multisite aggregation of p53 and implications for drug … · multisite aggregation of p53 and...
TRANSCRIPT

Multisite aggregation of p53 and implications fordrug rescueGuoZhen Wanga and Alan R. Fershta,1
aMRC Laboratory of Molecular Biology, PNAC Division, Cambridge CB2 0QH, United Kingdom
Contributed by Alan R. Fersht, February 6, 2017 (sent for review January 9, 2017; reviewed by David P. Lane and Karen H. Vousden)
Protein aggregation is involved in many diseases. Often, a uniqueaggregation-prone sequence polymerizes to form regular fibrils. Manyoncogenic mutants of the tumor suppressor p53 rapidly aggregate butform amorphous fibrils. A peptide surrounding Ile254 is proposed tobe the aggregation-driving sequence in cells. We identified severaldifferent aggregating sites from limited proteolysis of harvestedaggregates and effects of mutations on kinetics and products ofaggregation. We present a model whereby the amorphous nature ofthe aggregates results from multisite branching of polymerizationafter slow unfolding of the protein, which may be a common featureof aggregation of large proteins. Greatly lowering the aggrega-tion propensity of any one single site, including the site of Ile254,by mutation did not inhibit aggregation in vitro because aggrega-tion could still occur via the other sites. Inhibition of an individual siteis, accordingly, potentially unable to prevent aggregation in vivo.However, cancer cells are specifically killed by peptides designed toinhibit the Ile254 sequence and further aggregation-driving se-quences thatwe have found. Consistent with our proposedmechanismof aggregation, we found that such peptides did not inhibit aggrega-tion of mutant p53 in vitro. The cytotoxicity was not eliminated byknockdown of p53 in 2D cancer cell cultures. The peptides causedrapid cell death, much faster than usually expected for p53-mediatedtranscription-dependent apoptosis. There may also be non-p53 tar-gets for those peptides in cancer cells, such as p63, or the peptidesmay alter other interactions of partly denatured p53 with receptors.
amyloid | mechanism | misfolding | disease
The tumor suppressor p53 is inactivated by mutation in a sub-stantial number of tumors (1–3). Some 30–40% of those on-
cogenic mutants are simply destabilized by mutations in its coredomain. Those mutants are temperature-sensitive, having a WTstructure at lower temperatures, but melt at close to body tem-perature or below and rapidly aggregate (4–6). Protein aggrega-tion occurs in many diseases (7–9). The best mechanisticallycharacterized examples involve the polymerization of aggregation-prone peptides (10, 11) or small proteins to give well-defined fi-brils based on a regular repeat structure (12–18). The fibrillaraggregates have a characteristic cross-β X-ray diffraction patternand bind such dyes as Congo Red and Thioflavine T (ThT) (16).The kinetics of aggregation usually follow a nucleation-growthmechanism, with very slow nucleation (19–21). WT p53 itself ag-gregates at body temperature (4–6, 22–24), and the oncogenicdestabilized mutants aggregate even faster to give amorphousstructures that display the characteristic diffraction pattern andbind those diagnostic dyes (23, 25), although under certain con-ditions, such as very high pressure, they will generate regular fibrils(23, 26). The mechanism of initiation of aggregation of p53 differsfrom the usually studied examples. Two molecules of the coredomain of p53 extensively unfold (27) and then combine, on amuch shorter time scale than in classical diseases of fibril forma-tion. Mutant and WT protein form mixed aggregates in a cross-reaction and coaggregation process rather than by seeding byalready aggregated molecules because the initiation of aggregationis relatively fast even in WT protein (27, 28).The core domain of p53 is built around a β-sandwich, com-
prising two antiparallel β-sheets of four strands (S1, S3, S8, and
S5) and five strands (S10, S9, S4, S7, and S6) that pack againsteach other across an extended hydrophobic core (29). Theβ-sandwich serves as a scaffold for formation of a DNA-bindinginterface by two large loops, L2 and L3, and a loop–sheet–helixmotif, containing loop L1 (113–123), a short β-sheet composedof β-hairpin S2–S2′ and the C-terminal residues of the extendedstrand S10, and α-helix H2 (278–289) (30, 31). Xu et al. (32) haveproposed residues 251–257 (ILTIITL) in β-strand S9 to be thekey aggregation-prone sequence. The mutation I254R is reportedto decrease greatly the aggregation propensity of the isolatedpeptide in model studies and to inhibit heterologous aggrega-tion-promoting activities of p53 (32). In contrast, Vousden andcoworkers (33) find the 251–257 region of p53 is not required forp63 or p73 binding in cancer cells. Neither the I254R mutation inWT p53 nor the deletion of 251–312 or 251–257 in p53R175Hinhibits their binding to p63 or p73, and even p53I254R itself canpromote invasion in a p53-null cancer cell line.The inhibition of aggregation of p53 may be a route for rescuing
mutant p53 in cancer cells (5). We initially tackled the rescue bystabilizing the folded conformation against unfolding, first by a ge-neric approach using a peptide that binds to native p53 (34) andmutants (35) and then by targeting a specific mutant where themutation forms a druggable cavity, Y220C. Its melting temperature israised, and its aggregation is slowed in vitro by small mutant-specificmolecules that bind in the cavity (36, 37) and rescue Y220C in cancercell lines (25, 38, 39). Eisenberg and coworkers (40) have taken adifferent approach of inhibiting the aggregation process, per se, witha peptide that caps the specific exposed aggregation-prone sequence,centered on Ile254, rather than stabilizing the native structure.A ϕ-value analysis reveals that many of the β-strands in the
cores of two associating molecules of the core domain are sub-stantially unfolded in the initiation of aggregation (27). Wesuspected that more than one of those strands could be involvedin pairing during aggregation. We searched the whole p53 coredomain sequence for additional aggregation-prone sites by bothcomputer algorithms and limited proteolysis of the harvested
Significance
Destabilized mutants of the tumor suppressor p53 are inacti-vated by self-aggregation in a substantial number of tumorsand may also coaggregate with and inactivate WT p53 andfamily members. We found in vitro that self-aggregation pro-ceeded via a network of multiple aggregation-prone sites inp53, and inhibition of an individual site did not inhibit aggre-gation. Nevertheless, peptides designed to be complementaryto various aggregation sequences and inhibit their polymeri-zation can specifically kill cancer cells and be potential anti-cancer drugs. We found that those peptides can also functionby p53-independent routes in cancer cell cultures, implyingfurther therapeutic targets.
Author contributions: G.W. and A.R.F. designed research; G.W. performed research; G.W.and A.R.F. analyzed data; and G.W. and A.R.F. wrote the paper.
Reviewers: D.P.L., A*Star; and K.H.V., CRUK Beatson Institute.
The authors declare no conflict of interest.1To whom correspondence should be addressed. Email: [email protected].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1700308114/-/DCSupplemental.
E2634–E2643 | PNAS | Published online March 14, 2017 www.pnas.org/cgi/doi/10.1073/pnas.1700308114

aggregate to identify regions that are buried in the aggregate. Wemutated those sites to inhibit their aggregation tendency andmeasured the kinetics of aggregation. Knocking out by mutationthe aggregation propensity of Ile254 and other individual sites didnot prevent aggregation from the native state in vitro. The initialaggregate did not elongate by forming regular repeating of aunique peptide as found in the most frequently studied examples,where well-ordered fibrils are formed, but a variety of alternativeheterologous or homologous pairings of β-strands were involved.
ResultsSequences of p53 Involved in Aggregation.WT p53 core domain. We used limited proteolysis of harvestedaggregates from WT p53 core domain (WTC) to analyze whichparts of the structure are resistant to proteases after aggregationfrom the native state at 37 °C in vitro. The large clumps of WTCaggregate were highly protease-resistant. Treatment of nativesoluble WTC by proteinase K for 9 h gave only small peptides(Fig. S1A). In contrast, similar proteolysis of the aggregateharvested after incubation of WTC overnight at 37 °C still gaveresidual full-length protein and peptides larger than 5 kDa(Fig. S1B).To reveal the sequences protected in the initial stages of aggre-
gation, we harvested the aggregate after 2 h of incubation at 37 °C,which is sufficient time for near-maximal binding of ThT but earlyin the maturation of the aggregate to large clumps. This aggregatewas more readily proteolyzed than the aggregate from overnightincubation. After 4 h of digestion with 1:50 (wt/wt) p53/proteinaseK, only a small fraction of the aggregate remained. Mass spec-troscopy (MS) revealed that the main peaks lower than 3 kDa in theremaining aggregate were 108–114 (S1L1), 137–146 (S3), 146–160(S4), 126–136 (S2S2′), and 248–257 (S9).We then analyzed in more detail a time course of tryptic digestion
of the aggregate formed after 2 h of incubation at 37 °C (identicalresults were found after 1-h and 3-h incubations). After digestion ofthe aggregate for defined times, we separated the remaining aggre-gate from the supernatant and analyzed both phases by MS. Peptides249–267 (2,069 Da, S9), 214–248 (3,880 Da, S7S8), 182–196 (1,609Da, H1S5), 193–213 (2,097 Da), and 203–213 (1,428 Da) were notpresent in the supernatant after 24 h of digestion with 1:100 (wt/wt)trypsin (Fig. 1A), although they were present in the main peaks in thedigested native soluble WTC (Fig. 1B). Only after a further 24 h ofdigestion by 1:50 (wt/wt) trypsin did 2,069 Da, 1,609 Da, and 3,880Da become three of the main peaks released into the supernatant(Fig. 1C). The cleavage pattern indicated that those peptides wereburied in the aggregate or formed relatively inflexible structures that
were initially inaccessible to trypsin. Thus, S5, S7, S8, and S9 were allinvolved in the aggregate of WTC.After 48 h of digestion, the peptides that were much more
abundant in the remaining aggregate (Fig. 1D) than in the super-natant (Fig. 1C) were 249–267 (2,069 Da, S9), 102–120 (2,090 Da,S1), 182–209 (3,169 Da, S5S6), 182–213 (3,688 Da, S5S6), 176–202(3,037 Da, H1S5), 102–132 (3,356 Da, S1L1S2), and 92–132 (4,301Da, S1L1S2). There were far fewer accessible tryptic cleavage sitesin the WTC aggregate (Fig. 1D) than in the native protein (Fig. 1B),and most of the inaccessible cleavage sites (Fig. 1D) resided in thepeptides in the insoluble aggregate. The much larger peak of S9 inthe remaining aggregate was consistent with a previous finding (32)that this peptide is a major site for aggregation.Peptides 249–267 (2,069 Da, S9) plus peptides 214–248 (3,880
Da, S7S8), 102–110 (1,079 Da, S1), 140–156 (1,855 Da, S3), 182–196 (1,609 Da, S5), and 165–174 (1,215 Da, L2) were present inboth remaining aggregate and supernatant after 48 h of di-gestion. Their presence in both phases was not due to poorsolubility because digestion of the same concentration of nativeWTC did not result in any aggregate during the digestion processand the intensity of their corresponding MS peaks in the su-pernatant increased with digestion. The presence in both phaseswas not caused by the aggregate dissolving and being digested inthe supernatant: More than 97% of the aggregate remained in-soluble after 48 h of incubation without protease.The most likely reason for the presence of those peptides in
the residual core structure and being released into solution isthat the aggregate is heterogeneous: In some part of the aggre-gate, a particular sequence is highly buried, whereas it is onlyloosely buried in other parts. Further, there were multiple ag-gregation sites in addition to the aggregation site of S9.WTCI254D. The mutation I254D in the middle of the S9 aggre-gation-prone sequence greatly reduces its intrinsic propensity toaggregate. However, the mutated protein still forms an amor-phous aggregate from the native state at 37 °C (Fig. S2) andrapidly forms a ThT-binding state and scattering particles (Fig.2). The harvested aggregate from I254D was also protease-resistant. After 6 h of digestion by trypsin, most of the nativesoluble I254D was digested to peptides of Mr lower than about2 kDa, except for peptide 214–248 (3,880 Da), which does not con-tain a tryptic cleavage site. In contrast, there was only partial digestionof the aggregate of I254D after 2 h of incubation at 37 °C, and thepeptides in the remaining aggregate were mainly of Mr greaterthan 2 kDa. Major peaks that were enriched in the remainingaggregate relative to the supernatant were similar to those peaksfrom the digestion of WTC aggregate, with common peaks inthe MS peaks in regions of 182–213 (3,688 Da, 3,169 Da; S5S6),
0
20
40
60
80
100
1000 2000 3000 4000 5000
38802071
1855
1079
1609
2097
(249-267)
0
20
40
60
80
100
1000 2000 3000 4000 5000
% In
tens
ity
1855
1079
20241371
1215 3037 0
20
40
60
80
100
1000 2000 3000 4000 5000
10791609
18552069
3880
1215
1371
0
20
40
60
80
100
1000 2000 3000 4000 5000
1079 1855
2069
3880
160920971428
13711215
2024
(214-248)
(182-196)
(197-213)
(249-267)
m/z
% In
tens
ity
0
20
40
60
80
100
1000 2000 3000 4000 5000m/z
2069
3880
3169
1855
16091079
1215 3688335630374301
2090
(249-267)
(182-209)(182-213)
EA
B D
0
20
40
60
80
100
1000 2000 3000 4000 5000m/z
3688
2090 3037
33563169 4301388018551079
2677 39832809171014383064
(102-120)
(182-213)
(268-282)
(176-202)
(102-132)
C
F
Fig. 1. Limited proteolysis of native WTC, WTC aggregate, native WTCI254D,and WTCI254D aggregate by trypsin. Mass spectra of supernatant of WTCaggregate after the initial 24-h digestion (A), native WTC after the initial 24-hdigestion (B), supernatant of WTC aggregate after the second 24 h of di-gestion (C), and remaining aggregate of WTC aggregates after second 24 h ofdigestion (D). WTC aggregate was from a 2-h incubation at 37 °C. NativeWTCI254D (E) and the remaining aggregate of WTCI254D aggregate (F) aftera 6-h digestion.
0
20
40
60
80
100
0 50 100 150 200 250 300
4 µM6 µM8 µM10 µM
ThT
fluor
esce
nce
0
20
40
60
80
100
0 50 100 150 200 250
3 µM6 µM9 µM12 µM
ThT
fluor
esce
nce
0
20
40
60
80
100
0 50 100 150
4 µM6 µM8 µM10 µM
ThT
fluor
esce
nce
5
10
15
20
25
30
35
0 5 10 15
1 µM2 µM3 µM4 µM
ThT
fluor
esce
nce
Time (min)
0
100
200
300
400
500
600
700
800
0 40 80 120 160 200 240 280 320
1 µM2 µM3 µM4 µM
Sca
tter 5
00 n
m
Time (min)
EA
B D
0
100
200
300
400
500
600
700
800
0 100 200 300 400 500 600
1 µM2 µM3 µM4 µM
Sca
tter 5
00 n
m
Time (min)
C
F
Fig. 2. Aggregation of WTC, WTCI254D, and WTCI254R from the nativestate. Time course of aggregation of WTC (A) and WTCI254D (B) at 37 °Cmonitored by ThT binding, WTCI254D (C) and WTCI254R (E) at 30 °C by ThTbinding, and WTCI254D (D) and WTCI254R (F) at 30 °C by light scattering.
Wang and Fersht PNAS | Published online March 14, 2017 | E2635
BIOCH
EMISTR
YPN
ASPL
US

102–120 (4,301 Da, 3,356 Da, 2,090 Da; S1), and 176–202 (3,037 Da,H1S5) (Fig. 1 E and F). Further, minor peaks corresponding topeptides 268–282 (1,710 Da, S10H2), 157–181 (3,064 Da, 2,809Da), 140–174 (3,983 Da), and 133–156 (2,677 Da, S2′S3) were alsoenriched in the remaining aggregate of WTCI254D. Digestion ofthe aggregate after overnight incubation also showed the samemajor and minor peaks enriched in the remaining aggregate as forproteolysis of the 2-h aggregate. These data provide further evi-dence that there are multiple aggregation-prone sequences andthat S9 is not necessary for aggregation to occur.Full-length p53. Most of the fragments found in the aggregate of coredomain were present in the aggregates of wild-type full-length p53(WTFL) andWTFLG245S (Fig. 3 and Table 1). The first three mainsegments contributing to the aggregation process (249–267, 182–213,and 268–282) were also the highly aggregation-prone segments wefound in the aggregation of denatured p53 mutants. Aggregation ofthe full-length protein was thus similar to aggregation of the coredomain. Further, a small amount of the C-terminal fragment 373–393(2,377 Da, 373–393; 2,594 Da, 373–395), which is known to bind tothe core domain, was buried in the aggregation core.
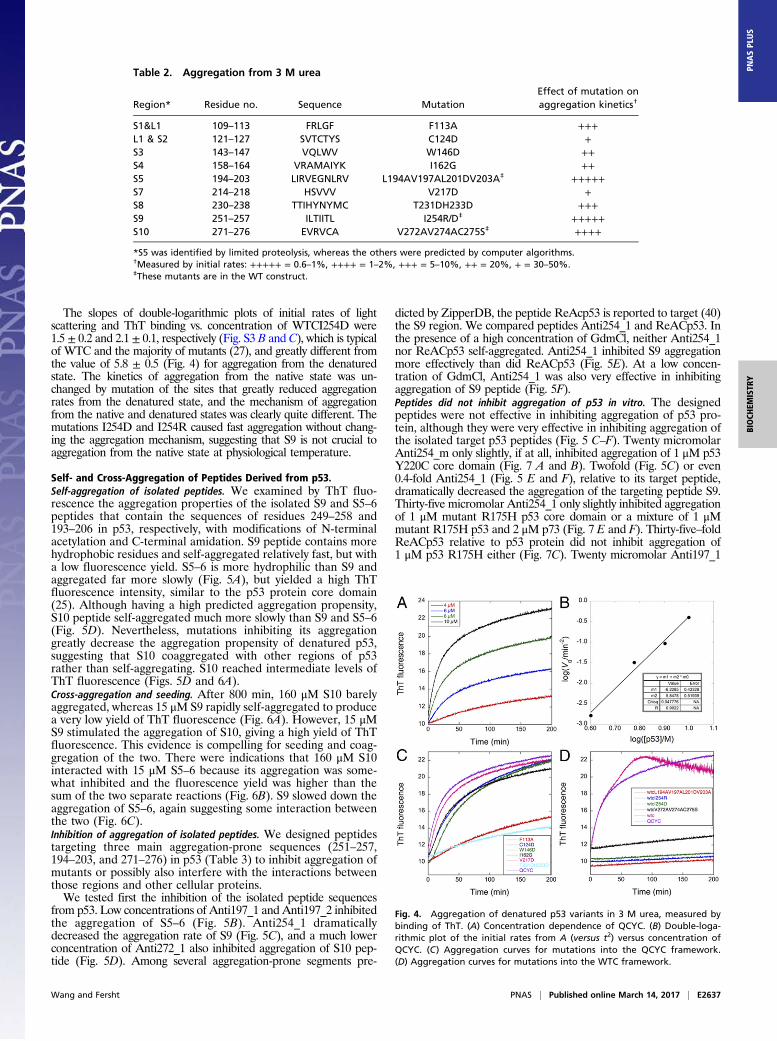
Effects of Mutating Aggregation-Prone Sites in Denatured p53.Usingthe programs Tango (41), Waltz (42), Aggrescan (43), FoldAmyloid(44), and Amylpred2 (45), we identified several aggregation-pronesequences (Table 2). We lowered the aggregation propensity ofeach sequence by introducing mutations that were modeled to becompatible with the crystal structure (46) and are predicted to beeffective using the above programs. Mutation to Asp or Arg shouldbe the most effective; however, because of steric constraints in thefolded protein, these changes were not always possible, and so wemade multiple, conservative mutations in each sequence instead. S5was not predicted to be a site, but we mutated it because it wasidentified by limited proteolysis to be protected in the aggregate.We determined the kinetics of aggregation of the core domain
of p53 from binding of ThT (Fig. 4A) after denaturation in 4 Murea at 0 °C and then diluting the denaturant to 3 M at 37 °C.The curve for aggregation of our standard mutant core domainY220C (QCYC) (27) had an exponential fast phase. A double-logarithmic plot of the initial rate (measured against t2) vs. theconcentration of [QCYC] (Fig. 4B) should give as its slope thenumber of molecules in the nucleus plus 2 for homogeneousnucleation (47). The slope was 5.8 ± 0.5, in contrast to 1–2 foundfor thermal aggregation of WT and mutants (27).Mutations in S1 (F113A), S2 (C124D), S3 (W146D), S4 (I162G),
S7 (V217D), and S8 (T231DH233D) (Fig. 4C) had small to signifi-cant lowering of rates (Table 2). Mutations in S5 (WTCL194A-V197AL201DV203A), S9 (WTCI254D, WTCI254R), and S10
(WTCV272AV274AC275S) had very large effects (Fig. 4D andTable 2), and there was little, if any, light scattering after overnightincubation in 3 M urea. Accordingly, S9 and S5 are highly ag-gregation-prone regions, followed by S10, S8, and S1 regions.Many regions within the p53 sequence are very aggregation-proneand can contribute to aggregation of p53 if they are exposed.
Urea Dependence of Aggregation. WTCI254D did not visibly ag-gregate after incubation for 24 h in 3 M urea. However, the rateincreased greatly at lower urea concentrations, and a logarithmicplot of the initial rate against [urea] had a slope of −2. The dena-tured WT p53 core domain and mutant QCYC were much lesssensitive to the concentration of urea, with a corresponding slope ofonly −0.69 (Fig. S3A). The effects of mutation on aggregation de-creased with decreasing concentrations of urea as the rates greatlyincreased (Fig. S3A). The sensitivity of rate constants to the con-centration of denaturant reflects the change in solvent-accessiblesurface area as a protein-folding reaction proceeds: The more thearea buried, the greater is the slowing down of the rate. Mutationof I254 to D254 was the most sensitive, indicating that the rate-determining step in aggregation involving the region surroundingI254 buried more surface area than in the other regions probed.
Effects of Mutation on Aggregation from the Native State. We an-alyzed, among other mutations of aggregation-prone sites, I254Dand I254R in S9, which greatly affected aggregation from thedenatured state and are reported to inhibit totally its gain offunction in cells (32). The structure of WTC is destabilized bymutations of I254D and I254R at 10 °C, by 6.15 kcal/mol and 4.28kcal/mol, respectively, and decreases in their melting temperatures(Tms) to 37.8 ± 0.1 °C and 39.7 ± 0.1 °C from 45.8 ± 0.1 °C, re-spectively. Mutating I254R and I254D increased the aggregationrate of WTC (Fig. 2) but retained the apparent two-step sequentialfirst-order kinetics (25) for the binding of ThT. At 37 °C, aggre-gation rate constants for WTC at 12 μM were 0.027 ± 0.001 min−1
and 0.79 ± 0.16 min−1, respectively, whereas the aggregationrate constants for 4 μM WTCI254D are 0.85 ± 0.02 min−1 and4.3 ± 0.5 min−1. Even at 30 °C, the aggregation rate constants ofWTCI254D (k1 = 0.069 ± 0.001 min−1, k2 = 0.33 ± 0.01 min−1)andWTCI254R (k1= 0.0255± 0.0001 min−1, k2= 0.38± 0.02 min−1)at 10 μM were still high. The amplitude of light scattering was notdiminished by mutation of I254 (Fig. 2 D and F), with the valuesbeing typical of WTC and all other mutants, and the aggregate ofWTCI254D had similar morphology to aggregates of WTC (Fig. S2).
Fig. 3. Limited proteolysis of WTFL aggregate and WTFLG245S aggregateby trypsin. Mass spectra of supernatant of WTFL aggregate after digestion(A), the remaining aggregate of WTFL aggregates after digestion (B), su-pernatant of FLG245S aggregates after digestion (C), and the remainingaggregate of FLG245S aggregates after digestion for 76 h (D).
Table 1. Regions protected against proteolytic degradation
Peptide regions enriched inremaining aggregate*
Effect onaggregation† RegionWTC WTCI254D WTFL FLG245S
249–267 — 249–267 249–267 +++++ S9182–213 182–213 182–213 182–213 +++++ H1S5S6— 268–282 268–282 268–282 +++++ S10H2102–120 102–120 102–120 102–120 +++ S1L1— — 284–292 284–292 H2140–174 140–174 140–174 140–174 ++ S3S4L2— — 373–393 373–393 C-term— 133–156 — — S2′S3
*Aggregates from aggregation of the proteins at 37 °C were digested bytrypsin for 48 h, 6 h, 76 h, and 76 h, respectively, for WTC, WTCI254D, WTFLand FLG245S at 20 °C. Peptides are shown in boldface: major peaks in thespectra. For WTFL and WTFLG245S, peptides are listed according to relativesize of peaks from large to small in MS spectra of the remaining aggregateafter digestion for 76 h.†Effect on aggregation is based on the aggregation rates of denatured ag-gregation-inhibition mutants in the corresponding region. Measured by ini-tial rates: +++++ = 0.6–1%, ++++ = 1–2%, +++ = 5–10%, ++ = 20%, + =30–50%.
E2636 | www.pnas.org/cgi/doi/10.1073/pnas.1700308114 Wang and Fersht
The slopes of double-logarithmic plots of initial rates of lightscattering and ThT binding vs. concentration of WTCI254D were1.5 ± 0.2 and 2.1 ± 0.1, respectively (Fig. S3 B and C), which is typicalof WTC and the majority of mutants (27), and greatly different fromthe value of 5.8 ± 0.5 (Fig. 4) for aggregation from the denaturedstate. The kinetics of aggregation from the native state was un-changed by mutation of the sites that greatly reduced aggregationrates from the denatured state, and the mechanism of aggregationfrom the native and denatured states was clearly quite different. Themutations I254D and I254R caused fast aggregation without chang-ing the aggregation mechanism, suggesting that S9 is not crucial toaggregation from the native state at physiological temperature.
Self- and Cross-Aggregation of Peptides Derived from p53.Self-aggregation of isolated peptides. We examined by ThT fluo-rescence the aggregation properties of the isolated S9 and S5–6peptides that contain the sequences of residues 249–258 and193–206 in p53, respectively, with modifications of N-terminalacetylation and C-terminal amidation. S9 peptide contains morehydrophobic residues and self-aggregated relatively fast, but witha low fluorescence yield. S5–6 is more hydrophilic than S9 andaggregated far more slowly (Fig. 5A), but yielded a high ThTfluorescence intensity, similar to the p53 protein core domain(25). Although having a high predicted aggregation propensity,S10 peptide self-aggregated much more slowly than S9 and S5–6(Fig. 5D). Nevertheless, mutations inhibiting its aggregationgreatly decrease the aggregation propensity of denatured p53,suggesting that S10 coaggregated with other regions of p53rather than self-aggregating. S10 reached intermediate levels ofThT fluorescence (Figs. 5D and 6A).Cross-aggregation and seeding. After 800 min, 160 μM S10 barelyaggregated, whereas 15 μM S9 rapidly self-aggregated to producea very low yield of ThT fluorescence (Fig. 6A). However, 15 μMS9 stimulated the aggregation of S10, giving a high yield of ThTfluorescence. This evidence is compelling for seeding and coag-gregation of the two. There were indications that 160 μM S10interacted with 15 μM S5–6 because its aggregation was some-what inhibited and the fluorescence yield was higher than thesum of the two separate reactions (Fig. 6B). S9 slowed down theaggregation of S5–6, again suggesting some interaction betweenthe two (Fig. 6C).Inhibition of aggregation of isolated peptides. We designed peptidestargeting three main aggregation-prone sequences (251–257,194–203, and 271–276) in p53 (Table 3) to inhibit aggregation ofmutants or possibly also interfere with the interactions betweenthose regions and other cellular proteins.We tested first the inhibition of the isolated peptide sequences
from p53. Low concentrations of Anti197_1 and Anti197_2 inhibitedthe aggregation of S5–6 (Fig. 5B). Anti254_1 dramaticallydecreased the aggregation rate of S9 (Fig. 5C), and a much lowerconcentration of Anti272_1 also inhibited aggregation of S10 pep-tide (Fig. 5D). Among several aggregation-prone segments pre-
dicted by ZipperDB, the peptide ReAcp53 is reported to target (40)the S9 region. We compared peptides Anti254_1 and ReACp53. Inthe presence of a high concentration of GdmCl, neither Anti254_1nor ReACp53 self-aggregated. Anti254_1 inhibited S9 aggregationmore effectively than did ReACp53 (Fig. 5E). At a low concen-tration of GdmCl, Anti254_1 was also very effective in inhibitingaggregation of S9 peptide (Fig. 5F).Peptides did not inhibit aggregation of p53 in vitro. The designedpeptides were not effective in inhibiting aggregation of p53 pro-tein, although they were very effective in inhibiting aggregation ofthe isolated target p53 peptides (Fig. 5 C–F). Twenty micromolarAnti254_m only slightly, if at all, inhibited aggregation of 1 μM p53Y220C core domain (Fig. 7 A and B). Twofold (Fig. 5C) or even0.4-fold Anti254_1 (Fig. 5 E and F), relative to its target peptide,dramatically decreased the aggregation of the targeting peptide S9.Thirty-five micromolar Anti254_1 only slightly inhibited aggregationof 1 μM mutant R175H p53 core domain or a mixture of 1 μMmutant R175H p53 and 2 μM p73 (Fig. 7 E and F). Thirty-five–foldReACp53 relative to p53 protein did not inhibit aggregation of1 μM p53 R175H either (Fig. 7C). Twenty micromolar Anti197_1
Fig. 4. Aggregation of denatured p53 variants in 3 M urea, measured bybinding of ThT. (A) Concentration dependence of QCYC. (B) Double-loga-rithmic plot of the initial rates from A (versus t2) versus concentration ofQCYC. (C) Aggregation curves for mutations into the QCYC framework.(D) Aggregation curves for mutations into the WTC framework.
Table 2. Aggregation from 3 M urea
Region* Residue no. Sequence MutationEffect of mutation onaggregation kinetics†
S1&L1 109–113 FRLGF F113A +++L1 & S2 121–127 SVTCTYS C124D +S3 143–147 VQLWV W146D ++S4 158–164 VRAMAIYK I162G ++S5 194–203 LIRVEGNLRV L194AV197AL201DV203A‡ +++++S7 214–218 HSVVV V217D +S8 230–238 TTIHYNYMC T231DH233D +++S9 251–257 ILTIITL I254R/D‡ +++++S10 271–276 EVRVCA V272AV274AC275S‡ ++++
*S5 was identified by limited proteolysis, whereas the others were predicted by computer algorithms.†Measured by initial rates: +++++ = 0.6–1%, ++++ = 1–2%, +++ = 5–10%, ++ = 20%, + = 30–50%.‡These mutants are in the WT construct.
Wang and Fersht PNAS | Published online March 14, 2017 | E2637
BIOCH
EMISTR
YPN
ASPL
US

could effectively inhibit aggregation of its target peptide 15 μMS5–6(Fig. 5B), although neither 20 μMAnti197_1 itself nor together withAnti254_1 inhibited aggregation of 1 μM p53 Y220C core domain(Fig. 7D). The peptides were unable to decrease the initial aggre-gation rate of p53. The lack of inhibition is consistent with ourfindings that the rate-determining steps in aggregation of p53 areearly unfolding events and that there are multiple aggregation sitesafter initial unfolding.
Cytotoxicity Toward Cancer Cells with Structural Mutants of p53. Weexamined the effects of the peptides on cancer cell lines containingWT p53 or destabilized structural mutant p53Y220C, p53V143A, orR175H. All of the designed peptides targeting the three majorregions had higher cytotoxicity toward the gastric cancer cellline NUGC3 (Y220C+/+) than toward NUGC4 (WT+/+) (Fig. 8).N-methylated peptides without a cell-penetrating sequence weresignificantly more cytotoxic toward NUGC3 than toward NUGC4(Fig. 8A). The more hydrophobic peptide Anti254_m was moreeffective than the other two sequences, possibly because with neg-ative charged residues and low hydrophobicity, Anti197_m andAnti272_m were less cell-penetrating.Anti254_1 was cytotoxic at concentrations lower than 30 μM
toward NUGC3 and MKN1 (V143A+/+) (Fig. 8B), althoughbeing much less toxic toward NUGC4. Anti197 also effectively
destroyed cancer cells harboring mutant p53 with either poly-arginine (Fig. 8C) or Xentry cell-penetrating sequence (Fig. 8E).Two consecutive 24-h treatments with 8 μM Anti197_1 wereeven more effective than 24-h treatment with 16 μM Anti197_1.Anti254_1, Anti197_1, and Anti197_2 showed higher cytotoxicitytoward breast cancer cell lines SKBR3 (R175H+/+) than towardMCF7 (WT+/+) (Fig. 8D). Anti254_2, with a proline to interruptthe formation of the β-sheet, had much higher toxicity towardNUGC3 than toward NUGC4, although it was much less effec-tive than Anti254_1 at concentrations higher than 20 μM. Pep-tides targeting region 272 also showed some cytotoxicity (Fig. 8G and H). Anti272_1 is very sensitive to the presence of serumand is effective only at high concentrations, possibly because ofits unmodified N-terminus (48).We changed the cell-penetrating sequence of Anti254_1 to
poly-arginine (Anti254_3) as in ReACp53 for better comparison.Anti254_3 was much more cytotoxic than ReACp53 toward cancercells (Fig. 9). The IC50s of Anti254_3 with NUGC3 and NUGC4cells were 4.2 ± 0.1 μM and 6.5 ± 0.2 μM, respectively, whereas theIC50s for ReACp53 were 17.4 ± 0.4 μM and 17.2 ± 0.6 μM, re-spectively. ReACp53 did not show selectivity between NUGC3(Y220C+/+) and NUGC4 (WT+/+), whereas Anti254_3 demon-strated selectivity toward the mutant. The presence of 10% heat-inactivated serum in our experiments would have led to highervalues of IC50s than reported (40).
Synergistic and Additive Effects of Cytotoxic Peptides.Pairwise synergy and additivity. There were synergistic and additiveeffects between the peptides targeting the 254 region and pep-tides targeting the 197 and 272 regions. Fifty micromolarAnti254_m had minor toxicity to NUGC3, which was greatlyenhanced by the addition of 5 μM or 10 μM Anti197_1 (Fig.10A). Combination of Anti254_1 and Anti197_1 greatly en-hanced the effect of either alone on both NUGC3 and MKN1cell lines (Fig. 10B). The combination index (CI) is ∼1, based onthe Chou–Talalay method (49), indicating that the effects wereadditive rather than synergistic. Combinations of Anti254_1 andAnti197_2 had much greater effects than the sums of the effectsof each peptide on NUGC3 (Fig. 10C) and MKN1 (Fig. 10D)cells. The combination between 10 μM Anti254_1 and 50 μMAnti197_2 (CI is around 0.8) is synergistic, and the combination of15 μM Anti254_1 and 25 μM Anti197_2 shows even strongersynergy (CI = 0.55).Western blots showed that treatment of NUGC3 cells with
Anti254_1 slightly up-regulated p21 levels, whereas incubationwith Anti197_2 up-regulated p21 more so (Fig. 10 E and F). Thecombination of these two peptides consistently up-regulated thep21 level in both cell lines. This up-regulation of p21 was de-tectable within 3 h in MKN1 cells (Fig. 10G). Further, combi-nation of Anti254_1 and Anti197_2 decreased levels of MDM2
6
7
8
9
10
11
0 20 40 60 80 100 120
62 µM S962 µM S9+25.4 µM ReACp5362 µM S9+25.4 µM Anti254_162 µM S9+46 µM ReACp5362 µM S9+46 µM Anti254_1
3.5
4
4.5
5
5.5
6
6.5
0
50
100
150
200
250
300
350
0 20 40 60 80 100
15 µM S9 15 µM S5-6
ThT
fluor
esce
nce
7.5
8
8.5
9
9.5
10
10.5
11
0 50 100 150 200 250
15 µM S915 µM S9 + 30 µM Anti254_1
0
50
100
150
200
250
300
350
400
0 40 80 120 160
15 µM S5-615 µM S5-6 + 20 µM Anti197_115 µM S5-6 + 40 µM Anti197_115 µM S5-6 + 20 µM Anti197_2
Time (min)
ThT
fluor
esce
nce
0
20
40
60
80
100
120
0 50 100 150 200 250 300 350
480 µM S10480 µM S10+160 µM Anti272_1
Time (min)
3.5
4.0
4.5
5.0
5.5
6.0
6.5
7.0
0 5 10 15 20 25 30 35 40
15 µM S9
15 µM S9+10 µM Anti254_1
Time (min)
A C E
B D F
Fig. 5. Aggregation of S9, S10, and S5–6 peptides and its inhibition in standardbuffer of 25 mMKPi, 150 mMNaCl, 1 mM TCEP, and 22 μMThT, unless indicatedotherwise, at pH 7.2 and various concentrations of GdmCl. (A) Fifteen micro-molar S9 peptide and 15 μM S5–6 peptide (+0.18 M GdmCl). (B) Inhibition byAnti197_1 and Anti197_2 of aggregation of 15 μM WT S5–6 peptide in buffer +0.18 M GdmCl. (C) Anti254_1 on 15 μM S9 in buffer + 1.2 M GdmCl.(D) Anti272_1 on aggregation of S10 in buffer + 0.37 M GdmCl. (E) Anti254_1and ReACp53 on 62 μM S9 peptide in buffer + 1.9 M GdmCl and 20 μM ThT.(F) Fifteen micromolar S9 peptide in buffer + 0.18 M GdmCl. (The peptides weredissolved in high concentrations of GdmCl to prevent self-aggregation, which ledto varying concentrations of GdmCl on mixing and dilution.)
0
50
100
150
200
0 20 40 60 80 100 120 140 160
15 µM S5-615 µM S9 15 µM S5-6+ 15 µM S9
Time (min)
0
20
40
60
80
100
120
140
160
0 200 400 600 800
15 µM S9160 µM S10160 µM S10+15 µM S9
ThT
fluor
esce
nce
Time (min)
0
50
100
150
200
250
300
0 50 100 150 200
15 µM S5-6160 µM S10160 µM S10+15 µM S5-6
Time (min)
CA B
Fig. 6. Cross-aggregation and seeding of S9, S10, and S5–6 peptides.Coaggregation of 15 μM S9 and 160 μM S10 (A), 15 μM S5–6 and 160 μM S10(B), and 15 μM S5–6 and 15 μM S9 (C) in buffer of 25 mM KPi, 150 mM NaCl,22 μM ThT, 1 mM TCEP, and 0.3 M GdmCl (pH 7.2) at 37 °C.
Table 3. Sequences of designed peptides
Peptide name Peptide sequence* Target
Anti254_m ILTIITL-NH2 S9Anti197_m1 LIRVEGNLRV-NH2 S5–6Anti197_m2 LIRVEGNLRV-NH2 S5-6Anti272_m1 EVRVCA-NH2 S10Anti272_m2 EVRVCA-NH2 S10Anti254_1 LCLRRRPIARIIRLL-NH2 S9Anti254_2 R10PILTPITLR-NH2 S9Anti254_3 R10PIARIIRLL-NH2 S9Anti197_1 R10GPHLIRREARYRAEYL S5–6Anti197_2 LCLRPHLIRREARYRAEY S5–6Anti272_1 LCLRPNSFEVRACA-NH2 S10Anti272_2 LCLRPNSFERRVCA-NH2 S10
N-termini of the unmethylated peptides are acetylated except Anti272_1.R10, 10 arginines.*Underlined residues are N-methylated.
E2638 | www.pnas.org/cgi/doi/10.1073/pnas.1700308114 Wang and Fersht

in both MKN1 and NUGC3 cell lines. Levels of the apoptosisregulator Bax did not change in either combination or separatetreatment of these two peptides. A decrease in p53 levels wasdetectable after 9 h of incubation with the combination of bothpeptides (Fig. 10 E and F), and levels of p53 dramatically de-creased after treatment for 24 h (Fig. 10G), indicative of deg-radation of aggregated p53.Anti272_1 had little effect on MKN1 cancer cells at concen-
trations lower than 400 μM, but it significantly enhanced the effectof Anti254_1 (Fig. S4A). Eighteen micromolar Anti254_1 by itselfreduced the viability of MKN1 by 80%, whereas the combinationof 50 μM Anti272_1 and 18 μM Anti254_1 decreased the viabilityto 46%, demonstrating synergism (CI = 0.88). However, furtherincreasing the concentration of Anti272_1 undermined the syn-ergy, indicating antagonism also exists. The antagonism may comefrom interactions between the two peptides. Synergy and its im-pairment at higher concentrations of Anti272_1 were also found at21 μM Anti254_1. Anti272_1 had little effect on NUGC3 at
concentrations lower than 200 μM, but there was enhancement ofthe effects of Anti254_1 (Fig. S4B).Anti272_2 at 200 μM had little effect on MKN1 cells, but greatly
enhanced the effect of 14 μM Anti254_1 in a dose-dependentmanner (Fig. S4C). At 200 μM, Anti272_2 had synergy with 14 μMAnti254_1 (CI = 0.8), inducing an 80% decrease in viability ofMKN1 cells. Fifty micromolar Anti272_2 also enhanced the cyto-toxicity of 15 μM Anti254_1 toward NUGC3 (Fig. S4D).Triple-wise synergy and additivity. Anti272_2 and Anti197_2 did notshow synergistic or additive effects at the concentrations tested; how-ever, combination of Anti254_1, Anti272_2, and Anti197_2 was moreeffective than a single peptide or combination of two (Fig. S4D and E)with both NUGC3 and MKN1 cell lines. There was synergy of cyto-toxicity of the three peptides in combination (CI = 0.8) toward MKN1cancer cells. Combination of 100 μMAnti272_2 and 12 μMAnti254_1decreased their viability to ∼80%. Addition of a further 50 μMAnti197_2 dramatically decreased the viability to ∼14% (Fig. S4E).
Peptides and Peptide Combinations Selectively Kill Cancer Cells.Anti254_1 had far less effect on a cancer cell line harboring acontact mutant of p53 SW480 (R273H) and a cancer cell line withno p53 (H1299) (Fig. S5A) than on human cancer cell lines con-taining structural mutants of p53, NUGC3, and SKBR3 (Fig. 8 Band D). It was least effective on noncancerous human fibroblastcell line WI38 (Fig. S5A), demonstrating selective toxicity towardcancer cells. Anti272_2 at concentrations less than 400 μM wastoxic to NUGC3 cells but showed no toxicity to WI38. It did notenhance the effect of Anti254_1 on WI38 (Fig. S5B) at concen-trations that greatly enhanced the effect of Anti254_1 on NUGC3cells (Fig. S4D). There was no synergistic effect of Anti254_1 andAnti272_2 or Anti197_2 on H1299 cells (Fig. S5C) at the con-centrations at which there was synergy for cytotoxicity towardNUGC3 cells (Fig. 10C and Fig. S4D) and MKN1 cells (Fig. 10D).The combination of 15 μM Anti254_1 and 100 μM Anti272_2 wasselectively toxic toward NUGC3 cells, although lacking toxicitytoward normal WI38 fibroblast cells (Fig. S5D). The combinationof 10 μM Anti254_1 and 50 μM Anti197_2 also had high cytotoxicselectivity toward cancer cells (Fig. S5D).Dependence of cytotoxicity of peptides on p53. Although Anti254_1had higher toxicity toward cancer cell lines containing structuralmutants of p53 than toward cancer cell lines containing a contactmutant or lacking p53 (Fig. 8 and Fig. S5), it was still significantlytoxic to the latter two cancer cell lines (Fig. S5A), indicating a majorp53-independent effect of Anti254_1, especially at concentrationshigher than 14 μM. A better control is to compare two closely iso-genic cell lines. To check further the p53 dependency of Anti254_1,Anti197_1, and other peptides, we knocked down p53 using p53-targeted small interfering RNA (siRNA) and compared their via-bility with the same cells that had been treated with nontargetedsiRNA (Fig. 11). Near-complete deletion of p53 (Fig. S6) didsomewhat reduce the effect of Anti254_1 (Fig. 11 A–C) andAnti197_1 (Fig. 11D) on both sets of NUGC3 and MKN1 cell lines.However, there was still a major p53-independent effect ofAnti254_1 and Anti197_1, especially at concentrations higher than
A
D
C E
B F
Fig. 7. Peptides not effectively inhibiting aggregation of p53 proteins. In-hibition effect of Anti254_m on aggregation of 1 μM p53 Y220C mutant at34 °C monitored by light scattering (A) and ThT binding (B). Thirty-five mi-cromolar ReACp53 (C) and 35μM Anti254_1 (E) on aggregation of 1 μMp53R175H at 37 °C. (D) Anti197_1 and Anti254_1 on aggregation of 1 μMp53 Y220C. (F) Thirty-five micromolar Anti254_1 on aggregation of themixture of 1 μM p53R175H and 2 μM p73 at 37 °C.
0
20
40
60
80
100
120
140
0 75 200 400 600 800Anti272_1 (µM)
0
20
40
60
80
100
120
0 12.5 25 50 75 150Anti197_2 (µM)
0
20
40
60
80
100
120
0 8 12 16 8 (48 h)Anti197_1 (µM)
replace withfresh at 24h
0
20
40
60
80
100
120
0 7 14 21 28Anti254_1 (µM)
0
20
40
60
80
100
120
Viab
ility
(%)
0
20
40
60
80
100
120
0 10 15 20 30
Viai
bilit
y (%
)
Anti254_2 (µM)
A B C D
0
20
40
60
80
100
120
0
20
40
60
80
100
120
0 50 100 200 400Anti272_2 (µM)
F G HE
Fig. 8. Peptides selectively kill cancer cells harboring mutant p53. Viabilitywas measured after treatments with peptides Anti254_m (A); Anti254_1 (B);Anti197_1 (C); Anti254_1, Anti197_1, and Anti197_2 (D); Anti254_2 (E);Anti197_2 (F); Anti272_1 (G); and Anti272_2 (H) on cancer cells harboringmutant p53 (NUGC3, MKN1, SKBR3) and cancer cells harboring WT p53(NUGC4, MCF-7) for 24 h unless otherwise indicated.
0
20
40
60
80
100
120
0 2 4 6 8 10 12 14 16
NUGC4NUGC3
Via
bilit
y (%
)
0
20
40
60
80
100
120
0 5 10 15 20 25 30 35 40
NUGC4NUGC3
Via
bilit
y (%
)
A B
Fig. 9. Relative cytotoxicity of Anti254_3 and ReACp53. Viability wasmeasured after treatment with Anti254_3 (A) and ReAcp53 (B) for 48 h.
Wang and Fersht PNAS | Published online March 14, 2017 | E2639
BIOCH
EMISTR
YPN
ASPL
US

15 μM for Anti254_1, which is consistent with observations of thepeptides on the other cell lacking p53 or harboring a contact mutant.There was significant p53-independent cytotoxicity of combi-
nations of 15 μM Anti254_1 and 100 μM Anti272_2 or 50 μMAnti197_2 (Fig. 11E), as there was with Anti254_2 towardNUGC3 cells. The cytotoxicity toward MKN1 cells was com-pletely independent of p53 (Fig. 11G). The transfection reagentlipofectamine and nontargeting siRNA weakened the effect ofAnti197_2, making it ineffective at concentrations lower than150 μM. At high concentrations of Anti197_2, knocking downp53 actually enhanced the cytotoxicity (Fig. 11F). Becausetreatment of the transfection reagent greatly affected the efficacyof Anti272_2, we did not test further its effect using siRNA.Similar to Anti254_1, ReACp53 also had a significant p53-independent effect (Fig. 11 B and C).Peptides did not increase native p53 or reduce denatured p53 in cells. Totest further whether the peptides affected the status of p53, wetreated NUGC3 cells with 12 μM Anti254_1 or 120 μMAnti197_2 for 6 h. In contrast to the reported effect of ReACp53(40), immunostaining showed no dramatic change in the level ofp53 in different states (Fig. 12). We detected a great decrease inthe denatured p53 level, which is similar to the effect ofReACp53; however, this decrease only happened when the cellnuclei were shrinking, indicating they were dying (Fig. S7).mRNA levels of p53 target genes. Consistent with our Western blotresults (Fig. 10E), mRNA quantification by real-time PCR showedthat treatment of NUGC3 cells with Anti254_1 slightly up-regulatedlevels of the cyclin-dependent kinase inhibitor p21, whereas in-cubation with Anti197_2 up-regulated p21 more so (Fig. 13). Thep53 upregulated modulator of apoptosis (PUMA) was up-regulatedto a very high level (Fig. 13). PUMA up-regulation normally willinduce apoptosis; however, the PUMA up-regulation did not lead tocells dying through apoptosis. Anti197_2 induced caspase 3/7 acti-vation to some extent, but no caspase 3/7 activation was detectedfor Anti254_1 treatment and carbobenzoxy-valyl-alanyl-aspartyl-[O-methyl]-fluoromethylketone (z-VAD) could not inhibit thecytotoxicity of the peptides toward NUGC3 cancer cells (Fig.14). Necrostatin-1 could not inhibit its cytotoxicity either (Fig.14C), so Anti254_1-induced NUGC3 cancer cell death is notthrough necroptosis.We conclude that there is significant p53-independent cytotoxicity
of the peptides in our cell lines that are on 2D plates and the pres-ence of mutant p53 enhances the toxicity somewhat in some cases.
DiscussionDenatured p53 Contains Several Aggregation-Prone Sequences. Manyproteins will form amorphous aggregates when their denaturedstate in urea is rapidly diluted into lower concentrations of de-naturant, and p53 is no exception. Its aggregate formed from itsdenatured state in 3 M urea contained sequences that were highlyprotected against controlled proteolysis. They contained a siteidentified only by limited proteolysis (S5) and sites that werepredicted to be prone to form fibrillar aggregates, among whichwere S7 and S10 in addition to those regions predicted by ZipperDB(40) (Table 2). Mutation of several of those sites to make themless aggregation-prone inhibited greatly the overall rate of aggre-gation in 3 M urea (Table 2). One of those sequences, residues251–257 (ILTIITL) in β-strand S9, is proposed to be the key ag-gregation-prone sequence (32). Indeed, mutating I254 to R or Ddecreased the rate of aggregation in 3 M urea by an estimatedgreater than 104-fold (extrapolated from Fig. S3A). Very largedecreases were found for mutations in S5 and S10, and significantdecreases were found elsewhere (Table 2). There is clearly not aunique aggregation sequence in the denatured state of p53, but acooperative network of such sites.Sequences involved in aggregation of p53 from its native state. Usinglimited proteolysis in vitro, we found four main sequences pro-tected in the aggregate core formed on incubation of the nativestate at 37 °C (Table 1): 249–267 (S9), 182–213 (H1S5S6), 268–282 (S10H2), and 102–120 (S1L1), which are sequences thatoverlap with those sequences found to be aggregation-prone inthe denatured state. Mutating those sequences to be less ag-gregation-prone did not inhibit p53 aggregation in vitro; instead,they destabilized p53 and made it aggregate faster by increasingthe rate of unfolding, just as did the I254D and I254R mutations.The slow initial steps in aggregation from the native state are
the sequential unfolding of two molecules of p53, followed byrapid polymerization to form ThT-binding oligomers, which thenslowly rearrange to large aggregates (27, 28). The denaturedstates of all of the mutants aggregated far more rapidly in theabsence of urea (Fig. S3A) than did the native state. Mutation ofI254D greatly speeds up the rate of unfolding. Further, the rateof aggregation of denatured p53 is 500- to 1,000-fold faster in
0
20
40
60
80
100
120
0 10 15 21
Viab
ility
(%)
Anti254_1 (µM)
NUGC3 24 h
0
20
40
60
80
100
120
0 10 15 21 10 15 21
Anti254_1 (µM) ReACp53 (µM)
p53(+/+) Si non-targeting Sip53
NUGC3 48 h
0
20
40
60
80
100
120
0 15 18 24 10 15 21
Anti254_1 (µM) RecACp53 (µM)
MKN1 48 h
0102030405060708090
100
24 h 48 h 24 h 48 h
Viab
ility
(%)
NUGC3 MKN1
Anti197_1
0
20
40
60
80
100
120
50 75 150 75 150 75 150 75 15024 h 48 h 24 h 48 h
Anti197_2 (µM)
NUGC3 MKN1
0
20
40
60
80
100
NUGC3 MKN1
Anti254_2
A B C
GFD E
0
20
40
60
80
100
Fig. 11. Effects of p53 knockdown by siRNA on cytotoxicity of peptides. Afterknockdown for 48 h, cancer cells were washed with PBS. Then, NUGC3 cellswere incubated with Anti254_1 for 24 h (A); NUGC3 cells (B) and MKN1 cells(C) were incubated with Anti254_1 and ReACp53 for 48 h; NUGC3 and MKN1cells were incubated with 12 μM Anti197_1 for 24 h and 48 h (D); NUGC3 cellswere incubated with 100 μM Anti272_2, the combination of 15 μM Anti254_1and 100 μM Anti272_2, and the combination of 15 μM Anti254_1 and 50 μMAnti197_2 for 24 h (E); NUGC3 and MKN1 cells were incubated with variousconcentrations of Anti197_2 for 24 h or 48 h (F); and NUGC3 and MKN1 cellswere incubated with 30 μM Anti254_2 for 24 h (G).
NUGC4 (WT +/+) NUGC3 (Y220C +/+) MKN1 (V143A +/+)
0
20
40
60
80
100
120
Via
bili
ty(
%)
0
20
40
60
80
100
Via
bili
ty (
%)
0
20
40
60
80
100
Via
bili
ty (
%)
0
20
40
60
80
100
Via
bili
ty (
%)
A C
B
G
F
p53
-actin
p21
3h 5h 21h
MDM2
Anti254_17 µ M
Anti197_50 µ M
MKN1
--
+ ++ +-
- --
++
E
p53
p21
5h 9h
MDM2
Anti254_12 µ M
Anti197_75 µ M
NUGC3
--
+ +- ++
-
-actin
--
+ +- ++
-
MKN1
Bax
p53
-actin
p21
MDM2
Anti254_17 µ M
Anti197_50 µ M
-
-
+ +
- ++
-
D
Fig. 10. Peptides targeting the 197 region enhance the effect of peptidestargeting the 254 region. Anti197_1 enhances the effect of N-methylatedpeptide (A) and Anti254_1 (B). Anti197_2 enhances effect of Anti254_1 onNUGC3 (C) and MKN1 (D) cancer cells. Western blot of the combinationeffect of Anti254_1 and Anti197_2 on NUGC3 cells (E) and MKN1 (G) fordifferent lengths of time and on MKN1 cells after treatment for 9 h (F).
E2640 | www.pnas.org/cgi/doi/10.1073/pnas.1700308114 Wang and Fersht

water than in 3 M urea, so that even alternative, less aggregation-prone sequences can aggregate sufficiently fast. Accordingly,although mutating I254 to D254 virtually eliminated aggregationof p53 in 3 M urea (Fig. 4), it actually speeded up aggregation, asdid R254, in the absence of urea at 37 °C and neutral pH, whichare conditions close to physiological (Fig. 2), and produced anaggregate of similar morphology to WT p53 (Fig. S2).Scheme for multisite aggregation. The kinetic and analytical resultsmay be summed up in Fig. 15 in a mechanism that may be a basisfor other proteins with multiple aggregation-prone sites. Onemolecule of p53 unfolds, partially or fully, to expose its aggre-gation-prone sequences (25, 27, 28, 50). Another molecule isinduced to unfold to give the elongation-competent state. Ag-gregation then occurs by elongation and cross-linking, either byhomologous pairing of sequences or by combination of homol-ogous and heterologous pairing. If one of the aggregation sites iseliminated by mutation, then there are sufficient remaining sitesfor aggregation to proceed. We conclude that elimination of asingle aggregation-prone site will not prevent aggregation of p53in vitro because of the presence of other aggregation-prone sites.Implications for inhibiting aggregation in vivo. Because eliminating theaggregation propensity of the major aggregation-prone site sur-rounding Ile254 or any of the other aggregation-prone sites didnot inhibit aggregation of p53 in vitro, we wondered how a singlepeptide, ReACp53, that caps the buried Ile254 region in the nativestate could be effective in rescuing mutant p53 in cancer cells (40).
Transthyretin, for example, has two aggregation-prone sites andrequires two peptides, one to cap each and one to inhibit aggre-gation (51). To investigate the possibility of inhibiting aggregationin cancer cells, we designed peptides targeting three different se-quences that drive aggregation of p53. They inhibited in vitro theself-aggregation of isolated peptides from p53 that contained theaggregation-prone sequences, but not the aggregation of full-length p53. Nevertheless, the peptides preferentially killed cancercells carrying destabilized structural mutants of p53. The peptideAnti254_3 that targeted the S9 (251–257) region inhibited the self-aggregation of the isolated S9 peptide in vitro and in killing cancercells to the same extent but at a fourfold lower concentration thanthe reported peptide ReACp53 (40). Peptides targeting the S9region acted synergistically in cytotoxicity with peptides targetingother amyloidogenic regions. Both Anti254_1 itself and the syn-ergistic combinations could selectively kill cancer cells at con-centrations not affecting normal cells. At first sight, it seems acompelling story that peptides and peptide combinations that capamyloidogenic sequences are anticancer drug leads, and the ap-proach of targeting different aggregation-driving regions of p53represents a promising anticancer strategy. However, we could notverify that p53 was the target. In vitro, the peptides did not inhibitthe aggregation of full-length p53, consistent with our observationsthat knocking out amyloidogenic sequences by mutagenesis doesnot inhibit aggregation. We could effectively knock down the ex-pression of the p53 gene using siRNA in cell lines but still couldnot eliminate, for example, the selective cytotoxicity of Anti254_1.At high concentrations, Anti254_1 killed cancer cells harboring astable contact mutant of p53 or even cancer cells lacking p53.There are many peptides that have anticancer effects. In par-
ticular, amphiphilic peptides, especially those amphiphilic peptidescontaining long cationic tails, selectively bind to cancer cells andcause cell death by cytoplasmic membrane disruption, whereasothers trigger apoptosis via death receptor or mitochondrialpathways (52, 53). It should be noted, however, that a version ofReAcp53 with a shuffled sequence is not cytotoxic (40). We did a
DAPI Pab240 (unfolded p53) Merge
Con
trol
DAPI Pab1620 (native p53) Merge
Con
trol
DAPI Pab240 (unfolded p53) Merge
Con
trol
DAPI Pab1620 (native p53) Merge
Con
trol
Ant
i197
_2_1
20 µ
MA
nti1
97_2
_120
µ M
Ant
i254
_1 1
2 µ
MA
nti2
54_1
12
µ M
Fig. 12. Anti254_1 treatment induces little change in the status of p53.NUGC3 cells were treated with 12 μM Anti254_1 for 6 h before fixation andimmunostaining. Pab240 was used to detect unfolded p53, whereas Pab1620was used to detect p53 in the native status. (Scale bar, 20 μm.)
-1
1
3
5
7
9
11
13
Rel
ativ
e ra
tio
of
mR
NA
leve
l
p21
MDM2
PUMA
NOXA
Fig. 13. Effect of peptides on a subset of p53 transcriptional targets. mRNAlevels of p53 target genes were measured by real-time PCR after a 4-htreatment. In NUGC3 cells, p21, Noxa, and Puma are all up-regulated, es-pecially when treated with combined Anti254_1 and Anti197_1. The MDM2level was not greatly affected.
0
50
100
150
200
Control 0.5 h 1.5 h 3 h
Rel
ativ
e L
um
ines
cen
ce (
%)
NUGC4NUGC3
050
100150200250300350400Caspase 3/7 ActivityA B
0
20
40
60
80
100
120
Via
bili
ty (
%)
0102030405060708090
100
Via
bili
ty (
%)
C D
Fig. 14. Effect of Anti254_1 and Anti197_2 on caspase 3/7 activity. Caspase3/7 activity was induced by treatment with 21 μMAnti254_1 for different times(A) and 150 μM Anti197_1 for 6 h (B). (C) Fifty micromolar Necrostatin-1 (Nec-1), 50 μM inactive necrostatin-1 (Nec-1-inact), and 100 μM carbobenzoxy-valyl-alanyl-aspartyl-[O-methyl]-fluoromethylketone (z-VAD-fmk; z-VAD) show littleinhibition on the activity of Anti254_1. (D) One hundred micromolar z-VADattenuates the effect of Anti197_2. Cell viability was measured after 24 h ofincubation. For treatment in the presence of Nec-1, Nec-1-inact, or z-VAD, cellswere treated with Nec-1, Nec-1-inact, or z-VAD for 3–4 h before addition ofAnti254_1 or Anti197_2, and the reported viability is relative to treatment withonly Nec-1, Nec-1-inact, or z-VAD, respectively. The viability of NUGC3 cellsafter treatment with Nec-1, Nec-1-inact, or z-VAD alone was 70 ± 4%, 91 ±5%, and 92 ± 3%, respectively, relative to the DMSO control.
Wang and Fersht PNAS | Published online March 14, 2017 | E2641
BIOCH
EMISTR
YPN
ASPL
US

preliminary analysis of the mechanism of cell death induced bythe peptides. The results were inconsistent with p53-inducedapoptosis and consistent with known cytotoxic pathways of an-ticancer peptides. Anti254_1 killed cancer cells with a half-timeof about 1 h, which is far faster than is usually found in p53-induced, transcription-dependent apoptosis (Fig. S8 A–C), and itinduces a rapid decrease of mitochondrial membrane potentialwithin 1 h (Fig. S8D). Mitochondrial p53 translocation can alsoinduce fast transcription-independent apoptosis. However, wedid not find translocation of p53 to mitochondria after a 30-mintreatment of 21 μM Anti254_1 (Fig. S7). This fast cell death isnot via activating caspase 3/7, but is likely to occur via necrosis,although leading to cell nucleus condensation (Fig. 14 and Fig.S7). Similarly, necrostatin-1 and pan-caspase inhibitor show onlypartial inhibition on toxicity of ReACp53 (40).In contrast to our finding of multiple aggregation sites and
that mutating I254 to R or D speeds up aggregation, Xu et al.(32) report only the finding of the I254-based sequence onproteolysis of harvested aggregates of p53, and find that muta-tion of I254R prevents aggregation, abrogates oncogenic gain offunction, and inhibits binding of mutant p53 with p63/p73.However, others report p53I254R does bind to p63/p73, has gainof function, and promotes invasion (33).How do the peptides act? There is little doubt that ReACp53 (40),and now other peptides that target further amyloidogenic sequenceswithin oncogenic mutant p53, are indeed cytotoxic toward cancercells. The question now is how they act. There is not a simple ex-planation for several reasons, including that they may work simul-taneously by multiple routes and that there are pleiotropic effectson mutating p53, including both loss and gain of function. Anexample of multiple routes is a small alkylator that covalentlybinds to mutant p53 in some cases, stabilizes it, and rescues itsactivity in some cancer cell lines, but kills other cancer cell lines viaa reactive oxygen species (54). Similarly, several small moleculesappear to reactivate p53 but, instead, cause cell death by otherroutes (3). There is major p53-independent activity in several celllines we tested, but that finding does not preclude possible majorp53-dependent activity in other cell lines or in tumors. Do thepeptides prevent loss of function by directly inhibiting aggrega-tion? If p53 aggregates in cells by the same process as in vitro, thenthe peptides are unlikely to function by inhibiting its aggregationfrom the native state. However, there may be as yet unknowneffects of chaperones in vivo, perhaps by binding to partly dena-tured states. Do they affect gain of function? Mutant p53 may beoncogenic because of gain of function by interacting with otherproteins or DNA. For example, peptides may function partly byscreening the interaction between p53 and the transactivation in-hibition domain of alpha isomers of p63/p73 (55), Indeed, a pre-liminary experiment knocking down p63 showed cell death wasdependent on p63 (Fig. S9). Peptides might affect the oncogenicinterplay between Nrf2 and mutant p53 (56). There is evidence foractivity by more classical mechanisms of disruption of cell mem-branes and other processes, especially at high concentrations. Wemust emphasize that our results in cancer cell lines parallel the re-sults found in a study by Soragni et al. (40), and that experiments inconventional 2D cultures as used by us may overestimate nonspecific
cytotoxicity compared with 3D cultures and solid tumors. However,consistent with our results in cancer cells, our in vitro data do pointto non-p53 entities as being important targets of the antiaggregationpeptides, as well as possibilities of their interactions with p53.
MethodsPrediction of Aggregation Sites. Aggregation sites were predicted using theprograms Tango (41), Waltz (42), Aggrescan (43), FoldAmyloid (44), andAmylpred2 as well as by a consensus method combining the first three andNetCSSP, amyloid mutants, Pafig, amyloidogenic pattern, SecStr, averagepacking density, β-strand contiguity, and hexapeptide conformational en-ergy (45). Sequences predicted to be aggregation-prone by at least sixprograms were chosen to be the aggregation sites. A threshold of 90% andpH 7.0 were used when predicted by Waltz. For prediction by FoldAmyloid,the expected number of contacts within 8 Å was used as a scale; the aver-aging frame and reliable frame are both 5, and the cutoff value was 21.4.
Kinetics of Aggregation. Aggregation kinetics of native p53 variants weremonitored by light scattering and ThT fluorescence using a Horiba FluoroMax-3spectrophotometer as described previously (25). For aggregation of denaturedp53 variants, proteins were denatured in 4 M urea on ice overnight beforemeasurements. Experiments were generally performed with a protein con-centration of 10 μM in 25 mM potassium or sodium phosphate (pH 7.2),150 mMNaCl, 1 mM Tris(2-carboxyethyl)phosphine (TCEP), 5% (vol/vol) DMSO,20 μM ThT, and 3 M urea, and were monitored as for the native proteins. Totest the effect of antiaggregation peptides on aggregation of S9 peptide(Acetyl-RPILTIITLE-amide), S5–6 peptide, and S10 peptide, S9, S5–6, or S10peptide in 6 M GdmCl was diluted into buffer finally containing 25 mM po-tassium phosphate (pH 7.2), 150 mM NaCl, 1 mM TCEP, 22.7 μM or 20 μM ThT,and certain concentrations of GdmCl and antiaggregation peptides as in-dicated. Antiaggregation peptide stocks were prepared with sterilized Milli-Qwater. To test the initial aggregation rate of denatured p53 mutants in dif-ferent concentrations of urea, 3 μM p53 mutant was diluted into the abovebuffer with various concentrations of urea at 37 °C.
Limited Proteolysis. WTC, WTCI254D, WTFL, and WTFLG245S were incubated at37 °C usually for 2 h (also at 1 and 3 h for WTC and overnight for WTCI254D) in25mMpotassium phosphate (pH 7.2), 150 mMNaCl, 1 mMTCEP, and 5% (vol/vol)DMSO. Aggregates were harvested by centrifugation at 4 °C at 15,682 × g for30 min and washed once with proteolysis buffer [20 mM Tris·HCl (pH 7.4),1 mM TCEP] to remove residual phosphate buffer and soluble p53. Limitedproteolysis of either the aggregates or soluble native states of WTC,WTCI254D, WTFL, and WTFLG245S with trypsin was carried out in 20 mMTris·HCl (pH 7.4) and 1 mM TCEP at 20 °C. The enzyme/substrate (E/S) ratio forWTC and WTCI254D was 1:50 (wt/wt), and the enzyme/substrate ratio forWTFL and WTFLG245S was 1:100 (wt/wt). At a specified time, the remainingaggregate was separated from supernatant by centrifugation at 15,682 × gfor 30 min. The reaction was quenched with acetic or trifluoroacetic acid. Theremaining aggregate was dissolved in 70% (vol/vol) acetonitrile/3% (vol/vol)trifluoroacetic acid. Mass determination was performed using aMALDI-TOFmassspectrometer (Voyager-DE Pro; Applied Biosystems). Proteolysis by proteinaseK of native WTC and aggregate of WTC was carried out using an E/S ratio of1:22.5 (wt/wt) at 20 °C. The reaction was quenched by heating at 90 °C for 5 min.Mass and sequence determination of the resulted peptides was performedusing bothMALDI-TOF and Ultraflex III MALDI-TOF/TOF (Bruker Daltonics) MS.
Cell Lines and Culture Conditions. NUGC3 (p53-Y220C+/+), NUGC4 (WT p53+/+),and MKN1 (p53-V143A+/+) cells were obtained from the Japan Health Sci-ence Research Resources Bank, and they were maintained in RPMI medium.SKBR3 was purchased from the American Type Culture Collection, and MCF7(WT p53+/+), SW480, and WI38 fibroblast cells were maintained in DMEM. Allof the media were supplemented with 10% FBS and 1% penicillin/strepto-mycin (10,000 U/mL penicillin, 10,000 μg/mL streptomycin). The FBS was heat-inactivated. Other cell lines were cultured in RPMI 1640 GlutaMAX mediumwith the same concentration of serum and antibiotics. All cell cultures weremaintained at 37 °C and in 5% CO2 in a humidified incubator.
Cell Viability Assay. Cells (7,500 cells per well) were seeded in 96-well plates andcultured to about 60% confluence on the second day. Then, old medium wasreplaced by new medium with peptides or DMSO control. When test peptideswere combined, peptides were added to the cells simultaneously. After 24-htreatment, except if indicated otherwise, cell viability was assessed by mea-suring the intracellular levels of ATP using a Cell Titer-Glo Luminescent CellViability Assay Kit (Promega) according to the manufacturer’s instructions.
Fig. 15. Schematic mechanism for aggregation of p53. (A) Aggregationprocess. (B) Aggregation could not be blocked by only targeting one ag-gregation-driving region.
E2642 | www.pnas.org/cgi/doi/10.1073/pnas.1700308114 Wang and Fersht

Immunofluorescence. Cells were treated with peptides or DMSO control forthe indicated time and were then washed with PBS and fixed with 4% (vol/vol)paraformaldehyde for 10 min at room temperature. After being rinsed withPBS, cells were permeabilized with 0.5% (wt/vol) Triton X-100 in PBS for 5 minand blocked with 2% (wt/vol) BSA or 5% goat serum. The primary antibodieswere incubated overnight at 4 °C, and secondary antibody goat anti-mouseDylight488 was diluted to 1:1,000 and incubated for 1 h. The following pri-mary antibodies were used: anti-p53 antibody Pab 1620 (Abcam) and anti-p53antibody Pab 240 (Santa Cruz Biotechnology). Hoechst 33342 (Cell Signaling)or DAPI and MitoTracker Red (Lonza) were used to stain the nucleus andmitochondria of cells, respectively. Images were acquired using a Leica TCS SP8confocal microscope.
Western Blots. Cell lysates were prepared with radioimmunoprecipitation assaybuffer (Sigma) containing a protease inhibitormixture (Roche) after treatment ofpeptides or DMSO control. The lysates were run on SDS/PAGE and transferredonto polyvinylidene fluoride membranes. Membranes were blocked for 1 h with5% (wt/vol) milk in Tris-buffered saline containing 0.1% Tween-20 (TBST) atroom temperature before immunoblotting. The membranes were incubated
with primary antibodies at 4 °C overnight. After three rinses with TBST, themembranes were stained with horseradish peroxidase-conjugated anti-mouseor anti-rabbit IgG (Dako) and imaged with ChemiDoc XRS (Bio-Rad). The fol-lowing primary antibodies were used: p53 DO-7 (Dako), p21 (Millipore),MDM2 (Abcam), Bax (Cell Signaling), and β-actin (Abcam).
p53 Knockdown by siRNA. Cancer cells were transfected with the siRNA di-rected against human p53 (Cell Signaling Technology), p63 (OriGene), ornontargeting negative control siRNA (Qiagen) using Lipofectamine RNAi-MAX (Life Technologies) according to the manufacturer’s protocol. The finalconcentration of the siRNAs was 10 nM.
ACKNOWLEDGMENTS. We thank Matthias Baud for synthesizing theN-methylated peptides, Frank Abendroth for help with synthesis of theAnti254_1 peptide, and the laboratory MS facility for help with MS experi-ments using Ultraflex III MALDI-TOF/TOF. We thank Drs. David Eisenberg andAlice Soragni for sharing with us unpublished data and for their insightfulcomments. This work was funded by European Research Council AdvancedGrant 268506 (P53LAZARUS to A.R.F.).
1. Freed-Pastor WA, Prives C (2012) Mutant p53: One name, many proteins. Genes Dev26(12):1268–1286.
2. Hainaut P, Hollstein M (2000) p53 and human cancer: The first ten thousand muta-tions. Adv Cancer Res 77:81–137.
3. Joerger AC, Fersht AR (2016) The p53 pathway: Origins, inactivation in cancer, andemerging therapeutic approaches. Annu Rev Biochem 85:375–404.
4. Bullock AN, et al. (1997) Thermodynamic stability of wild-type and mutant p53 coredomain. Proc Natl Acad Sci USA 94(26):14338–14342.
5. Bullock AN, Fersht AR (2001) Rescuing the function of mutant p53. Nat Rev Cancer1(1):68–76.
6. Bullock AN, Henckel J, Fersht AR (2000) Quantitative analysis of residual folding andDNA binding in mutant p53 core domain: Definition of mutant states for rescue incancer therapy. Oncogene 19(10):1245–1256.
7. Aguzzi A, O’Connor T (2010) Protein aggregation diseases: Pathogenicity and ther-apeutic perspectives. Nat Rev Drug Discov 9(3):237–248.
8. Koo EH, Lansbury PT, Jr, Kelly JW (1999) Amyloid diseases: Abnormal protein ag-gregation in neurodegeneration. Proc Natl Acad Sci USA 96(18):9989–9990.
9. Goedert M (2015) NEURODEGENERATION. Alzheimer’s and Parkinson’s diseases: The prionconcept in relation to assembled Aβ, tau, and α-synuclein. Science 349(6248):1255555.
10. Nelson R, et al. (2005) Structure of the cross-beta spine of amyloid-like fibrils. Nature435(7043):773–778.
11. Sawaya MR, et al. (2007) Atomic structures of amyloid cross-beta spines reveal variedsteric zippers. Nature 447(7143):453–457.
12. Sambashivan S, Liu Y, Sawaya MR, Gingery M, Eisenberg D (2005) Amyloid-like fibrilsof ribonuclease A with three-dimensional domain-swapped and native-like structure.Nature 437(7056):266–269.
13. Eisenberg D, Jucker M (2012) The amyloid state of proteins in human diseases. Cell148(6):1188–1203.
14. Nelson R, Eisenberg D (2006) Recent atomic models of amyloid fibril structure. CurrOpin Struct Biol 16(2):260–265.
15. Chiti F, Dobson CM (2006) Protein misfolding, functional amyloid, and human disease.Annu Rev Biochem 75:333–366.
16. Makin OS, Serpell LC (2005) Structures for amyloid fibrils. FEBS J 272(23):5950–5961.17. Westermark P, Engström U, Johnson KH, Westermark GT, Betsholtz C (1990) Islet
amyloid polypeptide: Pinpointing amino acid residues linked to amyloid fibril for-mation. Proc Natl Acad Sci USA 87(13):5036–5040.
18. Frare E, et al. (2006) Identification of the core structure of lysozyme amyloid fibrils byproteolysis. J Mol Biol 361(3):551–561.
19. Come JH, Fraser PE, Lansbury PT, Jr (1993) A kinetic model for amyloid formation inthe prion diseases: Importance of seeding. Proc Natl Acad Sci USA 90(13):5959–5963.
20. Harper JD, Lansbury PT, Jr (1997) Models of amyloid seeding in Alzheimer’s diseaseand scrapie: Mechanistic truths and physiological consequences of the time-dependent solubility of amyloid proteins. Annu Rev Biochem 66:385–407.
21. Knowles TP, Vendruscolo M, Dobson CM (2014) The amyloid state and its associationwith protein misfolding diseases. Nat Rev Mol Cell Biol 15(6):384–396.
22. Friedler A, Veprintsev DB, Hansson LO, Fersht AR (2003) Kinetic instability of p53 core domainmutants: Implications for rescue by small molecules. J Biol Chem 278(26):24108–24112.
23. Ano Bom AP, et al. (2012) Mutant p53 aggregates into prion-like amyloid oligomersand fibrils: Implications for cancer. J Biol Chem 287(33):28152–28162.
24. Butler JS, Loh SN (2003) Structure, function, and aggregation of the zinc-free form ofthe p53 DNA binding domain. Biochemistry 42(8):2396–2403.
25. Wilcken R, Wang G, Boeckler FM, Fersht AR (2012) Kinetic mechanism of p53 oncogenicmutant aggregation and its inhibition. Proc Natl Acad Sci USA 109(34):13584–13589.
26. Ishimaru D, et al. (2003) Fibrillar aggregates of the tumor suppressor p53 core do-main. Biochemistry 42(30):9022–9027.
27. Wang G, Fersht AR (2015) Mechanism of initiation of aggregation of p53 revealed byΦ-value analysis. Proc Natl Acad Sci USA 112(8):2437–2442.
28. Wang G, Fersht AR (2015) Propagation of aggregated p53: Cross-reaction andcoaggregation vs. seeding. Proc Natl Acad Sci USA 112(8):2443–2448.
29. Cho Y, Gorina S, Jeffrey PD, Pavletich NP (1994) Crystal structure of a p53 tumor suppressor-DNA complex: Understanding tumorigenic mutations. Science 265(5170):346–355.
30. Joerger AC, Allen MD, Fersht AR (2004) Crystal structure of a superstable mutant ofhuman p53 core domain. Insights into the mechanism of rescuing oncogenic muta-tions. J Biol Chem 279(2):1291–1296.
31. Cañadillas JM, et al. (2006) Solution structure of p53 core domain: Structural basis forits instability. Proc Natl Acad Sci USA 103(7):2109–2114.
32. Xu J, et al. (2011) Gain of function of mutant p53 by coaggregation with multipletumor suppressors. Nat Chem Biol 7(5):285–295.
33. Stindt MH, et al. (2015) Functional interplay between MDM2, p63/p73 and mutantp53. Oncogene 34(33):4300–4310.
34. Friedler A, et al. (2002) A peptide that binds and stabilizes p53 core domain: Chaperonestrategy for rescue of oncogenic mutants. Proc Natl Acad Sci USA 99(2):937–942.
35. Friedler A, et al. (2004) Structural distortion of p53 by the mutation R249S and its rescue bya designed peptide: Implications for “mutant conformation”. J Mol Biol 336(1):187–196.
36. Boeckler FM, et al. (2008) Targeted rescue of a destabilized mutant of p53 by an insilico screened drug. Proc Natl Acad Sci USA 105(30):10360–10365.
37. Kaar JL, et al. (2010) Stabilization of mutant p53 via alkylation of cysteines and effectson DNA binding. Protein Sci 19(12):2267–2278.
38. Liu X, et al. (2013) Small molecule induced reactivation of mutant p53 in cancer cells.Nucleic Acids Res 41(12):6034–6044.
39. Wilcken R, et al. (2012) Halogen-enriched fragment libraries as leads for drug rescueof mutant p53. J Am Chem Soc 134(15):6810–6818.
40. Soragni A, et al. (2016) A designed inhibitor of p53 aggregation rescues p53 tumorsuppression in ovarian carcinomas. Cancer Cell 29(1):90–103.
41. Fernandez-Escamilla AM, Rousseau F, Schymkowitz J, Serrano L (2004) Prediction ofsequence-dependent and mutational effects on the aggregation of peptides andproteins. Nat Biotechnol 22(10):1302–1306.
42. Maurer-Stroh S, et al. (2010) Exploring the sequence determinants of amyloid struc-ture using position-specific scoring matrices. Nat Methods 7(3):237–242.
43. Conchillo-Solé O, et al. (2007) AGGRESCAN: A server for the prediction and evaluationof “hot spots” of aggregation in polypeptides. BMC Bioinformatics 8:65.
44. Garbuzynskiy SO, Lobanov MY, Galzitskaya OV (2010) FoldAmyloid: A method of pre-diction of amyloidogenic regions from protein sequence. Bioinformatics 26(3):326–332.
45. Tsolis AC, PapandreouNC, IconomidouVA,Hamodrakas SJ (2013) A consensusmethod forthepredictionof ‘aggregation-prone’peptides inglobularproteins.PLoSOne8(1):e54175.
46. Schymkowitz J, et al. (2005) The FoldX web server: an online force field. Nucleic AcidsRes 33(Web Server issue):W382–W388.
47. Vitalis A, Pappu RV (2011) Assessing the contribution of heterogeneous distributions of olig-omers to aggregation mechanisms of polyglutamine peptides. Biophys Chem 159(1):14–23.
48. Brinckerhoff LH, et al. (1999) Terminal modifications inhibit proteolytic degradationof an immunogenic MART-1(27-35) peptide: Implications for peptide vaccines. Int JCancer 83(3):326–334.
49. Chou TC (2010) Drug combination studies and their synergy quantification using theChou-Talalay method. Cancer Res 70(2):440–446.
50. Wang G, Fersht AR (2012) First-order rate-determining aggregation mechanism ofp53 and its implications. Proc Natl Acad Sci USA 109(34):13590–13595.
51. Saelices L, et al. (2015) Uncovering the mechanism of aggregation of human trans-thyretin. J Biol Chem 290(48):28932–28943.
52. Boohaker RJ, Lee MW, Vishnubhotla P, Perez JM, Khaled AR (2012) The use of ther-apeutic peptides to target and to kill cancer cells. Curr Med Chem 19(22):3794–3804.
53. Zhao J, Hao X, Liu D, Huang Y, Chen Y (2015) In vitro characterization of the rapidcytotoxicity of anticancer peptide HPRP-A2 through membrane destruction and in-tracellular mechanism against gastric cancer cell lines. PLoS One 10(9):e0139578.
54. Bauer MR, Joerger AC, Fersht AR (2016) 2-Sulfonylpyrimidines: Mild alkylating agents withanticancer activity toward p53-compromised cells. Proc Natl Acad Sci USA 113(36):E5271–E5280.
55. Kehrloesser S, et al. (2016) Intrinsic aggregation propensity of the p63 and p73 TIdomains correlates with p53R175H interaction and suggests further significance ofaggregation events in the p53 family. Cell Death Differ 23(12):1952–1960.
56. Walerych D, et al. (2016) Proteasomemachinery is instrumental in a common gain-of-functionprogram of the p53 missense mutants in cancer. Nat Cell Biol 18(8):897–909.
Wang and Fersht PNAS | Published online March 14, 2017 | E2643
BIOCH
EMISTR
YPN
ASPL
US