p53-induced microrna-107 inhibits hif-1 and tumor ... - pnas · p53-induced microrna-107 inhibits...
TRANSCRIPT

P53-induced microRNA-107 inhibits HIF-1 andtumor angiogenesisMunekazu Yamakuchia,1, Craig D. Lottermanb,c,d, Clare Baob, Ralph H. Hrubane, Baktiar Karime, Joshua T. Mendellc,2,David Husof,2, and Charles J. Lowensteina,1
aDepartment of Medicine, Aab Cardiovascular Research Institute, University of Rochester School of Medicine and Dentistry, Rochester, NY 14642; andbDepartment of Medicine, cThe McKusick-Nathans Institute of Genetic Medicine, dDepartment of Pediatrics, eDepartment of Pathology, and fDepartment ofComparative Molecular Medicine and Pathobiology, The Johns Hopkins University School of Medicine, Baltimore, MD 21205
Edited by Patrick J. Paddison, Fred Hutchinson Cancer Research Center, Seattle, WA, and accepted by the Editorial Board February 22, 2010 (received forreview September 29, 2009)
The pathway involving the tumor suppressor gene TP53 can reg-ulate tumor angiogenesis by unclear mechanisms. Here we showthat p53 regulates hypoxic signaling through the transcriptionalregulation of microRNA-107 (miR-107). We found that miR-107 is amicroRNA expressed by human colon cancer specimens and regu-lated by p53. miR-107 decreases hypoxia signaling by suppressingexpression of hypoxia inducible factor-1β (HIF-1β). Knockdown ofendogenous miR-107 enhances HIF-1β expression and hypoxic sig-naling in human colon cancer cells. Conversely, overexpression ofmiR-107 inhibits HIF-1β expression and hypoxic signaling. Further-more, overexpression of miR-107 in tumor cells suppresses tumorangiogenesis, tumor growth, and tumor VEGF expression in mice.Finally, in human colon cancer specimens, expression of miR-107 isinversely associated with expression of HIF-1β. Taken togetherthese data suggest that miR-107 can mediate p53 regulation ofhypoxic signaling and tumor angiogenesis.
endothelial | hypoxia | PANK1 | cancer | nitric oxide
Many solid tumors require angiogenesis to grow beyond acertain size (1, 2). Tumor neovascularization is driven in
part by hypoxia, which stimulates tumor cell production ofangiogenic factors such as VEGF-A, basic FGF (bFGF), andplacental growth factor (PlGF) (1–8). The hypoxia induciblefactor-1 (HIF-1) heterodimer plays a critical role in hypoxicsignaling in tumors (8–15). Mutations in genes regulating theHIF-1 pathway might alter tumor cell responses to hypoxia andtumor cell survival.One of the genes most commonly mutated in human cancers is
TP53 (16–21). When induced by cellular stress such as DNAdamage or hypoxia, the tumor suppressor protein p53 normallyinhibits cell growth and stimulates apoptosis. However, muta-tions in p53 or other disruptions in the p53 pathway are asso-ciated with tumor growth and angiogenesis (20, 21). Severalstudies suggest that p53 can regulate hypoxic signaling, but thesignaling pathways are unknown (20, 22–24).MicroRNA (miRNA) are critical components of the p53 sig-
naling pathway. p53 activates transcription of a subset of miRNAthat in turn suppresses transcription of genes that regulateapoptosis, DNA repair, and cell-cycle progression (25–28).Because p53 also can regulate hypoxic signaling by unknownpathways, we hypothesized that miRNA can mediate p53 regu-lation of tumor angiogenesis. Accordingly, we searched formiRNA that are expressed by human colon cancer specimensand are regulated by p53 and then screened this subset formiRNA that regulate hypoxia signaling.
ResultsWe characterized miRNA expression in colon cancers harvestedfrom four patients at the JohnsHopkinsHospital, usingmicroarray(Table S1). Of the 30 miRNA most highly expressed in humancolon cancer specimens, we noted four miRNA that we and othershad identified previously as being induced by p53 (26–30). To
investigate the role of these miRNA in hypoxia signaling, we em-ployed a bioinformatics approach. We found that miR-107, one ofthe members of this set of miRNA regulated by p53, is a potentialregulator of HIF-1β. The HIF-1β and HIF-1α subunits form theheterodimer HIF-1, a transcription factor that mediates the tran-scriptional response to hypoxia (8–14, 31).We first characterized p53 transcriptional regulation of miR-
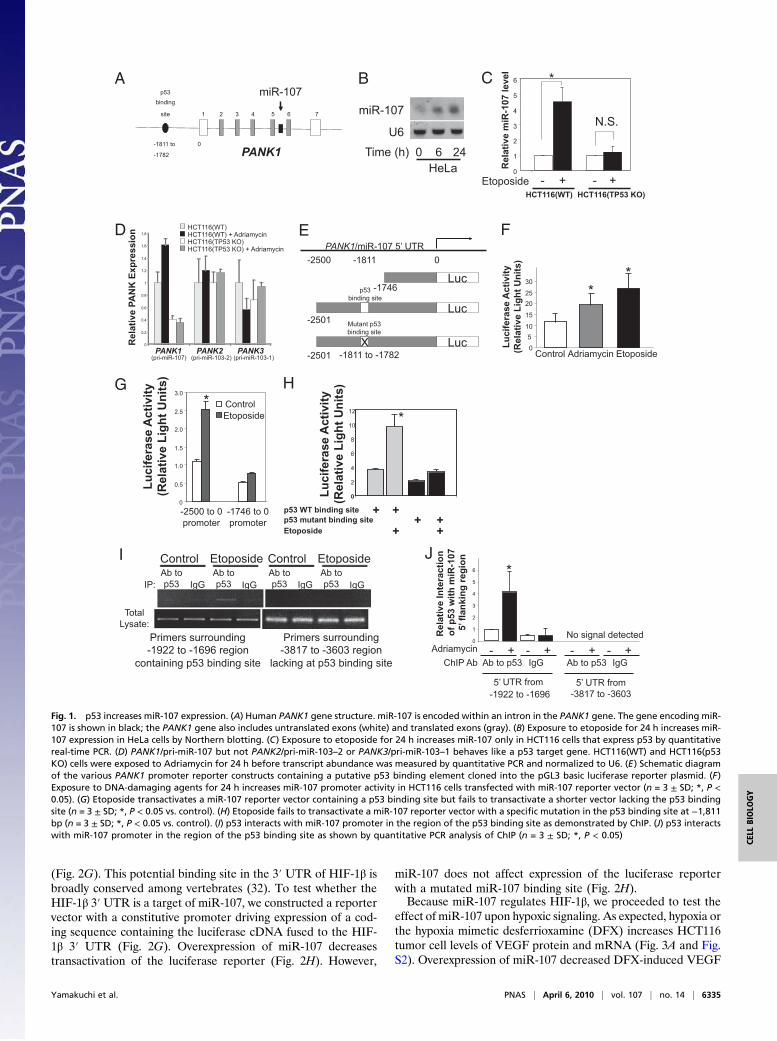
107. miR-107 is encoded within an intron of the gene for pan-tothenate kinase enzyme 1, PANK1. Computer analysis revealsa potential p53 binding site 1,811 bp upstream of the PANK1/miR-107 transcriptional start site (Fig. 1A). The DNA-damag-ing agent etoposide increased miR-107 levels in HeLa cells(Fig. 1B). Although etoposide also increased miR-107 in wild-type human colon cancer HCT116 cells [HCT116(WT)], eto-poside failed to increase miR-107 in HCT116 cells lacking bothp53 alleles [HCT116(p53 KO)] (Fig.1C). Expression of theparent gene PANK1 also is partly dependent upon p53 (Fig. 1Dand Fig. S1).To identify the regions upstream of PANK1/miR-107 that
mediate p53 induction, we made reporter constructs consistingof 2,500 bp upstream of the PANK1/miR-107 gene drivingexpression of firefly luciferase (Fig. 1E). Genotoxic stressincreases transactivation of the full-length reporter construct inHCT116(WT) cells (Fig. 1F). However, genotoxic stress fails toincrease expression of the reporter construct in HCT116(p53KO) cells (Fig. 1G). Mutation of the p53 binding element at−1,811 to −1,782 bp blunts the ability of etoposide to activateluciferase expression (Fig. 1H). ChIP confirms that p53 interactswith the 5′ UTR region from −1,922 to −1,696 bp containing thep53 binding site (Fig. 1 I and J). Taken together, these datasuggest that p53 directly regulates miR-107 expression.We next explored miR-107 regulation of HIF-1β. Over-
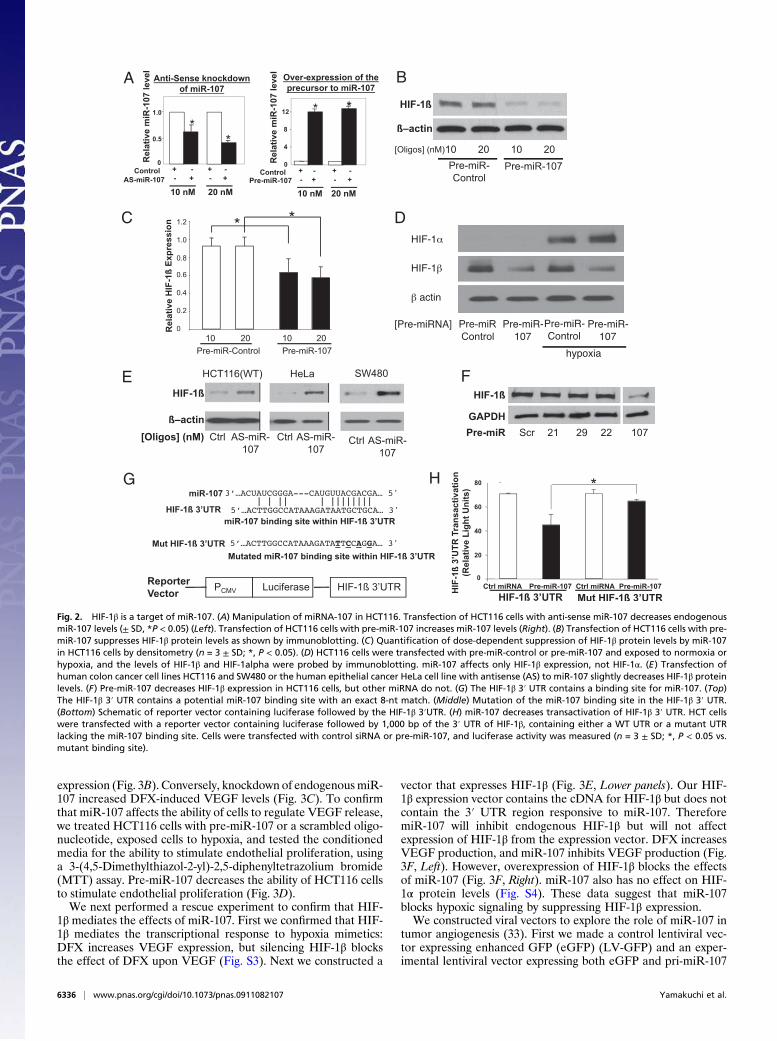
expression of the precursor to miR-107 (pre-miR-107) decreasesHIF-1β protein levels (Fig. 2 A–D). Also, miR-107 does notaffect the partner of HIF-1β, namely HIF-1α (Fig. 2D). Con-versely, knockdown of miR-107 increases HIF-1β expression in avariety of cell lines (Fig. 2E). Additionally, overexpression ofother miRNA does not affect HIF-1β (Fig. 2F).The 3′ UTR of HIF-1β contains a potential binding element
for miR-107 with an 8-nt match to the miR-107 seed region
Author contributions: M.Y., C.D.L., J.T.M., D.H., and C.J.L. designed research; M.Y., C.D.L.,C.B., B.K., and D.H. performed research; M.Y., R.H.H., J.T.M., and D.H. contributed newreagents/analytic tools; M.Y., B.K., J.T.M., D.H., and C.J.L. analyzed data; and M.Y. andC.J.L. wrote the paper.
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. P.J.P. is a guest editor invited by theEditorial Board.1Towhomcorrespondencemaybeaddressed.E-mail:[email protected] or [email protected].
2J.T.M. and D.H. contributed equally to this work.
This article contains supporting information online at www.pnas.org/cgi/content/full/0911082107/DCSupplemental.
6334–6339 | PNAS | April 6, 2010 | vol. 107 | no. 14 www.pnas.org/cgi/doi/10.1073/pnas.0911082107
(Fig. 2G). This potential binding site in the 3′ UTR of HIF-1β isbroadly conserved among vertebrates (32). To test whether theHIF-1β 3′ UTR is a target of miR-107, we constructed a reportervector with a constitutive promoter driving expression of a cod-ing sequence containing the luciferase cDNA fused to the HIF-1β 3′ UTR (Fig. 2G). Overexpression of miR-107 decreasestransactivation of the luciferase reporter (Fig. 2H). However,
miR-107 does not affect expression of the luciferase reporterwith a mutated miR-107 binding site (Fig. 2H).Because miR-107 regulates HIF-1β, we proceeded to test the
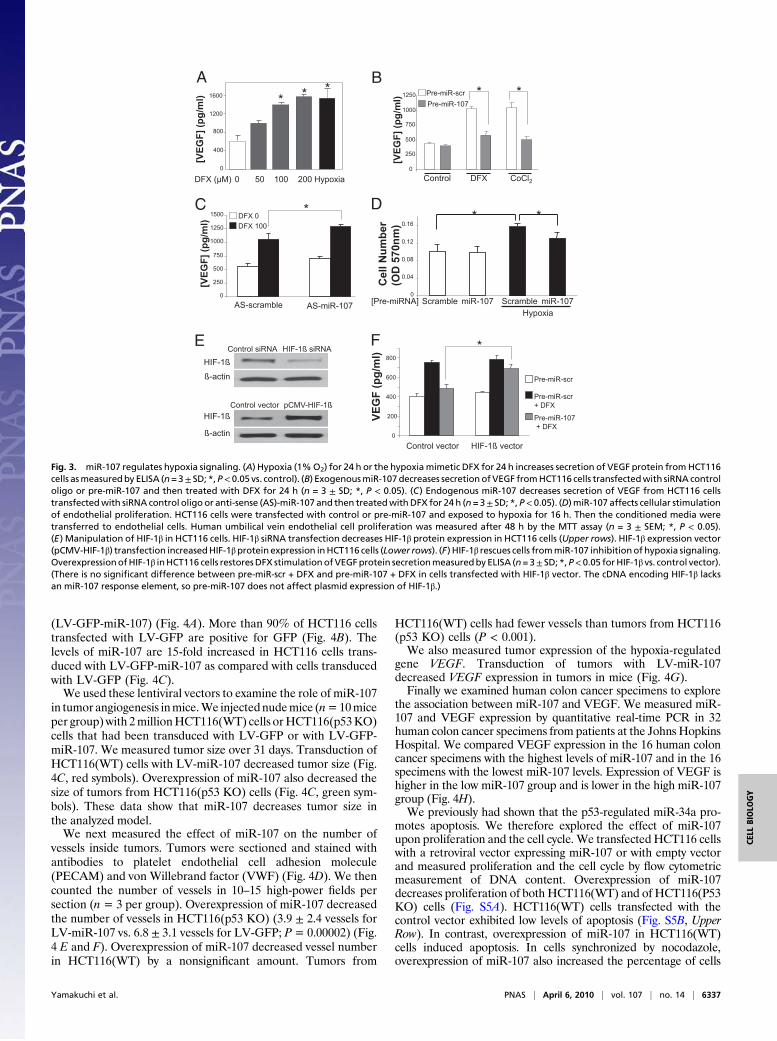
effect of miR-107 upon hypoxic signaling. As expected, hypoxia orthe hypoxia mimetic desferrioxamine (DFX) increases HCT116tumor cell levels of VEGF protein and mRNA (Fig. 3A and Fig.S2). Overexpression of miR-107 decreased DFX-induced VEGF
D
G H
I J
E F
PANK1
miR-107
0-1811 to
-1782
p53binding
site 1 2 3 4 5 6 7
HeLa
miR-107
6 240U6
Time (h) 0
1
2
3
4
5
6
HCT116(TP53 KO) HCT116(WT)
+-Etoposide +-
Re
lati
ve
miR
-10
7 l
ev
el
*
N.S.
0
0.2
0.4
0.6
0.8
1
1.2
1.4
1.6
1.8
Re
lati
ve
PA
NK
Ex
pre
ss
ion
PANK1 (pri-miR-107)
PANK2 (pri-miR-103-2)
PANK3 (pri-miR-103-1)
HCT116(WT)HCT116(WT) + AdriamycinHCT116(TP53 KO)HCT116(TP53 KO) + Adriamycin
Luc0-1811
PANK1/miR-107 5’ UTR -2500
Lucp53
binding site
-2501
-1746
LucX-1811 to -1782 -2501
Mutant p53binding site
Lu
cif
era
se
Ac
tiv
ity
(Re
lati
ve
Lig
ht
Un
its
)
*
05
10
15
2025
30
Control EtoposideAdriamycin
*
0
0.5
1.0
1.5
2.0
2.5
3.0
ControlEtoposide
-2500 to 0promoter
-1746 to 0promoter
Lu
cif
era
se
Ac
tiv
ity
(Re
lati
ve
Lig
ht
Un
its
)
*
0
2
4
6
8
10
12
Lu
cif
era
se
Ac
tiv
ity
(Re
lati
ve
Lig
ht
Un
its
)
Etoposide
p53 WT binding site
+p53 mutant binding site
+
+
++ +
*
Control EtoposideAb to p53 IgGIP:
Total Lysate:
Primers surrounding -1922 to -1696 region
containing p53 binding site
Primers surrounding-3817 to -3603 region
lacking at p53 binding site
Ab to p53 IgG
Control EtoposideAb to p53 IgG
Ab to p53 IgG
Adriamycin0
1
2
3
4
5
6
- + - +
Re
lati
ve
In
tera
cti
on
of
p5
3 w
ith
miR
-10
7
5’ fl
an
kin
g r
eg
ion
- + - +
5’ UTR from -1922 to -1696
5’ UTR from -3817 to -3603
No signal detected
*
ChIP Ab Ab to p53 Ab to p53 IgG IgG
A B C
Fig. 1. p53 increases miR-107 expression. (A) Human PANK1 gene structure. miR-107 is encoded within an intron in the PANK1 gene. The gene encoding miR-107 is shown in black; the PANK1 gene also includes untranslated exons (white) and translated exons (gray). (B) Exposure to etoposide for 24 h increases miR-107 expression in HeLa cells by Northern blotting. (C) Exposure to etoposide for 24 h increases miR-107 only in HCT116 cells that express p53 by quantitativereal-time PCR. (D) PANK1/pri-miR-107 but not PANK2/pri-miR-103–2 or PANK3/pri-miR-103–1 behaves like a p53 target gene. HCT116(WT) and HCT116(p53KO) cells were exposed to Adriamycin for 24 h before transcript abundance was measured by quantitative PCR and normalized to U6. (E) Schematic diagramof the various PANK1 promoter reporter constructs containing a putative p53 binding element cloned into the pGL3 basic luciferase reporter plasmid. (F)Exposure to DNA-damaging agents for 24 h increases miR-107 promoter activity in HCT116 cells transfected with miR-107 reporter vector (n = 3 ± SD; *, P <0.05). (G) Etoposide transactivates a miR-107 reporter vector containing a p53 binding site but fails to transactivate a shorter vector lacking the p53 bindingsite (n = 3 ± SD; *, P < 0.05 vs. control). (H) Etoposide fails to transactivate a miR-107 reporter vector with a specific mutation in the p53 binding site at −1,811bp (n = 3 ± SD; *, P < 0.05 vs. control). (I) p53 interacts with miR-107 promoter in the region of the p53 binding site as demonstrated by ChIP. (J) p53 interactswith miR-107 promoter in the region of the p53 binding site as shown by quantitative PCR analysis of ChIP (n = 3 ± SD; *, P < 0.05)
Yamakuchi et al. PNAS | April 6, 2010 | vol. 107 | no. 14 | 6335
CELL
BIOLO
GY
expression (Fig. 3B). Conversely, knockdown of endogenousmiR-107 increased DFX-induced VEGF levels (Fig. 3C). To confirmthat miR-107 affects the ability of cells to regulate VEGF release,we treated HCT116 cells with pre-miR-107 or a scrambled oligo-nucleotide, exposed cells to hypoxia, and tested the conditionedmedia for the ability to stimulate endothelial proliferation, usinga 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide(MTT) assay. Pre-miR-107 decreases the ability of HCT116 cellsto stimulate endothelial proliferation (Fig. 3D).We next performed a rescue experiment to confirm that HIF-
1β mediates the effects of miR-107. First we confirmed that HIF-1β mediates the transcriptional response to hypoxia mimetics:DFX increases VEGF expression, but silencing HIF-1β blocksthe effect of DFX upon VEGF (Fig. S3). Next we constructed a
vector that expresses HIF-1β (Fig. 3E, Lower panels). Our HIF-1β expression vector contains the cDNA for HIF-1β but does notcontain the 3′ UTR region responsive to miR-107. ThereforemiR-107 will inhibit endogenous HIF-1β but will not affectexpression of HIF-1β from the expression vector. DFX increasesVEGF production, and miR-107 inhibits VEGF production (Fig.3F, Left). However, overexpression of HIF-1β blocks the effectsof miR-107 (Fig. 3F, Right). miR-107 also has no effect on HIF-1α protein levels (Fig. S4). These data suggest that miR-107blocks hypoxic signaling by suppressing HIF-1β expression.We constructed viral vectors to explore the role of miR-107 in
tumor angiogenesis (33). First we made a control lentiviral vec-tor expressing enhanced GFP (eGFP) (LV-GFP) and an exper-imental lentiviral vector expressing both eGFP and pri-miR-107
A B
D
E F
G H
C
Fig. 2. HIF-1β is a target of miR-107. (A) Manipulation of miRNA-107 in HCT116. Transfection of HCT116 cells with anti-sense miR-107 decreases endogenousmiR-107 levels (± SD, *P < 0.05) (Left). Transfection of HCT116 cells with pre-miR-107 increases miR-107 levels (Right). (B) Transfection of HCT116 cells with pre-miR-107 suppresses HIF-1β protein levels as shown by immunoblotting. (C) Quantification of dose-dependent suppression of HIF-1β protein levels by miR-107in HCT116 cells by densitometry (n = 3 ± SD; *, P < 0.05). (D) HCT116 cells were transfected with pre-miR-control or pre-miR-107 and exposed to normoxia orhypoxia, and the levels of HIF-1β and HIF-1alpha were probed by immunoblotting. miR-107 affects only HIF-1β expression, not HIF-1α. (E) Transfection ofhuman colon cancer cell lines HCT116 and SW480 or the human epithelial cancer HeLa cell line with antisense (AS) to miR-107 slightly decreases HIF-1β proteinlevels. (F) Pre-miR-107 decreases HIF-1β expression in HCT116 cells, but other miRNA do not. (G) The HIF-1β 3′ UTR contains a binding site for miR-107. (Top)The HIF-1β 3′ UTR contains a potential miR-107 binding site with an exact 8-nt match. (Middle) Mutation of the miR-107 binding site in the HIF-1β 3′ UTR.(Bottom) Schematic of reporter vector containing luciferase followed by the HIF-1β 3′UTR. (H) miR-107 decreases transactivation of HIF-1β 3′ UTR. HCT cellswere transfected with a reporter vector containing luciferase followed by 1,000 bp of the 3′ UTR of HIF-1β, containing either a WT UTR or a mutant UTRlacking the miR-107 binding site. Cells were transfected with control siRNA or pre-miR-107, and luciferase activity was measured (n = 3 ± SD; *, P < 0.05 vs.mutant binding site).
6336 | www.pnas.org/cgi/doi/10.1073/pnas.0911082107 Yamakuchi et al.
(LV-GFP-miR-107) (Fig. 4A). More than 90% of HCT116 cellstransfected with LV-GFP are positive for GFP (Fig. 4B). Thelevels of miR-107 are 15-fold increased in HCT116 cells trans-duced with LV-GFP-miR-107 as compared with cells transducedwith LV-GFP (Fig. 4C).We used these lentiviral vectors to examine the role of miR-107
in tumor angiogenesis inmice.We injectednudemice (n=10miceper group)with 2millionHCT116(WT) cells orHCT116(p53KO)cells that had been transduced with LV-GFP or with LV-GFP-miR-107. We measured tumor size over 31 days. Transduction ofHCT116(WT) cells with LV-miR-107 decreased tumor size (Fig.4C, red symbols). Overexpression of miR-107 also decreased thesize of tumors from HCT116(p53 KO) cells (Fig. 4C, green sym-bols). These data show that miR-107 decreases tumor size inthe analyzed model.We next measured the effect of miR-107 on the number of
vessels inside tumors. Tumors were sectioned and stained withantibodies to platelet endothelial cell adhesion molecule(PECAM) and von Willebrand factor (VWF) (Fig. 4D). We thencounted the number of vessels in 10–15 high-power fields persection (n = 3 per group). Overexpression of miR-107 decreasedthe number of vessels in HCT116(p53 KO) (3.9 ± 2.4 vessels forLV-miR-107 vs. 6.8 ± 3.1 vessels for LV-GFP; P= 0.00002) (Fig.4 E and F). Overexpression of miR-107 decreased vessel numberin HCT116(WT) by a nonsignificant amount. Tumors from
HCT116(WT) cells had fewer vessels than tumors from HCT116(p53 KO) cells (P < 0.001).We also measured tumor expression of the hypoxia-regulated
gene VEGF. Transduction of tumors with LV-miR-107decreased VEGF expression in tumors in mice (Fig. 4G).Finally we examined human colon cancer specimens to explore
the association between miR-107 and VEGF. We measured miR-107 and VEGF expression by quantitative real-time PCR in 32human colon cancer specimens from patients at the Johns HopkinsHospital. We compared VEGF expression in the 16 human coloncancer specimens with the highest levels of miR-107 and in the 16specimens with the lowest miR-107 levels. Expression of VEGF ishigher in the low miR-107 group and is lower in the high miR-107group (Fig. 4H).We previously had shown that the p53-regulated miR-34a pro-
motes apoptosis. We therefore explored the effect of miR-107upon proliferation and the cell cycle. We transfected HCT116 cellswith a retroviral vector expressing miR-107 or with empty vectorand measured proliferation and the cell cycle by flow cytometricmeasurement of DNA content. Overexpression of miR-107decreases proliferation of both HCT116(WT) and of HCT116(P53KO) cells (Fig. S5A). HCT116(WT) cells transfected with thecontrol vector exhibited low levels of apoptosis (Fig. S5B, UpperRow). In contrast, overexpression of miR-107 in HCT116(WT)cells induced apoptosis. In cells synchronized by nocodazole,overexpression of miR-107 also increased the percentage of cells
A
0
400
800
1200
1600
0 50 100 200 DFX ( M)
[VE
GF
] (p
g/m
l) **
Hypoxia
*B
[VE
GF
] (p
g/m
l) Pre-miR-scr
Pre-miR-107
DFX CoCl2Control
* *
0
250
500
750
1000
1250
CDFX 0 DFX 100
[VE
GF
] (p
g/m
l)
AS-scramble AS-miR-107
*
0
250
500
750
1000
1250
1500
HIF-1ß siRNA Control siRNA
HIF-1ß ß-actin
pCMV-HIF-1ß Control vector HIF-1ß
ß-actin
E F
Pre-miR-scr
Pre-miR-scr + DFX
Pre-miR-107 + DFX
HIF-1ß vector Control vector
*
VE
GF
(p
g/m
l)
0
200
400
600
800
0
0.04
0.08
0.12
0.16
miR-107 [Pre-miRNA] Scramble Hypoxia
miR-107 Scramble
Ce
ll N
um
be
r
(OD
57
0n
m)
* * D
Fig. 3. miR-107 regulates hypoxia signaling. (A) Hypoxia (1%O2) for 24 h or the hypoxiamimetic DFX for 24 h increases secretion of VEGF protein fromHCT116cells asmeasuredby ELISA (n=3± SD; *,P< 0.05 vs. control). (B) ExogenousmiR-107 decreases secretion ofVEGF fromHCT116 cells transfectedwith siRNA controloligo or pre-miR-107 and then treated with DFX for 24 h (n = 3 ± SD; *, P < 0.05). (C) Endogenous miR-107 decreases secretion of VEGF from HCT116 cellstransfectedwith siRNA control oligo or anti-sense (AS)-miR-107 and then treatedwithDFX for 24 h (n=3± SD; *, P< 0.05). (D)miR-107 affects cellular stimulationof endothelial proliferation. HCT116 cells were transfected with control or pre-miR-107 and exposed to hypoxia for 16 h. Then the conditioned media weretransferred to endothelial cells. Human umbilical vein endothelial cell proliferation was measured after 48 h by the MTT assay (n = 3 ± SEM; *, P < 0.05).(E) Manipulation of HIF-1β in HCT116 cells. HIF-1β siRNA transfection decreases HIF-1β protein expression in HCT116 cells (Upper rows). HIF-1β expression vector(pCMV-HIF-1β) transfection increasedHIF-1βprotein expression inHCT116 cells (Lower rows). (F) HIF-1β rescues cells frommiR-107 inhibitionof hypoxia signaling.OverexpressionofHIF-1β inHCT116 cells restoresDFX stimulationofVEGFprotein secretionmeasuredby ELISA (n=3± SD; *,P<0.05 forHIF-1β vs. control vector).(There is no significant difference between pre-miR-scr + DFX and pre-miR-107 + DFX in cells transfected with HIF-1β vector. The cDNA encoding HIF-1β lacksan miR-107 response element, so pre-miR-107 does not affect plasmid expression of HIF-1β.)
Yamakuchi et al. PNAS | April 6, 2010 | vol. 107 | no. 14 | 6337
CELL
BIOLO
GY

in G1 (Fig. S5B, Lower Row). These data suggest that miR-107 inpartmediates p53 regulation of the cell cycle. Taken together, thesedata also suggest that miR-107 can regulate tumor progression bytwo distinct mechanisms, regulation of the cell cycle and control ofhypoxic signaling.
DiscussionThe major finding of our study is that miR-107 regulates hypoxicsignaling (Fig. 4I). Several aspects of our study are intriguing.First, although it is widely assumed that HIF-1 signaling isregulated primarily through the HIF-1α subunit, our data suggestthat regulation of HIF-1β also may modulate hypoxic responses.
In addition, our studies suggest that specific miRNA can mod-ulate the cellular response to hypoxia. Our data reinforce thework of others who have shown that hypoxia can alter theexpression of miRNA and that miRNA can alter HIF-1αexpression in vitro (34, 35). Others have shown that, comparedwith WT tumors, tumors that lack HIF-1α are poorly vascular-ized but are faster growing, perhaps because of a loss ofdependency upon neovascularization (36, 37).Folkman (1) and others (38–40) proposed that some tumors can
undergo an angiogenic switch, during which avascular tumorsrecruit new blood vessels, increasing the supply of oxygen andnutrients andpermitting further tumorgrowth.Tumor angiogenesis
A B
C D
5’LTR cPPT CMV eGFP H1 WPRE 3’LTR
Pri-miR-107 Cel
l num
ber
0
40
80
120
160
LV-GFP Control 200
0
40
80
120
160
200
100 101 102 103 104 100 101 102 103 104
GFP Fluorescence GFP Fluorescence R
elat
ive
miR
-107
Exp
ress
ion
0
4
8
12
16
HCT116(TP53 KO) HCT116(WT)
* * LV-GFP
LV-GFP
LV-GFP-miR-107
LV-GFP-miR-107
0
200
400
600
800
1000
1200
1400
1600
0 5 10 15 20 25 30 35
Time (days)
Tum
or V
olum
e (c
ubic
mm
)
HCT116(WT) & LV-GFP
HCT116(WT) & LV-GFP-miR-107
HCT116(TP53 KO) & LV-GFP
HCT116(TP53 KO) & LV-GFP-miR-107
* * * *
*
* * *
* *
E F G HCT116(WT) & LV-GFP
HCT116(WT) & LV-GFP-miR-107
HCT116(TP53 KO) & LV-GFP
HCT116(TP53 KO) & LV-GFP-miR-107
0
2
4
6
8
10
12 * * N
umbe
r of v
esse
ls
per h
igh
pow
ered
fiel
d
LV-GFP LV-GFP- miR-107
LV-GFP LV-GFP- miR-107
Tumors from HCT116(WT)
Tumors from HCT116(TP53 KO)
Rel
ativ
e VE
GF
mR
NA
expr
essi
on
0
0.2
0.4
0.6
0.8
1.0
1.2 * *
LV-GFP LV-GFP- miR-107
LV-GFP LV-GFP- miR-107
Tumors from HCT116(WT)
Tumors from HCT116(TP53 KO)
I Hypoxia
p53 HIF-1
Angiogenesis
miR-107
VEGF
Low miR-107 High miR-107
miR-107VEGF
Rel
ativ
e ex
pres
sion
* H
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
Fig. 4. miR-107 decreases tumor volume and vessels. (A) Schematic of LV-GFP-miR-107. (B) GFP fluorescence of HCT116 cells transduced with control media(Left) or with lentivirus expressing GFP (Right) analyzed by FACS. (C) LV-GFP-miR-107-transduced cells (solid bars) contain 10-fold more miR-107 than LV-GFP–infected cells (white bars) by quantitative real-time PCR (n = 3–10 ± SD; *, P < 0.05). (D) miR-107 decreases tumor volume. Nude mice were injected with 2million HCT116 cells after transduction with lentiviral vectors. Four different HCT116 cell types were injected: HCT116(WT) and LV-GFP; HCT116(WT) and LV-miR-107; HCT116(p53 KO) and LV-GFP; and HCT116(p53 KO) and LV-miR-107. Tumor dimensions were measured for 31 d. Expression of miR-107 decreases thesize of HCT116(WT) and HCT116(P53 KO) tumors (n = 10; *, P < 0.05 vs. LV-miR-107). (E) miR-107 decreases the density of blood vessels in tumors, as assessedby immunohistochemistry for VWF. (F) miR-107 decreases the number of tumor blood vessels, as measured by counting the number of vessels per high-powerfield for 12–15 fields for three tumors in each of the four groups (n = 3 ± SD; *, P < 0.001). (G) miR-107 decreases expression of the hypoxia-regulated geneVEGF. Quantitative real-time PCR was used to analyze expression of VEGF in tumors from nude mice implanted with HCT116 cells transduced with lentiviralvectors (n = 3–10 ± SD; *, P < 0.05). (H) miR-107 is inversely related to VEGF expression in human colon cancer. We measured miR-107 expression and VEGFexpression by quantitative real-time PCR in 32 specimens of colon cancer harvested from patients at the Johns Hopkins University School of Medicine. Wegrouped the 32 human colon cancer specimens into the 16 specimens with the lowest expression of miR-107 and the 16 specimens with the highest expressionof miR-107 (n = 16 ± SD; *, P < 0.0001). VEGF expression is higher in the low miR-107 group than in the high miR-107 group (n = 16 ± SD; *, P < 0.03). (I)Schematic for miR-107 regulation of hypoxia signaling.
6338 | www.pnas.org/cgi/doi/10.1073/pnas.0911082107 Yamakuchi et al.

may be driven not only by the tumor microenvironment but also bytumor and stromal mutations. One of the genes most commonlymutated in human cancers is TP53 (16–18) (19), and mutations inTP53 are associatedwith an increase in tumor angiogenesis (20, 21).Some studies suggest that p53 regulates hypoxic signaling and thattumor cells lacking TP53 are resistant to hypoxia (20, 22–24); con-ventional explanations for this phenomenon include decreasedapoptosis and senescence, and our data suggest an additionalmechanism, namely increased levels of HIF-1 signaling. Our dataprovide one pathway through which p53 can regulate hypoxic sig-naling and tumor angiogenesis. Cancerswithmutations inTP53 andincreased hypoxia signaling may be more susceptible to anti-angiogenic therapy than tumors with an intact p53 pathway (20, 41).
Materials and MethodsCell Culture. HCT116 (p53 WT and p53 KO) cells were a gift from BertVogelstein, Baltimore, MD.
Transfection. HCT116 cells were transfected using siPORT NeoFX (AppliedBiosystems) or Lipofectamine 2000 (Invitrogen) with precursor miRNA or withantisense miRNA (0–20 nM) and were harvested 48 or 72 h later.
Quantitative Real-Time PCR. To analyze miRNA expression, TaqManMicroRNAassays (Applied Biosystems) were used to quantify levels of mature miRNAsfollowing the manufacturer’s instructions. The primers for quantitative real-time PCR and the PCR mix were purchased from Applied Biosystems.
Plasmid Construction. The 3′ UTR of HIF-1β (960 bp) containing the HIF-1β-miR-107 response element was cloned into pMIR-REPORT Luciferase vector(Applied Biosystems). For reporter assays, the 5′ UTR region of Pank1/miR-107 extending upstream from the transcriptional start site (0 bp) to −2,500bp was amplified from genomic human DNA and cloned into the pGL3promoter plasmid (Promega).
Cell Proliferation Assay. Cell proliferation was measured byMTT assay. Briefly,we plated each group of cells (1 × 104) on 96-well plates, added MTT, andincubated the cells for 3 h. The cells were dissolved by adding HCl/SDS, andabsorbance was measured at 570 nm.
Apoptosis Analysis. HCT116 cells were transfected with precursor miRNA orscrambled oligonucleotides as a control. After a further incubation of 72 h,the cells were harvested, stained with propidium iodide and anti-annexin-Vantibody, and analyzed by FACS.
Lentivirus Construction and Transduction. Third-generation lentiviral vectorswere a gift of I. M. Verma (La Jolla, CA) (33). The lentiviral vectors wereconstructed and prepared according to previously published protocols (33).HCT116 cells were transduced with lentiviral vectors at an MOI of 10, andexpression of GFP was confirmed by FACS.
Murine Tumor Model. Tumors were implanted into mice as described pre-viously (42). Briefly, nude mice were anesthetized and injected in the flankwith 2 million HCT116 cells. Mice were divided into four groups of 10 mice:group 1 received HCT116(WT) transduced with LV-GFP; group 2 receivedHCT116(WT) transduced with LV-miR-107; group 3 received HCT(p53 KO)transduced with LV-GFP; and group 4 received HCT116(p53 KO) transducedwith LV-miR-107. Tumor diameters were measured with an electronic caliperover 31 d. Mice were killed at 31–38 d, tumors were harvested, and sectionsof tumors were stained for PECAM and VWF. The number of vessels per highpowered field was counted in 12–15 fields for three tumors in each of thefour groups.
ACKNOWLEDGMENTS.We thank Dr. Bert Vogelstein for providing the HCT116cells, Dr. I. M. Verma for providing the lentiviral vectors, and Dr. Scott Kernfor providing human colon cancer specimens. This work was supported bythe National Institutes of Health (C.J.L., J.T.M., and D.H.) and the AmericanHeart Association (M.Y.).
1. Folkman J (1971) Tumor angiogenesis: Therapeutic implications. N Engl J Med 285:1182–1186.
2. Ferrara N, Kerbel RS (2005) Angiogenesis as a therapeutic target. Nature 438:967–974.3. Ferrara N (2002) VEGF and the quest for tumour angiogenesis factors. Nat Rev Cancer
2:795–803.4. Hicklin DJ, Ellis LM (2005) Role of the vascular endothelial growth factor pathway in
tumor growth and angiogenesis. J Clin Oncol 23:1011–1027.5. Kerbel RS (2005) Therapeutic implications of intrinsic or induced angiogenic growth
factor redundancy in tumors revealed. Cancer Cell 8:269–271.6. Kerbel RS (2008) Tumor angiogenesis. N Engl J Med 358:2039–2049.7. Kaelin WG, Jr (2008) The von Hippel-Lindau tumour suppressor protein: O2 sensing
and cancer. Nat Rev Cancer 8:865–873.8. Kaelin WG, Jr, Ratcliffe PJ (2008) Oxygen sensing by metazoans: The central role of
the HIF hydroxylase pathway. Mol Cell 30:393–402.9. Maxwell PH, et al. (1997) Hypoxia-inducible factor-1 modulates gene expression in
solid tumors and influences both angiogenesis and tumor growth. Proc Natl Acad SciUSA 94:8104–8109.
10. Ryan HE, Lo J, Johnson RS (1998) HIF-1 alpha is required for solid tumor formation andembryonic vascularization. EMBO J 17:3005–3015.
11. Zhu H, Bunn HF (2001) Signal transduction. How do cells sense oxygen? Science 292:449–451.
12. Semenza GL (2003) Targeting HIF-1 for cancer therapy. Nat Rev Cancer 3:721–732.13. Pugh CW, Ratcliffe PJ (2003) Regulation of angiogenesis by hypoxia: Role of the HIF
system. Nat Med 9:677–684.14. Safran M, Kaelin WG, Jr (2003) HIF hydroxylation and the mammalian oxygen-sensing
pathway. J Clin Invest 111:779–783.15. Kung AL, Wang S, Klco JM, Kaelin WG, Livingston DM (2000) Suppression of tumor
growth through disruption of hypoxia-inducible transcription. Nat Med 6:1335–1340.16. Prives C, Hall PA (1999) The p53 pathway. J Pathol 187:112–126.17. Oren M (2003) Decision making by p53: Life, death and cancer. Cell Death Differ 10:
431–442.18. Vogelstein B, Lane D, Levine AJ (2000) Surfing the p53 network. Nature 408:307–310.19. Vogelstein B, Kinzler KW (2004) Cancer genes and the pathways they control. Nat
Med 10:789–799.20. Yu JL, Rak JW, Coomber BL, Hicklin DJ, Kerbel RS (2002) Effect of p53 status on tumor
response to antiangiogenic therapy. Science 295:1526–1528.21. Teodoro JG, Parker AE, Zhu X, Green MR (2006) p53-mediated inhibition of
angiogenesis through up-regulation of a collagen prolyl hydroxylase. Science 313:968–971.
22. Blagosklonny MV, et al. (1998) p53 inhibits hypoxia-inducible factor-stimulatedtranscription. J Biol Chem 273:11995–11998.
23. Ravi R, et al. (2000) Regulation of tumor angiogenesis by p53-induced degradation ofhypoxia-inducible factor 1alpha. Genes Dev 14:34–44.
24. Sano M, et al. (2007) p53-induced inhibition of Hif-1 causes cardiac dysfunction duringpressure overload. Nature 446:444–448.
25. Xi Y, Shalgi R, Fodstad O, Pilpel Y, Ju J (2006) Differentially regulated micro-RNAs andactively translated messenger RNA transcripts by tumor suppressor p53 in coloncancer. Clin Cancer Res 12:2014–2024.
26. Chang TC, et al. (2007) Transactivation of miR-34a by p53 broadly influences geneexpression and promotes apoptosis. Mol Cell 26:745–752.
27. Raver-Shapira N, et al. (2007) Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Mol Cell 26:731–743.
28. He L, et al. (2007) A microRNA component of the p53 tumour suppressor network.Nature 447:1130–1134.
29. Bommer GT, et al. (2007) p53-mediated activation of miRNA34 candidate tumor-suppressor genes. Curr Biol 17:1298–1307.
30. He L, He X, Lowe SW, Hannon GJ (2007) microRNAs join the p53 network—anotherpiece in the tumour-suppression puzzle. Nat Rev Cancer 7:819–822.
31. Wang GL, Semenza GL (1995) Purification and characterization of hypoxia-induciblefactor 1. J Biol Chem 270:1230–1237.
32. Bartel DP (2009) MicroRNAs: Target recognition and regulatory functions. Cell 136:215–233.
33. Tiscornia G, Singer O, Verma IM (2006) Production and purification of lentiviralvectors. Nat Protoc 1:241–245.
34. Kulshreshtha R, et al. (2007) A microRNA signature of hypoxia. Mol Cell Biol 27:1859–1867.
35. Taguchi A, et al. (2008) Identification of hypoxia-inducible factor-1 alpha as a noveltarget for miR-17-92 microRNA cluster. Cancer Res 68:5540–5545.
36. Carmeliet P, et al. (1998) Role of HIF-1alpha in hypoxia-mediated apoptosis, cellproliferation and tumour angiogenesis. Nature 394:485–490.
37. Yu JL, et al. (2001) Heterogeneous vascular dependence of tumor cell populations.Am J Pathol 158:1325–1334.
38. Algire GH, et al. (1950) Vascular reactions of normal and malignant tissues in vivo. III.Vascular reactions of mice to fibroblasts treated in vitro with methylcholanthrene. JNatl Cancer Inst 11:555–580.
39. Greenblatt M, Shubi P (1968) Tumor angiogenesis: Transfilter diffusion studies in thehamster by the transparent chamber technique. J Natl Cancer Inst 41:111–124.
40. Warren BA, Shubik P (1966) The growth of the blood supply to melanoma transplantsin the hamster cheek pouch. Lab Invest 15:464–478.
41. Kerbel RS (2006) Antiangiogenic therapy: A universal chemosensitization strategy forcancer? Science 312:1171–1175.
42. Nanda A, et al. (2006) Tumor endothelial marker 1 (Tem1) functions in the growthand progression of abdominal tumors. Proc Natl Acad Sci USA 103:3351–3356.
Yamakuchi et al. PNAS | April 6, 2010 | vol. 107 | no. 14 | 6339
CELL
BIOLO
GY