novel derivatives of ascomycin through...
TRANSCRIPT

Indian Journal of Chemistry Vol. 38B, October 1999, pp. 1159 - 1164
Novel derivatives of ascomycin through photochemistry t
Murty ARC Bulusu*, Ewald Haidl , Gerhard Schulz, Peter Waldstatten & Maximilian Grassberger,
Novartis Forschungsinstitut GmbH, Brunnerstrasse 59, A-I23S Vienna, Austria
Received 27 July /998; accepted 23 December /998
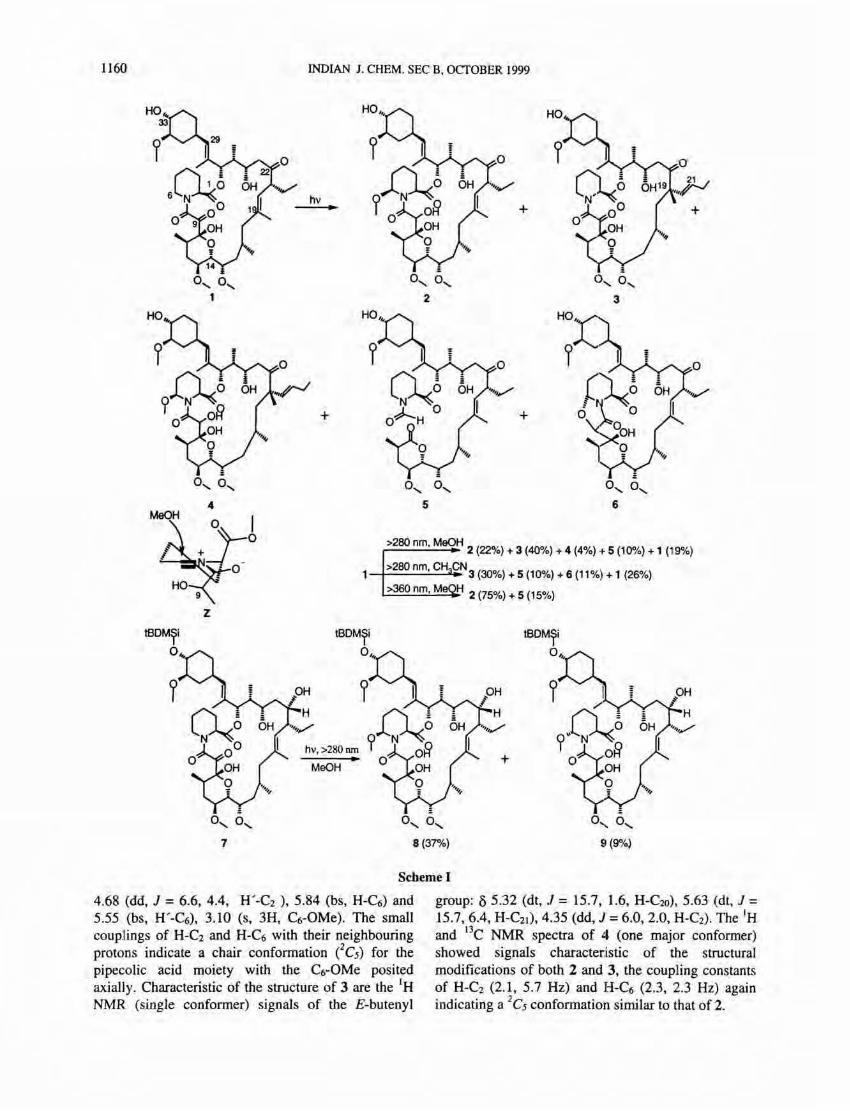
Irradiation of ascomycin 1 leads to three reaction pathways: (a) [I ,3)-sigmatropic shift of the aJJylic carbonyl group lead,ing to the ring-contracted products 3 and 4, (b) electron-transfer from the amide nitrogen to the carbonyl group fo llowed by reorganisation leading to a zwitterionic intermediate Z which undergoes either intramolecular cyclisation (leading to. 6 in CH3CN) or addition of methanol (leading to 2 in MeOH), and, (c) decarbonylative fragmentation resulting in the acyclic lactone 5. The effects of solvent, wavelength, and the substrate-srtucture on the product distribution have been examined. Simple transformations of the photoproducts leading to novel derivatives are presented.
The macrolactam ascomycin 1 1, (Figure 1) is a fermentation product originally isolated from Streptomyces hygroscopicus var. ascomyceticus due to its antifungal activities. Recent preclinical2 and clinical studies with two ascomycin derivatives, SDZ 281-240: and SDZ ASM 981 ,4 revealed that this class of substance has a high therapeutic potential for the treatment of inflammatory skin diseases, such as atopic dermatitis, allergic contact dermatitis, and psoriasis. These results stimulated the search for novel ascomycin derivativess and their biological evaluation. The analogs studied so far are limited with respect to the structural variations because of the inaccessibility, in particular, of the pipecolic acid and CWC22 regions, for derivatisation through routine organic methods. The pipecolic acid and C W C22 regions are involved in bindingS to macrophilin and calcineurin, respectively, and are termed the binding and effector domains. The presence of a novel tricarbonyl system and an allylic ketone functionality in these domains prompted us to study the photochemistry of this molecule towards synthesising new and novel derivatives. It is anticipated that the partial rigidity imposed on the molecule by the 23-membered macrolactam structure would lead to good regio- and stereoselectivities.
Irradiation of 1 in methanol under He for 2 hr at room temperature using pyrex filtered UV light led to
t Dedicated to Professor M. V. George on the occasion of his 701h
birthday.
R=
o "
HO ....... ("j 33 ~
o I
Ascomycin 1
Fh!:ure 1
o
ASM 981
6-~-methoxy-9-dihydroascomycin 2 (22 %), the butenyl derivatives 3 (40 %) and 4 (4 %), and the lactone 5 (10 %); in addition, unchanged 1 (19 %) was recovered (Scheme I). The structures6 of the products were arrived at on the basis of spectral data. The 13C NMR spectrum of 2 (3: 1 mixture of amide bond rotamers) showed the absence of C9=O and the presence of new signals at 8 75 .24 and 69.51 (C9); 80.49 and 84.32 (C6) and a fourth OCH3-signal. The lH NMR signals characteristic of 2 are: 8 4.42 (bs, 2H, H-C9 and HO-C9), 5.45 (d. J = 3.6 Hz, H-C2) and
1160
°
z
7
INDIAN 1. CHEM. SEC B, ocrOBER 1999
hv ..
+
hv, >280 nrn .. MeOH
+
! : 0, 0, 0, 0, 2 3
? H0:C
° ? °
+
>280nm MeOH r------'-' -. 2 (22%) + 3 (40%) + 4 (4%) + 5 (10%) + 1 (19%)
1 >280 nm, CH CN 3 (30%) + 5 (10%) + 6 (11%) + 1 (26%)
>360 nm, M H 2 (75%) + 5 (15%)
0, 0,
8 (37%)
Scheme I
+
? I /'
?~~
~Ofl OH
° 0 ...... 0,
~I (9%)
4.68 (dd, J = 6.6, 4.4, H'-C2 ), 5.84 (bs, H-C6) and 5.55 (bs, H'-C6), 3.10 (s, 3H, C6-0Me). The small coupUngs of H-C2 and H-C6 with their neighbouring protons indicate a chair conformation eC5) for the pipecolic acid moiety with the C6-OMe posited axially. Characteristic of the structure of 3 are the IH NMR (single conformer) signals of the E-butenyl
group: 05.32 (dt, .T = 15.7, 1.6, H-C20), 5.63 (dt, J = 15.7,6.4, H-C21), 4.35 (dd, J = 6.0, 2.0, H-C2). The IH and I3C NMR spectra of 4 (one major conformer) showed signals characteristic of the structural modifications of both 2 and 3, the coupling constants of H-C2 (2.1, 5.7 Hz) and H-C6 (2.3, 2.3 Hz) again indicating a 2C5 conformation similar to that of 2.

BULUSU et al. : NOVEL DERIVATIVES OF ASCOMYCIN THROUGH PHOTOCHEMISTRY 1161
The fonnation of 2 could be rationalised through the intennediacy of the zwitterion Z fonned through electron transfer ftom the amide nitrogen to the C9=O, followed by electron reorganisation and proton transfer.7 Of the two possible confonnations of Z, the one shown is expected to be of lower energy than the alternative one wherein the ester group would be in the semiequatorial position resulting in strong 1,2-steric repulsions with the neighbouring group on the planar nitrogen. Attack of MeOH from the ~-face of Z (chair transition state, resulting in 2) is favoured over that from the a-face (boat transition state). It appears that the 1,3-diaxial interactions between the incoming MeOH and the CI-ester in the chair transition state are not of much significance. The transfonnation of 1 to 3 could be rationalised as a photoallowed [1,3]sigmatropic shift. Molecular models and X-ray crystal structure of 15 indicate an ideal allignment of the bonds involved for a a\ + n\ mode of interaction, which is expected to result in ~-methyl and E-butenyl configurations. The fonnation of only one isomer 3 with E-butenyl chain led us to tentatively assign the configuration of the Cl9-methyl group as ~. No epimerisation at C22 in the recovered starting material could be detected thus indicating a concerted migration pathway for the fonnation of 3. Finally, the acyclic derivative 5 could be arising through decarbony lative fragmentation.
The above mechanistic considerations led us to study the photoreaction in a nonnucleophilic solvent to look at the fate of the intennediate Z. Indeed, irradiation of 1 in CH3CNIH20 (4:1) led to the butenyl compound 3 (30%), the lactone 5 (10%) and the oxazolidinone 6 (11 %); in addition, unchanged 1 (26%) was recovered. Characteristic of 6 are the IH_ NMR signals at 8 4.29 (d, J = 1.4, H-C9), 4.61 (d, J = 7.0, H-C2), 5.24-5.28 (m, J 6•9 = 1.4, J6.5e = 4.2, J6.5a = 10.3, H-C6). The couplings of H-C6 and H-C2 clearly point to a chair confonnation of the 6-membered ring eC5), with the new C6-0 bond posited equatorially. Clearly, in the absence of an external nucleophile the intennediate Z undergoes intramolecular cyclisation through the higher energetic boat-like transition state involving a-attack by C9-oxygen, ~-attack being much less favourable because of the strain it would involve. The fonnation of 6-f3-methoxy-analog 2 stereoselectively, and none of the oxazolidinone 6 in the irradiation of 1 in methanol, shows that ~-attack on Z by an external nucleophile (chair transition state) is preferred over a-attack either intramolecularly or by an external nucleophile (both involving boat
transition states). The photoproduct 2 features a heteroacetal
functionality which has potential for further modifications in the amino acid moiety. The low conversion, the poor yield of 2 and the difficulty in its purification prompted us to optimise the reaction further. To this end we looked at the photochemistry of 7 wherein the C22=O is reduced to the alcohol, thus, hoping to channel the reaction exclusively in the required direction.
Irradiation of 7 in MeOH at 0 DC for 10 hr under He using pyrex filtered light resulted in a mixture of the 6-methoxy derivatives 8 (37%) and 9 (9%) as the only products; the lower isolated yields are due to losses during repeated chromatographic purification. Clearly, the transfonnation of the trigonal C22 to a tetrahedral center made the macrolactam ring more flexible thus .reducing the stereoselectivity of the attack by MeOH. ,Nevertheless, the ~-attack is still predominant over the a-attack. It is gratifying to note that there arise no further competing reactions.
Encouraged by the above results, and, in view of the necessity of the presence of C22=O for biological activities,5 we looked at the possibility of improving the functional group selectivity not through modification of the substrate, but rather through modification of the reaction conditions. As the tricarbonyl chromophore is expected to absorb at longer wavelengths compared to the allylic carbonyl, selective excitation of the fonner chromophore appeared to be a promising method. Indeed, irradiation of 1 in MeOH using filtered UV light (>360 nm, aq. NaBr-Pb(N03h filter) for 8 hr resulted in total consumption of the starting material leading to 2 (75%) and 5 (15%) as the only products isolated. Analysis of the crude reaction mixture on HPLC also gave similar yields, and, no other products could be detected. The reaction could be carried out conveniently in large scale (40 g) and the required 2 could be freed off 5 through a short chromatographic filtration followed by crystallisation (CH2Chpentane). It may be mentioned here that in a semiquantitative study, oxygen did not show any effect on the reaction, thus indicating noninvolvemennt of the triplet state in the reaction.
The intact tricarbonyl moiety is crucial for the biological activity of 1.5 Thus, 2 was oxidized8
selectively using 0.5 equiv Cu(OAc)2IMeOHJ02 giving 6-~-methoxyascomycin 10 (40%) and the ester 11 (28%) a product of consecutive reaction (benzilic acid type rearrangement) of 10 with MeOH (Scheme II) ;

1162
I
8
INDIAN J. CHEM. SEC B, OCTOBER 1999
o o
+
• = 0, 0 ,
10 11
0.5 equiv. CU(OACh, O2, MeOH, 25 °C .----...!.----.:....-..:.£.:..~---=----- 10 (40%) + 11 (28%) + 2 (20%)
5 equiv. CU(OAC)2' 02' THF, reflux 2 -I----:....---=---=--=:.-~------ 10 (75%)
cat CU(OAC)2' cat pyridine, 0 2' CH2CI2, 25°C 10 (90%)
9 4
~ 1) Cu(II), 0 2 2) aq HCI / CH3CN
! 1) Cu(II), 0 2 2) aq HCI / CH3CN
~ Cu(II) , 02
I I H H
°
° ?~~ II"/~
~, """
0, 0 , 0 , 0,
12 13 14
tBOMyi
I ° I ° ~~
H
""1/ ~~H "'"''
0, 0, 0,
15 16 17
Scheme II

BULUSU et al. : NOVEL DERIV A TIVES OF ASCOMYCIN THROUGH PHOTOCHEMISTRY 1163
in addition, unchanged 2 (20%) was recovered. Using THF as solvent and employing 5 equivalents of Cu(OAch under refluxing conditions with continuous oxygen bubbling, good yields of 10 (75 %) could be obtained in one gram scale and the unwanted consecuti ve reaction could be circumvented; runs wi th catalytic amounts of CU(OAC)2 were not successful. The reaction, however, was difficult to scale up due to slow reaction and irreproducible yields in larger scales. In addition, considerable amounts of peroxides were formed in the reaction. Finally, we found that using 5 mol % Cu(OAch or cupric 2-ethylhexanoate/5 mol% pyridine/02-balloonl4A-MSieves/CH2ChI16 hr the reaction cou ld be run catalytically and safely (no perox ides detected) at room temperature in large scales (30 g) providing 10 in 90% yield. A control run employing 5 mol% cupric 2-ethylhexanoate and omitting pyrid ine, resulted in ca. 30% reaction after 20 days, thus indicating the role of pyridine in the high catalytic tu rnover apparently through its ligation. To our knowledge, this represents a very mild and efficient variation compared to the reported methods for the oxidation of a -hydroxy ketones us ing copper salts.8 The compounds 12 and its C6-epimer 13 could be prepared analogously starting fro m 8 and 9 through oxidation and desi lylation. Oxidation of 4 provided 14. Lastly, as a proof of the structure, the disubstituted double bond in 3 was hydrogenated (PdlCIH2IEtOAc) selectively giving 15.
Finally, it may be mentioned that irradiations of 24-desoxyascomycin 16 (prepared through PTSHcatalysed dehydration of 1, followed by controlled catalytic hydrogenation using PdlC) and 33-tBDMS-24-desoxy-22-dihydroascomyci n 17 in methanol led to mixtures consisting mainl y of the corresponding oxazolidinones (isomers) and only small amounts of the 6-methoxy-9-dihydroderivatives (isomers). It appears that in the absence of the C24-OH group the macrolactam ring (especially the pipecolic ac id ring) is much more fl ex ible resulting in further loss of selectivity in product formation.
Brief comment may be made here on the conforma·· tion of the pipecolic ac id ri ng which is important because o f its ability to fit into the binding pocket of the macrophilin-12 protein and also its pivotal effect on the conformation of the macrol actam ring and, hence, its effect on biulogical activity. The 6-B-methoxy analogs 10, 12 and 14, the ringcontracted analogs 3 and 15, and , the oxazolidinone 6, all showed (' H MR coupling constants and nOe experiments in CDCI) the presence of a 2C5
conformation for the pipecolic acid similar to that observed with ascomycin (both in free form and in its complex with macrophilin-12 binding protein).5 Interestingly, the 6-a-methoxy analog 13 showed a 5C2 conformation thus positing the C6-0Me group axially presumably for avoiding the repuls ive i:;teractions with the amide substituent in the alternative 2C5 conformation (pseudoallylic effect).
In summary , we have shown that irradiation of ascomycin leads to novel derivatives with modificatio ns in the binding and effector domains of the molecule, which are not accessible through routi ne methods. The regio and stereoselecti viti es could be modified by modifying the substrate, the solvent, and the wavelength employed, thus providing a useful method for synthesis of these derivatives in good yie lds. The scope of the photoreaction and the chemistry of 2 are currently under investigation . The biological activ it ies will be reported elsewhere.
References
Arai T , Koyoma Y, Suenaga T & Honda H, J Antibiot Ser A,
i 5, 1962, 23 l. 2 a) G rassberger M, Me ingassner J G, StUtz A & Stiitz P.
Patent DE 3:838-035 (Sandoz).
b) Meingassner 1 G & Stii tz A, J In vest DermalOl, 98, 1992, 85 l. c) 19th Collegium Internationale Allergologicum, Capri , Italy, May, 3 _7'h, Abstr. S 144.
3 Rappersberger K, Meingassner J, Fialla R, Fbdinger D, Sternichy B, Rauch S, Putz E, StUtz A & Wol ff K, J In vest Dermatol , 106, 1996,70 1.
4 a) Meingassner J, Grassberger M, Fahmgruber H, Moore H,
Schuurrnan H & Stiitz A, Brit J Dermatol, 137, 1997. 568. b) Me ing<;lssner 1 G, Fllhrngruber H, Bavandi A & Grassberger M, Australasian J Dermatol, abs tracts of the 19th
world congress of dermatology 15-20th June, 1997, Sydney , Australia, Abstr. Nr. 5258. c) Van Leent E J M, Graber M, Thurston M, Wagenaar A,
Spuls P I & Bos J D, Australasian J Dermatol, 38(Suppl. 2),
1997, 234. d) Mrowietz U, Brautigam M, Graber M, Thurston M.
Wagenaar A, Weidinger G & Christophers E, Australasian J Derllla lOl , 38 (Suppl. 2), 1997,44. e) Queille- Roussel C, Graber M, Wagenaar A, Thurston M, Lachapelle J M, Decroix 1, De Cuyper Ch & Ortonne J P,
Australasian J Dermatol, 38 (Suppl. 2),1997,55. 5 Reviews: a) Be lshaw P J , Meyer S D, Johnson D D, Romo D,
Ikeda Y, Andrus M, Albcrg D G, Schulz L W, Clardy J &
Schreiber S L, Synlell , 1994,38 1. b) Stlitz A, Grassberger M A, Baumann K, Edmunds A J F, Hiestand P, Me ingassner J G, Nussbaumer P, Schuler W & Zenkc G. in : Perspectives in Medicinal Chemistry, edi ted by B Testa , E Kyburz, W Fuhrer and R Giger, (Verlag Helv Chim Acta, Basle and VCH , Wei nhei m), 1993, Chapl. 27, 427.

ll64 INDIAN J. CHEM. SEC B, OCTOBER 1999
c) Grassberger M A & Baumann K, Current Opinion in Therapeutic Patents, 1993,93l.
d) Rosen M K & Schreiber S L, Angew Chern Int Edn Engl, 31,1992,384.
6 All the compounds are fully characterised using lH NMR (500 MHz), l3C NMR (125 MHz) and mass spectral data and elementary analysis. The lH and l3C-signal assignments for representative compounds were made through C-H correlation spectra and lH-decoupling experiments.
7 a) Chesta C A & Whitten D G, J Arn Chern Soc, 114, 1992, 2188. b) Aoyama H, Hasegawa T, Watabe M, Shiraishi H & Omote Y, J Org Chern, 43, 1978,419.
8 a) Sasaki K, Chern Pharm Bull, 9, 1961, 684.
b) Gleiter R, Kraemer R, Imgartinger H & Bissinger C, J Org Chern,1, 1992,252.
c) Seko N, Yoshino K, Yokota K & Tsukamoto G, Chern Pharm BuIl,39, 1991,651.
d) Eistert B, Schneider H & Wollheim R, Chern Ber, 92, 1959,2061.
e) Biel J H, J Arn Chern Soc, 77, 1955, 2250.
f) Macaione D P & Wentworth S E, Synthesis, 1974,716.
g) Gardner T S, Wenis E & Lee 1, J Org Chern, 23, 1958, 823 . h) Kratzer R H, Paciorek K L & Karle D W, J Org Chern, 41(12),1976,2230.