molecular dynamics and density functional theory...
TRANSCRIPT

Molecular Dynamics and Density Functional Theory (DFT)

What do we need?
• An account in “pemfc” cluster:
Host name: pemfc.chem.sfu.ca
I will take care of that. This can be usually a common account for all of you but please at the beginning of your work every group makes its own directory.
• pemfc cluster is a linux based cluster, therefore you should know some linux commands. I will give you a list of important linux commands later
• A package called VASP (Vienna Ab-initio Simulation Package).http://cms.mpi.univie.ac.at/vasp/
(it is installed in pemfc cluster, you just have to run it J )
• The following packages: 1. “putty.exe” a open shell program for SSH under the windows.
With that you can connect to pemfc cluster and run the programs/commandsof the cluster.
2. “psftp.exe” a open shell program for ftp to transfer your files from your computer to the pemfc cluster and vise versa.you can download them from the following address (they are free):http://www.chiark.greenend.org.uk/~sgtatham/putty/download.html
3. “rasmol” it is a graphic interface packege in which you can see the position of the atoms in your systemyou can download it from the following address:http://www.bernstein-plus-sons.com/software/rasmol/
(please download the latest version under the windows
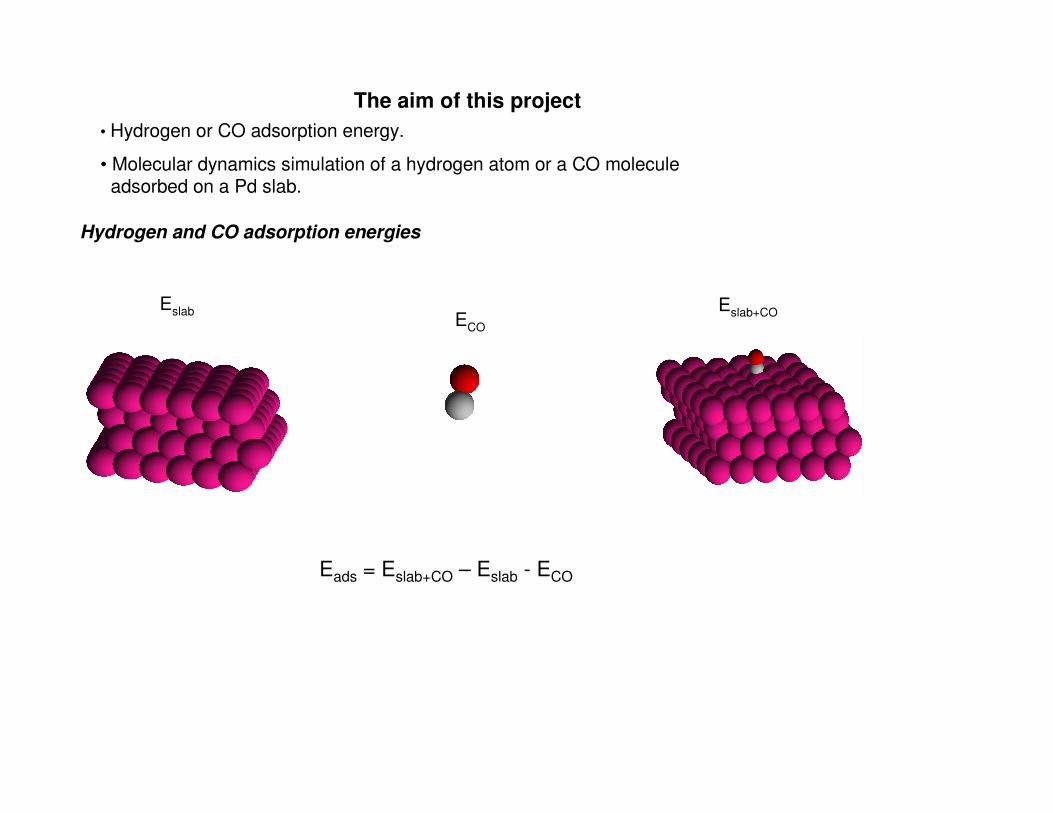
The aim of this project
• Hydrogen or CO adsorption energy.
• Molecular dynamics simulation of a hydrogen atom or a CO molecule adsorbed on a Pd slab.
Hydrogen and CO adsorption energies
Eslab Eslab+COECO
Eads = Eslab+CO – Eslab - ECO

Ab-initio total energy calculation of a system of nucleons and electrons
. Electrons -> quantum particles -> Wavefunction -> Charge density n(r) -> Schrödinger eq.
Nucleons -> classical particles -> Point charge -> Newton law
Ab-initio -> No experimental data in needed as an input for the calculation
nucleon(classic)
electrons(quantum)
In principal electrons and nucleons are very small therefore we have to consider them as quantum particles
Approximation: 1. nucleons are much bigger compare to electrons -> nucleons as classical particles
Approximation 2: When we are solving the Schrödinger Eq. for electrons the nuclei are regarded as a fixedcharges.
Introduction:

Velocity = 0
Ground state charge density
n0(r)
Velocity ≠ 0
Charge densityn(r)
mion >> melec
Approximation 2:
We can always assume that the electrons are in ground state even when nucleons are in motion
Velect >> Vion
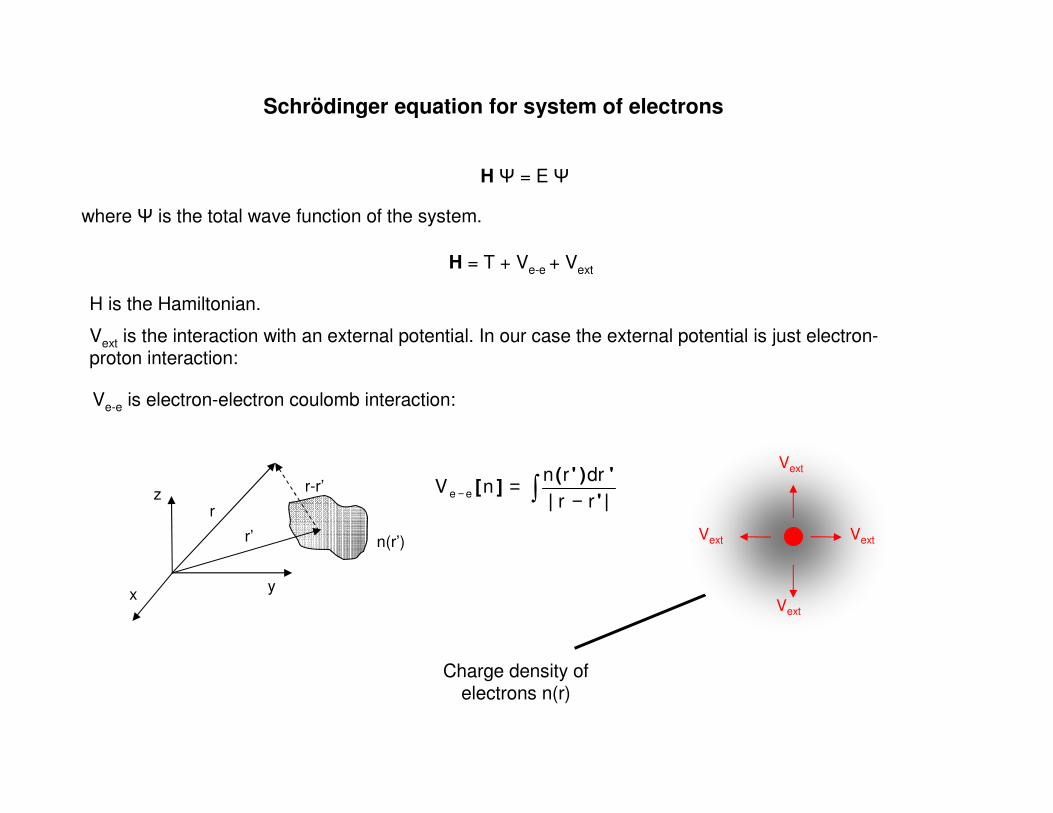
where is the total wave function of the system.
H = E
H = T + Ve-e + Vext
H is the Hamiltonian.
∫ −=−
|'|
')'(][
rr
drrnnV ee
n(r’)
xy
z
r’
r
r-r’
Charge density of electrons n(r)
Vext
Vext
Vext
Vext
Schrödinger equation for system of electrons
Vext is the interaction with an external potential. In our case the external potential is just electron-proton interaction:
Ve-e is electron-electron coulomb interaction:
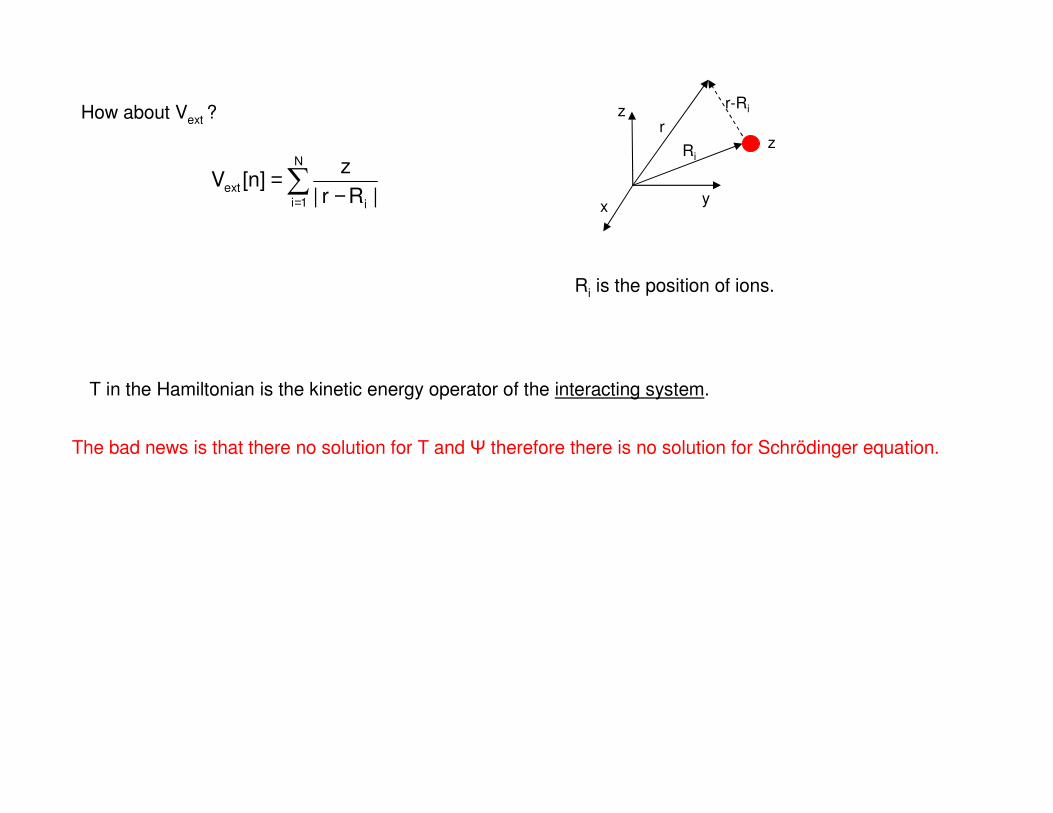
∑= −
=N
1i i
ext|Rr|
z]n[V
z
xy
z
Ri
r
r-Ri
Ri is the position of ions.
T in the Hamiltonian is the kinetic energy operator of the interacting system.
The bad news is that there no solution for T and therefore there is no solution for Schrödinger equation.
How about Vext ?
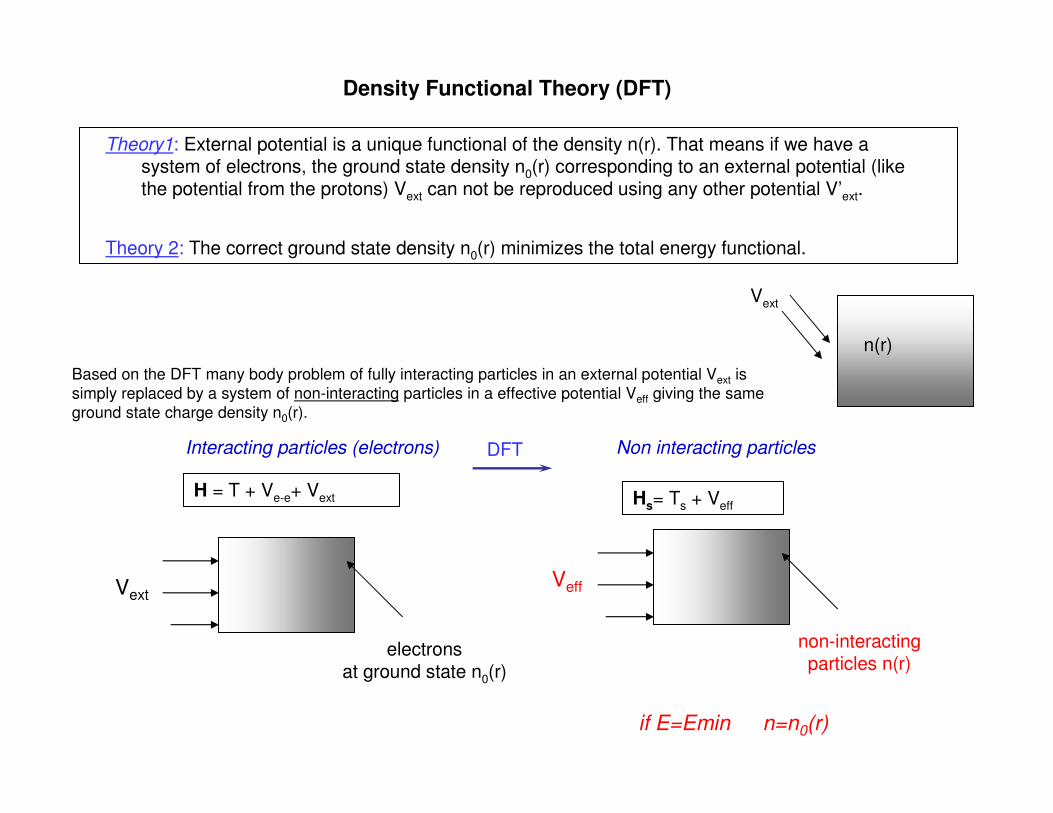
H = T + Ve-e+ Vext Hs= Ts + Veff
Interacting particles (electrons) Non interacting particles
Density Functional Theory (DFT)
Theory1: External potential is a unique functional of the density n(r). That means if we have a system of electrons, the ground state density n0(r) corresponding to an external potential (like the potential from the protons) Vext can not be reproduced using any other potential V’ext.
Theory 2: The correct ground state density n0(r) minimizes the total energy functional.
DFT
Based on the DFT many body problem of fully interacting particles in an external potential Vext is
simply replaced by a system of non-interacting particles in a effective potential Veff giving the same
ground state charge density n0(r).
Vext
electronsat ground state n0(r)
Veff
non-interactingparticles n(r)
if E=Emin à n=n0(r)
Vext
n(r)

Veff = Ve-e + Vext + Vxc
The effective potential Veff is:
Vxc : The extra term which contains all the energy contribution which were not taking into
account in the transition from the interacting system to the non-interacting system. Vxc is
called exchange-correlation potential
Ts is now the kinetic operator of a non-interacting system with N particles which can be easily given by:
2
2N
1i
2
sdr
d
m2T ∑
=
−= h

The exchange-correlation potential can be expressed as:
dn
]n[dE]n[V xc
xc =
where Exc[n] is the exchange-corralation energy.
The above DFT is formally exact, but it is impossible to find a exact from for Vxc
dr)r(n)n(]n[E xcxc ∫ ε=
where xc(n) is the energy of a homogeneous electron gas and it is known. Thereforethe Exc[n] and hence Vxc[n] are all known.
Various approximations have been introduced in the course of the years to improve LDA. The Generalized Gradient Approximation (GGA) is one of those approximations which is more or less commonly accepted to be an improvement over LDA.
In GGA approximation the Vxc depends not only on charge density n(r) but also on the magnitude of the gradient of the charge density || n∇
The most common approach is the Local Density Approximation LDA
Vxc [n] à in GGA approximation à [ ]||, nnVxc ∇

Implementation
Veff[n] = Ve-e[n] + Vext[n] + Vxc[n]
In order to calculate to total energy of the system of electrons we need to solve the following Schrödinger equation.
Hs = Eel (Ts + Ve-e[n] + Vext[n] + Vxc[n]) = Eel
(Ts + Ve-e[n] + Vext[n] + Vxc[n]) i = i i , i=1,….,N
∑=
ϕ=N
1i
2i ||)r(n
The effective potential depends on the density, the density
depends on Kohn-Sham orbitals ( i) and Kohn-Sham orbitals depends on the effective
potential!!
The result of solving the above self consistent equation are the total energy of the electrons:
∑ε=i
ielE
and Kohn-Sham orbitals i and charge density n(r)
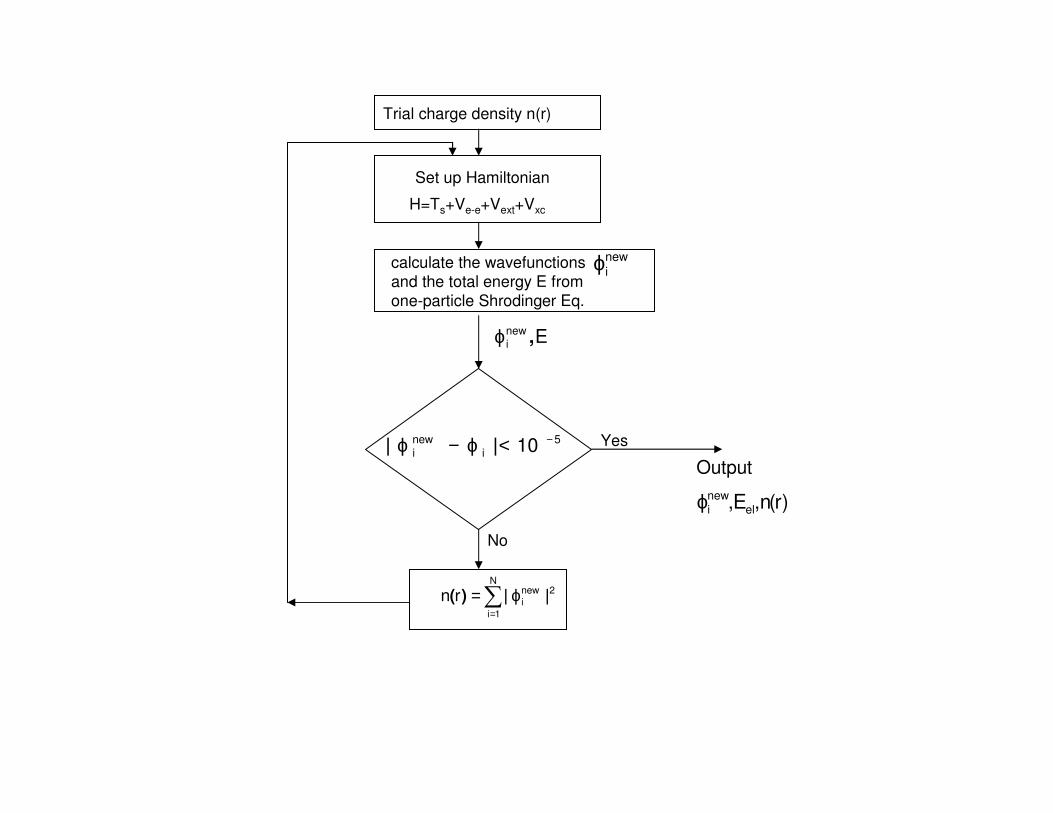
Trial charge density n(r)
Set up Hamiltonian
H=Ts+Ve-e+Vext+Vxc
calculate the wavefunctions
and the total energy E from
one-particle Shrodinger Eq.
∑=
ϕ=N
1i
2newirn ||)(
Enewi ,ϕ
newiϕ
5
i
new
i 10 −<ϕ−ϕ ||
No
Yes
)r(n,E, elnewiϕ
Output

Basis set-Plane wave
In this section we are going to briefly explain how we can solve the Kohn-Sham equation
and obtain the wavefunctions and energies. If we multiply both sides of the Kohn-Sham Eq.? by *i(r) and take an integral from
both sides we gets:
)r.ikexp(.C)r(1k
k,ii ∑∞
==ϕ
One way to solve the Kohn-Sham equation is to expand the wavefunction i in a
plane wave basis set. Using plane wave as a basis set has some advantages and some disadvantage. The advantage of using the plane wave is that it
which help us to simplify the above Kohn-Sham equation.
( ) dr)r()r(dr)r(]n[V]n[V]n[V)r(dr).r(dr
d).r(
m2i
*
iiixcextee
*
ii2
2*
i
2
∫∫∫ ϕϕε=ϕ++ϕ+ϕϕ−−
h
where mathematically the sum is over a infinite number of plane wave but of course in practice we see that it already converges for a large number of plane wave. By
substituting this equation into the above Shrodinger equation and using the
following mathematical principal:( ) ( ) 'kkdrr'ikexp.rikexp δ=⋅⋅∫
We end up with a matrix with Nmax x Nmax elements where Nmax is the number of
the plane waves. Nmax depends very much to the size of your system. i.e. number of atoms in your
system and also the elements you use. it can be as big as 1e+5 number of plane
waves. In order to get i and i from the matrix we need to diagonal it which of
course needs a powerful computer or even several computers working as parallel.
Reminding: When i and i are calculated in the next iteration a new charge density
should be constructed and again a new matrix should be diagonalized. Usually 10-30 iterations in needed to get the total energy of the system self consistently.

Total energy of the system
Etot = Eel + Eion
where Eion is just the coulomb interaction between ions
∑≠ −
=ji,j,i
2ji
2
ion|RR|
ZE
After we have calculated the total energy we can then calculate the force on each ion in the system
i
toti
dR
dEF =
Ri
Rj
xy
z
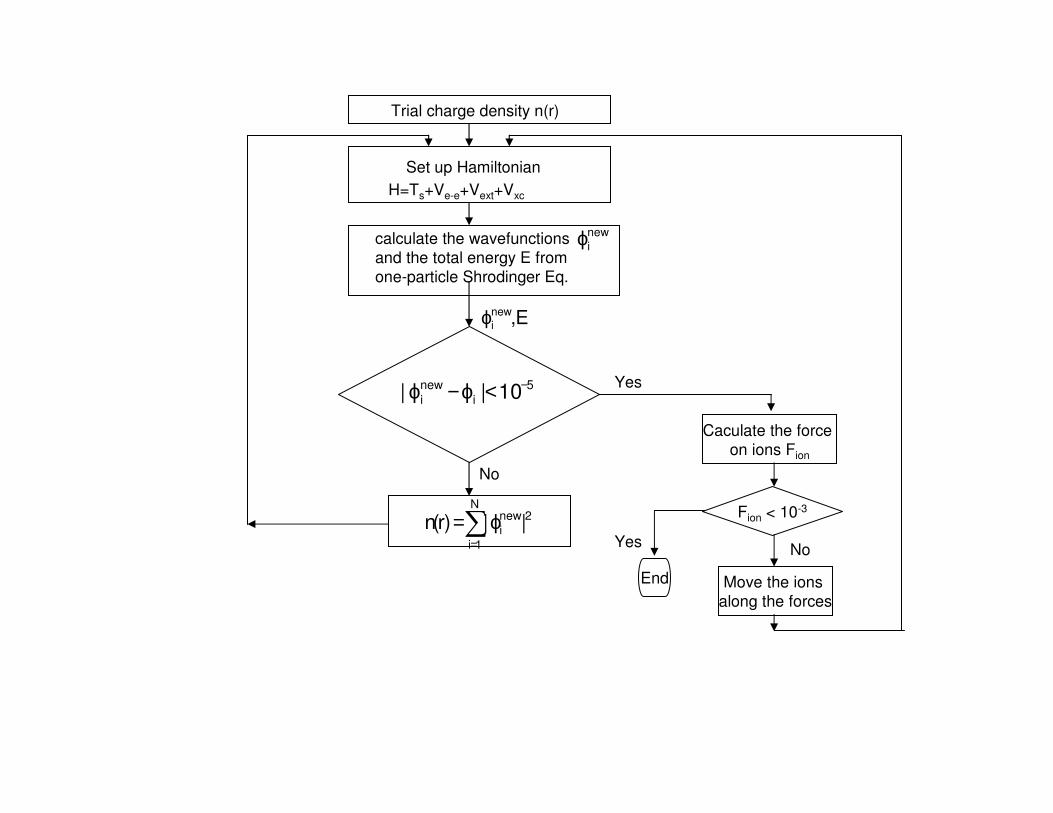
Trial charge density n(r)
Set up Hamiltonian
H=Ts+Ve-e+Vext+Vxc
calculate the wavefunctions
and the total energy E from
one-particle Shrodinger Eq.
∑=
ϕ=N
1i
2newi ||)r(n
E,new
iϕ
newiϕ
5
i
new
i 10|| −<ϕ−ϕ
No
Yes
Caculate the force
on ions Fion
Fion < 10-3
Yes
End Move the ions
along the forces
No
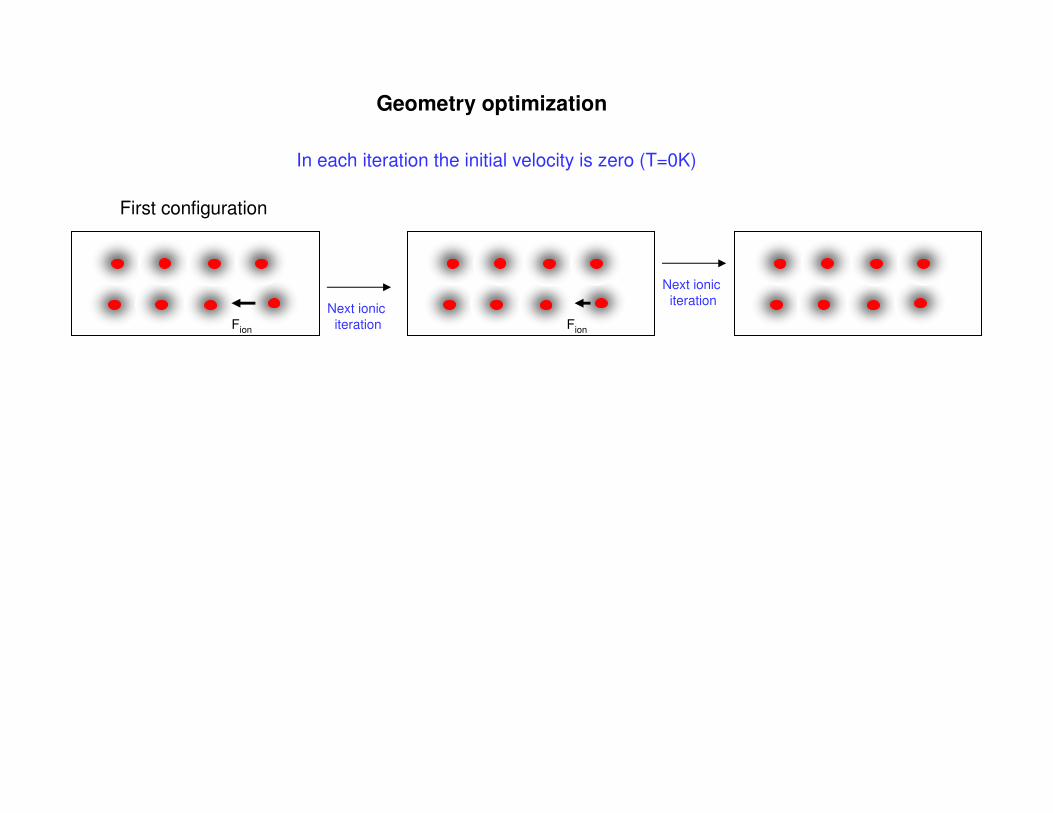
Geometry optimization
Fion Fion
Next ionic
iteration
Next ionic
iteration
In each iteration the initial velocity is zero (T=0K)
First configuration

Molecular dynamics
Fion Fion
Next ionic
iteration i=2
Next ionic
iteration i=3
In each iteration the initial velocity is not zero. It is calculated according to the temperature of the system
First configuration
Fion FionFion
i=4i=5i=6