p53 is a transcriptional activator of the muscle-specific...
TRANSCRIPT

p53 Is a Transcriptional Activator of the Muscle-specificPhosphoglycerate Mutase Gene and Contributesin Vivo to the Control of Its Cardiac Expression1
Pilar Ruiz-Lozano, Mary L. Hixon, Mark W. Wagner,Ana I. Flores, Shuntaro Ikawa, Albert S. Baldwin, Jr.,Kenneth R. Chien, and Antonio Gualberto2
Departments of Physiology and Biophysics [P. R-L., M. W. W., A. I. F.,A. G.] and Genetics [M. L. H.], Case Western Reserve University Schoolof Medicine, Cleveland, Ohio 44106; Department of Medicine andCenter for Molecular Genetics, University of California at San Diego,California 92093 [P. R-L., K. R. C.]; Department of Cell Biology, IDAC,Tohoku University, Sendai, Japan 980 [S. I.]; and LinebergerComprehensive Cancer Center and Department of Biology, Universityof North Carolina at Chapel Hill, North Carolina 27599 [A. S. B.]
AbstractThe role that the p53 tumor suppressor gene productplays in cellular differentiation remains controversial.However, recent evidence indicates that p53 isrequired for proper embryogenesis. We have studiedthe effect of p53 on the expression mediated by thepromoter of the rat muscle-specific phosphoglyceratemutase gene (M-PGAM), a marker for cardiac andskeletal muscle differentiation. Experiments involvingtransient transfection, mobility shift assay, and site-directed mutagenesis demonstrated that p53specifically binds and transactivates the M-PGAMpromoter. The p53-related proteins p51A and p73L alsotransactivated M-PGAM. Moreover, stable expressionof a p53 dominant mutant in C2C12 cells blocked theinduction of M-PGAM expression during the myoblastto myotube transition and the ability of p53, p51A, andp73L to transactivate the M-PGAM promoter. Inaddition, impaired expression of M-PGAM wasobserved in a subset of p53-null animals in heart andmuscle tissues of anterior-ventral location. Theseresults demonstrate that p53 is a transcriptionalactivator of M-PGAM that contributes in vivo to thecontrol of its cardiac expression. These data supportprevious findings indicating a role for p53 in cellulardifferentiation.
IntroductionThe differentiation of cardiac muscle cells is a process that isbeginning to be understood in detail. Cardiogenesis begins
with a commitment of mesodermally derived progenitor cellsto the myocyte lineage in response to endodermal signals,followed by the formation of the primordial heart tube (1, 2).Organogenesis then proceeds through a series of involutionsof the heart tube and the onset of septation, chamber for-mation, and the acquisition of regional-specific properties ofatrial, ventricular, and conduction system cells (3). This proc-ess progresses during the embryonic life and is completedearly after birth. Each step in cellular differentiation is char-acterized by the expression of a specific set of molecularmarkers. The transcriptional control of these genes dependsupon the synchronized action of cardiac-specific and ubiq-uitous transcription factors (3).
We isolated previously the rat M-PGAM3 subunit (4, 5).M-PGAM encodes a dimeric metabolic enzyme and resem-bles the MCK gene in its timing and pattern of developmentalexpression (6). It is, therefore, not surprising that both genescontain similar DNA regulatory elements that control theirspecific expression in skeletal and cardiac muscle (4, 5).Previous studies have shown that MCK also contains p53-responsive elements (7–10). An MCK p53 site was shown tomediate p53-responsiveness when subcloned into a heter-ologous minimal promoter (8). Transactivation of MCK byp53 can be inhibited by MDM2 (11), a protein frequentlyamplified in human sarcomas (12). Importantly, although ithas been shown that p53 binds and transactivates the MCKpromoter, little is presently known about the role of p53 in theactivation of this or other muscle-specific genes during myo-cyte differentiation.
We have investigated the ability of p53 to regulate tran-scription from the M-PGAM promoter in rat neonatal car-diocytes, C2C12 cells, and SAOS cells. We show that p53and p53-related proteins transactivate the rat M-PGAM pro-moter. Moreover, we identified a p53-responsive element inthe M-PGAM promoter. This DNA element contains a con-sensus p53 DNA binding site that is highly homologous (86%identity) to that located in the MCK enhancer. Mobility shiftassays detected binding of endogenous rat cardiac p53 andpurified human p53 to the M-PGAM p53 site. In addition,mutagenesis of the M-PGAM p53 site blocked the transac-tivation of the M-PGAM promoter by p53 in C2C12 cells anddecreased its expression in rat neonatal cardiocytes. Impor-tantly, reduced M-PGAM expression was observed by in situhybridization and Northern analysis in a subset of p53-nullanimals. Strikingly, differences were observed specifically inmuscle tissues of anterior and ventral location, such astongue or heart. These results demonstrate that p53 directly
Received 10/15/98; revised 2/11/99; accepted 3/8/99.The costs of publication of this article were defrayed in part by thepayment of page charges. This article must therefore be hereby markedadvertisement in accordance with 18 U.S.C. Section 1734 solely to indi-cate this fact.1 This work was supported in part by American Heart Association Grant9750205N (to A. G.) and NIH Grants AI35098 (to A. S. B.) and HL46345 (toK. R. C.).2 To whom requests for reprints should be addressed, at Department ofPhysiology and Biophysics, Case Western Reserve University School ofMedicine, 10900 Euclid Avenue, Cleveland, OH 44106-4970. Phone:(216) 368-3400; Fax: (216) 368-3658; E-mail: [email protected].
3 The abbreviations used are: M-PGAM, muscle-specific phosphoglycer-ate mutase gene; MCK, muscle creatine kinase gene; CAT, chloramphen-icol acetyltransferase; CMV, cytomegalovirus; ANF, atrial natruiretic factorgene; MLC2v, myosin light chain 2v gene; EMSA, electrophoretic mobility-shift assay; p.c., postcoitum; GAPDH, glyceraldehyde 3-phosphate de-hydrogenase.
295Vol. 10, 295–306, May 1999 Cell Growth & Differentiation

interacts with and transactivates the M-PGAM promoter andsupport a role for this protein as a regulator of gene expres-sion during muscle differentiation.
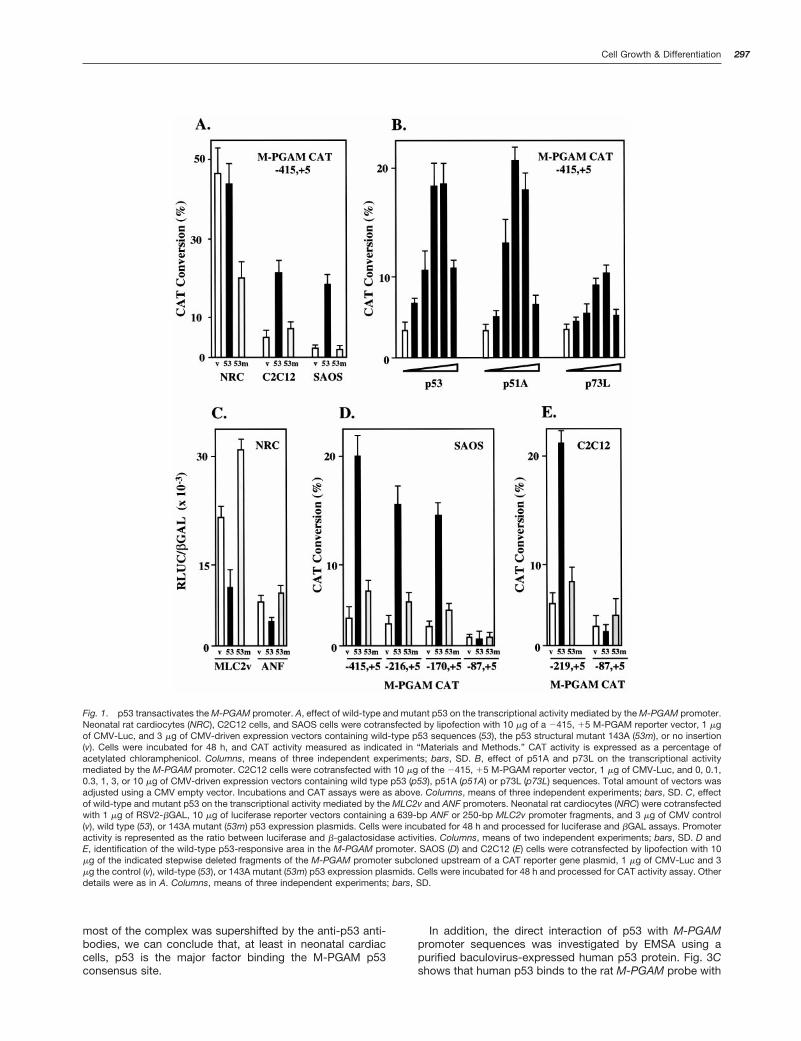
Resultsp53 Transactivates the Rat M-PGAM Gene Promoter. Totest the effect of p53 on the transcription mediated by theM-PGAM promoter, we transfected primary rat neonatal car-diocytes, C2C12 cells, and SAOS cells with a reporter plas-mid containing 2400- to 15-bp sequences of the M-PGAMpromoter subcloned upstream of the CAT gene (plasmidM-PGAM CAT). We have previously shown that this pro-moter fragment accounts for most of the M-PGAM promoterstrength and mediates its muscle-specific expression (5).M-PGAM CAT was cotransfected in combination with CMV-driven expression vectors containing no insert, human wild-type p53, or a dominant mutant p53 cDNA sequences innormal rat neonatal cardiocytes, C2C12 cells, or SAOS cells.C2C12 is a myoblast cell line that carries wild-type p53 (13),whereas SAOS is a human osteosarcoma cell line that lacksboth p53 alleles (14). Cells were then incubated for 48 h,harvested, and processed for the assay of CAT enzymeactivity. Fig. 1A shows that the basal expression mediated byM-PGAM was higher in neonatal cardiocytes than in C2C12or SAOS cells. Cotransfection of M-PGAM CAT with thewild-type p53 expression vector did not alter M-PGAM CATexpression in neonatal cardiocytes. However, cotransfectionof M-PGAM CAT with the mutant p53 expression vector inneonatal cardiocytes originated a 60% decrease in CATactivity. In contrast, cotransfection of M-PGAM CAT with thewild-type p53 expression vector in C2C12 and SAOS cellsresulted in strong activation of M-PGAM CAT activity. Notransactivation was observed when M-PGAM CAT was co-transfected with a mutant p53 expression plasmid in C2C12and SAOS cells (Fig. 1A). Titration experiments indicated thatmaximal transactivation by p53 was reached using 3 mg ofthe CMV-p53 expression plasmid (Fig. 1B). As a whole, thesedata indicated that the transcriptional activity mediated bythe M-PGAM promoter is regulated by p53. The lack oftransactivation of M-PGAM by wild-type p53 in neonatalcardiocytes suggested that the level of endogenous p53protein in neonatal cardiocytes saturates a putativeM-PGAM p53-responsive promoter element. The inhibitionof M-PGAM CAT activity obtained by the overexpression ofthe mutant p53 form supported this hypothesis. It has pre-viously been shown that structural p53 mutant proteins maywork as dominant negative mutants, inhibiting the transcrip-tional activity of the wild-type protein (8, 15–17). Intriguingly,although C2C12 cells contain wild-type p53, M-PGAM wastransactivated by cotransfection with the CMV-p53 vector inthese cells. These results suggested that endogenous p53 isnot transcriptionally active in C2C12 cells at basal condi-tions. Cotransfection of wild-type p53 with a 22 kb to 15 bpM-PGAM CAT construct in C2C12 cells did not result in ahigher level of transcriptional activation by p53, indicatingthat upstream sequences are not required for full transacti-vation of the M-PGAM promoter by this protein (data notshown). Also, the presence of wild-type p53 protein in neo-natal cardiocytes and C2C12 was confirmed by immunopre-cipitation with antibody PAb 246 (data not shown).
It has been recently shown that members of a family ofproteins, referred as p51, p63, or p73, which show significantsequence similarity with p53, are able to transactivate p53gene targets such as the p21cip, Bax, MDM2, cyclin G,
GADD45, and IGF-BP3 promoters as well as p21 and RGCp53 site reporter constructs (18–25). We investigated theability of two members of this family, p51A (p51A/p63g) andp73L (p63a/p51B/p73L), to transactivate the M-PGAM pro-moter in C2C12 cells. Fig. 1B shows that p51A and, at alower intensity, p73L were able to transactivate M-PGAM.These experiments supported further the hypothesis thatM-PGAM is a p53 gene target. Although the regulation ofM-PGAM expression by multiple members of this family ofproteins deserves further investigation, here we have fo-cused our attention on the regulation of M-PGAM by p53, thebest-characterized member of this family.
As an experimental control, we tested the ability of p53 totransactivate two muscle-specific reporter constructs con-taining a 639-bp ANF promoter (26) or a 250-bp MLC2vpromoter (27). A repression of the transcription mediated bythese promoters was observed (Fig. 1C). Thus, the activationof M-PGAM by p53 was specific.
The Rat M-PGAM Gene Promoter Contains a Wild-Type p53-responsive Element. To identify an M-PGAMpromoter element that could mediate the effects of p53, weassayed a series of M-PGAM CAT deletion mutants by co-transfection with the wild-type p53 expression vector. Be-cause the effect of p53 on M-PGAM in primary neonatalcardiocytes may be masked by the presence of endogenousp53 protein, these assays were performed in SAOS cells. Theresult of these experiments is shown in Fig. 1D. The M-PGAM CAT deletion mutants defined a promoter fragmentbetween 2172- and 287-bp sequences that mediates re-sponsiveness to p53. Similar results were obtained withC2C12 cells (Fig. 1E). Inspection of the 2172- to 287-bppromoter sequences revealed the presence of a consensusp53 binding site at positions 2116- to 290-bp. Interestingly,this site is strikingly homologous to the MCK and RGC p53sites (Fig. 2) and contains two TGCCT (pentamers) motifs(28–30). Also, an additional imperfect pentamer, TGCCA,was found five nucleotides upstream of this site (data notshown). This finding strongly suggested that p53 directlyinteracts with the M-PGAM promoter.
p53 Directly Interacts with the M-PGAM Gene Pro-moter. A specific interaction of p53 with the M-PGAM pro-moter was investigated by EMSA using the oligonucleotideprobe (duplex) tcgacTGCCACTGGTTGCCTGCCTCTGC-CTG (M-PGAM, pentamer motifs underlined) and nuclearextracts prepared from neonatal rat hearts. One major nu-cleoprotein complex band was observed that was effectivelycompeted by a mass excess of an oligonucleotide containingthe p53 MCK site but not by oligonucleotides containingSp1- or nuclear factor kB-binding sites (Fig. 3A, p53, arrow-head). Thus, the formation of this complex was p53 sitespecific. The presence of p53 in this band was then con-firmed using anti-p53 monoclonal antibodies. A supershiftwas observed with the addition to the EMSA reactions of theanti-p53 antibodies PAb 421 (31) and DO-1 (32), which rec-ognize wild-type p53 associated with DNA (Ref. 33; Fig. 3B).However, PAb 240, a monoclonal antibody that recognizesmutant p53 (34), had no effect (Fig. 3B). In summary, theseexperiments demonstrated that endogenous rat heart p53binds specifically to the M-PGAM p53 consensus site. Asmall amount of nucleoprotein complex was not supershiftedby the PAb 421 and DO-1 antibodies. This result suggestedthat either some of the p53 in the complex was not recog-nized by the antibodies, as has been shown by others (33), orother proteins were present in this band. However, because
296 Regulation of Muscle Gene Expression by p53
most of the complex was supershifted by the anti-p53 anti-bodies, we can conclude that, at least in neonatal cardiaccells, p53 is the major factor binding the M-PGAM p53consensus site.
In addition, the direct interaction of p53 with M-PGAMpromoter sequences was investigated by EMSA using apurified baculovirus-expressed human p53 protein. Fig. 3Cshows that human p53 binds to the rat M-PGAM probe with
Fig. 1. p53 transactivates the M-PGAM promoter. A, effect of wild-type and mutant p53 on the transcriptional activity mediated by the M-PGAM promoter.Neonatal rat cardiocytes (NRC), C2C12 cells, and SAOS cells were cotransfected by lipofection with 10 mg of a 2415, 15 M-PGAM reporter vector, 1 mgof CMV-Luc, and 3 mg of CMV-driven expression vectors containing wild-type p53 sequences (53), the p53 structural mutant 143A (53m), or no insertion(v). Cells were incubated for 48 h, and CAT activity measured as indicated in “Materials and Methods.” CAT activity is expressed as a percentage ofacetylated chloramphenicol. Columns, means of three independent experiments; bars, SD. B, effect of p51A and p73L on the transcriptional activitymediated by the M-PGAM promoter. C2C12 cells were cotransfected with 10 mg of the 2415, 15 M-PGAM reporter vector, 1 mg of CMV-Luc, and 0, 0.1,0.3, 1, 3, or 10 mg of CMV-driven expression vectors containing wild type p53 (p53), p51A (p51A) or p73L (p73L) sequences. Total amount of vectors wasadjusted using a CMV empty vector. Incubations and CAT assays were as above. Columns, means of three independent experiments; bars, SD. C, effectof wild-type and mutant p53 on the transcriptional activity mediated by the MLC2v and ANF promoters. Neonatal rat cardiocytes (NRC) were cotransfectedwith 1 mg of RSV2-bGAL, 10 mg of luciferase reporter vectors containing a 639-bp ANF or 250-bp MLC2v promoter fragments, and 3 mg of CMV control(v), wild type (53), or 143A mutant (53m) p53 expression plasmids. Cells were incubated for 48 h and processed for luciferase and bGAL assays. Promoteractivity is represented as the ratio between luciferase and b-galactosidase activities. Columns, means of two independent experiments; bars, SD. D andE, identification of the wild-type p53-responsive area in the M-PGAM promoter. SAOS (D) and C2C12 (E) cells were cotransfected by lipofection with 10mg of the indicated stepwise deleted fragments of the M-PGAM promoter subcloned upstream of a CAT reporter gene plasmid, 1 mg of CMV-Luc and 3mg the control (v), wild-type (53), or 143A mutant (53m) p53 expression plasmids. Cells were incubated for 48 h and processed for CAT activity assay. Otherdetails were as in A. Columns, means of three independent experiments; bars, SD.
297Cell Growth & Differentiation

a similar affinity than to other oligonucleotide probes con-taining consensus p53 sites from the RGC or MCK genes.Moreover, binding of p53 to the M-PGAM probe was com-peted by a mass excess of an oligonucleotide containing aconsensus p53 site (MCK) but not by an unrelated sequence(Fig. 3D). In summary, these experiments confirmed that p53specifically interacts with the M-PGAM promoter.
p53 Is Required for Full Activation of the M-PGAMPromoter in Rat Cardiocytes. To determine whether theM-PGAM p53 consensus binding site was responsible forthe transactivation of the M-PGAM promoter by wild-typep53, we created an M-PGAM reporter construct with pointmutations at the two consensus p53 pentamer motifs,namely Dp53 M-PGAM CAT. The native and mutantM-PGAM CAT plasmids were transfected in rat neonatalcardiocytes and cells incubated and processed for the assayof CAT activity. Mutation of the consensus pentamer motifsdecreased the transcriptional activity mediated by the M-PGAM promoter in neonatal cardiocytes by ;65% (Fig. 4A).In addition, we cotransfected C2C12 cells with the native ormutant M-PGAM CAT reporters and wild-type or mutant p53expression vectors. The results of these experiments, shownin Fig. 4B, indicated that mutagenesis of the M-PGAM p53binding site blocks the transactivation of this promoter bywild-type p53. Thus, these experiments confirmed that theM-PGAM promoter contains a consensus p53 binding sitethat mediates the transactivation of this promoter by p53.Moreover, these experiments demonstrate that theM-PGAM-responsive element is constitutively active in pri-mary rat neonatal cardiocytes but not in C2C12 myoblasts.
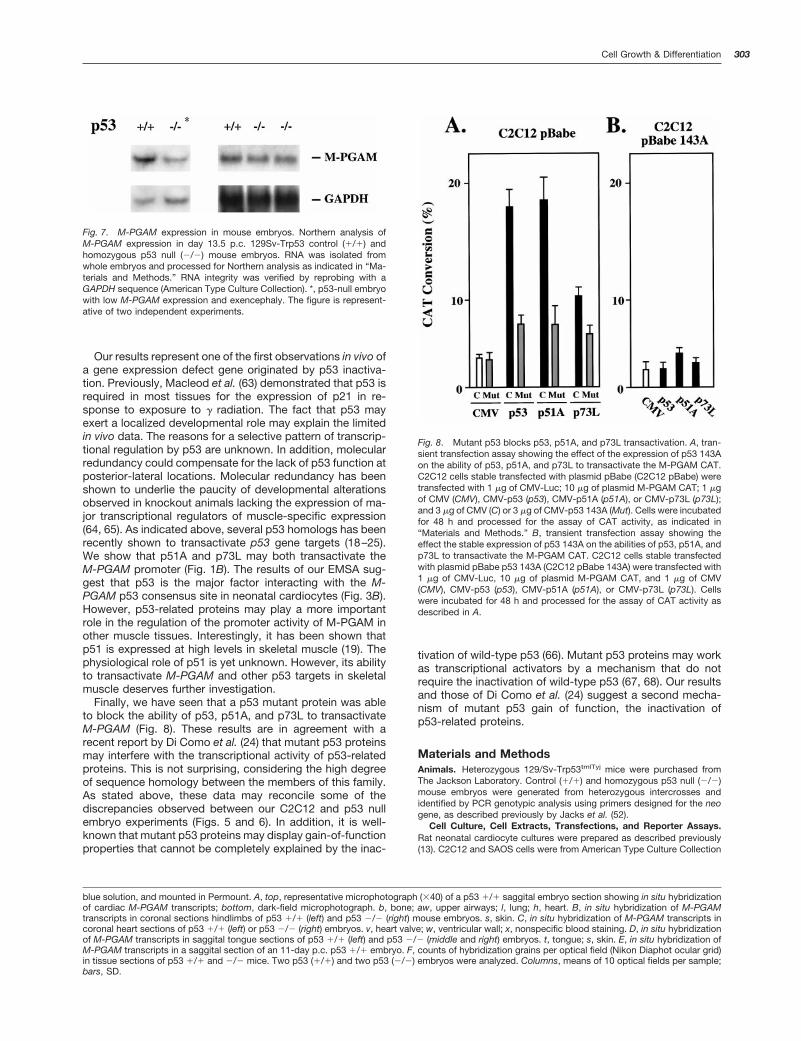
p53 Regulates the Expression of the M-PGAM GenePromoter in Vivo. The experiments described above sug-gested that p53 function might be important for the regula-tion of M-PGAM expression in vivo. To begin to elucidate therole that p53 may play in the transcriptional control of M-PGAM in vivo, we investigated the effect of the expression ofa dominant mutant p53 protein on M-PGAM expression inC2C12 cells. For that purpose, we generated a cell line ofmutant p53-expressing C2C12 cells by the stable transfec-tion of these cells with a retroviral-based vector containing aselectable marker and p53 143A cDNA sequences. Controlcells were generated by the stable transfection of an emptyvector. Metabolic labeling and immunoprecipitation using ananti-mutant p53 specific antibody demonstrated stable ex-pression of the mutant p53 protein in C2C12 cells (Fig. 5A).A similar procedure was employed previously by Soddu et al.(13) to demonstrate the requirement of wild-type p53 func-tion for the transcriptional activity of a reporter plasmid con-taining multiple copies of the p53 RGC site. Control andmutant p53-expressing C2C12 cells were then induced todifferentiate by incubation for 2–4 days in low serum mediumsupplemented with insulin and transferrin (13). The results ofthese experiments, shown in Fig. 5B, demonstrate that mu-tant p53 blocks the induction of endogenous M-PGAM ex-pression during myocyte differentiation. As an experimental
control, the expression of MCK and ANF in these RNA ex-tracts was also investigated. A modest effect of mutant p53on MCK expression was observed only at early incubationtimes (Fig. 5C), suggesting the existence of some differencesin the control of the expression of M-PGAM and MCK duringmuscle differentiation (35). ANF expression was barely de-tectable in these extracts and was not affected by mutantp53 (data not shown).
Moreover, when C2C12 myoblasts were transfected withthe native or D53 mutant M-PGAM CAT reporters and theninduced to differentiate as above, the mutagenesis of the p53site originated a dramatic decrease in the induction of CATexpression (Fig. 5D). These results demonstrated that inter-action of p53 or p53-related proteins with M-PGAM is re-quired for the induction of the activity of this promoter duringthe myoblast to myotube transition in C2C12 cells. However,experiments of gel shift assay showed no changes in proteinbinding to the M-PGAM p53 site during this period (data notshown). Thus, other processes, such as protein-protein in-teraction, may be implicated in the regulation of M-PGAMtransactivation by p53 and related factors during myocytedifferentiation.
Subsequent experiments investigated the role that p53plays in the control of M-PGAM expression in vivo usingNorthern analysis and in situ hybridization of gene tran-scripts. Control and p53 null mouse embryos at day 13.5 p.c.were employed in these studies. This stage was selectedbecause it represents the period of maximal expression ofp53 in mouse embryonic development (36–38). Crosses be-tween mice heterozygous for a targeted mutation in exon 5 ofp53 on the inbred 129/sv genetic background (129/Sv-Trp53tmlTyj mice) yielded 17% homozygous mutant offspring(6 of 34 embryos). A preliminary Northern analysis revealedno significant differences in the level of whole-animal M-PGAM expression between a control and a p53-null embryos(data not shown). M-PGAM expression was then investi-gated by in situ hybridization. This technique was used be-cause it provided the advantage to study M-PGAM expres-sion in an individual and tissue-specific fashion. Fig. 6Ashows a typical muscle-specific distribution of M-PGAMtranscripts found in control embryos. M-PGAM mRNA wasenriched in heart cavities, eluding the valvular system, anddiaphragm, whereas no expression was found in bone orlung. Similarly, M-PGAM transcripts accumulated at tongue,limb, and intercostal muscles (data not shown). Control ex-periments using a sense M-PGAM riboprobe resulted inbackground hybridization levels over these tissue sections(data not shown). The expression of M-PGAM in p53-nullmice was characterized by a similar muscle-specific distri-bution, indicating that p53 does not affect the tissue speci-ficity of this transcript (Fig. 6, B–D, and data not shown).Three of five p53-null embryos studied had no alterations inM-PGAM expression (60%). However, in two of these em-bryos (40%), reduced M-PGAM expression was detected.Strikingly, lower levels of M-PGM expression were evi-denced in muscle organs of anterior-ventral but not in laterallocations. Although a small reduction (,30%) in M-PGAMexpression was observed in embryonic hindlimb muscles(Fig. 6, B and E), M-PGAM hybridization was reduced by 40and 65%, respectively, at heart and tongue muscles relatedto their wild type p53 littermates (Fig. 6, C, D, and F). Thisdefective pattern of M-PGAM expression was not observedin any of the control embryos studied (six in total; data notshown). Importantly, the lower M-PGAM expression in thesep53 null animals was not due to organ hypoplasia. Nuclei
Fig. 2. Consensus p53 sites in the M-PGAM, MCK, and RGC genes.M-PGAM sequences were from the rat M-PGAM gene positions 2116 to290 bp. MCK and RGC sequences were as described previously (8, 15).
298 Regulation of Muscle Gene Expression by p53

counts per optical field were similar in day 13.5 p.c. heartsand tongues of control and p53 null embryos with low M-PGAM expression (not shown). In addition, M-PGAM expres-sion was first detected in control animals at day 11 p.c. inheart and tail muscles (Fig. 6E). This localized expressionspread to lateral muscles by day 13.5 p.c. (Figure 6B). There-fore, because all p53-null animals demonstrated relativelynormal M-PGAM expression in hindlimb muscles, it is highlyunlikely that a defective M-PGAM expression in heart wasoriginated by deferred development.
To confirm these results, new crosses between heterozy-gous 129/Sv-Trp53tmlTyj mice were carried out yielding a16.6% homozygous mutant offspring (3 of a total of 18embryos at day 13.5 p.c.). Northern analysis of whole em-bryo M-PGAM transcripts demonstrated a significant reduc-
tion in M-PGAM expression in one of three p53-null embryosobtained (Fig. 7). No change in M-PGAM expression wasfound in six heterozygous p53 mutant embryos studied (datanot shown). In summary, these experiments showed a re-duced M-PGAM expression in a subset of p53-null animals.These data demonstrated that p53 contributes to the regu-lation of M-PGAM expression in vivo and support previousobservations that indicate a role for p53 in mouse embryo-genesis (37, 39).
The fact that partial penetrance was observed in p53 nullanimals whereas a more dramatic decrease in M-PGAMexpression was observed in C2C12 cells that stably expressa mutant p53 protein, prompted us to investigate the possi-bility that mutant p53, in addition to inactivate wild-type p53,may interfere with the ability of other p53-related proteins to
Fig. 3. p53 binds the ratM-PGAM promoter. A, EMSAshowing the binding of neonatalrat heart p53 to an M-PGAM pro-moter probe. Nuclear extractswere prepared from rat neonatalcardiocytes as indicated in “Ma-terials and Methods.” Five mg ofnuclear extracts were assayed byEMSA using 0.2 ng of a M-PGAMp53 site oligonucleotide probeand 10 mg of poly (dI z dC):(dI z dC)in buffer B. Extracts were incu-bated for 30 min with 40 ng of theindicated oligonucleotides priorto the addition of the M-PGAMprobe. B, supershift assay identi-fying p53 bound to an M-PGAMprobe. EMSA reactions were asabove. One mg of the respectiveantibodies was added to the re-action, and extracts were incu-bated for 1 h at 37°C. C, EMSA ofpurified human baculovirus ex-pressed human p53 using a se-ries of DNA probes. Binding reac-tions were prepared as in A. D,EMSA of purified human baculo-virus-expressed human p53 us-ing 0.2 ng of the M-PGAM probeand 0, 0.1, 1, or 10 ng of theindicated oligonucleotide com-petitor. For a description of theseoligonucleotides probes andother experimental details, see“Materials and Methods.” Figuresshow experiments that are repre-sentative of at least two assays.
299Cell Growth & Differentiation

transactivate the M-PGAM promoter. To test this hypothesis,C2C12 cells were cotransfected with the M-PGAM CAT re-porter plasmids and CMV-driven expression vectors con-taining p53, p51A, or p73L sequences, alone or in the pres-ence of a p53 143A expression vector. Interestingly, mutantp53 blocked in part the transactivation of M-PGAM by p53,p51A, and p73L (Fig. 8A). Moreover, these proteins failed totransactivate the M-PGAM promoter in C2C12 cells stablyexpressing p53 143A (Fig. 8B). These results demonstratethat, at least in transient transfection assays, mutant p53may interfere with the ability of p51A and p73L to activateM-PGAM expression. These results are in agreement withrecent data demonstrating that mutant p53 blocks the tran-scriptional activity of p73 isoforms (24). These data suggestthe possibility that p53-related proteins may compensate forsome p53 functions in p53 null animals and may reconcilesome of the differences observed between C2C12 cells ex-pressing mutant p53 and p53-null embryos.
DiscussionWe have found that the expression mediated by the ratM-PGAM promoter is regulated by p53. The activation byp53 of the activity of an M-PGAM CAT reporter constructwas demonstrated in C2C12 and SAOS cells (Fig. 1A). Trans-fection of wild-type p53 in rat neonatal cardiocytes did notalter M-PGAM CAT activity (Fig. 1A). However, the expres-
sion of a dominant mutant p53 form in these cells inhibitedM-PGAM-mediated expression. The fact that some mutantp53 proteins behave in a dominant negative fashion, inhib-iting the transactivation of coexpressed or endogenous wild-type p53, is well documented (40–42). In view of these data,we interpreted that the overexpression of mutant p53 proteinin neonatal cardiocytes inhibited the endogenous wild-typep53 that constitutively transactives the M-PGAM promoter.This hypothesis was supported by the reduction of the tran-scriptional activity mediated by the M-PGAM promoter inneonatal cardiocytes by the mutagenesis of the M-PGAMp53 site (Fig. 4, see below). Moreover, current experiments inour laboratory indicate that overexpression of the p53 tran-scriptional inhibitor protein MDM2 (11) in neonatal cardio-cytes represses M-PGAM expression in a p53 site-depen-dent manner (data not shown). The fact that p53 protein isexpressed in neonatal cardiocytes has been shown previ-ously (36). Also, gel shift assays demonstrated that p53 is themajor factor in neonatal hearts binding a p53 consensus sitein the M-PGAM promoter (Fig. 3).
Maximal transactivation of M-PGAM in C2C12 was ob-served with 1–3 mg of a p53 expression vector (Fig. 1B).Similar concentrations of p51A and p73L expression vectorswere required for maximal M-PGAM promoter activity. Loweramounts of p53 and p53-related proteins have been shownto transactivate p53 gene targets in other cell types (24).However, in agreement with our results, relatively large con-centrations of p53 were used by others to determine theability of p53 to transactivate gene targets in muscle cells (7,8). We have seen also that 1–3 mg of a p53 expression vectorwere required in C2C12 to obtain maximal transactivation ofreporter plasmids containing the RGC p53 site (PG13) or ap21 promoter fragment (not shown). These results indicatethat M-PGAM is as responsive to p53 transactivation asother p53 targets. We hypothesize that the differences ob-served with the results obtained in other cell types are orig-inated by the lower transfection efficiency of muscle cells.
A wild-type p53-responsive promoter fragment in the M-PGAM promoter was defined by cotransfection of a wild-type p53 expression vector and a series of M-PGAM CATdeletion mutants in C2C12 and SAOS cells (Fig. 1, B and C).Inspection of this DNA sequence revealed the presence oftwo TGCCT pentamers motifs at positions 2116 to 290 bpwith an additional imperfect pentamer TGCCA 5 nucleotidesupstream. This putative p53 binding site was found to bestrikingly homologous to the MCK and RGC p53 sites (Fig. 2).Importantly, mutagenesis of this site decreased the activityof M-PGAM in neonatal cardiocytes and blocked the trans-activation of the M-PGAM promoter by wild-type p53 inC2C12 cells (Fig. 4). These experiments demonstrated thatthe rat M-PGAM promoter contains a wild-type p53 consen-sus site that mediates the transactivation of M-PGAM by thisprotein. A similar sequence, with 13 of 17 identical nucleo-tides, was found in the human M-PGAM promoter, suggest-ing that this element is well conserved (data not shown).Whether the human M-PGAM promoter is also responsive top53 should be the object of further investigation. EMSAdetected the binding of endogenous rat cardiac p53 andpurified baculovirus-expressed human p53 to the p53 M-PGAM site (Fig. 3). Importantly, these experiments demon-strated that p53 directly interacts with M-PGAM promotersequences. The fact that rat and human p53 are both able tobind with high affinity the rat M-PGAM promoter in a specific
Fig. 4. Mutagenesis of the M-PGAM p53 binding site blocks p53 trans-activation of the M-PGAM promoter. A, transient transfection assay show-ing the effect of the mutagenesis of the p53 site on the transcriptionmediated by the M-PGAM promoter in rat neonatal cardiocytes. Cellswere transfected with 1 mg of CMV-Luc and 10 mg of plasmids M-PGAMCAT (WT) or M-PGAMp53D CAT (D53), incubated for 48 h and processedfor the assay of CAT activity as indicated in “Materials and Methods.” B,transient transfection assay showing the effect of the mutagenesis of thep53 site on the transactivation of M-PGAM by p53. C2C12 cells werecotransfected with 1 mg of CMV-Luc, 10 mg of reporter plasmids M-PGAMCAT (WT), or M-PGAMp53D CAT (D53) and 3 mg of expression plasmidsCMV (CMV), CMV-p53 (CMV-p53), or CMV-p53 143 A (CMV-p53m). Cellswere incubated for 48 h and processed for the assay of CAT activity, asindicated in “Materials and Methods.” Column, means of three experi-ments; bars, SD.
300 Regulation of Muscle Gene Expression by p53

manner indicated that the interaction between p53 and M-PGAM is well conserved.
Finally, a role for p53 in the transcriptional regulation ofM-PGAM was demonstrated in C2C12 cells (Fig. 5) and inmouse embryos (Figs. 6 and 7). Strikingly, a decrease inM-PGAM expression was observed in p53-null mouse em-bryos in heart and tongue but not at limb muscles, indicatingthat p53 contributes to the control of M-PGAM expression inmuscle tissues of anterior-ventral location. Studies of p53expression during mouse embryogenesis indicated high lev-els of p53 mRNA in all tissues (38). At late stages of devel-opment, p53 expression becomes more pronounced in cellsundergoing differentiation (38). A similar scenario has beenobserved during chicken embryogenesis (43), supporting thehypothesis that p53 plays a role in tissue-specific differenti-ation. Multiple studies have implicated the p53 tumor sup-pressor gene during differentiation in vitro (13, 14, 44–50).These results were disputed by the absence of developmen-tal alterations initially reported in p53 null animals (51, 52).However, recent evidence has indicated the presence ofneural tube and cranio-facial malformations in a subset ofp53-null animals (37, 39, 53). These data underscore the factthat p53 may be important in normal development as well asin tumorigenesis. Sah et al. (37) hypothesized that, during
neural tube closure, p53 could have a role in mediating cellcycle arrest to limit cell proliferation or to prepare cells for adifferentiation event. Our results support the hypothesis thatp53 may have a role as a positive regulator of muscle celldifferentiation in vivo. These results are not in contradictionto the well-known cell cycle regulatory properties of p53. Thegrowth suppressor properties of p53 are well substantiatedby the ability of this protein to inhibit the proliferation ofcultured tumor cells (54), prevent neoplastic transformationin vitro (55–59), and inhibit the formation of tumors in animalmodels (24). Importantly, unlike in skeletal muscle, wherecellular proliferation and a differentiated phenotype are mu-tually exclusive (60), the increase in cardiac mass duringembryonic life arises predominantly from the proliferation ofmononucleated differentiated cardiomyocytes (61). Terminaldifferentiation, with irreversible withdrawal from the cell cy-cle, does not take place in cardiomyocytes until shortly afterbirth (62). Our results indicate that p53 may function in theheart as a regulator of a specific set of genes associated withphenotypic differentiation rather than as a growth suppres-sor. This conclusion is supported by recent data of Soddu etal. (13) indicating that, in the C2C12 model, inactivation ofp53 function affects cell differentiation but not cell cyclearrest.
Fig. 5. Expression of a mutant p53 form blocks the increase in M-PGAM expression during the terminal differentiation of C2C12 cells. A, immunopre-cipitation of mutant p53 with antibody PAb 240 in C2C12 cells infected with vector pBabe (no insert, pBabe) or vector pBabe p53 143A (p53143A). Cellswere incubated with radiolabeled methionine, harvested, and lysed; PAb 240-reactive p53 was immunoprecipitated; and immunoprecipitates were resolvedby SDS-PAGE and exposed to a PhosphorImager screen (Molecular Dynamics). B, Northern analysis of M-PGAM expression in C2C12 stable transfectedwith plasmids pBabe (pBabe, no insert) or pBabe p53 143A (p53143A). Cells (3 3 104 cells/cm2) were incubated in serum-free DMEM supplemented with10 mg/ml insulin and 5 mg/ml transferrin for 2–4 days (13). RNA was isolated and processed for Northern analysis, as indicated in “Materials and Methods.”RNA integrity was verified by reprobing with a GAPDH sequence (American Type Culture Collection). The figure is representative of two experiments. C,Northern analysis of MCK expression in the C2C12 RNA extracts used in part B. MCK sequence probe was from American Type Culture Collection. D, effectof the mutagenesis of the p53 M-PGAM site on the activation of M-PGAM CAT during the terminal differentiation of C2C12 cells. C2C12 cells werecotransfected with 1 mg of CMV-Luc and 10 mg of reporter plasmids M-PGAM CAT (WT) or M-PGAMp53D CAT (D53). Cells were incubated for 72 h in DMEMwith 10% fetal bovine serum (10% Serum) or in serum-free DMEM supplemented with 10 and 5 mg/ml transferrin (No Serum), harvested, and processedfor the assay of CAT activity as indicated in “Materials and Methods.” Column, means of three experiments; bars, SD.
301Cell Growth & Differentiation
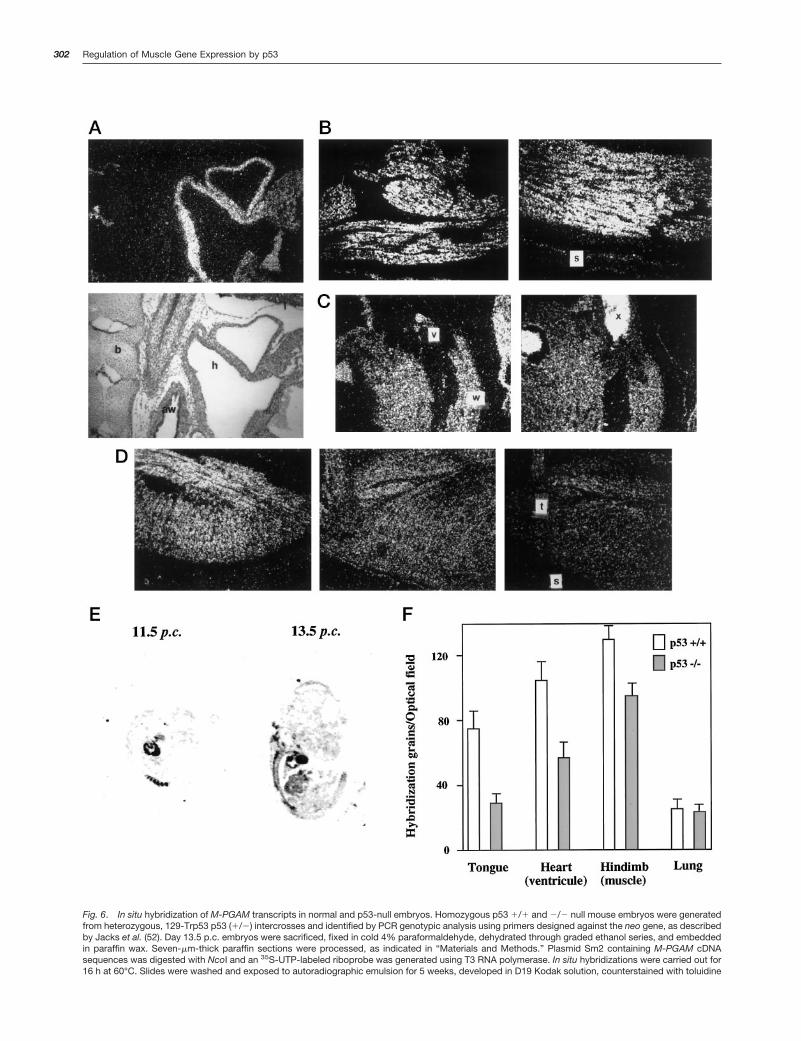
Fig. 6. In situ hybridization of M-PGAM transcripts in normal and p53-null embryos. Homozygous p53 1/1 and 2/2 null mouse embryos were generatedfrom heterozygous, 129-Trp53 p53 (1/2) intercrosses and identified by PCR genotypic analysis using primers designed against the neo gene, as describedby Jacks et al. (52). Day 13.5 p.c. embryos were sacrificed, fixed in cold 4% paraformaldehyde, dehydrated through graded ethanol series, and embeddedin paraffin wax. Seven-mm-thick paraffin sections were processed, as indicated in “Materials and Methods.” Plasmid Sm2 containing M-PGAM cDNAsequences was digested with NcoI and an 35S-UTP-labeled riboprobe was generated using T3 RNA polymerase. In situ hybridizations were carried out for16 h at 60°C. Slides were washed and exposed to autoradiographic emulsion for 5 weeks, developed in D19 Kodak solution, counterstained with toluidine
302 Regulation of Muscle Gene Expression by p53
Our results represent one of the first observations in vivo ofa gene expression defect gene originated by p53 inactiva-tion. Previously, Macleod et al. (63) demonstrated that p53 isrequired in most tissues for the expression of p21 in re-sponse to exposure to g radiation. The fact that p53 mayexert a localized developmental role may explain the limitedin vivo data. The reasons for a selective pattern of transcrip-tional regulation by p53 are unknown. In addition, molecularredundancy could compensate for the lack of p53 function atposterior-lateral locations. Molecular redundancy has beenshown to underlie the paucity of developmental alterationsobserved in knockout animals lacking the expression of ma-jor transcriptional regulators of muscle-specific expression(64, 65). As indicated above, several p53 homologs has beenrecently shown to transactivate p53 gene targets (18–25).We show that p51A and p73L may both transactivate theM-PGAM promoter (Fig. 1B). The results of our EMSA sug-gest that p53 is the major factor interacting with the M-PGAM p53 consensus site in neonatal cardiocytes (Fig. 3B).However, p53-related proteins may play a more importantrole in the regulation of the promoter activity of M-PGAM inother muscle tissues. Interestingly, it has been shown thatp51 is expressed at high levels in skeletal muscle (19). Thephysiological role of p51 is yet unknown. However, its abilityto transactivate M-PGAM and other p53 targets in skeletalmuscle deserves further investigation.
Finally, we have seen that a p53 mutant protein was ableto block the ability of p53, p51A, and p73L to transactivateM-PGAM (Fig. 8). These results are in agreement with arecent report by Di Como et al. (24) that mutant p53 proteinsmay interfere with the transcriptional activity of p53-relatedproteins. This is not surprising, considering the high degreeof sequence homology between the members of this family.As stated above, these data may reconcile some of thediscrepancies observed between our C2C12 and p53 nullembryo experiments (Figs. 5 and 6). In addition, it is well-known that mutant p53 proteins may display gain-of-functionproperties that cannot be completely explained by the inac-
tivation of wild-type p53 (66). Mutant p53 proteins may workas transcriptional activators by a mechanism that do notrequire the inactivation of wild-type p53 (67, 68). Our resultsand those of Di Como et al. (24) suggest a second mecha-nism of mutant p53 gain of function, the inactivation ofp53-related proteins.
Materials and MethodsAnimals. Heterozygous 129/Sv-Trp53tmlTyj mice were purchased fromThe Jackson Laboratory. Control (1/1) and homozygous p53 null (2/2)mouse embryos were generated from heterozygous intercrosses andidentified by PCR genotypic analysis using primers designed for the neogene, as described previously by Jacks et al. (52).
Cell Culture, Cell Extracts, Transfections, and Reporter Assays.Rat neonatal cardiocyte cultures were prepared as described previously(13). C2C12 and SAOS cells were from American Type Culture Collection
Fig. 8. Mutant p53 blocks p53, p51A, and p73L transactivation. A, tran-sient transfection assay showing the effect of the expression of p53 143Aon the ability of p53, p51A, and p73L to transactivate the M-PGAM CAT.C2C12 cells stable transfected with plasmid pBabe (C2C12 pBabe) weretransfected with 1 mg of CMV-Luc; 10 mg of plasmid M-PGAM CAT; 1 mgof CMV (CMV), CMV-p53 (p53), CMV-p51A (p51A), or CMV-p73L (p73L);and 3 mg of CMV (C) or 3 mg of CMV-p53 143A (Mut). Cells were incubatedfor 48 h and processed for the assay of CAT activity, as indicated in“Materials and Methods.” B, transient transfection assay showing theeffect the stable expression of p53 143A on the abilities of p53, p51A, andp73L to transactivate the M-PGAM CAT. C2C12 cells stable transfectedwith plasmid pBabe p53 143A (C2C12 pBabe 143A) were transfected with1 mg of CMV-Luc, 10 mg of plasmid M-PGAM CAT, and 1 mg of CMV(CMV), CMV-p53 (p53), CMV-p51A (p51A), or CMV-p73L (p73L). Cellswere incubated for 48 h and processed for the assay of CAT activity asdescribed in A.
blue solution, and mounted in Permount. A, top, representative microphotograph (340) of a p53 1/1 saggital embryo section showing in situ hybridizationof cardiac M-PGAM transcripts; bottom, dark-field microphotograph. b, bone; aw, upper airways; l, lung; h, heart. B, in situ hybridization of M-PGAMtranscripts in coronal sections hindlimbs of p53 1/1 (left) and p53 2/2 (right) mouse embryos. s, skin. C, in situ hybridization of M-PGAM transcripts incoronal heart sections of p53 1/1 (left) or p53 2/2 (right) embryos. v, heart valve; w, ventricular wall; x, nonspecific blood staining. D, in situ hybridizationof M-PGAM transcripts in saggital tongue sections of p53 1/1 (left) and p53 2/2 (middle and right) embryos. t, tongue; s, skin. E, in situ hybridization ofM-PGAM transcripts in a saggital section of an 11-day p.c. p53 1/1 embryo. F, counts of hybridization grains per optical field (Nikon Diaphot ocular grid)in tissue sections of p53 1/1 and 2/2 mice. Two p53 (1/1) and two p53 (2/2) embryos were analyzed. Columns, means of 10 optical fields per sample;bars, SD.
Fig. 7. M-PGAM expression in mouse embryos. Northern analysis ofM-PGAM expression in day 13.5 p.c. 129Sv-Trp53 control (1/1) andhomozygous p53 null (2/2) mouse embryos. RNA was isolated fromwhole embryos and processed for Northern analysis as indicated in “Ma-terials and Methods.” RNA integrity was verified by reprobing with aGAPDH sequence (American Type Culture Collection). *, p53-null embryowith low M-PGAM expression and exencephaly. The figure is represent-ative of two independent experiments.
303Cell Growth & Differentiation

(Manassas, VA). Cells were cultured in DMEM plus 10% dialyzed fetalbovine serum (Life Technologies, Inc., Grand Island, NY) with penicillin (10units/ml) and streptomycin (10 units/ml). C2C12-pBabe and -pBabe 143Acells were generated by transfection of C2C12 cells with plasmids pBabeor pBabe p53 143A, followed by selection in medium supplemented with3 mg/ml puromycin. Polyclonal populations at passages 1–3 were used.Nuclear extracts were prepared as described previously (68). Extractswere aliquoted, quickly frozen in liquid N2, and stored at 280°C. Fortransfection, C2C12 cells were incubated as above until 80% confluencyand transfected by the lipofectamine method following the recommenda-tions of the manufacturer (Life Technologies, Inc.). Neonatal cardiocyteswere transfected by the calcium phosphate method as described (5). Asa control for transfection efficiency, cells were cotransfected with 1 mg ofCMV promoter-driven luciferase vector (69). Transfection efficiency wasalso independently monitored using a CMV-CAT plasmid (85% CAT con-version). 48 h after transfection, cells were lysed by 50 ultrasonic vibrationsat 4°C using a Branson sonifier at a setting of 3. Equal amounts ofluciferase activity units, as determined by a luminescence assay (Pro-mega, Madison, WI), were then analyzed for CAT activity using TLC(Baker, Phillipsburg, NJ). Chromatography plates were scanned using aPhosphorImager screen and quantified using ImageQuant software (Mo-lecular Dynamics). Alternatively, chloramphenicol acetyltransferase activ-ity was measured using the Fluor diffusion assay (70). CAT activity wasexpressed as a percentage of acetylated chloramphenicol. Luciferase andb-galactosidase activities were measured as described previously (71).
Plasmids. The CAT reporter plasmids containing stepwise deletedfragments of the rat M-PGAM gene were as described previously (4).Plasmid M-PGAM CAT contains 2415 to 15 M-PGAM sequences. Site-directed mutagenesis was performed by PCR, as described previously(72). The M-PGAMp53D CAT plasmid contains 2415 to 15 bp M-PGAMsequences with the substitution (double strand, p53 sites underlined)CTGCAGCCTCGGTAC for TGCCTGCCTCTGCCT at positions 2105 to290 bp. The ANF and MLC2v reporter vectors were as described previ-ously (26, 27, 73). The CMV enhance-promoter-driven expression vectorscontaining wild-type p53 or the structural mutant p53 143A form; thereporter plasmid PG13, containing 13 copies of a wild type p53 consensusbinding site; and the p21 promoter luciferase reporter plasmid were allgifts from Dr. B. Vogelstein (Johns Hopkins University, Baltimore, MD;Refs. 74 and 75). The pBabe p53 143A plasmid containing p53 mutant143A sequences and a puromycin selectable marker was generated bythe subcloning of a p53 mutant 143A cDNA fragment originated by BamHIdigestion of plasmid CMV-p53 143A into the retroviral-based vectorpBabe (76). The p51A and p73L expression vectors were generated bysubcloning p51A and p73L cDNA sequences into pCMV (19).
EMSAs. The oligonucleotide probe sequences used were as follows(double-stranded): RGC, tcgacCTTGCCTGGACTTGCCTGG, with the p53consensus site at the ribosomal gene cluster gene (75); MCK, TGGC-CGGGGCCTGCCTCTCTCTGCCTCTGA, with the p53 binding site at theMCK enhancer (8); C/EBP, tcgacAAGTTGAGAAATTTG, with the C/EBPconsensus site at the 422 (aP2) promoter (77); Sp1I, CGGGACTGGG-GAGTGGCGAGCCCTC, Sp1II, CAGGGAGGCGTGGCCTGGGCGG-GACTGGGG, and kB, tcgacGCTGGGGACTTTCCAGGG, with the Sp1I,Sp1II and 39 kB sites, respectively, at the HIV1 long terminal repeat (78).DNA oligonucleotides were prepared with an Applied Biosystems 391EPDNA synthesizer using the phosphoramidite method and purified usingSep-Pak C18 cartridges (Waters Associates, Milford, MA). Gel shift mo-bility assays were prepared as follows: in a 10-ml reaction volume con-taining buffer B [20 mM HEPES (pH 7.5), 100 mM KCl, 0.2 mM ZnCl2, 1 mM
DTT, 1 mM phenylmethylsulfonyl fluoride, and 5% glycerol], 10 mg ofpoly(dI z dC):(dI z dC), and 5 mg of nuclear extract protein per reaction.Incubation time was 30 min at 20°C, unless otherwise indicated. Equalamounts of cardiac protein extracts were assayed in each condition asdetermined by the Bradford assay (Bio-Rad). When antibodies wereadded to the reaction mix, the total incubation time was 1 h at 20°C.Mouse monoclonal antibodies anti-p53 PAb 240, 246, 421, and 1801 werepurchased from Santa Cruz Biotechnology (Santa Cruz, CA). Purifiedbaculovirus-expressed human p53 protein was as described previously(14). Oligonucleotide probes used in the binding assays were labeled withT4 polynucleotide kinase (NEB) and [g-32P] ATP (.4500 Ci/mmol; Amer-sham, Piscataway, NJ). Labeled probes were purified using Pharmaciaspun columns according to the directions of the manufacturer. Oligonu-cleotide competition experiments were carried out with a fixed concen-tration of probe and 200-fold excess of nonlabeled competitor. Reactionswere loaded into a 5% nondenaturating polyacrylamide gels previouslyprerun for 15 min at 200 V. Electrophoresis was performed at 20 V/cm in22 mM Tris-borate buffer with 0.5 mM EDTA. Gels were dried and exposed
to film overnight at 270°C with an intensifying screen. Alternatively, driedgels were scanned using a PhosphorImager screen and analyzed usingImageQuant software (Molecular Dynamics, Sunnyvale, CA).
Immunoprecipitations. Antibodies were purchased from Santa CruzBiotechnology, except anti-b-actin, which was from Sigma Chemical Co.(St. Louis, MO). In immunoprecipitation studies, 2 3 107 cells werewashed twice for 10 min in 10 ml of methionine-free DMEM and incubatedfor 4 h at 37°C in 5 ml of new medium with 2.5 mCi of [35S]methionine(1175 Ci/mmol; NEN). Cells were collected by centrifugation and lysed in1 ml of immunoprecipitation buffer: PBS containing 1% Triton X-100,0.1% SDS, 1 mM sodium orthovanadate, 1 mM DTT, and 1 mM phenyl-methylsulfonyl fluoride, followed by centrifugation at 1500 3 g for 10 min.Cell lysates were incubated in the presence of 1 mg of the correspondingantibody at 4°C for 4 h followed by incubation for 1 h with proteinA/G-agarose (Santa Cruz Biotechnology). Immunoprecipitates were col-lected by centrifugation at 5000 3 g for 10 min, washed five times with 1ml of immunoprecipitation buffer, resuspended in SDS-PAGE samplebuffer, boiled for 5 min, and electrophoresed by 15% PAGE. Dried gelswere exposed to a PhosphorImager screen and analyzed using Image-Quant software (Molecular Dynamics).
Northern Blot Analysis. RNA was isolated from 13.5 p.c. mouseembryos or C2C12 cells using Trizol reagent (Life Technologies, Inc.). ForNorthern analysis, 30 mg of total RNA were electrophoresed in a 1.3%agarose/formaldehyde gel, visualized using ethidium bromide, transferredto nitrocellulose filters (Amersham), fixed by UV cross-linking, and bakedat 80°C for 1 h. For hybridizations, 3 3 106 cpm/ml of a random primed32P-labeled NcoI/SmaI M-PGAM cDNA fragment was used as a probe (5).The MCK and GAPDH sequences used for the generation of probes werefrom American Type Culture Collection. The ANF probe was as describedpreviously (73). Filters were hybridized at 42°C in 40% formamide with 63SSC, 23 Denhardt’s solution, 0.1% SDS, and 0.1 mg/ml denaturatedsalmon sperm DNA for 4 h, washed in 0.23 SSC at 60°C, and exposed toautoradiography film. Radioactive bands were also quantified using aPhosphorImager screen and ImageQuant software (Molecular Dynamics).
In Situ Hybridizations. On day 13.5 p.c., embryos were sacrificed,fixed in cold 4% paraformaldehyde, dehydrated through graded ethanolseries, and embedded in paraffin wax. Seven-mm-thick paraffin sectionswere mounted in polylysine-pretreated slides. Tissue sections were thendewaxed, rehydrated, and treated with acetic anhydride. Specimens werethen dehydrated and dried. In situ hybridizations were performed accord-ing to the method described in Lyons et al. (79). A Sm2 genomic fragmentof M-PGAM (5) was digested with NcoI and a 35S-UTP-labeled riboprobewas generated using T3 RNA polymerase. Hybridizations were carried outfor 16 h at 60°C. Slides were then washed and exposed to Ilford autora-diographic emulsion for 5 weeks, developed in D19 Kodak solution,counterstained with 0.2% toluidine blue solution, and mounted in Per-mount. Alternatively, slides were exposed for 3 days to Kodak Biomaxfilm.
AcknowledgmentsWe thank J. Jacobberger, J. Nagy, G. Pons, M. Rico, and B. Vogelstein forreagents and suggestions.
References1. Schultheiss, T. M., Xydas, S., and Lassar, A. B. Induction of aviancardiac myogenesis by anterior endoderm. Development (Camb.), 121:4203–4214, 1995.
2. Sugi, Y., and Lough, J. Anterior endoderm is a specific effector ofterminal cardiac myocyte differentiation of cells from the embryonic heartforming region. Dev. Dyn., 200: 155–162, 1994.
3. Olson, E. N., and Srivastava, D. Molecular pathways controlling heartdevelopment. Science (Washington DC), 272: 671–676, 1996.
4. Nakatsuji, Y., Hidaka, K., Tsujino, S., Yamamoto, Y., Mukai, T., Yanagi-hara, T., Kishimoto, T., and Sakoda, S. A single MEF-2 site is a majorpositive regulatory element required for transcription of the muscle-spe-cific subunit of the human phosphoglycerate mutase gene in skeletal andcardiac muscle cells. Mol. Cell. Biol., 12: 4384–4390, 1992.
5. Ruiz-Lozano, P., de Lecea, L., Buesa, C., Perez de la Osa, P., LePage,D., Gualberto, A., Walsh, K., and Pons, G. The gene encoding rat phos-phoglycerate mutase subunit M: cloning and promoter analysis in skeletalmuscle cells. Gene, 147: 243–248, 1994.
304 Regulation of Muscle Gene Expression by p53

6. Adamson, E. D. Isoenzyme transitions of creatine phosphokinase, al-dolase and phosphoglycerate mutase in differentiating mouse cells. J.Embryol. Exp. Morphol., 35: 355–367, 1976.
7. Weintraub, H., Hauschka, S., and Tapscott, S. J. The MCK enhancercontains a p53 responsive element. Proc. Natl. Acad. Sci. USA, 88:4570–4571, 1991.
8. Zambetti, G. P., Bargonetti, J., Walker, K., Prives, C., and Levine, A. J.Wild-type p53 mediates positive regulation of gene expression through aspecific DNA sequence element. Genes Dev., 6: 1143–1152, 1992.
9. Tamir, Y., and Bengal, E. p53 protein is activated during muscle dif-ferentiation and participates with MyoD in the transcription of musclecreatine kinase gene. Oncogene, 17: 347–356, 1998.
10. Zhao, J., Schmieg, F. I., Logsdon, N., Freedman, D., Simmons, D. T.,and Molloy, G. R. p53 binds to a novel recognition sequence in theproximal promoter of the rat muscle creatine kinase gene and activates itstranscription. Oncogene, 13: 293–302, 1996.
11. Momand, J., Zambetti, G. P., Olson, D. C., George, D., and Levine,A. J. The mdm-2 oncogene product forms a complex with the p53 proteinand inhibits p53-mediated transactivation. Cell, 69: 1237–1245, 1992.
12. Oliner, J. D., Kinzler, K. W., Meltzer, P. S., George, D. L., andVogelstein, B. Amplification of a gene encoding a p53-associated proteinin human sarcomas. Nature (Lond.), 358: 80–83, 1992.
13. Soddu, S., Blandino, G., Scardigli, R., Coen, S., Marchetti, A., Rizzo,M. G., Bossi, G., Cimino, L., Crescenzi, M., and Sacchi, A. Interferencewith p53 protein inhibits hematopoietic and muscle differentiation. J. CellBiol., 134: 193–204, 1996.
14. Chen, P. L., Chen, Y. M., Bookstein, R., and Lee, W. H. Geneticmechanisms of tumor suppression by the human p53 gene. Science(Washington DC), 250: 1576–1580, 1990.
15. Kern, S. E., Pietenpol, J. A., Thiagalingam, S., Seymour, A., Kinzler,K. W., and Vogelstein, B. Oncogenic forms of p53 inhibit p53-regulatedgene expression. Science (Washington DC), 256: 827–830, 1992.
16. Harvey, M., Vogel, H., Morris, D., Bradley, A., Bernstein, A., andDonehower, L. A. A mutant p53 transgene accelerates tumour develop-ment in heterozygous but not nullizygous p53-deficient mice. Nat. Genet.,9: 305–311, 1995.
17. Farmer, G., Bargonetti, J., Zhu, H., Friedman, P., Prywes, R., andPrives, C. Wild-type p53 activates transcription in vitro. Nature (Lond.),358: 83–86, 1992.
18. Schmale, H., and Bamberger, C. A novel protein with strong homol-ogy to the tumor suppressor p53. Oncogene, 15: 1363–1367, 1997.
19. Osada, M., Ohba, M., Kawahara, C., Ishioka, C., Kanamaru, R., Katoh,I., Ikawa, Y., Nimura, Y., Nakagawara, A., Obinata, M., and Ikawa, S.Cloning and functional analysis of human p51, which structurally andfunctionally resembles p53. Nat. Med., 4: 839–843, 1998.
20. Trink, B., Okami, K., Wu, L., Sriuranpong, V., Jen, J., and Sidransky,D. A new human p53 homologue. Nat. Med., 4: 747–748, 1998.
21. Bian, J., and Sun, Y. p53CP, a putative p53 competing protein thatspecifically binds to the consensus p53 DNA binding sites: a third memberof the p53 family? Proc. Natl. Acad. Sci. USA, 94: 14753–14758, 1997.
22. Jost, C. A., Marin, M. C., and Kaelin, W. G., Jr. p73 is a humanp53-related protein that can induce apoptosis. Nature (Lond.), 389: 191–194, 1997.
23. Kaghad, M., Bonnet, H., Yang, A., Creancier, L., Biscan, J. C., Valent,A., Minty, A., Chalon, P., Lelias, J. M., Dumont, X., Ferrara, P., McKeon, F.,and Caput, D. Monoallelically expressed gene related to p53 at 1p36, aregion frequently deleted in neuroblastoma and other human cancers.Cell, 90: 809–819, 1997.
24. Di Como, C. J., Gaiddon, C., and Prives, C. p73 function is inhibitedby tumor-derived p53 mutants in mammalian cells. Mol. Cell. Biol., 19:1438–1449, 1999.
25. Senoo, M., Seki, N., Ohira, M., Sugano, S., Watanabe, M., Inuzuka, S.,Okamoto, T., Tachibana, M., Tanaka, T., Shinkai, Y., and Kato, H. Asecond p53-related protein, p73L, with high homology to p73. Biochem.Biophys. Res. Commun., 248: 603–607, 1998.
26. Harris, A. N., Ruiz-Lozano, P., Chen, Y. F., Sionit, P., Yu, Y. T., Lilly,B., Olson, E. N., and Chien, K. R. A novel A/T-rich element mediates ANFgene expression during cardiac myocyte hypertrophy. J. Mol. Cell. Car-diol., 29: 515–525, 1997.
27. Zhu, H., Garcia, A. V., Ross, R. S., Evans, S. M., and Chien, K. R. Aconserved 28-base-pair element (HF-1) in the rat cardiac myosin light-
chain-2 gene confers cardiac-specific and a-adrenergic-inducible expres-sion in cultured neonatal rat myocardial cells. Mol. Cell. Biol., 11: 2273–2281, 1991.
28. Wang, Y., Reed, M., Wang, P., Stenger, J. E., Mayr, G., Anderson,M. E., Schwedes, J. F., and Tegtmeyer, P. p53 domains: identification andcharacterization of two autonomous DNA-binding regions. Genes Dev., 7:2575–2586, 1993.
29. Pavletich, N. P., Chambers, K. A., and Pabo, C. O. The DNA-bindingdomain of p53 contains the four conserved regions and the major muta-tion hot spots. Genes Dev., 7: 2556–2564, 1993.
30. Bargonetti, J., Manfredi, J. J., Chen, X., Marshak, D. R., and Prives, C.A proteolytic fragment from the central region of p53 has marked se-quence-specific DNA-binding activity when generated from wild-type butnot from oncogenic mutant p53 protein. Genes Dev., 7: 2565–2574, 1993.
31. Banks, L., Matlashewski, G., and Crawford, L. Isolation of human-p53-specific monoclonal antibodies and their use in the studies of humanp53 expression. Eur. J. Biochem., 159: 529–534, 1986.
32. Vojtesek, B., Bartek, J., Midgley, C. A., and Lane, D. P. An immuno-chemical analysis of the human nuclear phosphoprotein p53. New mono-clonal antibodies and epitope mapping using recombinant p53. J. Immu-nol. Methods, 151: 237–244, 1992.
33. Hupp, T. R., Meek, D. W., Midgley, C. A., and Lane, D. P. Regulationof the specific DNA binding function of p53. Cell, 71: 875–886, 1992.
34. Gannon, J. V., Greaves, R., Iggo, R., and Lane, D. P. Activatingmutations in p53 produce a common conformational effect. A monoclonalantibody specific for the mutant form. EMBO J., 9: 1595–1602, 1990.
35. Andres, V., Cusso, R., and Carreras, J. Distribution and developmen-tal transition of phosphoglycerate mutase and creatine phosphokinaseisozymes in rat muscles of different fiber-type composition. Differentia-tion, 41: 72–77, 1989.
36. Kim, K. K., Soonpaa, M. H., Daud, A. I., Koh, G. Y., Kim, J. S., andField, L. J. Tumor suppressor gene expression during normal and patho-logic myocardial growth. J. Biol. Chem., 269: 22607–22613, 1994.
37. Sah, V. P., Attardi, L. D., Mulligan, G. J., Williams, B. O., Bronson,R. T., and Jacks, T. A subset of p53-deficient embryos exhibit exen-cephaly. Nat. Genet., 10: 175–180, 1995.
38. Schmid, P., Lorenz, A., Hameister, H., and Montenarh, M. Expressionof p53 during mouse embryogenesis. Development (Camb.), 113: 857–865, 1991.
39. Armstrong, J. F., Kaufman, M. H., Harrison, D. J., and Clarke, A. R.High-frequency developmental abnormalities in p53-deficient mice. Curr.Biol., 5: 931–936, 1995.
40. Unger, T., Mietz, J. A., Scheffner, M., Yee, C. L., and Howley, P. M.Functional domains of wild-type and mutant p53 proteins involved intranscriptional regulation, transdominant inhibition, and transformationsuppression. Mol. Cell. Biol., 13: 5186–5194, 1993.
41. Hachiya, M., Chumakov, A., Miller, C. W., Akashi, M., Said, J., andKoeffler, H. P. Mutant p53 proteins behave in a dominant, negative fashionin vivo. Anticancer Res., 14: 1853–1859, 1994.
42. Chen, J. Y., Funk, W. D., Wright, W. E., Shay, J. W., and Minna, J. D.Heterogeneity of transcriptional activity of mutant p53 proteins and p53DNA target sequences. Oncogene, 8: 2159–2166, 1993.
43. Louis, J. M., McFarland, V. W., May, P., and Mora, P. T. The phos-phoprotein p53 is down-regulated post-transcriptionally during embryo-genesis in vertebrates. Biochim. Biophys. Acta, 950: 395–402, 1988.
44. Feinstein, E., Gale, R. P., Reed, J., and Canaani, E. Expression of thenormal p53 gene induces differentiation of K562 cells. Oncogene, 7:1853–1857, 1992.
45. Gerwin, B. I., Spillare, E., Forrester, K., Lehman, T. A., Kispert, J.,Welsh, J. A., Pfeifer, A. M., Lechner, J. F., Baker, S. J., Vogelstein, B., etal. Mutant p53 can induce tumorigenic conversion of human bronchialepithelial cells and reduce their responsiveness to a negative growthfactor, transforming growth factor b-1. Proc. Natl. Acad. Sci. USA, 89:2759–2763, 1992.
46. Kastan, M. B., Radin, A. I., Kuerbitz, S. J., Onyekwere, O., Wolkow,C. A., Civin, C. I., Stone, K. D., Woo, T., Ravindranath, Y., and Craig, R. W.Levels of p53 protein increase with maturation in human hematopoieticcells. Cancer Res., 51: 4279–4286, 1991.
47. Shaulsky, G., Goldfinger, N., and Rotter, V. Alterations in tumor de-velopment in vivo mediated by expression of wild-type or mutant p53proteins. Cancer Res., 51: 5232–5237, 1991.
305Cell Growth & Differentiation

48. Shaulsky, G., Goldfinger, N., Peled, A., and Rotter, V. Involvement ofwild-type p53 in pre-B-cell differentiation in vitro. Proc. Natl. Acad. Sci.USA, 88: 8982–8986, 1991.
49. Radinsky, R., Fidler, I. J., Price, J. E., Esumi, N., Tsan, R., Petty, C. M.,Bucana, C. D., and Bar-Eli, M. Terminal differentiation and apoptosis inexperimental lung metastases of human osteogenic sarcoma cells bywild-type p53. Oncogene, 9: 1877–1883, 1994.
50. Woodworth, C. D., Wang, H., Simpson, S., Alvarez-Salas, L. M., andNotario, V. Overexpression of wild-type p53 alters growth and differenti-ation of normal human keratinocytes but not human papillomavirus-expressing cell lines. Cell Growth Differ., 4: 367–376, 1993.
51. Donehower, L. A., Harvey, M., Slagle, B. L., McArthur, M. J., Mont-gomery, C. A., Jr., Butel, J. S., and Bradley, A. Mice deficient for p53 aredevelopmentally normal but susceptible to spontaneous tumours. Nature(Lond.), 356: 215–221, 1992.
52. Jacks, T., Remington, L., Williams, B. O., Schmitt, E. M., Halachmi, S.,Bronson, R. T., and Weinberg, R. A. Tumor spectrum analysis in p53-mutant mice. Curr. Biol., 4: 1–7, 1994.
53. Kaufman, M. H., Kaufman, D. B., Brune, R. M., Stark, M., Armstrong,J. F., and Clarke, A. R. Analysis of fused maxillary incisor dentition inp53-deficient exencephalic mice. J. Anat., 191: 57–64, 1997.
54. Baker, S. J., Markowitz, S., Fearon, E. R., Willson, J. K., andVogelstein, B. Suppression of human colorectal carcinoma cell growthby wild-type p53. Science (Washington DC), 249: 912–915, 1990.
55. Vogelstein, B., and Kinzler, K. W. p53 function and dysfunction. Cell,70: 523–526, 1992.
56. Finlay, C. A., Hinds, P. W., and Levine, A. J. The p53 proto-oncogenecan act as a suppressor of transformation. Cell, 57: 1083–1093, 1989.
57. Eliyahu, D., Michalovitz, D., Eliyahu, S., Pinhasi-Kimhi, O., and Oren,M. Wild-type p53 can inhibit oncogene-mediated focus formation. Proc.Natl. Acad. Sci. USA, 86: 8763–8767, 1989.
58. Michalovitz, D., Halevy, O., and Oren, M. Conditional inhibition oftransformation and of cell proliferation by a temperature-sensitive mutantof p53. Cell, 62: 671–680, 1990.
59. Levine, A. J., Momand, J., and Finlay, C. A. The p53 tumour suppres-sor gene. Nature (Lond.), 351: 453–456, 1991.
60. Holtzer, H., Schultheiss, T., Dilullo, C., Choi, J., Costa, M., Lu, M., andHoltzer, S. Autonomous expression of the differentiation programs of cellsin the cardiac and skeletal myogenic lineages. Ann. N.Y. Acad. Sci., 599:158–169, 1990.
61. Rumyantsev, P. P., and Borisov, A. DNA synthesis in myocytes fromdifferent myocardial compartments of young rats in norm, after experi-mental infarction and in vitro. Biomed. Biochim. Acta, 46: S610–S615,1987.
62. Clubb, F. J., Jr., and Bishop, S. P. Formation of binucleated myocar-dial cells in the neonatal rat. An index for growth hypertrophy. Lab. Invest.,50: 571–577, 1984.
63. Macleod, K. F., Sherry, N., Hannon, G., Beach, D., Tokino, T., Kinzler,K., Vogelstein, B., and Jacks, T. p53-dependent and independent expres-sion of p21 during cell growth, differentiation, and DNA damage. GenesDev., 9: 935–944, 1995.
64. Zhang, W., Behringer, R. R., and Olson, E. N. Inactivation of themyogenic bHLH gene MRF4 results in up-regulation of myogenin and ribanomalies. Genes Dev., 9: 1388–1399, 1995.
65. Lassar, A., and Munsterberg, A. Wiring diagrams: regulatory circuitsand the control of skeletal myogenesis. Curr. Opin. Cell Biol., 6: 432–442,1994.
66. Levine, A. J., Wu, M. C., Chang, A., Silver, A., Attiyeh, E. F., Lin, J., andEpstein, C. B. The spectrum of mutations at the p53 locus. Evidence fortissue-specific mutagenesis, selection of mutant alleles, and a “gain offunction” phenotype. Ann. N.Y. Acad. Sci., 768: 111–128, 1995.
67. Frazier, M. W., He, X., Wang, J., Gu, Z., Cleveland, J. L., and Zambetti,G. P. Activation of c-myc gene expression by tumor-derived p53 mutantsrequires a discrete C-terminal domain. Mol. Cell. Biol., 18: 3735–3743,1998.
68. Gualberto, A., Hixon, M. L., Finco, T. S., Perkins, N. D., Nabel, G. J.,and Baldwin, A. S., Jr. A proliferative p53-responsive element mediatestumor necrosis factor a induction of the human immunodeficiency virustype 1 long terminal repeat. Mol. Cell. Biol., 15: 3450–3459, 1995.
69. Gualberto, A., LePage, D., Pons, G., Mader, S. L., Park, K., Atchison,M. L., and Walsh, K. Functional antagonism between YY1 and the serumresponse factor. Mol. Cell. Biol., 12: 4209–4214, 1992.
70. Newman, J. R., Morency, C. A., and Russian, K. O. A novel rapidassay for chloramphenicol acetyltransferase gene expression. Biotech-niques, 5: 444–448, 1987.
71. Vincent, C. K., Gualberto, A., Patel, C. V., and Walsh, K. Differentregulatory sequences control creatine kinase-M gene expression in di-rectly injected skeletal and cardiac muscle. Mol. Cell. Biol., 13: 1264–1272, 1993.
72. Hemsley, A., Arnheim, N., Toney, M. D., Cortopassi, G., and Galas,D. J. A simple method for site-directed mutagenesis using the polymerasechain reaction. Nucleic Acids Res., 17: 6545–6551, 1989.
73. Knowlton, K. U., Baracchini, E., Ross, R. S., Harris, A. N., Henderson,S. A., Evans, S. M., Glembotski, C. C., and Chien, K. R. Co-regulation ofthe atrial natriuretic factor and cardiac myosin light chain-2 genes duringa-adrenergic stimulation of neonatal rat ventricular cells. Identification ofcis sequences within an embryonic and a constitutive contractile proteingene which mediate inducible expression. J. Biol. Chem., 266: 7759–7768, 1991.
74. el-Deiry, W. S., Tokino, T., Velculescu, V. E., Levy, D. B., Parsons, R.,Trent, J. M., Lin, D., Mercer, W. E., Kinzler, K. W., and Vogelstein, B.WAF1, a potential mediator of p53 tumor suppression. Cell, 75: 817–825,1993.
75. Kern, S. E., Kinzler, K. W., Bruskin, A., Jarosz, D., Friedman, P.,Prives, C., and Vogelstein, B. Identification of p53 as a sequence-specificDNA-binding protein. Science (Washington DC), 252: 1708–1711, 1991.
76. Morgenstern, J. P., and Land, H. Advanced mammalian gene transfer:high titre retroviral vectors with multiple drug selection markers and acomplementary helper-free packaging cell line. Nucleic Acids Res., 18:3587–3596, 1990.
77. Christy, R. J., Yang, V. W., Ntambi, J. M., Geiman, D. E., Landschulz,W. H., Friedman, A. D., Nakabeppu, Y., Kelly, T. J., and Lane, M. D.Differentiation-induced gene expression in 3T3–L1 preadipocytes:CCAAT/enhancer binding protein interacts with and activates the promot-ers of two adipocyte-specific genes. Genes Dev., 3: 1323–1335, 1989.
78. Nabel, G., and Baltimore, D. An inducible transcription factor acti-vates expression of human immunodeficiency virus in T cells. Nature(Lond.), 326: 711–713, 1987.
79. Lyons, G. E., Micales, B. K., Schwarz, J., Martin, J. F., and Olson,E. N. Expression of mef2 genes in the mouse central nervous systemsuggests a role in neuronal maturation. J. Neurosci., 15: 5727–5738,1995.
306 Regulation of Muscle Gene Expression by p53