part 1, section 3 applications of stenotrophomonas...
TRANSCRIPT

PART 1, SECTION 3
Applications of Stenotrophomonas maltophilia Lipase in the
Preparation of Enantiomerically Enriched Compounds

Part I, Section 3, Resolution of 2-arylpropanoates:Results
In the previous section, we have described the characterization of a new lipase
from Stenotrophomonas maltophilia. In this section, we describe the applications of
Stenotrophomonas maltophilia in the preparation of enantiomerically enriched
compounds on preparative scale. We decided to explore biocatalysis by whole cells of
Stenotrophomonas maltophilia for the following reasons:
(i) The lipase has been shown to be present on the surface of the cells of
Stenotrophomonas maltophilia (Section 1.2.5). The presence of an
enantioselective lipase on the surface of a cell is a highly desirable
property from practical point of view as it eliminates the need for
expensive isolation, purification and stabilization of the protein. Moreover,
the penetration of the substrate into the cell and excretion of the product
into the media is unlikely to be an issue when lipase is present on the
surface of the cell.
(ii) The disadvantages of using whole cell biocatalysis include reduction in
e. e. because of the presence of other intracellular enzymes of opposite or
relaxed specificity. But the lipase of Stenotrophomonas maltophilia has
been shown to be present on the surface of the cells, we, therefore,
envisaged that intracellular esterases, even if present, are unlikely to
compete with the surface bound enzyme.
Several publications in the recent past have described the expression of known
lipases on the surface of fusion proteins where several expression systems have been
developed to display lipases and other proteins on the surface of bacteria, fungi or
mammalian cells by appropriately fusing them to surface anchoring motifs. 132-139 The
outer membrane proteins have been widely used as anchoring motifs, because they
have unique membrane-spanning structures. Several membrane proteins including
OmpA, OprF, OmpS, invasin, LamB, PhoE, OmpC and FadL have been used as
anchoring motifs. 139'147
'148
Traditional substrates, 2-methyl and 2-hydroxy substituted aromatic esters
were selected for testing the substrate specificity of the enzyme. In addition, aliphatic
ester, ethyl2-chloropropanoate was also tested.
1.3.1 Stenotrophomonas maltophilia catalyzed kinetic resolution of 2-
arylpropanoates: formation of enantiomerically pure (S)-acids
We started our investigation by studying the biocatalyzed hydrolysis of methyl
2-( 4-isobutylphenyl)propanoate (ibuprofen methyl ester; 39). Stenotrophomonas
38

Part 1, Section 3, Resolution of2-arylpropanoates:Results
maltophilia was grown in medium comprising of peptone (1.0%) and beef extract
(0.5%) at 30 oc for I8-22 has described in experimental section. Cells were harvested
by centrifugation and washed with phosphate buffer (50 mM, pH 7.0). The washed
cells were resuspended in IOO mL of same buffer at a concentration of 0.1 g/mL. To
the cell suspension, racemic methyl 2-( 4-isobutylphenyl)propanoate (39) at 20 mM
concentration was added and the contents incubated at 37 °C on an orbital shaker at
200 rpm. The reaction was stopped at 48% conversion (28 h; Scheme I4). Progress of
the reaction was monitored by TLC. pH of the reaction mixture was adjusted to 8.0
with IN NaOH. The remaining ester was removed by extraction with ethyl acetate.
The aqueous layer was acidified to pH 2.0 with IN HCl and extracted with ethyl
acetate to recover acid formed during the reaction. The organic layer was separated,
dried over sodium sulphate and evaporated to give 40 in 45% yield, which was
purified by column chromatography over silica-gel (200-400 mesh; ethyl
acetate/hexane, 2:3).
Scheme 14
39 (S)-40, e. e. - 97% (R)-39
The structure of 40 was in agreement with its 1HNMR spectral data. The
absence of singlet for -OCH3 protons at 8 3.64 in the 1HNMR of the product
confirmed that ester hydrolysis has occurred. 2-methyl protons and single proton at C-
2 appeared as doublet (J=7.2 Hz) at 8 1.55 and quartet (J=7.2 Hz) at 8 3.73,
respectively. Single methine proton and two methyl groups of the isobutyl were
present as multiplet at 8 1.84-1.97 and doublet (J=7.2 Hz) at 8 0.96, respectively. The
aromatic protons appeared as multiplet at 8 7.03-7.43, whereas methylene protons
appeared as doublet (J=7.2 Hz) at 8 2.51. Except for methine proton of isobutyl
group, all protons resonated slightly downfield as compared to ester 39. The presence
of acid was also confirmed by IR which showed carbonyl of carboxylic acid at I7I 0
cm-1• IR and 1HNMR of the product and racemic 2-(4-isobutylphenyl)propanoic acid
were indistinguishable.
39
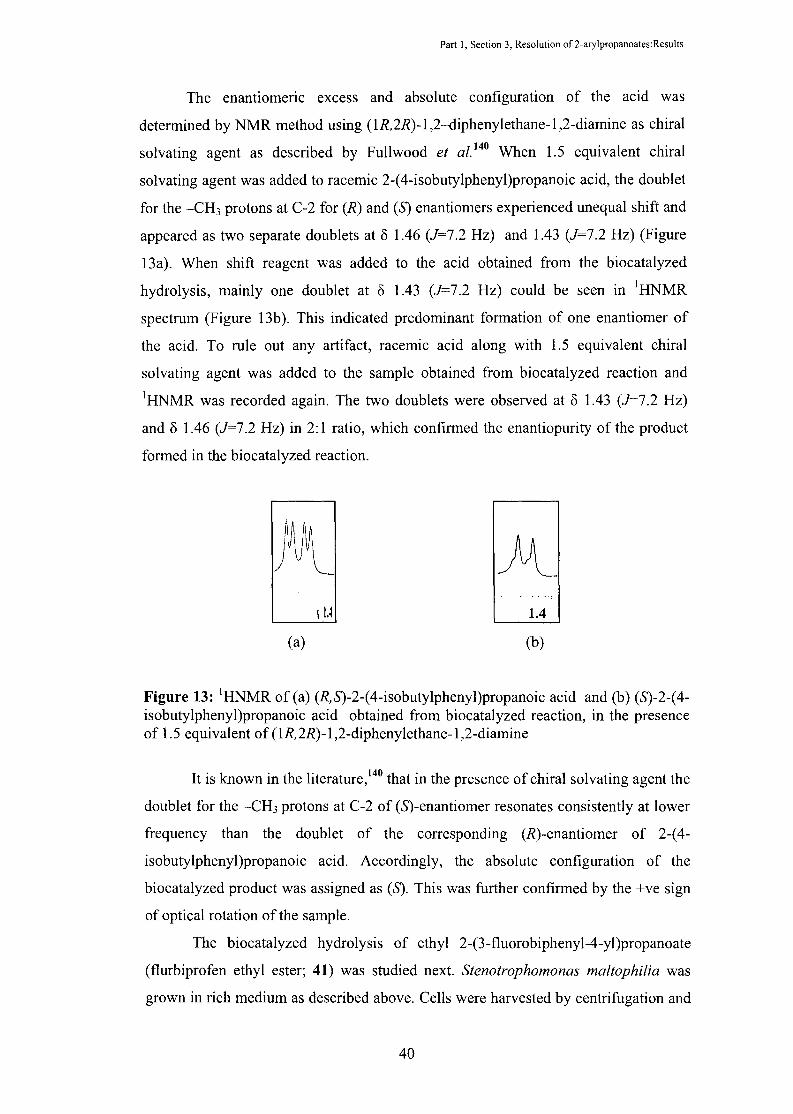
Part I, Section 3, Resolution of 2-arylpropanoates:Results
The enantiomeric excess and absolute configuration of the acid was
determined by NMR method using (1R,2R)-1 ,2--diphenylethane-1 ,2-diamine as chiral
solvating agent as described by Fullwood et a/. 140 When 1.5 equivalent chiral
solvating agent was added to racemic 2-(4-isobutylphenyl)propanoic acid, the doublet
for the -CH3 protons at C-2 for (R) and (S) enantiomers experienced unequal shift and
appeared as two separate doublets at 8 1.46 (J=7.2 Hz) and 1.43 (J=7.2 Hz) (Figure
13a). When shift reagent was added to the acid obtained from the biocatalyzed
hydrolysis, mainly one doublet at 8 1.43 (J=7.2 Hz) could be seen in 1HNMR
spectrum (Figure 13b ). This indicated predominant formation of one enantiomer of
the acid. To rule out any artifact, racemic acid along with 1.5 equivalent chiral
solvating agent was added to the sample obtained from biocatalyzed reaction and 1HNMR was recorded again. The two doublets were observed at 8 1.43 (J=7.2 Hz)
and 8 1.46 (J=7.2 Hz) in 2:1 ratio, which confirmed the enantiopurity of the product
formed in the biocatalyzed reaction.
1.4
(a) (b)
Figure 13: 1HNMR of(a) (R,S)-2-(4-isobutylphenyl)propanoic acid and (b) (S)-2-(4-isobutylphenyl)propanoic acid obtained from biocatalyzed reaction, in the presence of 1.5 equivalent of (1R,2R)-1 ,2-diphenylethane-1 ,2-diamine
It is known in the literature/ 40 that in the presence of chiral solvating agent the
doublet for the -CH3 protons at C-2 of (S)-enantiomer resonates consistently at lower
frequency than the doublet of the corresponding (R)-enantiomer of 2-( 4-
isobutylphenyl)propanoic acid. Accordingly, the absolute configuration of the
biocatalyzed product was assigned as (S). This was further confirmed by the +ve sign
of optical rotation of the sample.
The biocatalyzed hydrolysis of ethyl 2-(3-fluorobiphenyl-4-yl)propanoate
(flurbiprofen ethyl ester; 41) was studied next. Stenotrophomonas maltophilia was
grown in rich medium as described above. Cells were harvested by centrifugation and
40

Part I, Section 3, Resolution of2-arylpropanoates:Results
washed with phosphate buffer (50 mM, pH 7.0). The washed cells were resuspended
in 100 mL of same buffer at a concentration ofO.l g/mL. To the cell suspension, ethyl
2-(3-fluorobiphenyl-4-yl)propanoate (41) at 20 mM concentration was added and the
contents incubated at 37 °C in an orbital shaker at 200 rpm. The reaction was stopped
at 48% conversion after 29 h (Scheme 15). The desired acid 42 formed in the reaction
was separated from the ester according to the procedure described above for methyl 2-
( 4-isobutylphenyl)propanoate (39) and purified by column chromatography over
silica-gel (200-400 mesh; ethyl acetate/hexane, 1 :3).
Scheme 15
41 (S)-42, e.e. > 99% (R)-41
The structure of the product was in agreement with its 1 HNMR spectral data.
The absence of a triplet (J=7.2 Hz) at 8 1.27 and quartet (J=7.2 Hz) at 8 4.2
corresponding to ester group of 41 confirmed that the hydrolysis had occurred. 2-
methyl protons and single proton at C-2 appeared as doublet (J=7.2 Hz) at 8 1.55 and
quartet (J=7.2 Hz) at 8 3.80, respectively. The aromatic protons appeared as multiplet
at 8 7.40-7.53. The presence of acid was also confirmed by IR which showed carboxyl
carbonyl group at 1698 cm-1•
The e.e. and the absolute configuration of acid was determined by NMR
method using (1R,2R)-1 ,2-diphenylethane-1 ,2-diamine as chiral solvating agent as
described. 140 When 1.5 equivalent shift reagent was added to racemic 2-(3-
fluorobiphenyl-4-yl)propanoic acid, the doublet for the -CH3 protons at C-2 for (R)
and (S) enantiomers experienced unequal shift and appeared as two separate doublets
at 8 1.46 (J=7.2 Hz) and 1.48 (J=7.2 Hz) (Figure 14a). When shift reagent was added
to the acid obtained from the biocatalyzed hydrolysis, only one doublet at 8 1.46
(J=7.2 Hz) could be seen in 1HNMR spectrum (Figure 14b). This indicated
predominant formation of one enantiomer of the acid. To rule out any artifact,
racemic acid along with 1.5 equivalent solvating reagent was added to the sample
obtained from biocatalyzed reaction and 1 HNMR was recorded again. The two
41
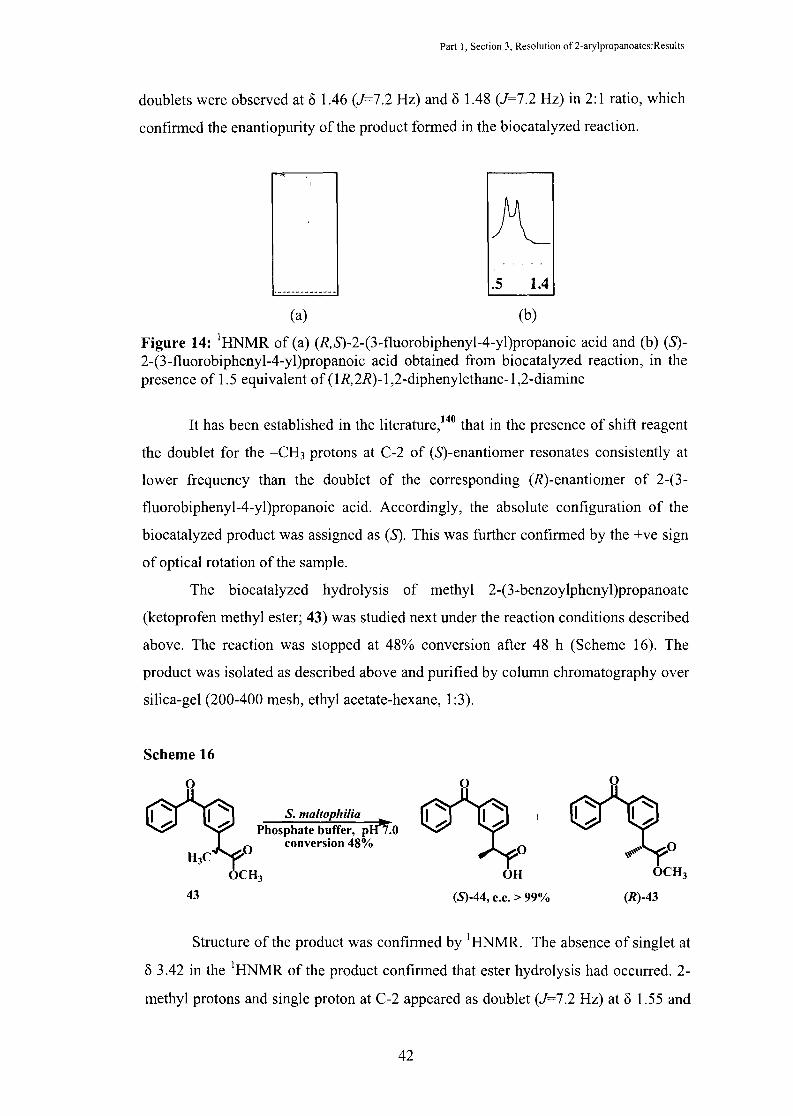
Part I, Section 3, Resolution of 2-arylpropanoates:Results
doublets were observed at o 1.46 (J=7.2 Hz) and o 1.48 (J=7.2 Hz) in 2:1 ratio, which
confirmed the enantiopurity of the product formed in the biocatalyzed reaction .
(a)
. s 1.4
(b)
Figure 14: 1HNMR of (a) (R,S)-2-(3-fluorobiphenyl-4-yl)propanoic acid and (b) (S)-2-(3-fluorobiphenyl-4-yl)propanoic acid obtained from biocatalyzed reaction, in the presence of 1.5 equivalent of (1R,2R)-1 ,2-diphenylethane-1 ,2-diamine
It has been established in the literature/40 that in the presence of shift reagent
the doublet for the -CH3 protons at C-2 of (S)-enantiomer resonates consistently at
lower frequency than the doublet of the corresponding (R)-enantiomer of 2-(3-
fluorobiphenyl-4-yl)propanoic acid. Accordingly, the absolute configuration of the
biocatalyzed product was assigned as (S). This was further confirmed by the +ve sign
of optical rotation of the sample.
The biocatalyzed hydrolysis of methyl 2-(3-benzoylphenyl)propanoate
(ketoprofen methyl ester; 43) was studied next under the reaction conditions described
above. The reaction was stopped at 48% conversion after 48 h (Scheme 16). The
product was isolated as described above and purified by column chromatography over
silica-gel (200-400 mesh, ethyl acetate-hexane, 1 :3).
Scheme 16
43
S. maltophilia Phosphate buffer, pi/7.o
conversion 48%
0 0
+
0
OH OCH3
(S)-44, e.e. > 99% (R)-43
Structure of the product was confirmed by 1 HNMR. The absence of singlet at
o 3.42 in the 1HNMR of the product confirmed that ester hydrolysis had occurred. 2-
methyl protons and single proton at C-2 appeared as doublet (J=7.2 Hz) at o 1.55 and
42

Part I, Section 3, Resolution of2-arylpropanoates:Results
quartet (J=7.2 Hz) at 8 3.78, respectively. The aromatic protons appeared as multiplet
at 8 7.19-7. 81. The presence of acid was also confirmed by IR which showed carboxyl
carbonyl group at 1697 crn-1.
The e.e. and absolute configuration of the acid was determined by NMR
rnethod. 140 When 1.5 equivalent (1R,2R)-1 ,2-diphenylethane-1 ,2-diarnine was added
to racemic 2-(3-benzoylphenyl)propanoic acid, the doublet for the -CH3 protons at C-
2 for (R) and (S) enantiomers experienced unequal shift and appeared as two separate
doublets at 8 1.43 (J=7.2 Hz) and 8 1.47 (J=7.2 Hz) (Figure 15a). When shift reagent
was added to the acid obtained from the biocatalyzed hydrolysis, only one doublet at 8
1.43 (J=7.2 Hz) could be seen in 1HNMR spectrum (Figure 15b). This indicated
predominant formation of one enantiomer of the acid. To rule out any artifact,
racemic acid along with 1.5 equivalent of chiral solvating agent was added to the
sample obtained from biocatalyzed reaction and 1HNMR was recorded again. The two
doublets were observed at 8 1.43 (J=7.2 Hz) and 8 1.47 (J=7.2 Hz) in 2:1 ratio, which
confirmed the enantiopurity of the product formed in the biocatalyzed reaction.
(a) (b)
Figure 15: 1HNMR of (a) (R,S)-2-(3-benzoylphenyl)propanoic acid and (b) (S)-2-(3-benzoylphenyl)propanoic acid obtained from biocatalyzed reaction, in the presence of 1. 5 equivalent of ( 1R, 2R)-1 ,2-diphenylethane-1 ,2-diamine
It has been established, 140 that in the presence of shift reagent the doublet for
the -CH3 protons at C-2 of (S)-enantiomer resonates consistently at lower frequency
than the doublet of the corresponding (R)-enantiomer of 2-(3-
benzoylphenyl)propanoic acid. Thus, the absolute configuration of the biocatalyzed
product was assigned as (S). This was further confirmed by the +ve sign of optical
rotation of the sample.
43

Part I, Section 3, Resolution of2-arylpropanoates:Results
1.3.2 Stenotrophomonas maltophilia catalyzed kinetic resolution of 2-
arylpropanoates: formation of enantiomerically pure (R)-acids
In the preceding section, we have described enantioselective formation of (S)
acids from the esters of 2-arylpropanoic acids. In this section, we describe the
formation of corresponding (R)-acids using the cells of same organism, but afte:r
treatment with acetone. Acetone treated cells were obtained by following method.
Stenotrophomonas maltophilia was grown as described in Section 1.3.1. The cells
were harvested by centrifugation and washed twice with phosphate buffer (pH 7.0, 50
mM). 10 g cells were then resuspended in 100 mL acetone and incubated at 30 °C for
30 min. Acetone was removed by filtration. Acetone treatment was repeated 4 times.
Finally, cells were dried under vacuum, lyophilized and stored at 4 °C.
As first example, biocatalyzed hydrolysis of methyl 2-( 4-isobutylphenyl)
propanoate (39) was studied. The acetone treated cells (1 g) were suspended in the
100 mL of phosphate buffer (pH 7.0, 50 mM). To the cell suspension, methyl 2-(4-
isobutylphenyl)propanoate (39) at 20 mM concentration was added and the contents
incubated at 37 °C in an orbital shaker at 200 rpm. The reaction was stopped at 48%
conversion after 22 h (Scheme 1 7). The desired acid was isolated, purified and
analyzed as described in Section 1.3 .1.
Scheme 17
Acetone treated S. maltophilij ~ IClf OCH3 Phosphate buffer, pH 7.0 : OH + OCH3
conversion 48% ~
39 (R)-40, e.e. > 99% (S)-39
The e.e. and absolute configuration of the acid was determined by NMR
method.140 As shown above (Section 1.3 .1 ), the 2-CH3 protons of racemic 40 in the
presence of 1.5 equivalent chiral solvating agent appeared as two doublets (J= 7.2
Hz) at 8 1.43 and 8 1.46 (Figure 16a) When shift reagent was added to the acid
obtained from the biocatalyzed hydrolysis, only one doublet (1=7.2 Hz) at 8 1.46
could be seen in 1HNMR spectrum (Figure 16b). This indicated predominant
formation of one enantiomer of the acid. To rule out any artifact, racemic acid along
with 1.5 equivalent chiral solvating agent was added to the sample obtained from
44

Part I, Section 3, Resolution of2-arylpropanoates:Results
biocatalyzed reaction and 1HNMR was recorded agam. The two doublets were
observed at o 1.43 (1=7.2 Hz) and o 1.46 (1=7.2 Hz) in 1:2 ratio, which confirmed the
enantiopurity of the product formed in the biocatalyzed reaction.
qf )I I \ ~ I v ~ ! 1 I I V'' ) l_
(a)
1.4
(b)
Figure 16: 1HNMR of(a) (R,S)-2-(4-isobutylphenyl)propanoic acid and (b) (R)-2-(4-isobutylphenyl)propanoic acid obtained from biocatalyzed reaction, in the presence of 1.5 equivalent of (1R,2R)-1 ,2-diphenylethane-1 ,2-diamine
As stated earlier, it has been established!40 that in the presence of shift reagent
the doublet for the -CH3 protons at C-2 of (S)-enantiomer resonates consistently at
lower frequency than the doublet of the corresponding (R)-enantiomer of 2-( 4-
isobutylphenyl)propanoic acid. Accordingly, the absolute configuration of the
biocatalyzed product was assigned as (R). This was further confirmed by the -ve sign
of optical rotation of the sample.
Biocatalyzed hydrolysis of ethyl 2-(3-fluorobiphenyl-4-yl)propanoate (41)
with acetone treated cells was studied next under the reaction conditions described
above (Scheme 18). The reaction was stopped at 48% conversion after 48 h. The
desired acid 42 was isolated and purified as described in Section 1.3.1. The structure
of the product was confirmed by 1 HNMR as described before.
Scheme 18
41
Acetone treated S. ma/tophilia._ Phosphate buffer, pH 7.0
conversion 48%
(R)-42, e. e. > 99% (S)-41
The e.e. and absolute configuration of the acid was determined by NMR
method. 140 As described above (Section 1.3 .1 ), the 2-CH3 protons of racemic 42 in the
presence of 1.5 equivalent chiral solvating agent appeared as two doublets (1=7.2 Hz)
at o 1.46 and o 1.48 (Figure 17a). When shift reagent was added to the acid obtained
45

Part I, Section 3, Resolution of 2-arylpropanoates:Results
from the biocatalyzed hydrolysis, only one doublet at 8 1.48 (J=7.2 Hz) could be seen
in 1HNMR spectrum (Figure 17b). To rule out any artifact, racemic acid along with
1.5 equivalent chiral solvating agent was added to the sample obtained from
biocatalyzed reaction and 1 HNMR was recorded again. The two doublets were
observed at 8 1.46 (J=7.2 Hz) and 8 1.48 (J=7.2 Hz) in 1:2 ratio, which confirmed the
enantiopurity of the product formed in the biocatalyzed reaction.
I '
r 0
' 1\
J 'J\ 1.5 .)
(a) (b)
Figure 17: 1HNMR of (a) (R,S)-2-(3-fluorobiphenyl-4-yl)propanoic acid and (b) (R)-2-(3-fluorobiphenyl-4-yl)propanoic acid obtained from biocatalyzed reaction, in the presence of 1.5 equivalent of (lR, 2R)-l ,2-diphenylethane-1 ,2-diamine
As already stated, it has been established, 140 that in the presence of shift
reagent the doublet for the -CH3 protons at C-2 of (S)-enantiomer resonates
consistently at lower frequency than the doublet of the corresponding (R)-enantiomer
of 2-(3-fluorobiphenyl-4-yl)propanoic acid. Accordingly, the absolute configuration
of the biocatalyzed product was assigned as (R). This was further confirmed by the -
ve sign of optical rotation of the sample.
The biocatalyzed hydrolysis of methyl 2-(3-benzoylphenyl)propanoate (43)
was studied next. Reaction conditions were same as described above, the reaction was
stopped at 48% conversion after 14 h (Scheme 19). The desired acid 44 was isolated
and purified as described in Section 1.3 .1. The structure of the product was confirmed
by 1 HNMR as described before.
Scheme 19
43
Acetone treated S. maltophilia,.
Phosphate buffer, pH 7.0 conversion 48%
46
+
0
H
(R)-44, e.e. > 99% (S)-43

Part I, Section 3, Resolution of2-arylpropanoates:Results
The e.e. and absolute configuration of the acid was determined by NMR
method. 14° From the NMR spectral data (Figure 18b) it was clear that only one
enantiomer of the acid was formed. As mentioned in literature, 140 in the presence of
shift reagent the doublet for the -CH3 protons at C-2 of (S)-enantiomer resonates
consistently at lower frequency than the doublet of the corresponding (R)-enantiomer
of 2-(3-benzoylphenyl)propanoic acid. Accordingly, the absolute configuration of the
biocatalyzed product was assigned as (R). This was further confirmed by the -ve sign
of optical rotation of the sample.
1.4 (a) (b)
Figure 18: 1HNMR of (a) (R,S)-2-(3-benzoylphenyl)propanoic acid and (b) (R)-2-(3-benzoylphenyl)propanoic acid obtained from biocatalyzed reaction, in the presence of 1.5 equivalent of (1R,2R)-1 ,2-diphenylethane-1 ,2-diamine
1.3.3 Formation of (R)-2-arylpropanoic acids in >99% e.e. at 100% conversion
with acetone treated cells of Stenotrophomonas maltophilia: Dynamic
Kinetic Resolution
As described in the preceding section, untreated whole cells of
Stenotrophomonas maltophilia enantioselectively hydrolyzed (S)-enantiomers of
methyl 2-( 4-isobutylphenyl)propanoate (39), ethyl 2-(3-fluorobiphenyl-4-
yl)propanoate (41) and methyl 2-(3-benzoylphenyl)propanoate (43), whereas acetone
treated cells of Stenotrophomonas maltophilia enantioselectively hydrolyzed (R)
enantiomers of these esters. In both the cases, the reaction was deliberately stopped at
<50% conversion to obtain high e.e. of the products. However, we were pleasantly
surprised to note that (i) the hydrolysis reaction of methyl 2-( 4-
isobutylphenyl)propanoate (39) with acetone treated cells proceeded to 100%
conversion and (ii) at 100% conversion, the e.e. of the product was >99% and the
configuration was (R). The reaction was done under same conditions as described
above for acetone treated cells, only time-period of the reaction was increased to 80-
90 h (Scheme 20). The e.e. and configuration of the product was determined as
described above. The other two esters, ethyl 2-(3-fluorobiphenyl-4-yl)propanoate
47
l
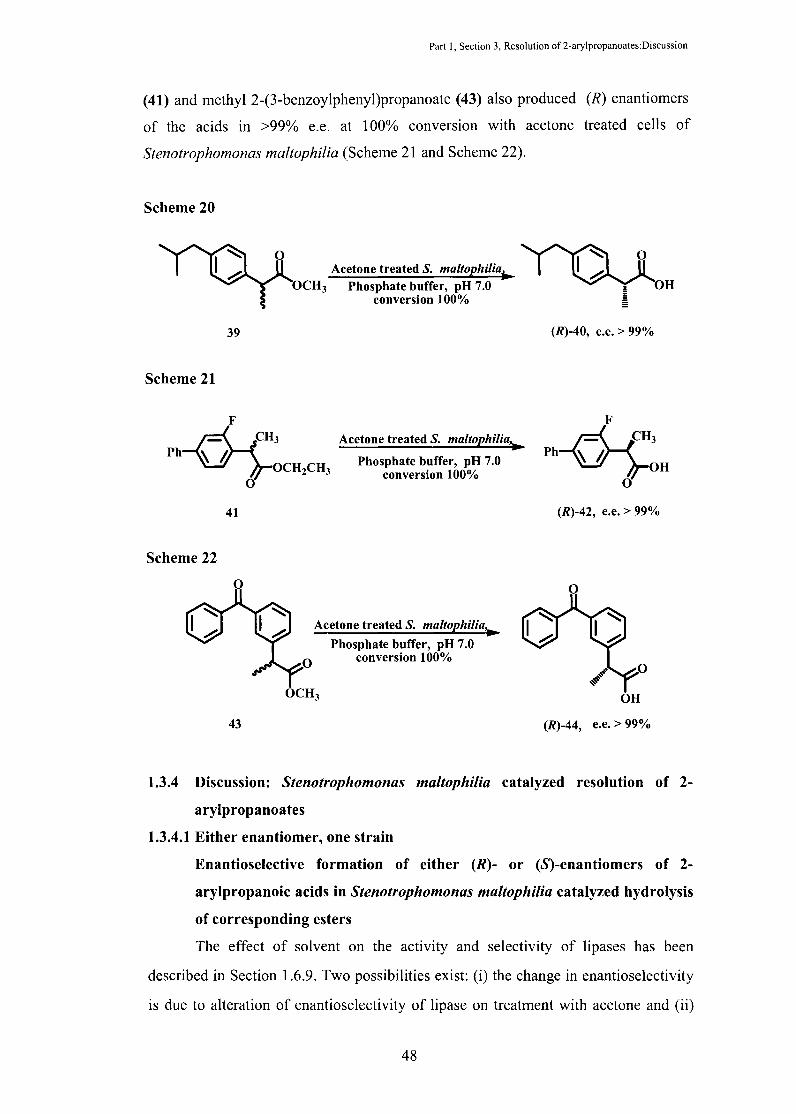
Part I, Section 3, Resolution of2-arylpropanoates:Discussion
(41) and methyl 2-(3-benzoylphenyl)propanoate (43) also produced (R) enantiomers
of the acids in >99% e.e. at 100% conversion with acetone treated cells of
Stenotrophomonas maltophilia (Scheme 21 and Scheme 22).
Scheme 20
39
Scheme 21
41
Scheme 22
43
Acetone treated S. maltophilia• OCH3 Phosphate buffer, pH 7.0
conversion 100%
Acetone treated S. maltophilia.._
Phosphate buffer, pH 7.0 conversion 100%
Acetone treated S. maltophilia•
Phosphate buffer, pH 7.0 conversion 100%
IGlJ_ : OH s 5
(R)-40, e.e. > 99%
(R)-42, e.e. > 99%
OH
(R)-44, e.e. > 99%
1.3.4 Discussion: Stenotrophomonas maltophilia catalyzed resolution of 2-
arylpropanoates
1.3.4.1 Either enantiomer, one strain
Enantioselective formation of either (R)- or (S)-enantiomers of 2-
arylpropanoic acids in Stenotrophomonas maltophilia catalyzed hydrolysis
of corresponding esters
The effect of solvent on the activity and selectivity of lipases has been
described in Section 1.6.9. Two possibilities exist: (i) the change in enantioselectivity
is due to alteration of enantioselectivity of lipase on treatment with acetone and (ii)
48

Part I, Section 3, Resolution of 2-arylpropanoates:Discussion
more than one enzyme with opposite selectivity are present but with greatly different
rates of hydrolysis. Acetone treatment inactivates enzyme for major activity, but has
very little or no effect on the enzyme for minor activity. But none of these appears to
be operative in our case for following reasons (i) the lipase has been purified as
described in Section 1.2.3. The pure lipase behaved in a manner similar to acetone
untreated cells of Stenotrophomonas maltophilia and exhibited enantioselectivity for
(S)-enantiomer. Treatment of pure enzyme with acetone did not result in loss of
activity or change in selectivity and (ii) the rate of reaction after treatment of
Stenotrophomonas maltophilia cells with acetone remained almost similar, only the
enantioselectivity was reversed.
The most likely explanation for the result is as follows. In Section 1.2.5 above,
the presence of a 22 kDa lipase on the surface of the cells of Stenotrophomonas
maltophilia was established by immunogold labelling studies, carried out with ultra
thin sections of Stenotrophomonas maltophilia. We have also confirmed that the pure
enzyme is enantioselective for the (S)-enantiomer of all the three esters of 2-
arylpropanoic acids tested.
Repeated treatment with acetone removes the surface bound (S)-selective
lipase from the cells. The presence of 22 kDa lipase in acetone washing was
confirmed by SDS-PAGE and activity assay. This observation is consistent with the
fact that the lipase can be extracted from the cells with 1M NaCl (Section 1.2.3). In
addition, the acetone treatment increases the penetration of substrates into the cells by
increasing the permeability of the outer membrane. This allows the substrate to
experience the activity of other intracellular enzymes. The formation of (R)
enantiomer of 2-arylpropanonic acid, in all probability is due to the presence of an
(R)-selective esterase in the cells of Stenotrophomonas maltophilia. In brief, (i)
untreated cells of Stenotrophomonas maltophilia produced (S)-acids due to the
presence of an (S)-selective lipase on the surface of the cells, (ii) Acetone treatment
removes this surface bound enzyme and increases the permeability of the outer
membrane and (iii) acetone treated cells produced (R)-acids due to the presence of an
intracellular (R)-selective esterase.
49

Part I, Section 3, Resolution of 2-arylpropanoates:Discussion
1.3.4.2 Formation of (R)-enantiomers at 100% conversion in Stenotrophomonas
maltophilia catalyzed dynamic kinetic resolution
Kinetic resolution of racemates can proceed to a maxtmum of 50%
conversion. In case of acetone treated cells high e.e. of acids was obtained at 1 00%
conversion. The yield of isolated product was 80-90%, which rules out the selective
degradation of one enantiomer. Another process called Dyanamic Kinetic Resolution
(DKR) is known in literature, in which enzymatic kinetic resolution is combined with
in situ racemization of substrate with a base, a metal, metal catalyst or an enzyme.
The details of DKR have been discussed in Section 1.6.2.
Another technique called deracemization reaction, which provides with a
route to obtain the enantiomerically pure compounds, theoretically in 100% yield
starting from the racemic mixture has been reported. In this reaction, synthesis of a
racemate is almost equal to the synthesis of the optically active compound, and this
concept is entirely different from the commonly accepted one in the asymmetric
synthesis. In this deracemization process, the chirality of either enantiomer of a
racemate is converted to the other antipode via a single or plural reactions, resulting in
an optically active compound starting from a racemic mixture as a whole. In total,
successful application of this process means that the synthesis of a racemate is almost
equal to the synthesis of an optically active compound. Most of the reported
deracemization reactions utilize oxidation-reduction process. A representative
example is the deracemization of secondary alcohols, the key being the combination
of two enzymes that catalyze the oxidation of one enantiomer followed by
enantioselective reduction to the antipode.149 Turner and co-workers reported the
deracemization of a-amino acids and a-methylbenzylamine combining the
enantioselective enzyme-catalyzed oxidation of the amine to imine and the non
enantioselective chemical reduction.150'151 There are only a few reports on the
enzymatic deracemization of a-substituted carboxylic acids with microorganisms.
Cordyceps militaris and Verticillium lecanii are capable of inverting the chirality of
the (R)-enantiomer of 2-arylpropanoic acids to the (S)-antipode, whereas,
Rhodococcus and Nocardia diaphanozonaria have opposite selectivity.152-155
50
Part I, Section 3, Resolution of2-arylpropanoates:Discussion
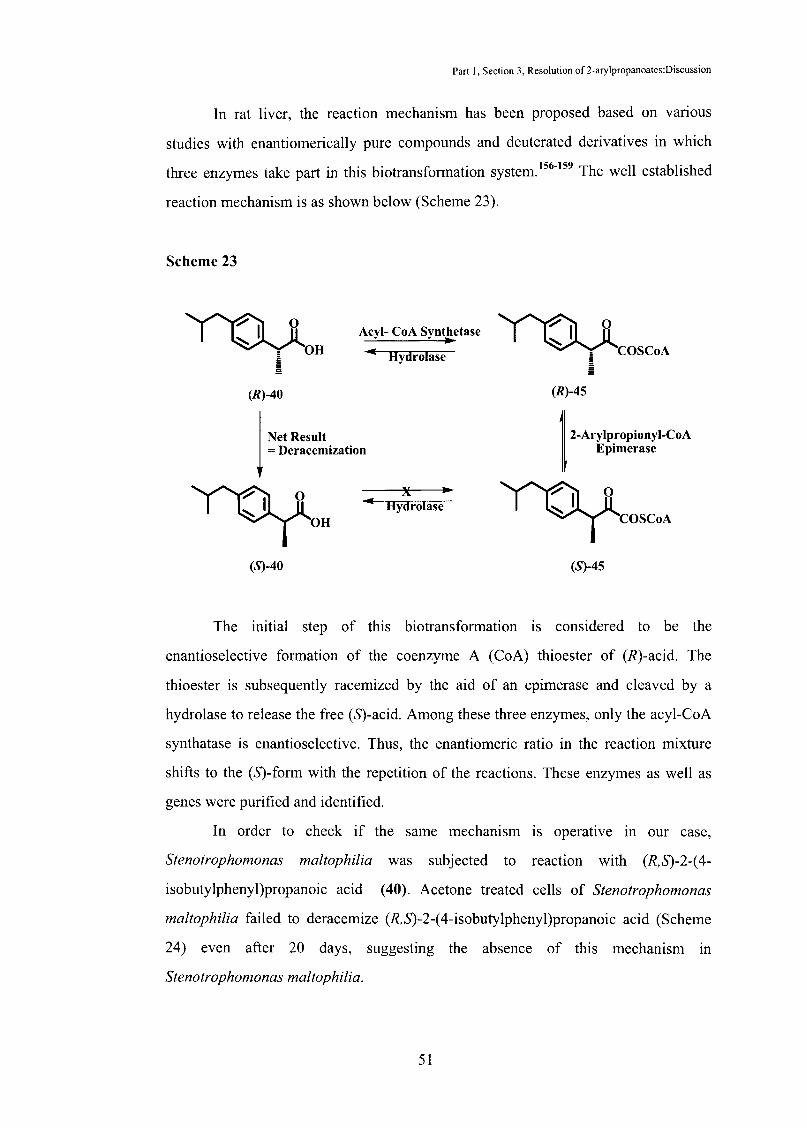
In rat liver, the reaction mechanism has been proposed based on various
studies with enantiomerically pure compounds and deuterated derivatives in which
three enzymes take part in this biotransformation system. 156-159 The well established
reaction mechanism is as shown below (Scheme 23).
Scheme 23
'Y'OJl : OH £ §
(R)-40
Acyl- CoA Synthetase
.,. Hydrolase
1 Net Result = Deracemization
X • .,. Hydrolase
(S)-40
~Q I ~COSCoA
I 5
(R)-45
2-Arylpropionyl-CoA Epimerase
(S)-45
The initial step of this biotransformation is considered to be the
enantioselective formation of the coenzyme A (CoA) thioester of (R)-acid. The
thioester is subsequently racemized by the aid of an epimerase and cleaved by a
hydrolase to release the free (S)-acid. Among these three enzymes, only the acyl-CoA
synthatase is enantioselective. Thus, the enantiomeric ratio in the reaction mixture
shifts to the (S)-form with the repetition of the reactions. These enzymes as well as
genes were purified and identified.
In order to check if the same mechanism ts operative m our case,
Stenotrophomonas maltophilia was subjected to reaction with (R,S)-2-( 4-
isobutylphenyl)propanoic acid (40). Acetone treated cells of Stenotrophomonas
maltophilia failed to deracemize (R,S)-2-(4-isobutylphenyl)propanoic acid (Scheme
24) even after 20 days, suggesting the absence of this mechanism in
Stenotrophomonas maltophilia.
51

Scheme 24
Part 1, Section 3, Resolution of mandelates:Results
-~~~~~~--~~~!ClA~ I o Acetone treated S. maltophilia,
OH Phosphate buffer, pH 7.0 20 days ! OH
(R,S)-40 (R)-40
The synthatase catalyzed thioester formation acyl-CoA requires the presence
of the free acid. Although no mechanistic studies have been done, we propose an
oxidation-reduction process via exomethylene type intermediate 46 for the
deracemization of esters (Scheme 25).
Scheme 25
~H ~~ ... SCoA
(S)-45 l Epim•ri,.Uon
Y'OJt :-.... ~ SCoA
(R)-45
Transesterification ~ R ~~~OCH3
(S)-39
Hydrolase .. X Y'OJt
~ OH
(R)-40
Oxidation
Reduction ~OCH3 46 CH2
! R<dn<tloo
1111111( Hydrolase ~ Q
I ~OCH3 (R)-39
However, transesterification of the ester group of 2-aryl propionates with acyl
CoA followed by epimerization and hydrolysis of CoA ester cannot be ruled out. The
results can also be explained based on DKR mechanisms, whereby the (S)-ester gets
racemized by a combination of oxidation-reduction sequence (Scheme 25).
1.3.5 Stenotrophomonas maltophilia catalyzed enantioselective resolution of 2-
hydroxyarylacetates (mandelates): formation of enantiomerically pure
(R)-2-hydroxyarylacetic acids (mandelic acids)
We started our investigation by studying the hydrolysis of ethyl mandelate
(47). Stenotrophomonas maltophilia was grown in medium comprising of peptone
(1.0%) and beef extract (0.5%). Cells were harvested by centrifugation and washed
with phosphate buffer (50 mM, pH 7.0). The washed cells were resuspended in the
52

Part I, Section 3, Resolution mandelates: Results
100 mL of same buffer at a concentration of 0.1 g/mL. To the cell suspension, ethyl
mandelate at 20 mM concentration was added and the contents incubated at 3 7 °C in
an orbital shaker at 200 rpm. The reaction was stopped at 48% conversion (34 h;
Scheme 14). Progress of the reaction was monitored by TLC. pH of the reaction
mixture was adjusted to 8.0 with 1N NaOH. The remaining ester was removed by
extraction with ethyl acetate. The aqueous layer was acidified to pH 2.0 with 1N HCl
and extracted with ethyl acetate to recover acid formed during the reaction. The
organic layer was separated, dried over sodium sulphate and evaporated to give acid
48 (Scheme 26).
Scheme 26
~O ~ __ S_te_n_ot_r_:op_h_o_m_o_na_s_m_a_l_to:....1Jh_,_·u_,a..,..
~ g Phosphate buffer, pH 7.0 conversion 100%
OH
G('cooH 47 (R)-48, e.e. >99%
The structure of the product was in agreement with its 1 HNMR spectral data.
The absence of a triplet (J=7.2 Hz) at 8 1.20 and quartet (J=7.2 Hz) at 8 4.24
corresponding to ester group of 47 confirmed that the hydrolysis had occurred. The
proton at C-2 appeared at 8 5.24 as a singlet. The aromatic protons appeared as
multiplet at 8 7.35-7.43.
The e.e. of the product was determined using (1R,2R)-1 ,2-diphenylethane-1 ,2-
diamine as chiral solvating agent. To the best of our knowledge, the use of this chiral
solvating agent for determination of the absolute configuration and e.e. of mandelic
acid and its derivatives has never been reported in literature. When 1.5 equivalent
shift reagent was added to racemic mandelic acid, the methine singlet at 8 5.24 for the
(R) and (S) enantiomers experienced unequal shifts and appeared as well separated
singlets at 8 4.80 and 8 4.78 (Figure 19a). When shift reagent was added to the acid
obtained from the biocatalyzed hydrolysis, only one singlet at 8 4.80 could be seen in 1HNMR spectrum (Figure 19b ). This indicated exclusive formation of one enantiomer
of the acid.
53

Part I, Section 3, Resolution mandelates: Results
r
5.0 5.0
(a) (b)
Figure 19: 1HNMR of (a) (R,S)-mandelic acid and (b) (R)-mandelic acid obtained from biocatalyzed reaction, in presence of 1.5 equivalent of (1R,2R)-1 ,2-diphenylethane-1 ,2-diamine
The absolute configuration of the acid was determined as follows: 1HNMR of
a commercial sample of (S)-mandelic acid was recorded in the presence of 1.5
equivalent chiral solvating agent. A singlet at 8 4. 78 appeared along with a minor
singlet at 8 4.80 as the commercial sample was not enantiomerically pure. Thus, the
singlet resonating at lower frequency can be assigned to (S)-enantiomer. To determine
the configuration of the acid obtained from the biocatalyzed hydrolysis, racemic
standard acid along with 1.5 equivalent chiral solvating agent was added to the sample
obtained from biotransformation and 1 HNMR of the sample was again recorded. The
singlets at 8 4.78 and 8 4.80 appeared in approximately 1:2 ratio, thus confirming the
formation of (R)-acid. In this case, the reaction did not stop at 50% conversion and
went on to complete conversion when allowed to run for longer time period. The acid
obtained after 100% conversion had (R)-configuration, with e.e. >99%.
The next substrate taken for the study was ethyl 4-chloromandelate (49).
Biocatalyzed reaction was performed under the conditions as described above. Acid
50 was obtained in 70% yield at 100% conversion (Scheme 27).
Scheme 27
(R)-50 (S)-50
R/S=9:1
54

Part I, Section 3, Resolution mandelaies: Results
The structure of the productwas in agreement with its 1HNMR spectral data.
The absence of a triplet at 8 1.22 (1=7.2 Hz) and quartet at 8 4.30 (1=7.2 Hz)
corresponding to ester group of 49 confirmed that the hydrolysis had occurred. The
me thine proton appeared as a singlet at 8 5.20 and the aromatic protons appeared as
multiplet at 8 7.36-8.31.
The e.e. of the product was determined using (1R,2R)-1 ,2--diphenylethane-1 ,2-
diamine as chiral solvating agent as described above. When shift reagent (1.5
equivalent) was added to racemic 4-chloromandelic acid, the methine singlet at 8 5.20
for (R) and (S) enantiomers experienced unequal shift and appeared as two singlets at
8 4.65 and 4.73 (Figure 20a). When shift reagent was added to the acid obtained from
the biocatalyzed hydrolysis, singlets in unequal ratio at 8 4.73 and 8 4.65 could be
seen in 1 HNMR spectrum (Figure 20b ). The major enantiomer resonated at higher
frequency and was tentatively assigned (R)-configuration in analogy with the results
obtained with ethyl mandelate.
5.0 5.0
(a) (b)
Figure 20: 1HNMR of (a) (R,S)-4-chloromandelic acid and (b) (R)-4-chloromandelic acid obtained from biocatalyzed reaction, in the presence of 1.5 equivalent of (1R,2R)-1 ,2-diphenylethane-1 ,2-diamine
Biocatalyzed hydrolysis of ethyl 4-methoxymandelate was studied next under
the reaction conditions as described above. Acid 52 was obtained in 75% yield at
100% conversion (Scheme 28).
The structure of the product was in agreement with its 1HNMR spectral data.
The absence of a triplet at 8 1.23 (1=7.2 Hz) and quartet at 8 4.28 (1=7.2 Hz)
corresponding to ester group of 51 confirmed that the hydrolysis had occurred. The
methine proton appeared as a singlet at 8 5.19 and the aromatic protons appeared as a
multiplet at 8 6.82-7.37.
55

Part I, Section 3, Resolution mandelates: Results
Scheme 28
OHOOE_t __ ==~~~~==~==-- mOHOOH Stenotrophomonas maltophilia
Phosphate buffer, pH 7.0 Me conversion 100% MeO
+
51 (R)-52 (S)-52
RIS=6:1
The e.e. of the product was determined using (1R,2R)-1 ,2-diphenylethane-1 ,2-
diamine as chiral solvating agent as described above. When shift reagent (1.5
equivalent) was added to racemic 4-methoxymandelic acid, the methine singlet at 8
5.19 for (R) and (S) enantiomers experienced unequal shift and appeared as two
singlets at 8 4.63 and 4.71 (Figure 21a). When shift reagent was added to the acid
obtained from the biocatalyzed hydrolysis, singlets in unequal ratio at 8 4.71 and 8
4.63 could be seen in 1HNMR spectrum (Figure 21b). The acid obtained after 100%
conversion was tentatively assigned (R)-configuration in analogy with the results of
ethyl mandelate.
115.0 I !i
(a) (b)
Figure 21: 1HNMR of (a) (R,S)-4-methoxymandelic acid and (b) (R)-4 methoxymandelic acid obtained from biocatalyzed reaction, in the presence of 1.5 equivalent of (1R,2R)-1 ,2-diphenylethane-1 ,2-diamine
Biotransformation of ethyl 2-chloromandelate with the whole cells of
Stenotrophomonas maltophilia was studied next under the reaction conditions described
above. Acid 54 was obtained in 77% yield at 100% conversion (Scheme 29).
Scheme 29
~OEt VH
53
Stenotrophomonas maltophilia • Phosphate buffer, pH 7.0
conversion 100%
56
~OH VH
+
(R)-54 R/S=4:1 (S)-54
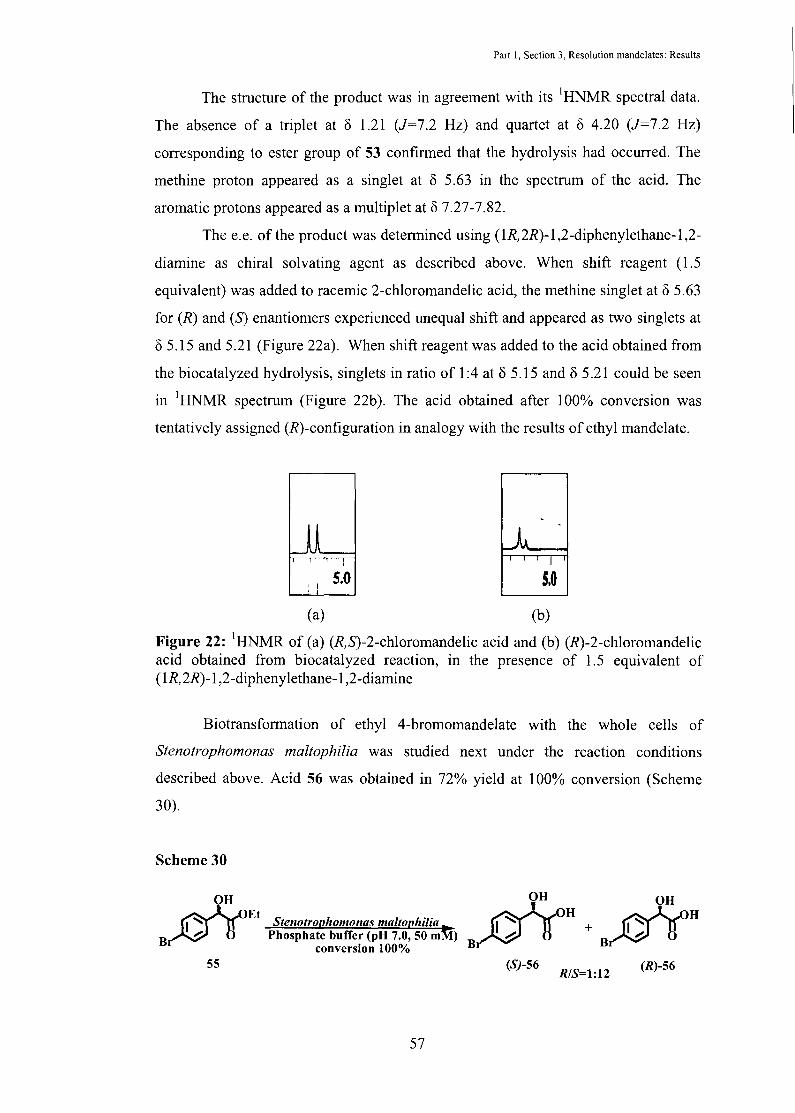
Part I, Section 3, Resolution mandelates: Results
The structure of the product was in agreement with its 1HNMR spectral data.
The absence of a triplet at 8 1.21 (1=7.2 Hz) and quartet at 8 4.20 (1=7.2 Hz)
corresponding to ester group of 53 confirmed that the hydrolysis had occurred. The
methine proton appeared as a singlet at 8 5.63 in the spectrum of the acid. The
aromatic protons appeared as a multiplet at 8 7.27-7.82.
The e.e. of the product was determined using (1R,2R)-1,2-diphenylethane-1,2-
diamine as chiral solvating agent as described above. When shift reagent (1.5
equivalent) was added to racemic 2-chloromandelic acid, the methine singlet at 8 5.63
for (R) and (S) enantiomers experienced unequal shift and appeared as two singlets at
8 5.15 and 5.21 (Figure 22a). When shift reagent was added to the acid obtained from
the biocatalyzed hydrolysis, singlets in ratio of 1:4 at 8 5.15 and 8 5.21 could be seen
in 1HNMR spectrum (Figure 22b). The acid obtained after 100% conversion was
tentatively assigned (R)-configuration in analogy with the results of ethyl mandelate.
: I s.o 5.0
(a) (b)
Figure 22: 1HNMR of (a) (R,S)-2-chloromandelic acid and (b) (R)-2-chloromandelic acid obtained from biocatalyzed reaction, in the presence of 1.5 equivalent of ( 1R, 2R)-1 ,2-diphenylethane-1 ,2-diamine
Biotransformation of ethyl 4-bromomandelate with the whole cells of
Stenotrophomonas maltophilia was studied next under the reaction conditions
described above. Acid 56 was obtained in 72% yield at 100% conversion (Scheme
30).
Scheme 30
~Et Stenotrophomonas maltophilia ~ B» B Phosphate buffer (pH 7.0, 50 mM)
conversion 100%
OH
+ ~H BM tl 55 (S)-56
RIS=1:12 (R)-56
57

Part I, Section 3, Resolution mandelates: Results
l-'"
( .I
____) '.'---
.0
(a) (b)
Figure 23: 1HNMR of (a) (R,S)-4-bromomandelic acid and (b) (S)-4-bromomandelic acid obtained from biocatalyzed reaction, in the presence of 1.5 equivalent of (lR,2R)-1 ,2-diphenylethane-1 ,2-diamine
The structure of the product was in agreement with its 1 HNMR spectral data.
The absence of a triplet at 8 1.22 (1=7.2 Hz) and quartet at 8 4.24 (1=7.2 Hz)
corresponding to ester group of 55 confirmed that the hydrolysis had occurred. The
methine proton appeared as a singlet at 8 5.18 in the spectrum of the acid. The
aromatic protons appeared as multiplet at 8 7.23-7 .50.
The e.e. of the product was determined using (1R,2R)-1 ,2-diphenylethane-1 ,2-
diamine as chiral solvating agent as described above. When shift reagent (1.5
equivalent) was added to racemic 4-bromomandelic acid, the methine singlet at 8 5.1
for (R) and (S) enantiomers experienced unequal shift and appeared as two singlets at
8 4.68 and 4.75 (Figure 23a). When shift reagent was added to the acid obtained from
the biocatalyzed hydrolysis, singlets in ratio of 1:12 at 8 4. 75 and 8 4.68 could be seen
in 1HNMR spectrum (Figure 23b). (S)-configuration was tentatively assigned to the
product ofbiocatalyzed reaction in analogy with the results of ethyl mandelate.
1.3.6 Acetone treated Stenotrophomonas maltophilia catalyzed resolution of 2-
hydroxyarylacetates (mandelates): formation of enantiomerically pure
(R)-2-hydroxyarylacetic acids (mandelic acids)
In 2-arylpropanoate series, untreated and acetone treated cells of
Stenotrophomonas maltophilia showed opposite enantioselectivity. Biocatalyzed
hydrolysis of mandelates 47, 49, 51, 53 and 55 with acetone treated cells was also
studied. The biocatalyzed reactions were performed as described in Section 1.3.5,
except that the acetone treated cells of Stenotrophomonas maltophilia, prepared as
described in Section 1.3.2 were used as biocatalyst in place of untreated cells. The
products were isolated, purified and analyzed as described in Section 1.3.5. The
results are summarized in Schemes 31-35 and Figures 24-28.
58
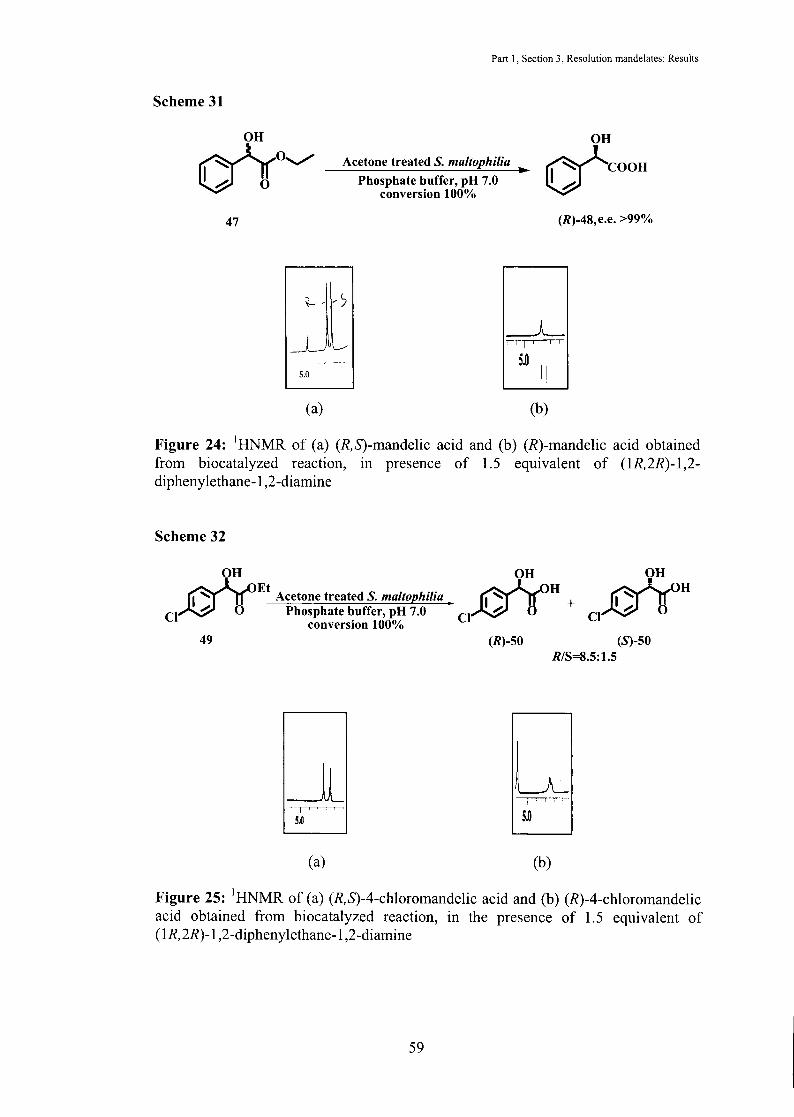
Scheme 31
~0~ VH
47
5.0
(a)
Part I, Section 3, Resolution mandelates: Results
OH
Acetone treated S. maltophilia cfcoon Phosphate buffer, pH 7.0
conversion 100%
(R)-48,e.e. >99%
___l_ .,....'T""""-,--
5.0 I!.
(b)
Figure 24: 1HNMR of (a) (R,S)-mandelic acid and (b) (R)-mandelic acid obtained from biocatalyzed reaction, in presence of 1.5 equivalent of (1R,2R)-1,2-diphenylethane-1 ,2-diamine
Scheme 32
OH OH
~Et Acetone treated S. maltophilia , ~H + Cl~ H Phosphate buffer, pH 7.0 Cl~ H
~H ~ H
conversion 100% Cl
49 (R)-50 (S)-50 R/S=8.5: 1.5
L 5.0
(a) (b)
Figure 25: 1HNMR of (a) (R,S)-4-chloromandelic acid and (b) (R)-4-chloromandelic acid obtained from biocatalyzed reaction, in the presence of 1.5 equivalent of ( 1 R, 2R)-I ,2-diphenylethane-1 ,2-diamine
59
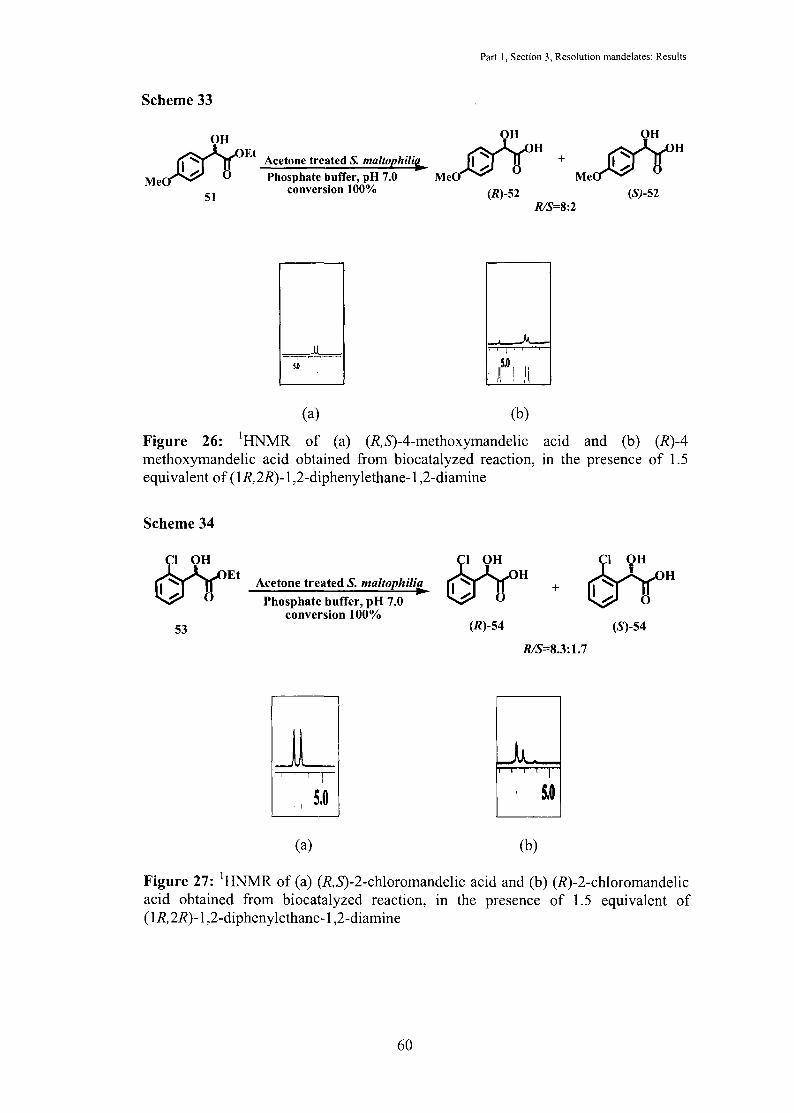
Scheme 33
Me mOH Et
.....:
51
Acetone treated S. maltophili(j. Phosphate buffer, pH 7.0
conversion 100%
(a)
Me
Part I, Section 3, Resolution mandelates: Results
mOHH
0
(R)-52 R/S=8:2
(b)
Figure 26: 1HNMR of (a) (R,S)-4-methoxymandelic acid and (b) (R)-4 methoxymandelic acid obtained from biocatalyzed reaction, in the presence of 1.5 equivalent of ( 1R, 2R)-1 ,2-diphenylethane-1 ,2-diamine
Scheme 34
53
Acetone treated S. maltophilia
Phosphate buffer, pH 7.0 conversion 100%
JL . I S,O
(a)
(R)-54
+
(S)-54
R/S=8.3:1.7
I
5.0
(b)
Figure 27: 1HNMR of (a) (R,S)-2-chloromandelic acid and (b) (R)-2-chloromandelic acid obtained from biocatalyzed reaction, in the presence of 1.5 equivalent of (1R,2R)-1 ,2-diphenylethane-1 ,2-diamine
60

Scheme 35
Acetone treated S. maltophilia. Phosphate buffer, pH 7.0
conversion 100%
.0
Part 1, Section 3, Resolution mandelates: Discussion
(R)-56 RIS=1:6
(S)-56
(a) (b) Figure 28: 1HNMR of (a) (R,S)-4-bromomandelic acid and (b) (S)-4-bromomandelic acid obtained from biocatalyzed reaction, in the presence of 1.5 equivalent of ( 1 R, 2R)-1 ,2-diphenylethane-1 ,2-diamine
In the mandelate series, untreated and acetone treated cells of
Stenotrophomonas maltophilia gave essentially similar results. In both the cases, the
biocatalyst showed selectivity for (R)-enantiomer, except in the case of 55, which
gave (S)-acid as the major product with treated as well as untreated cells. At 100%
conversion, (R)-enantiomer was produced exclusively (48) or predominantly (50, 52,
54). Only in case of ethyl mandelate (47) e.e. of more than 99% was obtained.
Substitution at ortho, meta or para position resulted in decrease in the e. e. to 66-70%.
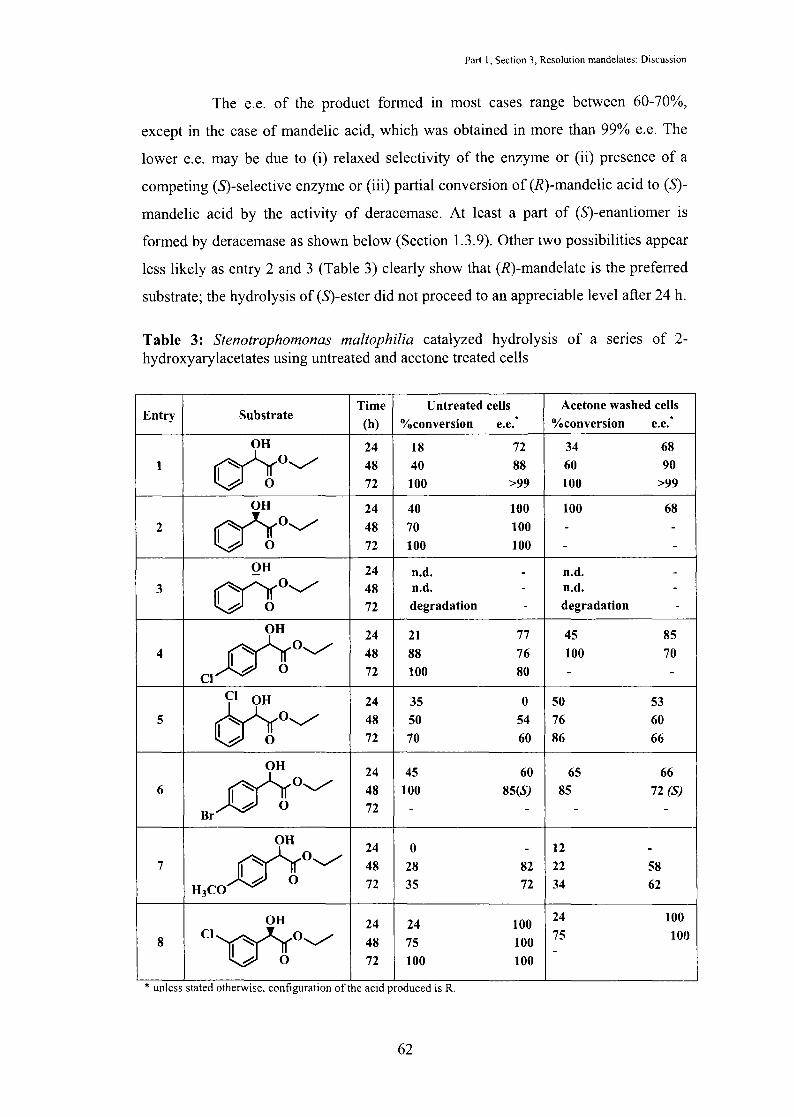
1.3. 7 Discussion: Stenotrophomonas maltophilia catalyzed resolution of
mandelates
Stenotrophomonas maltophilia catalyzed hydrolysis of a senes of ethyl
mandelate derivatives was studied using intact cells and acetone treated cells (Table
3). In all the cases studied, hydrolysis of mandelates resulted in the formation of (R)
acids, except in the case of ethyl 4-bromomandelate (Entry 6, Table 3). This is in
contrast to 2-arylpropanoates, where intact cells of Stenotrophomonas maltophilia
gave (S)-acids and acetone treated cells gave (R)-acids. Two possibilities exist, (i)
mandelates are not the substrates for surface bound lipase; all the activity for
mandelates is because of intracellular esterase and (ii) surface bound lipase has
opposite selectivity for mandelates. The pure lipase did not show any activity for ethyl
mandelate indicating that the activity is due to intracellular esterase.
61
Part I, Section 3, Resolution mandelates: Discussion
The e.e. of the product formed in most cases range between 60-70%,
except in the case of mandelic acid, which was obtained in more than 99% e.e. The
lower e.e. may be due to (i) relaxed selectivity of the enzyme or (ii) presence of a
competing (S)-selective enzyme or (iii) partial conversion of (R)-mandelic acid to (S)
mandelic acid by the activity of deracemase. At least a part of (S)-enantiomer is
formed by deracemase as shown below (Section 1.3.9). Other two possibilities appear
less likely as entry 2 and 3 (Table 3) clearly show that (R)-mandelate is the preferred
substrate; the hydrolysis of (S)-ester did not proceed to an appreciable level after 24 h.
Table 3: Stenotrophomonas maltophilia catalyzed hydrolysis of a series of 2-hydroxyarylacetates using untreated and acetone treated cells
Entry Substrate Time Untreated cell& Acetone washed cells
(h) 0/oconversion * 0/oconversion * e.e. e.e.
OH 24 18 72 34 68 1 cnro~ 48 40 88 60 90
0 72 100 >99 100 >99
OH 24 40 100 100 68 2 ~0~ 48 70 100 - -
0 72 100 100 - -OH 24 n.d. - n.d. -
3 0/ro~ 48 n.d. - n.d. -0 72 degradation - degradation -
OH 24 21 77 45 85 4 __o-)_t~ 48 88 76 100 70
0 72 100 80 - -Cl
Mo~ 24 35 0 50 53 5 48 50 54 76 60
0 72 70 60 86 66
OH 24 45 60 65 66 6 __o~~t~ 48 100 85(S) 85 72 (S)
0 72 - - - -Br
OH 24 0 12
d't~ - -
7 48 28 82 22 58 0 72 35 72 34 62 H3CO
OH 24 24 100 24 100
8 c•mo~ 48 75 100 75 100
I h o -72 100 100
* unless stated otherw1se, configuratiOn of the ac1d produced IS R.
62

Part 1, Section 3, Resolution mandelates: Discussion
The e.e. ofthe mandelic acid formed at 24 h (18% conversion) was 72% but
increased to >99% after 72 h (100% conversion) (Entry1, Table 3). Thus, e.e. of the
product increased with increase in conversion. This is possible if either (i) the (S)-acid
formed is deracemized to (R)-acid or (ii) (S)-acid is selectively degraded. When (R,S)
mandelic acids were incubated with cells of Stenotrophomonas maltophilia, a slow
degradation to benzoic acid and keto acid could be seen, when the crude product of
the reaction was analyzed by NMR. However, (R)-acid showed no degradation
product even after 1 00 h of incubation. This observation suggests that the increase in
e.e. may have occurred due to selective oxidation and degradation of (S)-acid.
The presence of chloro group at ortho, meta or para position of aromatic
ring of ethyl mandelate does not appear to have any significant effect on the rate of
the reaction but a decreased enantioselectivity was observed. The substrate with 4-
methoxy, however, resulted in greatly reduced rate of reaction (Entry 7, Table 3).
Substitution by a bromo group at para position showed very interesting behaviour.
Not only the rate ofreation was enhanced, the configuration ofthe product obtained at
100% conversion was (S) (Entry 6, Table 3).
The reaction with ethyl mandelate proceeded to 100% conversion, but the
e.e. of product remained very high. This is possible if either (S)-ester or (S)-acid is
selectively transformed to corresponding (R)-enantiomer. As described below
(Section 1.3.9), deracemization of racemic acid occurs, but very slowly compared to
rate of ester hydrolysis. Therefore, deracemization must be occurring at the stage of
ester. This was confirmed by HPLC analysis of the reaction on chiral column (Diacel
chiracel OB-H) (Figure 29). As the reaction proceeded the (R)-ester started depleting
till whole of it disappears, leaving behind only the (S)-ester (Figure 29b ). As the
reaction is continued further, conversion of (S)-ester to (R)-ester could be clearly seen.
The proposed mechanism for deracemization is shown in Scheme 36. The enzyme
responsible for deracemization of ethyl mandelate and 2-arylpropanoates may be
similar.
63

svt!OXldo211 ETHYL MANDALATE CONn AU
,,. ~ ! '
15<><>
m-
l"o I !
I I•"' I
i A ~ \ ' o'o 25.0
(a)
(c)
uv"" W\11..:217nm
I
I
J5a
Part I, Section 3, Presence of mandelate dehydrogenase inS. maltophilia
6 ~undo ESd ~thvtmandelale19hr
•2
o'o s.o 1oo 1s.o 20.o 2so 30o JS.o 4D.o 4s.o
(b) S-ethylmend•
·""'
00 5.0 10.0
(d)
WVL217nnj
•
I
-! '""
Figure 29: HPLC profiles of (a) (R,S)-ethyl mandelate (b) ethyl mandelate recovered from biocatalyzed reaction after 19 h (c) ethyl mandelate recovered from biocatalyzed reaction after >72 h and (d) (S)-ethyl mandelate prepared from (S)mandelic acid
Scheme 36
Reductase .. lf
Oxidase
(S)-47 57 (R)-47
1.3.8 Demonstration of the presence of mandelate dehydrogenase in the cells of
Stenotrophomonas maltophilia
1.3.8.1 Activity Staining (Zymography)
The presence of mandelate dehydrogenase has been implicated in the
deracemization of ethyl mandelate. To confirm the presence of a dehydrogenase in the
system, cells of Stenotrophomonas maltophilia were grown and proteins extracted
according to the procedure described in the experimental section. Polyacrylamide gel
electrophoresis, under non denaturing conditions at pH 7.5 was performed in 10%
(w/v) polyacrylamide gels according to Maurer (1968). 169 NADP dependent
dehydrogenase activity was detected in the gel using staining solution consisting of
64

Part I, Section 3, Mandelate dehydrogenase inS. maltophilia
phenazine methosulfate (0.05 mg/mL), nitroblue tetrazolium (0.3 mg/mL), NADP
(0.5 mM) and ethyl mandelate (20 mM).170 The gel was kept in the staining solution
overnight, when white band appeared on blue background. The zymogram clearly
shows the presence of a NADP dependent dehydrogenase in the protein (Figure 30).
Figure 30: Activity staining of the dehydrogenase: Stenotrophomonas maltophilia was grown in peptone (1 %) and beef extract (0.5%). Proteins were extracted by incubating cells with 1M NaCl and incubated overnight in a solution consisting of phenazine methosulfate (0.05 mg/mL), nitroblue tetrazolium (0.3 mg/mL), NADP (0.5 mM) and ethyl mandelate (20 mM). White band appeared on blue background.
1.3.8.2 Peptide Mass Fingerprinting (PMF) and bioinformatics studies
Non-denaturing PAGE of the purified protein was performed and the gel
was stained for dehydrogenase activity as described in detail above. White band
obtained from the zymogram was excised thoroughly and collected in eppendorf. The
gel pieces were dehydrated in 100% acetontrile ( 40 flL) for 5 min. After removing
acetonitrile, the gel pieces were destained with ammonium bicarbonate (20 mM in
30% acetonitrile) at 30 °C for 30 min. This destaining procedure was repeated four
times. Gel pieces were again dehydrated in 100% acetonitrile for 5 min and then dried
in speed-vac for 30 min. MS-grade trypsin solution (25 flL) was added to the finely
dried gel pieces and incubated at 3 7 °C for 36 h. After complete incubation, the
trypsinised sample was centrifuged and supernatant was collected. The proteolyzed
fragments obtained from the gel pieces were extracted twice with 60% acetonitrile in
0.1% trifluoroacetic acid and centrifuged again. This was combined with the
supernatant obtained above and concentrated to a final volume of 10 flL in speed-vac.
The sample so obtained was subjected for peptide mass fingerprinting. The peptide
masses obtained from peptide mass fingerprinting were searched against the proteins
65

Part I, Section 3, Mandelic acid deracemase
in MASCOT server at http//www.matrixscience.com. The database used was MSDB
20060831 (3239079 sequences; 1079594 700 residues). The search parameters used
were: Type of search: PMF, Enzyme: trypsin, Mass values: monoisotopic, Protein
mass: unrestricted, Peptide mass tolerance: ±1.2 Da, Peptide charge state: ±1.
Extensive search in MASCOT showed the masses matched with a NADP dependent
dehydrogenase from bacteria, Blastopireuella marina.
1.3.9 Stenotrophomonas maltophilia catalyzed deracemization of 2-
hydroxyarylacetic acids
Racemic mandelic acid (48), 4-chloromandelic acid (50) and 4-
methoxymandelic acid (52) were subjected to reaction with the acetone treated cells
of Stenotrophomonas maltophilia under the reaction conditions described in Section
1.3.6. The products were isolated, purified and analyzed as described in Section 1.3.5.
The results are summarized in Schemes 3 7-3 9 and Figures 31-3 3. In all the three case,
a conversion of (R)-enantiomer to (S)-enantiomer could be seen. The e.e. for the (S)
enantiomer was 60-65%. The configuration of the acid present in major amount after
7 days was found to be (S) in all the examples studied.
Scheme 37
~OH VH
(R,S)-48
5.0
(a)
Acetone treated S. maltophilia
Phosphate buffer, pH 7.0
OH
~OH ~ 0
(S)-48, e.e. 65%
4.t4o 4.7 4.()
(b)
Figure 31: 1HNMR of (a) (R,S)-mandelic acid and (b) Mandelic acid from biocatalyzed reaction, in the presence of 1.5 equivalent of (1R,2R)-1 ,2·diphenylethane-1 ,2-diamine
66

Scheme 38
Cl
~OH N H
(R,S)-50
(a)
Part I, Section 3, Resolution of 2-aryloxypropanoates: R1:sults
Acetone treated S. maltophilia
Phosphate buffer, pH 7.0 Cl
~OH ~ 0
(S)-50, e.e. 60%
(b)
Figure 32: 1HNMR of(a) (R,S)-4-chloromandelic acid and (b) 4-chloromandelic acid obtained from biocatalyzed reaction, in the presence of 1.5 equivalent of (lR, 2R)-l ,2-diphenylethane-1 ,2-diamine
Scheme 39
~OH N g
MeO
(R,S)-52
(a)
Acetone treated S. maltophilia
Phosphate buffer, pH 7.0
~OH ~ 0
MeO
(S)-52, e.e. 61%
5.0 II
(b)
Figure 33: 1HNMR of(a) (R,S)-4-methoxymandelic acid and (b) 4 methoxymandelic acid obtained from biocatalyzed reaction, in the presence of 1.5 equivalent of ( 1R, 2R)-1 ,2-diphenylethane-1 ,2-diamine
67

Part I, Section 3, Resolution of2-aryloxypropanoates: Results
Both (R) and (S) mandelic acid dehydrogenases are known in literature.160
The mechanism of deracemization is shown in Scheme 40. The formation of keto acid
and benzoic acid in the reaction was confirmed when crude product obtained from the
reaction mixture was analyzed by 1HNMR. In addition to mandelic acid, NMR
showed the presence of keto acid, 58 and benzoic acid, 66. Aromatic protons
corresponding to keto acid were present at 8 7.43-7.73 and 8 8.24-8.28 and that of
benzoic acid at 8 7.51-7.65 and 8 8.08-8.11.
Scheme 40
OH
cfcoou -.=::::O::xi::da::s::e ~ Reductase
(R)-48
0
c{coon 58
! ~OOH
v 66
Reductase 4 X
Oxidase
1.3.10 Stenotrophomonas
aryloxypropanoates
maltophilia catalyzed
OH
cfcoon S-48
hydrolysis of 2-
We started our investigation in this series by studying the hydrolysis of ethyl
2-(3-chlorophenoxy)propanoate (59). Stenotrophomonas maltophilia was grown in
medium comprising of peptone (1.0%) and beef extract (0.5%). Cells were harvested
by centrifugation and washed with phosphate buffer (50 mM, pH 7.0). The washed
cells were resuspended in the 100 mL of same buffer at a concentration of 0.1 g/ mL.
To the cell suspension, ethyl 2-(3-chlorophenoxy)propanoate (59) at 20 mM
concentration was added and the contents incubated at 3 7 °C in an orbital shaker at
200 rpm (Scheme 41). The product was isolated and purified as described in Section
1.3 .1. The biotransformation with the untreated cells of Stenotrophomonas
maltophilia proceeded very fast with 100% conversion of the substrate taking place in
8 h.
68

Part I, Section 3, Resolution of2-aryloxypropanoates: Results
The formation of acid 60 was confirmed by 1 HNMR_ The absence of triplet at
8 1.23 (1=7.2 Hz) and quartet at 8 4.21 (1=7.2 Hz) confirmed that the hydrolysis had
occurred. The -CH3 protons at C-2 appeared at 8 1.64 (1=7.2 Hz) as a doublet,
methine proton as a quartet (1=7.2 Hz) at 8 4.75 and aromatic protons as a multiplet at
8 6.89-7.26.
Scheme 41
Ck ,::-1
..0. Jl cn,cn, S. maltophilia Ck "'' ..0.. Jl.,H ~k "'1..0.. Jl H U -T U Phosphate buffer, pti 7.0 U T l U T U
59 conversion 100% (S)-60 (R)-60
SIR= 1:0.9
The e.e. and absolute configuration of the acid was determined by NMR
method using (1R,2R)-1,2-diphenylethane-1,2-diamine as chiral solvating agent as
described. 140 When 1.5 equivalent solvating reagent was added to racemic 2-(3-
chlorophenoxy)propanoic acid (60), the quartet for the methine proton for (R) and (S)
enantiomers experienced unequal shift and appeared as two quartets at 8 4.32 (J=7.2
Hz) and 4.4 (1=7.2 Hz) (Figure 34a). When shift reagent was added to the acid
obtained from the biocatalyzed hydrolysis, quartets at 8 4.32 (1=7.2 Hz) and 4.4
(1=7.2 Hz) in 1HNMR spectrum were observed in the ratio 1:0.9 (Figure 34b),
indicating that no resolution has occurred in this case.
4.0
(a) (b)
Figure 34: 1HNMR of (a) (R,S)-2-(3-chlorophenoxy)propanoic acid and (b) 2-(3-chlorophenoxy)propanoic acid obtained from biocatalyzed reaction, in the presence of 1.5 equivalent of (lR, 2R)-I ,2-diphenylethane-1 ,2-diamine
The next substrate chosen for the biocatalyzed hydrolysis in this category of
compounds was ethyl 2-(2,4-dichlorophenoxy)propanoate (61). The reaction was
69

Part I, Section 3, Resolution of2-aryloxypropanoates: Results
performed under the same reaction conditions described above. The product was
isolated and purified as described in Section 1.3 .1.
Scheme 42
(S)-62 SIR= 2:1
The structure of the product was in agreement with its 1HNMR spectral data.
The absence of triplet at 8 1.23 (J=7.2 Hz) and quartet at 8 4.17 (J=7.2 Hz) confirmed
that the hydrolysis had occurred. The -CH3 protons appeared at 8 1.69 as a doublet
(J=7.2 Hz), the methine proton appeared as a quartet at 8 4.68 (J=7.2 Hz) and
aromatic protons appeared as a multiplet at 8 6.65-7.39.
The e.e. and absolute configuration of the acid was analyzed by NMR method
using (1R,2R)-1 ,2-diphenylethane-1 ,2-diamine as chiral solvating agent as
described. 140 When 1.5 equivalent solvating reagent was added to racemic 2-(2,4-
dichlorophenoxy)propanoic acid, the quartet for the methine proton for (R) and (S)
enantiomers experienced unequal shift and appeared as two separate quartets at 8 4.42
(J=7.2 Hz) and 4.51 (J=7.2 Hz) (Figure 35a). When shift reagent was added to the
acid obtained from the biocatalyzed hydrolysis, quartets at 8 4.42 (J=7 .2 Hz) and 4.51
(J=7.2 Hz) in 1HNMR spectrum were observed in the ratio 2:1 (Figure 35b). The
quartet resonating at lower frequency in presence of chiral shift reagent has been
assigned to (S)-enantiomer.140
4.0 4.0
(a) (b)
Figure 35: 1HNMR of (a) (R,S)-2-(2,4-dichlorophenoxy)propanoic acid and (b) 2-(2,4-dichlorophenoxy)propanoic acid obtained from biocatalyzed reaction, in the presence of 1.5 equivalent of (1R, 2R)-1 ,2-diphenylethane-1 ,2-diamine
70
Part I, Section 3, Resolution of2-aryloxypropanoates: Results
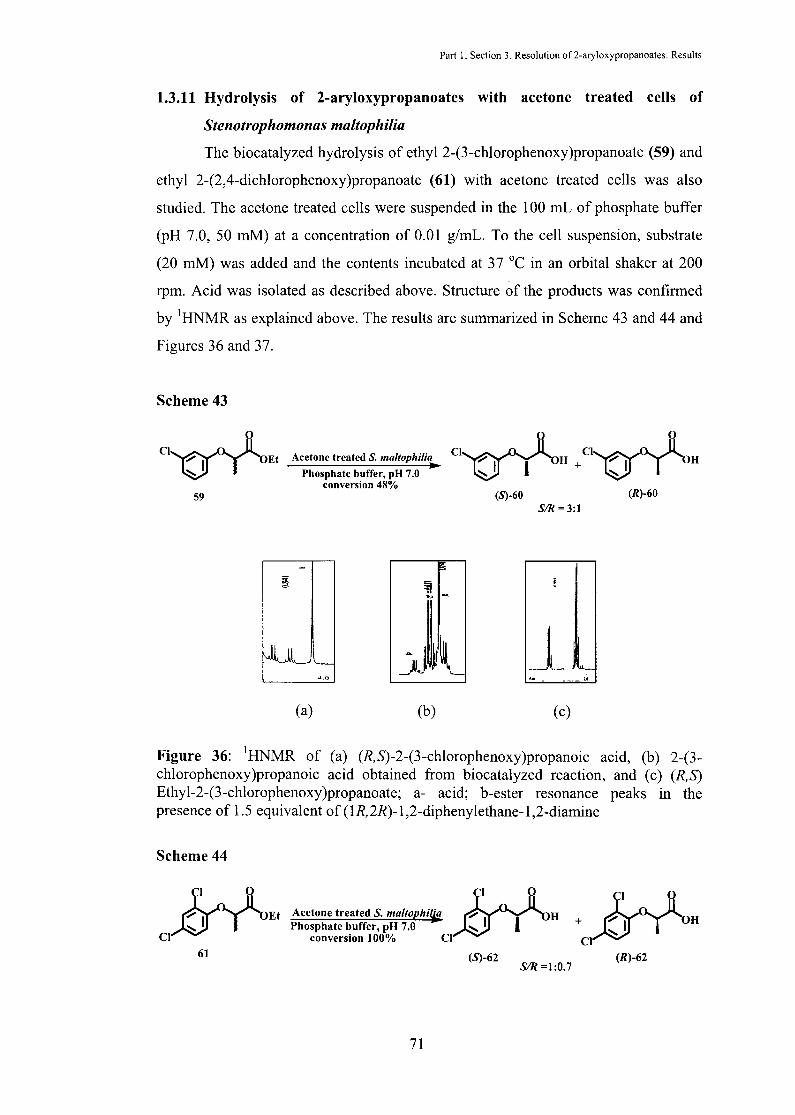
1.3.11 Hydrolysis of 2-aryloxypropanoates with acetone treated cells of
Stenotrophomonas maltophilia
The biocatalyzed hydrolysis of ethyl 2-(3-chlorophenoxy)propanoate (59) and
ethyl 2-(2,4-dichlorophenoxy)propanoate (61) with acetone treated cells was also
studied. The acetone treated cells were suspended in the 1 00 mL of phosphate buffer
(pH 7.0, 50 mM) at a concentration of 0.01 g/mL. To the cell suspension, substrate
(20 mM) was added and the contents incubated at 3 7 °C in an orbital shaker at 200
rpm. Acid was isolated as described above. Structure of the products was confirmed
by 1HNMR as explained above. The results are summarized in Scheme 43 and 44 and
Figures 36 and 37.
Scheme 43
ct ..... -..... ...o... ~ u T UEt
59
~
Acetone treated S. maltophilia
Phosphate buffer, pH 7.0 conversion 48%
~ "'
!li
-
Ct.... --.. ...0... ~ Cl...._ -..... ...0.... ~. UTUH+ UTUH (S)-60 (R)-60
SIR= 3:1
- ~~~ ~JlJ ·.__ _l~ 4.0
(a) (b) (c)
Figure 36: 1HNMR of (a) (R,S)-2-(3-chlorophenoxy)propanoic acid, (b) 2-(3-chlorophenoxy)propanoic acid obtained from biocatalyzed reaction, and (c) (R,S) Ethyl-2-(3-chlorophenoxy)propanoate; a- acid; h-ester resonance peaks in the presence of 1.5 equivalent of (1R,2R)-1,2-diphenylethane-1,2-diamine
Scheme 44
~Et CI
Acetone treated S. maltophilj: l...o... .1 H + ~I H Phosphate buffer, pH 7.0 ~ T U ~ I
conversion 100% Cl Cl 61 (S)-62
S/R=l:0.7 (R)-62
71

Part I, Section 3, Resolution of2-aryloxypropanoates: Discussion
4.0 "I 4.0
Figure 37: 1HNMR of (a) (R,S)-2-(2,4-dichlorophenoxy)propanoic acid and (b) 2-(2,4-dichlorophenoxy)propanoic acid obtained from biocatalyzed reaction, in the presence of 1.5 equivalent of (1R,2R)-1,2-diphenylethane-1,2-diamine
1.3.12 Discussion: Stenotrophomonas maltophilia catalyzed hydrolysis of 2-
aryloxypropanoates
High enantioselectivity was obtained in Stenotrophomonas maltophilia
catalyzed hydrolysis of 2-arylpropanoates, whereas 2-aryloxypropanoates gave poor
e.e. Acetone treated cells were (R)-selective for 2-arylpropanoates, but exhibited
preference for (S)-enantiomer of 2-aryloxypropanoates. The rates of reaction were
much higher with 2-aryloxypropanoates than 2-arylpropanoates. Theses results show
the marked influence of inserting an oxygen atom between aryl moiety and chiral
carbon on the rate of reaction as well as enantioselectivity.
Providing an explanation of results with whole cell system containing multiple
enzyme activities at best could be speculative. A possible explanation of results could
be as follows (i) acetone treated cells have a deracemase and a hydrolase activity, (ii)
deracemase has high selectivity for (S)-enantiomer in case of arylpropanoates but has
low or poor selecitivity for (R/S) enantiomer of aryloxypropanoates, (iii) an (S)
selective esterase, but with low or poor selectivity for (R)-enantiomers is present.
Alternatively, two esterases one selective for (S) and other selective for (R) may be
present, the rate of hydrolysis being higher for the (S)-selective than the (R)-selective
esterase. Thus, in case of 2-arylpropanoic acids (Scheme 45) (S)-ester is rapidly
transformed to (R)-ester by deracemase before any hydrolysis could occur. The ester
that remains at 48% conversion in acetone treated Stenotrophomonas maltophilia
catalyzed hydrolysis of 39 had (R)-configuration (data not shown), which confirms
much faster rate of deracemization compared to hydrolysis. (S)-selective esterase of
relaxed selectivity then hydrolyses (R)-ester at a slow rate. In case of 2-
aryloxypropanoates, the deracemase may have relaxed selectivity; therefore the
equilibrium is driven towards (S)-acid because of faster rate of reaction with (S)-ester
than (R)-ester (Scheme 46)
72

Part I, Section 3, Resolution of ethyl 2-chloropropanoate
Scheme 45
2-Arylpropanoic acid (overall (R)-acid with high e.e.)
(S)-ester kt
(R)-ester
l k, k2
k,l slow
(S)-acid (R)-acid
k1>>k2 and k3
Scheme46
2-Aryloxypropanoic acid (overall (S)-acid with poor e.e.)
(S)-ester kt (R)-ester
j ., k2
(S)-acid (R)-acid
kt::k2::k3, k3>k4
1.3.13 Stenotrophomonas maltophilia catalyzed hydrolysis of ethyl 2-chloro
propanoate
An aliphatic ester was taken as substrate to study substrate specificity of the
biocatalyst. Stenotrophomonas maltophilia was grown in medium comprising of
peptone (1.0%) and beef extract (0.5%). Cells were harvested by centrifugation and
washed with phosphate buffer (50 mM, pH 7.0). The washed cells were suspended in
the 100 mL of same buffer at a concentration of 0.1 g/ mL. To the cell suspension,
ethyl 2-ch1oropropanoate at 20 mM concentration was added and the contents
incubated at 37 °C for 2 h in an orbital shaker at 200 rpm (Scheme 47). The product
was isolated, purified and analyzed as described previously (Section 1.3 .1 ).
The e.e. and absolute configuration of the acid was analyzed by NMR method
usmg (1R,2R)-1,2-diphenylethane-1,2-diamine as chira1 solvating agent as
described. 140 When 1.5 equivalent chira1 solvating agent was added to racemic 2-
chloropropanoic acid, the quartet for the -CH proton at C-2 for (R) and (S)
enantiomers experienced unequal shift and appeared as two separate quartets at 8 3.95
(1=7.2 Hz) and 3.80 (1=7.2 Hz) (Figure 38a). When shift reagent was added to the
73

Part I, Section 3, Conclusions
acid obtained from the biocatalyzed hydrolysis, quartets at 8 3.95 (1=7.2 Hz) and 3.80
(1=7.2 Hz) in 1HNMR spectrum were observed in the ratio 1:0.95 (Figure 38b),
indicating that no resolution has occurred in this case.
Scheme 47
Cl
~~ 0
63
v u L fl. II
(a)
Stenotrophomonas maltophilia
Phosphate buffer, pH 7.0 conversion 100%
4.0
(b)
Cl
~OH 0
64, e.e. 0%
Figure 38: 1HNMR of (a) (R,S)-2-chloropropanoic acid and (b) 2-chloropropanoic acid obtained from biocatalyzed reaction, in the presence of 1.5 equivalent of (lR, 2R)-1 ,2-diphenylethane-1 ,2-diamine
1.3.14 Conclusions
In conclusion, we have demonstrated the preparation of either (R) or (S)
enantiomer in >99% e.e. of 2-arylpropanoic acids by kinetic resolution of their esters
using the same strain of microorganism, but used under different set of conditions.
Detailed studies revealed that the result is due to presence of two different enzymes
within the organism. We have also demonstrated Dynamic Kinetic Resolution of 2-
arylpropanoates to (R)-acids in >99% e.e. Mandelates were also resolved by dynamic
kinetic resolution process, but the e.e. of the acids produced was in the range of 62-
99%. 2-Aryloxypropanoates and ethyl 2-chloropropanoate were hydrolyzed, but
without any selectivity for enantiomers. In addition, we have demonstrated the
deracemization of mandelic acids, in which biocatalyst converted racemic mandelic
acids to (S)-mandelic acids in 60-65%. Overall, the results achieve significance due to
the fact that by using appropriate reaction conditions, we have been able to harness
multiple enzyme activities of one organism for the selective production of either
enantiomer.
74