microsatelliteinstability,apoptosis ...cancerres.aacrjournals.org/content/56/6/1374.full.pdf ·...
TRANSCRIPT

(CANCER RESEARCH 56, 1374-1381, March 15. I
Microsatellite Instability, Apoptosis, and Loss of p53 Function in Drug-resistantTumor Cells1
D. Alan Anthoney, Amanda J. Mcllwrath, William M. Gallagher, Angela R. M. Edlin,2 and Robert Brown3
CRC Department of Medical Oncology, CRC Bealson Laboratories, Garsciibe Estate. Switchback Road, Glasgow G6I I HI). Uniteti Kingdom
ABSTRACT
We have examined microsatellite instability and loss of p53 function inhuman tumor cell line models of acquired anticancer drug resistance. Weobserve acquisition of an RER+ phenotype in cell lines selected for resist
ance to cisplatin or doxorubicin. The majority of independent cisplatin-resistant sublines are RKR+, whereas the parental line shows no evidence
of microsatellite instability. Microsatellite mutations in random, nonse-Icriril subclones of a cisplatin-resistant line are observed in the absence offurther drug exposure, suggesting that the KIR' phenotype is a stable
phenotype rather than being transiently induced by DNA damage. Furthermore, a cisplatin-resistant derivative shows reduction in a G:T mis
match recognition activity compared to the parental line. Independentlines selected by multiple exposure to cisplatin show resistance factors ofup to 5-fold by clonogenic assay and have reduced cisplatin-induced
apoptosis. The resistant lines that are Ul K show evidence of loss ofpS.Vdependent functions, as measured by loss of radiation-induced G,arrest and reduced CIP I mRNA. Induced loss of p53 function by trans-fection of mutant TPS3 does not cause a detectable RER"1"phenotype. We
speculate that tolerance of DNA damage and expansion of cells with anKl l\ phenotype may select for reduced ability to engage apoptosis andloss of p53 function.
INTRODUCTION
Resistance to anticancer agents is a major problem in the successfultreatment of tumors. An initially sensitive tumor or cell line canacquire resistance to multiple anticancer agents after exposure to evena single agent (1,2). DNA is generally accepted as the main target formany of the clinically successful anticancer drugs (3). A variety ofmechanisms that affect the level of drug reaching DNA has beendemonstrated (4, 5); however, none of these are entirely satisfactory inexplaining the patterns of cross-resistance to agents such as cisplatin,
doxorubicin. and ionizing radiation. Recently, much attention hascentered on the ability of cells to engage an apoptotic response afterDNA damage as being an important mechanism that could explainacquired resistance to multiple anticancer agents (6). The TP53 geneis required for DNA damage-induced apoptosis in certain cell types
(7, 8), although this response can be reduced by altered expression ofmembers of the BCL2 and BAX gene family (9). Functional p53 is alsorequired for DNA damage-induced G, cell cycle arrest, as measuredby ionizing radiation-induced inhibition of replicative DNA synthesis(10, 11). This is most likely mediated by transcriptional transactiva-
tion of the CIPÃŒgene, encoding the cell cycle kinase inhibitor p21(12).
The main mechanism of cytotoxicity of the anticancer drug cisplatin is generally thought to be through induced DNA damage, primarilyin the form of intrastrand cross-links at the N-7 positions of adjacent
Received 10/11/95: accepted 1/16/95.The costs of publication of this article were defrayed in part by the payment of page
charges. This article must therefore be hereby marked adverti.'.enu'ni in accordance with
18 U.S.C. Section 1734 solely to indicate this fact.1This work was supported by the Cancer Research Campaign (United Kingdom) and
the Scottish Hospitals Endowment Research Trust. A. A.. W. G.. and A. E. were supportedby Cancer Research Campaign (United Kingdom) studentships.
-Present address: Louisiana State University. Medical Center. 1901 Perdido Street.New Orleans, LA 70112-1393.
'To whom requests for reprints should be addressed. Phone: 0141 330 4335: Fax:
0141 3304127; E-mail: [email protected].
guanine bases (13). The ability of cisplatin to form DNA cross-links
and to subsequently inhibit DNA replication has been related to itscytotoxic effects (14). Furthermore, it has been suggested that DNAreplication is necessary for cells to engage an apoptotic response aftercisplatin treatment (15). Doxorubicin. another effective anticancerdrug ( 16). intercalates into DNA and is believed to be cytotoxic dueto the induction of DNA double-strand breaks caused by inhibition ofDNA topoisomerase II (3), although recently, doxorubicin-inducedDNA cross-links have been suggested to correlate with chemothera-peutic effectiveness (17). Wild-type function of the TP53 gene is
necessary for induction of apoptosis in mouse thymocytes by topoisomerase II inhibitors (7).
Enhanced replicative bypass, defined as the ability of the replication complex of a cell to synthesize DNA past the site of DNAdamage, has been suggested to have a potential role in cisplatinresistance (18). This could lead to increased DNA damage tolerance,although the molecular basis of replicative bypass has yet to beelucidated. Tolerance to alkylating damage in human cell lines hasbeen shown in some cases to be associated with deficiency in strand-specific mismatch repair and high frequency of mutations in micro-satellite DNA sequences (microsatellite instability; Refs. 19-21).
Microsatellite instability has been observed in a variety of tumor types(22, 23), and such tumors have been described as having an RER 'phenotype (for replication error) (21 ). RER ' cells have been shown to
be deficient in mismatch repair and to have mutations in geneshomologous to yeast mismatch repair genes (24-26). Indeed, it has
been speculated that mismatch repair genes may serve as a generalsensor for genetic damage (19), since in bacteria mismatch repairrecognizes DNA damage induced by alkylating agents, UV, andcisplatin, as well as being involved in recombination repair (27, 28).
To address whether a RER ' phenotype may have a wider signifi
cance in resistance of tumor cells to agents other than monofunctionalalkylating agents, we have examined microsatellite instability in human tumor cells selected for resistance to cisplatin and doxorubicin.We have also explored an association between acquisition of anRER+ phenotype and loss of p53 function, which may result in
expansion of cells resistant to multiple DNA-damaging agents due to
reduced apoptosis.
MATERIALS AND METHODS
Cell Culture, Cell Cycle Analysis, and Drug Sensitivity Assays. The celllines A2780, A2780/CP70 (29), A2780/AD (30), MCF7. and MCF7/AD (31)were grown as monolayer cultures in RPM1 1640 with 109ÕPCS in 95%air/5% CO2 at 37°C. Independent, single-step, cisplatin-selected subîmes
(A2780/SCP1-10) were obtained by selection with a single exposure to 15 /J.M
cisplatin for 24 h. as described previously (32). The A2780 population usedwas of clonal origin to reduce any problems of heterogeneity in the cell line.Independent, multiply selected lines (A2780/MCP1-9) were obtained by seven
cycles of selection, with increasing concentrations of cisplatin. culminating ina final concentration of 15 /J.M for 24 h. A278()/mp53 are A2780 cellstranst'ected with and expressing a dominant-negative mutant p53 protein
(codon 143, Val to Ala), which have been shown previously to have lost anionizing radiation-induced G, arrest (111. All lines were free of Mycoplusnm
infection.Ionizing radiation-induced cell cycle arrest was determined by bromode-
oxyuridine labeling and propidium iodide staining after exposure ofexponen-
1374
Research. on February 2, 2019. © 1996 American Association for Cancercancerres.aacrjournals.org Downloaded from

MICROSATKI.I.ITK INSTABILITY AND DRUG RESISTANCE
(¡allygrowing cells to 2 Gy of -y-rays at room temperature using a '""Co source
as described previously (11). Clonogcnic drug sensitivity assays were performed hy seeding 10' cells into plates and exposing cells to 20 fiM cisplatin
for l h or leaving them untreated. Surviving colonies were counted after anadditional 10 days growth. Surviving fractions were calculated as the fractionof colonies after drug exposure compared to untreated cultures and the resistance factors calculated as the fold difference in surviving fraction from theparental line.
Microsatellite Analysis. The primers for amplification of the microsatel-
lite loci were obtained from Research Genetics (Huntsville. Alabama) or weresynthesized from published sequence data (33). Amplification was carried outusing a Touchdown PCR protocol (34) with an initial annealing temperature of64°Cand 25 cycles at a final temperature of 57°C.[a-12P]dCTP (Amersham,
Buckinghamshire. United Kingdom) was added to the reaction mixture, andthe products of the reaction were electrophoresed on a denaturing 6% poly-
acrylamide gel and detected by autoradiography. Mutations in the microsatellite loci were defined as an alteration in the size of the predominant amplifiedPCR product as compared to the parental line (Fig. 1).
Gel Retardation Assay. Cell extracts were prepared from A2780. A2780/cp70. and A2780/AD cells, and mismatch-specific gel retardation assays were
done as described previously (35. 36). Equal amounts of protein extracts wereincubated with 20 jil assay buffer [25 mM HEPES-KOH (pH 8.0). 0.5 mM
EDTA. 10% glycerol. 0.1 mM ZnCU and 0.5 mM DTT] containing 1 /xgpoly(dl-dC) and 40 tniol nonmismatched oligonucleotide per reaction. After a5-min incubation at room temperature. ':P-labeled mismatched or nonmis
matched oligonucleotide was added to each reaction and incubated for anadditional 20 min. The products of this reaction were separated on 6%nondenaturing polyacrylamide gels and detected by autoradiography.
Immunoblotting. Cell extracts were prepared by lysing exponentiallygrowing cells in l<7rNP40, 500 mM NaCl, 50 mM Tris (pH 7.5). and 1 mM DTT
in the presence of protease inhibitors. Protein concentrations were determinedby the Bio-Rad (Richmond. CA) protein assay. Immunoblotting was carriedout as described previously (37) using the p53 antibody Ab-6 (Oncogene
Science. Cambridge. MA) and visuali/ed by enhanced chemiluminesence; theintensity of the autoradiographie signal was quantified by laser densitometry.
Field Inversion Gel Electrophoresis. Exponentially growing cells weretreated with 40 /IM cisplatin for l h and harvested at various times aftertreatment. Samples were prepared for field inversion gel eleclrophoresis essentially as described previously (38). Cells were resuspended in PBS. and anequal volume of 2% low-melting-point agarose was added. Embedded cellswere incubated at 50°Cfor 48 h in 10 mw Tris-HCl (pH 7.6). 100 HIMEDTA.20 mM Nad. I7r SDS, and 1 mg/ml proteinase K. Approximately IO*1cells
were loaded into a well of a 1% agarose gel. Field inversion gel electrophoresisof samples was as described previously (39). DNA was subsequently transferred to Hybond-N nylon membrane (Amersham) by Southern blotting andhybridized with human DNA. which was '"P-labeled by random priming. This
allows visualization of total human DNA present on the Southern blot due tohybridization of human repetitive DNA sequences.
Detection of Apoptosis by Flow Cytometry. Apoptotic cells were detected as described previously (40). Exponentially growing cells were treatedwith 20 or 40 /xM cisplatin for l h or left untreated and harvested 72 h later.Attached and nonattached cells were collected and fixed in \r/t formaldehyde.Cells were rehydrated in PBS. and aliquots of 10'' cells were incubated for 30
min at 37°Cwith cacodylate buffer [0.2 M potassium cacodylate. 2.5 mM
Tris-HCl (pH 6.6). 2.5 mM CoCl,, 0.25 mg/ml BSA. 5 units terminal DNAtransferase/1011 cells, and 0.5 nmoles biotin-dUTP/101' cells]. After washing in
PBS. cells were incubated for 30 min at room temperature in the dark with 4XSSC and 0.1% Triton X-IOO containing 5% Marvel and 5 ng/ml fluorescein-ated-avidin. After an additional wash in PBS and 0.1% Triton X-100, cells
were resuspended in PBS and stained with propidium iodide. Cellular fluorescence was detected using a FACScan flow cytometer (Becton Dickinson, SanJose, CA).
Northern Analysis. RNA was extracted from cells using the TRIzol reagent method (GIBCO-BRL. Paisley, Scotland). Twenty /¿gof RNA was
separated on 1.4% agarose/2.2 M formaldehyde gels and transferred to Hybond-N nylon membranes (Amersham). The 2.1-kb CIPI mRNA transcriptwas detected using a human CIPI cDNA probe, pCEP-WAFl-S (41). RNA
loading was quantified by rehyhridization of the Northern blots to a GAPDHprobe (42). Hybridization was quantified by laser densitometric scanning ofintensity of the autoradiographic signal.
RESULTS
Mutations at Microsatellite Loci in Drug-resistant Cell Lines.Human tumor cell lines, selected in vitro for resistance to the anti-
cancer drugs cisplatin and doxorubicin. were analyzed for geneticalterations at 14 different polymorphic microsatellite loci compared totheir respective parental lines (Table I ). Mutations in the microsatellite loci were identified by a change in size of the PCR-amplified
allelic markers, caused by an alteration in the number of repeats of thebasic microsatellite sequence (Fig. 1). Mutations in microsatellite lociwere observed in the resistant tumor cell lines. The cisplatin-resistant
ovarian cell line A2780/cp7() showed new alÃelesat 8 of 12 of themicrosatellite loci studied. A doxorubicin-resistant derivative. A2780/AD, showed 2 of 11 new alÃeles, and the doxorubicin-resistant
MCF7/AD breast tumor cell line showed 6 of 13 microsatellite loci tobe mutated. In the examples of microsatellites examined, generallyonly one alÃelewas mutated at each locus.
Microsatellite instability has been shown to occur in human ovariancell lines and tumors (43); therefore, the new microsatellite alÃeles
Fig. 1. Mutations at the microsatellitc locusDI7S7V6 in A2780/cp70. Products of PCR amplification at the microsatellile locus D17S796 inDNA from A27X()/cp7(l subclones (Lanes 1-20)and the parent line (CP70) were analy/ed hy denaturing gel eleclrophoresis. Ltmes T, A, G, and C aresequencing ladders of M13pUC18 used to determine ihe relative size of PCR products.
T A G C 12345 6 78 9 10 11 12 13 14 15 16 17 18 19 20 CP70
1375
Research. on February 2, 2019. © 1996 American Association for Cancercancerres.aacrjournals.org Downloaded from
MICROSATELLITE INSTABILITY AND DRUG RESISTANCE
Table 1 Microsatellite mutation in drug-sensitive and -resistant cell lines
LocusCHRNB-1DI7S796DI7S26ÃŒDI7S856D17SSSÃŽDI751830175579DI7SI8ID17S801DI7SIV3D2S12JD2SI72D2SI2ID3S1298A2780A2780
A2780 cp70AD0
•¿�00000
•¿�0000000000oo000•0o*oD•D
DDDD DMCF-70.'.'.'•D00'-••.'•.'.'.'0MCF-7
AD••.'-•'
¡•'.'l.'••-•.'•»0
Each cell line was studied at the loci indicated. O, no microsatellite mutation detected. •¿�,microsatellite mutation present as compared to parental line. An asterisk (*) indicates the
loss of hetero/.ygosily at the locus, while those loci uninformative for loss of heterozygosity are indicated by a diagonal bar through the symbol. Ü,data unavailable.
Table 2 Fraction of random, ntmselected subclones showing microsatellile mutations
A2780subclonesA2780/cp70
subclonesA278()/mp53
subclonesCHRNB-10/15IS/200/13D17S1810/1515/190/14D17SI830/152/180/14D17S7960/1515/200/14D2S1720/15ND0/8D2S1110/1511/11NDD2S1230/1517/18NDD2S1210/15ND0/10D6S3I10/1517/180/13D6S2740/15NDNDD3S12960/1519/19ND
Random nonselected subcloncs were derived from the parental line A2780. the cisplatin-resistant derivative A2780/cp70, and mutant (codon 143. VAL (o ALA) p53 transfectanls.Denominator represents the number of clones producing assessable data. ND. loci where data were not obtained.
observed in the resistant derivatives may simply reflect an inherentinstability in the parental lines. To determine if the parental A2780cell line and the cisplatin-resistant derivative A2780/cp70 demonstrated inherent microsatellite instability, independent, random sub-
clones of A2780 and A2780/cp70 were derived, and microsatelliteloci were amplified in each subclone to look for mutations in alÃelesize (Table 2). No evidence of microsatellite mutation was observedat the 11 loci examined in 15 subclones of A2780. This indicates thatthe microsatellite instability observed in the A2780 drug-resistant
derivatives is not inherent to the A2780 cell line but has occurred inA2780/cp70 and A2780/AD as a consequence of the drug resistanceselection. All 20 subclones of A2780/cp70 showed multiple mutationsat the microsatellite loci examined (Fig. 1; Table 2). The A278()/cp70line and all the drug-resistant lines described were grown in the
absence of further drug exposure. Therefore, since new microsatellitemutations were observed in the A2780/cp70 subclones compared tothe A278()/cp70 line, the microsatellite instability is inherent to thiscell line and is manifested in the absence of cisplatin-induced DNA
damage.Deficiency of Mismatch Recognition Proteins in Cisplatin-re
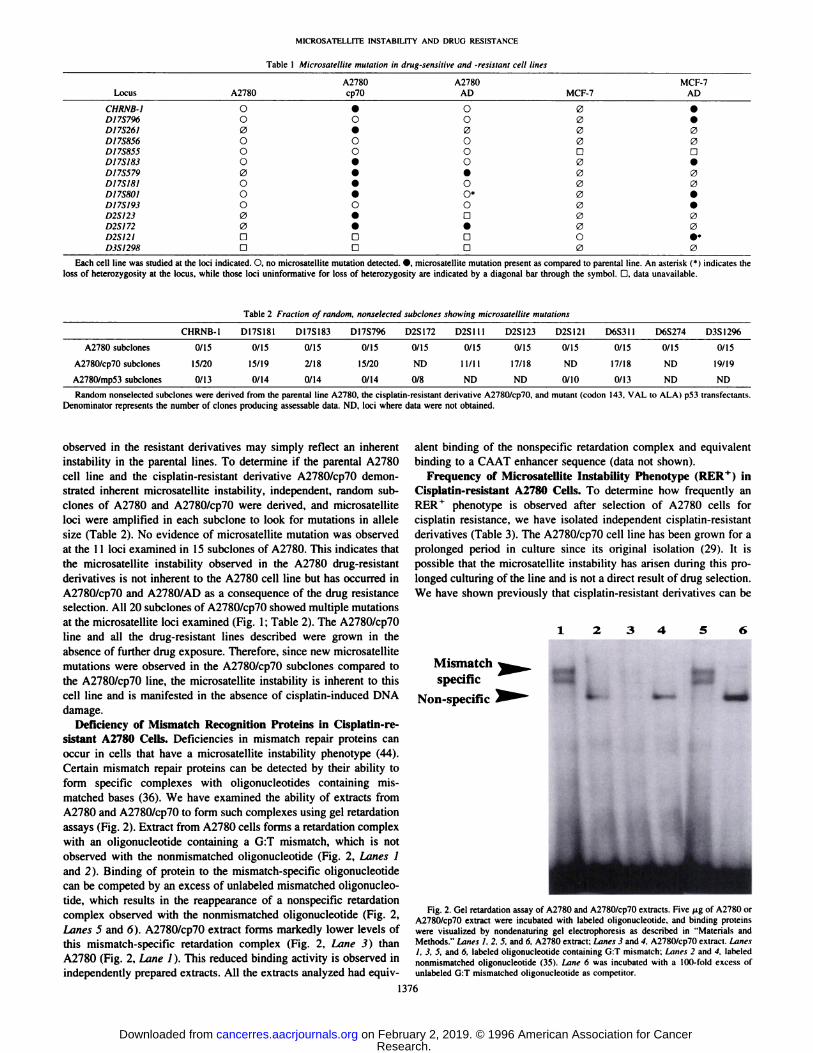
sistant A2780 Cells. Deficiencies in mismatch repair proteins canoccur in cells that have a microsatellite instability phenotype (44).Certain mismatch repair proteins can be detected by their ability toform specific complexes with oligonucleotides containing mismatched bases (36). We have examined the ability of extracts fromA2780 and A2780/cp70 to form such complexes using gel retardationassays (Fig. 2). Extract from A2780 cells forms a retardation complexwith an oligonucleotide containing a G:T mismatch, which is notobserved with the nonmismatched oligonucleotide (Fig. 2, Lanes 1and 2). Binding of protein to the mismatch-specific oligonucleotide
can be competed by an excess of unlabeled mismatched oligonucleotide, which results in the reappearance of a nonspecific retardationcomplex observed with the nonmismatched oligonucleotide (Fig. 2,Lanes 5 and 6). A2780/cp70 extract forms markedly lower levels ofthis mismatch-specific retardation complex (Fig. 2, Lane 3) than
A2780 (Fig. 2, Lane 1). This reduced binding activity is observed inindependently prepared extracts. All the extracts analyzed had equiv
alent binding of the nonspecific retardation complex and equivalentbinding to a CAAT enhancer sequence (data not shown).
Frequency of Microsatellite Instability Phenotype i kl U ' ) in
Cisplatin-resistant A2780 Cells. To determine how frequently anRER ' phenotype is observed after selection of A2780 cells for
cisplatin resistance, we have isolated independent cisplatin-resistant
derivatives (Table 3). The A2780/cp70 cell line has been grown for aprolonged period in culture since its original isolation (29). It ispossible that the microsatellite instability has arisen during this prolonged culturing of the line and is not a direct result of drug selection.We have shown previously that cisplatin-resistant derivatives can be
Mismatchspecific
Non-specific
Fig. 2. Gel retardation assay of A2780 and A2780/cp70 extracts. Five /ig of A2780 orA2780/cp70 extract were incubated with labeled oligonucleotide. and binding proteinswere visualized by nondenaturing gel electrophoresis as described in "Materials andMethods." Lanes I. 2. 5. and 6. A2780 extract; Lanes 3 and 4. A2780/cp70 extract. Lanes
1, 3. 5, and fi, labeled oligonucleotide containing G:T mismatch: Limes 2 and 4. labelednonmismatched oligonucleotide (35). Lane 6 was incubated with a 100-fold excess ofunlabeled G:T mismatched oligonucleotide as competitor.
1376
Research. on February 2, 2019. © 1996 American Association for Cancercancerres.aacrjournals.org Downloaded from

MICROSATELLITE INSTABILITY AND DRUG RESISTANCE
Table 3 Microxatellile mutaîionin cisplatin-resistant A2780 ceil lines"
CelllineSCPISCP2SCP3SCP4SCP5SCP6SCP7SCP8SCP9SCPK)MCP1MCP2MCP3MCP4MCP5MCP6MCP7MCP8MCP9CHRNB1O•0o••0o[
1C:,C.:C0D17S796OOoo0•000C:;:,:DO•00oD17SI81D0000C.:000•:0C;:~,:oD17S183O0o0C,0DLI0:;:0:oD6S31IO0(30000o00•;>o0000;•0DD6S26I00o0•0000D0000:0DaD6S274000C1CC(0CC1C:OD2S17200(ii:>•00:•••••C::)•D2S121DO00o•0000•••000a0OD2S111O00o0•0(01•••••C(0•D2SI23D[J(
!I11111Da1
1u•0••0•)••
" SCP1-10, single-step selected cisplatin-re.sistanl clones of A2780; MCPI-9, multiple-step selected cisplatin-resistant lines of A2780. O. no microsatellite mutation detected,
microsatellite mutation present as compared to parent A2780 line. D. data unavailable.
isolated by a single 24-h exposure to 15 JAMcisplatin; however, suchsubclones only show I- to 7-fold differences in cisplatin sensitivity
from the parental line and may not represent stable resistant clones(32). Nevertheless, such single-step selected clones will not have had
the opportunity to genetically diverge in culture any more than thenonselectively isolated clones of A2780 shown in Table 2. Tensingle-step, cisplatin-selected clones of A2780 (SCP1-10) were stud
ied for microsatellite mutation at 10 different loci. Three of 10 clonesshowed microsatellite mutation at one locus, while one clone, SCP6.showed mutation at 6 of the microsatellite loci. Since no microsatelliteinstability was observed in the random nonselected clones of A2780,the appearance of instability in some of the single-step selected clones
shows that this phenotype occurs as a direct result of cisplatin selection and is not due to genetic divergence in tissue culture.
Nine new cisplatin-resistant A2780 lines were also isolated by
repeated exposure to increasing concentrations of cisplatin up to afinal concentration of 15 ¿ÌM(MCPI-9). All of these lines were stably
resistant to cisplatin compared to the parental A2780 line, as measuredby clonogenic assay after growth in the absence of drug (Table 4).Eleven loci were studied for evidence of microsatellite mutation ineach of these resistant lines (Tables 3 and 4). Mutations were observedin eight of the nine multiply selected lines, with the number ofmutated loci ranging from one to four (Table 4). The observation ofmicrosatellite instability in the single-step and multiple-step cisplatin-
selected lines shows that this phenotype is selected frequently inindependent resistant derivatives, especially after multiple drugexposures.
Cisplatin-induced Apoptosis in Cisplatin-resistant Lines. Non-
random DNA fragmentation is widely used as a biochemical markerfor apoptosis, where the appearance of 50-kbp fragments of DNA is
often (but not always) followed by internucleosomal cleavage to180-200-bp integers (the DNA ladder: Ref. 45). We have analyzed
DNA fragmentation induced by cisplatin in the A2780 and A2780/cp70 lines as a measure of the propensity of these cells to undergo
Table 4 Characteri.'Hic.'i of A2780 (•¡.•iptatin-resislanllines
CelllineA2780A2780/cp7()A2780/MCPIA2780/MCP2A2780/MCP3A2780/MCP4A2780/MCP5A2780/MCP6A2780/MCP7A2780/MCP8A2780/MCP9Foldresistance"14.9
(±0.3)1.3
(±0.1)3.3
(±0.3)4.7
(±0.3)4.1
(±0.4)2.1
(±0.3)2.6
(±0.2)3.1
(±0.1)3.6
(±0.2)1.6(±0.1)Fraction
of mutantmicrosatelliteloci*0/148/123/104/114/113/112/112/110/111/103/9Percentage
ofapoptotic cells 72 h
after 40 fiMcisplatin'2214211872011Fraction
of cells inS-phase 24 h after2 Gy irradiation''0.541.050.941.030.981.20.621.00.640.820.71Relative
p53 Relative CIPIprotein level' mRNAlevel^+
1+
+ + + +0.29+
+0.52+
0.55+
+ +0.32+
0.58+
+0.33+
0.49-1-
0.95+
0.66+
0.56" Calculated from the clonogenic surviving fraction of cells after exposure to 20 /IM cisplatin for l h relative to parental A2780 cells. Each value is the mean of five experiments,
and the figures in parentheses are the SEM.* The fraction of loci showing a mutated alÃeleof the total number of loci studied.' The percentage of total cells undergoing apoptosis 72 h after treatment with 40 JIM cisplatin as detected by flow cytometry of fluorescently labeled DNA strand breaks. Values
shown are the means of duplicate experiments with 15,(KM)cells counted in each experiment.The fraction of cells in S phase after 2 Gy ionizing radiation relative to untreated controls. Values shown are the means of quadruplicate experiments with 20,(KH)cells counted
in each experiment.'' Relative levels of p53 protein as detected by Western blot analysis using 50 /ng of protein extracts. Approximate relative levels are indicated. In all cases of elevated p53. this
was identified as being nuclcarly localized by pf>3 immunocytochemistry.' Relative levels of CIP1 mRNA were determined by Northern blot analysis by hybridization to a CIPI cDNA probe and standardized by rehybridization against a glyceraldehyde-
3-phosphate dehydrogenase probe.
1377
Research. on February 2, 2019. © 1996 American Association for Cancercancerres.aacrjournals.org Downloaded from
MICROSATKU.ITE INSTABILITY ANI) DRl'C, RESISTANCE-:
A. 1 2 3 4 5 6 7 8 9 1011 12
WELL
•¿�300kbp
50kbp
48h 72h 96h 48h 72h 96h
B.A2780
1. A2780No Treatment
V
A2780/cp702. A2780
Cisplatin
200FL2-H
3. A2780/cp70No Treatment
200Fia-n
400
4. A278«/cp70
200FL2-H
200F12-H
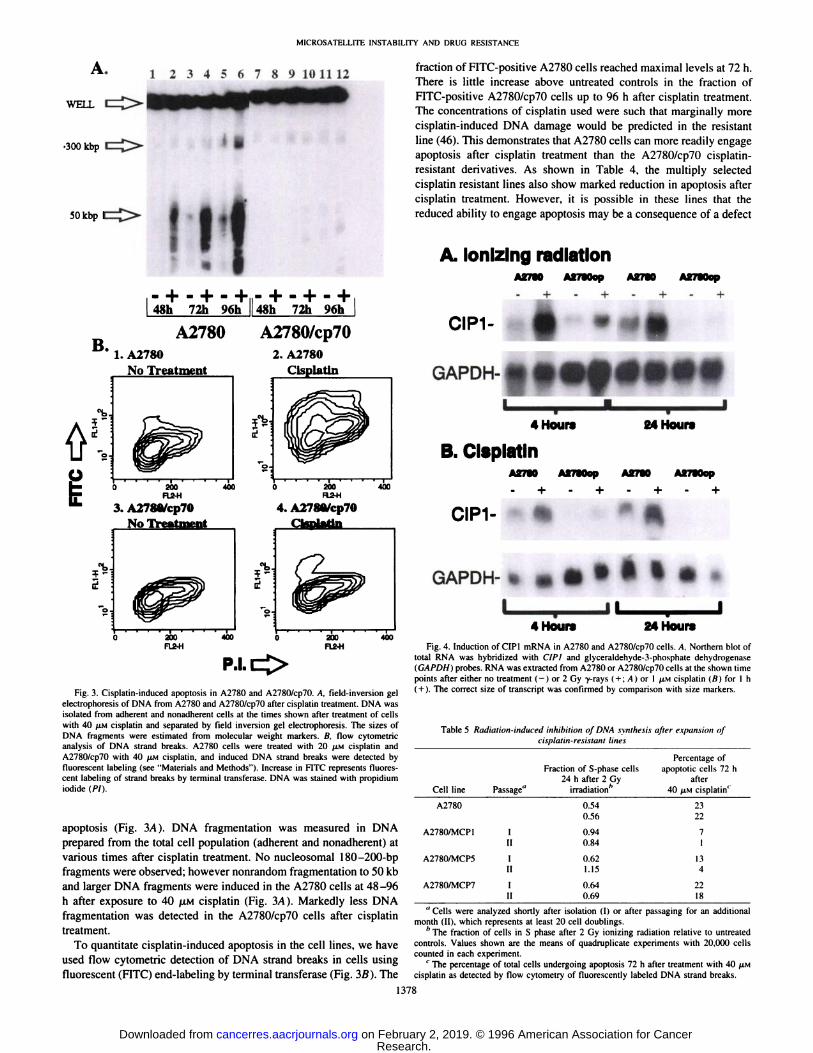
Fig. 3. Cisplatin-induced apoptosis in A2780 and A2780/cp70. A, field-inversion gel
electrophoresis of DNA from A2780 and A2780/cp70 after cisplatin treatment. DNA wasisolated from adherent and nonadherent cells at the times shown after treatment of cellswith 40 LAMcisplatin and separated by field inversion gel electrophoresis. The sizes ofDNA fragments were estimated from molecular weight markers. B, flow cytometricanalysis of DNA strand breaks. A2780 cells were treated with 20 /AMcisplatin andA2780/cp7() with 40 /AMcisplatin, and induced DNA strand breaks were detected byfluorescent labeling (see "Materials and Methods"). Increase in FITC represents fluores
cent labeling of strand breaks by terminal transferase. DNA was stained with propidiumiodide (PI).
apoptosis (Fig. 3/4). DNA fragmentation was measured in DNAprepared from the total cell population (adherent and nonadherent) atvarious times after cisplatin treatment. No nucleosomal 180-200-bp
fragments were observed; however nonrandom fragmentation to 50 kband larger DNA fragments were induced in the A2780 cells at 48-96
h after exposure to 40 ¿UMcisplatin (Fig. 3/4). Markedly less DNAfragmentation was detected in the A2780/cp70 cells after cisplatintreatment.
To quantitate cisplatin-induced apoptosis in the cell lines, we have
used flow cytometric detection of DNA strand breaks in cells usingfluorescent (FITC) end-labeling by terminal transferase (Fig. 3B). The
fraction of FITC-positive A2780 cells reached maximal levels at 72 h.
There is little increase above untreated controls in the fraction ofFITC-positive A2780/cp70 cells up to 96 h after cisplatin treatment.
The concentrations of cisplatin used were such that marginally morecisplatin-induced DNA damage would be predicted in the resistant
line (46). This demonstrates that A2780 cells can more readily engageapoptosis after cisplatin treatment than the A278()/cp70 cisplatin-
resistant derivatives. As shown in Table 4, the multiply selectedcisplatin resistant lines also show marked reduction in apoptosis aftercisplatin treatment. However, it is possible in these lines that thereduced ability to engage apoptosis may be a consequence of a defect
A. Ionizing radiationA2780 A2780ep A27BO A27Mep
CIP1-
GAPDH-
I —¿�•4 Hours 24 Hours
B. CisplatinA2780 A2780ep A27M A2780op
CIP1-
GAPDH- i
I4 Hour* 24 Hours
Fig. 4. Induction of CIP1 mRNA in A2780 and A2780/cp7() cells. A. Northern blot oftotal RNA was hybridized with CÕP1and glyceraldehyde-3-phosphate dehydrogenase(GAPDH) probes. RNA was extracted from A2780 or A2780/cp70 cells at the shown timepoints after either no trealment (-) or 2 Gy -y-rays ( + ; A) or 1 AIMCisplatin (ß)for l h
( + ). The correct size of transcript was confirmed by comparison with size markers.
Table 5 Radiation-induced inhibition of DNA synthesis after expansion ofcis¡ilÃtlin-re!ÃisÃanllìnea
CelllineA2780A2780/MCP1A2780/MCP5A2780/MCP7Passage"InininFraction
of S-phasecells24h after 2Gyirradiation''0.540.560.940.840.621.150.640.69Percentage
ofapoptoticcells 72hafter40
AIMcisplatin'2322711342218
"Cells were analyzed shortly after isolation (I) or after passaging for an additional
month (II), which represents at least 20 cell doublings.'' The fraction of cells in S phase after 2 Gy ionizing radiation relative to untreated
controls. Values shown are the means of quadruplicate experiments with 20.(XX) cellscounted in each experiment.
' The percentage of total cells undergoing apoptosis 72 h after treatment with 40 JIMcisplatin as detected by flow cytometry of"fluorescently labeled DNA strand breaks.
1378
Research. on February 2, 2019. © 1996 American Association for Cancercancerres.aacrjournals.org Downloaded from

MICROSATELLITE INSTABILITY AND DRUG RESISTANCE
in cisplutin delivery to the DNA target or in the signal that engages anupoptotic response, rather than a deficiency in apoptosis per se.Nevertheless, the line A2780/MCP7, which shows no detectable microsatellite instability, shows the greatest cisplatin-induced apoptosis.
Loss of p53 Function in the Cisplatin-resistant Derivatives of
A2780. p53 function has been shown to be necessary for DNAdamage-induced apoptosis and sensitivity of A2780 cells to ionizing
radiation and cisplatin (11. 47. 48). Ionizing radiation can induce G,and G2 cell cycle arrest of proliferating cells. The radiation-inducedG, arrest has been shown to be dependent on wild-type p53 function
(10). The A2780 cell line has a functional G, arrest after irradiation.as measured by inhibition of replicative DNA synthesis, and thisinhibition is lost after transfection of a dominant-negative mutant ofTP53 (11, 49). The A2780/cp70 line, despite having wild-type TP53
gene sequence (49). showed a reduction in G, arrest after irradiationcompared to the parental line (Table 4), suggesting that the p53protein is partially inactivated in these cells. As shown in Table 4, lossof ionizing radiation-induced inhibition of DNA synthesis is observedfrequently in cisplatin-resistant lines derived from A2780 cells. It is of
interest to note that the cell line A2780/MCP7, which showed nodetectable microsatellite instability and little alteration in cisplatin-
induced apoptosis, also showed no loss of p53 function as measuredby G, arrest. This line appears to have acquired resistance by adifferent type of mechanism from the other lines. One cell line,A2780/MCP5, which showed microsatellite instability, also stillshowed an apparent G, arrest after irradiation, and further characterization of this line will be returned to later.
Transcription of the CIP I gene is partially regulated by p53 and isnecessary for radiation-induced G, arrest (12). The A2780/cp70 cellshave 3-4-fold reduced CIP1 mRNA compared to the parental A2780
cells (Fig. 4). Both ionizing radiation and cisplatin induced increasedlevels of CIP I mRNA in the A2780 line. However, cisplatin did notdetectably induce CIPÃŒmRNA in the A2780/cp70 line (Fig. 4). Asshown in Table 4. the multiply selected cisplatin-resistant lines show
reduced CIPÃŒmRNA levels relative to the parental line. The lineA2780/MCP7. which shows no detectable microsatellite instabilityand no loss of G, arrest, shows the least reduction in CIPÃŒmRNA andhas levels very similar to the parental A2780 cells. All of the lineswith microsatellite instability show 2-5-fold reduction in CIPI
mRNA. Together these results are consistent with abrogated p53function in the majority of cisplatin-resistant lines that have an RER+
phenotype.We have shown previously that A2780/cp70 cells have elevated
constitutive levels of p53 protein compared to the parental A2780cells but that both lines express wild-type TP53 mRNA (49). Westernblot analysis revealed that the multiply selected cisplatin-resistant
lines had varying constitutive levels of p53 protein (Table 4). However, whereas some lines (A2780/MCP1. A2780/MCP3, and A2780/MCP5) exhibited elevated levels of p53 protein compared to theparental line A2780, many had no detectable difference, and none hadenhanced levels to the extent observed in the A2780/cp70 line. Wehave specifically examined loss of p53 function in the cisplatin-
resistant lines used in the present study, although loss of an apoptoticresponse can occur by other mechanisms (6). For instance, levels ofBCL2 and BAX (and other members of this gene family) modulate thepropensity of a cell to undergo apoptosis (9). The cisplatin-resistant
A2780/cp7() cells have increased levels of BCL2 protein compared tothe A2780 parental line (47), although this may reflect the negativetranscriptional regulation of BCL2 by wild-type p53 (50). We have
observed no difference compared to the parental line in levels ofBCL2 in the other multiply selected cisplatin-resistant A2780 lines
(data not shown).Loss of p53 function and consequent loss of G, arrest has been
shown to allow cells to become permissive for genetic changes, suchas DNA amplification (51). To examine whether loss of p53 functioncould confer an RER ' phenotype. microsatellite instability was ex
amined in A2780 cells transfected with a dominant-negative mutant
TP53 (A2780/mp53). These mutant p53 transfectants have beenshown to have lost a radiation-induced G, arrest present in the
parental cells (11). In 14 subclones of the mutant p53 transfectants, noevidence of microsatellite mutation was observed at any of the locistudied (Table 2). This would suggest that lack of G, arrest due toexpression of a dominant-negative mutant p53 does not confer mic
rosatellite instability in A2780 cells.Interestingly, one line, A2780/MCP5, shows an apparent radiation-
induced G | arrest but also shows markedly reduced CIP! mRNA(Table 4). This line had undergone expansion in culture prior toisolation of RNA and CIPÃŒmRNA analysis. Therefore, the cell cyclearrests were reexamined in this expanded cell population to examineif any loss of ability to G, arrest had occurred compared to the sameline shortly after selection. As shown in Table 5, the cisplatin-resistantderivative A2780/MCP1 still shows loss of radiation-induced inhibi
tion of DNA synthesis after further growth. The line MCP7, whichshows no microsatellite instability, still shows inhibition of DNAsynthesis after irradiation. However, the resistant line MCP5, whichoriginally showed a radiation-induced arrest, shows no evidence ofradiation-induced inhibition of DNA synthesis after further expansion
of the cells. Furthermore, the MCP1 and MCP5 lines both showreduced apoptosis induced by cisplatin after this growth period. Thisexpansion represents at least an additional 20 cell doublings. Thissuggests that loss of DNA damage-induced cell cycle arrest and lossof ability to engage cisplatin-induced apoptosis has occurred in thisRER+ cell line during expansion of the cells.
DISCUSSION
We observed microsatellite instability to be a common phenotype in tumor cell lines selected for resistance to the anticancerdrug cisplatin. An RER+ phenotype appears to be selected for
during the acquisition of resistance since: (a) 90% of independentmultiply cisplatin-selected lines are stably resistant and have mul
tiple mutations in microsatellites; (b) there is a low mutationfrequency at microsatellite loci in the parental line; (r) microsatellite instability is an inherent property of sublines of the resistantline A2780/cp70 in the absence of further drug exposure; and (d)there is deficiency in a G:T mismatch recognition protein in acisplatin-resistant line. A single selection of A2780 cells with 15
/UM cisplatin for 24 h, which gives a surviving fraction of1.7 X 10" 6 per viable cell (32), is sufficient to generate a line with
multiple microsatellite mutations. This would suggest that tolerance to cisplatin-induced DNA damage is concomitant with theacquisition of an RER+ phenotype and argues against resistance
occurring due to mutation in drug resistance genes arising duringgrowth of an RER+ mutator line. There is no significant difference
in the doubling times of the A2780 clones and the cisplatin-
resistant derivatives, suggesting that there has been no selection forsimply faster-growing clones. It is perhaps surprising that such ahigh frequency of clones with an RER+ phenotype is observed, if
the underlying genetic basis of this phenotype was recessive.However, it has been suggested recently that some mutations inmismatch repair genes may be dominant negatives (52). Alternatively, since tumor cells frequently demonstrate hemizygosity atautosomal loci, only one alÃelemay be required to be inactivated.
We also observed microsatellite instability in doxorubicin-resistant
lines, which together with previous observations of acquisition of anRER+ phenotype in alkylating agent-resistant cell lines (19, 20),
1379
Research. on February 2, 2019. © 1996 American Association for Cancercancerres.aacrjournals.org Downloaded from

MICROSATELLITE INSTABILITY AND DRUG RESISTANCE
suggests that decreased fidelity of DNA replication may be acommon feature of tumor cells resistant to DNA-damaging anti-
cancer drugs. The ability of the replication complex of a cell tosynthesize DNA past the site of DNA damage has been suggestedpreviously to play a role in cisplatin resistance (18). Changes inmismatch repair proteins or other components of the replicationcomplex involved in fidelity of DNA synthesis may allow bypassof the lesion during replication, leading directly to drug resistance.The cisplatin-resistant line A2780/cp70 has reduced activity of a
G:T mismatch recognition protein, which is also frequently absentin alkylating agent-tolerant cell lines that have an RER f phenotype
(19, 53). p53 function has been shown to be necessary for DNAdamage-induced apoptosis and sensitivity of A2780 cells to ion
izing radiation and cisplatin (11, 47, 48). It has been suggested thatDNA replication is necessary for cisplatin to engage an apoptoticresponse (15), perhaps by converting cisplatin cross-links intodouble-strand DNA breaks, which initiates a p53-dependent apop
totic response. Thus, in replicating RER cells with wild-type p53,
such as A2780, cisplatin may be particularly cytotoxic due toengagement of a p53-dependent apoptotic response. A2780 is
derived from an untreated patient and is relatively chemosensitivein vitro (11). Many cisplatin-sensitive tumor types express wild-type p53 (54), and there are preliminary reports of wild-type TP53
gene sequence correlating with carboplatin sensitivity of ovariantumors (55).
Cells selected for resistance to cisplatin, including the A2780/cp70cell line, demonstrate cross-resistance to multiple cytotoxic agents,
including ionizing radiation, melphalan, doxorubicin. etoposide, andmitoxantrone (1, 48). Failure of the cell to engage an apoptoticresponse after induced DNA damage can lead to resistance to suchmultiple agents (6). The cisplatin-resistant lines examined in thepresent study show reduced cisplatin-induced apoptosis. This reduced
apoptosis may be a consequence of a defect in cisplatin delivery to theDNA target, rather than a deficiency in the engagement of apoptosisper se. However, similar levels of cisplatin adducts are induced in theA2780 and A2780/cp70 lines at the concentrations of cisplatin used inthe present study (46). We also observed abrogated p53 function, asmeasured by radiation-induced G, arrest and reduced CIP! mRNA inall of the RER ' cisplatin-resistant lines studied. Thus, the resistant
lines may have acquired resistance to multiple agents due to loss ofp53-mediated apoptosis; furthermore, this loss of p53 function mayhave been selected for during expansion of cells with an RER4
phenotype.This hypothesis would also predict that chemoresponsive ovar
ian tumors would normally have an RER phenotype. However,after acquisition of an RER* phenotype due to anticancer drug
treatment, selection against cells capable of undergoing apoptosiswill occur during expansion of the RER+ tumor cell population.
This will lead to expansion of clones that have lost DNA damage-
induced apoptosis and which will, therefore, have acquired resistance to multiple chemotherapeutic drugs. Thus, of all the cisplatin-resistant RER * lines examined in the present study, the A2780/
cp70 line has been expanded in culture the longest and has the mostextreme phenotype of loss of an apoptotic response and loss of p53function. Furthermore, the resistant line A2780/MCP5, althoughinitially showing radiation-induced inhibition of DNA synthesis,after further growth in culture appears to lose this p53-mediated
response. Whether this model is relevant to the acquisition ofresistance in tumors should be readily testable, although care willhave to be taken that a clonal resistant tumor population is beingexamined to detect microsatellite instability.
ACKNOWLEDGMENTS
We thank Dr. D. Black (Glasgow. United Kingdom) for advice and helpwith the microsatellite analysis, Dr. P. Karran (London, United Kingdom) foradvice with the gel retardation assays, and Dr. C. Dive (Manchester, UnitedKingdom) for advice with flow cytometry. We also thank Prof. D. P. Lane(Dundee. United Kingdom) forp53 antibodies. Prof. B. Vogelslein (Baltimore,MD) for the plasmid pCEP-WAF-l-S, and Prof. R. F. Ozols and Dr. T. C.
Hamilton (Philadelphia, PA) for the cell lines A2780 and A2780/cp70.
REFERENCES
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
25.
Hamaguchi, K.. Godwin. A. K.. Yakushiji. M.. O'Dwyer, P. J.. Ozols, R. F.. and
Hamilton. T. C. Cross-resistance to diverse drugs is associated with primary cisplatinresistance in ovarian cancer cell lines. Cancer Res.. 53: 5225-5232. 1993.Kaye, S. B. The multidrug resistance phenotype. Br. J. Cancer. 58: 691-694. 1988.Epstein. R. J. Drug-induced DNA damage and tumour chemosensitivity. J. Clin.Oncol.. 8: 2062-2084. 1990.Andrews. P. A., and Howell. S. B. Cellular pharmacology of cisplatin: perspectiveson mechanisms of acquired resistance. Cancer Cells, 2: 35-43. 1990.Gottesman, M. M. How cancer cells evade chemotherapy. Cancer Res.. 53: 747-754,1993.Hickman. J. A. Apoptosis induced by anticancer agents. Cancer Metastasis Rev., 11:121-139, 1992.
Clarke. A. R., Purdie. C. A., Harrison, D. J.. Morris, R. G.. Bird, C. C.. Hooper. M. L.,and Wyllie, A. H. Thymocyle apoptosis induced by p53-dependem and independentpathways. Nature (Lund.). 3f>2: 849-852. 1993.Lowe. S. W.. Ruley. H. E., Jacks. T.. and Housman. D. E. p53-dependent apoptosismodulates the cytotoxicity of anticancer agents. Cell. 74: 957-967, 1993.Oltvai, Z. N., Milliman, C. L., and Korsmeyer. S. J. BCL-2 heterodimerizes in vivowith a conserved homolog. BAX. that accelerates programed cell death. Cell, 74:609-619, 1993.Kuerbilz. S. J., Plunkett, B. S.. Walsh. W. V., and Kastan, M. B. Wild-type p53 is acell cycle checkpoint determinant following irradiation. Proc. Nati. Acad. Sci. USA,Ä9:7491-7495, 1992.
Mcllwrath, A. J.. Vasey. P. A., Ross, G. M.. and Brown. R. Cell cycle arrests andradiosensitivity of human tumour cell lines: dependence on wild-type p53 for radio-sensitivity. Cancer Res., 54: 3718-3722, 1994.EI-Deiry. W. S.. Harper. J. W., O'Connor. P. M., Velculescu, V. E., Canman, C. E.,
Jackman. J.. Pietenpol. J. A.. Burrell, M.. Hill, D. E., Wang, Y.. Wiman, K. G.,Mercer, W. E., Kastan. M. B.. Kohn, K. W.. Elledge. S. J., Kinzler, K. W., andVogelstcin. B. WAF-l/CIP-1 is induced in p53-mediated G, arrest and apoptosis.Cancer Res., 54: 1169-1174, 1994.
Sherman. S. E., and Lippard, S. J. Structural aspects of platinum anticancer druginteractions with DNA. Chem. Rev., 87: 1153-1181. 1987.Heiger-Bernays. W. J.. Essigmann. J. M., and Lippard. S. J. Effect of the antitumordrug rf.v-diammincdichloroplatinum(II) and related platinum complexes on eukary-otic DNA replication. Biochemistry, 2V: 8461-8466. 1990.
Evans, D. L.. Tilby, M., and Dive. C. Differential sensitivity to the induction ofapoptosis by cisplatin in proliferating and quiescent immature rat thymocytes isindependent of the level of drug accumulation and DNA adduci formation. CancerRes., 54: 1596-1603, 1994.
Blum. R. H.. and Carter. S. K. Adriamycin: a new anticancer drug with significantclinical activity. Ann. Intern. Med.. SO: 249-267, 1974.Skladanowski, A., and Konopa, J. Relevance of interstrand DNA crosslinking induced by anthracyclines for their biological activity. Biochem. Pharmacol., 47:2279-2287. 1994.Mámenla. E. L., Poma. E. E., Kaufmann. W. K.. Delmastro, D. A., Grady. H. L., andChancy. S. G. Enhanced replicative bypass of platinum-DNA adducts in cisplatin-resislanl human ovarian carcinoma cell lines. Cancer Res.. 54: 3500-3505. 1994.Kai. A.. Thilly. W. G., Fang, W., Longley, M. J.. Li. G., and Modrich. P. Analkylaling-tolerant. mutator human cell line is deficient in strand-specific mismatchrepair. Proc. Nati. Acad. Sci. USA, 90: 6424-6428. 1993.
Aquilina. G.. Hess. P.. Branch. P.. MacGeoch. C.. Casciano. I.. Karran. P., andBignami. M. A mismatch recognilion defect in colon carcinoma confers DNAmicrosatellite instability and a mutalor phenotype. Proc. Nail. Acad. Sci. USA. 91:8905-8909. 1994.Parsons, R., Li, G. M.. Longley. M. J.. Fang. W. H.. Papadopoulos. N.. Jen, J.. de laChapelle. A.. Kin/.ler. K. W., Vogelstein. B., and Modrich, P. Hypermulahilily andmismatch repair deficiency in RER * tumor cells. Cell. 75.- 1227-1236, 1993.
Horii, A., Han. H. J.. Shimada. M.. Yanagisawa. A.. Kalo. Y., Ohta. H.. Yasui. W..Tañara.E., and Nakamura. Y. Frequent replication errors at microsatellile loci inlumors of patients with multiple primary cancers. Cancer Res., 54: 3373-3375, 1994.Loeb. L. A. Microsatellile inslabilily: marker of a mulalor phenolype in cancer.Cancer Res.. 54: 5059-5063. 1994.Leach, F. S.. Nicolaides, N. C.. Papadopoulos. N.. Liu. B.. Jen. J., Parsons. R.,Pellomaki. P., Sistonen. P., Aaltonen, L. A., and Nyslrom-Lahli. M. Mulalions of amutS homolog in hereditary nonpolyposis coloreclal cancer. Cell. 75: 1215-1225,
1993.Bronner. C. E.. Baker. S. M.. Morrison, P. T.. Warren. G.. Smith. L. G.. Lescoe,M. K., Kane. M., Earabino. C.. Lipford. J.. and Lindblom. A. Mutation in théDNAmismatch repair gènehomologue hMLH 1 is associated with hereditary non-polyposiscolon cancer. Nature (Lond.). 368: 258-261. 1994.
1380
Research. on February 2, 2019. © 1996 American Association for Cancercancerres.aacrjournals.org Downloaded from

MICROSATELLITE INSTABILITY AND DRUG RESISTANCE
26. Papadopoulos. N.. Nicolaides. N. C., Wei, Y.. Ruhen, S. M., Carter. K. C.. Rosen. 41.C. A.. Haselline. W. A.. Fleischmann. R. D.. Fräser.C. M., Adams. M. D.. Venter.J. C., Hamilton. S. R.. Petersen. G. M.. Watson. P.. Lynch. H. T., Peltomaki, P..Mecklin. J.. de la Chapelle. A.. Kinzler. K. W., and Vogelslein. B. Mutation of a mutL 42.homolog in hereditary colon cancer. Science (Washington DC). 263: 1625-1628,
1994.27. Feng. W. Y., Lee. E. H., and Hays. J. B. Recombinagenic processing of UV-light
photoproducts in nonreplicating phage DNA by the Escherichia coli methyl-directed 43.mismatch repair system. Genetics. 129: 1007-1020. 1991.
28. Fram, R. J.. Cusick. P. S.. Wilson. J. M.. and Marinus. J. M. Mismatch repair ofr/.v-diammincdichlnropluiinum ll-induced DNA damage. Mol. Pharmacol., 28: 44.51-55. 1985.
29. Behrens. B. C.. Hamilton. T. C.. Masuda. H.. Grotzinger. K. R., Whang-Peng, J..Lonie. K. G., Knutsen, T., McKoy, W. M.. Young. R. C.. and Ozols. R. F. Charac- 45.terisation of a r/.v-diamminedichloroplatinum II resistant human ovarian cancer cellline and its use in evaluation of platinum analogues. Cancer Res., 47: 414-418. 1987.
30. Rogan. A. M.. Hamilton. T. C.. Young, R. C.. Klecker. R. W.. and Ozols. R. F.Reversal of Adriamycin resistance by verapamil in human ovarian cancer. Science 46.(Washington DC). 224: 994-996. 1984.
31. Cowan. K. H.. Bastist. G.. Tulpule. A.. Sinha. B. K.. and Myers. C. E. Similarbiochemical changes associated with multidrug resistance in human breast cancercells and carcinogen-induced resistance to xenobiotics in rats. Proc. Nati. Acad. Sci. 47.USA, 83: 9328-9332, 1986.
32. McLaughlin. K., Stephens, I., McMahon, N., and Brown. R. Single step selection ofr/.ï-diamminedichloroplatinum(II) resistant mutants from a human ovarian carcinoma 48.cell line. Cancer Res., 51: 2242-2245, 1991.
33. Anderson, L. A.. Friedman. L.. Osborne-Lawrence, S., Lynch, E.. Weissenbach. J..Bowcock, A., and King, M. High density genetic map of the BRCA-1 region of 49.chromosome 17ql2-21. Genomics. 17: 618-623, 1993.
34. Don, R. H., Cox, P. T.. Wainwright, B. J.. Baker, K.. and Mattick. J. S. "Touchdown"
PCR to circumvent spurious priming during gene amplification. Nucleic Acids Res.. 50.19: 4008-4009. 1991.
35. Jiricny. J., Hughes, M.. Corman. N.. and Rudkin. B. B. A human 200-kDa proteinbinds selectively to DNA fragments containing G-T mismatches. Proc. Nail. Acad. 51.Sci. USA, 85: 8860-8864. 1988.
36. Griffin. S.. and Karran. P. Incision al DNA G.T mispairs by extracts of mammaliancells occurs preferentially at cytosine methylation sites and is not targeted by a 52.separate G.T binding reaction. Biochemistry. 32: 13032-13039. 1993.
37. Vojtesek, B.. Bartek. J.. Midgley. C. A., and Lane. D. P. An immunochemicalanalysis of the human nuclear phosphoprotein p53: new monoclonal antibodies and 53.epitope mapping using recombinant p53. J. Immunol. Methods, 151: 237-244. 1992.
38. Kokileva, L. Endogenous degradation of rat liver chromatin studied by agar gelelectrophoresis of nuclei. Mol. Biol. Reports. /.?: 139-143. 1989. 54.
39. Filipski. J.. Leblanc. J.. Youdale. T.. Sikorska. M.. and Walker, P. R. Periodicity ofDNA folding in higher order chromatin structures. EMBO J., 9: 1319-1327, 1990.
40. Gorczyca. W., Gong, J., Ardelt. B.. Tráganos. F.. and Darzynkiewicz. Z. The cell 55.cycle related differences in susceptibility of HL-60 cells to apoptosis induced byvarious antitumour agents. Cancer Res.. 53: 3186-3192, 1993.
El-Deiry. W. S.. Tokino. T.. Velculescu. V. E., Levy. D. B.. Parsons, R.. Trent, J. M..Lin, D.. Mercer. W. E.. Kinzler, K. W.. and Vogelstein. B. WAF-1. a potentialmediator of p53 tumour suppression. Cell, 75: 817-825, 1993.
Tso. J. Y., Sun. X.. Kao. T.. Reece, K. S.. and Wu. R. Isolation and characterisationof rat and human glyceraldehyde-3-phosphate dehydrogenase cDNAs: genomic complexity and molecular evolution of the gene. Nucleic Acids Res.. 13: 2485-2488.
1985.Orth. K., Hung. J.. Gazdar. A.. Bowcock, A., Mathis, J. M.. and Sambrook. J. Geneticinstability in human ovarian cancer cell lines. Proc. Nati. Acad. Sci. USA. 91:9495-9499, 1994.Parsons. R., Li, G.. Longley, M. J., Fang. W., Papadopoulos, N., Jen. J.. Chapelle. A..Kinzler. K. W.. Vogelstein. B., and Modrich, P. Hypermutability and mismatch repairdeficiency in RER * tumour cells. Cell. 75: 1227-1236, 1993.
Oberhammer. F.. Wilson, J. W.. Dive. C.. Morris. 1. D.. Hickman. J. A.. Wakeling.A. E., Walker, P. R., and Sikorska. M. Apoptotic death in epithelial cells: cleavage ofDNA to 300 and/or 50 kb fragments prior to or in the absence of internucleosomalfragmentation. EMBO J.. 12: 3679-3684, 1993.Parker, R. J., Eastman. A.. Bostick-Bruton. F., and Reed, E. Acquired cisplatinresistance in human ovarian cancer cells is associated with enhanced repair ofcisplatin-DNA lesions and reduced drug accumulation. J. Clin. Invest., 87: Ill-Ill.
1991.Eliopoulos. A. G., Kerr, D. J.. Herod. J.. Hodgkins. L.. Krajewski. S.. Reed, J. C., andYoung. L. S. The control of apoptosis and drug resistance in ovarian cancer: influenceof p53 and bcl-2. Oncogene. //: 1217-1228. 1995.Vasey, P. A.. Jenkins. S.. Bissett. D.. and Brown, R. p53-mediated multi-agent
resistance and approaches to circumvention in u human ovarian cell line. Br. J.Cancer, 71: 7. 1995.Brown, R., Clugston. C., Burns. P.. Edlin, A., Vasey, P., Vojtesek, B.. and Kaye, S. B.Increased accumulation of p53 in cisplatin-resistant ovarian cell lines. Int. J. Cancer,55: 678-684, 1993.Miyashita. T.. Harigai. M.. Hanada, M.. and Reed. J. C. Identification of a p53-dependent negative response element in the bcl-2 gene. Cancer Res.. 54: 3131-3135.
1994.Livingstone. W. R., White. A., Sprouse, J.. Livanose. E., Jacks, T.. and Tlsty, T. D.Altered cell cycle arrest and gene amplification potential accompany loss of wild-typep53. Cell, 70: 923-935, 1992.
Parsons, R.. Li, G.. Longley, M., Modrich. P.. Liu. B., Berk. T.. Hamilton, S. R.,Kinzler, K. W., and Vogelstein, B. Mismatch repair deficiency in phenotypicallynormal human cells. Science (Washington DC). 26«:738-740. 1995.
Branch. P.. Aquilina. G.. Bignami. M., and Karran. P. Defective mismatch bindingand a mutator phenotype in cells tolerant to DNA damage. Nature (Lond.). 362:652-654, 1993.
Peng. H.. Hogg. D.. Malkin, D., Bailey, D., Gallic, B. L., Bulbul, M., Jewett, M.,Buchanan. J.. and Goss. P. E. Mutations of the r>53 gene do not occur in testis cancer.Cancer Res., 53: 3574-3578, 1993.Al-Azraqi, A., Chapman. C.. Challen. C., SÃgalas,J., Aswaad. S., Sinha, D., Calven,A. H., and Lunec. J. p53 alterations in ovarian cancer as a determinant of response tocarboplatin. Br. J. Cancer, 69: 7, 1994.
1381
Research. on February 2, 2019. © 1996 American Association for Cancercancerres.aacrjournals.org Downloaded from

1996;56:1374-1381. Cancer Res D. Alan Anthoney, Amanda J. McIlwrath, William M. Gallagher, et al. Drug-resistant Tumor CellsMicrosatellite Instability, Apoptosis, and Loss of p53 Function in
Updated version
http://cancerres.aacrjournals.org/content/56/6/1374
Access the most recent version of this article at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://cancerres.aacrjournals.org/content/56/6/1374To request permission to re-use all or part of this article, use this link
Research. on February 2, 2019. © 1996 American Association for Cancercancerres.aacrjournals.org Downloaded from