molecular diagnostic methods nucleic acid hybridization technology probe is a labeled (with...
TRANSCRIPT


Molecular diagnostic methods

Nucleic Acid hybridization technology
• Probe is a labeled (with radioactive P32 or fluorescent ) single strand DNA that is complement with desired DNA or RNA
• Probe can be small (20-600 nt) or large (10-30kbp)
• Southern blot for detecting desired DNA (qualitative) and the copy number of it (quantitative) in sample

Nucleic Acid hybridization technology
• Northern blot for detecting desired RNA(qualitative) and the copy number of it (quantitative) in sample. Positive bands with lower MW can suggest a new mutated splicing site
• Nowadays RT-Real time PCRs have been used instead of NB
• Fluorescent InSitu Hybridization (FISH) with large fluorescent probes can detect some large deletions or duplication or translocations

Southern blot



DNA amplification
• In vivo amplification:Cloning• In vitro amplificationPCR

What is the PolymeraseChain Reaction?
• It’s a means of selectively amplifying a particular segment of DNA.
• It can be thought of as a molecular photocopier.

PCR• On different sources of DNA and wide range of
DNA amount e.g. single cell PCR in PGD• Real time PCR (Quantitative)• In PCR we can amplify known DNA sequence but
by cloning we can amplify known and unknown sequences
• PCR has limitation in length of DNA that can amplify. Usual enzymes can amplify 2-3 kbp but there are special kits to amplify up to 20-30kbp
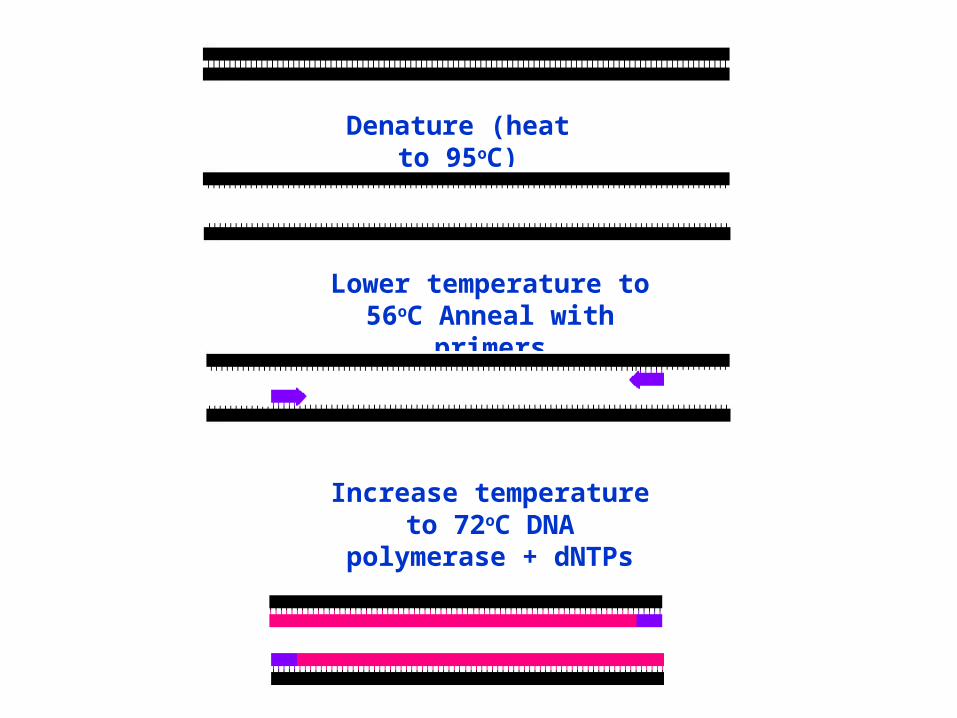
Denature (heat to 95oC)
Lower temperature to 56oC Anneal with primers
Increase temperature to 72oC DNA polymerase + dNTPs


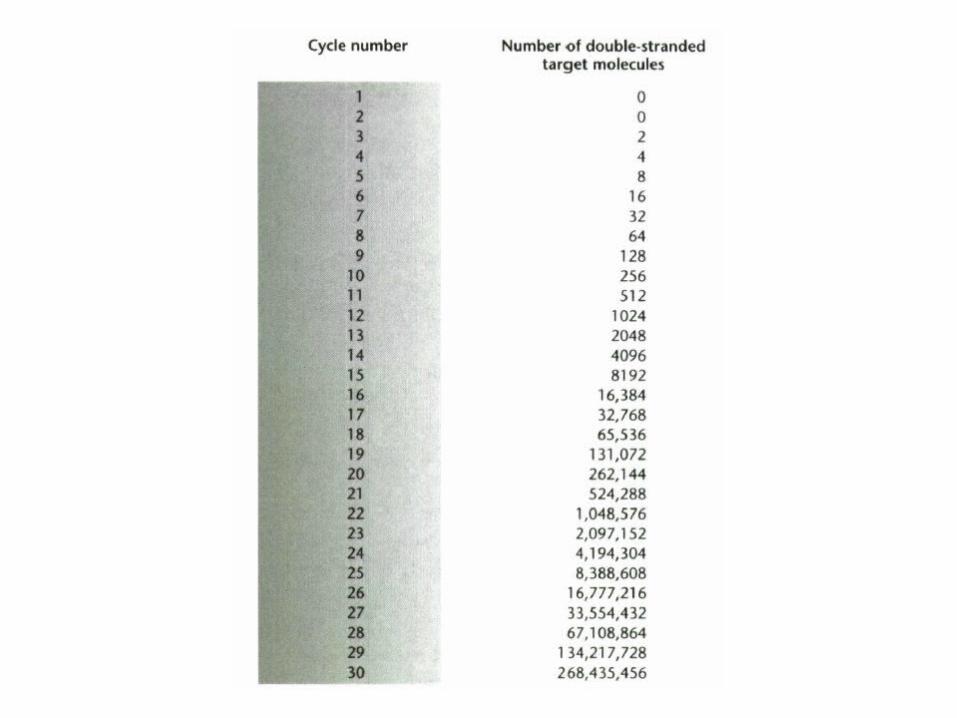
DNA copies vs Cycle number
0
500000
1000000
1500000
2000000
2500000
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23
Cycle number
DN
A c
op
ies

Applications of PCR
•Classification of organisms
•Genotyping•Mutagenesis•Mutation
detection•Sequencing•Cancer
research
•Detection of pathogens DNA
• fingerprinting•Drug discovery•Genetic
engineering •Pre-natal
diagnosis

The Basics of PCR Cycling
•30–35 cycles each comprising:
–denaturation (95°C), 30 sec.
–annealing (55–60°C), 30 sec.
–extension (72°C),time depends on
product size.

What’s in the Reaction?
• Template DNA• Reaction buffer (Tris, ammonium ions (and/or
potassium ions), magnesium ions, bovine serum albumin)
• Nucleotides (dNTPs)• Primers (Forward and Reverse)• DNA polymerase (usually Taq)

Thermal Cyclers
•PCR cyclers available from many suppliers.•Many block formats and multi-block systems.•Reactions in tubes or 96-well micro-titre plates.

RFLP
• A complete restriction site is between primers in middle of PCR product
• PCR products are digested by RE• Restriction fragment length analysis

ARMS(Amplification Refractory mutation System)
• We have wild type and mutant forward primers and a common reverse primer
• We have a pair of primer to amplify an internal control DNA fragment
• Three results may we havee


Three methods for cDNA synthesis

RT-PCR
• For detection of RNA viruses• For detection of gene
transcription(expression)• Real time RT-PCRFor quantitative evaluation of gene expressionFor determining virus titer

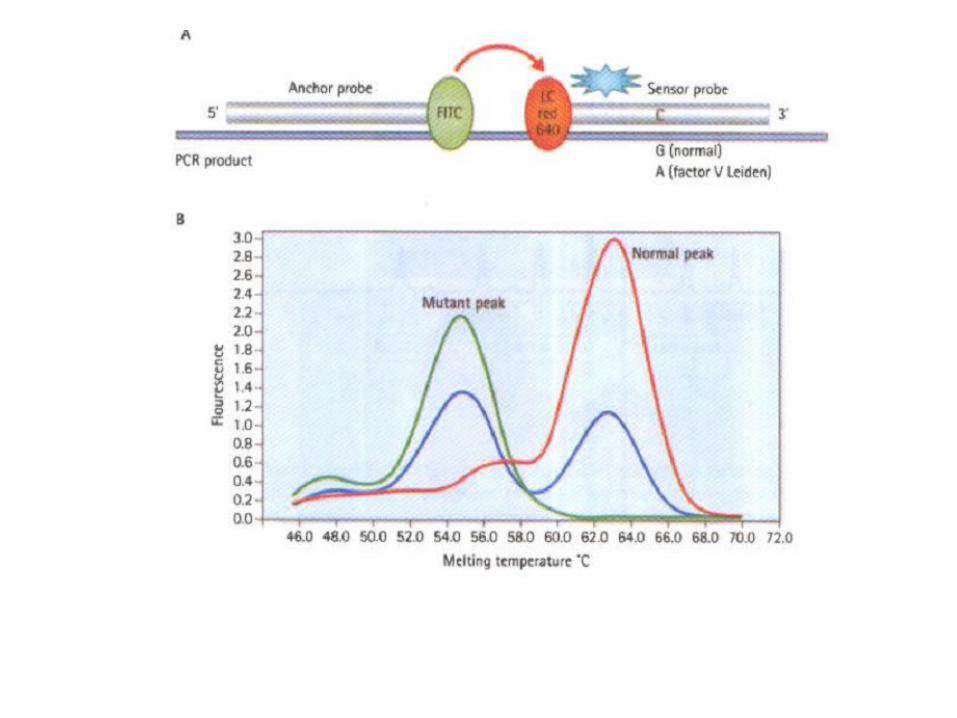
Real time PCR
• Syber green• Melting curve• High Resolution Melting (HRM)• Taq man probes

Real time PCR

TaqMan • The most widely used probe system is TaqManı. In this
system the probes used are oligonucleotides with a reporter fluorescent dye attached at the 5' end and a quencher dye at the 3' end. While the probe is intact. the proximity of the quencher reduces the fluorescence emitted by the reporter dye.
• If the target sequence is present. the probe anneals downstream from one of the primer sites. As the primer is extended. the probe is cleaved by the 5' nuclease activity of the Taqpolymerase . This cleavage of the probe separates the reporter dye from the quencher dye. thereby increasing the reporter-dye signal.

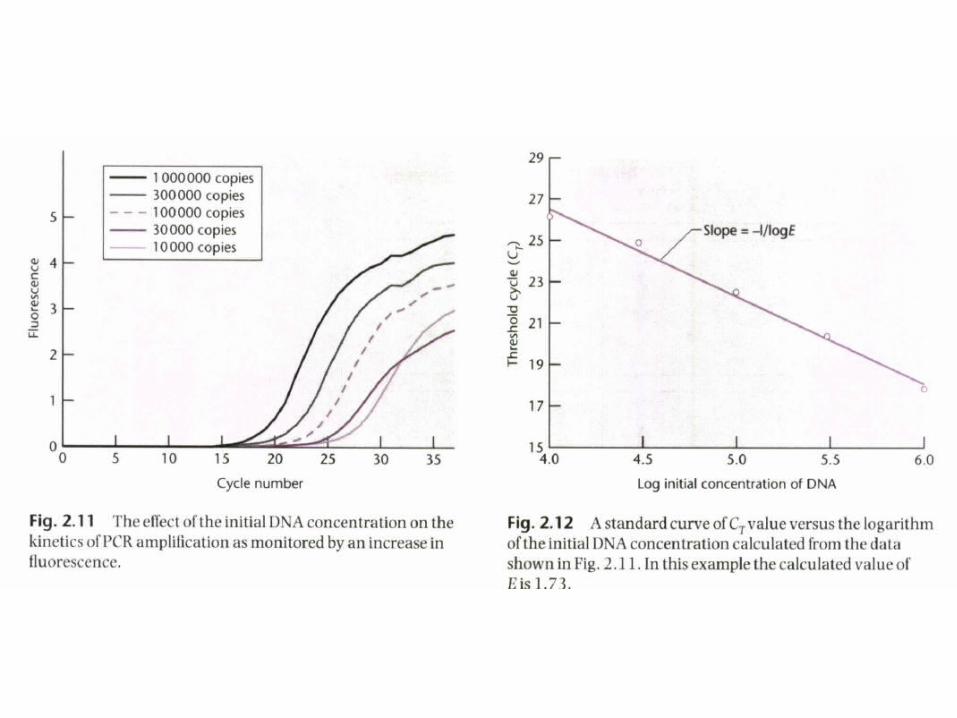
Standard curve
• Quantitation of the amount of target in unknown samples is achieved by preparing a standard curve. using different starting copy numbers of the target sequence.
• A major advantage of fluorescent probes is that they can be constructed using a number of different fluorophores each of which has a characteristic color.
• This means that it is possible to determine the relative amounts of two targets in a mixture provided that they have the same amplification efficiency


DNA microarray
• Differential display (comparison of gene experession)
• Mutation and SNP detectiont• Deletion and insertion detection with
CGH(Comparative Genomic Hybridization)• SNP-CGH for uniparental disomy detection

DNA microarray

Mutation Detection
• Detection of known and frequent mutationsARMS,RFLP,SSCP(also unknown)• Detection of unknown or infrequent
mutationsSequencing is a gold standard method in
mutation detection

Screening methods based on size of the PCR products
• Three nucleotides deletion in CF (p.phe508del) can be detected on PAGE
• Expansion of three nucleotides in HD (CAG repeat) gene
• Restriction fragment length polymorphism (RFLP)


RFLP

ARMS PCR

Oligo nucleotide ligation assay
• Probes for normal and mutant alleles are designed with different fluorescent dye.
• All exons of the desired gene e.g. CF are amplified by PCR and normal and mutant probes hybridized and probes that are completely match with related PCR product ligated with T4 DNA ligase and analyzed with fragment analyzer computerized equipment

Denaturating High Performance Liquid Chromatography(DHPLC)
• when heterozygote alleles are denaturated and renaturated some heterdouplex can be formed and these heteroduplex have shift in running in the column of DHPLC
• In the case of homozygote mutant we mix normal PCR product with homozygote mutant PCR product and after denaturation and renaturation there will be hetroduplex with running shift

Conformation Sensitive Capillary Electrophoresis (CSCE)
• In this method we use from flurescent detction in monitoring the running shift that is occurred in heteroduplex .Heteroduplexes have different movement speed and each PCR product labeled with different fluorescent dye and can be detected by capillary is faster than DHPLC electrophoresis

High Resolution Melting (HRM)
• This method is a kind of Real time PCR with intercalating agent with specific buffer and reagent that can discriminate two PCR products with only one nucleotide different by high resolution melting curve
• This method can be used for high throughput SNP or point mutation screening


Sequencing
• Sequencing is the gold standard method for mutation detection but is expensive
• Sanger method: Chain termination with dideoxy nucleotide triphosphates labeled with four different fluorescent dyes (which have not hydroxyl group on their 3’ carbon)


Mass Spectroscopy
• Mass Spectroscopy has high discrimination power and is usually used for protein analysis
• With MALDI-TOF (Matrix assisted laser desorption/Ionizing time of flight) technology we can analyze thousands DNA sample during several mins.
• One application for MALDI-TOF is high throughput SNP analysis

Polymorphisms• Single nucleotide polymorphism (SNP)With RFLP, HRM, ARMS PCR and other
methods. New high throughput DNA microarrayVariable number tandem repeat (VNTR) Unlike of SNP , VNTR has several allelesMinisatellites are VNTRs with 10-15 nt core and
are individual for each person. (DNA finger printing)

Polymorphism
• Short tandem repeat (STR) or microsatellites are repeats of two, three or four nucleotides and is individual for each person ( CA repeat)
• STRs are very polymorph in each site between different person
• VNTRs and STRs are usually used in identification and paternity tests

Follow a mutant gene with STR
• We can determine linkage between a mutant gene and one or some contiguous STR and use of them for PGD and PND by PCR
• In this way recombination can cause error in our diagnosis


PCR and detection of infectious diseases
• PCR can be used for detection of all infectious diseases. Although PCR is not a gold standard method in the majority of infectious diseases but it helps to rapid diagnosis and decision for treatment
• Serological and culture methods usually need long time • All bacterial, viral, fungal and parasitic infections can be
detected by PCR and there are several commercial Kit.• These kits are usually used in rapid response in out
breaks

PCR and detection of malignant diseases
• There are several PCR ,Real time and RT-Real time PCR for detection and following up of malignant diseases
• Translocation of t(9;22) can be detected by PCR in CML

Large deletions
• Southern blot analysis• Well-characterized deletions and
amplifications can be detected by PCR. However, the exact breakpoint sites of most deletions have not been determined.

Multiplex Ligation-dependent Probe Amplification (MLPA)
• For large deletion and deletion in unpredicted sites and duplication we can use MLPA
• In detection of aneupolidy e.g. trisomy 13,18 and 21 and monosomy turner. In this case probes are designed against STRs

Multiplex ligation-dependent probe amplification
• Analysis by MLPA is a suitable alternative that is also capable of detecting new deletions and amplifications.
• With MLPA, it is possible to perform a multiplex PCR reaction in which up to 45 specific sequences are simultaneously quantified.
• Amplification products are separated by sequence type electrophoresis As only one pair of PCR primers is used,

MLPA
• MLPA reactions result in a very reproducible gel pattern with fragments ranging from 130 to 490 bp.
• Comparison of this gel pattern with that obtained with a control sample indicates which sequences show an aberrant copy number



Recombinant Technology and its RISKS
• People have worried about the risks that GM micro organisms and plants will have for human and the environment
• International organism like NIH of USA and GMAG (Genetic manipulation advisory group) of UK developed some guide lines for genetic manipulation.

Bio safety
• Bio safety Levels• I• II• III• IV

Attenuation
• All GM organism should be attenuated or auxotroph to some amino acids or critical enzymes. With this strategy we can control the risks of GM micro organisms


Cloning and transformation

New Technologies
• Cloning and Cloning vectors• Restriction endonucleases• SOUTHERN, Northern and western bloting• Polymerase chain Reaction (PCR)• Sequencing methods

Vectors
• Vectors can replicate in host independently (REPLICON)PlasmidPhage and PhagmidCosmid up to 50 kbpPAC 100kbp BAC 200-300 kbpYAC has Autonomous replication sequences 1000kbp
Eukaryotic DNA that has repeatative fragments can be amplified with YAC
HAC unlimited

• YAC is suitable for long gene that are 2000-3000 kbp . Each gene is inserted in 4-5 YAC with overlap regions

A plasmid vector for cloning
• Contains an origin of replication, allowing for replication independent of host’s genome.
• Contains selective markers: Selection of cells containing a plasmid
• Blue-white screening• Contains a multiple cloning site (MCS)

Restriction Enzymes
• Before 1970 there was no method for cleaving DNA at discrete points.
• The only method where any degree of control could be exercised was the use of mechanical shearing.
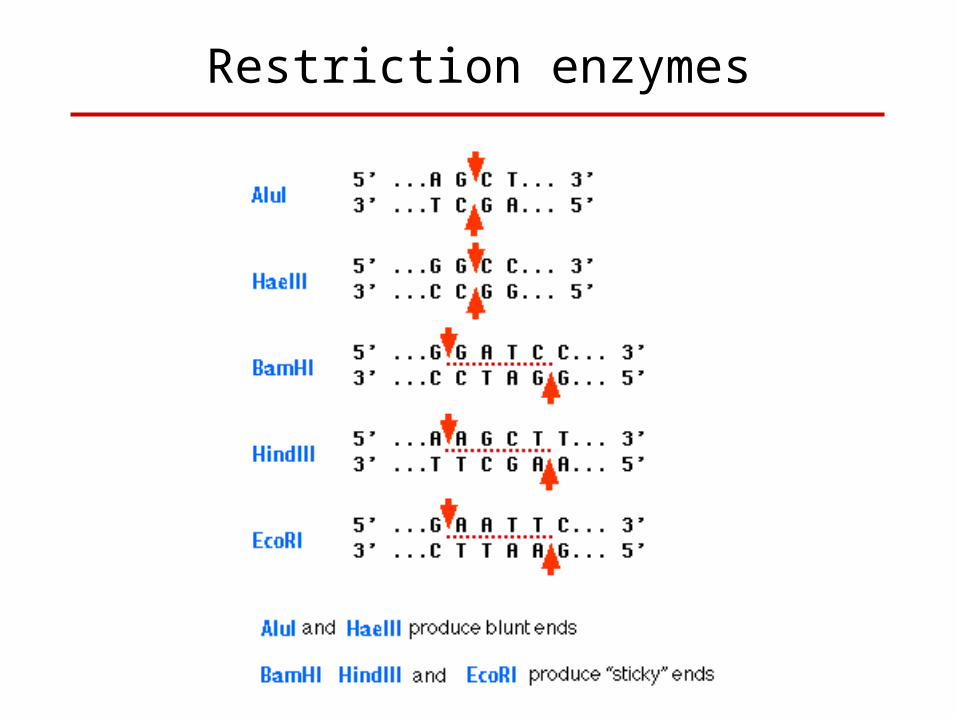
Restriction enzymes


• Blunt end like EcoRV• 5’ over hang like XhoI• 3’ Over hang like kpnI and PstI


• Cloning DNA Fragments with Protruding Ends• Directional CloningWith two different sticky end enzymeWith one sticky and another blunt enzyme• Cloning Blunt-ended DNA Fragments

Cloning Steps
• DNA fragment preparation by mechanical sheer stress or Restriction endonucleases or by PCR
• Digestion of PCR products or plasmids with RE:Blunt end DNA fragmentsSticky End or cohesive end• Ligation with T4 DNA ligase Enzyme

ligation
• Molar ratio of insert to vector: • Molar ratio ≥ 3-5

T4 DNA ligase
• The optimum temperature for ligation of nicked DNA is 37°C. but at this temperature the hydrogen-bonded join between the sticky ends is unstable.
• The optimum temperature for ligating the cohesive termini is in the range 4-15°C


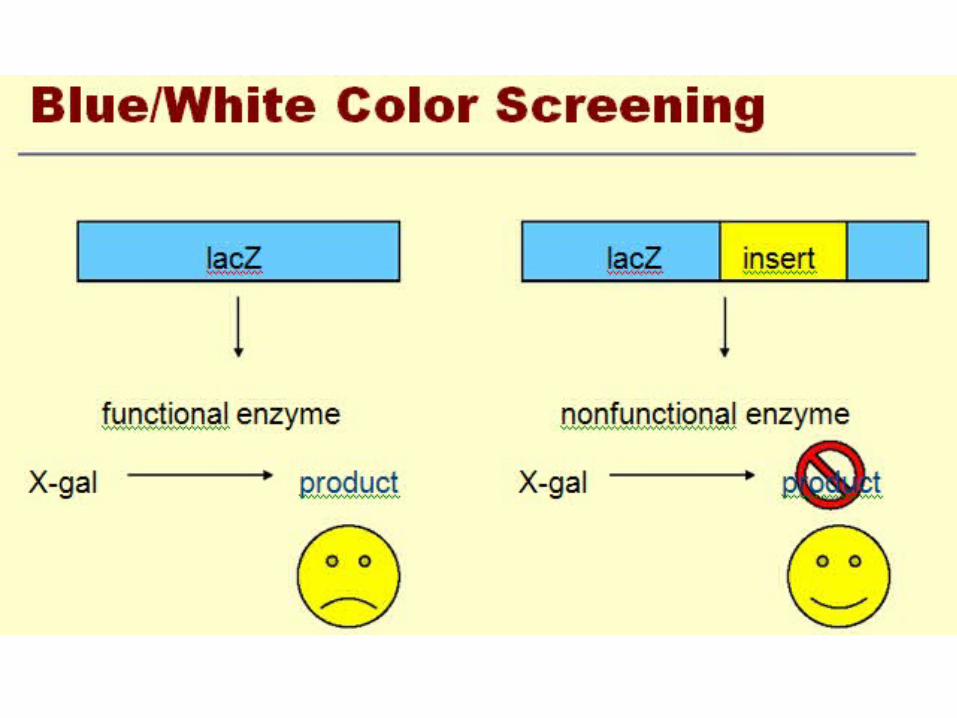
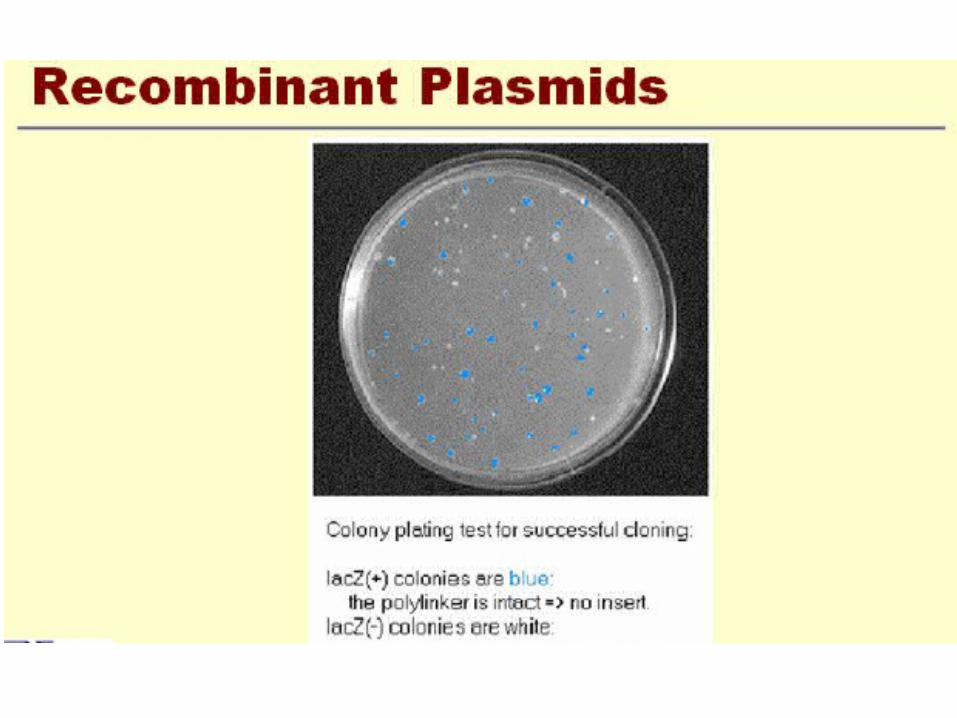
Screening methods
• Screening Bacterial Colonies Using X-gal and IPTG: α-Complementation
• Screening Bacterial Colonies by Hybridization:
• Screening Bacterial Colonies by antibiotics

Identifying Recombinant Plasmids by α-Complementation
• Many plasmid vectors (e.g., the puc series, Bluescript, pGem, and their derivatives) carry a short segment of E. coli DNA containing the regulatory sequences and the coding information for the first 146 amino acids of β-galactosidase.
• Embedded in the coding region is a polycloning site that maintains the reading frame and results in the harmless interpolation of a small number of amino acids into the amino-terminal fragment of β-galactosidase.
• Vectors of this type are used in host cells that express the carboxy-terminal portion of β-galactosidase.

blue/white screening
• Another QC test is blue/white screening by 5-bromo- 4-chloro-3-indolyl-β-D-galactoside (Xgal).
• This compound is colorless but on cleavage releases a blue indolyl derivative.
• Since Xgal is not an inducer of β-galactosidase, the non-substrate inducer isopropyl-β-D-thiogalactoside (lPTG) is also added to the medium. the plasmid usually carries the lacI gene and the first 146 codons of the lacZ gene,

X-GAL
• The activity of the enzyme can be assayed with a chromogenic substrate such as X-gal, which is converted by β-galactosidase into an insoluble dense blue compound



Identifying Recombinant Plasmids by Hybridization
• [In the mid-1960s, after Nygaard and Hall (1963) had shown that single-stranded DNA could be immobilized on nitrocellulose filters, Denhardt (1966) and Gillespie and Spiegelman (1965) demonstrated that nucleic acids fixed in this way could be detected with exquisite sensitivity by hybridization to radiolabeled probes.


Colony PCR (Screening)
• With specific primer for insert• With universal primer (e.g M13 Forward and
Reverse primers)• For selection direction with specific and
universal primer

Confirmation
• Digestion with restriction enzyme• PCR• Sequencing
•

Competent E. coli
• Preparation and Transformation of Competent E. coli using Calcium Chloride
• The Inoue Method for Preparation and Transformation of Competent E. Coli: "Ultra-Competent" Cells
• The Hanahan Method for Preparation and Transformation of Competent E. coli: High-efficiency Transformation

TRANSFORMATION OF COMPETENT E. COLI
• Chemical Methods• Most of the chemical methods currently used
for bacterial transformation are based on the observations of Mandel and Higa.
• This simple and robust procedure regularly generates between 105 and 106 transformed colonies of E. coli per 1µg of supercoiled plasmid DNA

Electroporation
• Plasmids ranging in size from 2.6 kb to 85 kb can be introduced with efficiencies ranging from 6 x 1010 transformants/µg of DNA to 1 x 107
transformants/µg DNA, respectively. This is 10-20 times higher than can be achieved with competent cells prepared by chemical methods. Transformation frequencies of this magnitude are especially useful when constructing large and highly complex cDNA libraries Electroporation works well with most commonly used laboratory strains of E. coli

Transformation of E. coli by Electroporation
• Preparing electrocompetent bacteria is considerably easier than preparing cells for transformation by chemical methods. Bacteria are simply grown to mid-log phase, chilled, centrifuged, washed extensively with ice-cold buffer or H20 to reduce the ionic strength of the cell suspension, and then suspended in an ice-cold buffer containing 10% glycerol. DNA may be introduced immediately into the bacteria by exposing them to a short high-voltage electrical discharge

Electroporation• Optimal efficiency is achieved using small
volumes of a dense slurry of bacteria (2 x 1010/ml) contained in specially designed cuvettes fitted with closely spaced electrodes. Electroporation is temperature-dependent and is best carried out at 0-4°c. The efficiency of transformation drops as much as 100-fold when electroporation is carried out at room temperature.

Electroporation
• The highest efficiency of transformation (colonies/µg input plasmid DNA) is obtained when the concentration of input DNA is high (1-10 µglml) and when the length and intensity of the electrical pulse are such that only 30-50% of the cells survive the procedure.

DNA library
• Genomic DNA library of human genomePlasmid DNA library 300000-400000 clonesYAC DNA library 13000-14000 clones• cDNA library