabolism to changes in cellular and environmental
parameters depends on the values of the metabolic fluxes and
possibly on the kinetic mechanisms of the enzymes. The
sensitivity of the distribution of the control coefficients on
the kinetic mechanisms used for the estimation of the
elasticities, suggests that uncertainty in the reaction mech-
anism and in the value of the elasticities of some enzymes,
contribute significantly to the uncertainty of control coeffi-
cients. Simulation studies using alternative kinetic mecha-
FIGURE 8 CCDFs of the control coefficients of flux through ADH with respect to glucose transport, PFK, and PYK activity. (a) Control of ethanol
production by three enzymes, transporter (dotted line), PFK (dashed line), and PYK (solid line), based on the kinetic mechanisms provided by Teusink et al.
(2000). (b) Control of ethanol production by the conserved pyridine nucleotides moiety (solid line) and adenylates moiety (dashed line) based on the kinetic
mechanisms provided by Teusink et al. (2000). (c) Control of ethanol production by three enzymes, transporter (dotted line), PFK (dashed line), and PYK
(solid line), for unknown kinetic mechanisms. (d) Control of ethanol production by the conserved pyridine nucleotides moiety (solid line) and adenylates
moiety (dashed line) for unknown kinetic mechanisms.
MCA under Uncertainty 3761
Biophysical Journal 87(6) 3750–3763

nisms will help us identify the enzymes whose kinetics
have the most significant effect on the uncertainty of the
predicted system properties. To address this problem
quantitatively we have developed a framework, based on
uncertainty propagation methodologies, that identifies the
relative contribution of the uncertainties in the elasticities of
each enzyme to the uncertainty in the estimation of the
control coefficients (F. Mu and V. Hatzimanikatis, un-
published). The enzymes with the greatest contribution in the
uncertainty of the control coefficients should be the subject
of experimental studies that will determine the details of their
kinetic mechanism for an accurate prediction of the network
properties.
Conserved moieties, enzyme channeling, futile cycles,
cofactor coupling, and regulatory interactions contribute
significantly to the complexity of metabolic networks.
Application of the proposed framework and the related
framework for the quantification of uncertainty propagation
(F. Mu and V. Hatzimanikatis, unpublished) to such complex
pathways will allow us to build a better understanding on
how these elements, e.g., levels of conserved moieties and
enzyme channeling, contribute to the values of the control
coefficients and to the uncertainty in estimating these values
since the framework requires no explicit knowledge of
kinetic parameters.
The MCA formalism allows the estimation of the response
of metabolic networks to relatively small changes in the
metabolic parameters. Estimation of the response of metab-
olism to large changes in the metabolic parameters will
require the use of detailed nonlinear models for the kinetics of
the enzymes in the pathway. However, at the vicinity of small
changes in the metabolic parameters, the results of both
formalisms will be equivalent and the distribution of the
control coefficients will remain unaffected, unless the non-
linear model operates near critical bifurcation points. An
MCA framework, like the one presented here, will provide
guidance for studies of nonlinear models under large changes
in the kinetic parameters. The ability of our framework to
capture the effect of the hypothesized kinetic mechanisms,
and the functional form of the corresponding elasticities, on
the estimated distributions of the control coefficients will
further provide a systematic method and a starting point for
the evaluation of the effects of uncertainty in the kinetic
mechanisms of the enzymes in nonlinear models.
The identifications of drug targets in metabolic diseases,
the metabolic engineering of industrial organisms, and the
identification of the genotype-to-phenotype relationship, are
some of the important applications of MCA (Cascante et al.,
2002). All of these systems involve a great degree of
uncertainty, they are subject to wide variations of their
extracellular environment, and their ultimate function is the
result of a population of individual cells. The ability of the
proposed framework to quantify system properties based on
a range of system parameters is ideal for studying the
properties of multicellular complex systems.
SUPPLEMENTARY MATERIAL
An online supplement to this article can be found by visiting
BJ Online at http://www.biophysj.org.
The authors are grateful for the financial support provided by the
Department of Energy (DE-AC36-99GO103), the National Aeronautics
and Space Administration (NAG 2-1527), and DuPont through a DuPont
Young Professor Award (to V.H.). L.W. received partial support by the
Chinese Government through the State Excellence Scholarship program for
students studying overseas.
REFERENCES
Almaas, E., B. Kovacs, T. Vicsek, Z. N. Oltvai, and A. L. Barabasi. 2004.Global organization of metabolic fluxes in the bacterium Escherichiacoli. Nature. 427:839–843.
Alves, R., and M. A. Savageau. 2000. Systemic properties of ensembles ofmetabolic networks: application of graphical and statistical methods tosimple unbranched pathways. Bioinformatics. 16:534–547.
Andersen, K. B., and K. von Meyenburg. 1977. Charges of nicotinamideadenine nucleotides and adenylate energy charge as regulatory pa-rameters of the metabolism in Escherichia coli. J. Biol. Chem. 252:4151–4156.
Atkinson, D. E. 1968. The energy charge of the adenylate pool asa regulatory parameter. Interaction with feedback modifiers. Biochem-istry. 7:4030–4034.
Bailey, J. E. 1991. Toward a science of metabolic engineering. Science.252:1668–1675.
Bailey, J. E. 1998. Mathematical modeling and analysis in biochemicalengineering: past accomplishments and future opportunities. Biotechnol.Prog. 14:8–20.
Ball, W. J., and D. E. Atkinson. 1975. Adenylate energy charge inSaccharomyces cerevisiae during starvation. J. Bacteriol. 121:975–982.
Berthiaume, F., A. D. MacDonald, Y. H. Kang, and M. L. Yarmush. 2003.Control analysis of mitochondrial metabolism in intact hepatocytes:effect of interleukin-1b and interleukin-6. Metab. Eng. 5:108–123.
Bowden, A. C. 1999. Metabolic control analysis in biotechnology andmedicine. Nat. Biotechnol. 17:641–643.
Cascante, M., L. G. Boros, B. Comin-Anduix, P. de Atauri, J. J. Centelles,and P. W. N. Lee. 2002. Metabolic control analysis in drug discovery anddisease. Nat. Biotechnol. 20:243–249.
Chapman, A. G., L. Fall, and D. E. Atkinson. 1971. Adenylate energycharge in Escherichia coli during growth and starvation. J. Bacteriol.108:1072–1086.
Fell, D. A., and H. M. Sauro. 1985. Metabolic control and its analysis.Additional relationships between elasticities and control coefficients.Eur. J. Biochem. 148:555–561.
Forster, J., I. Famili, P. Fu, B. O. Palsson, and J. Nielsen. 2003. Genome-scale reconstruction of the Saccharomyces cerevisiae metabolic network.Genome Res. 13:244–253.
Hatzimanikatis, V., and J. E. Bailey. 1996. MCA has more to say. J. Theor.Biol. 182:233–242.
Hatzimanikatis, V., and J. E. Bailey. 1997. Effects of spatiotemporalvariations on metabolic control: approximate analysis using (log)linearkinetic models. Biotechnol. Bioeng. 54:91–104.
Hatzimanikatis, V., C. A. Floudas, and J. E. Bailey. 1996. Analysis anddesign of metabolic reaction networks via mixed-integer linearoptimization. AICHE J. 42:1277–1292.
Heinrich, R., S. M. Rapoport, and T. A. Rapoport. 1977. Metabolicregulation and mathematical models. Prog. Biophys. Mol. Biol. 32:1–82.
Heinrich, R., and T. A. Rapoport. 1974. A linear steady-state treatment ofenzymatic chains. General properties, control and effector strength. Eur.J. Biochem. 42:89–95.
3762 Wang et al.
Biophysical Journal 87(6) 3750–3763

Heinrich, R., and S. Schuster. 1996. The Regulation of Cellular Systems.Chapman & Hall, New York.
Holzer, H., F. Lynen, and G. Schultz. 1956. Biochem. Z. 328:252–263[Determination of diphosphopyridine nucleotide/reduced diphosphopyr-idine nucleotide quotient in living yeast cells by analysis of constantalcohol and acetaldehyde concentrations].
Kacser, H., and J. A. Burns. 1973. The control of flux. Symp. Soc. Exp.Biol. 27:65–104.
Kanehisa, M., and S. Goto. 2000. KEGG: Kyoto encyclopedia of genes andgenomes. Nucleic Acids Res. 28:27–30.
Kholodenko, B. N., H. M. Sauro, and H. V. Westerhoff. 1994. Control byenzymes, coenzymes and conserved moieties. A generalisation of theconnectivity theorem of metabolic control analysis. Eur. J. Biochem.225:179–186.
Kholodenko, B. N., and H. V. Westerhoff. 1993. Metabolic channelling andcontrol of the flux. FEBS Lett. 320:71–74.
Klapa, M. I., J. C. Aon, and G. Stephanopoulos. 2003. Systematicquantification of complex metabolic flux networks using stable isotopesand mass spectrometry. Eur. J. Biochem. 270:3525–3542.
Krieger, C. J., P. Zhang, L. A. Mueller, A. Wang, S. Paley, M. Arnaud,J. Pick, S. Y. Rhee, and P. D. Karp. 2004. MetaCyc: a multiorganismdatabase of metabolic pathways and enzymes. Nucleic Acids Res. 32:D438–D442.
Papin, J. A., N. D. Price, S. J. Wiback, D. A. Fell, and B. O. Palsson. 2003.Metabolic pathways in the post-genome era. Trends Biochem. Sci.28:250–258.
Papoulis, A., and S. U. Pillai. 2002. Probability, Random Variables, andStochastic Processes. McGraw-Hill, Boston, MA.
Papoutsakis, E. T. 1984. Equations and calculations for fermentations ofbutyric-acid bacteria. Biotechnol. Bioeng. 26:174–187.
Petkov, S. B., and C. D. Maranas. 1997. Quantitative assessment ofuncertainty in the optimization of metabolic pathways. Biotechnol.Bioeng. 56:145–161.
Price, N. D., J. A. Papin, C. H. Schilling, and B. O. Palsson. 2003. Genomescale microbial in-silico models: the constraints-based approach. TrendsBiotechnol. 21:162–169.
Pritchard, L., and D. B. Kell. 2002. Schemes of flux control in a model ofSaccharomyces cerevisiae glycolysis. Eur. J. Biochem. 269:3894–3904.
Reder, C. 1988. Metabolic control theory: a structural approach. J. Theor.Biol. 135:175–201.
Reich, J. G. 1974. Near-equilibrium reactions and the regulation ofpathways. Symp. Biol. Hung. 18:159–171.
Reich, J. G., and E. E. Selkov. 1981. Energy Metabolism of the Cell.Academic Press, New York.
Reich, J. G., E. E. Selkov, T. Geier, and V. Dronova. 1976. Elementaryproperties of energy-regenerating pathways. Studia Biophys. 54:57–76.
Reijenga, K. A., J. L. Snoep, J. A. Diderich, H. W. van Verseveld, H. V.Westerhoff, and B. Teusink. 2001. Control of glycolytic dynamics byhexose transport in Saccharomyces cerevisiae. Biophys. J. 80:626–634.
Sauer, U., V. Hatzimanikatis, J. E. Bailey, M. Hochuli, T. Szyperski, andK. Wuthrich. 1997. Metabolic fluxes in riboflavin-producing Bacillussubtilis. Nat. Biotechnol. 15:448–452.
Schaaff, I., J. Heinisch, and F. K. Zimmermann. 1989. Overproduction ofglycolytic enzymes in yeast. Yeast. 5:285–290.
Schmidt, K., J. Nielsen, and J. Villadsen. 1999. Quantitative analysis ofmetabolic fluxes in Escherichia coli, using two-dimensional NMRspectroscopy and complete isotopomer models. J. Biotechnol. 71:175–189.
Schomburg, I., A. Chang, O. Hofmann, C. Ebeling, F. Ehrentreich, and D.Schomburg. 2002. BRENDA: a resource for enzyme data and metabolicinformation. Trends Biochem. Sci. 27:54–56.
Schuster, S. 1999. Use and limitations of modular metabolic controlanalysis in medicine and biotechnology. Metab. Eng. 13:232–242.
Schuster, S., and C. Hilgetag. 1995. What information about the conservedmoiety structure of chemical reaction systems can be derived from theirstoichiometry. J. Phys. Chem. 99:8017–8023.
Segel, I. H. 1975. Enzyme Kinetics: Behavior and Analysis of RapidEquilibrium and Steady State Enzyme Systems. Wiley, New York.
Stephanopoulos, G., and J. J. Vallino. 1991. Network rigidity and metabolicengineering in metabolite overproduction. Science. 252:1675–1681.
Teusink, B., J. Passarge, C. A. Reijenga, E. Esgalhado, C. C. van derWeijden, M. Schepper, M. C. Walsh, B. M. Bakker, K. van Dam, H. V.Westerhoff, and J. L. Snoep. 2000. Can yeast glycolysis be understoodin terms of in vitro kinetics of the constituent enzymes? Testingbiochemistry. Eur. J. Biochem. 267:5313–5329.
Thomas, S., and D. A. Fell. 1994. Metabolic control analysis—sensitivityof control coefficients to experimentally determined variables. J. Theor.Biol. 167:175–200.
Vallino, J. J., and G. Stephanopoulos. 1993. Metabolic flux distributions inCorynebacterium glutamicum during growth and lysine overproduction.Biotechnol. Bioeng. 41:633–646.
Varma, A., and B. O. Palsson. 1993a. Metabolic capabilities of Escherichiacoli. 1. Synthesis of biosynthetic precursors and cofactors. J. Theor. Biol.165:477–502.
Varma, A., and B. O. Palsson. 1993b. Metabolic capabilities of Escherichiacoli. 2. Optimal growth patterns. J. Theor. Biol. 165:503–522.
Vaseghi, S., A. Baumeister, M. Rizzi, and M. Reuss. 1999. In vivodynamics of the pentose phosphate pathway in Saccharomycescerevisiae. Metab. Eng. 1:128–140.
Westerhoff, H. V., and D. B. Kell. 1996. What biotechnologists knew allalong? J. Theor. Biol. 182:411–420.
Yarmush, M. L., and F. Berthiaume. 1997. Metabolic engineering andhuman disease. Nat. Biotechnol. 15:525–528.
MCA under Uncertainty 3763
Biophysical Journal 87(6) 3750–3763