molecular tools in diagnosis and characterization of infectious diseases
TRANSCRIPT

MOLECULAR TOOLS IN DIAGNOSIS AND CHARACTERIZATION OF
INFECTIOUS DISEASES
Tawheed Ahmad Shafi

Introduction
• Since the advent of the PCR, numerous applicationsin infectious diseases diagnostics have beendeveloped
• Several applications have been incorporated in theroutine diagonostic labs with a more user-friendly,cost-effective, and accurate profile.
• Realtime PCR allowed this transition of the scientifictechnology from basic research and reference centertesting into the mainstream clinical laboratories withthe ability to rapidly detect organisms such as groupB Streptococcus (GBS) and influenza virus

• Nucleic acid testing can be separated into amplified and
nonamplified methods.
• Nonamplified methods consist of DNA-labeled or RNA-labeled
probes that bind to the target nucleic acid and generate a signal
from the attached reporter molecule.
• Target amplification allows the use of multiple different types of
postamplification technologies to further characterize the
amplified targets of organism nucleic acids.
• A variety of nucleic acid methods are currently utilized for
detection/identification of organisms and their virulence
factors/resistance determinants.

History OF PCR
Great mind behind this PCR :Kary Banks Mullis Developed PCR in 1985 and was awarded nobel prize in
1993.
PCR machine otherwise called Thermocycler.
• 1983—Kary Mullis, a scientist working for the CetusCorporation was driving along US Route 101 in northernCalifornia when he came up with the idea for thepolymerase chain reaction.
• 1985—the polymerase chain reaction was introduced to thescientific community at a conference in October. Cetusrewarded Kary Mullis with a $10,000 bonus for hisinvention
• Later, during a corporate reorganization, Cetus sold thepatent for the PCR process to a pharmaceutical companyHoffmann-LaRoche for $300 million.

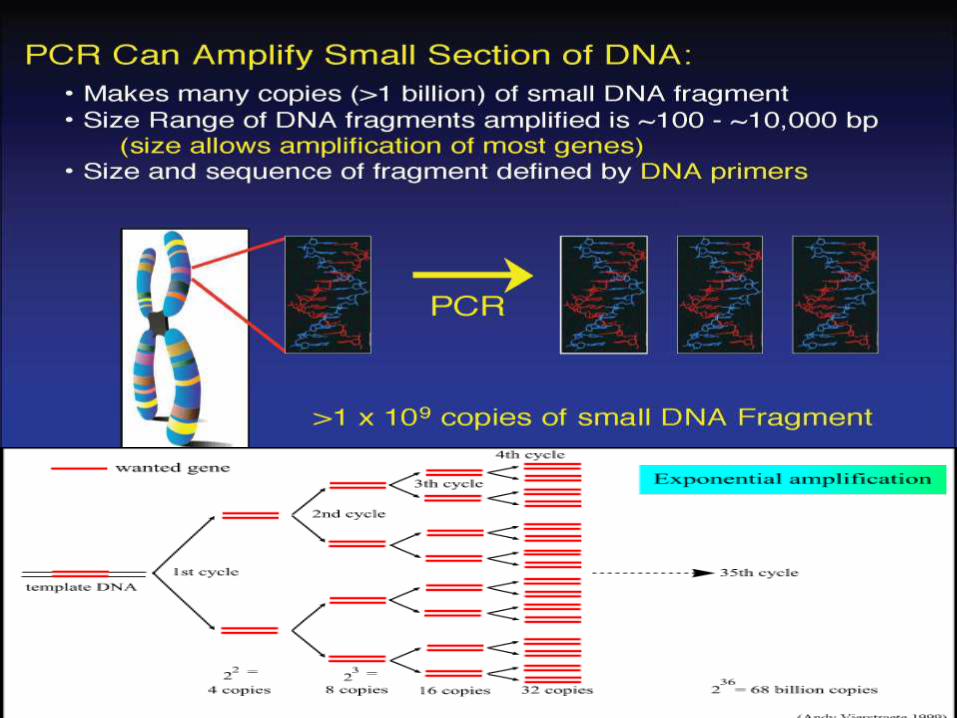
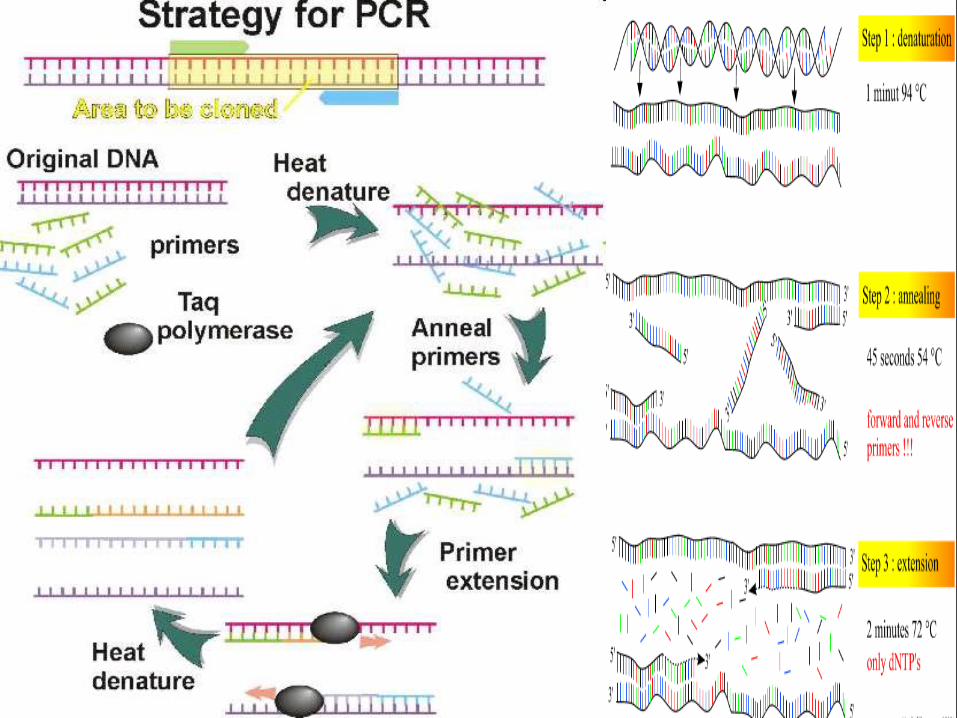
PCR a Revolution in Scienceamplify a single or few copies of a piece of DNA,generating millions or more copies of aparticular DNA sequence.
The method relies on, cycles of repeatedheating and cooling of the reaction for DNAmelting and enzymatic replication of the DNA.
Almost all PCR applications employ a heat-stable DNA polymerase, such as Taqpolymerase, an enzyme originally isolated fromthe bacterium Thermus aquaticus.

PCR Reagents• 1X Buffer
– 10mM Tris-HCl, 50mM KCl
• MgCl2– 1mM - 4mM (1.5mM)
• dNTPs– 200μM
• Primers– 100nM-1μM, 200nm (or less) for real time analysis
• DNA polymerase – Taq DNA polymerase is thermostable– 1-4 Units (1 unit)
• DNA– 10pg-1μg (20ng)
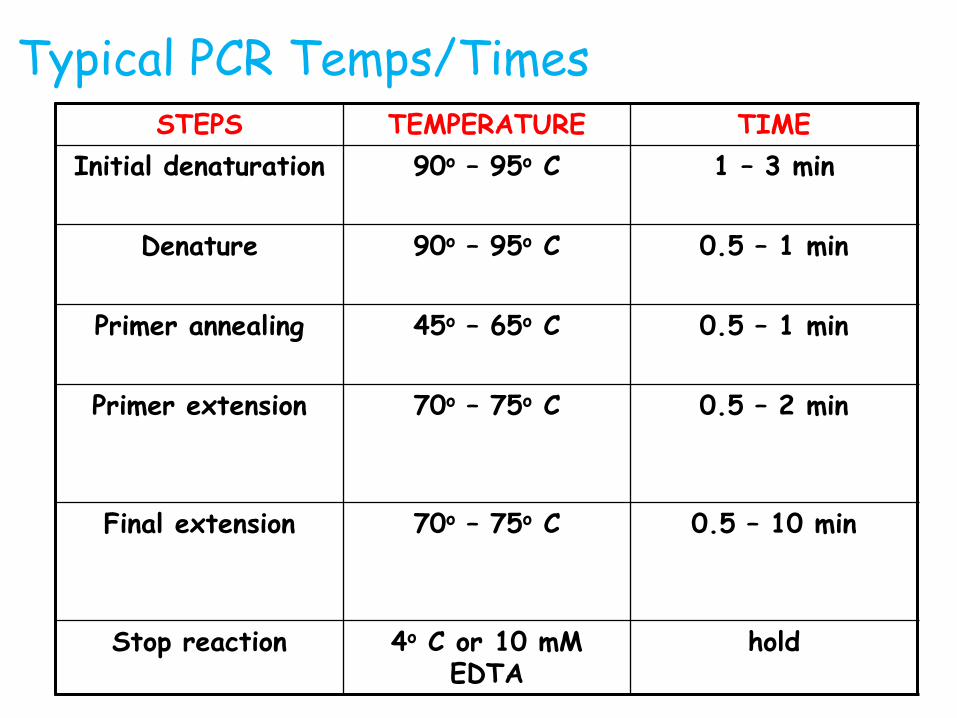
Typical PCR Temps/TimesSTEPS TEMPERATURE TIME
Initial denaturation 90o – 95o C 1 – 3 min
Denature 90o – 95o C 0.5 – 1 min
Primer annealing 45o – 65o C 0.5 – 1 min
Primer extension 70o – 75o C 0.5 – 2 min
Final extension 70o – 75o C 0.5 – 10 min
Stop reaction 4o C or 10 mM EDTA
hold
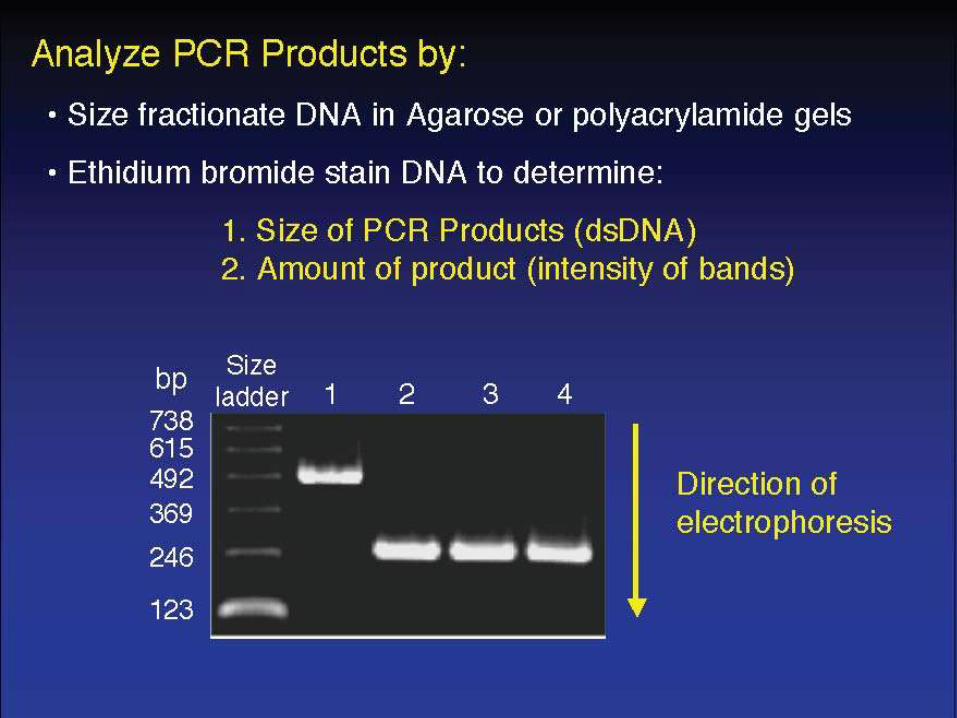
Variations of the PCR
• Colony PCR
• Nested PCR
• Multiplex PCR
• Hot Start PCR
• Inverse PCR
• Asymmetric PCR
• Long PCR
• Reverse Transcriptase PCR
• Real time PCR
• Touchdown PCR

Colony PCR: the screening of bacterial (E.Coli) or yeastclones for correct ligation or plasmid products.
Nested PCR: Involves two consecutive PCR reactions of 25 cycles. The first PCR uses primers external to the sequence of interest. The second PCR uses the product of the first PCR in conjunction with one or more nested primers to amplify the sequence within the region flanked by the initial set of primers.
Multiplex PCR: is a variant of PCR which enablssimultaneous amplification of many targets of interest in onereaction by using more than one pair of primers.
Hot start PCR: This is a technique that reduces non-specific amplification during the initial set up stages of the PCR. The technique may be performed manually by heating the reaction components to the melting temperature (e.g., 95°C) before adding the polymerase

Long PCR: Used to amplify DNA over the entire length up to 25kb ofgenomic DNA segments cloned.
Inverse PCR: Used to amplify DNA of unknown sequence that is adjacentto known DNA sequence.
Quantitative PCR: Product amplification w r t time, which is comparedwith a standard DNA.
Asymmetric PCR: preferentially amplifies one DNA strand in a double-stranded DNA template. It is used in sequencing and hybridization probingwhere amplification of only one of the two complementary strands isrequired.
Reverse Transcriptase PCR- First step of RT-PCR - "first strandreaction“-Synthesis of cDNA using oligo dT primers (37°C) 1 hr.“Secondstrand reaction“-Digestion of cDNA:RNA hybrid. Allows the detection ofeven rare or low copy mRNA sequences by amplifying its complementaryDNA.

Touchdown PCR (Step-down PCR):
a variant of PCR that aims to reduce nonspecific background by gradually lowering the annealing temperature as PCR cycling progresses.
The annealing temperature at the initial cycles is usually a few degrees (3-5 °C) above the Tm of the primers used,
while at the later cycles, it is a few degrees (3-5 °C) below the primer Tm.
The higher temperatures give greater specificity for primer binding, and
the lower temperatures permit more efficient amplification from the specific products formed during the initial cycles

Applications of PCR Methods
• Medical Diagnostics1) Diagnosis and characterisation of Infectious diseases:
- Detect presence of viral pathogens- Detect presence of pathogenic bacteria
2) Diagnosis and characterisation of genetic diseases3) Diagnosis and characterisation of Neoplasia
• Forensics1) Identify criminal suspects2) Paternity cases

Advances on PCR MethodsReal Time Assays
called “real-time PCR” because it allows to view the increase in the amount of DNA as it is amplified.
The Real Time assays are proving to better technologies Rapid Quantitative measurement Lower contamination rate Higher sensitivity Higher specificity Easy standardization
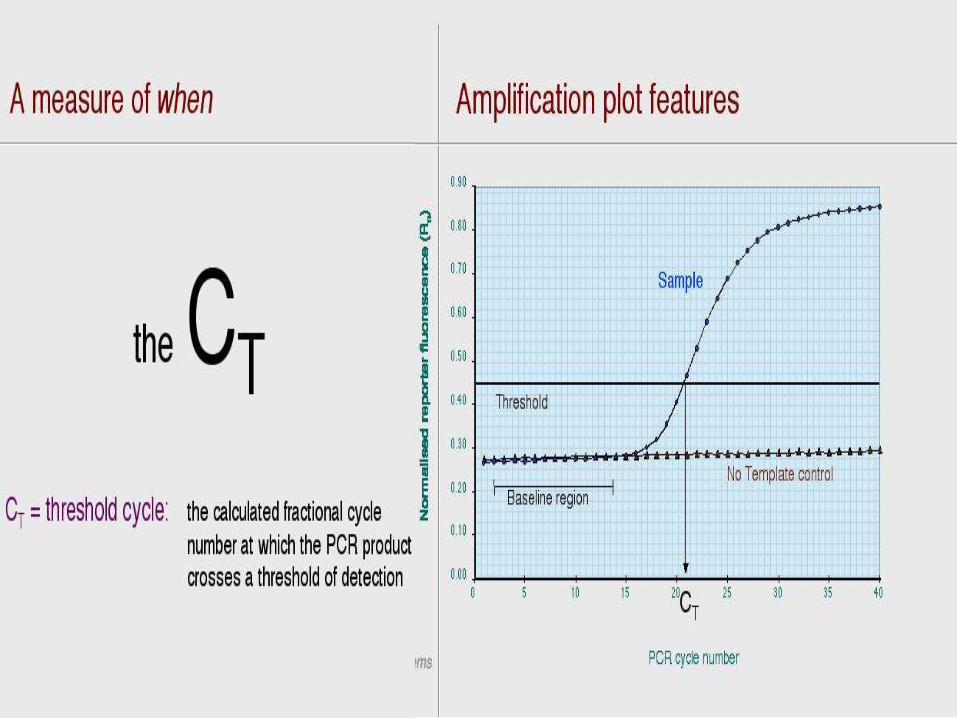

Real Time Reporters
• All real time PCR systems rely upon thedetection and quantization of fluorescentreporter, the signal of which increases in directproportion of the amount of PCR product in areaction.
REAL TIME PCR Cyber Green
• The simplest and economical reporter is thedouble strand DNA specific dye SYBR Green
• Called as Molecular Probe.
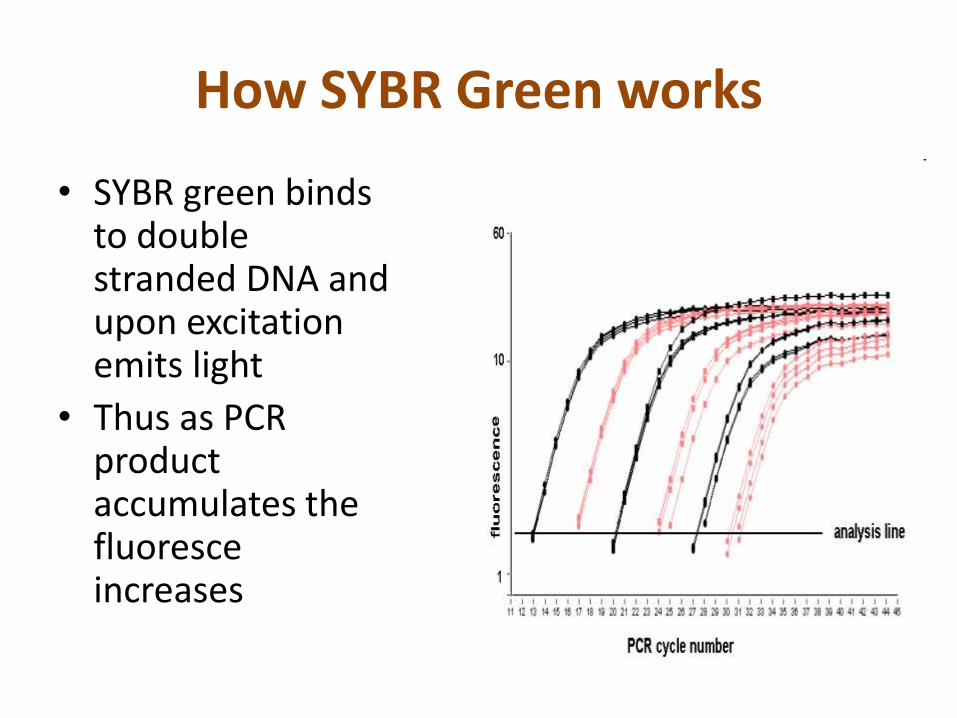
How SYBR Green works
• SYBR green binds to double stranded DNA and upon excitation emits light
• Thus as PCR product accumulates the fluoresce increases

Advantages
• Inexpensive
• Easy to Use
• Sensitive
Disadvantages
• SYBR green will bind to any double stranded DNA in a reaction, may result in an overestimation of the target concentration
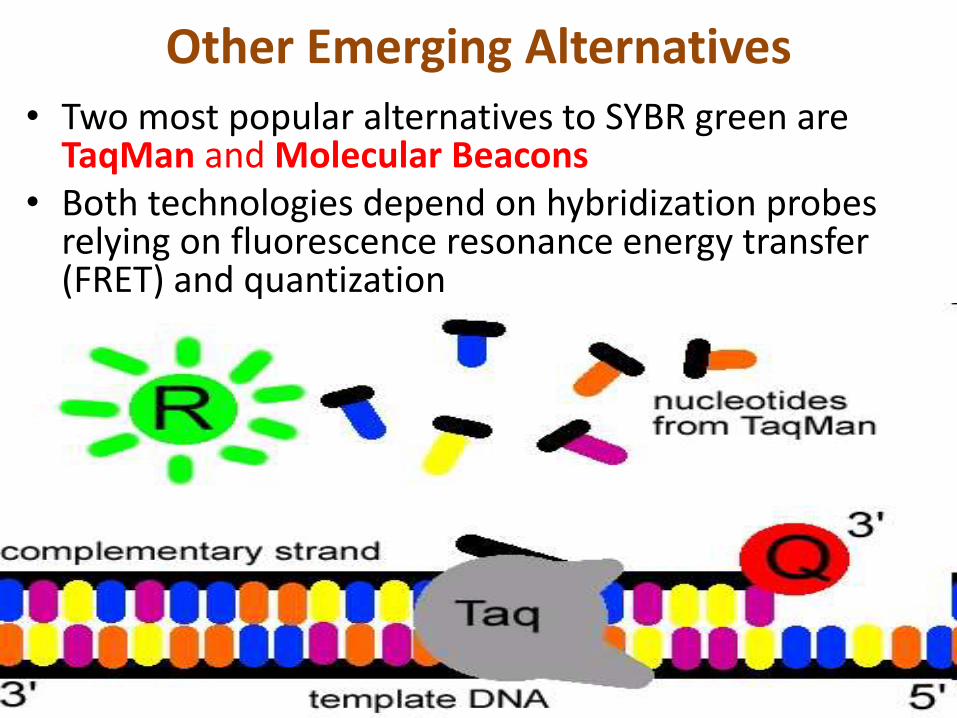
Other Emerging Alternatives• Two most popular alternatives to SYBR green are
TaqMan and Molecular Beacons• Both technologies depend on hybridization probes
relying on fluorescence resonance energy transfer (FRET) and quantization
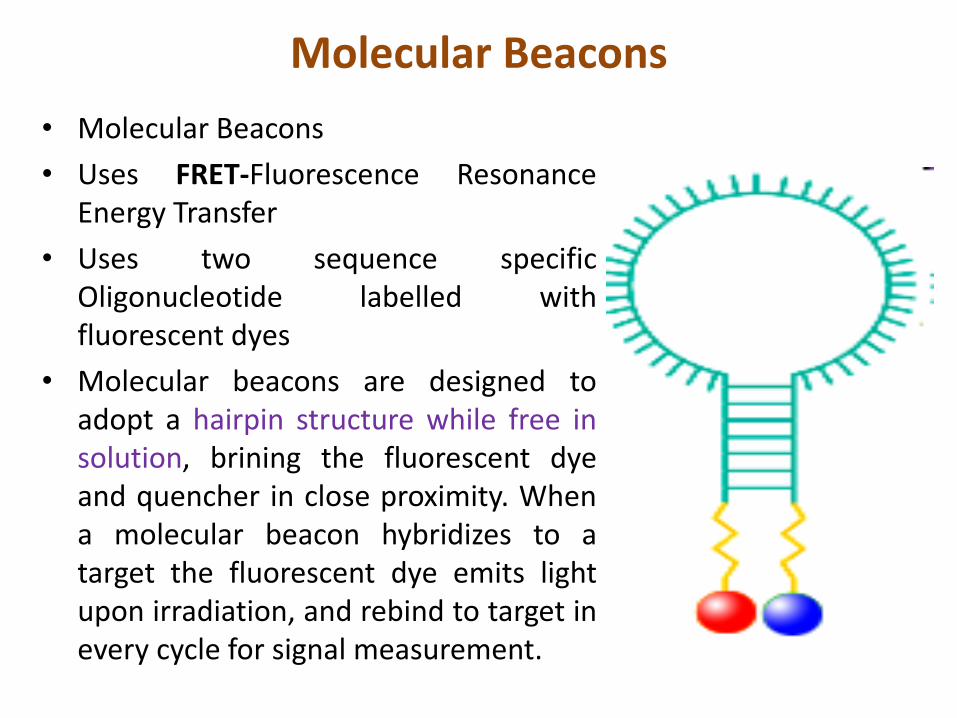
Molecular Beacons
• Molecular Beacons
• Uses FRET-Fluorescence ResonanceEnergy Transfer
• Uses two sequence specificOligonucleotide labelled withfluorescent dyes
• Molecular beacons are designed toadopt a hairpin structure while free insolution, brining the fluorescent dyeand quencher in close proximity. Whena molecular beacon hybridizes to atarget the fluorescent dye emits lightupon irradiation, and rebind to target inevery cycle for signal measurement.
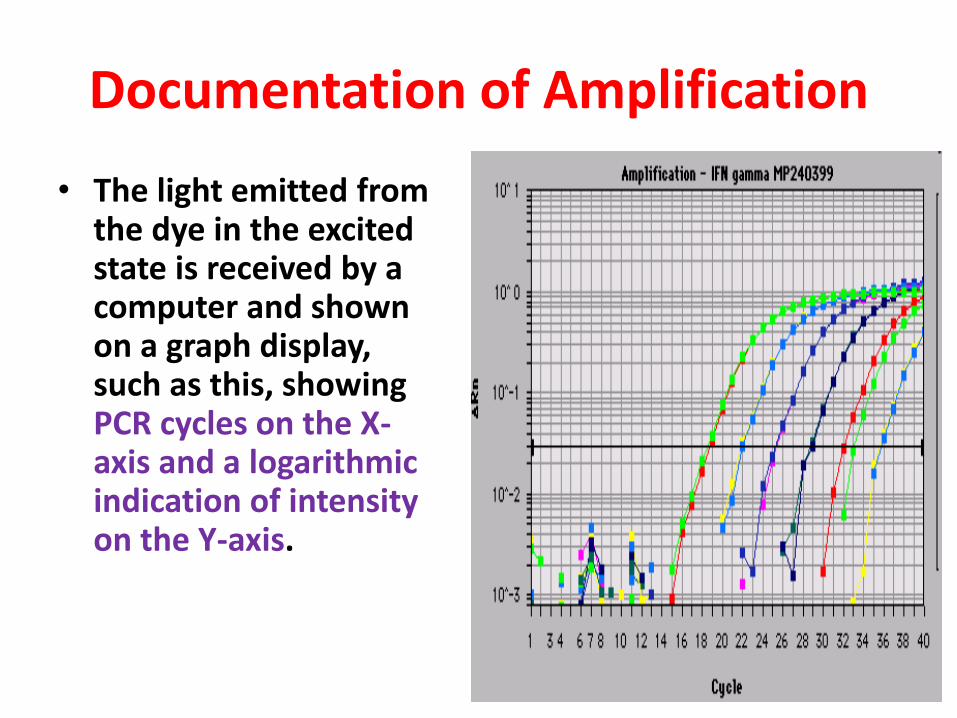
Documentation of Amplification
• The light emitted from the dye in the excited state is received by a computer and shown on a graph display, such as this, showing PCR cycles on the X-axis and a logarithmic indication of intensity on the Y-axis.

Applications • Some of the common real-time PCR assays that are
available include the tests for group A/B streptococcus,methicillin-resistant Staphylococcus aureus (MRSA) andinfluenza virus.
• There are numerous laboratory-developed realtime PCRtests, including assays for poorly cultivatable or atypicalorganisms (Bordetella pertussis, Legionella pneumophila,Mycoplasma pneumoniae, Chlamydia pneumophila), andthe herpes viruses
• Recent development of assays for Zygomycetes,Aspergillus, Candida sp., Pneumocystis jiroveci, andCoccidiodes show promise for addressing some of thecommon problems of analysis for these pathogens
Loop Mediated Isothermal Amplification (LAMP)
• Loop mediated isothermal amplification is asimple, rapid, specific and cost effective nucleicacid amplification method.
• The amplification proceeds at a constanttemperature using strand displacement reaction.
• Amplification and detection of gene can becompleted in a single step, by incubating themixture of samples, primers DNA polymerase andsubstrates at a constant temperature of 630c.

LAMP in Clinical Diagnosis
• LAMP technology proving to be ideal in detection of DNA orRNA of the pathogenic organisms
• Proving to be highly efficient in diagnosis of Viral and Bacterialinfections
• LAMP is capable of detecting the presence of pathogenicagents earlier than PCR
• A one step single tube real time accelerated reversetranscription loop mediated isothermal amplification (RT-LAMP) assays for rapid detection of some recently emergedviral pathogen eg West Nile, Dengue, Japanese encephalitisH5N1- highly pathogenic avian influenza.

Advantages of LAMP
• LAMP does not require an expensivethermocycler
• Amplification specificity is extremely high asLAMP requires 4/6 oligonucleotide primers
• Detection limit : LAMP≥ PCR
• Detection time : LAMP< PCR
• Visualization of DNA products by LAMP:
(a) Eye – turbidity, colour change
(b) Real Time Turbidimeter
(C) Electrophoresis

PCR is susceptible to hemoglobin, Ig and
Heparin
LAMP resists contamination of above
mentioned materials
LAMP can amplify parasite DNA from fresh
infected blood
LAMP can be done by using rather crude DNA
extracted by simple methods

Hybridisation
• Nucleic acid hybridization as a technique involves using alabeled nucleic acid probe, to bind with the target nucleic acids
• A probe labeled with detectable tracer is the prerequisite fordetermining a specific DNA sequence or gene in a sample orgenomic DNA by nucleic acid hybridization.
• The target nucleic acids to be analyzed are usually denatured,and then mixed with the labeled probe in the hybridizationsystem.

• The probe will bind to thesegment of nucleic acid withcomplementary sequenceunder proper conditions.
• The hybridization can beidentified by the detection ofthe tracer labeling the probe.
• Thus the existence or theexpression of specific genecan be determined.
CTGATGGTCATGAGCTGTCCGATCGATCATACAGGCTAGCTAGTA
ACAGGCTAGCTAGTA
Hybridization
ACAGGCTAGCTAGTA
nucleic acid probe
DNA from source “X”

Preparation And Labeling Of Nucleic Acid
Probes may be• single-stranded or
• double-stranded molecules
working probe must be single-stranded molecules.
The probes used in hybridization include
• oligonucleotide(15-50 nucleotides)
• genomic DNA fragment
• cDNA fragment and
• RNA.

Preparation And Labeling Of Nucleic Acid
• Probe is usually labeled with a detectable tracer, which iseither isotopic or non-isotopic. The purifiedoligonucleotide is labeled in vitro by using a suitableenzyme to add the labeled nucleotide to the end of theoligonucleotide.
• The labels in common use include radioactive (32P and 35S)and nonradioactive (digoxigenin, biotin, fluorescein)substances which are used to label dNTP.
• After hybridization, the location and the quantity of thehybrid molecules can be determined.

Hybridization Of Nucleic Acids(Southern blot hybridization)
• In Southern blot hybridization, the target DNA is digestedwith restriction endonucleases
• Following electrophoresis, the sample DNA fragments aredenatured in strong alkali, such as NaOH.
• The denatured DNA fragments are transferred to anitrocellulose or nylon membrane and become immobilizedon the membrane.
• The immobilized single-stranded target DNA sequences areallowed to interact with labeled single-stranded probe DNA.
• The probe will bind only to complementary DNA sequencesin the target DNA to form a target-probe heteroduplex.
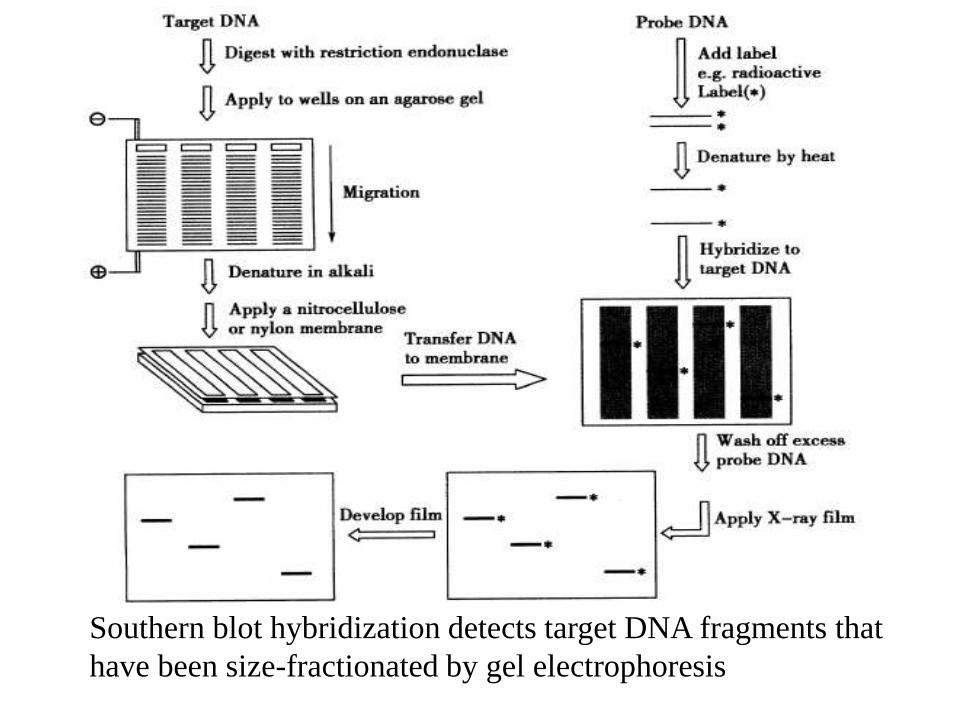
Southern blot hybridization detects target DNA fragments that
have been size-fractionated by gel electrophoresis

Widely applied in researches since its invention.
• Identification DNA from pathogenic
microorganism
• For analysis of gene expression
• Screening of recombinant plasmids
• Analysis of gene mutation

Typing
The process of differentiating strains based on theirphenotypic and genotypic differences is known as 'typing'.These typing methods are useful in:
hospital infection control epidemiological studies, and understanding the pathogenesis of infection.
In hospital settings they may be used to:determine whether a set of isolates obtained from onepatient represents a single infecting strain or multiplecontaminants.determine whether a series of isolates obtained over timerepresents relapse of an infection due to single strain orseparate episodes of disease due to different strains.
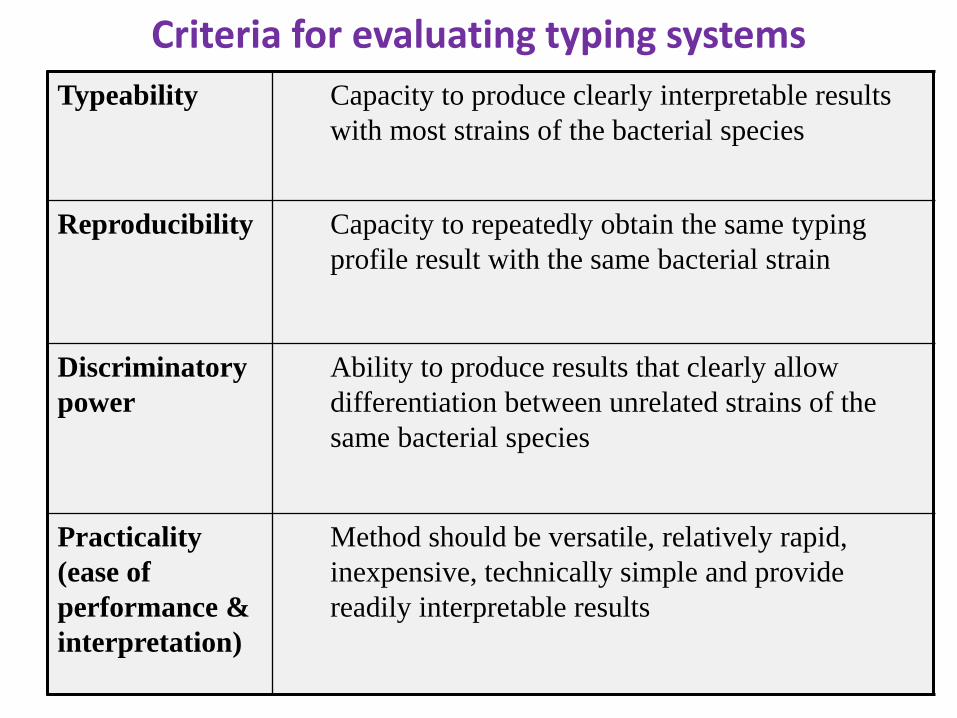
Criteria for evaluating typing systems
Typeability Capacity to produce clearly interpretable results
with most strains of the bacterial species
Reproducibility Capacity to repeatedly obtain the same typing
profile result with the same bacterial strain
Discriminatory
power
Ability to produce results that clearly allow
differentiation between unrelated strains of the
same bacterial species
Practicality
(ease of
performance &
interpretation)
Method should be versatile, relatively rapid,
inexpensive, technically simple and provide
readily interpretable results

Molecular Typing TechniquesRestriction analysis
Plasmid profiling
Restriction fragment length polymorphism (RFLP)
Ribotyping
Pulse Field Gel Electrophoresis (PFGE)
PCR amplification of particular genetic targetsAmplified fragment length polymorphism (AFLP)
Random Amplified Polymorphic DNA (RAPD)
Repetitive element PCR (Rep-PCR)
Variable number of tandem repeat (VNTR) analysis and
Multiple locus VNTR analysis (MLVA)
Sequencing-based methodsMultilocus sequence typing (MLST)
Single nucleotide polymorphism (SNPs)

Random Amplified Polymorphic DNA
(RAPD) PCR
• Shortly after Kary Mullis invented the Polymerase Chain Reaction
(PCR) it was realized that short primers would bind to several
locations in a genome and thus could produce multiple fragments
• Williams et al. (1990) developed Random Amplified Polymorphic
DNA (RAPD) a technique using very short 10 base primers to
generate random fragments from template DNAs
• RAPD fragments can be separated and used as genetic markers or a
kind of DNA fingerprint

• The primers can be designed without the experimenter having any
genetic information for the organism being tested.
• More than 2000 different RAPD primers can be available
commercially.
• Genomic DNA normally has complimentary sequences to RAPD
primers at many locations.
• If two of these locations are close to each other (<2000-3000bp),
and the sequences are in opposite orientation, the amplification will
be established. This amplified region is said as a RAPD locus
• Normally, a few (3-20) loci can be amplified by one single RAPD
primer.
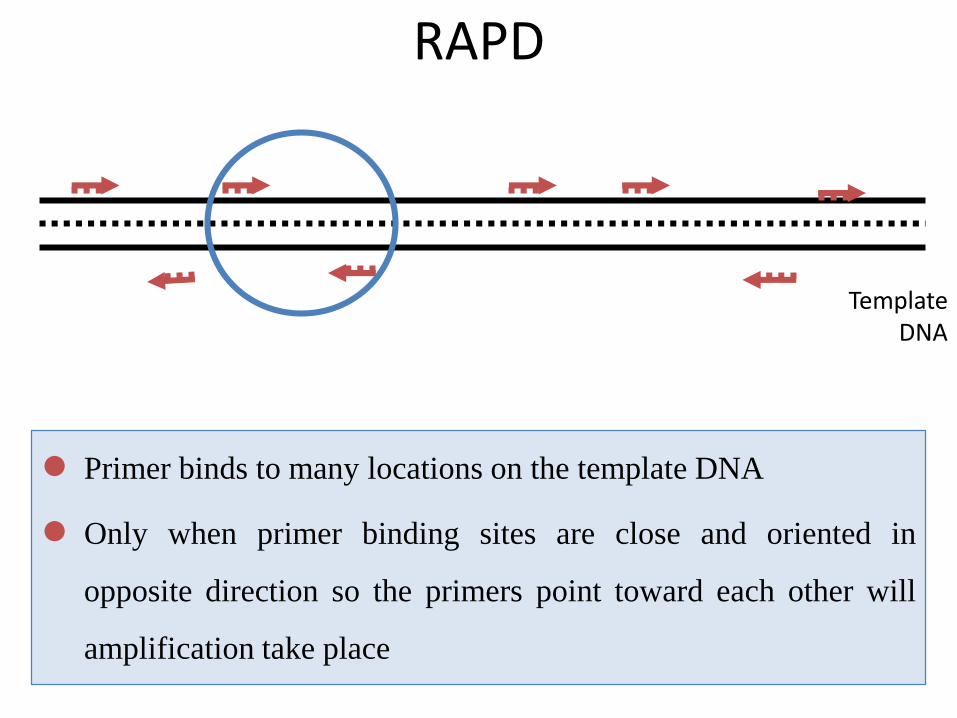
Template DNA
RAPD
Primer binds to many locations on the template DNA
Only when primer binding sites are close and oriented in
opposite direction so the primers point toward each other will
amplification take place
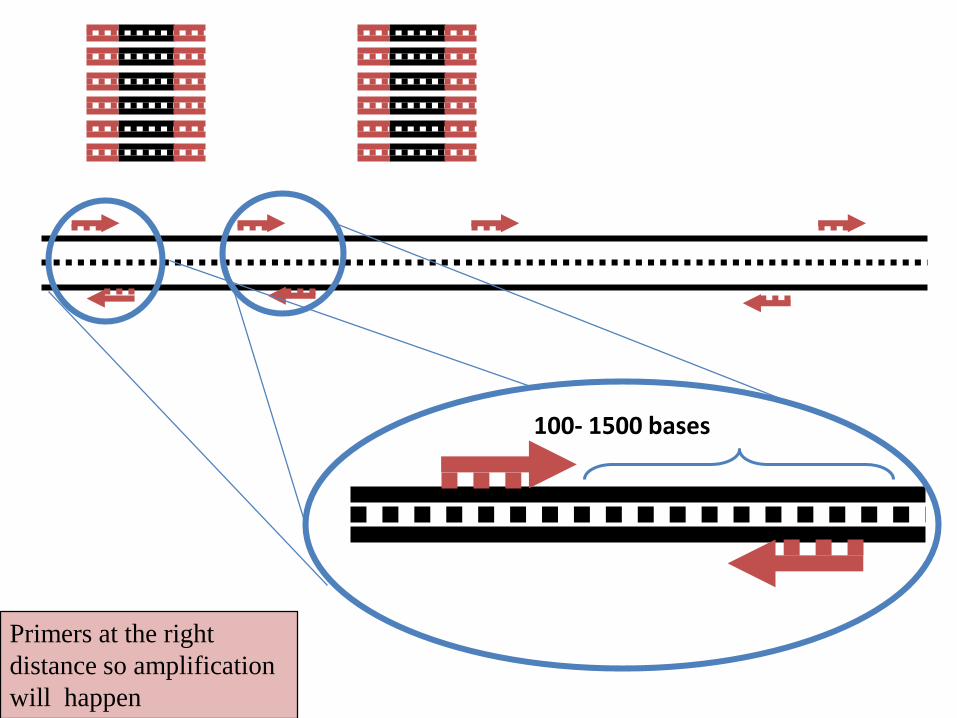
Primers at the right
distance so amplification
will happen
100- 1500 bases
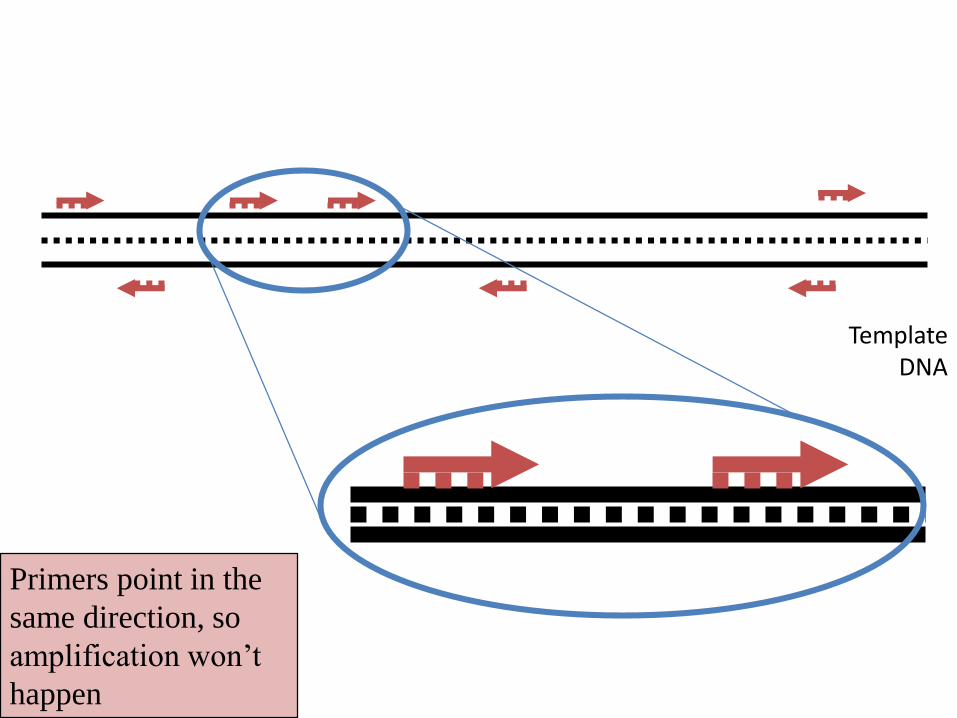
Primers point in the
same direction, so
amplification won’t
happen
Template DNA
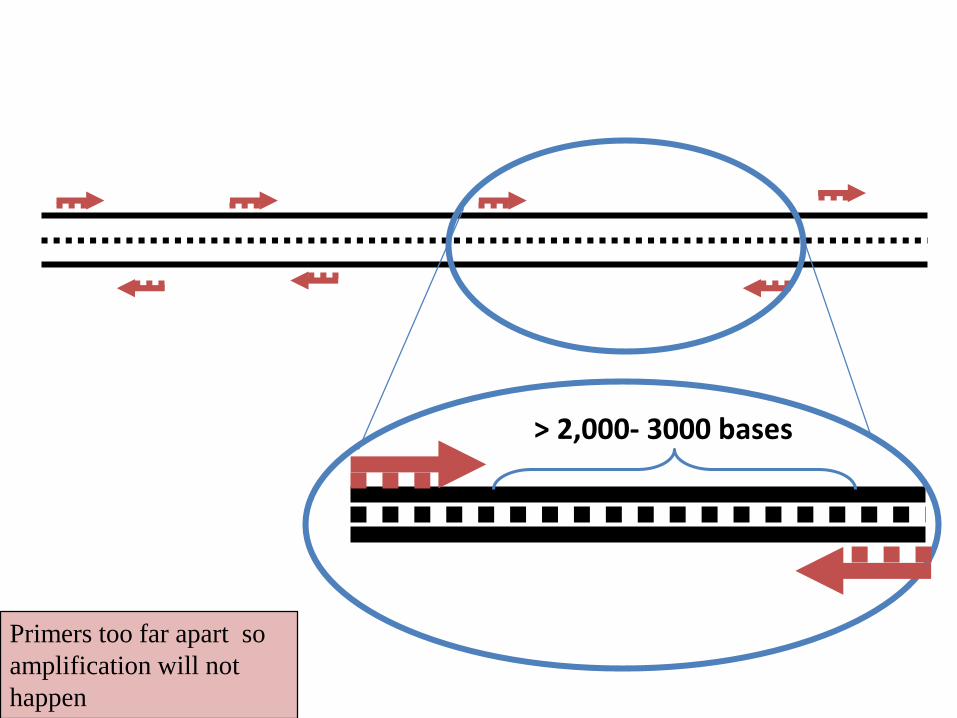
Primers too far apart so
amplification will not
happen
> 2,000- 3000 bases
RAPD
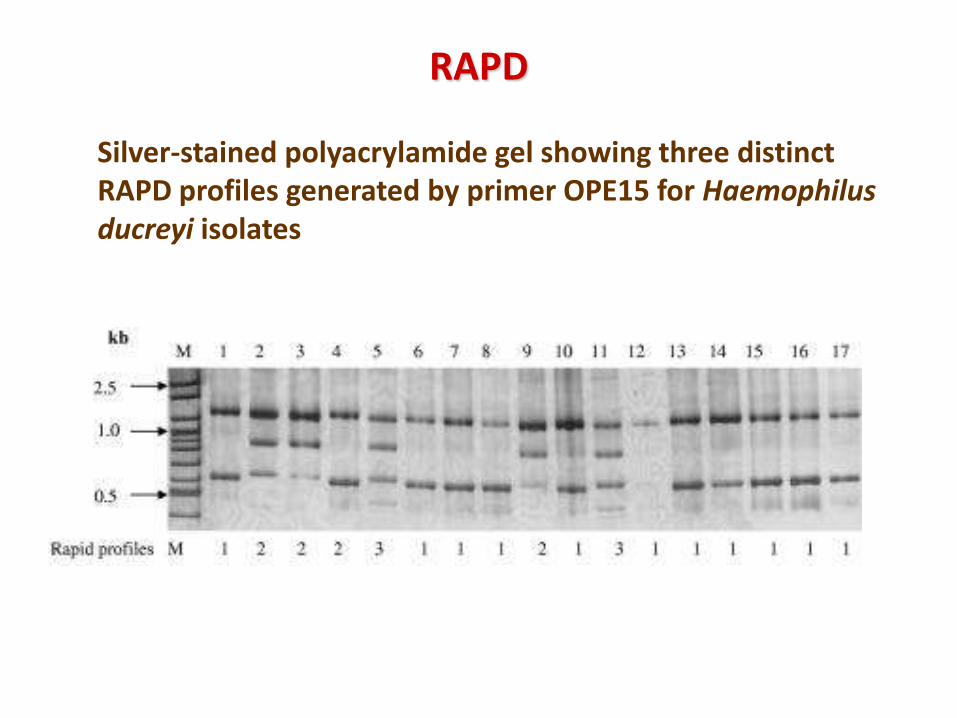
Silver-stained polyacrylamide gel showing three distinct RAPD profiles generated by primer OPE15 for Haemophilus ducreyi isolates

Applications
• Has been largely carried out for variability analysis and
individual-specific genotyping, but is less popular due to
problems such as poor reproducibility, faint or fuzzy products,
and difficulty in scoring bands, which lead to inappropriate
inferences.
• RAPDs have been used for many purposes, ranging from studies
at the individual level (e.g. genetic identity) to studies involving
closely related species.
• RAPDs have also been applied in gene mapping studies to fill
gaps not covered by other markers

Limitations
• PCR based technique, therefore quality and concentration of
template DNA, concentrations of PCR components, and the
PCR cycling conditions may greatly influence the outcome.
• Thus, the RAPD technique is notoriously laboratory dependent
and needs carefully developed laboratory protocols to be
reproducible.
• Mismatches between the primer and the template may result in
the total absence of PCR product as well as in a merely
decreased amount of the product. Thus, the RAPD results can
be difficult to interpret.

Restriction Fragment Length
Polymorphism (RFLP)
• RFLP is a technique in which organisms may be
differentiated by analysis of patterns derived from
cleavage of their DNA.
• If two organisms differ in the distance between sites
of cleavage of particular Restriction Endonucleases,
the length of the fragments produced will differ when
the DNA is digested.

• The similarity of the patterns generated can be used to
differentiate species (and even strains) from one another.
• This technique is mainly based on the special class of
enzyme i.e. Restriction Endonucleases.
• The variability of restriction sites have their origin in the
DNA rearrangements, point mutations within the restriction
enzyme recognition site sequences, insertions or deletions
within the fragments, and unequal crossing over
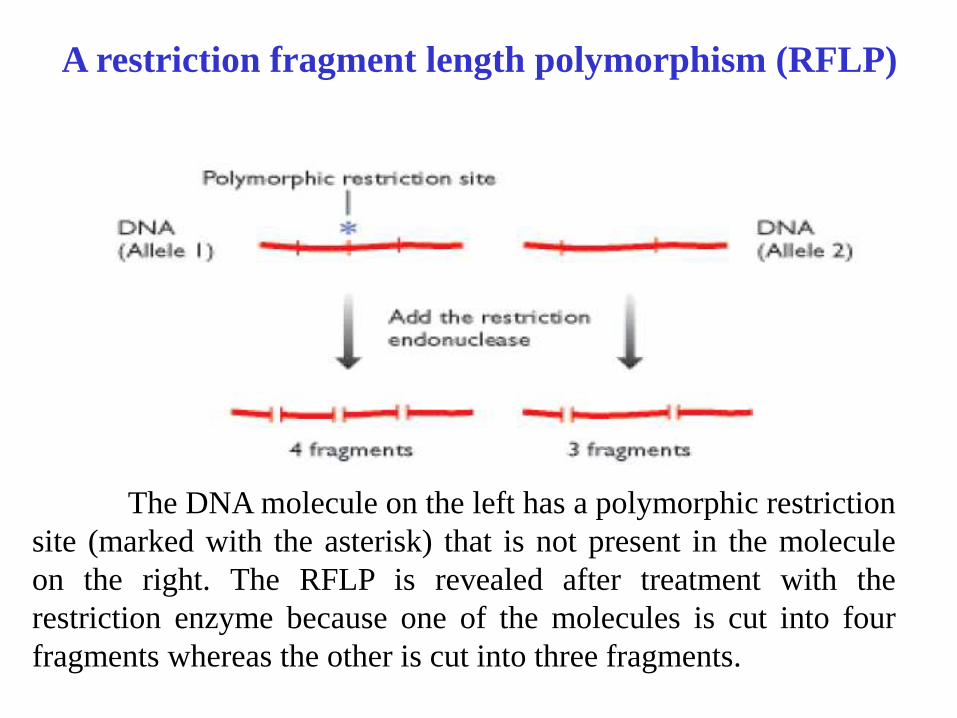
A restriction fragment length polymorphism (RFLP)
The DNA molecule on the left has a polymorphic restriction
site (marked with the asterisk) that is not present in the molecule
on the right. The RFLP is revealed after treatment with the
restriction enzyme because one of the molecules is cut into four
fragments whereas the other is cut into three fragments.

Two methods for scoring an RFLP :
(A)RFLPs can be scored by Southern hybridization.
The DNA is digested with the appropriate restriction enzyme and
separated in an agarose gel. The smear of restriction fragments is
transferred to a nylon membrane and probed with a piece of
DNA that spans the polymorphic restriction site. If the site is
absent then a single restriction fragment is detected (lane 2); if
the site is present then two fragments are detected (lane 3).
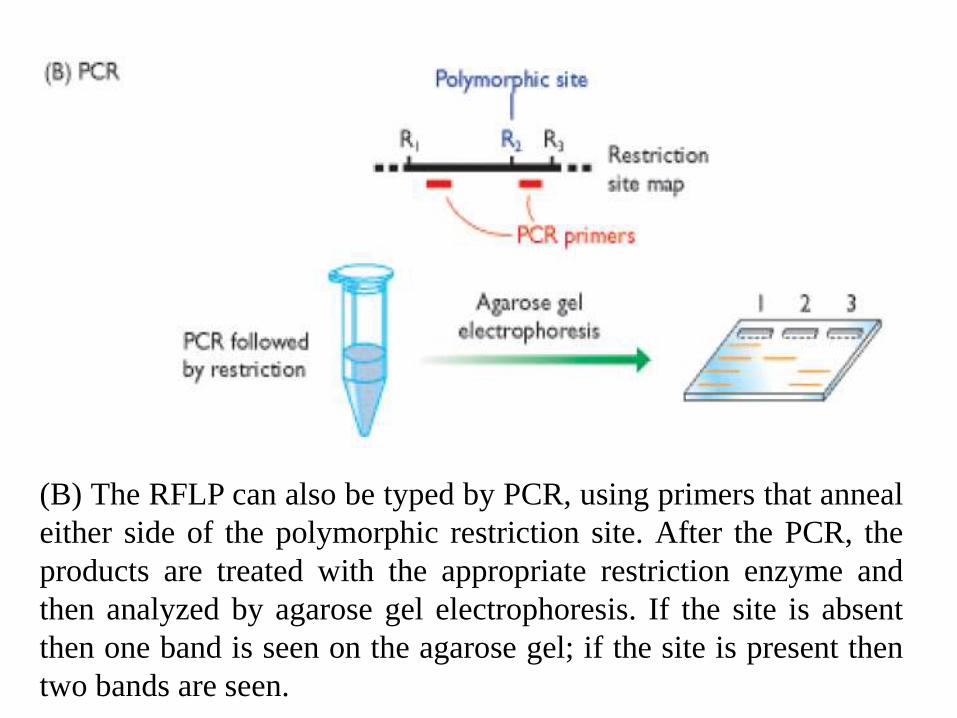
(B) The RFLP can also be typed by PCR, using primers that anneal
either side of the polymorphic restriction site. After the PCR, the
products are treated with the appropriate restriction enzyme and
then analyzed by agarose gel electrophoresis. If the site is absent
then one band is seen on the agarose gel; if the site is present then
two bands are seen.

Applications:
• RFLPs can be applied in diversity and phylogenetic
studies ranging from individuals within populations or
species, to closely related species.
• RFLPs have been widely used in gene mapping studies
because of their high genomic abundance due to the ample
availability of different restriction enzymes and random
distribution throughout the genome
• RFLP markers were used for the first time in the
construction of genetic maps

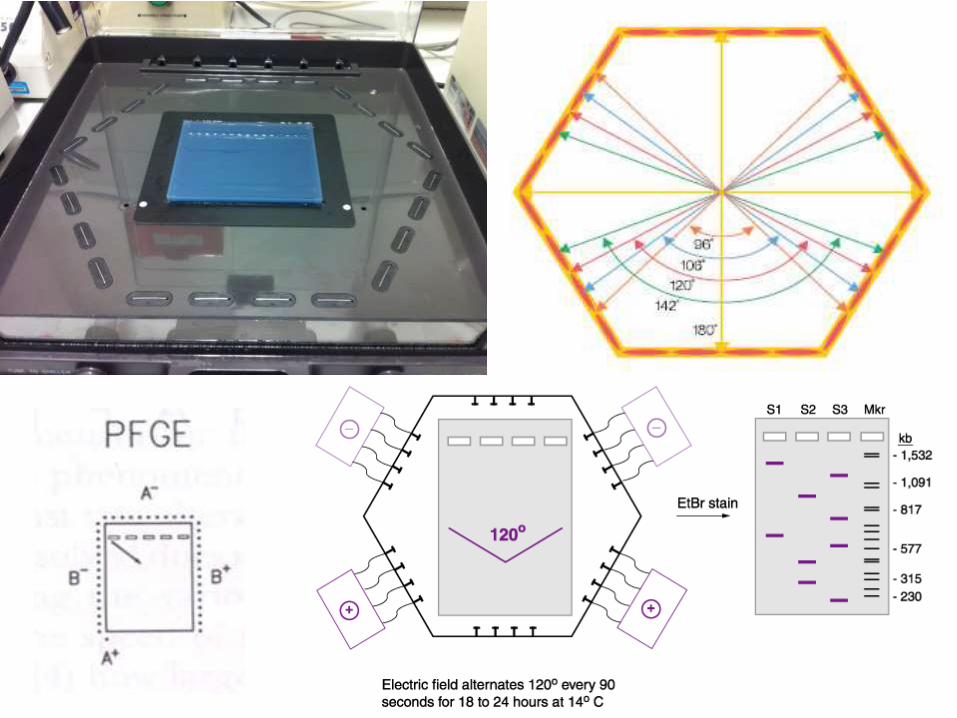
Pulsed field gel electrophoresis (PFGE)
Conventional gel electrophoresis techniques:
separates DNA fragments from 100 to 200 bp to 50 kilobase pairs (kb)
only
DNA(>50kb) cant be separated by this method.
In 1982, Schwartz introduced the concept that DNA molecules larger
than 50 kb can be separated by using two alternating electric fields.
In conventional gels, the current is applied in a single direction (from top
to bottom).
But in PFGE, the direction of the current is altered at a regular interval.

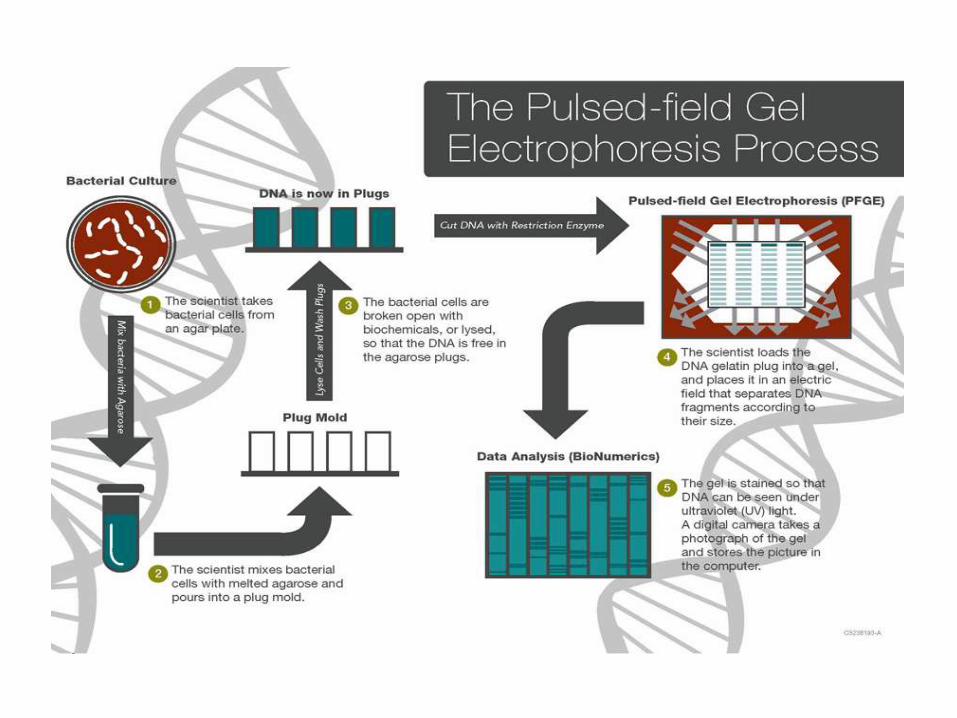
Pulsed-field gel electrophoresis is based on the digestion ofbacterial DNA with restriction endonucleases that recognizefew sites along the chromosome, generating large DNAfragments (30-800 Kb)
The basis for PFGE separation is the size-dependent time-associated reorientation of DNA migration achieved byperiodic switching of the electric field in different directions.
The DNA fragments will form a distinctive pattern of bandsin the gel, which can be analyzed visually and electronically.
Bacterial isolates with identical or very similar band patternsare more likely to be related genetically than bacterial isolateswith more divergent band patterns.
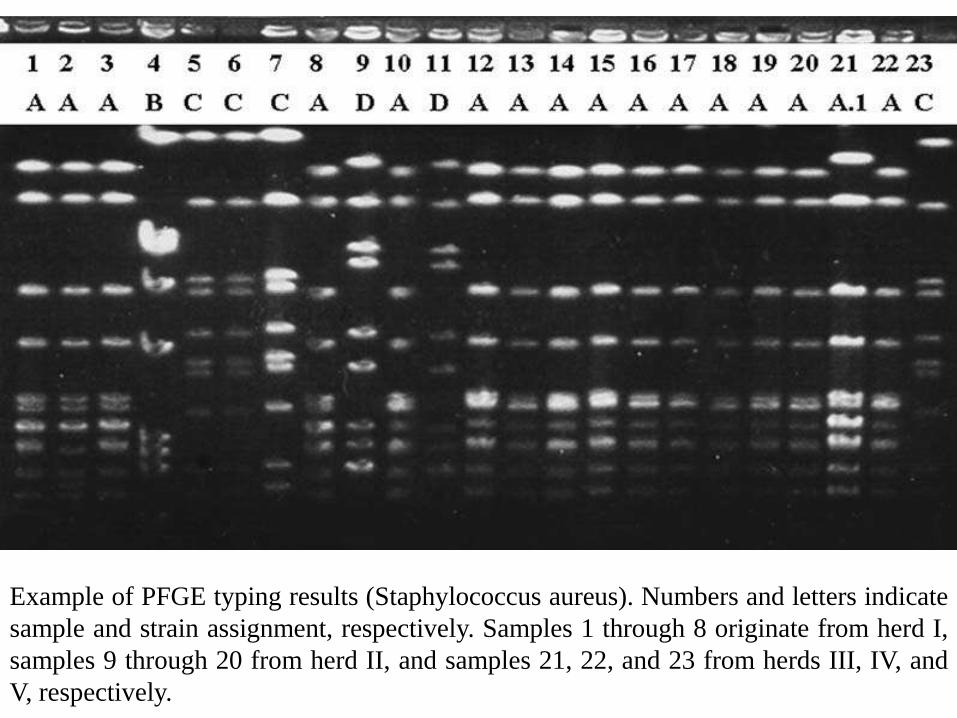
Example of PFGE typing results (Staphylococcus aureus). Numbers and letters indicate
sample and strain assignment, respectively. Samples 1 through 8 originate from herd I,
samples 9 through 20 from herd II, and samples 21, 22, and 23 from herds III, IV, and
V, respectively.

Advantages of PFGE
PFGE has proved to be an efficient method forgenome size estimation
In PFGE DNA fragments obtained by usingendonucleases produce a discrete pattern ofbands useful for the fingerprinting and physicalmapping of the chromosome.
The PFGE technique is useful to establish thedegree of relatedness among different strains ofthe same species.

Applications of PFGE
• PFGE is used for epidemiological studies of pathogenic organisms.
• PFGE is often employed to track pathogens, such as Salmonella, Shigella,
Escherichia coli (including O157), Campylobacter, and Listeria species
• PFGE has remarkable discriminatory power and reproducibility. It is
currently considered the strain typing method of choice for many
commonly encountered pathogens.
• PFGE has proven extremely powerful in the analysis of large DNA
molecules from a variety of sources including intact chromosomal DNAs
from fungi, parasitic protozoa and specifically fragmented genomes of
bacteria and mammal.

LIMITATIONS OF PFGE
• Time consuming (2-4 days)
• Requires a trained and skilled technician.
• Pattern results vary slightly between technicians.
• Don’t really know if bands of same size are same
pieces of DNA.
• Not applicable for all organisms.
• The choice of restriction enzyme may be important to
optimize the results

Conclusion
• The future of the molecular diagnostics of infectious
diseases will undoubtedly be focused on a marked
increase in the amount of information detected with
remarkably simplified, rapid platforms that will need
complex software analysis to resolve the data for use in
clinical decision-making.

THANK YOU