overview of clinical research - gcp guidelines
TRANSCRIPT

Clinical Research Overview
“GCP Guidelines” Syed Junaid Rehan
QA Manager e-Compliance, Novartis CRA - Norton Audits Inc, USA
IPPCR – National Institute of Health, USA M.Phil. Quality Management, Hamdard University
M.Sc. Food Science & Technology, University of Karachi
SJR/ GCP Presentation / workshop Liaquat College of Medicine & Dentistry

Overview of Clinical Research & History
Informed Consent
Human Subject’s Rights, Safety, and Welfare Protection;
Regulations
GCP - Good Clinical Practices;
Data Integrity, Misconduct and Fraud;
Adverse Event
Topics
SJR/ GCP / LCMD

Patient-Oriented Research Research conducted with human subjects (or on material of human origin such as tissues, specimens and cognitive phenomena) for which an investigator (or colleague) directly interacts with human subjects…includes: Development of new technologies Mechanisms of human disease Therapeutic interventions Clinical Trials
Epidemiologic and Behavioral Studies Outcomes Research and Health Services Research
Definition of Clinical Research
*From NIH Director’s Panel on Clinical Research, 1996 SJR/ GCP / LCMD

Drug
A drug is defined as:
• (A) articles recognized in the official USP, HPUS or NF or any supplement to any of them;
• (B) articles intended for use in the diagnosis, cure, mitigation, treatment, or prevention of disease in man or other animals;
• (C) articles (other than food) intended to affect the structure or any function of the body of man or other function of the body of man or other animals…..
“Defining a drug”
SJR/ GCP / LCMD

PHASE 1 PHASE 2 PHASE 3
First in Man Safety and Tolerability Pharmacokinetics
Proof of Concept Dose Ranging Safety/PK in Special Populations and Risk Factors
Large Multi-centered Usually Placebo-Controlled Usually replicated Primary data to support marketing approval in NDA
SJR/ GCP / LCMD

“Then Daniel said to the steward… Test your servants for ten days; let us be given vegetables to eat and water to drink. Then let our appearance and the appearance of the youths who eat the king’s rich food be observed by you, and according to what you see, deal with your servants. So he harkened to them in this matter; and tested them for ten days. At the end of ten days it was seen that they were better in appearance and fatter in flesh than all the youths who ate the king’s rich food. So the steward took away their rich food and the wine they were to drink, and gave them vegetables.”
History of Clinical Trials
Daniel 1:11 – 16 c. 530 BC SJR/ GCP / LCMD

History Rich in Trials and Tribulation
Ensuring Subject’s Research Protection Rights, Risks, and Welfare
Nuremberg Code Kefauver Amendments Declaration of Helsinki National Research Act Belmont Report The Common Rule
“Ethics and Research”
SJR/ GCP / LCMD

Nuremberg Code - 1948
“ Voluntary Consent”
• A well-known chapter in the history of research with human subjects opened on December 9, 1946, when an American military tribunal opened criminal proceedings against 23 leading German physicians and administrators for their willing participation in war crimes and crimes against humanity.
• Among the charges were that German physicians conducted medical experiments on thousands of concentration camp prisoners without their consent. Most of the subjects of these experiments died or were permanently crippled as a result.
• As a direct result of the trial, the Nuremberg Code was established in 1948, stating that "The voluntary consent of the human subject is absolutely essential," making it clear that subjects should give consent and that the benefits of research must outweigh the risks.
• Although it did not carry the force of law, the Nuremberg Code was the first international document which advocated voluntary participation and informed consent.
SJR/ GCP / LCMD

Kefauver Amendments - 1962
“Ethics become
Law”
• US History:
– Thalidomide • Late 1950s, thalidomide was approved as a sedative in Europe. • It was not approved in the United States by the FDA. • The drug was prescribed to control sleep and nausea throughout pregnancy,
but it was soon found that taking this drug during pregnancy caused severe deformities in the fetus.
• Many patients did not know they were taking a drug that was not approved for use by the FDA, nor did they give informed consent.
• Some 12,000 babies/patients were born with severe deformities due to thalidomide.
– Tuskegee Syphilis Study (1932-1972) • Conducted by the U.S. Public Health Service. • Six hundred low-income African-American males, 400 of whom were
infected with syphilis, were monitored for 40 years. • Free medical examinations were given; however, subjects were not told
about their disease. • Even though a proven cure (penicillin) became available in the 1950s, the
study continued until 1972 with participants being denied treatment. • In some cases, when subjects were diagnosed as having syphilis by other
physicians, researchers intervened to prevent treatment. • Many subjects died of syphilis during the study.
SJR/ GCP / LCMD

Declaration of Helsinki - 1964
“A call for Independence”
• Issues outlined in the Declaration of Helsinki include:
– Research with humans should be based on the results from laboratory and animal experimentation
– Research protocols should be reviewed by an independent
committee prior to initiation – Informed consent from research participants is necessary – Research should be conducted by medically/scientifically
qualified individuals – Risks should not exceed benefits
SJR/ GCP / LCMD

National Research Act - 1974
• In Summary:
– Nazi atrocities in World War II drew attention to the lack of international standards on research with human subjects and led to the formulation of the Nuremberg Code.
– The thalidomide disaster led to the adoption of the "Kefauver Amendments" to the Food, Drug and Cosmetic Act, requiring drug manufacturers to prove to the FDA the effectiveness of their products before marketing them.
– The Declaration of Helsinki is the basis for Good Clinical Practices used today.
– The Tuskegee Syphilis Study is probably the worst case of unethical human subjects research in the history of the United States.
– The National Research Act codified the requirement that human
subjects in research must be protected and set the stage for the issuance of the Belmont Report.
“Human Subject’s Protection”
SJR/ GCP / LCMD

The Belmont Report - 1979
“Foundation for Regulations”
• Beneficence:
– Human subjects should not be harmed
– Research should maximize possible benefits and minimize possible harms.
• Justice:
– The benefits and risks of research must be distributed fairly.
• Respect for persons:
– Individuals should be treated as autonomous agents
– Persons with diminished autonomy are entitled to
protection. SJR/ GCP / LCMD

The Common Rule - 1981
• The Main Elements of the Common Rule include :
– requirements for assuring compliance by research institutions;
– requirements for researchers obtaining and documenting informed consent;
– requirements for Institutional Review Board (IRB) membership, function, operations, review of research, and record keeping;
– additional protections for certain vulnerable research subjects-- pregnant women, prisoners, and children
“Additional Protections”
SJR/ GCP / LCMD

1898: William Osler
“To deliberately inject a poison of known
high degree of virulency into a human being,
unless you obtain a man’s sanction…is criminal.”
(In response to an oral presentation by Giuseppe Sanarelli on discovery of
the etiologic agent of yellow fever)
History of Informed Consent
1849-1919
SJR/ GCP / LCMD

“…how to provide each patient with a reasonable understanding of his role in a study project and the means for obtaining evidence of such understanding and consent.”
1962:
Kefauver-Harris amendment to Food and Drug Act stipulating subjects must be told whether a drug is being used for investigational purposes
Surgeon General requires peer review (IRBs) for all PHS grants.
1967:
FDA required all new drug sponsors obtain informed consent for use of investigational drugs in humans.
Informed Consent
Minutes of Medical Board MEDICAL BOARD
Tuesday, June 9, 1953, 3:00 p.m. Chairman: Dr. Luther Terry
SJR/ GCP / LCMD

The study involves research of an unproven drug, the purpose of the research
How long the participant will be expected to participate in the study
What will happen in the study
Possible risks/benefits to the participant
Participation is voluntary and that participants can quit the study at any time without penalty or loss of benefits to which they are otherwise entitled.
SJR/ GCP / LCMD
Under GCP, the FDA Requires That People be Informed

Federal Regulations
TITLE 21 – Food and Drug
CHAPTER I – Food and Drug
Administration
Department of Health and Human Services
SUBCHAPTER D – Drugs for Human Use
Investigational New Drug Application
21Code of Federal Regulations
Part 312
FDA’s Compliance Policy
Programs
7348.811 CHAPTER 48
Bioresearch Monitoring
Clinical Investigators
OCTOBER 1, 1997
Guidance for Industry
Good Clinical Practices:
Consolidated Guidance
(ICH-E6)
International Conference on
Harmonization
April 1996
Regulations & Guidelines
SJR/ GCP / LCMD

Clinical Trial Oversight
“Safety Nets”
Clinical Investigator
Sponsor
CRO
IRB
FDA Sponsor/ CRO
IRB Clinical Investigator
Subject & Consumer Protection
“Umbrella of Protection SJR/ GCP / LCMD

Clinical Trial Oversight
Understanding the roles defined in the FD&C Act
The Sponsor The CRO
“The Sponsor”
SJR/ GCP / LCMD

Clinical Trial Oversight
• Understanding the roles defined in the FD&C Act
– The Institutional Review Board
– Synonym: Ethics Committee
“IRB”
SJR/ GCP / LCMD

Clinical Trial Oversight
Understanding the roles defined in the FD&C Act
The Clinical Investigator Synonym: Principal Investigator or Researcher
The Study Coordinator
“Clinical Investigator”
SJR/ GCP / LCMD
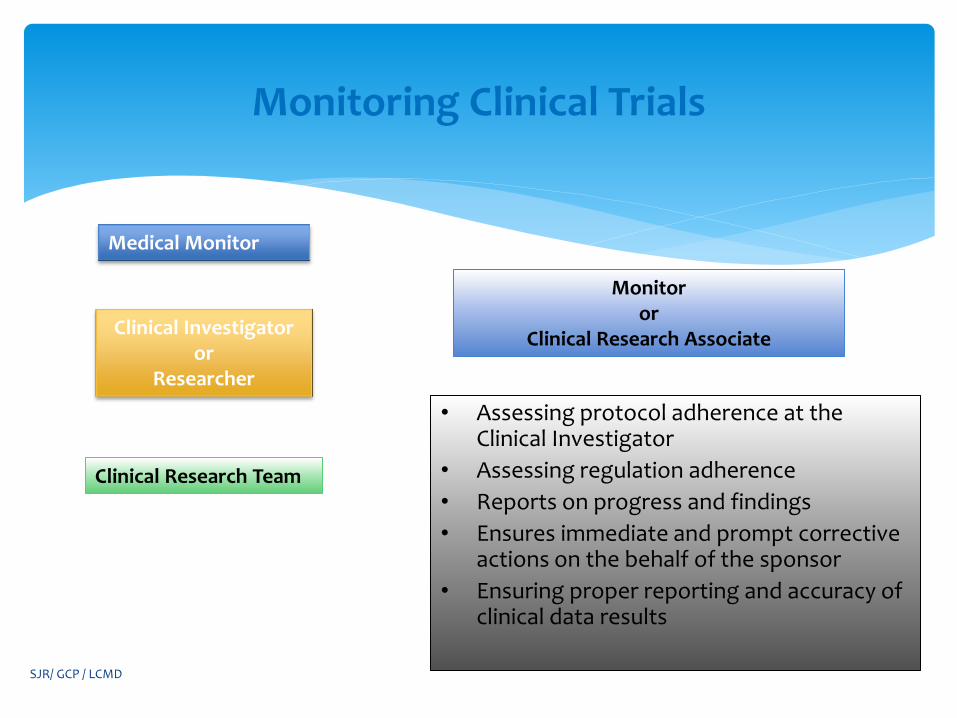
Monitoring Clinical Trials
Medical Monitor
Clinical Research Team
Clinical Investigator or
Researcher
Monitor or
Clinical Research Associate
• Assessing protocol adherence at the Clinical Investigator
• Assessing regulation adherence
• Reports on progress and findings
• Ensures immediate and prompt corrective actions on the behalf of the sponsor
• Ensuring proper reporting and accuracy of clinical data results
SJR/ GCP / LCMD

Goal of Monitoring
Clinical Investigator’s
Data
Sponsor/CRO Database
Monitor’s Role
Monitor: ◘Source verify clinical data ◘Request clarifications ◘Submit paper case report forms or ◘Approve electronic submissions
SJR/ GCP / LCMD

Inspection Procedures
The inspections conducted should be sufficient in scope to determine the clinical investigator's practices for each point identified:
– Authority and Administration
– Protocol
– Subjects’ records
– Other study records
– Consent of human subjects
– Institutional review board (IRB)
– Sponsor
– Test article accountability
– Records retention
– Electronic records and signatures
SJR/ GCP / LCMD

Monitoring / Auditing Skills
Global Skills Presented
– Communication Skills
– Interviewing Skills
– Observation Skills
– Data Techniques
– Writing Skills
– Organizational Skills
– Legal Expertise
SJR/ GCP / LCMD

ICH-GCP Guideline – E6 Good Clinical Practice
A given standard for the:
– Design
– Conduct
– Performance
– Monitoring
– Auditing
– Recording
– Analysis
– Reporting of clinical trials
SJR/ GCP / LCMD

Provides....
assurance that the data and reported results are credible and accurate, and that the rights, integrity and confidentiality of trial subjects are Protected.
What it Does ?
SJR/ GCP / LCMD

WHY???
• Increase protection of human research subjects
• Ensure the integrity of the clinical investigation
• Ensure the approval of safe and effective products
SJR/ GCP / LCMD

Are mainly focused on the protection of human rights in clinical trial.
Provide assurance of the safety of the newly developed compounds.
Provide standards on how clinical trials should be conducted.
Define the roles and responsibilities of
Clinical sponsors,
Clinical research investigators,
Clinical Research Associates, and
Monitors.
Good Clinical Practice Guidelines
SJR/ GCP / LCMD

Good Clinical Practice Guidelines
Data Integrity
Protection of Subject’s
Rights
Compliance with GCPs provide public assurance that the rights and safety of participants in human subject research are protected and that the data that arises from the study is credible
They are defined as an international ethical and scientific standard for designing, conducting, recording and reporting trials that involve the participation of human subjects
SJR/ GCP / LCMD

Composition of ICH
ICH Co-Sponsors
United States
Japan
European Union
ICH Observers
Canada
WHO
European Free Trade Area
IFPMA
International Federation of
Pharmaceutical
Manufacturers Ass.
(represents 56 countries) * Pharmaceutical Research & Manufacturers of America
SJR/ GCP / LCMD

1 Clinical trials should be conducted in accordance with the ethical principles that have their origin in the Declaration of Helsinki, and that are consistent with GCP and the applicable regulatory requirements 2 Before a trial is initiated, foreseeable risks and inconveniences should be weighed against the anticipated benefit for the individual trial subject and society. A trial should be initiated and continued only if the anticipated benefits justify the risks 3 The rights, safety, and well-being of the trial subjects are the most important considerations and should prevail over interests of science and society 4 The available non clinical and clinical information on an investigational product should be adequate to support the proposed clinical trial
SJR/ GCP / LCMD
Thirteen core principles of GCP Guidance

5 Clinical trials should be scientifically sound, and described in a clear, detailed protocol 6 A trial should be conducted in compliance with the protocol that has received prior institutional review board (IRB)/independent ethics committee (IEC) approval/favorable opinion 7 The medical care given to, and medical decisions made on behalf of, subjects should always be the responsibility of a qualified physician or, when appropriate, of a qualified medical health practitioner. 8 Each individual involved in conducting a trial should be qualified by education, training, and experience to perform his or her respective tasks 9 Freely given informed consent should be obtained from every subject prior to clinical trial participation
SJR/ GCP / LCMD
Thirteen core principles of GCP Guidance

10 All clinical trial information should be recorded, handled, and stored in a way that allows its accurate reporting, interpretation, and verification 11 The confidentiality of records that could identify subjects should be protected, respecting the privacy and confidentiality rules in accordance with the applicable regulatory requirements 12 Investigational products should be manufactured, handled, and stored in accordance with applicable good manufacturing practice (GMP). They should be used in accordance with the approved protocol 13 Systems with procedures that assure the quality of every aspect of the trial should be implemented
SJR/ GCP / LCMD
Thirteen core principles of GCP Guidance

Data Integrity
The integrity of these data touches every part of business and operations we are constantly creating and processing data throughout the entire product life cycle which often more than 10 years. It is relevant during preclinical in research, clinical trial, registration, manufacturing, testing, release & distribution.
Data Generates at every Step. SJR/ GCP / LCMD

Actual – complete – factual – concise – precise
Proper storage & accessibility
Mishandling of data – however intentional can result in misleading interpretation of Clinical trial results, wrong dosage of medicine, malfunction of medical products, Drug Shortages or interruption of therapists for patients and reputation damage to organization.
Data Integrity
SJR/ GCP / LCMD

Misconduct
deliberate or repeated noncompliance
does not include honest error or honest differences of opinion.
Generally has elements of safety concerns.
• Mismanagement of responsibilities
• Intentional wrongdoing; specifically :
– deliberate violation of a law or standard
• Improper professional behavior
SJR/ GCP / LCMD

Fraud Can be combined with misconduct charges
• Intentional perversion of truth in order to induce another to part with something of value or to surrender a legal right
– An act of deceiving or misrepresenting
• A person who is not what he or she pretends to be
FALSIFICATION OF DATA!!
PHS - PLAGIARISM!!!
FDA’s Definition of Research Fraud
SJR/ GCP / LCMD

Acts of Fraud
– Fabrication and Manufactured data
– Falsification (False Information)
– Forgery
– False statements
– Altered data
– Omitted data
– Plagiarism
SJR/ GCP / LCMD

Adverse Event (AE):
Any untoward medical occurrence in a patient, consumer, or clinical trial subject administered a product, which does not necessarily have a causal relationship
with this treatment.
An adverse event can therefore be any unfavorable and unintended sign (e.g. an abnormal laboratory finding), symptom, or disease temporally associated with the use of a product, whether or not considered related to the product.
Adverse Event
SJR/ GCP / LCMD

In addition, all cases of drug-drug interaction, pregnancy (with or without outcome), exposure during breastfeeding, paternal exposure, LOE, overdose, drug abuse and misuse, drug maladministration or accidental exposure and dispensing errors are collected and databased even if no adverse event has been reported.
Adverse Event
SJR/ GCP / LCMD

Serious Adverse Event
An adverse event is any undesirable experience associated with the use of a medical product in a patient. The event is serious and should be reported to FDA when the patient outcome is:
• Death • Life-threatening • Hospitalization (initial or prolonged) • Disability or Permanent Damage • Congenital Anomaly/Birth Defect • Required Intervention to Prevent Permanent Impairment or Damage
(Devices)
SJR/ GCP / LCMD

Other Serious (Important Medical Events) Report when the event does not fit the other outcomes, but the event may jeopardize the patient and may require medical or surgical intervention (treatment) to prevent one of the other outcomes. Examples include allergic bronchospasm (a serious problem with breathing) requiring treatment in an emergency room, serious blood dyscrasias (blood disorders) or seizures/convulsions that do not result in hospitalization. The development of drug dependence or drug abuse would also be examples of important medical events.
Serious Adverse Event
SJR/ GCP / LCMD

SJR/ GCP / LCMD
It is estimated that only 5 in 5,000 compounds that enter preclinical testing make it to human testing, and only 1 of those 5 may be safe and effective enough to reach pharmacy shelves.

Tools for Understanding Legal Requirements
• International Conference on Harmonization (ICH- GCP) – E6 • FD&C Act of 1938 • Code of Federal Regulations (CFR) • Guidance and Information Documents:
– Good Clinical Practice in FDA-Regulated Clinical Trials: http://www.fda.gov/oc/gcp/default.htm
– Information Sheets: http://www.fda.gov/oc/gcp/guidance.html
– Dear Doctor Letters – Educational Outreach
SJR/ GCP / LCMD

References:-
• http://www.fda.gov/
• http://en.wikipedia.org/wiki/ICH-GCP
• NIH • www.NIDCR.nih.gov
• www.NIAID.nih.gov
• NIH IPPCR Training Modules
• NAI Training Modules for CRA
• University/research institute sites
UTHSC – www.uth.tmc.edu
SJR/ GCP / LCMD

Thanks...
Q&A
SJR/ GCP / LCMD