pharmacoepidemiology used for · pdf filepharmacoepidemiology used for pharmacovigilance...
TRANSCRIPT

Châtenay-Malabry January 2013 1
Pharmacoepidemiology
used
for pharmacovigilance
Stéphanie Tcherny-Lessenot,
MD MSc MPHPharmacoepidemiologistGlobal
Pharmacovigilance & Epidemiology,
Sanofi R&D
23
January 2013

2
Objectives
●Objectives of the training are to:●
Provide
some
background on Pharmacoepidemiology
(PE)
●
Provide
the current
European
Regulatory
context
relative to Pharmacovigilance (PV)
●
Connect
Pharmacoepidemiology
to Pharmacovigilance

3
Agenda
●
Background on pharmacoepidemiology●
Current
European
PV regulations
●
Place of pharmacoepidemiology
in pharmacovigilance●
Illustrative cases
●
Conclusion

4
What is pharmacoepidemiology
(PE)?
Genetic Epidemiology Pharmacoepidemiology
Cardiovascular Epidemiology
Cancer Epidemiology
Environmental EpidemiologyNutrition
Epidemiology
Epidemiology
is the study of the distribution and determinants of diseases in populations
Pharmacoepidemiology
is the study of the use of and the effects of drugs in large numbers of people
Ref: Textbook of Pharmacoepidemiology
Brian L. Strom and Stephen E. Kimmel-
John Wiley & Sons , Ltd

5
What are Epidemiological Studies?
= Observational Studies= Non-interventional Studies= No treatment assignment by a third party (decision only by patient and physician)= In real life setting
●
Main types●
Prospective
new data collection●
Retrospective
use of existing data●
Cross-sectional
one shot data
●
Epidemiologic study designs:●
Cohort
study●
(nested) Case-control
study●
Case-crossover study●
…●
Main data sources●
Field studies:
specifically performed to address specific needs, and study
participants recruited
for the purpose
of the study●
Databases studies: Study
subjects
are identified
in the existing
databases
where
data are usually
collected
for administrative purpose; thus, not specific
to the study
6
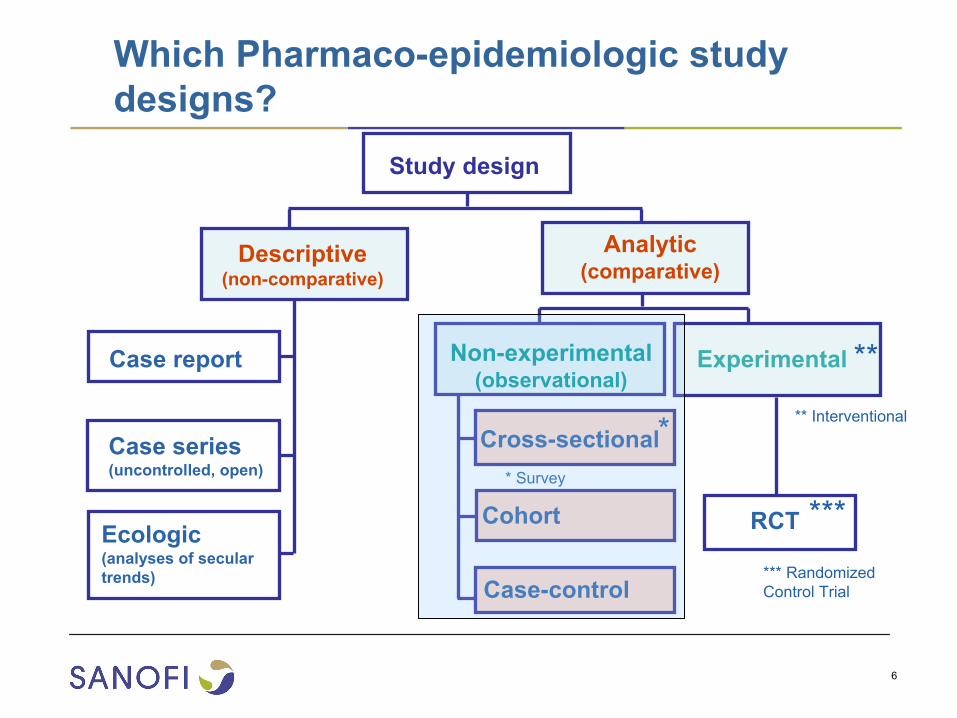
Which Pharmaco-epidemiologic study designs?
Case report
Case series (uncontrolled, open)
Ecologic (analyses of secular
trends)
Descriptive (non-comparative)
Cross-sectional
Cohort
Non-experimental (observational)
RCT
Experimental
Analytic (comparative)
Case-control
Study design
** Interventional** Survey
**
*** Randomized Control Trial
***

7
+ Rx+ RxOutcome?Outcome?
ExposureExposure
Main designs in (pharmaco-)epidemiology
OutcomeOutcomeCase-controlExposure?Exposure?
Cross-sectional
cohort

8
Cohort
study
●
A group or groups of individuals to be followed forward and defined
on ●
Status of exposure
to a specified risk factor for a disease (e.g., drug)●
Status of disease
presence●
Exposure is defined at time of cohort entry and is assessed before outcome
●
Drug
registry
: is a cohort of patients defined by exposure to the drug●
Provide information about:●
Incidence data of outcome(s) of interest●
Disease course between exposed and non-exposed patients●
Possible delayed drug effect of newly marketed drug
+ Rx+ Rx
Cohort (prospective)
Outcome?Outcome?
ExposureExposure TimeTime retrospective (if existing data)

9
Cross-sectional
survey
●
Exposure and disease status are assessed simultaneously at a specific time point
among individuals in a well-defined population
●
Provide information about:●
Frequency and characteristics of a disease●
Health status and health care needs●
Prevalence of disease or other health outcomes in certain occupations
●
Conditions of use of a drug
(i.e. at treatment initiation)
Cross-sectional (one snapshot)
Cross-sectional

10
Case-control study
●
Goal is to
compare
cases with a disease
to controls without the disease, looking for differences in antecedent exposures
Cases
Controls
Exposed?
Exposed?
●Provide information about:●Multiple causes (number of exposures) of a single disease●Allow to study rare
diseases
Case-control (retrospective)

11
Advantages
& Disadvantages
of
PE study designs
Design Advantages Disadvantages
Cross-sectional ● Logistically easier & faster● Less expensive
●
Not always possible to distinguish whether the exposure precedes or follows the disease since exposure and disease status are measured at the same point in time● No longitudinal data
Cohort ● Can study multiple outcomes● Can study uncommon exposures● Selection bias less likely● Unbiased exposure data
● Possibly biased outcome data● More expensive●
If done prospectively, may take years to complete
Case-control ● Can study multiple exposures● Can study uncommon diseases● Logistically easier & faster● Less expensive
● Control selection problematic● Possibly biased exposure data

12
Different
databases
available
●
Medical claims or hospital administrative databases●
US: PharMetrics, Premier, LabRx
●
Population-based registries●
Danish and Swedish registry
●
Automated medical records●
UK: THIN, GPRD
●
US: Department of Defense, GE

13
Agenda
●
Background on Pharmacoepidemiology●
Current
European
PV regulations
●
Place of Pharmacoepidemiology
in Pharmacovigilance●
Illustrative cases
●
Conclusion

14
PV & RMP Regulations are linked to crises!

15
Pharmaceutical regulation evolution since 50 years: toward risk management…
2002 2004 2006 2007 20102005 2009
ICH E2E
PDUFA III
CIOMS VI: Management of safety information from Clinical Trials (not regulatory)
• Regulation n°726/2004 -
art 6• Directive 2004/27/EC -
art 8(3)
FDAAA –
PDUFA IV (REMS)
Guidelines on Risk Management Systems
All EU-RMP regulations now in VOL9A, Part I, Section 3
EU -
RMP Template
2008
Guideline on Risk Management ATMPs
2012
New EU Pharmacovigilance legislation
ICH:
International Conference
on Harmonisation; CIOMS: Council for international organizations of Medical Sciences; ATMP: Advanced Therapy
Medicinal
Product; REMS: Risk
Evaluation and Mitigation Strategy; PDUFA: Prescription Drug User Fee Act;FDAAA: Food and Drug Administration Amendments Act
Draft REMS guidance
•Premarketing Risk Assessment•Good PV Practices & PE•Development and Use of
Risk Minimization Action Plans

16
●
Mar 2004: EU directive 2001/83/CE amended by the EU directive 2004/27/CE (EU directive 2001/83/CE amended by the EU directive 2004/27/CE (art 8(3))art 8(3))
•
Introduction of the inclusion of “A detailed description of the pharmacovigilance and, where appropriate, of the risk management system”
within a Market Authorization (MA) application
●
Dec 2004: ICH E2E Pharmacovigilance Planning (CHMP/ICH/5716/3)ICH E2E Pharmacovigilance Planning (CHMP/ICH/5716/3)
•
Intended to aid in planning pharmacovigilance
activities
(e.g., preparation for early post-marketing period of a new drug)
•
Main focus on Safety Specifications and Pharmacovigilance
Plan
that might be submitted at the time of MA application
●
Nov 2005: Guideline on Risk Management Systems (CHMP/96268/2005)Guideline on Risk Management Systems (CHMP/96268/2005)
•
Form of the risk management system, a RMP, should be presented to CAs●
Jan 2007: VolVol
9A 9A ““Rules Governing Medicinal Products in the European UnionRules Governing Medicinal Products in the European Union””•
Legal Basis of the MAH’s
Obligations for
Human Pharmacovigilance
●
Nov 2008: VolVol
9A updated with details on Post9A updated with details on Post--Authorization Safety StudiesAuthorization Safety Studies
(PASS),
defined as:
•
“pharmacoepidemiological
study or clinical trial
carried out in accordance with the terms of a marketing authorization […] This includes all company-sponsored studies conducted within the EU, and those conducted outside the EU as part of a Risk Management Plan,
where the investigation of safety
is among study objectives.”
European regulations

17
European
regulations Situations when an EU-RMP is required
●
EU-RMP should
be
submitted
with the application for a new marketing authorisation
for●
Any
product containing
a new active substance●
A similar
biological
medicinal
product●
A generic/hybrid
medicinal
product where a safety concern
requiring
additional
risk minimisation activities has been identified with the reference medicinal
product●
with an application for a pediatric use marketing authorization●
with an application involving
a significant
change in a marketing authorisation (e.g. new dosage form, new route of administration, new manufacturing
process of a biotechnologically-derived
product, significant
change in indication including pediatric indication) unless
it has been agreed
with the Competent
Authority
that submission is not required
●
On request from a Competent
Authority
(both pre-and post-
authorisation)●
On the initiative of a MAA/MAH when they identify
a safety concern
with a medicinal
product at any stage of its life cycle
Refs: Guideline on Risk Management Systems
(CHMP/96268/2005)
incorporated in Vol
9A, Part I, Section 3
in Mar 2007; Updated
Vol9A
in Sep 2008

18
●
Dec 2010: New EU PV legislationNew EU PV legislation
in force in July 2012 ●
“Conditional”
Marketing Authorisation granted with conditions and deadlines
Penalties in case of non-compliance (Suspension or revocation of MA or Financial penalties)
●
Revised & broader definition of PASS
“Any study relating to an authorised medicinal product conducted with the aim of identifying, characterising or quantifying a safety hazard, confirming the safety profile of the medicinal product, or of measuring the effectiveness of risk management measures”
●
MAH can be required to conduct also
Post-Authorisation Efficacy (PAES) studies*:
To collect data in everyday medical practice (observational)
Request at any time in the product life cycle from initial MA
European regulations New!
* Ref
: Amendment
EU Directive 2010/84/E-
31 Dec 2010

19
New EU PV legislation
in force in July 2012 PhV
Risk Assessment Committee
(PRAC)
●
PRAC
= New scientific committee at the EMA= New scientific committee at the EMA
with a key role in the pharmacovigilance assessments
●
Scope: All
medicinal products (RMP, PSUR, PASS, signal detection, Urgent Union Procedure)●
Replaces the CHMP Pharmacovigilance Working Party (PhWP)●
At the same level
as the CHMP●
Composition:
Representatives from MS with competence in PhV
and risk assessment
6 independent scientific experts appointed by the EU Commission
Representatives from Healthcare Professionals (1+1) and Patient associations (1+1)●
Decision making
●
Final responsibility for opinion on the risk-benefit remains with CHMP (centralised
products)
or CMDh
(other products);●
CHMP or CMDh
should rely on the recommendations of the PRAC
CHMP: Committe
for human
medicinal
products; CMDh: Committe
for
mature products
New!

20
New EU PV legislation Impact on Post-Marketing activities (PSURs, RMPs, PASS,
PAES…)●●
Systematic development of postSystematic development of post--authorization non interventional (realauthorization non interventional (real--life) life) studiesstudies●
For safety (risk) as well as efficacy (benefit) => effectiveness●
Pharmacoepidemiology methods (databases, cohorts, registries…)●
Link with the ENCePP
program from the EMA●
New regulatory process for endorsement
●●
Repetitive Benefit EvaluationRepetitive Benefit Evaluation●
Medical “efficacy”
expertise to be maintained and resourced during the whole life-cycle
●●
Development of methods for assessing effectiveness of minimizatiDevelopment of methods for assessing effectiveness of minimizationon●
Accurate and detailed ongoing evaluation of exposure●
Develop methods for drug utilization studies
●●
ProactivityProactivity
in signal detection and labeling updatesin signal detection and labeling updates

21
Agenda
●
Background on Pharmacoepidemiology●
Current
European
PV regulations
●
Place of Pharmacoepidemiology
in Pharmacovigilance●
Illustrative cases
●
Conclusion

22
Safety obtained during Clinical Development is only the tip of the iceberg…
At the time of clinical development
Post-marketing surveillance
BenefitsBenefitsIntrinsic ToxicityIntrinsic Toxicity
Medication errorsMedication errorsPoor compliancePoor compliance
Wrong populationWrong populationRisk perceptionRisk perception
Efficacy+
Usage
+Condition of use
RisksRisks
Product defectsProduct defects
Tolerance
BenefitsBenefitsIntrinsic ToxicityIntrinsic Toxicity
Medication errorsMedication errorsPoor compliancePoor compliance
Wrong populationWrong populationRisk perceptionRisk perception
Efficacy+
Usage
+Condition of use
RisksRisks
Product defectsProduct defects
Tolerance
BenefitsBenefits
RisksIntrinsic Toxicity
RisksIntrinsic Toxicity
Efficacy
ToleranceBenefitsBenefits
RisksIntrinsic Toxicity
RisksIntrinsic Toxicity
Efficacy
Tolerance

23
Limited safety data at time of drug approval
●
Limited number of people exposed in RCTs●
Some type of patients may have never been enrolled
●
Lack of detection of uncommon/rare or delayed adverse drug reactions (ADRs)
●
ADRs
specific to high risk populations due to misuse of the drugs by prescribers or patients
●
Causal inference may be difficult to draw
PharmacoepidemiologyPharmacoepidemiology
cancan
contributecontribute
information about information about drugdrug
safetysafety
and and effectivenesseffectiveness
in real life setting in real life setting thatthat
isis
not not availableavailable
fromfrom
premarketingpremarketing
studiesstudies

24
Post approval Safety data
Pharmacovigilance Plan
[Risk assessment]
Continuous characterization of safety profile &
detection of newly emerging safety signals or risks
Routine Routine pharmacovigilancepharmacovigilance•Collection of ADRs•Specific forms for documentation•Expedited Reporting•Signal detection•Analysis•PSURs•DSURs
(if applicable)
Additional PV activitiesAdditional PV activities•Active surveillance (Sentinel sites (eg, OMOP), intensive monitoring schemes or )•PE studies (Case control, Cohort…)•Clinical trials•Nonclinical studies

25
Post approval Risk Minimization Activities
Prevent or minimize
safety risks
Evaluation of the need for risk minimization
Routine risk minimization:Routine risk minimization:• Legal status • Pack size• Package leaflet (PIL)• Labeling (SPC, USPI,…)
Additional Risk Minimization activities:Additional Risk Minimization activities:• Communication plan• Reminder tools• Restricted prescription • Drug registry

26
Risk Management is an iterative process! Risk assessment, Risk minimization & its effectiveness
:Sa
fety
Ris
kA
sses
smen
t
Routine: labeling
Routine: pharmacovigilance activities including PS(U)Rs
Additional: PV plan to identify and characterize specific risks, e.g.:• PASSPASS
(i.e., pharmacoepidemiology or non-clinical investigations)• Clinical studies• Others
Drug utilization studiesDrug utilization studies
(e.g., using existing databases or prescription survey)
Additional:Communication and education (e.g., Continuous Medical Education)Reminder tools (e.g. physician brochure or patient card)Restricted prescription (e.g., specialists only prescribers) Drug registry
EffectivenessA
ssessment
Min
imiz
atio
n A
ctiv
ities

27
Final outcomesindicators
Process indicators
Effectiveness of minimization measures
1.
Implementation of measures-
Measures by increasing level of constraints
-
Communication plan-
Reminder tools (i.e. prescriber checklist)-
Restricted prescription-
Mandatory drug registry-
1st assessment:
success of implementation
2.
Assessment of effectivenesswhat is the IMPACT?
Clinical knowledge
Score
Clinical actions
Appropriate use
ADR occurrence or severity
Frequency of events

28
In Summary: Main
features of post-marketing studies
●●
ObjectivesObjectives●
Safety (risk assessment and characterization)•
Causal relationship for a potential risk•
Quantification (incidence)•
Preventability: risk factors, early detection, markers & surrogates●
Other objectives•
Efficacy (benefit)•
Use (drug utilization studies) •
Cost/effectiveness
●●
““RealReal--lifelife””●
Real conditions of use: per labeling, distribution system, and reimbursement determined by the local health care organization
●
Observational setting, opposed to controlled/randomized clinical
trial situations●
Risk assessment in conditions of minimization
●●
Methods = (pharmaco)Methods = (pharmaco)--epidemiologyepidemiology●
Epidemiology designs and methods to be developed

29
Some
other
practical
aspects/questions
●●
WhenWhen
do do wewe
have to have to startstart
studiesstudies? For how long?? For how long?●
Quickly
after
launch●
Usually
several
years, until
there
is
enough
evidence
on drug
use
●●
WhereWhere
do do wewe
have to have to implementimplement
studiesstudies??●
A diversified
set of countries (i.e. at
least two
European
countries), and where
requested
or needed
as per local country
●●
WhatWhat
isis
the the reasonablereasonable
goal to goal to reachreach??●
Practical
goals to be
set, based
on labeling, past
experience, and literature
(perfect
use probaly
never
observed)
●
Goals to be
agreed
with
regulators
before
starting
the study
●●
WhatWhat
do do wewe
do do withwith
studystudy
resultsresults??●
Submission
to regulatory
agencies
(i.e.
at
time of RMP updates and/or PSUR updates)●
Results
to be
interpreted
cautiously, considering
limitations of data sources & bias

30
Agenda
●
Background on Pharmacoepidemiology●
Current
European
PV regulations
●
Place of Pharmacoepidemiology
in Pharmacovigilance●
Illustrative cases
●
Conclusion

31
Example of specific PV plan to investigate risk of hepatic risk of hepatic injuryinjury
and Risk Minimization Plan to minimize this risk
Specific
PV plan
Elevation of transaminases
enzymes (hepatotoxicity
?)
Standard PV:
ADRs Enhanced PV: use of specific form for fast capture and best evaluation of severe/serious hepatic cases
Pharmaco-epi
studies:In databases (Pharmo
in NL, Saskatchewan in Canada, GPRD in the UK, HMOs in the US) to assess risk of liver injury
Clinical studies: to increase patient exposure enhancing profile understanding /Real-life pragmatic CTMechanistic studies
(in-vitro, pharmaco-genetic, Proteonomics/ metabonomics
study)
Hepatic surveillance programme
(HSP): Voluntary although target all patients on Drug X to assess risk of liver injury
Specific
Minimization
plan
Labeling: monthly monitoring of liver enzymes (stopping rules)ContraindicationLegal status (prescription by specialists only)Limitation of prescription size (30 days)
Educational program:Introductory letter to prescribers, prescriber’s guide, patient information brochure, prescriber’s pocket card, patient card
Reminder
tools: prescriber
check list
for LFT monitoringControlled
dispensing: patients with
specific
genotyping
not authorized
to receive
drug
(if applicable)
Effectiveness
of tools
(PE studies)Cross-sectionnal
surveys
(market
research)Drug utilization
study
using
databasesDrug utilization
data through
HSP

32
Example of Nested Case Control Study Using US claims databases to evaluate severe liver injury
associated with drug x
Cases (N = 414) Controls
(N = 40,572)
Age (mean±SD), years 65.1 (11.3) 65.6 (10.8)
Sex
(women) 134 (32.4%) 12,887 (31.8%)
CHF 187 (45.2%) 11,143 (27.5%)
Diabetes 139 (33.6%) 9,620 (23.7%)
Hypertension 314 (75.8%) 29,187 (71.9%)
Stroke 43 (10.4%) 3,210 (7.9%)
AMI 37 (8.9%) 2,833 (7.0%)
Tx
duration
of drug
x(median, min, max), days
44 (0, 2274) 0 (0, 3272)
Tx
duration, others
in class 0 (0, 2475) 90 (0, 3826)
Cases Controls Crude
ORAdjusted
OR(95%CI)
Drug X vs. not
184 /230
11,782 /28,790
1.8 1.7 [1.3, 2.2]
Patients treated with drug X have 1.7 Patients treated with drug X have 1.7 times higher risk of severe liver injury times higher risk of severe liver injury than those not treated with drug Xthan those not treated with drug X

33
Example of retrospective cohort using databases to evaluate severe hepatic injury
associated with telithromycin
●Data sources: ●
Ingenix Proprietary Research Database
●
PHARMetrics Integrated Outcome Database
●Design: retrospective cohort●Applications:
●
The studies were part of the telithromycin
Risk Management Plan
commitment●
The results were presented to the FDA Advisory Committee in December 2006

34
Crude and Adjusted Risk Ratios of Severe Hepatic Injury
Crude Adjusted*
Risk ratio 95% C.I. Risk ratio 95% C.I.
AUG ** 1.00 N/A 1.00 N/A
CLA 2.00 0.82 –
4.85 1.95 0.80 –
4.73
MOX 2.90 1.17 –
7.19 2.58 1.04 –
6.43
TEL 1.37 0.51 –
3.71 1.44 0.53 –
3.89
*
Covariates age, sex, prior history of liver disease, and Charlson Index were adjusted in the GEE model
**
Augmentin (AUG) was used as a reference group in the GEE modelCLA=clarithromycin; MOX= moxifloxacin; TEL= telithromycin

35
96,7 98,9 99,798,5
49,143,1
64,2
49,1
0
10
20
30
40
50
60
70
80
90
100
Switzerland (N=273) Germany (N=369)
%
No use in CHF classIV/unstable III
No concomitant use ofCYP3A4 inhibitors
Creatinine plannedwithin one week
Creatinine plannedwithin one month
Example of evaluation of drug utilization using cross- sectional survey
Cross-sectional
surveys
to measure
prescribers’
understanding
of labeling
recommendations
and subsequent
compliance
to
contraindications
and laboratory
monitoring of Drug X

36
Example of evaluation of drug utilization
using
US claims database
Patient profile at
initiation of Drug X
Contraindication
N % [95%CI]
Diagnosis of AF/AFL 1,712 94.1 [92.9, 95.1]
Diagnosis of hypertension 1,352 74.3 [72.2, 79.6]
Diagnosis of diabetes 361 19.8 [18.0, 21.7]
Diagnosis of stroke 118 6.5 [5.4, 7.7]
Diagnosis of AMI 85 4.7 [3.7, 5.7]
≥ 1 risk factors* 1,454 79.9 [78.0, 81.7] *including hypertension, diabetes, stroke, AMI, or aged > 70 years
Prevalence of AF/AFL and cardiovascular risk factors (N = 1,820)
Worsening /hospitalized CHF # of cases % [95%CI]
Within 0-30 days prior to drug x prescription 70 3.8 [3.0, 4.8]
Prevalence of worsening / hospitalized CHF within 0-30 days before drug x prescription (N =1,820)

37
Agenda
●
Background on Pharmacoepidemiology●
Current
European
PV regulations
●
Place of Pharmacoepidemiology
in Pharmacovigilance●
Illustrative cases
●
Conclusion

38
●
Post-marketing evaluation methods are needed in the domain of Pharmacovigilance:●
To increase knowledge on drug safetyknowledge on drug safety
in real life setting ●
To increase knowledge on drug utilizationknowledge on drug utilization
in real life setting ●
To address new PV regulations and increasing requirements address new PV regulations and increasing requirements regarding PV assessment using pharmacoepidemiology
methods
A specific competency in A specific competency in PharmacoEpidemiologyPharmacoEpidemiology
is required is required
The European Network of Centres
for Pharmacoepidemiology and Pharmacovigilance
(ENCePP)•
Project led by the
European Medicines Agency (EMA)•
Goal: strengthen the post-authorisation
monitoring of medicinal products in Europe by facilitating the conduct of multi-centre, independent, post-authorisation
studies focusing on safety and on benefit/risk
http://www.encepp.eu/
Conclusion

39
Thank you!Thank you!
ありがとうありがとう !!謝謝謝謝 !!
DankeDanke!!GraciasGracias!!
MerciMerci!!

Châtenay-Malabry January 2013 40
Back-ups

41
Advantages
& Disadvantages
of
data sources
Data sources Advantages Disadvantages
Field studies ●
All data specific
to the study
needs
may
be
collected
and recorded in a consistent way for each patients (usually based on a study protocol) ● General population based●Timely information available for patients
●Regulatory
requirements
of general
AE collection, not specific
to the study●Participation rates may
be
low, questionable
representativeness●
Limited number
of study
subjects
may
be
pratically
planned
for a study●Selection
bias: (study
subjects
enrolled
may
be
different
from
the target
population)●voluntary
patients patient most
frequently
seen
in consultation «
well-
treated
»
patients●More expensive
and time-
consuming

42
Advantages
& Disadvantages
of
data sources
Data sources Advantages Disadvantages
Database
studies ●
Data already
collected, avoid
recall
bias●
Usually
large population based
data ●
All patients in the database
are included
(limited
selection
bias)●
More cost-efficient and less
time-consuming
● Lag
time of data availability●Data limited
to what’s
available
in the database●Difficult
to get
a sufficient
number
of study
subjects, if the exposure
of interest
is
uncommon●Data specific
to the database
population, may
not be
generalizable

43
ICH and CIOMS
●
ICH E2E -
Pharmacovigilance Planning (PVP)●
Tripartite harmonized guideline finalized (step 4) in November 2004●
Intended to aid in planning pharmacovigilance activities (eg, preparation for early post-marketing period of a new drug)
●
Main focus on Safety Specifications and Pharmacovigilance Plan that might be submitted at the time of license application
●
CIOMS VI -
Management of Safety Information from Clinical Trials (2005)●
Basis of Risk Management during development
ICH: International Conference
on Harmonisation; CIOMS: Council for international organizations of Medical Sciences

44
New EU PV legislation Extract from the original text
New Article 22a•
After
the granting
of a marketing authorisation, the national competent
authority
may
impose an obligation on the marketing authorisation
holder: •
(a) to conduct
a post-authorisation
safety
study
if there
are concerns
about the risks
of an authorised
medicinal
product. If the same
concerns
apply
to more than
one medicinal
product, the national competent
authority
shall, following
consultation with
the Pharmacovigilance Risk
Assessment
Committee, encourage the marketing authorisation
holders
concerned
to conduct
a joint post-authorisation
safety
study; •
(b) to conduct
a post-authorisation
efficacy
study
when
the understanding
of the disease
or the clinical
methodology
indicate
that
previous
efficacy
evaluations
might
have to be
revised
significantly. The obligation to conduct
the post-
authorisation
efficacy
study
shall
be
based
on the delegated
acts
adopted
pursuant
to Article 22b while
taking
into
account
the scientific
guidance referred
to in Article 108a.
•
The imposition of such
an obligation shall
be
duly
justified, notified
in writing, and shall
include
the objectives and timeframe
for submission
and conduct
of the study.
»
* Ref
: Amendment
EU Directive 2010/84/E-
31 Dec 2010