our dermatology online original article aa novel nonsense ... · two different research groups have...
TRANSCRIPT

Our Dermatology Online
© Our Dermatol Online 2.2018 110
How to cite this article: Chourabi M, H’mida-Ben Brahim D, Bonnard C, Aounallah A, Yu Ng A, Tohari S, Venkatesh B, Saad A, Boussofara L, Reversade B, Denguezli M. A novel nonsense ATP2C1 mutation causes Hailey-Hailey disease in a Tunisian family. Our Dermatol Online. 2018;9(2):110-113.Submission: 29.09.2017; Acceptance: 06.01.2018 DOI: 10.7241/ourd.20182.1
A novel nonsense ATP2C1 mutation causes A novel nonsense ATP2C1 mutation causes Hailey-Hailey disease in a Tunisian familyHailey-Hailey disease in a Tunisian familyMarwa Chourabi1,2, Dorra H’mida-Ben Brahim2, Carine Bonnard1, Amina Aounallah3, Alvin Yu Ng4, Sumanty Tohari4, Byrappa Venkatesh4, Ali Saad2, Lobna Boussofara3, Bruno Reversade1, Mohamed Denguezli3
1 Laboratory of Human Genetics and Embryology, Institute of Medical Biology, A*STAR, Singapore, 2Laboratory of Human Cytogenetic, Molecular Genetics and Reproductive Biology, Farhat Hached University Hospital, Sousse, Tunisia, 3Department of Dermatology and Venerology, Farhat Hached University Hospital, Sousse, Tunisia, 4 Institute of Molecular and Cell Biology, A*STAR, Singapore
Corresponding author: Prof. Mohamed Denguezli, E-mail: [email protected]
INTRODUCTION
Hailey-Hailey disease (HHD), or benign familial chronic pemphigus, which was described by the Hailey brothers in 1939, is characterized by recurrent vesicles and erosions, usually affecting the neck, axillae and groins [1]. HHD is an autosomal dominant blistering skin disorder that manifests in the 3rd to 4th decade of life with a prevalence of 1 in 50,000 [1,2]. Vesicular, itchy and painful lesions on an erythematous base characterize the rash and affect the flexural areas of the skin. Friction, sweating, heat, stress, UV exposure and cutaneous infections trigger the rash. The disease has a fluctuating course with remissions
and exacerbations [3]. In rare instances, the rash may become generalized and the skin lesions may even develop into squamous cell carcinoma [4]. Penetrance in adults affected by HHD is complete but expressivity is variable [2]. Histopathological features show a widespread loss of cell-to-cell adhesion (acantholysis) in the suprabasal layer of the epidermis. Ultrastructural studies demonstrate perinuclear aggregation of keratin intermediate filaments, which have retracted from the desmosomal plaques in the acantholytic cells [5,6].
Two different research groups have linked HHD to mutations in the gene ATP2C1, which encodes the human secretory pathway Ca2+/Mn2+ ATPase
ABSTRACT
Background: Hailey-Hailey disease (HHD) is an autosomal dominant blistering skin disorder that manifests in the third to fourth decade of life. The ATP2C1 has been identified as the pathogenic gene of this disease since 2000. Materials and Methods: We report here a three generations Tunisian pedigree, where almost all males are severely affected and present with complete penetrance of HHD, while only one female shows a mild disease’s phenotype in her fourth decade. A molecular study using Whole exome sequencing and direct sequencing was performed to this family. Results: By whole exome sequencing and direct DNA sequencing, a novel nonsense mutation in ATP2C1 (c.2698A>T; p.Lys900Ter) was identified in all patients, supporting that alterations in ATP2C1 are causative of HHD. Unexpectedly, this mutation was found in one female who was initially not diagnosed for HHD. Our observations would be in line with incomplete penetrance and variable expressivity between male and female of this disease, or evidence for genetic modifiers. Conclusion: We report here a novel nonsense heterozygous mutation in ATP2C1 gene in 5 patients with HHD. Interestingly, one woman carries the nonsense ATP2C1 mutation but displays a mild phenotype of HHD. This could indicate a variation in pattern and expressivity between male and female developing HHD phenotype which should be considered when providing genetic counselling to family members carrying such mutations.
Keywords:ATP2C1; Hailey-Hailey disease; benign familial chronic pemphigus.
Original Article

www.odermatol.com
© Our Dermatol Online 2.2018 111
(hSPCA1) [7,8]. SPCA1 is ubiquitously expressed in human tissues with the highest abundance in keratinocytes [9]. SPCA1 localizes to the Golgi-apparatus and controls its Ca2+ stores along with SERCA transporters. In primary keratinocytes, the role of SPCA1 is more significant than in other cell types [10,11], explaining why only the skin is affected in HHD patients. In this report we investigated a three generations Tunisian pedigree where males are severely affected with HHD. By whole exome sequencing followed by Sanger sequencing, we identified a novel nonsense mutation (c.2698A>T; p.Lys900Ter) in the ATP2C1 gene that causes Hailey-Hailey phenotype in this family
MATERIALS AND METHODS
Subject and informed consent
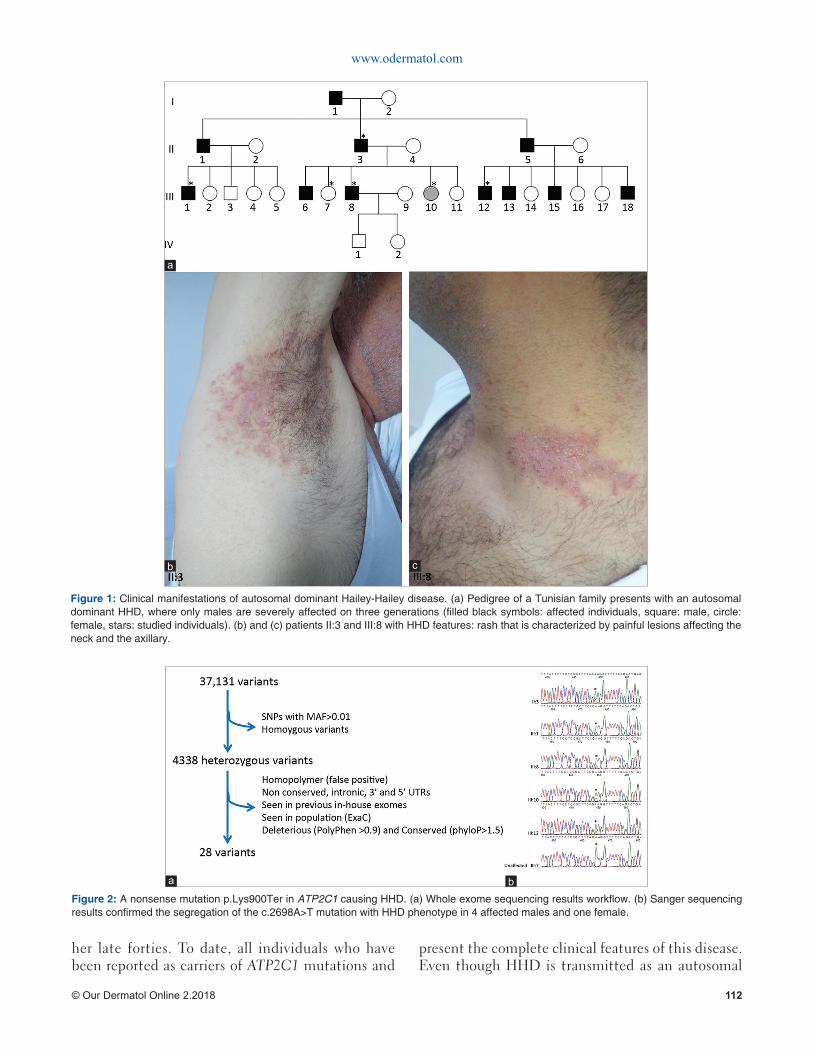
We performed molecular studies in a Tunisian family with HHD where males are severely affected in three generations (Fig. 1a). After obtaining the informed written consent and IRB approval (NUS IRB 10-051), blood samples were taken from four affected males and two unaffected females. Genomic DNA (gDNA) was extracted using Qiagen® kit.
Whole exome sequencing
For mutation analysis, 1ug of gDNA of proband III:8 was used for exome capture with Ion TargetSeqTM Exome and Custom Enrichment Kit. The exome library was prepared on an Ion OneTouch System and sequenced on an Ion Proton instrument (Life Technologies, Carlsbad, CA, USA) using one ION PI chip. Sequence reads were aligned to the human reference genome [Human GRCh37 (hg19) build] using Torrent Mapping Alignment Program (TMAP) from the Torrent Suite (v4.2.1). The variants were called using the Torrent Variant Caller (TVC) plugin (v4.2.1) and were annotated using the “annotate single sample variants” workflow, including the associated gene, variant location, quality-score, coverage, predicted functional consequences, protein position and amino acid changes, SIFT [12], PolyPhen2 [13], and Grantham [14] prediction scores, phyloP [15]
conservation scores and 5000 genomes Minor Allele Frequencies. Annotated variants were filtered for common SNPs using the ClinVar “common and no known medical impacts” database (ftp://ftp.ncbi.nlm.nih.gov/pub/clinvar/vcf_GRCh37/) and the Exome
Aggregate Consortium (ftp://ftp.broadinstitute.org/pub/ExAC_release/release0.2/). Variants were next compared to an in-house database of 485 previously sequenced samples, and those that were present in more than 1% of the previously sequenced samples were removed.
Sanger sequencing
Sanger sequencing was performed using 2 different sets of primers (Table 1) to verify the segregation of the identified ATPC2 nonsense mutation. After amplification, products were purified and sequenced on DNA sequencing system (model 3730XL; ABI).
RESULTS AND DISCUSSION
For all the affected members from this family (Fig. 1a) the disease began within the third decade of life. All affected individuals exhibited the typical clinical features of HHD with late onset of the rash characterized by itchy and painful lesions on an erythematous base and affect the flexural areas of the skin (Figs 1b and c). All the patients mentioned that friction, heat or sweating exacerbated the disease and the symptoms were worse during summer or if they were under stress. It is noteworthy that HHD penetrance is complete in adults affected with a variable expressivity.
To look for the causative mutation, 1ug of gDNA of patient III:8 was used for exome capture with Ion TargetSeqTM Exome and Custom Enrichment Kit. Whole exome sequencing of this proband generated a total of 14.5 Gb with an average read length of 155 bp. An average coverage of 188X was achieved across the exome, with 96% of the targeted sequences covered at ≥20X. A total of 37,131 variants were identified across protein-coding exons, UTRs, splice sites and flanking introns. After applying a series of filters and following an autosomal dominant mode of inheritance, a final set of 28 heterozygous variants were selected (Fig. 2a), including a deleterious mutation in ATP2C1, known as the causative gene of HHD. By Sanger sequencing, we confirmed that the nonsense mutation c.2698A>T (p.Lys900Ter) was heterozygous in all affected male members and unexpectedly in the female III:10 who was initially not diagnosed for HHD (Fig. 2b). This result prompted us to re-examine this individual over time, and noted that she has started developing a milder HHD phenotype, less severe than her male siblings and father despite the fact that she is in
www.odermatol.com
© Our Dermatol Online 2.2018 112
her late forties. To date, all individuals who have been reported as carriers of ATP2C1 mutations and
present the complete clinical features of this disease. Even though HHD is transmitted as an autosomal
Figure 1: Clinical manifestations of autosomal dominant Hailey-Hailey disease. (a) Pedigree of a Tunisian family presents with an autosomal dominant HHD, where only males are severely affected on three generations (fi lled black symbols: affected individuals, square: male, circle: female, stars: studied individuals). (b) and (c) patients II:3 and III:8 with HHD features: rash that is characterized by painful lesions affecting the neck and the axillary.
a
b c
Figure 2: A nonsense mutation p.Lys900Ter in ATP2C1 causing HHD. (a) Whole exome sequencing results workfl ow. (b) Sanger sequencing results confi rmed the segregation of the c.2698A>T mutation with HHD phenotype in 4 affected males and one female.
a b

www.odermatol.com
© Our Dermatol Online 2.2018 113
dominant disease, variation in the pattern and the severity of symptoms within the same family have been reported [2]. The variability of age of onset and the different expressivity of the phenotype that we report here may provide evidence for genetic modifiers as well as for environmental factors such as sun, heat, stress and friction that affect the HHD development [2].
CONCLUSION
We report here a novel nonsense heterozygous mutation in ATP2C1 gene in 5 HHD-patients supporting that alterations in the human secretory pathway Ca2+/ Mn2+ ATPase are causative for this disease. It is worth noting that in this family one woman carries the nonsense ATP2C1 mutation but displays a mild phenotype of HHD. Our observations would thus be in line with variation in pattern and expressivity between male and female developing Hailey-Hailey disease phenotype or evidence for other modifiers. This aspect should be considered when providing genetic counselling to family members carrying such mutations.
ACKNOWLEDGEMENTS
We are grateful to the family’s members for their contribution and participation in our study. We thank Sihem Sassi for her help on sample collection. M. Chourabi is a PhD student in the University of Monastir in Tunisia and is funded by an A*STAR Research Attachment Program. B. Reversade is a fellow of the Branco Weiss Foundation, an A*STAR Investigator and EMBO Young Investigator. This work was funded by the Skin Research Institute of Singapore and a Strategic Positioning Fund on Genetic Orphan Diseases from A*STAR, Singapore.
REFERENCES
1. Hailey H. Familial benign chronic pemphigus; report thirteen years after fi rst observation of a new entity. South Med J. 1953;46:763-5.
2. Burge SM. Hailey-Hailey disease: the clinical features, response to treatment and prognosis. Br J Dermatol. 1992;126:275-82.
3. Kellermayer R. Hailey-Hailey disease as an orthodisease of PMR1 deficiency in Saccharomyces cerevisiae. FEBS Lett. 2005;579:2021-5.
4. Chave TA, Milligan A. Acute generalized Hailey-Hailey disease. Clin Exp Dermatol. 2002;27:290-2.
5. Metze D, Hamm H, Schorat A, Luger T. Involvement of the adherens junction-actin fi lament system in acantholytic dyskeratosis of Hailey-Hailey disease. A histological, ultrastructural, and histochemical study of lesional and non-lesional skin. J Cutan Pathol. 1996;23:211-22.
6. Hashimoto K, Fujiwara K, Harada M, Setoyama M, Eto H. Junctional proteins of keratinocytes in Grover’s disease, Hailey-Hailey’s disease and Darier’s disease. J Dermatol. 1995;22:159-70.
7. Hu Z, Bonifas JM, Beech J, Bench G, Shigihara T, Ogawa H, et al. Mutations in ATP2C1, encoding a calcium pump, cause Hailey-Hailey disease. Nat Genet. 2000;24:61-5.
8. Sudbrak R, Brown J, Dobson-Stone C, Carter S, Ramser J, White J, et al. Hailey-Hailey disease is caused by mutations in ATP2C1 encoding a novel Ca(2+) pump. Hum Mol Genet. 2000;9:1131-40.
9. Callewaert G, Parys JB, De Smedt H, Raeymaekers L, Wuytack F, Vanoevelen J, et al. Similar Ca(2+)-signaling properties in keratinocytes and in COS-1 cells overexpressing the secretory-pathway Ca(2+)-ATPase SPCA1. Cell Calcium. 2003;34:157-62.
10. Behne MJ, Tu CL, Aronchik I, Epstein E, Bench G, Bikle DD, et al. Human keratinocyte ATP2C1 localizes to the Golgi and controls Golgi Ca2+ stores. J Invest Dermatol. 2003;121:688-94.
11. Missiaen L, Raeymaekers L, Dode L, Vanoevelen J, Van Baelen K, Parys JB, et al. SPCA1 pumps and Hailey-Hailey disease. Biochem Biophys Res Commun. 2004;322:1204-13.
12. Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073-81.
13. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248-9.
14. Grantham R. Amino acid difference formula to help explain protein evolution. Science. 1974;185:862-4.
15. Pollard KS, Hubisz MJ, Rosenbloom KR, Siepel A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 2010;20:110-21.
Table 1: Primers used for Sanger sequencing.Primer SequenceSet1 Forward 5’ CACACAATTAGGTCCATTCTCCA 3’
Set1 Reverse 5’ TCATTCCTCACACCTACACAG 3’
Set2 Forward 5’ AGCGAATTCTCACTAATTGACCA 3’
Set2 Reverse 5’ TTCTGCTATTTGGTCAGACTGA 3’
Copyright by Marwa Chourabi, et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.Source of Support: M. Chourabi is a PhD student in the University of Monastir in Tunisia and is funded by an A*STAR Research Attachment Program. B. Reversade is a fellow of the Branco Weiss Foundation, an A*STAR Investigator and EMBO Young Investigator. This work was funded by the Skin Research Institute of Singapore and a Strategic Positioning Fund on Genetic Orphan Diseases from A*STAR, Singapore.Confl ict of Interest: None declared.