pharmacology review…intro?
DESCRIPTION
Pharmacology Review…Intro?. Pharmacokinetics Pharmacodynamics Drug Interactions Tolerance. Pharmacokinetics. Pharm - drug , kinetics - movement The interaction of drugs with the body - PowerPoint PPT PresentationTRANSCRIPT

1
Pharmacology Review…Intro?
• Pharmacokinetics
• Pharmacodynamics
• Drug Interactions
• Tolerance

2
Pharmacokinetics
• Pharm - drug, kinetics - movement
• The interaction of drugs with the body
• Primarily: Absorption, distribution and elimination of drugs and the factors that affect this process(s).

3
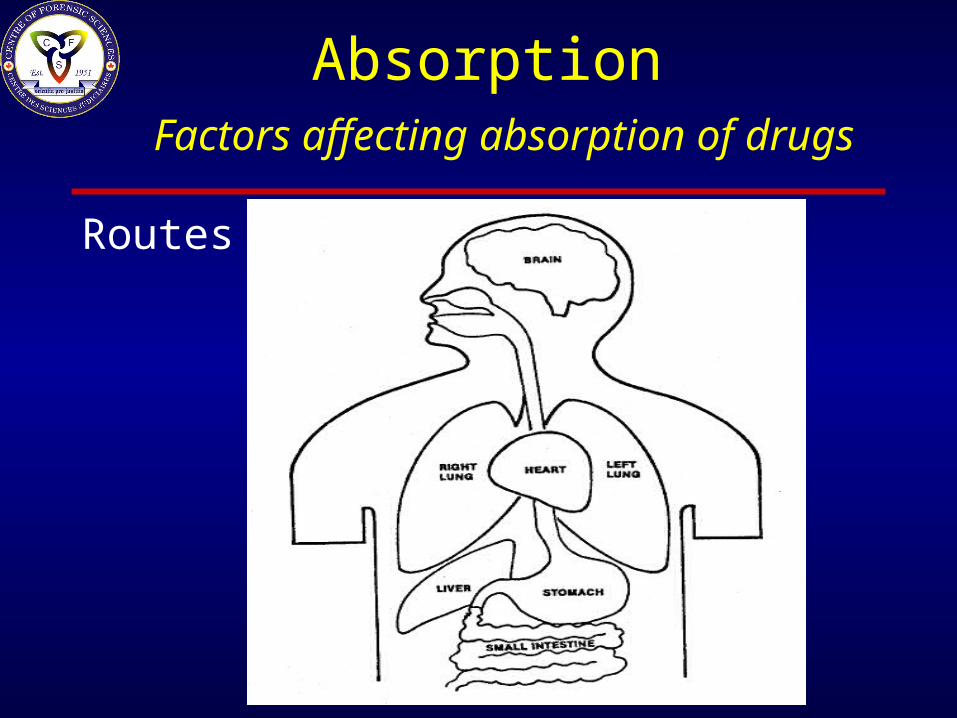
Absorption
Factors affecting absorption of drugs
– Route of Administration
– Physical properties of the drug and the tissues
(organs)

4
Absorption Factors affecting absorption of drugs
Routes of Administration– Topical: Skin & Eyes– Oral: Gastrointestinal & Sublingual – Rectal– Nasal– Inhalational (Lungs)– Parenteral: Intra- venous or muscular (IV, IM)– Intrathecal

5
Absorption Factors affecting absorption of drugs
Physical properties of the drug and the tissues (organs)
• Solubility of the drug; solubility coefficient– Lipophilic versus Hydrophilic
• pH of the solvent (drug is the solute) – Degree of Ionization (pKa)
• Concentration of the drug

6
Absorption Factors affecting absorption of drugs
Physical properties of the drug and the tissues (organs)
• Permiability of the tissue and size of the drug molecule
• Surface area of the tissue/organ• Blood supply (vasculature) of the route
tissue/organ
7
Absorption Factors affecting absorption of drugs
Routes

8
Absorption Factors affecting absorption of drugs
Liver Circulation
IV - - - -GI Tract
Liver

9
Absorption Factors affecting absorption of drugs
Routes of Administration– Topical: Skin & Eyes– Oral: Gastrointestinal & Sublingual – Rectal– Nasal– Inhalational (Lungs)– Parenteral: Intra- venous or muscular (IV, IM)– Intrathecal

10
Distribution
• Most drugs are not distributed evenly throughout the body
• Circulatory system is the primary route for distribution
• Physico-chemical properties of the drug will determine its distribution

11
DistributionFactors that affect Distribution
• Solubility (Polarity & Lipophilicity)– distribution in water versus fat compartments
• Binding to serum albumin proteins
• pKa
• Molecular size

12
• Relationship of Dose to Drug Concentration depends upon the Volume
• Drug concentrations are measured in blood (serum) only, but drugs are usually distributed into other compartments as well
DistributionVolume of Distribution
C = D
V

13
• Volume of Distribution therefore refers to the apparent distribution from blood alone
• Blood has a very high water content
• Blood represents only about 6% of the total body water.
DistributionVolume of Distribution
C = D
Vd

14
• Volume of Blood, Interstitial Fluid and Tissue Fluid = Total Body Water
–Plasma = 2.8 L (4%)–Interstitial Fluid = 9.2 L (12 L; 20%)–Intracellular Fluid = 30L (42 L; 60%)
• TBW: Males = 60-70 % / Females 50-60 %
DistributionVolume of Distribution
C = D
Vd

15
• A drug which is poorly distributed beyond the blood will have a lower Vd than a drug which distributes throughout the body, which will have a lower Vd from a drug that distributes in fat
• Barbiturates : Vd << 1 L/kg
• Ethanol : Vd = 0.53 L/kg
• THC: Vd = 4 to 14 L/kg
DistributionVolume of Distribution
C = D
Vd

16
DistributionFactors that affect Distribution
• Solubility (Polarity & Lipophilicity)– distribution in water versus fat compartments
• fat soluble drugs pass through membranes and low water tissues more rapidly
• may be distributed/stored in fat
• may be poorly taken up in the blood/need a carrier

17
DistributionFactors that affect Distribution
• Binding to serum albumin proteins
• Drug distribution to other compartments is reduced and slowed
• eg. Barbiturates. ~ 99% protein bound.
• Apparent distribution is in 2.8 L; blood concentrations are much higher than target tissues (eg. Brain cells).

18
DistributionFactors that affect Distribution
• Solubility (Polarity & Lipophilicity)– distribution in water versus fat compartments
• Binding to serum albumin proteins
• pKa
• Molecular size

19
DistributionFactors that affect Distribution
• pKa (Ionization)
• CH3-CH3-COOH CH3-CH3-COO-
• Ionized forms are more polar, thus retarded by cell membranes, but more soluble in water

20
DistributionFactors that affect Distribution
• Molecular Size
• Small molecules (Ethanol) CH3-CH20H pass rapidly through cell membranes without uptake mechanisms
• Larger molecules (Mannitol) are retarded and may be excluded from some compartments

21
Elimination
• Definition
• Removal of the drug by either – metabolism (conversion) to another subtance– excretion of the drug in unchanged form

22
EliminationRoutes
• Liver
• Kidney
• Lungs
• Sweat
• Hair

23
EliminationLiver
• Major site of metabolism
• Enzymatic
• Type I - modification of the drug by hydrolysis, methylation, oxidation etc.
• Type II - conjugation to a large polar molecule (eg glucuronidation)

24
EliminationLiver
• “Goal” to make the drug more – soluble in water (excretion via Kidneys)– soluble in bile (excretion via feces).
• Liver takes 100% of blood from stomach and intestines and 40% total circulation
• Major metabolic and de-toxifying organ

25
EliminationKidneys
• Major site of water and salt balance and nitrogen (urea) elimination.
• Filters the blood
• Polar drugs and metabolites will pass into the urine with water
• Ionization status will also affect excretion

26
Minor Routes of Elimination
• Lungs– Drug must be volatile (eg. Inhaled anaesthetics
and alcohols)
• Sweat– polar, water soluble drugs will pass into the sweat
• Hair– deposition from growing end, drugs sequestered
in dead hair cells outside of body

27
Elimination Kinetics
• Enzymatic elimination of drugs primarily First Order Kinetics (Michaelis-Menton)
C =Coe-kt
• Elimination half life t 1/2 = 0.693/k

28
Elimination Kinetics (Michaelis-Menton)
Actual change in concentration over time
Logarithimic change in concentration over time

29
Elimination Kinetics
•Elimination is dependent upon concentration, but almost 97% will be eliminated after 5 half-lives
Initial concentration 1 mg% One half life 0.5 mg% Two half lives 0.25 mg%Three half lives 0.125 mg% Four half lives 0.0625 mg% Five half lives 0.03125 mg% or 0.96875 mg% (97%) eliminated

30
Non-Enzymatic Breakdown
• Some drugs are unstable at physiological pH or in either acid or base conditions
• These drugs will breakdown to products over time (the equivalent of metabolism and elimination)
• The problem for Forensic Toxicology is that these processes can occur postmortem and especially in-vitro.

31
Non-Enzymatic BreakdownExample Cocaine
• Cocaine is metabolized to ecgonine methylester (EME) by an esterase in the blood (pseudocholinesterase)
• Cocaine also breaks down over time to Benzoylecgonine (BE), especially at physiological and alkaline pH

32
Non-Enzymatic BreakdownExample Cocaine
• Preservatives (chemicals which bind enzymes and prevent postmortem enzymatic changes and tissue breakdown) will stop the metabolism of cocaine to EME, but not the breakdown of cocaine to BE.
• Thus some or all of the BE detected in a sample may have been cocaine prior to death

33
Steady State
• At therapeutic concentrations drugs taken over a period of time attain Css Steady state concentration
Blo
od c
once
ntra
tion
Time
Mean Css

34
Kinetics in Overdose
• Absorption, Distribution and especially elimination of drugs can change in overdose
• Large amounts of drugs in the stomach cause ‘concretions’ which reduces absorption

35
Kinetics in Overdose
• Distribution may be altered, for example protein binding sites in the blood may become saturated - Vd may change
• Elimination kinetics may change; often dramatically, when enzymes become saturated and enter zero-order kinetics, an increase in dose will now lead too much greater blood concentrations with an altered half-life

36
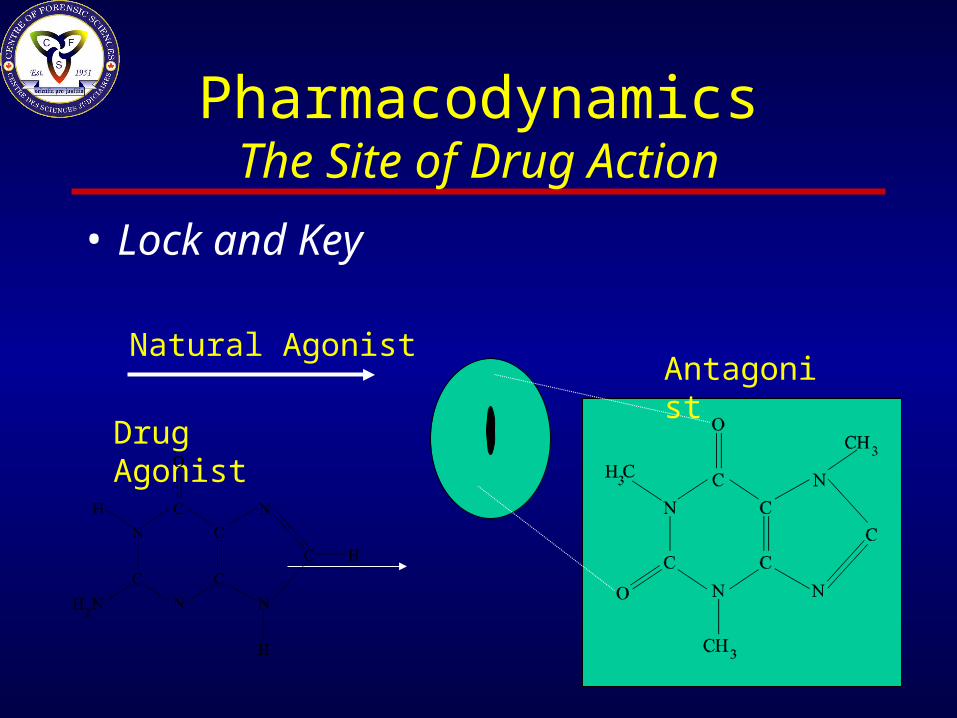
PharmacodynamicsThe Site of Drug Action
• Agonist -a molecule that ‘fits’ a receptor by either its spacial and/or ionic properties
• Antagonist - a molecule which blocks a receptor by either binding to a non-activating site or physically blocking the site (or causing only very weak activity).
• Most drugs mimic natural agonists
37
PharmacodynamicsThe Site of Drug Action
• Lock and Key
Natural Agonist
Drug Agonist
Antagonist

38
PharmacodynamicsThe Site of Drug Action
• Antagonist effects
• Blocking metabolism - re-uptake inhibitors allow the natural molecule (cocaine - dopamine; Prozac etc - serotonin) to remain longer at the site of action
• May have beneficial or detrimental effects
• May also act as an antidote

39
PharmacodynamicsThe Site of Drug Action
• Some drug families are based on receptor binding eg. Opioids- related to morphine; all opioids bind to one or more of the three classes of opioid receptors
• Receptors may activate inhibitory pathways; thus agonists may produce inhibitory responses while antagonists may produce excitatory responses (e.g. GABA pathways).

40
Drug - Drug InteractionsKinetic Effects
• Drugs may be metabolised by the same enzyme system(s).
• Consequence: Induction or inhibition of metabolic pathways by one drug resulting in increasing or decreasing blood concentrations of the other drug (Has implications for the drug with the lowest therapeutic index/greatest disease impact).

41
Drug - Drug InteractionsExample Warfarin
• Warfarin is taken as an anticoagulant to combat thrombosis (blood clots) which may break free and damage the heart or lungs.
• Warfarin is highly protein bound
• Warfarin is metabolised by P450 enzyme system (more about this later).

42
Drug - Drug InteractionsExample Warfarin
• Phenobarbital is also highly protein bound and induces the P450 system.
• Consequence of someone on warfarin who takes a course of phenobarbital:– Increased free warfarin offset by increased
metabolism; net result is increased dose of warfarin needed to achieve anticoagulation.
– If dose not lowered at end of phenobarbital treatment, bleeding due to warfarin may result

43
Drug - Drug Interactions Kinetic Effects
• Drug Interactions may also be beneficial, e.g. probenecid blocks movement of penicillin into the urine resulting in higher blood concentrations at lower doses and more effective antibiotic treatment.

44
Drug - Drug InteractionsTarget Effects
• Drugs may have similar (synergistic) effects (e.g. sedation) which may be additive (similar recptors) or supra-additive (two or more targets) or infra-additive (similar receptors; agonist and antagonist)
A+B = C
A+B > C
A+B < A or B

45
Drug - Drug InteractionsPharmacokinetics
• Absorption– Chemical incompatability of two drugs– Prevention of absorption or delay (e.g.
decreased gastric motility by opioids)
• Distribution– Changes in protein binding– Inhibition in Liver uptake– Change in circulatory flow

46
Drug - Drug InteractionsPharmacokinetics
• Elimination– Enzyme Induction– Enzyme inhibition or competition– Inhibition of recirculation from the bile– Changes in Renal processes
• acidic urine facilitates the uptake of bases (and vice versa) into the urine
• Reabsorption of some drugs back into the circulation may be blocked

47
Tolerance
• Two primary aspects of tolerance are Metabolic and Functional
• Metabolic (primarily hepatic).
• Functional (CNS)

48
Tolerance
Metabolic• Dispositional• Kinetic• Primarily Liver• Can involve all drugs• Change in [blood]• enzyme induction• no abstinance
(withdrawl) syndrome
Functional• CNS• Dynamic (receptor)• Brain• CNS drugs only• No Change in [blood]• learning-like/change
in excitation status• abstinance syndrome

49
Tolerance
• Cross tolerance for drugs using the same
• Tolerance level is moderate (up to 35% increase in dose)
• Cross tolerance for drugs with the same effect
• Tolerance level is high (up to 200% increase in dose)

50
Tolerance Stimulants
• Functional tolerance develops rapidly and large increases in dose can occur over one drug session e.g. crack cocaine binges
• however metabolic tolerance dose not develop as rapidly; deaths due to non-CNS effects, especially cardiac or vascular and/or thermoregulatory causes are common.

51
ToleranceCNS Depressants
• Functional tolerance develops more slowly and is lost over a shorter period than acquisition.
• One major problem is respiratory depression; the respiratory centre is located in the medulla and most if not all sedatives depress respiration - tolerance to this effect, especially for opioids, dose not occur

52
References
• Principles of Medical Pharmacology (Kalant & Roschlau eds.) 6th ed., Oxford Publ., 1998.
• Goodman and Gilman’s The Pharmacological basis of Therapeutics (Hardman & Limbird eds-in-chief) 9th ed., McGraw-Hill, 1996