mutant viral polymerase in the transition of virus to error catastrophe identifies a critical site...
TRANSCRIPT

doi:10.1016/j.jmb.2005.09.022 J. Mol. Biol. (2005) 353, 1021–1032
Mutant Viral Polymerase in the Transition of Virus toError Catastrophe Identifies a Critical Site forRNA Binding
Armando Arias1, Ruben Agudo1, Cristina Ferrer-Orta2
Rosa Perez-Luque2, Antero Airaksinen1, Emiliana Brocchi3
Esteban Domingo1*, Nuria Verdaguer2 and Cristina Escarmıs1
1Centro de Biologıa Molecular“Severo Ochoa” (CSIC-UAM)Cantoblanco, E-28049 MadridSpain
2Institut de Biologıa Molecularde Barcelona (CSIC), ParcCientific de Barcelona, JosepSamitier 1-5, E-08028 BarcelonaSpain
3Istituto ZooprofilatticoSperimentale della Lombardia edell’Emilia, 25125 BresciaItaly
0022-2836/$ - see front matter q 2005 E
Abbreviations used: FMDV, foot-virus; 3D, FMDV-coded RNA-depease; BSA, bovine serum albumin; mantibody.
E-mail address of the [email protected]
A foot-and-mouth disease virus (FMDV) polymerase (3D) with amino acidreplacements G118D, V239M and G373D (triple DMD mutant) wasobtained from a molecular clone derived from a virus population treatedwith ribavirin, in the transition to error catastrophe (virus extinctionthrough lethal mutagenesis). DMD 3D was expressed in Escherichia coli,purified, and its activity compared with that of wild-type enzyme andmutant enzymes with either replacement G118D, G118A or D338A (thelatter affecting the catalytic motif YGDD), generated by site-directedmutagenesis. No differences among the enzymes were noted in theirinteraction with monoclonal antibodies specific for the FMDV polymerase.Mutant enzymes with G118D or G118A showed a 100-fold decrease inpolymerization activity relative to wild-type 3D, using poly(A)/oligo(dT)15
and poly(A)/VPg as template-primers, under several reaction conditions.As expected, the activity of 3D with D338A was undetectable (!0.01 timesthe value for wild-type 3D). DMD and the G118 mutants showed impairedbinding to template-primer RNA whereas the D338A mutant showed abinding similar to wild-type 3D. Transfection of cells with FMDV RNAencoding DMD 3D resulted in selection of revertant viruses thatmaintained only substitutions V239M and G373D. Consistently, wheninfectious transcripts encoded 3D with either G118D, G118A or D338A,viruses with reversions to the wild-type sequence were isolated. Theimplication of G118 in template–primer binding is supported by thelocation of this residue in the template-binding groove of the FMDVpolymerase. In addition to identifying an amino acid residue that is criticalfor the binding of polymerase to RNA, the results document the presence ofdefective genomes in the transition of virus to error catastrophe.
q 2005 Elsevier Ltd. All rights reserved.
Keywords: viral quasispecies; lethal mutagenesis; RNA-dependent RNApolymerase; foot-and-mouth disease virus; reversion
*Corresponding authorIntroduction
Picornavirus replication is catalyzed by a virus-coded RNA-dependent RNA polymerase termed3D.1,2 Recently we have described the three-
lsevier Ltd. All rights reserve
and-mouth diseasendent RNA polymer-Ab, monoclonal
ing author:
dimensional structure of foot-and-mouth diseasevirus (FMDV) 3D unliganded and bound to atemplate-primer RNA decanucleotide, and haveidentified critical amino acid residues involved inviral RNA synthesis.3 More recently, the 3D ofFMDV O1Kaufbeuren has been characterized bio-chemically.4 Replication of FMDV, as that of otherRNA viruses, is error-prone, with mutation rates of10K3 to 10K5 misincorporations per nucleotidecopied.5,6 This low copying fidelity results inrapid generation of dynamic mutant spectra termedviral quasispecies.7–11During RNA replication,
d.

1022 Polymerase Defective in RNA Binding
mutant RNAs are continuously generated andsubjected to a process of competition and selectionof the most fit mutant distributions.6–11 A conse-quence of such a quasispecies dynamics is theexistence of an error threshold for template copyingabove which the genetic information cannot bemaintained. Violation of the error threshold resultsin virus extinction, as documented with severalvirus–host systems in cell culture and in vivo.11–13
Virus extinction through enhanced mutagenesis,also termed lethal mutagenesis, is currently studiedas a potential new antiviral strategy. However, thebiochemical basis of viral extinction remains largelyunexplored.
Our laboratory has investigated lethal muta-genesis of FMDV by 5-fluorouracil, 5-azacytidineand ribavirin (1-b-D-ribofuranosyl-1, 2, 4-triazole-3-carboxamide).14–18 The transition of FMDV towardsextinction involved decreases of 102 to 103-fold inspecific infectivity,19 without modification of thegenomic consensus nucleotide sequence.20 Pre-extinction FMDV RNA (the RNA of the viralpopulation subjected to mutagenesis that precedesthe population from which infectivity or virus-specific RT-PCR amplifiable material can no longerbe retrieved) interfered with the infectivity ofstandard RNA cotransfected into the same cells.19
It was proposed that both the decrease of infectivityand the interfering activity were due to expressionof abnormal or inactive FMDV proteins, but a directproof of the presence of functionally defective geneproducts in the transition towards FMDV extinctionwas lacking. Here, we show that an FMDVpolymerase (3D) isolated from a molecular cloneof an FMDV population subjected to ribavirin-induced mutagenesis includes three amino acidsubstitutions, and has a drastically impairedenzyme activity in nucleotide polymerization andin binding to template-primer RNA. Gel mobility-shift analysis with this enzyme, with a viablerevertant, and with additional 3D mutants con-
3D are detailed in Materials and Methods and by Ferrer-Ostudy were purified using the same procedure and showedbrilliant blue staining. (b) Standard polymerization assay uFMDV 3D. Reaction conditions and measurement of [a-32P]U3D, Mg2C, poly(A) and oligo(dT)15 are required for activitincorporation).
structed by site-directed mutagenesis, has identi-fied Gly118 as essential for FMDV 3D binding toRNA. The critical implication of this conservedamino acid in the binding of 3D to RNA is alsosupported by its location in the 3D molecule and itsinteractions with neighbor residues, as indicated bythe three-dimensional structure of FMDV 3Dunliganded and bound to template-primer.3
Results
Characterization of the FMDV polymerase (3D)
The polymerase (3D) of FMDV C-S8c1 wasexpressed in Escherichia coli, purified by Ni-NTAchromatography as described,3 and activityrequirements analyzed (Figure 1). A UTP incorpor-ation assay with poly(A)/oligo(dT)15 as template-primer, and a gel mobility-shift, RNA binding assay,were used to determine Mn or Mg ion concen-tration–activity profiles at a fixed temperature, andthe temperature–activity profile at a fixed Mg ionconcentration (Figure 2). These experiments servedto establish standard polymerization and RNAbinding assays to compare wild-type FMDVC-S8c1 and mutant polymerases, as detailed inMaterials and Methods. The results show a parallelvariation of polymerization and RNA bindingactivities, except for Mn ion concentrations above1.5 mM, at which polymerization was impairedrelative to RNA binding (Figure 2(b)). To ascertainthe specificity of the UTP incorporation assay, theinhibition of 3D activity by monoclonal antibodies(mAbs) that recognize different epitopes on theenzyme was measured, and the VPg uridylationactivity was tested. All 3D-specific mAbs reactedpositively with 3D in ELISA (Figure 3). However,while mAbs that bind to the N-terminal region of3D (mAbs 3B9, 3H11, 3F12 and 4B12) caused 80% orhigher inhibition of UTP incorporation activity,
Figure 1. Purification and activityof FMDV polymerase (3D)expressed in E. coli. (a) SDS-PAGEof 3D at different stages of purifi-cation. Extract of E. coli BL21 pLysScontaining plasmid pET-28a3Dpol,prepared from a culture untreated(lane 1) or treated (lane 2) withIPTG. Lane 3, protein purified byNi-NTA chromatography. Lane M,molecular mass markers. The gelwas stained with Coomassie brilli-ant blue R-250 in a solution ofmethanol and acetic acid asdescribed.23 The different stepsinvolved and fold-purification of
rta et al.3 All mutant polymerases used in the presentO95% purity according to SDS-PAGE and Coomassie
sing poly(A)/oligo(dT)15 as template-primer and purifiedTP incorporation are detailed in Materials and Methods.
y (left column, DE81 filter used to measure [a-32P]UTP

Figure 2. RNA polymerizationand RNA binding activities ofFMDV 3D. Polymerization activity(filled circles) was measured usingpoly(A)/oligo(dT)15 as the tem-plate-primer. RNA binding activity(open squares) was determined bygel mobility-shift assays usingsym/sub RNA decanucleotide26
as template-primer. Polymer-ization and RNA binding assayswere carried out as detailed inMaterials and Methods, withdifferent concentrations of (a)Mg(CH3COO)2, (b) MnCl2, at37 8C, or at different temperatures,(c) using 5 mM Mg(CH3COO)2.Activities are represented as thepercentage of the maximum valueobtained in each assay series.Values are the average of at leastthree determinations; standarddeviations are given.
Polymerase Defective in RNA Binding 1023
mAbs that map around residues 129 to 143 of 3D(mAbs 2D10 and 2D11) did not cause significantinhibition (Figure 4). These results are consistentwith the location of the epitopes in the three-dimensional structure of 3D, since mAbs 4B12 and3B9, which recognize residues 9–28 of 3D, can blockthe template entry channel, and mAbs 3F12 and3H11, which recognize residues 29–38 and 49–68,respectively, can affect the nucleotide entry site. Incontrast, the epitopes defined by mAbs 2D10 and2D11 are located at an exposed region that is distantfrom the catalytic site and RNA-binding domain(Figure 5).
Purified 3D was active in uridylating syntheticVPg in a time-dependent manner but was not activeon unrelated peptides containing a Tyr residue
chosen at random from a peptide library (Figure 6).The uridylated product was sensitive to trypsin andendopeptidase Glu-C, and yielded VPgpU uponRNase treatment (Figure 6(b)). Thus, FMDV 3Dexpressed in E. coli and purified by affinity chro-matography has polymerase activity that is specifi-cally inhibited by some anti-3D mAbs, and canuridylate cognate VPg also in a specific manner.
A mutant polymerase defective in RNA binding
FMDV subjected to lethal mutagenesis byreplicating in the presence of mutagenic agentsalone or in combination with antiviral inhibitorsproduces complex mutant spectra prior to virusextinction.14,15,17–19 Treatment of cells persistently
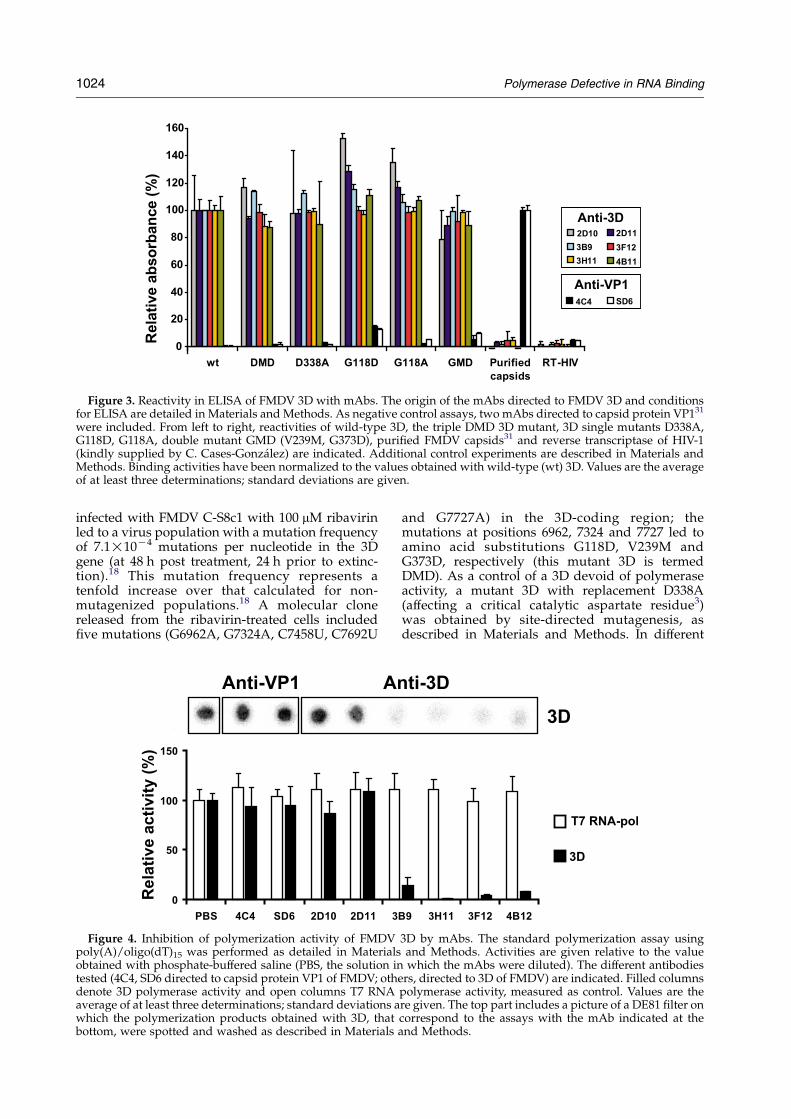
Figure 3. Reactivity in ELISA of FMDV 3D with mAbs. The origin of the mAbs directed to FMDV 3D and conditionsfor ELISA are detailed in Materials and Methods. As negative control assays, two mAbs directed to capsid protein VP131
were included. From left to right, reactivities of wild-type 3D, the triple DMD 3D mutant, 3D single mutants D338A,G118D, G118A, double mutant GMD (V239M, G373D), purified FMDV capsids31 and reverse transcriptase of HIV-1(kindly supplied by C. Cases-Gonzalez) are indicated. Additional control experiments are described in Materials andMethods. Binding activities have been normalized to the values obtained with wild-type (wt) 3D. Values are the averageof at least three determinations; standard deviations are given.
1024 Polymerase Defective in RNA Binding
infected with FMDV C-S8c1 with 100 mM ribavirinled to a virus population with a mutation frequencyof 7.1!10K4 mutations per nucleotide in the 3Dgene (at 48 h post treatment, 24 h prior to extinc-tion).18 This mutation frequency represents atenfold increase over that calculated for non-mutagenized populations.18 A molecular clonereleased from the ribavirin-treated cells includedfive mutations (G6962A, G7324A, C7458U, C7692U
Figure 4. Inhibition of polymerization activity of FMDVpoly(A)/oligo(dT)15 was performed as detailed in Materialsobtained with phosphate-buffered saline (PBS, the solution intested (4C4, SD6 directed to capsid protein VP1 of FMDV; othedenote 3D polymerase activity and open columns T7 RNAaverage of at least three determinations; standard deviations awhich the polymerization products obtained with 3D, thatbottom, were spotted and washed as described in Materials
and G7727A) in the 3D-coding region; themutations at positions 6962, 7324 and 7727 led toamino acid substitutions G118D, V239M andG373D, respectively (this mutant 3D is termedDMD). As a control of a 3D devoid of polymeraseactivity, a mutant 3D with replacement D338A(affecting a critical catalytic aspartate residue3)was obtained by site-directed mutagenesis, asdescribed in Materials and Methods. In different
3D by mAbs. The standard polymerization assay usingand Methods. Activities are given relative to the valuewhich the mAbs were diluted). The different antibodies
rs, directed to 3D of FMDV) are indicated. Filled columnspolymerase activity, measured as control. Values are there given. The top part includes a picture of a DE81 filter oncorrespond to the assays with the mAb indicated at theand Methods.

Figure 5. Stereo view of a ribbon diagram of the structure of the FMDV polymerase (3D). The structure highlights theamino acid stretches that include the epitopes recognized by mAbs 4B12 and 3B9 (residues in blue), 3F12 (red), 3H11(yellow), 2D10 and 2D11 (magenta). The positions of the RNA template and the incoming NTP are shown as a referencein dark and light green, respectively. Structure based on Ferrer-Orta et al.3
Polymerase Defective in RNA Binding 1025
polymerization assays the DMD mutant showed nodetectable activity, indistinguishable from back-ground levels shown by the inactive D338Apolymerase. The activity of the wild-type C-S8c13D was 70 to 1640-fold higher than that of mutantDMD using different in vitro assays. Despite theabsence of activity, purified 3D mutants DMD andD338A were recognized by 3D-specific monoclonalantibodies in ELISA (Figure 3) and Western blotassays (data not shown).
To identify the step of the RNA synthesis processaffected in mutant DMD, gel retardation of sym/sub RNA by increasing amounts of wild-type andmutant 3Ds was measured (Figure 7). Binding ofDMD 3D to RNA was not detectable (0.08(G0.09)%retarded molecules as compared to 40.5(G5.6)% forwild-type 3D), using 1800 nM 3D (Figure 7). Thisidentifies RNA binding as a defect of DMD 3D inRNA synthesis.
3D revertants to G118 regain RNA binding
To identify the residue(s) responsible of theactivity defect of DMD 3D, transcripts of cDNA ofFMDV encoding 3D mutants with replacementsG118D, or G118A, or V239M, or D338A, or V239Mand G373D (double mutant), or G118D and V239M(double mutant), or G118D and V239M and G373D(triple DMD mutant), were transfected into BHK-21cells. RNA from the virus produced at 72 h aftertransfection (complete cytopathology) was ampli-fied, and the relevant 3D-coding region sequencedas described in Materials and Methods. The 3Dsubstitutions included in the parental infectioustranscripts were maintained in progeny virus in allcases except for transcripts encoding 3D replace-ments G118A, G118D or D338A, which showed adelay in cytopathology, and produced revertantviruses in all cases. The DMD mutant reverted toGly in position 118 but maintained replacementsV239M and G373D. The progeny double mutantV239M/G373D regained in vitro RNA binding,polymerization and VPg uridylation activities,
that reached levels similar to those of wild-type3D (Table 1 and Figure 7). Both the cell transfectionexperiments and in vitro assays with purified 3Dagree, indicating an essential role for G118 in virusreplication.
Structural interpretation
The three-dimensional structure of the complexbetween FMDV 3D and a template-primer RNA3
provides a structural interpretation of the role ofG118 in RNA binding, and of the deleterious effectof substitutions G118D and G118A. G118 is locatedin a loop connecting helix a4 and strand b3 in thefingers subdomain (Figure 8(a)). The loop formspart of the template entry channel and someneighboring residues within this loop; in particular,A116 and T115 directly participate in hydrogenbonded contacts with the sugar phosphate back-bone of the RNA template3 (see also Discussion).Thus, the molecular environment of G118 suggeststhat its replacement by any amino acid shouldimpair the binding of 3D to RNA.
Discussion
We have recently reported the expression andpurification of the FMDV C-S8c1 polymerase (3D)and solved the crystal structure of the enzyme bothalone and in complex with a template-primermolecule.3 Here, we describe different in vitroactivities (poly(U) synthesis, VPg-uridylation,RNA-binding) displayed by the purified enzyme.C-S8c1 3D can uridylate VPg specifically, with theonly requirement being poly(A). In contrast, therecently characterized O1Kaufbeuren enzyme4
required both the presence of the cre (cis replicatingelement) stem–loop molecule as template2 and the3CD polypeptide to uridylate the FMDV VPgin vitro. The poly(A) molecule as template was notenough to uridylate VPg.4 Further investigationsare needed to clarify this difference.

Figure 6. Analysis of the products of VPg uridylationby FMDV 3D. The standard uridylation reaction and theanalysis of the products by SDS-PAGE were performed asdescribed in Materials and Methods. (a) At the indicatedtimes, 15 ml of the reaction mixture was fractionated bySDS-PAGE. (b) Samples of uridylated VPg (after 120 minreaction) were either untreated, or digested at 37 8C for1 h with trypsin (400 ng/ml), endopeptidase Glu-C(100 ng/ml) or RNase A (200 ng/ml) and analyzed bySDS-PAGE. (c) Control uridylation reactions in theabsence of 3D or VPg and use of peptides a (YTGTTTYTASARGDLAHLTTTHARHL), b (AVRMKRAELYCPRPIL), or c (ATYYFSDLEIAVTHT) (negative controls)substituting for VPg. Bands corresponding to mono-, di-,and poly-uridylated forms of VPg are indicated.
1026 Polymerase Defective in RNA Binding
A total of 12 residues in the fingers domain ofFMDV 3D, located between alpha helices 4 and 7,have been shown to be directly involved in bindingto RNA by crystallographic analysis of FMDV 3D ina complex with template-primer.3 G118 does notdirectly participate in contacts with the RNAmolecule. This residue is located in loop a4-b3that forms the entrance of the template entry groove
(Figure 8(a)). G118, with torsion angles fZ74 andjZK158, is folded in a hydrogen bonded turnconformation, with its carbonyl group contactingthe backbone amino group of Trp121 within thesame loop, and its amino group hydrogen bondedto the main-chain oxygen atom of V186 from theloop connecting strand b7 and helix a7. In addition,the carbonyl group of the neighboring P117 ishydrogen bonded to the amino group of L184,located also in the b7-a7 loop (Figure 8(b)). Thestructure of the region uses both the torsionalflexibility of the Gly backbone and the small sizeof this amino acid, leaving no room to accommo-date any side-chain at this position. Thus, substi-tution of glycine by any other bulkier amino acidwould introduce crashing contacts with neighbor-ing residues (Figure 8(c)), not only with residues inthe same loop but also with some residues in loopb7-a7, and large conformational rearrangements arepredicted to occur in order to prevent the sterichindrances. These changes would probably affectthe size and the shape of the template entry grooveand, in consequence, the RNA binding.
It is interesting to note that G118 and P120, bothlocated in the same loop, are residues strictlyconserved in picornaviruses. In PV, it has beendescribed that P120 is essential for polymeraseactivity.21 As in the case of G118, P120 is not directlycontacting the RNA molecule. It seems that bothresidues play a critical structural role, stabilizingthe loop in a conformation that allows RNAbinding.
The highly deleterious or lethal effect of substi-tution G118D in FMDV replication has beendocumented not only by undetectable binding toRNA and polymerization activities (both withpoly(A)/oligo(dT)15 and poly(A)/VPg as tem-plate-primers) but also by biological studies.Electroporation of a transcript of a cDNA represent-ing the entire FMDV genome, with a 3D encodingsubstitutions G118D, V239M and G373D resulted inthe rescuing of mutant FMDV that had revertedD118 to G118 but maintained V239M and G373D in3D. Either the DMD mutant permitted someresidual RNA synthesis activity inside the cellsand revertants were selected and rapidly becamedominant, or the initial RNA transcripts of thecDNA FMDV copy contained low levels of 3Dmutants, including 3D with G118. This latterpossibility is likely in view of the limited copyingfidelity of DNA-dependent RNA polymerases usedto produce the FMDV cDNA transcripts. Althoughthe former mechanism cannot be ruled out, it isunlikely that the severity of the RNA binding defectof DMD 3D would allow any RNA synthesis tooccur in the intracellular environment. If it did, themutant transcripts would qualify as quasi-infectious, as proposed by Agol and colleagues.22
The critical implication of G118 in polymerasefunction was further confirmed by constructing3D with replacement G118D or G118A. Again, these3Ds were inactive in RNA binding and polymer-ization assays, and cDNA transcripts encoding the

Figure 7. Gel mobility-shift assay for analysis of binding of mutant 3Ds to sym/sub RNA. The decanucleotide sym/sub RNA26 was labelled with 32P at its 5 0-end, incubated with increasing concentrations of 3D (or BSA as negativecontrol), and analyzed as described in Materials and Methods. (a) Gel shift patterns obtained with wild-type and severalmutant 3Ds. (b) Percentage of sym/sub-3D complex as a function of protein (3D or BSA) concentration. Free andcomplexed sym/sub were measured as detailed in Materials and Methods.
Polymerase Defective in RNA Binding 1027
FMDV genome with such mutant 3Ds reverted inall cases to yield a virus with the wild-type 3D.FMDV with a 3D with substitutions V239M andG373D (those that accompanied G118D in mutantDMD) produced normal yields, and the purified 3Dshowed RNA binding and polymerization activitiessimilar to those of the wild-type 3D.
The isolation of the triple DMD mutant fromribavirin-treated FMDV is consistent with thedecrease of specific infectivity of pre-extinction
Table 1. Polymerization and VPg-uridylylation activities of w
Polymerization (poly(A)/oligo(dT)15)a
3D Mg2C Mn2C
wt 159.68G28.67 (100) 328.50G68.DMD 0.41G1.47 (0.3) 0.20G0.5D338A 0.64G1.66 (0.4) 0.88G1.9G118D 0.22G0.79 (0.1) 1.58G2.3G118A 0.01G0.41 (0.01) 1.78G0.4M239V/G373D 148.06G33.87 (93) 271.25G77.
a The polymerization assay was the standard assay described in M1.2 mM Mn2C (second column). Specific activities are expressed as pmthe average of at least three determinations; standard deviations and pgiven.
b The VPg-uridylylation assay was the standard assay described inVPg molecules mono- and di-uridylylated (first column) and polPhosphorimager. Specific activities are expressed as pmol of UMP incoat least six determinations; standard deviations, and percentage of ac
RNA.14–16,19 This is one of several modifications(decrease in viral load, increase in mutant spectrumcomplexity with an invariant consensus genomicsequence) that accompany the transition of viruspopulations towards extinction.14–16,19,20,32 The con-cept of error catastrophe has been extensivelysupported by theory12,33,34and by experimentalstudies with many RNA viruses.14,35–39 MutantDMD could belong to a class of genomes that playa role in lethal defection by interfering with the
ild-type and mutant FMDV 3Ds
VPg uridylylationb
VPg-U(U) VPg-poly(U)
78 (100) 0.069G0.038 (100) 5.973G0.259 (100)1 (0.1) K0.006G0.013 (!11.3) 0.015G0.026 (0.3)4 (0.3) K0.005G0.008 (!3.2) 0.039G0.024 (0.7)1 (0.5) K0.005G0.006 (!0.5) 0.016G0.015 (0.3)6 (0.5) 0.001G0.004 (1.4) K0.003G0.021 (!0.3)49 (83) 0.068G0.022 (98) 3.900G0.429 (65)
aterials and Methods, using either 5 mM Mg2C (first column) orol of UMP incorporated per minute and microgram of 3D, and areercentage of activity (in parentheses) relative to wild-type 3D, are
Materials and Methods, using Mn2C (0.6 mM) as the divalent ion.y-uridylylated (second column) have been quantified using arporated per minute and microgram of 3D, and are the average oftivity (in parentheses), relative to the wild-type 3D, are given.

Figure 8. Stereo views of diagrams of the structure of the FMDV polymerase (3D) with indication of the positions ofG118 and D118. (a) Ribbon diagram in which the main-chain residues in the loops (a3-b4 and b7-a7) located at theentrance of the template groove are shown with sticks in blue. G118 is highlighted in red and the RNA template is shownin green. (b) Close-up of loops a3-b4 and b7-a7 with amino acids explicitly labelled. Intra and inter-loop hydrogen bondsare indicated with broken lines. (c) Same close-up view as in (b) in which substitution G118D (in red) has beenintroduced into the structure. The crashing contacts between the aspartic acid side-chain and the main-chain atoms ofP117, L184 and P185 are apparent. Based on Ferrer-Orta et al.3
1028 Polymerase Defective in RNA Binding
replication of standard FMDV genomes co-existingin the same mutant spectrum or, alternatively, DMDFMDV could be a representative of a dead-end classof genomes prompting a complete arrest ofreplication.19,23 We are carrying out experiments totry to distinguish between these possibilities.
Materials and Methods
Molecular cloning, expression and purification ofFMDV 3D
Procedures for the molecular cloning of the FMDV
genomic region encoding the wild-type polymerase (3D),in plasmid pET-28a, IPTG-induction of E. coli, cell lysis,and enzyme purification by chromatography throughNi-NTA have been described.3 The enzyme was O95%pure as judged by polyacrylamide gel electrophoresisanalysis and Coomassie brilliant blue staining; it had aspecific activity of 160(G27) units/mg of protein (at leastten determinations with several enzyme preparations); aunit of activity is the amount of enzyme that catalyzes theincorporation of 1 pmol of UMP per minute usingpolyriboadenylate as template and oligo(dT)15 as primer(poly(A)/oligo(dT)15).
The expression vector pET-28a including the wild-typeFMDV 3D is termed pET-28a3Dpol. A 3D with amino acidreplacements G118D, V239M and G373D (triple DMDmutant) was present in the mutant spectrum of FMDV

Table 2. Primers used to amplify or mutagenize the FMDV 3D gene
Primers Sequencea Position in FMDV sequenceb
1 GGGTTGATCGTTGATACCAGAGA 6610–66322 CCAATTGTGATGTTTGGCGGCCGCTGCGTCGCCGCACACGGCGTTC 8043–79983 GAACCGCACCCCATGGGGTTGATCGTTGATACCAGAG 6595–66314 GCCACCACGATGTCGGCTCCATAGG 7637–76135 CCATGATCTCCTATGGAGCCGACATC 7604–76296 TTCATGGCATCGCTGCAGTGG 7370–73507 GAAACGCCGCGGTGCACTTATC 6984–70058 TGTGGAAGTGTCTTTTGAGGAAAG 7783–77609 GCGACAAAGGTTTTGTTCTTGGTC 7718–774110 TAATACGACTCACTATAGGG T7 primer11 GAACTCTTCCAGGTCAGAAGGC 5677–569912 TCCGCAGACATCTTTGTGTCT 6815–679513 CACCGCACCTGCCCTTCCCTG 6951–697114 CGTCGACAATGCGAGTCTTGCCG 7156–7137
a Sequence of the oligonucleotides used in this study. Letters in bold are nucleotides changed with respect to the FMDV C-S8c1genomic sequence to introduce restriction enzyme sites, or a substitution.
b Numbering is as used by Escarmıs et al.30
Polymerase Defective in RNA Binding 1029
C-S8c1 during a persistent infection of BHK-21 cells, after48 h treatment with 100 mM ribavirin.18 DNA from thisclone was amplified with Pfu DNA polymerase(Promega) and primers 1 and 2 (Table 2). The ampliconwas digested with HindIII (position 6667) and NotI(position 8020), and ligated to plasmid pET-28a3Dpol;the latter was previously digested with the sameenzymes, and treated with shrimp alkaline phospha-tase.24 The resulting expression plasmid is termed pET-28aDMD.
The expression plasmid encoding 3D with substitutionD338A (affecting the catalytic motif YGDD, 3D positions336–339) was prepared by site-directed mutagenesisduring PCR amplification using the pair of primers 3and 4 and the pair of primers 5 and 2 (Table 2). Theamplicons were shuffled, digested with HindIII (position6667) and NotI (position 8020) and ligated to vector pET-28a3Dpol, previously digested with the same enzymesand treated with shrimp alkaline phosphatase. Theresulting plasmid is termed pET-28aD338A. Expressionplasmids encoding 3D with amino acid substitutionsG118A, or G118D, or V239M and G373D (double mutant)were obtained by amplifying the correspondinginfectious plasmids (described below) with primers 3and 6 (to yield the desired substitutions in G118), anddigested with HindIII (position 6667) and SalI (position7150), or with primers 7 and 2 (to yield double mutantV239M, G373D), and digested with NotI (position 8020)and SalI (position 7150). In all cases, the digestedamplicon was ligated to pET-28a3Dpol, which had beendigested with the same enzymes and treated with shrimpalkaline phosphatase.
The presence of all desired mutations was confirmed bysequencing an amplicon from each final plasmid. Therecombinant plasmids were used to express 3D in E. coli,and the enzyme was purified as reported.3 The mutantenzymes were O95% pure as judged by polyacrylamidegel electrophoresis analysis and Coomassie brilliant bluestaining.
Construction of FMDV containing mutations in 3Dpol:transfection assays
The expression plasmid pET-28aDMD, encoding the 3Dtriple mutant DMD, was amplified with primers 7 and 8(Table 2). The plasmid containing a cDNA copy of the
entire FMDV genome (pMT28)25 was amplified withprimers 9 and 10. These two amplicons were shuffledusing primers 9 and 8 as external primers. The resultingDNA fragment was digested with ClaI (position 7004)and NdeI (position 8140) and ligated to pMT28,previously digested with the same restriction enzymesand treated with shrimp alkaline phosphatase. Aftertransformation of E. coli DH-5a competent cells, severalcolonies were isolated. Since the mutation that leads toG373D is within primer 10, two mutants were isolated:one containing substitutions V239M and G373D, andanother containing only V239M. These two plasmids andpMT28 were used as alternative vectors for the cloning ofDNA encoding substitution G118D. The amplicon frompET-28aDMD (amplified with primers 7 and 8) wasshuffled with an amplicon obtained from pMT28 withprimers 11 and 12, using primers 11 and 8 as externalprimers. The amplicons and the vectors were digestedwith ClaI (position 7004) and RsrII (position 5839) andligated; before ligation, the digested vectors were treatedwith shrimp alkaline phosphatase. After transformationof competent E. coli DH-5a cells, and analysis of severalcolonies, plasmids containing mutations encoding V239Malone; V239M and G373D; G118D alone; G118D andV239M; G118D, V239M and G373D (DMD mutant) wereisolated. To obtain FMDV with substitution D338A,plasmid pET-28aD338A was amplified with primers 1and 8. The amplicon obtained was shuffled with theamplicon obtained from pMT28 with primers 9 and 10,using 1 and 10 as external primers. The resultingamplicon was digested with ClaI (position 7004) andNdeI (position 8140) and ligated to pMT28, previouslydigested with the same enzymes and treated with shrimpalkaline phosphatase. FMDV with substitution G118Awas constructed using a mutagenic oligonucleotide(oligonucleotide 13, Table 2). Plasmid pMT28 wasamplified with primers 13 and 14. This amplicon wasused as a long primer to amplify pMT28 in conjunctionwith primer 12. The resulting amplicon was digested withClaI (position 7003) and RsrII (position 5838) and ligatedto pMT28, previously digested with the same enzymesand treated with shrimp alkaline phosphatase. Thepresence of all mutations was confirmed by sequencingan amplicon from each final plasmid. The plasmidscontaining the complete FMDV genome mutated in the3Dpol were linearized by digestion with NdeI (position8140) and transcribed with SP6 RNA polymerase.

1030 Polymerase Defective in RNA Binding
Transfection of the infectious RNAs was carried out withLipofectin, as specified by the manufacturer. Afterapproximately 72 h, the cytopathic effect was completeand the virus was analyzed.
Poly(U) synthesis assay
To test UMP incorporation directed by poly(A)/oligo(dT)15 template-primer, the standard reactionmixture included 30 mM Mops (pH 7.0) (Sigma), 33 mMNaCl, 5 mM Mg(CH3COO)2, 40 ng/ml of poly(A) (averageof 300 residues; Amersham Pharmacia), 2.36 mMoligo(dT)15 (Life Technologies), 500 mM [a-32P]UTP(0.01 mCi/ml; 20 mCi/mmol, Amersham), and 0.4 to0.8 mM 3D; 46 ml of a mixture of all components except 3Dwas prewarmed at 37 8C for 2 min, and the reaction wasstarted by adding 4 ml of 3D (in 50 mM Tris–HCl (pH 7.5),100 mM NaCl, 1 mM EDTA, 10% (v/v) glycerol). Thestandard reaction was carried out for 5 min at 37 8C, andstopped by adding 10 ml of 500 mM EDTA. The reactionmixture was spotted onto DE81 filter paper (Whatman)and the UTP that was not incorporated into RNA wasremoved by washing the filter three times with an excessof 0.2 M Na2HPO4; the filter was then rinsed in ethanoland dried for 15 min at 55 8C. The radioactivity incorpor-ated was measured either by Phosphorimager analysis(BAS1500, Fuji) or in a scintillation counter. Severalcontrol assays indicated that both procedures gaveidentical activity measurements. The assays with Mn2C
were carried out in the same way except that the reactionmixture included 1.2 mM MnCl2 instead of 5 mMMg(CH3COO)2.
VPg uridylation assay
A synthetic peptide with the sequence of VPg1 ofFMDV C-S8c1 (GPYAGPLERQRPLKVRAKLPRQE) wasprepared by solid-phase peptide synthesis, purified byG25 Sephadex chromatography and HPLC, and analyzedby mass spectrometry. The uridylation mixture contained30 mM Mops (pH 7.0), 33 mM NaCl, 0.6 mM MnCl2,40 ng/ml of poly(A) (average of 300 residues), 150 mMVPg1 (synthetic peptide), 0.4 mg/ml of BSA (Boehringer),8% glycerol, 50 mM [a-32P]UTP (0.01 mCi/ml; 200 mCi/mmol) and 0.4 to 0.8 mM 3D; 46 ml of a mixture of allcomponents except 3D were prewarmed at 37 8C for2 min, and the reaction was started by adding 4 ml of 3D(in 50 mM Tris–HCl (pH 7.5), 100 mM NaCl, 1 mM EDTA,10% glycerol). The standard reaction was carried out for30 min at 37 8C; it was stopped either by adding 10 ml of500 mM EDTA or by heating at 90 8C for 5 min. Thereaction products were analyzed by SDS-PAGE in a Tris–Tricine buffer (stacking 4% (w/v) acrylamide, resolving16% (w/v) acrylamide, 10% (v/v) glycerol).
Gel mobility-shift assays
An RNA sym/sub decanucleotide (sequence 5 0-GCAUGGGCCC-3 0)26 was end-labeled using [g-32P]ATP(300 Ci/mmol; Amersham) and polynucleotide kinase(NEB) as described,24 and purified by G25 Sephadexchromatography (Mini Quick Spin Oligo Columns,Roche). The RNA-binding mixture included 20 nM[g-32P]sym/sub, 100 mM Mops (pH 7.0), 25 mM MgCl2,20 mM NaCl, 5% (w/v) polyethylenglycol and increasingconcentrations (0 to 1.8 mM) of 3D. The mixtures wereloaded onto a non-denaturing 10% polyacrylamide gel inTB buffer (85 mM Tris–HCl, 85 mM borate, pH 8.0) and
electrophoresed at 100 V, 4 8C for 1 h in TB buffer. The gelwas fixed in 10% (v/v) ethanol, 10% (v/v) acetic acid for5 min, and dried for 45 min at 80 8C in a vacuum geldryer. The radioactivity in free sym/sub and in the sym/sub–3D complex was measured using a Phosphorimager(BAS 1500, Fuji), subtracting background values.
Monoclonal antibodies
To obtain monoclonal antibodies (mAbs) specific forFMDV 3D, constructs with 3D of FMDV O1 Switzerland1965 fused to an N-terminal polyhistidine tag wereexpressed in E. coli using standard procedures,27 andpurified by Ni-NTA affinity chromatography. A Balb/Cmouse was immunized by three injections (one monthapart) each with 50 mg of 3D, previously polymerized byglutaraldehyde treatment; for the first inoculation, theantigen was administered subcutaneously in Freund’scomplete adjuvant; the second and third (booster)administrations were intraperitoneal in saline solution.Splenocytes were collected three days after the lastinjection, and fused to myeloma cells as detailed else-where.27 For the screening of hybridoma cultures, 48synthetic peptides representing FMDV 3D were preparedas 20-mers overlapping in ten amino acids, as detailedelsewhere28 (kindly provided by A. Douglas, Belfast).Indirect ELISA was performed with 3D or syntheticpeptides adsorbed to the plates; then supernatants ofhybridoma cultures were added, followed by incubationwith peroxidase-labeled anti-mouse immunoglobulinsand with hydrogen peroxide and O-phenyldiamine(H2O2/OPD) (Sigma). mAbs selected for further studywere: 2D10 (whose epitope maps between 3D residues129–148); 2D11 (residues 129–138); 3B9 (residues 9–28);3F12 (residues 29–38); 3H11 (residues 49–68); and 4B12(residues 9–18).
The ELISA procedure used to test the reactivity of wild-type and mutant 3Ds with mAbs is that described29 withminor modifications. Briefly, 3D (80 ng in 100 ml of PBS)was incubated overnight in microtiter ELISA plates. Aftersaturation with 5% BSA in PBS, 100 ml of a solutioncontaining a non-saturating amount of mAb was added tothe plates and incubated for 1 h at room temperature.Then the plates were incubated with peroxidase-con-jugated goat anti-mouse IgG (Bio Rad; 1:2500 dilution) for1 h at room temperature. The bound antibody wasquantified by reaction with H2O2/OPD, and the reactionwas stopped with 2 M H2SO4; absorbance was measuredat 492 nm.
Inhibition of 3D activity by monoclonal antibodies
mAbs were purified from ascitic fluid by chromato-graphy through protein A-Sepharose.29 Fractions contain-ing purified antibodies were pooled and dialyzed againstPBS. A mixture of mAb, 3D and BSA was pre-incubatedfor 15 min at 37 8C in PBS, and 8 ml of this mixture wasadded to 42 ml of the reaction mixture (prewarmed for2 min at 37 8C); final concentrations were 30 mM Mops(pH 7.0), 45 mM NaCl, 5 mM Mg(CH3COO)2, 2.36 mMoligod(T)15, 40 ng/ml of poly(A) (average of 300 residues),15 nM 3D, 340 nM mAb, 0.4 mg/ml BSA, 0.16!PBS. Toexclude the possibility that the inhibition could be due tothe presence of RNase in the mAbs, 40 ng/ml of ribosomalRNA and mAb were incubated for 30 min in the samebuffer used for the activity assay; no evidence of RNAdegradation was obtained, as judged by agarose gelelectrophoresis of treated versus untreated ribosomal

Polymerase Defective in RNA Binding 1031
RNA. Also, a number of mAbs not directed to 3D andpurified by the same procedure did not inhibit poly-merase activity. Furthermore, the inhibition specificity ofmAbs was tested with T7 RNA polymerase in a reactionmixture that included pMT1 DNA (250 ng) (containingresidues 409 to 930 of the FMDV genome in pBluescriptSKC (Fermentas)), 500 mM NTPs, 10 nM T7 RNApolymerase, 340 nM mAb, 0.4 mg/ml of BSA, and PBS0.16! in 40 mM Tris–HCl (pH 7.9), 6 mM MgCl2, 10 mMDTT, 10 mM NaCl, 2 mM spermidine. The reaction wasfor 30 min at 37 8C. No inhibition by any mAb wasobserved.
Acknowledgements
This work was supported by Grants BMC2001-1823-C02-01 of Comision Interministerial deCiencia y Tecnologıa, 08.2/0015/2001 of Comuni-dad Autonoma de Madrid and Fundacion R. Areces(to E.D.) and Grant BIO2002-00517 of ComisionInterministerial de Ciencia y Tecnologıa (to N.V.).A.A. is supported by a predoctoral I3P fellowshipfrom Ministerio de Educacion y Ciencia. R.A. issupported by a predoctoral fellowship from theComunidad Autonoma de Madrid. C.F.-O. issupported by a predoctoral fellowship from theMinisterio de Ciencia y Tecnologıa.
References
1. Cameron, C. E., Gohara, D. W. & Arnold, J. J. (2002).Poliovirus RNA-dependent RNA polymerase (3Dpol):structure, function and mechanism. In MolecularBiology of Picornaviruses (Semler, B. L. & Wimmer, E.,eds), pp. 255–267, ASM Press, Washington, DC.
2. Paul, A. V. (2002). Possible unifying mechanism ofpicornavirus genome replication. In Molecular Biologyof Picornaviruses (Semler, B. L. & Wimmer, E., eds),pp. 227–246, ASM Press, Washington, DC.
3. Ferrer-Orta, C., Arias, A., Perez-Luque, R., Escarmis,C., Domingo, E. & Verdaguer, N. (2004). Structure offoot-and-mouth disease virus RNA-dependent RNApolymerase and its complex with a template-primerRNA. J. Biol. Chem. 279, 47212–47221.
4. Nayak, A., Goodfellow, I. G. & Belsham, G. J. (2005).Factors required for the uridylylation of the foot-and-mouth disease virus 3B1, 3B2, and 3B3 peptides by theRNA-dependent RNA polymerase (3Dpol) in vitro.J. Virol. 79, 7698–7706.
5. Batschelet, E., Domingo, E. & Weissmann, C. (1976).The proportion of revertant and mutant phage in agrowing population, as a function of mutation andgrowth rate. Gene, 1, 27–32.
6. Drake, J. W. & Holland, J. J. (1999). Mutation ratesamong RNA viruses. Proc. Natl Acad. Sci. USA, 96,13910–13913.
7. Domingo, E., Sabo, D., Taniguchi, T. & Weissmann, C.(1978). Nucleotide sequence heterogeneity of an RNAphage population. Cell, 13, 735–744.
8. Eigen, M. & Schuster, P. (1979). The Hypercycle. APrinciple of Natural Self-Organization, Springer, Berlin.
9. Eigen, M. (1996). On the nature of virus quasispecies.Trends Microbiol. 4, 216–218.
10. Domingo, E., Biebricher, C., Eigen, M. & Holland, J. J.(2001). Quasispecies and RNA Virus Evolution: Principlesand Consequences, Landes Bioscience, Austin.
11. Domingo, E. (2005). Microbial evolution andemerging diseases. In Emerging Neurological Infections(Power, C. & Johnson, R. T., eds), Marcel Dekker, NewYork, NY. In the press.
12. Eigen, M. (2002). Error catastrophe and antiviralstrategy. Proc. Natl Acad. Sci. USA, 99, 13374–13376.
13. Anderson, J. P., Daifuku, R. & Loeb, L. A. (2004). Viralerror catastrophe by mutagenic nucleosides. Annu.Rev. Microbiol. 58, 183–205.
14. Sierra, S., Davila, M., Lowenstein, P. R. & Domingo, E.(2000). Response of foot-and-mouth disease virus toincreased mutagenesis: influence of viral load andfitness in loss of infectivity. J. Virol. 74, 8316–8323.
15. Pariente, N., Sierra, S., Lowenstein, P. R. & Domingo,E. (2001). Efficient virus extinction by combinationsof a mutagen and antiviral inhibitors. J. Virol. 75,9723–9730.
16. Pariente, N., Airaksinen, A. & Domingo, E. (2003).Mutagenesis versus inhibition in the efficiency ofextinction of foot-and-mouth disease virus. J. Virol.77, 7131–7138.
17. Pariente, N., Sierra, S. & Airaksinen, A. (2005). Actionof mutagenic agents and antiviral inhibitors on foot-and-mouth disease virus. Virus Res. 107, 183–193.
18. Airaksinen, A., Pariente, N., Menendez-Arias, L. &Domingo, E. (2003). Curing of foot-and-mouth diseasevirus from persistently infected cells by ribavirininvolves enhanced mutagenesis. Virology, 311,339–349.
19. Gonzalez-Lopez, C., Arias, A., Pariente, N., Gomez-Mariano, G. & Domingo, E. (2004). Preextinction viralRNA can interfere with infectivity. J. Virol. 78,3319–3324.
20. Gonzalez-Lopez, C., Gomez-Mariano, G., Escarmıs,C. & Domingo, E. (2005). Invariant consensussequence in the transition to error catastrophe. Infect.Genet. Evol 5, 366–374.
21. Thompson, A. A. & Peersen, O. B. (2004). Structuralbasis for proteolysis-dependent activation of thepoliovirus RNA-dependent RNA polymerase.EMBO J. 23, 3462–3471.
22. Gmyl, A. P., Pilipenko, E. V., Maslova, S. V., Belov,G. A. & Agol, V. I. (1993). Functional and geneticplasticities of the poliovirus genome: quasi-infectiousRNAs modified in the 5 0-untranslated region yield avariety of pseudorevertants. J. Virol. 67, 6309–6316.
23. Grande-Perez, A., Lazaro, E., Lowenstein, P.,Domingo, E. & Manrubia, S. C. (2005). Suppressionof viral infectivity through lethal defection. Proc. NatlAcad. Sci. USA, 102, 4448–4452.
24. Sambrook, J. & Russell, D. W. (2001). MolecularCloning: A Laboratory Manual, 3rd edit., Cold SpringHarbor Laboratory Press, Cold Spring Harbor, NY.
25. Garcıa-Arriaza, J., Manrubia, S. C., Toja, M., Domingo,E. & Escarmıs, C. (2004). Evolutionary transitiontoward defective RNAs that are infectious bycomplementation. J. Virol. 78, 11678–11685.
26. Arnold, J. J. & Cameron, C. E. (2000). Poliovirus RNA-dependent RNA polymerase (3D(pol)). Assembly ofstable, elongation-competent complexes by using asymmetrical primer-template substrate (sym/sub).J. Biol. Chem. 275, 5329–5336.
27. Brocchi, E., Gamba, D., Poumarat, F., Martel, J. L. & DeSimone, F. (1993). Improvements in the diagnosis of

1032 Polymerase Defective in RNA Binding
contagious bovine pleuropneumonia through theuse of monoclonal antibodies. Rev. Sci. Technol. 12,559–570.
28. Borrego, B., Garcia-Ranea, J. A., Douglas, A. &Brocchi, E. (2002). Mapping of linear epitopes on thecapsid proteins of swine vesicular disease virus usingmonoclonal antibodies. J. Gen. Virol. 83, 1387–1395.
29. Mateu, M. G., Andreu, D. & Domingo, E. (1995).Antibodies raised in a natural host and monoclonalantibodies recognize similar antigenic features of foot-and-mouth disease virus. Virology, 210, 120–127.
30. Escarmıs, C., Davila, M., Charpentier, N., Bracho, A.,Moya, A. & Domingo, E. (1996). Genetic lesionsassociated with Muller’s ratchet in an RNA virus.J. Mol. Biol. 264, 255–267.
31. Mateu, M. G., Martınez, M. A., Capucci, L., Andreu,D., Giralt, E., Sobrino, F. et al. (1990). A single aminoacid substitution affects multiple overlapping epi-topes in the major antigenic site of foot-and-mouthdisease virus of serotype C. J. Gen. Virol. 71, 629–637.
32. Grande-Perez, A., Gomez-Mariano, G., Lowenstein,P. R. & Domingo, E. (2005). Mutagenesis-induced,large fitness variations with an invariant arenavirusconsensus genomic nucleotide sequence. J. Virol. 79,10451–10459.
33. Swetina, J. & Schuster, P. (1982). Self-replication witherrors. A model for polynucleotide replication.Biophys. Chem. 16, 329–345.
34. Eigen, M. & Biebricher, C. K. (1988). Sequence spaceand quasispecies distribution. In RNA Genetics(Domingo, E., Ahlquist, P. & Holland, J. J., eds), vol.3, pp. 211–245, CRC Press, Boca Raton, FL.
35. Holland, J. J., Domingo, E., de la Torre, J. C. &Steinhauer, D. A. (1990). Mutation frequencies atdefined single codon sites in vesicular stomatitis virusand poliovirus can be increased only slightly bychemical mutagenesis. J. Virol. 64, 3960–3962.
36. Loeb, L. A., Essigmann, J. M., Kazazi, F., Zhang, J.,Rose, K. D. & Mullins, J. I. (1999). Lethal mutagenesisof HIV with mutagenic nucleoside analogs. Proc. NatlAcad. Sci. USA, 96, 1492–1497.
37. Crotty, S., Maag, D., Arnold, J. J., Zhong, W., Lau, J. Y.,Hong, Z. et al. (2000). The broad-spectrum antiviralribonucleoside ribavirin is an RNA virus mutagen.Nature Med. 6, 1375–1379.
38. Grande-Perez, A., Sierra, S., Castro, M. G., Domingo,E. & Lowenstein, P. R. (2002). Molecular indetermina-tion in the transition to error catastrophe: systematicelimination of lymphocytic choriomeningitis virusthrough mutagenesis does not correlate linearly withlarge increases in mutant spectrum complexity. Proc.Natl Acad. Sci. USA, 99, 12938–12943.
39. Domingo, E. (2005). Virus entry into error catastropheas a new antiviral strategy. Virus Res. 107, 115–228.
Edited by J. Karn
(Received 4 July 2005; received in revised form 1 September 2005; accepted 9 September 2005)Available online 26 September 2005