openmm library - prace research infrastructure · openmm library “one ring to rule ... namd amber...
TRANSCRIPT

OpenMM library“One ring to rule them all”
https://simtk.org/home/openmm

The OpenM(olecular)M(echanics) API
https://simtk.org/home/openmm
GromacsNAMD
AMBER ???
LAMMPS
HOOMD
PrestoCHARMM
Tinker
* Abalone * ACEMD * ADUN * Ascalaph * COSMOS * Desmond * Culgi * ESPResSo * GROMOS * GULP * Hippo * Kalypso MD * LPMD * MacroModel * MDynaMix * MOLDY * Materials Studio * MOSCITO * ProtoMol * RedMD * YASARA * ORAC * XMD

Goals of OpenMM
l Complete libraryl provide what is most neededl easy to learn and use
l Fast, general, extensiblel optimize for speedl hide hardware specificsl support new hardwarel add new force fields, integration methods etc.

l Available APIs in C++, Fortran95, Pythonl Advantages
l Object orientedl Modularl Extensible
l Disadvantagesl Programs written in other languages need to invoke it
through a layer of C++ code

Features
• Electrostatics: cut-off, Ewald, PME• Implicit solvent• Integrators: Verlet, Langevin, Brownian, custom• Thermostat: Andersen• Barostat: MonteCarlo• Others: energy minimization, virtual sites
5

The OpenMM Architecture
OpenMM Public API
OpenMM Implementation Layer
OpenMM Low Level API
CUDA/OpenCL/Brook/MPI etc.
Public Interface
Platform Independent Code
Platform Abstraction Layer
Computational Kernels

Public API Classes (1)
l Systeml A collection of interaction particlesl Defines the mass of each particle (needed for
integration)l Specifies distance restraintsl Contains a list of Force objects that define the
interactionsl OpenMMContext
l Contains all state information for a particular simulation− Positions, velocities, other parmeters

Public API Classes (2)
l Forcel Anything that affects the system's behavior: forces,
barostats, thermostats, etc.l A Force may:
− Apply forces to particles− Contribute to the potential energy− Define adjustable parameters− Modify positions, velocities, and parameters at the start of
each time stepl Existing Force subclasses
− HarmonicBond, HarmonicAngle, PeriodicTorsion, RBTorsion, Nonbonded, GBSAOBC, AndersenThermostat, CMMotionRemover...

Public API Classes (3)
l Integratorl Advances the system through timel Only fixed step size integrators are supportedl LeapFrog, Langevin, Brownian Integrators
l Statel A snapshot of the state of the systeml Immutable (used only for reporting)l Creating a state is the only way to access positions,
velocities, energies or forces.

Example
• Create a System System system ;for (int i = 0; i < numParticles; ++i) system.addParticle(mass[i]);for (int i = 0; i < numConstraints; ++i) system.addConstraint(constraint[i].atom1, constraint[i].atom2, constraint[i].distance);
• Add Forces to it HarmonicBondForce* bonds = new HarmonicBondForce;for (int i = 0; i < numBonds; ++i) bonds->addBond(bond[i].atom1, bond[i].atom2, bond[i].length, bond[i].k);system.addForce(bonds);// ... add Forces for other force !eld terms.

Example (continued)
• Simulate it
LangevinIntegrator integrator(297.0, 1.0, 0.002); // Temperature, friction, // step sizeOpenMMContext context(system, integrator);context.setPositions(positions);context.setVelocities(velocities);integrator.step(500); // Take 500 steps
• Retrieve state information
State state = context.getState(State::Positions | State::Velocities);for (int i = 0; i < numParticles ; ++i) cout << state.getPositions()[i] << endl;

Some algorithms for stream computing in OpenMM

l O(N2) (all-vs-all) algorithml O(N) algorithml Ewald summation (O(N3/2))l Particle-Mesh Ewald (PME) (O(N*logN)
Algorithms for calculation oflong-range interactions in MD

MD simulations on GPUs
l Algorithms need to be reconsidered from the ground up
l Fast on CPU != Fast on GPU
Slow on CPU != Slow on GPU

nonbonded interactions inmolecular systems
l Divide into short-range and long-range interactionsl Calculate short-range analyticallyl Use approximations for the long-range

O(N2) (all-vs-all) algorithm on GPUs

efficient implementation ofO(N2) algorithm would...
l minimize global memory accessl avoid thread synchronizationl take advantage of symmetry

algorithm
• Group atoms into blocks of 32 • Interactions divide into 32x32
tiles • Each tile is processed by a
group of 32 threads – Load atom coordinates and
parameters into shared memory – Each thread computes
interactions of one atom with 32 atoms
– Use symmetry to skip half the tiles
32 64 96 ...
32
64
96
...
0 0
Atoms
Atoms

algorithm (cont.)
• Each thread loops over atoms in a different order – Avoids con#icts between threads
• No explicit synchronization needed – Threads in a warp are always synchronized
Atoms
Threads

O(N) algorithm on GPUs

why this is hard
• Traditional O(N) methods (e.g. neighbor lists) are slow on GPUs – Out of order memory access
for i = 1 to numNeighbors
load coordinates and parameters for neighbor[i]
compute force
store force for neighbor[i]
• The inner loop contains non-coalesced memory access!
slow!
slow!
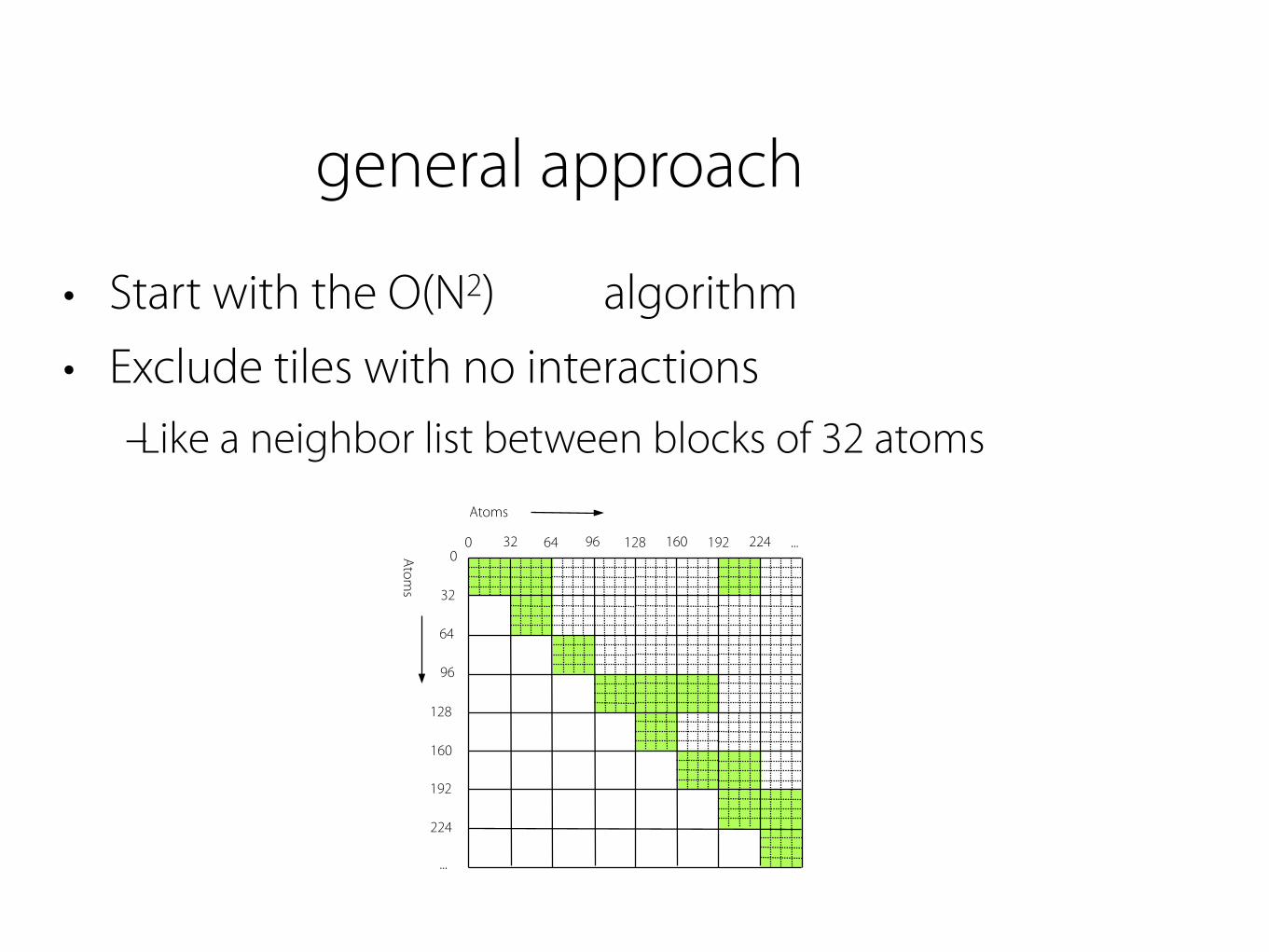
general approach
• Start with the O(N2) algorithm • Exclude tiles with no interactions
– Like a neighbor list between blocks of 32 atoms
64 128 192 ...
64
128
192
...
0 0
Atoms
Atoms
96 160 224 32
96
160
224
32

• Compute an axis aligned bounding box for the 32 atoms in each block
• Calculate the distance between bounding boxes
identifying tiles with interactions

solvent molecules
• Solvent molecules must be ordered to be spatially coherent – Otherwise bounding boxes will be very large
• Arrange along a space !lling curve • Reorder every ~100 time steps

performance of this algorithm
• Much faster than O(N2) for large systems • Performance scales linearly
But • Computes many more interactions than
really required –
few (or none!) are within the cutoff Computes all 1024 interactions in a tile, even if

Finer Grained Neighbor List
• For each tile with interactions: – Compute distance of each atom in one block
from the bounding box of the other block – Set a #ag for each atom

Force Computation for Tile for i = 1 to 32
if (hasInteractions[i])
compute interaction with atom i
• All 32 threads must loop over atoms in the same order
} on each thread
Atoms
Threads
– Requires a reduction to sum the forces
– For a few atoms, this is still much faster
– For many atoms, better to just compute all interactions

benchmarks

Tiling circles is difficult
serial computing stream computing
Two issues:
• You need a lot of cubes to cover a sphere
• All interactions beyond the cut-off need to be zero
Para2010 – p. 20/22
Tiling

The art of calculating zeros
0 2000 4000 6000 8000 10000
pair interactions per µs
GPU 3k, rc=1.5, buffered
GPU 48k, rc=1.5, buffered
CPU, rc=1.5, no water force loops
CPU, rc=1.5
CPU, rc=0.9
zeros
Para2010 – p. 21/22
Calculating zeros!

Ewald summation method

Ewald summation method O(N2) algorithm

Ewald summation methodO(N3 / 2) algorithm
for each wave vector K CosSum = 0 SinSum = 0
for each atom i StructureFactor_Real(i) = charge(i) * cos(K.coord(i)) StructureFactor_Imag(i) = charge(i) * sin(K.coord(i)) CosSum += StructureFactor_Real(i) SinSum += StructureFactor_Imag(i) for each atom i force(i) = CosSum * StructureFactor_Imag(i) – SinSum * StructureFactor_Real(i)

CalculateEwaldForces_kernel()
// Each thread is processing// one of the atoms
atom = threadIdx.x + blockIdx.x * blockDim.x;
// Load atom data force = Force(atom) coord = Coord(atom) // Loop over all wave vectors.// Compute the force contribution of this wave vector. for each wave vector K
// calculate again (!) the structure// factor for this wave vector structureFactor.x = cos(K.coord(i)) structureFactor.y = sin(K.coord(i))
// Load the Cosine/Sine Sum from memory cosSinSum = cSim.pEwaldCosSinSum[K]; force += charge(i) * (cosSinSum.x*structureFactor.y- cosSinSum.y*StructureFactor.x)
// Record the force on the atom. Force(atom) = force
CalculateCosSinSums_kernel()
// Each thread is processing// one of the wave vectors K
K = threadIdx.x + blockIdx.x * blockDim.x;
// Compute the sum for that K #oat2 sum = make_#oat2(0.0f, 0.0f );
// Read atom datafor each atom i sum.x += charge(i) * cos(K.coord(i)) sum.y += charge(i) * sin(K.coord(i))
// Store the sum in global memory Sum(K) = sum;
// Compute the contribution to the energy. energy += sum * sum
// Store energy in global memory Energy(K) = energy
Ewald O(N3 / 2) algorithm on GPUs

Particle-Mesh Ewald (PME) method

Particle-Mesh Ewald method
1) calculate scaled fractionals
2) calculate B-spline coefficient
3) “smear” the charges over the grid points from spline-coefficient 4) execute forward FFT
5) calculate the reciprocal energy 6) execute backward FFT
7) calculate force gradient

CPUspreading of charges
GPUgathering of charges
l sort the atoms before gather !
Particle-Mesh Ewald method

OpenMM GPU weak scaling
Water box on a Nvidia Telsa2050
3k 6k 12k 24k 48k 96k
# atoms
0
0.2
0.4
0.6µ
s/a
tom
/ste
p real space
F reduction
q spread/gather
3D FFT
Note: real space cut-off 1.5 nm, on a CPU around 0.9 nm
Para2010 – p. 18/22

GPU - CPU comparison
Nvidia Tesla 2050 vs Intel Core i7 920 (2.66 GHZ, 4 threads)
GPU 3k GPU 48k CPU rc=1.5 CPU r
c=0.9
0
0.5
1µ
s/a
tom
/ste
preal space
F reduction
q spread/gather
3D FFT
real space cost ! r3c
FFT cost ! spacing!3
for constant accuracy: spacing = rc/a
total cost: Cpp r3c + CFFT a3 r!3
c
Para2010 – p. 19/22

Integrating Gromacs and OpenMM

Gromacs & OpenMM in practice

Gromacs & OpenMM in practice
• GPUs supported in Gromacs 4.5mdrun ... -device “OpenMM:Cuda”

Gromacs & OpenMM in practice
• GPUs supported in Gromacs 4.5mdrun ... -device “OpenMM:Cuda”
• Same input files, same output files

Gromacs & OpenMM in practice
• GPUs supported in Gromacs 4.5mdrun ... -device “OpenMM:Cuda”
• Same input files, same output files
• Only subset of features supported

Gromacs & OpenMM in practice
• GPUs supported in Gromacs 4.5mdrun ... -device “OpenMM:Cuda”
• Same input files, same output files
• Only subset of features supported
• No shortcuts taken on the GPU:
• At least same accuracy as on the CPU (<1e-6)

Gromacs-OpenMM• run as
$ mdrun -device “OpenMM:platform=Cuda,memtest=off,deviceid=0,force-device=yes"
• options
• platform: only CUDA, potentially in future OpenCL too
• memtest: check the GPU memory for error, default 15s
• deviceid: select the GPU card for simulation (for multi-device systems)
• force-device: run on unsupported but CUDA capable low-end cards

PME Reaction-fieldImplicit All-vs-all
GPU performance over x86 CPU

PME Reaction-fieldImplicit All-vs-all
GPU performance over x86 CPU
0
500
1000
1500
Quad x863.2 GHz
ns/day
32 46 230470

PME Reaction-fieldImplicit All-vs-all
GPU performance over x86 CPU
0
500
1000
1500
Quad x863.2 GHz
ns/day
32 46 230470
C20500
300
600
900
1200
1500
2966
810
1450

PME Reaction-fieldImplicit All-vs-all
Fermi (C20) performance over C10BPTI (~21k atoms)Villin (600 atoms, implicit)

PME Reaction-fieldImplicit All-vs-all
Fermi (C20) performance over C10
0
500
1000
1500
C1060
ns/day
13 35 460790
BPTI (~21k atoms)Villin (600 atoms, implicit)

PME Reaction-fieldImplicit All-vs-all
Fermi (C20) performance over C10
0
500
1000
1500
C1060
ns/day
13 35 460790
C20500
300
600
900
1200
1500
2966
810
1450
BPTI (~21k atoms)Villin (600 atoms, implicit)

Conclusions
• Only a subset of the features are supported in OpenMM but they would cover most users needs
• Better integration expected to come
• Useful for implicit solvent simulations