mouth dissolving tablets of...
TRANSCRIPT

Drug Invention Today ISSN: 0975-7619 Research Article
www.ditonline.info
Corresponding Author: Sonia Dhiman, Chitkara College of Pharmacy, Chitkara University, Rajpura 140401, Patiala, Punjab, India; e-mail: <[email protected]> Received 04-08-2012; Accepted 11-09-2012 September, 2012 Drug Invention Today, 2012, 4(9), 455-464 455
Mouth Dissolving Tablets of Antiallergic Drug - Design and Optimization Using Response Surface Methodology Sonia Dhiman*1 , Surender Verma2, S.K Singh3 1Chitkara College of Pharmacy, Chitkara University, Chandigarh-Patiala NH-64, Rajpura 140401, Patiala, Punjab. 2Institute of Pharmaceutical Sciences, Kurukshetra University, Kurukshetra 134116, Haryana, India. 3Department of Pharmaceutical Sciences, G.J.U.S&T, Hissar-125 001, India.
INTRODUCTION Recent advances in Novel Drug Delivery System (NDDS) aim to enhance safety and efficacy of drug molecule by formulating a convenient dosage form for administration and to achieve better patient compliance. One such approach is "Mouth Dissolving Tablet”. Mouth dissolving tablets can be defined as a solid oral dosage form which when placed upon tongue, disintegrates or dissolves rapidly usually within a minute, without the need of water or chewing. The aim of the present study was to develop a mouth dissolving tablet of an anti-allergic drug, Levocetirizine dihydrochloride with texture acceptable to patients and with sufficient structural integrity by using direct compression method. Levocetirizine dihydrochloride is pharmacologically active enantiomer of cetirizine hydrochloride, which is a racemic mixture of R-and S-cetirizine. It comes under second-generation piperazine derivative, potent H-1 selective antihistaminic or antiallergic drug classification, with an advantage of having fewer side effects. [1] As compare to cetirizine, levocetirizine dihydrochloride has a two fold higher affinity for human H-1 receptors and is believed to be rapidly and extensively absorbed. [2] Mannitol and Microcrystalline cellulose were incorporated as a diluent. Mannitol was used to improve palatability and to impart sweet taste, but mannitol-containing tablets does not rapidly dissolve in saliva because it is difficult for water to penetrate into the tablets due to its low porosity. Therefore, microcrystalline cellulose was also used along with mannitol, as it has a disintegrant effect. [3] Most pharmaceutical dosage forms for oral administration, such as tablets and hard gelatin capsules, are formulated to be swallowed or chewed. Elderly people and children sometimes have difficulties swallowing or chewing such dosage forms. This problem is more serious to patients who are bedridden[4]. This difficulty in swallowing or dysphasia is currently
affecting approximately 55% of the general population. [5] Drinking water plays an important role in the swallowing of oral dosage forms. Often times people experience inconvenience in swallowing conventional dosage forms such as tablet when water is not available, in the case of the motion sickness (kinetosis) and sudden episodes of coughing during the common cold, allergic condition and bronchitis. [6] These problems can be resolved by means of tablets that disintegrate rapidly in the mouth, known as mouth dissolving tablets. The disintegrated mass can slide down smoothly along the esophagus with the help of saliva, so even people who have swallowing or chewing difficulties can take it with ease. Here drug is absorbed from the mouth, pharynx and esophagus as the saliva passes down into the stomach which results in elimination of first pass metabolism. [7] Mouth dissolving drug delivery systems have started gaining popularity and acceptance as new drug delivery systems, because they are easy to administer and lead to better patient compliance. Mouth dissolving tablets are also useful in cases when local action in the mouth is desirable such as local anaesthetic for toothaches, oral ulcers, cold sores and to deliver sustained release multiparticulate system to those patients who are not able to swallow intact sustained action tablets. [8] Response surface methodology (RSM) is one of the popular methods in the development and optimization of drug delivery systems. Based on the principles of design of experiments (DOE), the methodology involves the use of various types of experimental designs, generation of polynomial mathematical relationships and mapping of the response over the experimental domain to select the optimum formulation. [9, 10,
11] Different types of RSM designs available for statistical optimization of the formulations are Central Composite
The objective of present study was to formulate and evaluate mouth dissolving tablets of Levocetirizine dihydrochloride using combination of two different superdisintegrants Crospovidone and Sodium Starch Glycolate and to optimize the disintegration time, wetting time, water absorption ratio and drug release profile (10 min) using response surface methodology. Methods: A central composite design for 2 factors at 3 levels each was employed to systematically optimize disintegration time, wetting time, water absorption ratio and drug release profile (10 min). A total number of thirteen formulations were prepared by the use of direct compression method as per the standard experimental design protocol. Crospovidone and Sodium Starch Glycolate were taken as the independent variables. Results: Response surface plots and contour plots were drawn, and optimum formulations were selected by feasibility and grid searches. Polynomial mathematical models, generated for various response variables using multiple linear regression analysis, were found to be statistically significant (P < .01). Conclusions: It was concluded that combination of superdisintegrants (Crospovidone and Sodium Starch Glycolate) could be successfully used for the preparation of mouth dissolving tablets of Levocetirizine dihydrochloride. Key words: Mouth Dissolving Tablets, Levocetirizine Dihydrochloride, Superdisintegrants, Disintegration time, Response Surface Methodology, Central Composite Design.

Dhiman et al.: Mouth Dissolving Tablets of Antiallergic Drug - Design and Optimization Using Response Surface Methodology
September, 2012 Drug Invention Today, 2012, 4(9), 455-464 456
Design (CCD), [12, 13] 3-level factorial design, Box Behnken design [13] and D-optimal design. [14] Central composite design (CCD) is a response surface which provides information on direct effects, pair wise interaction effects and curvilinear variable effects and is widely used for formulation and process optimization in the field of pharmaceutics. [15] Various types of CCD are Face centered central composite design, Rotatable central composite design, Circumscribed central composite design and Inscribed central composite design. A face centered composite design (FCCD) results when the both factorial and star points in a CCD possess the same positive and negative distance from the centre. [16] It is a very efficient and flexible method which provides complete understanding of experiment variable effects and overall percentage error within the least number of experimental runs. [2] Therefore, face-centered Central Composite Design was found to be a very suitable tool for process optimization of mouth dissolving tablets in this study. MATERIALS AND METHODS Levocetirizine Dihydrochloride obtained from Vardhman Pharmaceuticals (Paonta Sahib), Sodium Starch Glycolate was
obtained as gift sample from Maple Biotech Pvt. Ltd (Pune) and Crospovidone from Macleod Pharmaceuticals (Baddi). Sodium Saccharin, Microcrystalline Cellulose, Mannitol, Magnesium stearate and Talc used were of analytical reagent grade. Preparation of Mouth dissolving tablets Levocetirizine dihydrochloride mouth dissolving tablets were prepared using combination of two different superdisintegrants, crospovidone and sodium starch glycolate by direct compression method according to the formula shown in Table 1. By using the standard experimental design protocol, a total number of thirteen formulations were fabricated. All ingredients were weighed accurately and passed through 60-mesh sieve separately and collected. They were mixed thoroughly and finally compressed into tablets after lubrication with talc and magnesium stearate using 6 mm punch in Fluid Pack 8 station Mini Rotary tablet compression machine (Fluid Pack Machinery, Ahmedabad, India). All the preparations were stored in airtight containers at room temperature for further study. [Table 1]
Table 1: Composition of Mouth dissolving tablet of Levocetirizine Dihydrochloride
Ingredients (mg) Formulation Code F1 F2 F3 F4 F5 F6 F7 F8 F9 F10 F11 F12 F13 Levocetirizine dihydrochloride
5 5 5 5 5 5 5 5 5 5 5 5 5
Crospovidone 0 0 0 2 2 2 4 4 4 2 2 2 2 Sodium Starch Glycolate
4 5 6 4 5 6 4 5 6 5 5 5 5
Sodium saccharin 2 2 2 2 2 2 2 2 2 2 2 2 2 Mannitol 10 10 10 10 10 10 10 10 10 10 10 10 10 Microcrystalline Cellulose
126 125 124 124 123 122 122 121 120 123 123 123 123
Magnesium stearate
1 1 1 1 1 1 1 1 1 1 1 1 1
Talc 2 2 2 2 2 2 2 2 2 2 2 2 2 Total Weight 150 150 150 150 150 150 150 150 150 150 150 150 150 Table 2: Factor Combination as per the Chosen Experimental Design
Formulation Code Coded Factor Levels X1 X2
F1 -1 -1 F2 -1 0 F3 -1 +1 F4 0 -1 F5 0 0 F6 0 +1 F7 +1 -1 F8 +1 0 F9 +1 +1
F10 0 0 F11 0 0 F12 0 0 F13 0 0
Translation of coded levels in actual units Coded level -1 0 +1
X1 : Crospovidone (mg) 0 2 4 X2 : Sodium Starch Glycolate (mg)
4 5 6
Evaluation of Mouth Dissolving Tablets [2] All the batches of formulated Levocetirizine dihydrochloride mouth dissolving tablets were subjected to the following quality control tests:
Weight variation The weight variation test is carried out in order to ensure uniformity in the weight of tablets in a batch. The total weight of 20 tablets from each formulation was determined and the

Dhiman et al.: Mouth Dissolving Tablets of Antiallergic Drug - Design and Optimization Using Response Surface Methodology
September, 2012 Drug Invention Today, 2012, 4(9), 455-464 456
average was calculated. The individual weights of the tablets were also determined accurately and the weight variation was calculated. Thickness Thickness of tablets was determined using Vernier caliper. Three tablets from each batch were used, and an average value was calculated. The mean ± standard deviation values of thickness were calculated. Tablet Hardness Hardness of tablet is defined as the force applied across the diameter of the tablet in the order to break the tablet. The resistance of the tablet to chipping, abrasion or breakage under condition of storage transformation and handling before usage depends on its hardness. Hardness of the tablet of each formulation was determined using Pfizer hardness tester. Three tablets from each formulation batch were tested randomly and the average reading was noted. Friability Friability is measured of mechanical strength of tablets. Friability test is carried out to access the ability of the tablet to withstand abrasion during packaging, handling and transport. Roche friabilator was used to determine the friability by following procedure. A pre weighed 20 tablets were placed in the friabilator. Friabilator consist of a plastic-chamber that revolves at 25 rpm, dropping the tablets at a distance of 6 inches with each revolution. The tablets were rotated in the friabilator for 4 minutes for 100 revolutions. At the end of test, tablets were dusted and reweighed; the loss in the weight of tablet is the measure of friability and is expressed in percentage as; Initial wt. of tablets – Final wt. of tablets % Friability = ————————————————— x 100
Initial wt. of tablets Disintegration Time The test was carried out on 6 tablets using the disintegration apparatus. Phosphate buffer (pH 6.8) maintained at 37ºC ± 2ºC was used as a disintegration media and the time taken for complete disintegration of the tablet with no palpable mass remaining in the apparatus was measured (Figure 7). Wetting Time
A piece of tissue paper folded twice was placed in a small petri dish containing 6ml. of distilled water. A tablet was carefully placed on the surface of the paper and the time required for water to reach the upper surface of the tablet was noted as the wetting time. Less is the wetting time, indicates more porous the tablet. Water Absorption Ratio A piece of tissue paper folded twice was placed in a small petridish containing 6ml. of distilled water. A pre-weighed tablet was carefully placed on the surface of the paper and when the water reaches the upper surface of the tablet, the
wetted tablet was taken and weighed again. Water absorption ratio ‘R’ was determined using the equation, R = 100 (Wb-Wa) / Wa Where, Wa is weight of tablet before water absorption and Wb is weight of tablet after water absorption. Drug Content The drug content was determined by triturating 10 tablets; the powder equivalent to 5 mg of drug was accurately weighed and dissolved in 100 ml of phosphate buffer pH 6.8. The solution was filtered, suitably diluted and assayed for drug content, using UV Spectrophotometer at max 231 nm. In Vitro Drug Release Studies The dissolution rate was studied using USP dissolution apparatus II (Paddle type). The dissolution medium taken was 900 ml of phosphate buffer (pH 6.8), at a temperature of 37±1°C and at rotational speed of 50 rpm. From each formulation 3 tablets were tested. 10 ml of aliquots of samples were withdrawn at specific time intervals i.e. 2, 4, 6, 8, 10, 15, 20, 25, 30 min. and was filtered. UV Spectrophotometer determined the absorbance of the samples at max 231nm. Absorbance for the sample withdrawn was recorded and % drug release at different time intervals were plotted as cumulative percent drug release versus time (min) curve. Optimization by Experimental Design A central composite design (CCD) was employed using Design Expert Software (Version 7.1.6, Stat- Ease Inc, Minneapolis, MN) as per the standard protocol. The amount of Crospovidone (X1) and Sodium Starch Glycolate (X2) were selected as the factors, studied at 3 levels each. The central point (0, 0) was studied in quintuplicate. All other formulation and processing variables were kept invariant throughout the study. Table 2 summarizes an account of the 13 experimental runs studied, their factor combinations, and the translation of the coded levels to the experimental units employed during the study. Disintegration time, wetting time, water absorption ratio and % drug release (10 min) were taken as the response variables. The dependent and independent variables selected are also shown along with their low, medium and high levels, which were selected based on the results from preliminary experimentation. Polynomial models including interaction and quadratic terms were generated for all the response variables using multiple linear regression analysis (MLRA) approach. Statistical validation of the polynomial equation was established on the basis of ANOVA provision in the Design Expert Software. Various feasibility and grid searches were conducted to find the composition of optimum formulations. Also, the 3-D response surface graphs and 2-D contour plots were constructed using the output files generated. Numerical optimization using the desirability approach was employed to locate the optimal settings of the formulation variables to obtain the desired response. An optimized formulation was developed by setting constraints on the dependent and independent variables and the formulation developed was than evaluated for the various response properties. The resultant experimental values obtained were compared with those predicted by the mathematical models generated.
457
Dhiman et al.: Mouth Dissolving Tablets of Antiallergic Drug - Design and Optimization Using Response Surface Methodology
September, 2012 Drug Invention Today, 2012, 4(9), 455-464 456
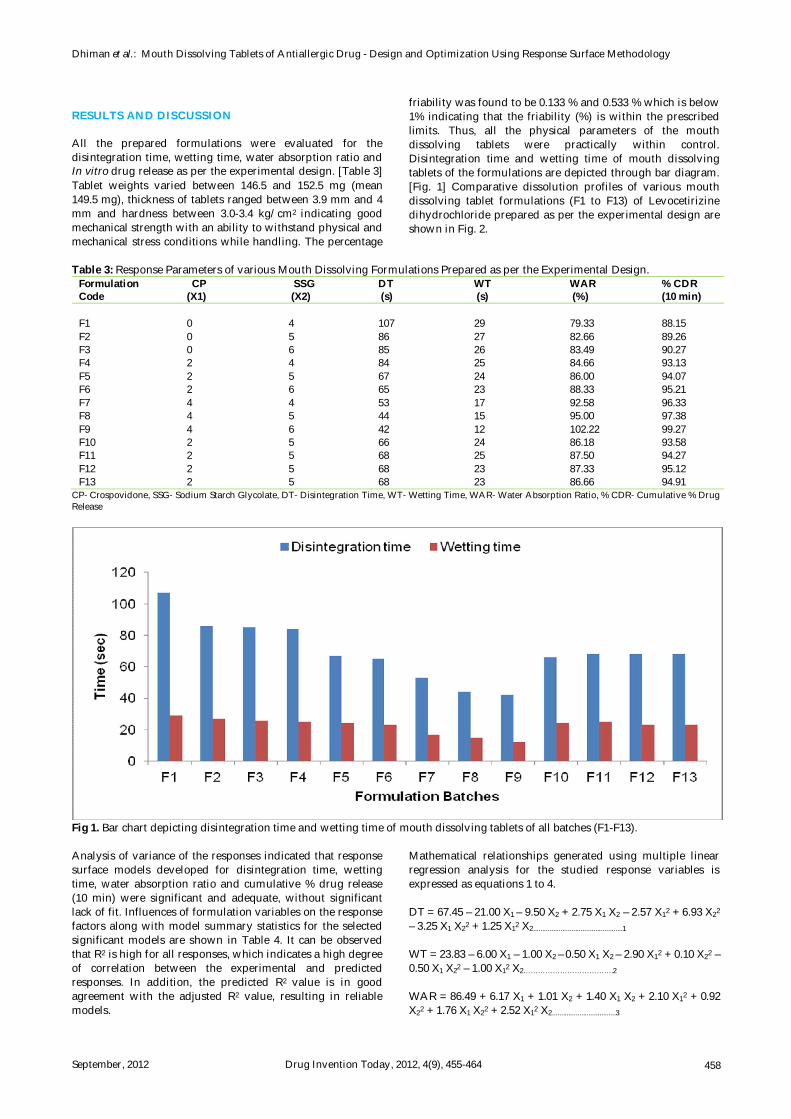
RESULTS AND DISCUSSION All the prepared formulations were evaluated for the disintegration time, wetting time, water absorption ratio and In vitro drug release as per the experimental design. [Table 3] Tablet weights varied between 146.5 and 152.5 mg (mean 149.5 mg), thickness of tablets ranged between 3.9 mm and 4 mm and hardness between 3.0-3.4 kg/cm2 indicating good mechanical strength with an ability to withstand physical and mechanical stress conditions while handling. The percentage
friability was found to be 0.133 % and 0.533 % which is below 1% indicating that the friability (%) is within the prescribed limits. Thus, all the physical parameters of the mouth dissolving tablets were practically within control. Disintegration time and wetting time of mouth dissolving tablets of the formulations are depicted through bar diagram. [Fig. 1] Comparative dissolution profiles of various mouth dissolving tablet formulations (F1 to F13) of Levocetirizine dihydrochloride prepared as per the experimental design are shown in Fig. 2.
Table 3: Response Parameters of various Mouth Dissolving Formulations Prepared as per the Experimental Design.
Formulation Code
CP (X1)
SSG (X2)
DT (s)
WT (s)
WAR (%)
% CDR (10 min)
F1
0
4
107
29
79.33
88.15
F2 0 5 86 27 82.66 89.26 F3 0 6 85 26 83.49 90.27 F4 2 4 84 25 84.66 93.13 F5 2 5 67 24 86.00 94.07 F6 2 6 65 23 88.33 95.21 F7 4 4 53 17 92.58 96.33 F8 4 5 44 15 95.00 97.38 F9 4 6 42 12 102.22 99.27 F10 2 5 66 24 86.18 93.58 F11 2 5 68 25 87.50 94.27 F12 2 5 68 23 87.33 95.12 F13 2 5 68 23 86.66 94.91
CP- Crospovidone, SSG- Sodium Starch Glycolate, DT- Disintegration Time, WT- Wetting Time, WAR- Water Absorption Ratio, % CDR- Cumulative % Drug Release
Fig 1. Bar chart depicting disintegration time and wetting time of mouth dissolving tablets of all batches (F1-F13).
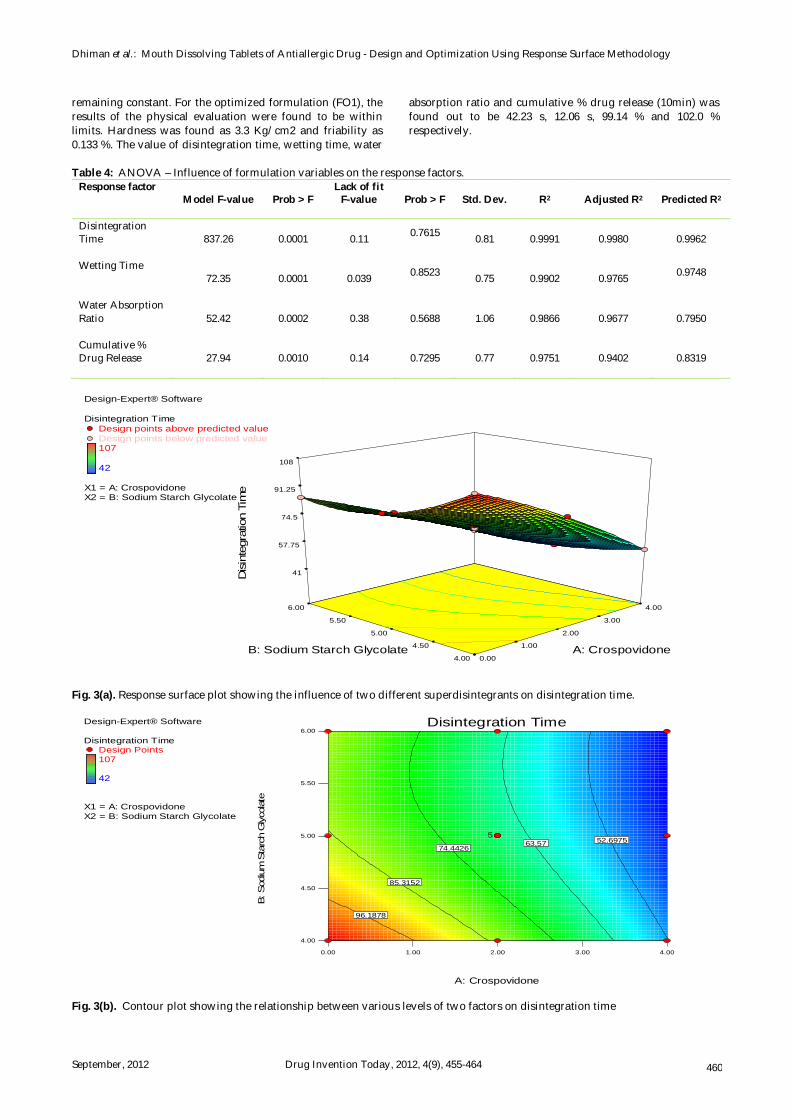
Analysis of variance of the responses indicated that response surface models developed for disintegration time, wetting time, water absorption ratio and cumulative % drug release (10 min) were significant and adequate, without significant lack of fit. Influences of formulation variables on the response factors along with model summary statistics for the selected significant models are shown in Table 4. It can be observed that R2 is high for all responses, which indicates a high degree of correlation between the experimental and predicted responses. In addition, the predicted R2 value is in good agreement with the adjusted R2 value, resulting in reliable models.
Mathematical relationships generated using multiple linear regression analysis for the studied response variables is expressed as equations 1 to 4. DT = 67.45 – 21.00 X1 – 9.50 X2 + 2.75 X1 X2 – 2.57 X12 + 6.93 X22 – 3.25 X1 X22 + 1.25 X12 X2.................................................1 WT = 23.83 – 6.00 X1 – 1.00 X2 – 0.50 X1 X2 – 2.90 X12 + 0.10 X22 – 0.50 X1 X22 – 1.00 X12 X2……………………………….2
WAR = 86.49 + 6.17 X1 + 1.01 X2 + 1.40 X1 X2 + 2.10 X12 + 0.92 X22 + 1.76 X1 X22 + 2.52 X12 X2...................................3
458

Dhiman et al.: Mouth Dissolving Tablets of Antiallergic Drug - Design and Optimization Using Response Surface Methodology
September, 2012 Drug Invention Today, 2012, 4(9), 455-464 456
% CDR = 94.29 + 4.06 X1 + 0.46 X2 + 0.21 X1 X2 –1.08 X12 + 0.35 X22 + 0.24 X1 X22 + 0.81 X12 X2........................................................................................4 All the polynomial equations were found to be statistically significant (P < 0.0002), as determined using ANOVA, as per the provision of Design Expert 7.1 Software. The values obtained for main effects of each factor in Equations 1 to 4 reveal that Crospovidone individually, has rather more pronounced effect on the values of disintegration time, wetting time, water absorption ratio and % CDR (10min) respectively. At a given set of factor levels, however, these higher-order polynomials yield results as the net effect of all the coefficient terms contained in the polynomial. In Equation 1 and 2, the results of multiple linear regression analysis showed that both the coefficients X1 and X2 bear a negative sign. Therefore, increasing the concentration of either Crospovidone or Sodium starch glycolate is expected to decrease the disintegration time and wetting time. However, the effect of Crospovidone seems to be more pronounced as compared with that of Sodium starch glycolate in cases, disintegration time and wetting time, as revealed by the response surface and the mathematical model. Equation 3 and 4 revealed that both main factors independently exerted a
significant positive influence on the Water absorption ratio and Cumulative % drug release respectively. Fig. 5 and Fig. 6 shows that the water absorption ratio and cumulative % drug release vary in somewhat linear fashion with increase in the amount of each superdisintegrant. The effect of increase in X1 seems to be more pronounced as compared with that of X2. The 3-dimensional response surface plots for the studied response properties viz., disintegration time, wetting time, water absorption ratio and cumulative % drug release (10 min) are shown in Fig. 3 to Fig. 6 respectively. To develop a optimum formulation with the desired responses, a numerical optimization technique using the desirability approach was employed. Selection was based on the constraints set at appropriate limits and importance like attaining minimum disintegration time and wetting time with high water absorption ratio and cumulative % drug release. Out of the suggested solutions by the software (Design Expert 7.1 software) the solution having highest desirability was selected. The solution showed the concentration of the optimum formulation as, Crospovidone, 4 mg, and Sodium Starch Glycolate, 6 mg and the values of response variables given by the software for this concentration was found out to be, disintegration time- 42.06 s, wetting time- 12.03 s, water absorption ratio-101.79 % and % cumulative drug release at 10min-99.10%.
Fig. 2. Comparative dissolution profiles of various mouth dissolving tablet formulations (F1 to F13) of Levocetirizine dihydrochloride prepared as per the experimental design.
So, for the optimum formulation, tablets were prepared by using 4 mg of Crospovidone and 6 mg of Sodium Starch
Glycolate, all other excipients were same (as shown in Table 1), the method of manufacturing and all other factor were
459
Dhiman et al.: Mouth Dissolving Tablets of Antiallergic Drug - Design and Optimization Using Response Surface Methodology
September, 2012 Drug Invention Today, 2012, 4(9), 455-464 456
remaining constant. For the optimized formulation (FO1), the results of the physical evaluation were found to be within limits. Hardness was found as 3.3 Kg/cm2 and friability as 0.133 %. The value of disintegration time, wetting time, water
absorption ratio and cumulative % drug release (10min) was found out to be 42.23 s, 12.06 s, 99.14 % and 102.0 % respectively.
Table 4: ANOVA – Influence of formulation variables on the response factors.
Response factor Model F-value Prob > F
Lack of fit F-value
Prob > F Std. Dev. R2 Adjusted R2 Predicted R2
Disintegration Time
837.26 0.0001 0.11 0.7615 0.81 0.9991 0.9980 0.9962
Wetting Time
72.35 0.0001 0.039 0.8523 0.75 0.9902 0.9765 0.9748
Water Absorption Ratio
52.42 0.0002 0.38 0.5688 1.06 0.9866 0.9677 0.7950
Cumulative % Drug Release
27.94 0.0010 0.14 0.7295 0.77 0.9751 0.9402 0.8319
Design-Expert® Software
Disintegration TimeDesign points above predicted valueDesign points below predicted value107
42
X1 = A: CrospovidoneX2 = B: Sodium Starch Glycolate
0.00
1.00
2.00
3.00
4.00
4.00
4.50
5.00
5.50
6.00
41
57.75
74.5
91.25
108
Disin
tegr
atio
n Ti
me
A: Crospovidone B: Sodium Starch Glycolate
Fig. 3(a). Response surface plot showing the influence of two different superdisintegrants on disintegration time.
Design-Expert® Software
Disintegration TimeDesign Points107
42
X1 = A: CrospovidoneX2 = B: Sodium Starch Glycolate
0.00 1.00 2.00 3.00 4.00
4.00
4.50
5.00
5.50
6.00Disintegration Time
A: Crospovidone
B: S
odiu
m S
tarc
h G
lyco
late
52.697563.5774.4426
85.3152
96.1878
5
Fig. 3(b). Contour plot showing the relationship between various levels of two factors on disintegration time
460

Dhiman et al.: Mouth Dissolving Tablets of Antiallergic Drug - Design and Optimization Using Response Surface Methodology
September, 2012 Drug Invention Today, 2012, 4(9), 455-464 456
Design-Expert® Software
Wetting TimeDesign points above predicted valueDesign points below predicted value29
12
X1 = A: CrospovidoneX2 = B: Sodium Starch Glycolate
0.00
1.00
2.00
3.00
4.00
4.00
4.50
5.00
5.50
6.00
12
16.5
21
25.5
30
Wet
ting
Tim
e
A: Crospovidone B: Sodium Starch Glycolate
Fig. 4(a). Response surface plot showing the influence of two different superdisintegrants on wetting time.
Design-Expert® Software
Wetting TimeDesign Points29
12
X1 = A: CrospovidoneX2 = B: Sodium Starch Glycolate
0.00 1.00 2.00 3.00 4.00
4.00
4.50
5.00
5.50
6.00Wetting Time
A: Crospovidone
B: S
odium
Sta
rch
Glyco
late
14.8678
17.701120.534523.367826.2011 5
Fig. 4(b). Contour plot showing the relationship between various levels of two factors on wetting time.
Design-Expert® Software
Water Absorption RatioDesign points above predicted valueDesign points below predicted value102.24
79.33
X1 = A: CrospovidoneX2 = B: Sodium Starch Glycolate
0.00
1.00
2.00
3.00
4.00
4.00
4.50
5.00
5.50
6.00
78
84.25
90.5
96.75
103
Wat
er A
bsor
ptio
n R
atio
A: Crospovidone B: Sodium Starch Glycolate
Fig. 5(a). Response surface plot showing the influence of two different super disintegrants on water absorption ratio.
461

Dhiman et al.: Mouth Dissolving Tablets of Antiallergic Drug - Design and Optimization Using Response Surface Methodology
September, 2012 Drug Invention Today, 2012, 4(9), 455-464 456
Design-Expert® Software
Water Absorption RatioDesign Points102.24
79.33
X1 = A: CrospovidoneX2 = B: Sodium Starch Glycolate
0.00 1.00 2.00 3.00 4.00
4.00
4.50
5.00
5.50
6.00Water Absorption Ratio
A: Crospovidone
B: S
odium
Sta
rch
Glyco
late
83.270587.0889
90.9072
94.7255
98.5439
5
Fig. 5(b). Contour plot showing the relationship between various levels of two factors on water absorption ratio.
Design-Expert® Software
% CDRDesign points above predicted valueDesign points below predicted value99.27
88.15
X1 = A: CrospovidoneX2 = B: Sodium Starch Glycolate
0.00
1.00
2.00
3.00
4.00
4.00
4.50
5.00
5.50
6.00
87
90.25
93.5
96.75
100
% C
DR
A: Crospovidone B: Sodium Starch Glycolate
Fig. 6(a). Response surface plot showing the influence of two different superdisintegrants on cumulative % drug release (10 min).
Design-Expert® Software
% CDRDesign Points99.27
88.15
X1 = A: CrospovidoneX2 = B: Sodium Starch Glycolate
0.00 1.00 2.00 3.00 4.00
4.00
4.50
5.00
5.50
6.00% CDR
A: Crospovidone
B: S
odiu
m S
tarc
h G
lyco
late
90.0583 91.911793.765
95.6183
97.4717
5
Fig. 6(b). Contour plot showing the relationship between various levels of two factors on cumulative % drug release (10 min)
462

Dhiman et al.: Mouth Dissolving Tablets of Antiallergic Drug - Design and Optimization Using Response Surface Methodology
September, 2012 Drug Invention Today, 2012, 4(9), 455-464 456
Fig. 7. Diagram showing various steps involved in the disintegration of the tablet of the optimized batch.
For the validation of results, percentage error was found out. Upon comparison of the observed responses with that of the anticipated responses, the prediction error varied between -2.098 % and 0.404 %. The closeness of the predicted and observed values indicates validity of derived equations for the dependent variables. The low values of the prediction error for the response parameters establish the high forecasting ability of response surface methodology. In pharmaceutical practice development and design of an optimized drug delivery products normally encompasses various goals and steps. From the past times, this task has been attempted through number of trial and error, supplemented with the previous experience, knowledge, and wisdom of the formulator. Optimization of a various pharmaceutical formulations or processes using this traditional approach involves changing one variable at a time. Using this methodology, the solution of a specific problematic formulation characteristic and development can certainly be achieved, but attainment of the true optimal composition is never achieved and for improvement in one characteristic, one has to trade off for degeneration in another. This ancient approach of development of a drug product or process has been proved to be not only uneconomical in terms of time, money and effort, but also unfavorable to fix errors, unpredictable and at times, even unsuccessful. On the other hand, the modern optimization approaches like systematic Design of Experiments (DOE) viz. central composite design, are extensively used in the development of various kinds of drug delivery devices to improve such irregularities. Such modern systematic approaches are far more advantageous as compare to traditional techniques, because they require few experiments to achieve an optimum formulation, make problem tracing and rectification quite easier, reveal drug/polymer interactions, simulate the product performance, and comprehend the process to assist in better formulation development and subsequent scale-up. Optimization techniques using DOE represent accurate and cost-effective analytical tools to yield the "better solution" to a particular "problem״ with an ease. Based on the results of DOE using central composite design, it was concluded that combination of superdisintegrants (Crospovidone and
Sodium Starch Glycolate) could be successfully used for the preparation of mouth dissolving tablets of Levocetirizine dihydrochloride. Superdisintegrants used in the concentration of 4mg and 6 mg for Crospovidone and Sodium Starch Glycolate respectively, proved to have less disintegration time and good dissolution profile. High degree of prognosis obtained using RSM corroborates that a 2-factor CCD is quite efficient in optimizing drug delivery systems that exhibit nonlinearity in response(s).
ACKNOWLEDGEMENT The authors are grateful to Dr. Madhu Chitkara, Vice-Chencellor, Chitkara University, Rajpura, Patiala, Punjab, India and Dr. Ashok Chitkara, Chairman, Chitkara Educational Trust, Chandigarh, India, for support and institutional facilities.
REFERENCES
1. Chaudhari GN, KhachaneVS. DeshmukhVP. Bhamre NB. Taste masking pharmaceutical composition containing levocetirizine. US Patent 20060083786, (2004).
2. Dhiman S, Verma S, Singh SK. Design, development, and optimization of mouth dissolving tablets of levocetirizine dihydrochloride using central composite design. Acta Pharmaceutica Sciencia, 2011; 53:363-375.
3. Koizumi KI, Watanabe Y, Morita K, Utoguchi N, Matsumoto M. New method for preparing high porosity rapidly saliva soluble compressed tablets using mannitol with camphor, a subliming material. Int J Pharm. 1997, 152:127–131.
4. Bagul US, Bagul NS, Kulkarni MS, Sawant SD, Gujar KN, Bidkar AA. Manufacturing Technologies for Mouth Dissolving Tablets. Pharmaceutical Reviews, 2006, 4(3).
5. Reddy LH, Ghosh BR. Fast dissolving drug delivery systems: A review of the literature. Ind J Pharm Sci, 2002, 64(4): 331-336.
6. Hirani JJ, Rathod DA, Vadalia KR. Orally disintegrating tablets: A review. Tropical J Pharma Research, 2009, 8 (2): 161-172.
7. Khan S., Kataria P. Taste masking of Ondansetron Hydrochloride by polymer carrier system and formulation of rapid disintegrating tablets. AAPS PharmSciTech, 2007, 8 (2): E1-E7.
463

Dhiman et al.: Mouth Dissolving Tablets of Antiallergic Drug - Design and Optimization Using Response Surface Methodology
September, 2012 Drug Invention Today, 2012, 4(9), 455-464 456
8. Sammour OA, Hammad MA, Megrab NA, Zidan AS. Formulation and optimization of mouth dissolve tablets containing rofecoxib solid dispersion. AAPS Pharm. Sci. Tech. 2006, 7(2): E48-55.
9. Sánchez-Lafuente C, Furlanetto S, Fernández-Arévalo M, Alvarez-Fuentes J, Rabasco A.M., Faucci M.T., Pinzauti S., Mura P. Didanosine extended-release matrix tablets: optimization of formulation variables using statistical experimental design.Int. J. Pharm. 2002, 237(1-2):107-18
10. Singh B, Chakkal SK, Ahuja N. Formulation and optimization of controlled release mucoadhesive tablets of atenolol using response surface methodology. AAPS Pharm. Sci. Tech,2006, 7(1): E1-3.
11. Singh B, Mehta G, Kumar R, Bhatia A, Ahuja N. Katare OP. Design, development and optimization of nimesulide-loaded liposomal systems for topical application. Curr Drug Drug Deliv. 2005, 2(2):143-53.
12. Box GEP, Wilson KB. On the experimental attainment of optimum conditions. J. Roy. Stat. Soc. B, 1951, 13:1-38.
13. Boza A, Cruz Y, Jordan G, Jauregui-Haza U, Aleman A, Caraballo I. Statistical optimization of a sustained-release matrix tablet of lobenzarit disodium. Drug Dev Ind Pharm. 2000, 26:1303–1307.
14. Chopra S, Patil V, Motwani SK. Release modulating hydrophilic matrix systems of losartan potassium: Optimisation of formulation using statistical experimental design, Eur. J. Pharm. Biopharm. 2007, 66: 73–82.
15. Krogars K, Heinamaki J, Vesalahti J, Marvola M, Antikainen O, Yliruusi J. Extrusion-spheronization of pH-sensitive polymeric matrix pellets for possible colonic drug delivery. Int. J. Pharm, 2000, 199(2):187-94.
16. Doornbos DA, Haan P. Optimization Techniques in formulation and Processing. In: Swarbrick J, Boylan JC, editors. Encyclopedia of Pharmaceutical technology. Vol. 11. New York: Marcel Dekker; 1995.p.77-160.
Source of support: Nil, Conflict of interest: None Declared.
464