multiple sequence alignment jarno tuimala. scoring matrices
TRANSCRIPT

Multiple sequence alignment
Jarno Tuimala

Scoring matrices

Uses of matrices• Sequence alignment• Database searches• Phylogenetics
Distances between sequencesAs evolutionary models
• For amino acids: PAM, Blosum, JTT…• For DNA: IUB… (match 1.9, mismatch 0)
• For evolutionary work, matrices are replaced by mathematical models, while working with DNA sequence data
AdeniiniAdeniini GuaniiniGuaniini
SytosiiniSytosiini TymiiniTymiiniMuu
nnet
tu k
uvis
ta: h
ttp://
ww
w.b
igch
alk.
com
/cgi
-bin
/Web
Obj
ects
/WO
Port
al.w
oa/w
a/H
WC
DA
/file
?file
id=
1837
3&fl
t=ga

An example of a DNA matrix
•For local alignments with this matrix, gap opening -16 and extension of -4 are typically used.

Sequence alignment

How to align sequences
• On paper / with computer– Description of alignment for computer:
• scoring matrix• gap penalties
• Aligning is not objective – Check the results computer gives you!
• Alignments can be used for – searching conserved sequence areas– searching point mutations– studying evolution of genes and species

Gap penalties• Gap are evolutionarily expensive.
– Opening is more costly than extension– Affine gap model
• Mathematically– P = c + gd– P is the total gap penalty– c is gap opening penalty– d is extension penalty– g is the (lenght of the gap - 1)
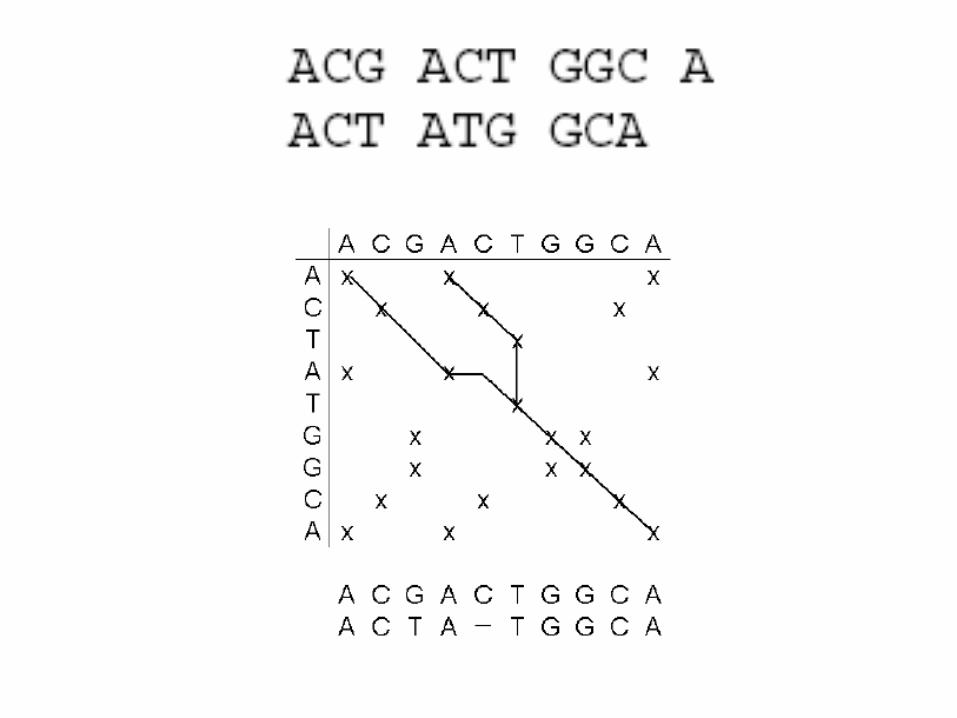

How to calculate an alignment score?
• match: +4
• mismatch: -5
• gap opening: -16
• gap extension: -4
• 4+4+(-4)+4+(-16)+4+4+4+4+4 = 12

Multiple sequence alignment(MSA)

What is MSA?• MSA is an alignment generated from
three or more sequences.
• MSA is usually a global alignment, i.e., the aim is to align homologous residues (nucleotides or amino acids) in columns across the length of the whole sequences.
A--GT
AC-GT
ACGGT
-CGGT

Alignability of sequences• If the similarity of sequences drops too low,
sequences can’t be reliably aligned (accuracy drops below acceptable).– For proteins <20% similarity– For DNA <~75% similarity
• This cut-off is called twilight zone.
• In other words, twilight zone marks the sequence similarity below which the observed similarity is mainly due to random variation, and not due to evolution.

MSA and dynamic programming
• There are methods that can produce the optimal alignment (in terms of gap penalties and scoring matrices), but they are computationally very heavy.– Program MSA uses dynamic programming
• In practise, dynamic programming would be good for up to about 10 sequences, and is not usually used for MSA.– But for pairwise alignment it can be used.

MSA methods
• There are two popular methods to perform a multiple sequence alignment:– Progressive alignment
• Clustal (ClustalW and ClustalX), Pileup…• Clustal is the most commonly used alignment
program
– Iterative alignment• SAGA…
• We will review the Pileup method first

Progressive alignment

Progressive alignment
• Produce pairwise alignment between all the sequences you want to align with MSA.– Dynamic programming, ktup-methods, dot matrix
method…(you choose it)
• Produce a “guide tree” on the basis of the pairwise distances calculated from pairwise alignments.– UPGMA, neighbor joining (you choose it)
• Produce an MSA using the guide tree.– Sequences are aligned in the same order as the
guide tree instructs.
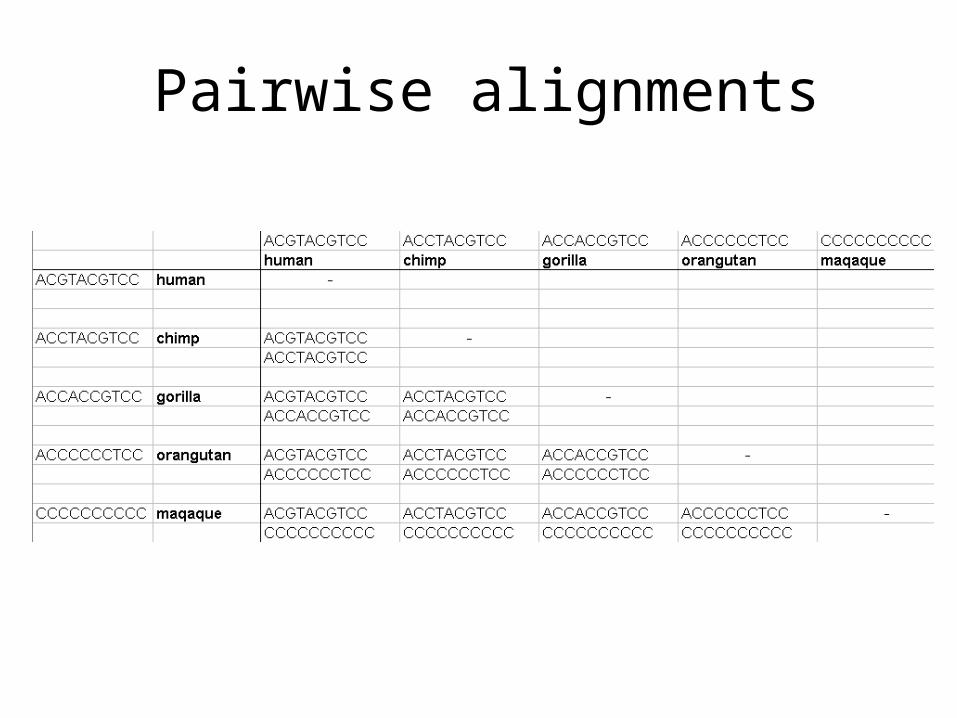
Pairwise alignments

Pairwise distances
No. of nucl. diffs.
Absolute distance, used in Pileup/ Clustal
JC-distance

UPGMA
• Unweighted Pair Group Method with Arithmetic mean
• One of the fastest and tree construction methods
• Used in Pileup (GCG package)
• Clustal uses neighbor joining, but calculating NJ tree is much more demanding; thus, UPGMA is demonstrated here
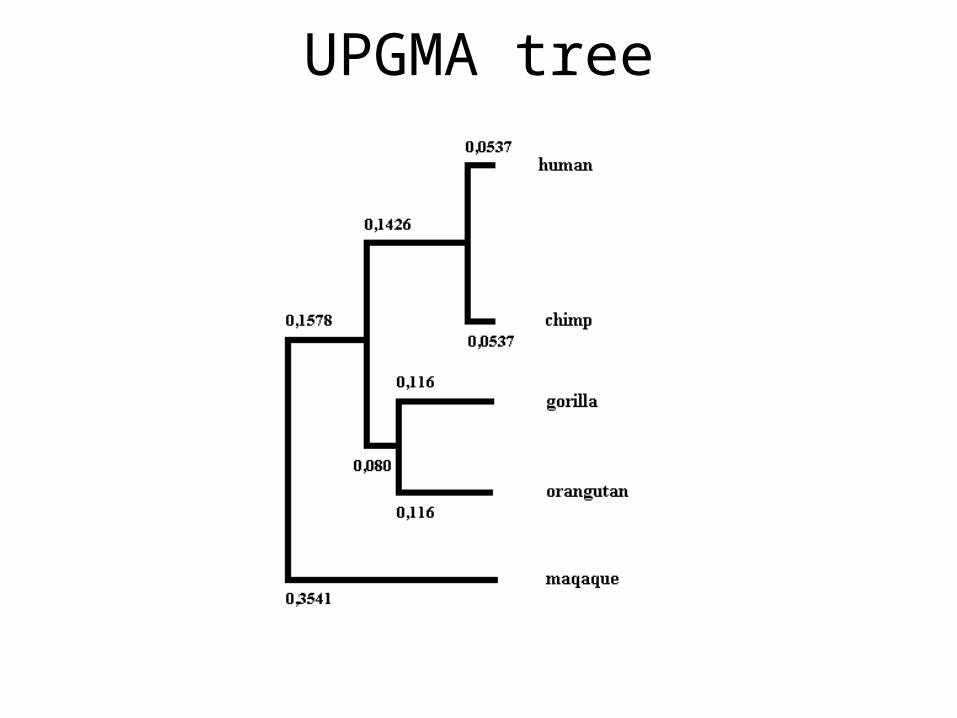
UPGMA tree

Constructing MSA
human ACGTACGTCCchimp ACCTACGTCCgorilla ACCACCGTCCorangutan ACCCCCCTCCmaqaque CCCCCCCCCC
human ACGTACGTCCchimp ACCTACGTCC
gorilla ACCACCGTCCorangutan ACCCCCCTCC
human ACGTACGTCC
chimp ACCTACGTCC
gorilla ACCACCGTCC
orangutan ACCCCCCTCC

Score of alignment• 1234• ACGT match=1• ACGA mismatch=0• AGGA
• 1: A-A + A-A + A-A = 1+1+1 = 3• 2: C-C + C-G + C-G =1+0+0 = 1• 3: G-G + G-G + G-G = 1+1+1 = 3• 4: T-A + T-A + A-A = 0+0+1 =1
• S(alignment) = S(1) + S(2) + S(3) + S(4) = 3+1+3+1 = 8• The higher the score, the better the alignment

Progressive alignment - pros and cons
• Pros– Fast– Quite accurate
• Cons– Once gaps are opened they can never be
closed• Errors in the alignment of the first few sequences
can have catastrophic effects on the whole alignment

Muscle – both progressive and iterative
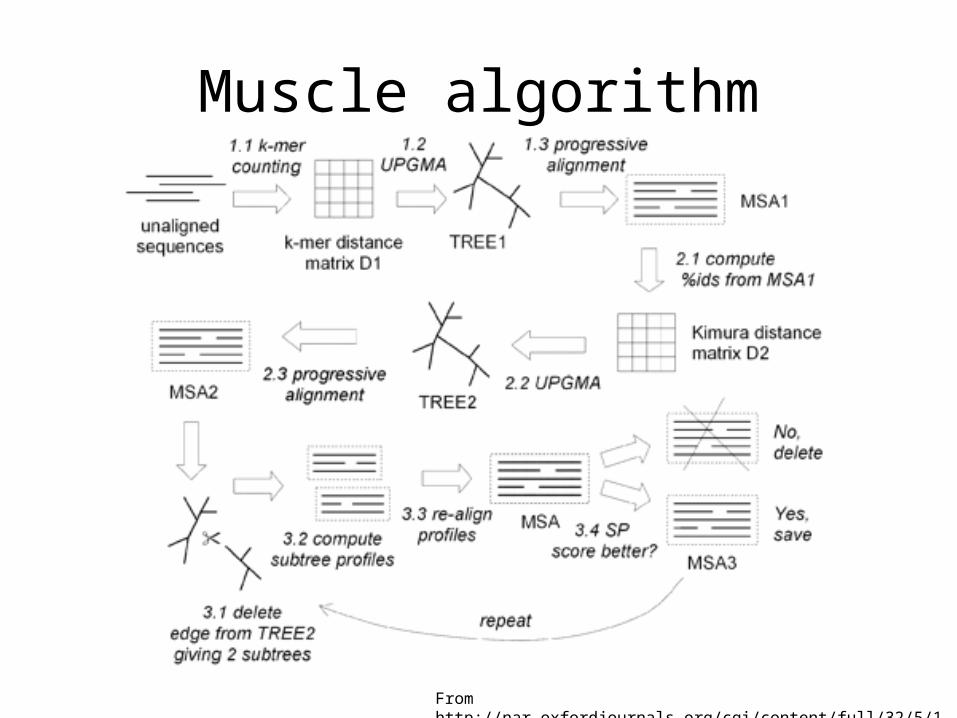
Muscle algorithm
From http://nar.oxfordjournals.org/cgi/content/full/32/5/1792/GKH340F2

Muscle – comparison results
• As fast as Clustal, but at the same time:
• As accurate as T-COFFEE!– T-COFFEE was previously the most accurate
alignment method (or software) available