one pot reactions of 3-oxo-2,3-diarylpropanals:...
TRANSCRIPT

142
CHAPTER 6
One Pot Reactions of 3-Oxo-2,3-Diarylpropanals:
Synthesis of Functionalized Nicotinonitriles
6.1. Introduction
Though synthetic organic chemistry has developed in a fascinating way over
the past decades its public image has been deteriorated due to the fear that chemistry
could negatively influence the ecological balance.
The synthesis of complex
molecules are traditionally performed by a sequence of separate reaction steps-each
step requiring its own conditions, reagents, solvents and catalysts. After the
completion of each reaction the solvent and the waste products are removed and
discarded, and the intermediate product is separated and purified. Now,
environmental and economic pressures are forcing the chemical community to
search for more efficient ways of performing chemical transformations. The use of
one-pot strategies would allow the minimization of waste; thus making the waste
management more economical, as compared to stepwise reactions, the amount of
solvents, reagents, adsorbents and energy would be dramatically decreased. In
addition the amount of labor involved would go down. Thus these reactions would
allow an ecologically and economically favorable production.
6.2. One-pot reactions for the synthesis of functionalized
heterocycles
A facile and high yielding synthesis of 1,2,3,4-tetrahydroquinoline-3-
carboxylic acids 5 from ortho-dialkylaminoaldehydes 1 and Meldrum’s acid 2 in a
one-pot two step method was reported by Ryabukhin and coworkers. In this

143
method, the reaction mixture was first heated for 12 hrs at 60 oC for the
intermediate to form and then raised the temperature to 100 oC for the
accomplishment of hydroxylation and decarboxylation (Scheme 1).1
O
N R
R
O O
OO
TMSCl
DMF, rt
N R
R
OO
OO rt or 60 0C
NR
R
O
OO
ON
R
COOH
R
TMSCl
DMF, rt
2
1
3 4 5
Scheme 1
A high yielding one-pot procedure for the synthesis of 2-substituted
benzimidazoles 8 from substituted o-phenylenediamines 6 and esters 7, using
microwave irradiation technique was described by Jing and coworkers.2 This
method is the first general method for the synthesis of this kind (Scheme 2).
NH2
NH2
RCOOC 2H5+2 HCl / Glycol
Microwave N
NR
X
X
6 7 8
Scheme 2
Tormyshev and coworkers have reported the synthesis of
2-aminothiophene-3-carboxylates 10 in good to moderate yields by the one-pot
Gewald reaction of aryl alkyl ketones 9 with ethyl cyanoacetate and elemental
sulfur in the presence of morpholine acetate and excess morpholine (Scheme 3).3

144
X
Oethyl cyanoacetate
morpholine, MeCO2H, sulfur
55 oC 36 h S NH2
O
O
X
9 10
Scheme 3
A simple and convenient one-pot synthesis of benzimidazoles and
benzoxazoles 14 from carboxylic acids 11 and o-phenylenediamine or 2-aminophenol
12 using N,N-dimethylchlorosulphitemethaniminium chloride 13 as the condensing
agent was reported by Kaul et al. The reaction proceeded through the formation of
activated carboxylic acid species, which react with o-phenylenediamine or 2-
aminophenol to yield acylated product, which cyclizes to benzimidazole or
benzoxazole in the presence of an acid (Scheme 4).4
R COOH
NH2
XH
N C
H3C
H3C O S
O
Cl
Cl
X
N
R+
0 oC 15 min
rt 6 hrX = NH, O
13
11 12 14
Scheme 4
A one-pot three component uncatalyzed conversion of activated C-H
compounds such as 4-hydroxycoumarin 15, 4-hydroxy-6-methylpyrone 16, 1,3-
dimethylbarbituric acid 17, 1,3-dimethyl-6-amino uracil 18 or dimedone 17 with p-
substituted benzaldehydes 20 and aliphatic nitriles 21 to pyran annulated
heterocyclic systems 22, 23, 24, 25 and 26 has been efficiently performed in water
as a green solvent. The reaction proceeds by consecutive Knoevenagel reaction,
Michael addition followed by a cyclization reaction to afford the products in good
yields (Scheme 5).5

145
OH
X
CN
R
+
O O
OH
H3C
O
OCN
NH2
H3C O
H
X
H2O, 80 oC
O O
OH
H2O, 80 oC
O O
O
NH2
R
X
N
N
O
O OH
CH3
H3C
N
N
O
O
CH3
H3C
O NH2
CN
NN
N
NH2
CN
O
O
CH3
H3C
N
N
O
O NH2
CH3
H3C
H2O, 80 oC
H2O, 80 oC
H2O, 80 oC
OO
O
CN
NH2
O
R = CN or CO2Et
X = H, NO2, OCH3
15 16
17
18
19 20 21
22 23
24
25
26
Scheme 5
A solvent-free condition under microwave irradiation technique was
developed for the synthesis of some useful basic heterocyclic systems by Heravi
and co-workers. 1,2,4-Triazines 32 & 33 were prepared from the condensation of
thiosemicarbazide 27 with diketones in the above mentioned condition. Reaction
of thiosemicarbazide 27 with dimethyl acetylenedicarboxylate (DMAD) and
diethyl acetylendicarboxylate (DEAD) in a solventless system under microwave
irradiation gave 3-mercapto-6-(methoxycarbonylmethylen)-1,2,4-triazine-5-one 28
and 3-mercapto-6-(ethoxycarbonylmethylen)-1,2,4-triazine-5-one 29 respectively.

146
2-Hydrazinothiazolon-4-one 30 was prepared from the condensation reaction of
thiosemicarbazide with chloroacetic acid under microwave irradiation in a
solventless system. In all the above mentioned reactions, the reaction time required
is minimum compared to the conventional reaction conditions (Scheme 6).6
H2NNH
S
NH2
O
O
H
H
HNN
NS
ClCH 2COOH
N
SHN
H2N
ODEAD
HNNH
HN
COOEt
H
S O
HNN
N CH3
CH3
S
CH3COCOCH 3
PhCOCOPh
HNN
N Ph
Ph
S
HNNH
HN
COOMe
H
S O
DMAD
27
28
29
30
31
33
32
Scheme 6
A mild and facile one-pot method was developed for the synthesis of 5-
methyl-[1,2,4]-triazoles 38 by Lindstrom and coworker. The synthesis involves the
treatment of N-alkyl substituted acetamides 34 with oxalyl chloride to form 35
which was then treated with either electron rich or electron poor aryl or
heterocyclic hydrazides 36 to afford the products 38 in good yields (Scheme 7).7

147
O
NH
R1
Cl
NR1
oxalylchloride2, 6-lutidine
DCM, 0 oC, 40 min
R2
O NH
NH2 rt, 1-5 h
NR1
HN
NH
O
R2
NaHCO3 saturated
reflux 100 oC, 1-3 hN N
N R2
R1
3435
36
3738
Scheme 7
A novel one-pot synthesis of cytotoxic and antiviral agents 3-(2-oxo-2H-
chromen-3-yl)-[1,3,4]thiadiazino[2,3-b]-quinazolin-6(2H)-ones 41 in high yields
has been developed by microwave-induced heterocyclization of 3-amino-2-
mercapto-3H-quinazolin-4-one 39 when irradiated in presence of 3-(2-
bromoacetyl) chromen-2-ones 40 in ethanol and anhydrous potassium carbonate.
The same reaction was repeated with conventional heating methods and the
reaction required much more time compared to the microwave technique (Scheme
8).8
N
N
O
SH
NH2 O O
O
Br
R1
R2
R3
R4 N
N
O
S
N
OO
R1
R2
R3
R4
+K2CO3 / MW / 400 W / 4-6 min
EtOH / K2CO3 / reflux / 4-6 h
39 40 41
Scheme 8
2-Oxazolidinones 43 was prepared from phenyl-2-hydroxyalkyl selenides
42 by a novel, convenient, efficient, three component one-pot reaction. The
reaction was carried out by treating phenyl-2-hydroxyalkyl selenides with benzoyl
isocyanate and subsequent oxidation and reduction of the resulting compound

148
followed by hydrolysis with hydrochloric acid solution and the product was
isolated in moderate to good yields (Scheme 9).9
PhSe
OHR1
R2
1. BzNCO, THF, rt, 15-17 h
2. MCPBA, K2HPO4, rt, 6-8 h
3. 4N HCl, 65 oC, 4h
O NH
R2R1
O
42 43
Scheme 9
Ketene-S,S-acetals 44 are effectively used for the generation of highly
substituted thiophene derivatives. In this method, one-pot reaction of electron
withdrawing group activated 2-methylene-1,3-dithiols 44 was treated with primary
amines, resulting in a ring opened intermediate which was cyclized in the presence
of the amine. By this reaction highly substituted thiophene derivatives 45 were
obtained in good yields. This reaction represents a new alternative method for the
preparation of multisubstituted thiophenes 45 (Scheme 10).10
R2
O
R1
SS S
R1R2
R3HN
O
R3NH2
MeCN 80 oC, 2-4 h
44 45
Scheme 10
A three component one-pot reaction was developed for the synthesis of
polysubstituted indeno[1,2-b]quinolines 49 using water as the solvent and
toluenesulfonic acid as the catalyst. In this reaction aldehydes 47 were treated with
various enaminones 48 and 1,3-indanedione 46 at 150 oC under microwave
irradiation condition. Products were isolated in moderate to excellent yields. This

149
one-pot three component reaction requires only minimal reaction time and simple
work-up procedure (Scheme 11).11
O
O
R CHO
R1HN
O
R2
R2
N
O O
R2
R2
R1
R
+ +
p-TsOH / H2O
MWI, 150 oC, 2-7 h
46 47 48 49
Scheme 11
A one-pot microwave mediated reaction between a wide variety of
propargylamine 50 and variety of aldehydes 51 by condensation followed by aza-
Claisen rearrangement for the synthesis of a wide variety of N-substituted pyrroles
52. For the reaction, different kinds of propargylamines 50 were treated with a
wide variety of aldehydes 51 in DMF with the presence of molecular sieves and the
reaction was conducted at 200oC for 30 min by microwave. By the reaction, an
enynamine intermediate was formed, which is converted by a [3,3]-pericyclic
rearrangement to iminoallene intermediate and undergoes subsequent cyclization to
give the substituted pyrroles in good yields (Scheme 12).12
R1
R2 NR3
O
HR4
N
H
R1
R3
R4
R2
DMF
Microwave-200 oC
30 min
+
50 51 52
Scheme 12
One-pot synthesis of highly substituted pyridine derivatives were described
by Rodinovskaya and co-workers. In this synthesis 4-di(tri)fluoromethyl-3-

150
cyanopyridine-2(1H)-thiones 57 were synthesized from R-methyl(methylene)
ketones 53 by a Claisen condensation with di(tri)fluoroacetate 54 followed by an
immediate Thorpe-Guareschi reaction of the intermediate diketone 55 formed with
cyanothioacetamide. By the reaction, 4-di(tri)fluoromethyl-3-cyanopyridine-2(1H)-
thiones 57 were synthesized in good yields without isolation and purification of the
intermediate formed (Scheme 13).13
R1
R2 O
CXF 2
ONa
N S
CN
CXF 2
R1
R2
H
NCS
NH2
R1
OR2
F2XC CO2EtHeat / EtOHAcOH
EtONa
Et2O, 0 oC
+
X = F or H
56
53 54 55 57
Scheme 13
6-Phenanthridinones and their heterocyclic analogues 60 were synthesized
through a one-pot procedure based on consecutive Pd-catalysed aryl-aryl and N-
aryl coupling from iodoarenes 58 with electron releasing substituents at the ortho
position and amides 59 of o-bromoarene and heteroarenecarboxylic acids. By the
reaction, the products 6-phenanthridinones and analogues 60 were isolated in
moderate to good yields (Scheme 14).14
I
R1
O
NHR 2
Br
Ar
O
NAr
R2
R1+
Pd(OAc)2 / TFP,
K2CO3, DMF or CH 3CN
58 59 60
Scheme 14

151
Highly substituted 3-chloro-1H-indole-2-carboxaldehydes 62 were prepared
in moderate to good yields by the one-pot reaction of various substituted 2-[(N-
carboxymethyl)amino]-5-alkylbenzoic acids 61 using Vilsmeier-Haack reagent.
This method offers a direct method for the conversion of diacids into indole
derivatives. When the nitrogen atom of the diacid 61 was replaced by oxygen or
sulfur, went the reaction proceeded smoothly and the products isolated were
benzofuran or benzothiophene derivatives (Scheme 15).15
R COOH
NH
COOH
R
NH
CHO
Cl
POCl3 / DMF
90 o
C, 4-6h
61 62
Scheme 15
Typically the reactions of active methylene compounds with Vilsmeier-
Haack reagent afford β-chloromethyleneiminium salts or β-chlorovinylaldehydes,
which have been recognized as useful intermediates in heterocyclic synthesis.16
Under Vilsmeier-Haack reaction condition, ketones 63 could effectively be
transformed into chlorovinamidinium salt intermediate which reacted with
malononitrile under Vilsmeier-Haack condition followed by cyclization and
aromatization to get the arylchloronicotinonitrile 64 in good yields (Scheme 16). 17
O
R1
R2
NR1
R2
Cl
CN
1. POCl3 / DMF, rt. 12 h
2. CNCH2CN, 90o C, 2 h
3. Aq. K2CO3
63 64
Scheme 16
Biginelli reaction for the synthesis of functionalized thiopyrimidone
derivatives 67 were modified by changing the conventional heating method with

152
microwave irradiation. In this reaction, urea or thiourea 65 was treated with ethyl
acetoacetate 66 and aldehydes 47 and the products 67 were isolated in good yields.
In the same way the Hantzsch synthesis for the synthesis of dihydropyridones 69
was also modified by treating aldehydes 47 and ethyl acetoacetate 66 with
ammonium acetate 68 and changing the conventional heating method with
microwave irradiation and the products 69 were isolated in good yields (Scheme 17
& 18).18
ArCHO
O O
OEt+ +
S
H2N NH2 NH
NH
S
Ar
EtO 2C
H3C
Microwave
47 66 65 67
Scheme 17
ArCHO
O O
OEt+ +
NH
CH3
Ar
EtO 2C
H3C
Microwave
2 eq.
NH4OAc
CO2Et
47 66 68 69
Scheme 18
Simple and efficient method for the synthesis of pyran annulated
heterocyclic systems 72 was developed by a three component condensation of
aldehydes 47, alkylnitriles 70 and coumarin 71. The reactions were carried out in
the presence of N,N,N',N'-tetramethylguanidinium trifluoroacetate (TMGT) as an
ionic liquid, which does not require any other reagent or organic solvent and the
products 72 were isolated in good yields (Scheme 19).19

153
CHO
X
RCH 2CN
O
OH
OO
O
O X
NH2
R
TMGT++
47 70 71 72
Scheme 19
The reaction of 6-aminouracils 73 with 2-oxoindolin-3-
ylideneacetophenones 74 afforded pyrimido[5,4:5'6']pyrido-[2,3-b]indole-2,4-
diones 75 via a regiospecific Michael addition, followed by cyclization. In the
same way the reaction of 6-aminouracil 73 with 2-oxoindolin-3-
ylidenemalononitriles 76 give rise to regiospecific formation of spiro-indolin-2-
one-3,5’-pyrido[2,3-d]pyrimidines 77 (Scheme 20 & 21).20
N
O
O
R1
R4
N
N
R3
O
R2
O
H2N
N
O
R1
R4
N
N
R3
O
R2O
NH
Ethanol
Reflux+
73 74 75
Scheme 20
N O
R1
N
N
CH3
O
CH3
O
H2N
Ethanol
Reflux+
NC
CN
N
O
HN
NN
O
OH3C
CH3
NC
H2N
R1
76 73 77
Scheme 21

154
As a continuation to these studies, it was of our interest to investigate the
reactions of 3-oxo-2,3-diarylpropanals with malononitrile under Vilsmeier-Haack
reaction condition for the synthesis of functionalized nitrogen heterocycles. The
results of our investigations are described in the following sections.
6.3. Results and discussions
6.3.1. Reactions of 3-oxo-2,3-diarylpropanals with malononitrile under
Vilsmeier-Haack reaction condition: Synthesis of 2-chloro-5,6-
diarylnicotinonitriles
The 3-oxo-2,3-diphenylpropanal 78 was treated with malononitrile in the
presence of Vilsmeier-Haack reagent, following the reported procedure (see
reference 17). In the above reaction, although diphenyl-2-chloronicotinonitrle was
expected to be formed, -chloroenone was formed in good yields. It was also
observed that the reaction is not proceeded to pyridine formation once -
chloroenone is formed in the reaction. As malononitrile is readily converted to
corresponding enamine derivative under Vilsmeier-Haack reaction condition, the
optimization studies provided unsatisfactory results only. However when 3-oxo-
2,3-diphenylpropanal 78a was treated with malononitrile 79 in the presence of 1.5
equivalent of Vilsmeier-Haack reagent at 700C for 3hrs, 2-chloro-5,6-
diphenylnicotinonitrile 80a was obtained in 27% yields. The yield of the reaction
was further improved to 55% by the use of excess malononitrile (4 equivalents) in
the above reaction (Scheme 22).
O
OH
DMF / POCl3 N Cl
CN70 0C
NC CN+
78a 79 80a
Scheme 22

155
2-Chloro-5,6-diphenylnicotinonitrile 80a was characterized on the basis of
IR, 13
C NMR, 1H NMR and GCMS spectra. In the IR spectrum of 2-chloro-5,6-
diphenylnicotinonitriles 80a the aromatic C-H stretchings were observed at 3045
and 3031 cm-1
respectively. The CN (cyano) group stretching was observed at
2221 cm-1
. The C=N bond stretching was observed at 1442 cm-1
and the C=C
stretching was observed at 1566 cm-1
respectively. The C-Cl bond stretching was
observed at 700 cm-1
. The GCMS spectrum of the compound 80a has given the
molecular ion peak at m/z 290 along 292 as (M+2)+ peak. The base peak, observed
at m/z 255 may be due to the fragment generated after the expulsion of the chlorine
atom from 2-chloro-5,6-diphenylnicotinonitrile 80a. Other major m/z peaks
observed in GCMS spectrum were 227, 212, 201, 189, 178, 149, 127, 113, 100, 77
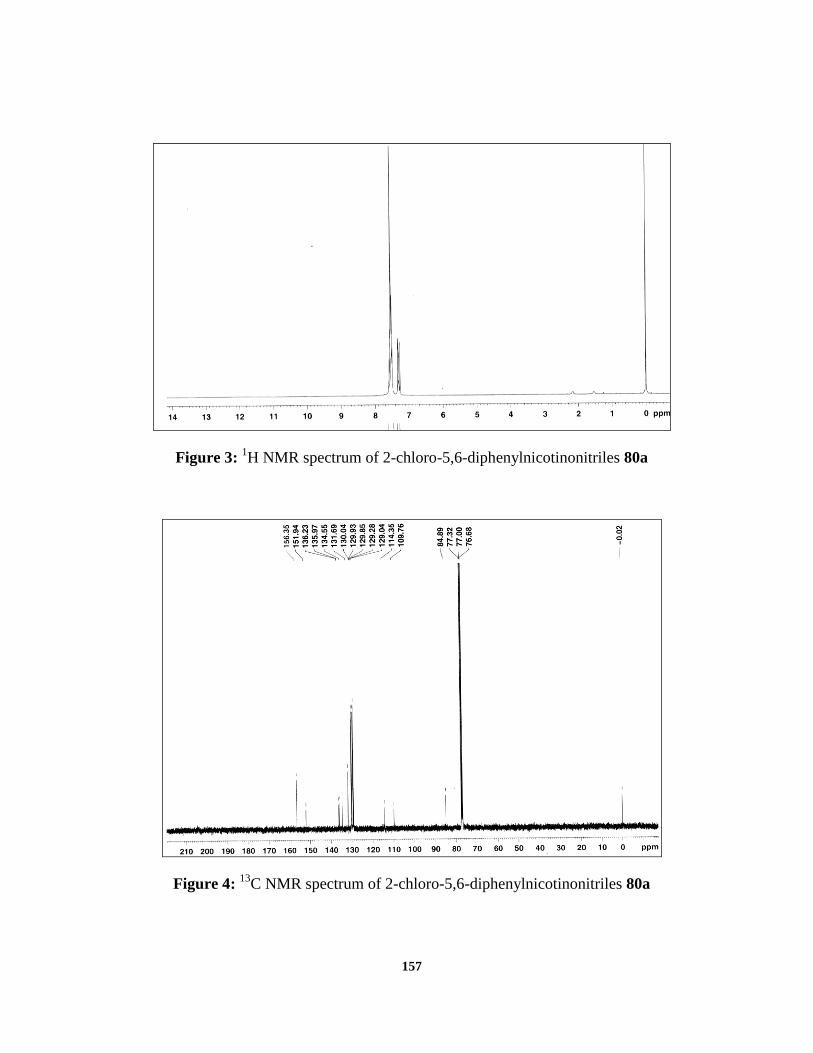
etc. The proton NMR spectrum of 2-chloro-5,6-diphenylnicotinonitrile showed
two multiplets at δ 7.6-7.45 (9H) and 7.35-7.28 (2H) shows the presence of
aromatic hydrogen atoms. The hydrogen atom at the para position in the newly
formed pyridine ring must give singlet peak in the NMR, but the peak is merged
with the multiplets of the aromatic protons of the two phenyl rings. In the carbon-
13 NMR spectrum, 2-chloro-5,6-diphenylnicotinonitrile 80a showed peaks at δ
156.35, 151.94, 136.23, 135.97, 134.55, 131.69, 130.04, 129.93, 129.85, 129.28,
129.04, 109.76 and 84.84 respectively. The CN carbon was observed at δ 114.35
ppm. The CHN elemental analysis of the compound 80a has given the percentage
of various elements presented as carbon-74.38, hydrogen-3.84 and Nitrogen-9.68 is
found to be agreeing with the calculated value Carbon-74.36, Hydrogen-3.81 and
Nitrogen-9.64 confirming the molecular formula C18H11ClN2.

156
Figure 1: IR spectrum of 2-chloro-5,6-diphenylnicotinonitriles 80a
50 75 100 125 150 175 200 225 250 275
500e3
1000e3
1500e3
2000e3
255
40
227100
11351
29077
149212201
189127 1788763252167
279 295
Figure 2: GCMS spectrum of 2-chloro-5,6-diphenylnicotinonitriles 80a
157
Figure 3: 1H NMR spectrum of 2-chloro-5,6-diphenylnicotinonitriles 80a
Figure 4: 13
C NMR spectrum of 2-chloro-5,6-diphenylnicotinonitriles 80a

158
On the basis of our investigations on the reaction of 2-oxo-3,6-
diphenylpropanal 78a under Vilsmeier-Haack reaction condition we propose a
suitable mechanism for the formation of 2-chloro-5,6-diphenylnicotinonitrile 80a
as follows. Initially, the 2-oxo-3,6-diphenylpropanal 78a underwent
chloromethylation on its oxygen atom to get an intermediate 81, which could be
electrophilically added to malononitrile 79, following an addition-elimination
mechanism to get 80a. The intramolecular addition of the nitrogen atom on the
nitrile group to the carbonyl group in the molecule, followed by the addition of
chloride ion to the iminium ion and aromatization could result in the formation of
2-chloro-5,6-diphenylnicotinonitrile (Scheme 23).
O
OH
DMF / POCl3
N
Cl
CN
O
OH
NNC
CN
H
CN
OHN
N
- DMF
- DMF, -H
78a
81a80a
O
OH
NC CNNN
Cl
H
H
Cl
H
O
N N
H
N
Cl HCl
H
-Cl
Scheme 23
The reaction was extended to other substituted propanals and found that the
5,6-diaryl-2-chloronicotinonitriles are formed in moderate yields (Scheme 24).

159
O
OH
DMF / POCl3 N Cl
CN70 0CR1
R2
R1
R2
NC CN+
78a-f 79 80a-f
Scheme 24
Table 1: Synthesis of 2-chloro-5,6-diarylnicotinonitrile from 2-chloro-5,6-
diarylnicotinonitriles and malononitrile
80 R1 R2 Yields ( % )
a H H 55
b p-Cl H 35
c p-OMe H 45
d p-Br H 36
e p-CH3 p-OMe 12
f p-CH3 o-Cl Trace amount
6.3.2. Reactions of 3-oxo-2,3-diarylpropanals with cyanoacetamide under
Vilsmeier-Haack reaction conditions: Synthesis of 2-chloro-5,6-
diarylnicotinonitriles
Next we treated 3-oxo-2,3-diarylpropanals 78 with cyanoacetamide 82
under Vilsmeier-Haack reaction condition at 70 °C. The reaction was monitored
by TLC and found that it is completed within 12 hours. 2-Chloro-5,6-
diphenylnicotinonitrile 80 was isolated as pale yellow crystalline solid in 70 %
yields (Scheme 25). The product was characterized on the basis of common

160
spectroscopic methods and the reaction was extended to other propanals to get
substituted nicotinoinitriles in good yields.
O
OH
R1
R2 1. POCl3 / DMF, rt., 12 h
2. Aq. K2CO3
N
CN
ClR1
R2
OCN
NH2
+
78 82 80
Scheme 25
Table 2 Synthesis of 2-chloro-5,6-diarylnicotinonitriles from 2-chloro-5,6-
diarylnicotinonitriles and cyanoacetamide
80 R1 R2 Yields ( % )
a H H 70
b p-Cl H 77
c p-OMe H 80
d p-Br H 90
e p-CH3 p-OMe 67
Mechanism of the reaction is explained as follows: As in the earlier case
cyanoacetamide 82 is added to 3-oxo-2,3-diarylpropanals 78 under Vilsmeier-
Haack condition to get an intermediate 83. The iminoalkylation on the amide
oxygen atom followed by chlorination and intramolecular cyclization of the
enamine to keto carbonyl group could result in the formation of 2-chloro-5,6-
diarylnicotinonitriles 80 in good to excellent yields.

161
O
OH
R1
R2
1. POCl3 / DMF rt., 2 h
N
CN
ClR1
R2
O
OH
R1
R2
HN
NH2
O
CN
O
H
R1
R2
CN
H2N O
O
H
R1
R2
CN
H2N O N
O
H
R1
R2
CN
H2N Cl
OH
CN
HN
Cl
-DMF
Cl
8378
80
R1
R2
H
N
HCl
-DMF
N
HCl
Scheme 26
6.4. Conclusion
Substituted halopyridines are important as substrates for coupling reactions
for the synthesis of functionalized heterocyclic compounds and natural product
synthesis. We could develop a facile, one-pot reaction for the synthesis of 2-
chloro-5,6-diarylnicotinonitriles from diarylpropanals, which are otherwise difficult
to synthesis.
6.5. Experimental
Melting points were determined on a Buchi 530 melting point apparatus and
were uncorrected. The IR spectra were recorded by KBr pellet method on a
Shimadzu FTIR 470 spectrometer and the frequencies are reported in cm-1
. Mass
spectra were recorded on a GCMS Shimadzu 5050 model instrument. 1H NMR

162
spectra were recorded on a Bruker EM 400 MHz spectrometer using CDCl3 as the
solvent. 13
C NMR spectra were recorded on a Bruker EM 400 MHz spectrometer
using CDCl3 as the solvent. Both 13
C NMR and
1H NMR values are expressed as δ
(ppm). CHN analyses were done on a Shimadzu CHNS analyzer.
All reagents were commercially available and were purified before use. All
the solvents used were dried and distilled under reduced pressure. All the purified
compounds gave single spot upon TLC analyses on silica gel 7GF using an ethyl
acetate/hexane as eluent. Iodine vapors or KMnO4 in water was used as the
developing agent for TLC.
6.5.1. Reaction of 3-oxo-2,3-arylpropanals with malononitrile
General procedure
The Vilsmeier-Haack reagent was prepared by the slow addition of POCl3
(1.395 ml, 15 mmol) to DMF (12 ml, 150 mmol) at 0oC followed by stirring at
room temperature for 15 min. Appropriate amount of 3-oxo-2,3-diarylpropanal (10
mmol) and malononitrile (3.87 gm, 58.7mmol) were added to this reagent. The
reaction mixture was stirred at room temperature followed by stirring at 70 oC for 3
hours and poured into ice cold saturated potassium carbonate solution (120ml).
The crude product was extracted with ethyl acetate (3 X 50 ml). The combined
organic layer was evaporated to afford the crude product. The crude product was
purified by column chromatography over silica gel using 2% ethyl acetate in
hexane as eluent. The pure products were isolated, and melting points were
recorded and characterized using IR, GCMS, 1H NMR and
13C NMR spectra and
elemental analyses.

163
N
CN
Cl
C18H11ClN2
Mol. Wt.: 290.75
2-Chloro-5,6-diphenylnicotinonitrile 80a was obtained
by the reaction between 3-oxo-2,3-diphenylproanal 78a
(10 mmol, 2.24 g) and 4 equivalent of malononitrile 79
(3.87 gm, 58.7mmol) in presence of POCl3 (1.395 ml, 15
mmol) and DMF (12 ml, 150 mmol) at 70oC as a pale
yellow crystalline substance. Yield = 55 % (1.6 g);
Melting point = 168-170oC; IR (KBr, υmax) = 3045
(aromatic C-H), 3031 (aromatic C-H), 2221 (CN), 1566
(C=C), 1548 (C=N), 765 (C-Cl), 700 (C-Cl) cm-1
; GCMS
m/z = 292 (M+2)+, 291 (M+1)
+, 290 (M)
+, 255 (base
peak), 227, 212, 201, 189, 149, 127, 113, 99, 77; 1H NMR
(400 MHz, CDCl3) δ = 7.6-7.45 (9H, m, ArH), 7.35-7.28
(2H, m, ArH); 13
C NMR (400 MHz, CDCl3) δ = 156.35,
151.94, 136.23, 135.97, 134.55, 131.69, 130.04, 129.93,
129.85, 129.28, 129.04, 114.35 (CN), 109.76, 84.89; Anal.
Calcd. data of C18H11ClN2: Carbon-74.38; Hydrogen-3.84
and Nitrogen-9.68. Found: Carbon-74.5; Hydrogen-4.1;
Nitrogen-9.8.
N
CN
Cl
Cl
C18H10Cl2N2
Mol. Wt.: 325.19
2-Chloro-6-(4-chlorophenyl)-5-phenylnicotinonitrile
80b was obtained by the reaction between 3-(4-
chlorophenyl)-3-oxo-2-phenylpropanal 78b (2.58 g, 10
mmol) and 4 equivalent of malononitrile 79 (3.87 gm,
58.7mmol) in presence of POCl3 (1.395 ml, 15 mmol) and
DMF (12 ml, 150 mmol) at 70oC as a pale yellow powder.
Yield = 35 % (1.14 g); Melting point = 120-122oC; IR
(KBr, υmax) = 3168 (aromatic C-H), 3080 (aromatic C-H),
2219 (CN), 1641 (C=C), 1554 (C=N), 1535 (C=N), 700

164
(C-Cl), 639 (C-Cl) cm-1
; GCMS m/z = 326 (M+2)+, 324
(M)+, 323, 299, 297, 281, 278, 253, 245, 218, 207, 190,
178, 164, 139, 113, 103, 77(base peak); 1H NMR (400
MHz, CDCl3) δ = 7.95-7.85 (3H, m, ArH), 7.44-7.2 (7H,
m, ArH); 13
C NMR (400 MHz, CDCl3) δ = 143.35,
139.52, 135.04, 129.53, 129.51, 128.89, 128.86, 128.7,
128.67, 128.08, 127.34, 126.84; Anal. Calcd. data of
C18H10Cl2N2: Carbon-66.48, Hydrogen-3.10 and Nitrogen-
8.61. Found: Carbon-66.53; Hydrogen-3.2; Nitrogen-8.68.
N
CN
Cl
MeO
C19H13ClN2O
Mol. Wt.: 320.77
2-Chloro-6-(4-methoxyphenyl)-5-phenylnicotinonitrile
80c was obtained by the reaction between 3-(4-
methoxyphenyl)-3-oxo-2-phenylpropanal 78c (2.54 g, 10
mmol) and 4 equivalent of malononitrile 79 (3.87 gm,
58.7mmol) in presence of POCl3 (1.395 ml, 15 mmol) and
DMF (12 ml, 150 mmol) at 70oC as a pale yellow
crystalline substance. Yield = 45 % (1.44 g); Melting
point = 200-202oC; IR (KBr, υmax) = 2215 (CN), 1599
(C=C), 1553 (C=N), 709 (C-Cl) cm-1
; GCMS m/z = 322
(M+2)+, 321 (M+1)
+, 320 (M)
+, 305, 294, 285 (base peak),
270, 253, 242, 227, 214, 189, 165, 158, 139, 121, 113, 93,
88, 77; 1H NMR (400 Mhz, CDCl3) δ = 7.669-7.5 (4H, m,
ArH), 7.46 (2H, d, j = 8.4 Hz, ArH), 7.4-7.2 (m, 2H, ArH),
7.01 (2H, d, j = 8.4 Hz, ArH), 3.916 (3H, s, OMe); 13
C
NMR (400 MHz, CDCl3) δ = 162.55, 157.05, 152.33,
135.12, 134.8, 132.22, 129.96, 129.74, 129.22, 128.49,
114.67, 114.39 (CN), 110.08, 83.75, 55.67; Anal. Calcd.
data of C19H13ClN2O: Carbon-71.14; Hydrogen-4.08 and

165
Nitrogen-8.73. Found: Carbon-71.19; Hydrogen-4.1;
Nitrogen-8.78.
N
CN
Cl
Br
C18H10BrClN2
Mol. Wt.: 369.64
2-Chloro-6-(4-bromophenyl)-5-phenylnicotinonitrile
80d was obtained by the reaction between 3-(4-
bromophenyl)-3-oxo-2-phenylpropanal 78d (3.03 g, 10
mmol) and equivalent of malononitrile 79 (3.87 gm,
58.7mmol) in presence of POCl3 (1.395 ml, 15 mmol) and
DMF (12 ml, 150 mmol) at 70 oC as a pale yellow
crystalline substance. Yield = 36 % (1.32 g); Melting
point = 178-180 oC; IR (KBr, υmax) = 3085 (aromatic
C-H), 3064 (aromatic C-H), 3028 (aromatic C-H), 2221
(CN), 1677 (C=C), 1583 C=N), 703 (C-Cl) cm-1
; GCMS
m/z = 370 (M+2)+, 368 (M)
+, 336, 335, 333, 332, 291,
289, 255, 254 (base peak), 253, 227, 201, 176, 175, 151,
127, 113, 100, 88, 77; 1H NMR (400 MHz, CDCl3)
δ = 7.679 (2H, d, j = 8.4 Hz, ArH), 7.57-7.5 (3H, m, ArH),
7.49 (1H, s, ArH), 7.37 (2H, d, j = 8.4 Hz, ArH), 7.34-7.26
(2H, m, ArH); 13
C NMR (400 MHz, CDCl3) δ = 155.8,
150.22, 136.28, 134.97, 134.31, 132.39, 131.44, 130.07,
129.74, 129.34, 126.52, 114.17 (CN), 109.64, 85.55; Anal.
Calcd. data of C18H10BrClN2: Carbon-58.49; Hydrogen-
2.73 and Nitrogen-7.58. Found: Carbon-58.5; Hydrogen-
2.8; Nitrogen-7.6.

166
N
CN
Cl
H3C
MeO
C20H15ClN2O
Mol. Wt.: 334.80
2-Chloro-5-(4-methoxyphenyl)-6-(4-
methylphenyl)nicotinonitrile 80e was obtained by the
reaction between 2-(4-methoxyphenyl)-3-(4-
methylphenyl)-3-oxopropanal 78e (2.68 g, 10 mmol) and
4 equivalent of malononitrile 79 (3.87 gm, 58.7mmol) in
presence of POCl3 (1.395 ml, 15 mmol) and DMF (12 ml,
150 mmol) at 70 oC as a pale yellow crystalline substance.
Yield = 12 % (0.401 g); Melting point = 172-174 oC; IR
(KBr, υmax) = 3033 (aromatic C-H), 3003 (aromatic C-H),
2218 (CN), 1608 (C=C), 1546 (C=N), 738 (C-Cl), 665 (C-
Cl) cm-1
; GCMS m/z = 336 (M+2)+, 335 (M+1)
+, 334 (M)
+,
319, 299, 284, 278, 269, 256, 241, 226, 207, 191, 189, 163,
152, 128, 119, 99, 91; 1H NMR (400 MHz, CDCl3) δ =
7.556 (1H, s, ArH), 7.371 (2H, d, j = 8.4 Hz, ArH), 7.314
(2H, d, j = 8.4 Hz, ArH), 7.261 (1H, s, ArH), 7.211 (2H, d, j
= 8 Hz, ArH), 7.024 (2H, d, j = 8 Hz, ArH), 3.868 (3H, s,
OMe), 2.463 (3H, s, -CH3); 13
C NMR (400 MHz, CDCl3) δ
= 160.69, 157.58, 151.96, 142.55, 135.22, 133.64, 131.25,
130.14, 129.66, 127.06, 114.6 (CN), 110.18, 84.4, 55.32,
21.55; Anal. Calcd. data of C20H15ClN2O: Carbon-71.75;
Hydrogen-4.52 and Nitrogen-8.37. Found: Carbon-71.78;
Hydrogen-4.8; Nitrogen-8.41.
6.5.2. Reaction of 3-oxo-2,3-diarylpropanals with cyanoacetamide
General procedure
The Vilsmeier-Haack reagent was prepared by the slow addition of POCl3
(3.7 ml, 40 mmol) to DMF (31 ml, 400 mmol) at 0 oC followed by stirring at room

167
temperature for 15 min. Appropriate amount of 3-oxo-2,3-diarylpropanal (10
mmol) and cyanoacetamide (0.84 g, 10 mmol) were added to this reagent. The
reaction mixture was stirred at room temperature followed by stirring at 70 oC for
12 hours and poured into ice cold saturated potassium carbonate solution (120ml).
The crude product was extracted with ethyl acetate (3 X 50 ml). The combined
organic layer was evaporated to afford the crude product. The crude product was
purified by column chromatography over silica gel using 2% ethyl acetate in
hexane as eluent. The pure products were isolated, and melting points were
recorded and characterized using IR, GCMS, 1H NMR and
13C NMR spectra.
N
CN
Cl
C18H11ClN2
Mol. Wt.: 290.75
2-Chloro-5,6-diphenylnicotinonitriles 80a was obtained
by the reaction between 3-oxo-2, 3-diphenylproanal 78a
(10 mmol, 2.24 g) and 1 equivalent of cyanoacetamide
(0.84 g, 10 mmol) in presence of POCl3 (3.7 ml, 40
mmol) and DMF (31 ml, 400 mmol) at 70oC as a pale
yellow crystalline substance. Yield = 70 % (2.03 g);
Melting point = 168-170oC
N
CN
Cl
Cl
C18H10Cl2N2
Mol. Wt.: 325.19
2-Chloro-6-(4-chlorophenyl)-5-phenylnicotinonitriles
80b was obtained by the reaction between 3-(4-
chlorophenyl)-3-oxo-2-phenylpropanal 78b (2.58 g, 10
mmol) and 1 equivalent of cyanoacetamide (0.84 g, 10
mmol) in presence of POCl3 (3.7 ml, 40 mmol) and DMF
(31 ml, 400 mmol) at 70oC as a pale yellow powder.
Yield = 77 % (2.51 g); Melting point = 120-122oC

168
N
CN
Cl
MeO
C19H13ClN2O
Mol. Wt.: 320.77
2-Chloro-6-(4-methoxyphenyl)-5-phenylnicotinonitriles
80c was obtained by the reaction between 3-(4-
methoxyphenyl)-3-oxo-2-phenylpropanal 78c (2.54 g, 10
mmol) and 1 equivalent of cyanoacetamide (0.84 g, 10
mmol) in presence of POCl3 (3.7 ml, 40 mmol) and DMF
(31 ml, 400 mmol) at 70oC as a pale yellow crystalline
substance. Yield = 80 % (2.56 g); Melting point = 200-
202oC
N
CN
Cl
Br
C18H10BrClN2
Mol. Wt.: 369.64
2-Chloro-6-(4-bromophenyl)-5-phenylnicotinonitriles
80d was obtained by the reaction between 3-(4-
bromophenyl)-3-oxo-2-phenylpropanal 78d (3.03 g, 10
mmol) and 1 equivalent of cyanoacetamide (0.84 g, 10
mmol) in presence of POCl3 (3.7 ml, 40 mmol) and DMF
(31 ml, 400 mmol) at 70oC as a pale yellow crystalline
substance. Yield = 90 % (3.32 g); Melting point = 178-
180oC
N
CN
Cl
H3C
MeO
C20H15ClN2O
Mol. Wt.: 334.80
2-Chloro-5-(4-methoxyphenyl)-6-(4-
methylphenyl)nicotinonitriles 80e was obtained by the
reaction between 2-(4-methoxyphenyl)-3-(4-methylphenyl)-
3-oxopropanal 78e (2.68 g, 10 mmol) and 1 equivalent of
cyanoacetamide (0.84 g, 10 mmol) in presence of POCl3
(3.7 ml, 40 mmol) and DMF (31 ml, 400 mmol) at 70oC
as a pale yellow crystalline substance. Yield = 67 % (2.24
g); Melting point = 172-174oC.

169
6.6. References
1 Ryabukhin, S. V.; Plaskon, A. S.; Volochnyuk, D. M.; Pipko, S. E.;
Tolmachev, A. A.; Synthetic communications, 2008, 38, 3032-3043.
2 Jing, X.; Zhu, Q.; Xu, F., Ren, X.; Li, D.; Yan, C.; Synthetic communications,
2006, 36, 2597.
3 Tormyshev, V. M.; Trukhin, D. V.; Rogozhnikova, O. Y.; Mikhalina, T. V.;
Troitskaya, T. I.; Flinn, A.; Synlett, 2006, No. 16, 2559-2564.
4 Kaul, S.; Kumar, A.; Sain, B.; Bhatnagar, A. K.; Synthetic Communications,
2007, 37, 2457-2460.
5 Shaabani, A.; Samadi, S.; Rahmati, A.; Synthetic Communications, 2007,
37, 491-499.
6 Heravi, M. M.; Nami, N.; Oskooie, H. A.; HekmatShoar, R.; Phosphorus,
Sulfur and Silicon, 2006, 181, 87-91.
7 Lindstrom, J.; Johansson, M. H.; Synthetic Communications, 2006, 36,
2217-2229.
8 Kumar, V. N.; De Clercq, E.; Rajitha, B.; Phosphorus, Sulfur and Silicon,
2007, 182, 273-279.
9 Sheng, S.-R.; Luo, H.-R.; Huang, Z.-Z.; Sun, W.-K.; Liu, X.-L.; Synthetic
Communications, 2007, 37, 2693-2699.
10 Liang, F.; Li, D.; Zhang, L.; Gao, J.; Liu, Q.; Org. Lett.; 2007, 9(23), 4845-
4848.
11 Tu, S.-J.; Jiang, B.; Zhang, J.-Y.; Jia, R. H.; Zhang, Y.; Yao, C.-S.; Org.
Biomol. Chem., 2006, 4, 3980-3985.
12 Bremner, W. S.; Organ, M. G.; J. Comb. Chem., 2008, 10, 142-147.

170
13 Rodinovskaya, L. A.; Shestopalov, A. M.; Gromova, A. V.; Shestopalov, A.
A.; J. Comb. Chem., 2008, 10, 313-322.
14 Ferraccioli, R.; Carenzi, D.; Rombola, O.; Catellani, M.; Org. Lett., 2004,
6(25), 4759-4762.
15 Majo, V. J.; Perumal, P. T.; J. Org. Chem.; 1996, 61, 6523-6525.
16 Arnold, Z.; Zemlicka, J.; Collect. Czech. Chem. Commun., 1959, 24, 2385.
17 Asokan, C. V.; Anabha, E. R.; Thomas, A. D.; Jose, A. M.; Lethesh, K. C.;
Prasanth, M.; Krishnaraj, K. U.; Tetrahedron Letters, 2007, 48, 5641-5643.
18 Kidwai, M.; Saxena, S.; Mohan, R.; Venkataramanan, R.; J. Chem. Soc.,
Perkin Trans І, 2002, 1845-1846.
19 Shaabani, A.; Samadi, S.; Badri, Z.; Rahmati, A.; Catalysis Letters, 2005,
104(1-2), 39-43.
20 El-Ahl, A.-A., S.; Synthetic Communications, 2000, 30(12), 2223-2231.