pharmacovigila final 2
TRANSCRIPT

DRUG –DRUG INTERACTIONS(PHARMACOKINETICS AND DYNAMICS)
The mechanisms by which drug interactions alter drug distribution include (1) competition for plasma protein binding, (2) displacement from tissue binding sites, and (3) alterations in local tissue barriers, eg, P-glycoprotein inhibition in the blood-brain barrier.
Although competition for plasma protein binding can increase the free concentration (and thus the effect) of the displaced drug in plasma, the increase will be transient owing to a compensatory increase in drug disposition.
The clinical importance of protein binding displacement has been overemphasized; current evidence suggests that such interactions are unlikely to result in adverse effects. Displacement from tissue binding sites would tend to transiently increase the blood concentration of the displaced drug.

When drug interactions associated with protein binding displacement and clinically important effects have been studied, it has been found that the displacing drug is also an inhibitor of clearance, and it is the change in clearance of the unbound drug that is the relevant mechanism explaining the interaction.
Concurrent administration of two nephrotoxic drugs can produce kidney damage even though the dose of either drug alone may have been insufficient to produce toxicity. Furthermore, some drugs can enhance the organ toxicity of another drug even though the enhancing drug has no intrinsic toxic effect on that organ.

• These estimates are intended to indicate simply whether or not the interaction will occur and do not always mean that the interaction is likely to produce an adverse effect.
• Whether the interaction occurs and produces an adverse effect or not depends upon
(1) the presence or absence of factors that predispose to the adverse effects of the drug interaction (diseases, organ function, dose of drugs, etc) and
(2) awareness on the part of the prescriber, so that appropriate monitoring can be ordered or preventive measures taken.
Sulfinpyrazone: [HP] Decreased uricosuric effect of sulfinpyrazone (interaction unlikely with less than 1.5 g of salicylate daily).Rifampin :Induction of hepatic microsomal drug-metabolizing enzymes

• Phenytoin ;Induces hepatic microsomal drug metabolism. Susceptible to inhibition of metabolism by CYP2C9 and, to a lesser extent, CYP2C19. Corticosteroids: [P] Decreased serum corticosteroid levels. Doxycycline: [P] Decreased serum doxycycline levels.Theophylline: [NE] Decreased serum theophylline levels.Verapamil: [NE] Decreased serum verapamil levels.Chloramphenicol: [P] Increased serum phenytoin.
• Most NSAIDs are highly bound to plasma proteins. Phenylbutazone may inhibit hepatic microsomal drug metabolism (also seems to act as enzyme inducer in some cases). Phenylbutazone may alter renal excretion of some drugs.ACE inhibitors: [P] Decreased antihypertensive response. Furosemide: [P] Decreased diuretic, natriuretic, and antihypertensive response to furosemide.Hydralazine: [NE] Decreased antihypertensive response to hydralazine.

• Monoamine oxidase inhibitors: [P] Hypertensive reaction (carbidopa prevents the interaction).Papaverine: [NE] Inhibits antiparkinsonism effect. Phenothiazines: [P] Inhibits antiparkinsonism effect. Phenytoin: [NE] Inhibits antiparkinsonism effect. Pyridoxine: [P] Inhibits antiparkinsonism effect (carbidopa prevents the interaction).
• Disulfiram Inhibits hepatic microsomal drug-metabolizing enzymes. Inhibits aldehyde dehydrogenase.Benzodiazepines: [P] Decreased metabolism of chlordiazepoxide and diazepam but not lorazepam and oxazepam.
• Chloramphenicol Inhibits hepatic drug-metabolizing enzymes. Phenytoin: [P] Decreased phenytoin metabolism.Sulfonylurea hypoglycemics: [P] Decreased sulfonylurea metabolism.

• Carbamazepine Induction of hepatic microsomal drug-metabolizing enzymes. Susceptible to inhibition of metabolism, primarily by CYP3A4.Cimetidine: [P] Decreased carbamazepine metabolism.Clarithromycin: [P] Decreased carbamazepine metabolism. Corticosteroids: [P] Increased corticosteroid metabolism.
• Allopurinol ;Inhibits hepatic drug-metabolizing enzymes.Anticoagulants, oral: [NP] Increased hypoprothrombinemic effect. Azathioprine: [P] Decreased azathioprine detoxification resulting in increased azathioprine toxicity.
• The disease to be treated may be drug-induced, or drugs being taken may lead to interactions with drugs to be prescribed. Maintain a high index of suspicion regarding drug reactions and interactions. Know what other drugs the patient is taking.


Pharmacokinetic interactions between drugs arise if one drug changes the absorption, distribution, metabolism, or excretion of another drug, thereby altering the concentration of active drug in the body.drugs can inhibit or induce hepatic P450 enzymes.
Toxicokinetics is the quantitation of the time course of toxicants in the body during the processes of absorption, distribution, biotransformation, and excretion or clearance of toxicants. In other words, toxicokinetics is a reflection of how the body handles toxicants as indicated by the plasma concentration of that xenobiotic at various time points .The end result of these toxicokinetic processes is a biologically effective dose of the toxicant.
Toxicodynamics refers to the molecular, biochemical, and physiological effects of toxicants or their metabolites in biological systems.These effects are result of the interaction of the biologically effective dose of the ultimate (active) form of the toxicant with a molecular target

Adverse toxic reactions• Toxicology is the study of poisons.Poisons are chemical/physical
agents that produce adverse responses in biological organisms• Is far from being unified
– Unwanted, adverse, side or toxic…effects/reactions– Effects (of drugs) vs reaction (of patients)
• adverse drug reaction (WHO def.) = unintended and noxious (harmful) response that occurs at normal doses of the drug used for prophylaxis, diagnosis and treatment of diseases– A, B, C, D, E – CLASSIFICATION !!!– They often require change of dose/dosage schedule or drug
withdrawal. – Sometimes Side effects (collateral effects) are distinguished
– the weak form of the adverse effect which is unpleasant but generally acceptable. The marked changes in dosage schedule or drug withdrawal are usually not necessarily.
– E.g. weak sedation with H1-antihistamines, constipation with opioids, dry mouth with antimuscarinics
• Attention! The term side effects is often used as a synonym to adverse effects.

• Toxic drug reaction– Unintended, primarily harmful and reactions occurring at high
(supratherapeutic) doses and/or after long treatment (acute or chronic overdose).
– Toxic effects are often associated with morphologic changes which might be irreversible.
– Reasons:• Iatrogenic intoxication – medication error, critical situations when high drug
doses are needed• Non-compliance and patients errors (multiple pharm. prep. with same active
drug), self-administration (overdose) in children• Suicidal attempts (antidepressants...)
– Paracelsus: only the dose makes the difference between the drug and poison
– Precise preclinical characterization of toxic drug effects is a mandatory part of the request for approval of the drug for clinical investigation and the same applies for final approval for its clinical use
Adverse toxic reactions terminology

Drug toxicity can occur on many different time scales. Acute toxicity results from a single exposure to a drug, with adverse effects resulting in minutes to hours. Examples of acute toxicity are the massive hepatic necrosis that can occur after a single toxic dose of acetaminophen and exacerbations of acute bronchoconstriction in patients with aspirin-intolerant asthma. Many immune-mediated adverse effects occur within hours to days after administration of the drug.
Chronic toxicity, on the other hand, refers to an adverse effect of a drug that occurs over a prolonged period of time.Long-term treatment with dopamine receptor antagonists for schizophrenia can result in tardive dyskinesia, an unfortunate on-target adverse effect that results from the critical role of dopamine as a neurotransmitter in the motor cortex.Hormone replacement therapy for postmenopausalwomen is another important example of chronic toxicity.While the administration of estrogens significantly reduces several of the effects of menopause (e.g., hot flashes, vaginal atrophy, and skin thinning), continued activation of the estrogen receptor pathway can lead to endometrial cancer.

• Are induced by high single doses or long-term therapy leading to high cumulative doses.
– Doses/duration of treatment is supratherapeutic!
• The safety for therapeutic use is defined by TI – Drugs with low TI values are approved to get in to the clinical
practice only in the case of life-saving indications where risks do not overweight the benefits
• They can be induced and manifested by – As extremely escalated therapeutic effects (e.g., overdose with
anticoagulant drugs induce life-threatening bleeding– By totally different mechanisms and symptoms with no
relationship to pharmacological action • Covalent interactions often occur with destruction of biomolecules
and histopathological findings which might be irreversible
Toxic drug effects

Toxic effects
• Possible molecular consequences of the drug-induced toxicity– ROS production (often the metabolite is reactive radical)
with subsequent oxidative damage to biomolecules (lipids, proteins, DNA)
– Ca2+ overload – activation of Ca-dependent proteases, Ca accumulation in mitochondria and impact on MPTP – depolarization of mitochondria
– Impaired ATP production– Direct impact on gene expression– Activation of proteolytic cascades – Triggering of apoptosis
Prevention: reduction of individual dose, number of individual dosage forms, monitoring of pharmacotherapy

• Treatment: – Non-specific treatment: to prevent or reduce further
drug absorption, to accelerate drug elimination and support of vital functions
– Specific treatment: with antidotes taking advantage of specific antagonisms (mostly pharmacological)
Evaluation of toxic effects of drugs
• Overlap of pharmacology and toxicology• Paracelsus postulate• MUST involve in vivo testing on experimental animals
– In vitro testing has only limited values for regulatory purposes
• Indispensable part of preclinical files of each drug which should be– Approved for testing on human beings– Approved for use in clinical practice

• Acute toxicity studies (TD50, LD50 – TI determination), subchronic toxicity studies (90 days) and chronic toxicity studies (at least 1 year)
– Choice of animal species, strain, age, sex is of critical importance – Control group – receives only drug vehicle, otherwise all must be same as in the tested group– Animal randomization into the groups (tested and control)– After repeated administration testing – the investigators look for the signs of drug
accumulation, link to toxicokinetics
• Evaluated parameters: general toxicity – e.g., changes in appearance, behavior, weight gain…
• Identification of target organ toxicities using histopathological an biochemical, hematological approaches
Drugs and organ toxicity
• Nephrotoxicity– Aminoglycosides, cyclosporin, ACE-inhibitors, NSAIDs, cisplatin, amphotericin B,
paracetamol• Hepatotoxicity
– Paracetamol, isoniazid, halothan, methotrexate• Neurotoxicity – vinca alcaloids• Ototoxicity – gentamicin, furosemide • Cardiotoxiicty
– anthracyclines, trastuzumab, tytosinkinase inhibitors, catecholamines– digoxin, antiarrhythmics
• GIT-toxicity – NSAIDs, cytostatics• Phototoxicity – piroxicam, diclofenac a sulfonamides,
hydrochlorothiazide

• Special attention must be paid on elderly patients and patients with prior kidney disease
• Renal function biomarker: creatininemiaAminoglycosides: active (saturable) transport into the tubularcells
– ROS production, lysosomal enlargement and phospholipids inside, apoptosis
– tubular toxicity– Reduced glomerular filtration, increased creatinineamia, and
blood urea – renal failure!– Once daily– Special risks in newborn (esp. Immature)
• Tubular toxicity also in: cisplatin, vankomycin• Endotelial toxicity: cyclosporin, tacrolimus• Decreased renal perfusion (due to the vasoconstriction):
NSAIDs, cyclosporin, tacrolimus, amphotericine B• Crystaluria: sulfonamides, acyclovir
Drugs and organ toxicity- nephrotoxicity

Drugs and organ toxicity - nephrotoxicity
NSAIDs:1. Single high dose induced acute renal failure with oligouria (due to
the vasoconstriction and drop in GF)2. Chronic analgesic nephropathy– papillary necrosis, chronic
interstitial nephritis (ischemia?). Irreversibility !!!3. Interstitial nephritis (rare) – increased creatininemia with proteinuria
(reversible, return to normal after 1-3 months
• Cyclosporin– Acute reversible renal dysfunction (due to the vasoconstriction)– Acute vasculopathy (non-inflammatory injury to arterioles and
glomerulus)– Chronic nephropathy with interstitial fibrosisRenal hypertension is frequent!!!!
• Cisplatin – acute and chronic renal failure (focal necrosis in in multiple segments of the nephron)
• Paracetamol – in overdose: necrosis of cells of proximal tubules• ACE-inhibitors – in higher doses, esp. captopril, in bilateral stenosis
of renal artery – risk of severe acute renal failure

• The most important case: paracetamol overdose– Paracetamol is a very safe drug in normal doses (OTC drug)– However, in overdose (10-15g in a healthy adults) it leads to
life-threatening hepatotoxicity and nephrotoxicity• Responsible is a reactive metabolite N-acetyl-p-
benzoquinon imin, which oversaturates its detoxification metabolism based on conjugation with GSHThis triggers a severe oxidative stress in hepatocytes which results in to the damage of biomolecules and necrotic cell death of hepatocyte
Risk factors – age (more likely in children), alcoholism, liver disease in anamnesis
• Treatment: acetylcysteine i.v. ASAP – donates SH to reduce GSH depletion in the liver
Risk of hepatocellular necrosis also in: halothan and isoniazid
Hepatic cirrhosis/fibrosis – methotrexate after long-term use
Cholestatic hepatitis: chlorpomazine, estrogens, cyclosporin
Drugs and organ toxicity - hepatotoxicity
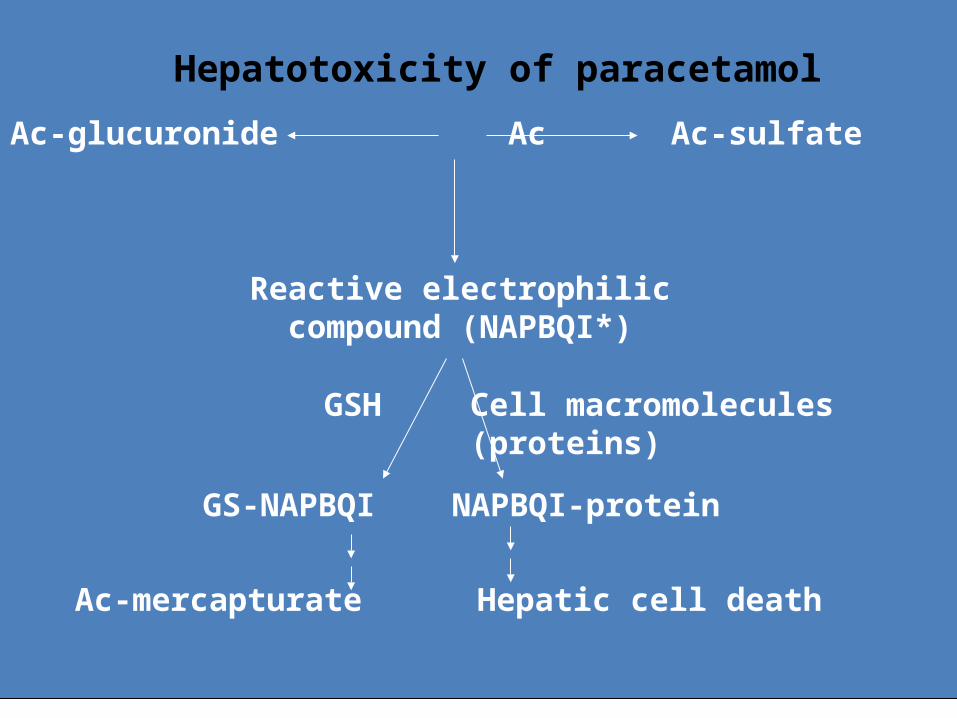
Ac-glucuronide Ac Ac-sulfate
Reactive electrophiliccompound (NAPBQI*)
GSH Cell macromolecules(proteins)
GS-NAPBQI NAPBQI-protein
Ac-mercapturate Hepatic cell death
Hepatotoxicity of paracetamol

• Impaired cardiac function – induction of arrhythmias– Most of antiarrhythmics have also proarhythmogenic effects– DAD after digoxin – bigeminias, trigeminias etc– Methylxantins – theophylline– Tricyclic antidepressants - amitriptyline– Drug-induced long QT syndrome - predisposition for
polymorphic ventricular arrhythmias of the torsade de point type, which may be fatal
• Safety pharmacology- QT interval testing in new drugs• Reason for drug withdrawal from market in many cases:
cisapride, terfenadine…• The risk is present in some currently prescribed drugs:
drugs in psychiatry: some antidepressants and antipsychotics, macrolides, fluoroquinolones…
Drugs and organ toxicity - cardiotoxicity

Drugs and organ toxicity - cardiotoxicity• Induction of cardiomyopathy and/or chronic heart failure • Anthracyclines, trastuzumab, tyrosinkinase inhibitors (sunitinib),
tacrolimus, reverse transcriptase inhibitors– Anthracycline cardiotoxicity
• Acute – mostly subclinical ECG changes• Subacute – myocarditis-pericarditis (rarely seen)• Chronic (within 1 year) • Delayed (late onset, 1-20 years after the chemotherapy)
– Chronic and delayed forms depend on the cumulative dose
– Options for prevention: pharmacological cardioprotection with dexrazoxane, targeted distribution of anthracyclines (liposomes)
– Mechanism of toxic action? The classic „ROS and iron“ hypothesis but it is rather multifactorial and less sure

• Pulmonary Toxicity Inhalation of silica (mining and quarrying area)causes fibrosis or formation of collagenous tissue. If large, can impair respiration.
• Asbestosis: may cause lung cancer.• Cigarette smoke has polycyclic aromatic hydrocarbons such a benzo(a)pyrene which is metabolized by cytochrome P450. The metabolites may trigger cancer.
Reproductive toxicity• A large number of chemicals are toxic to the male or female reproductive system. Can cause decreased sperm count.Some environmental chemicals such as dichlorodiphenyltrichloroethane (DDT),DDE etc can mimic human estrogen. They are called“environmental estrogens”. In wildlife they cause hermaphroditic fish and other reproductive anomalies (sex reversal) in alligators.
Late 1970-early 1980s--->Chemicals can alter functions of the immune system. Such toxicity is called Immunotoxicity.Several of these chemicals are environmental pollutants: organo metals, halogenated hydrocarbons, carbamates, organophosphates.Chemicals can alter the immune functions at very low concentrations--doses that do not produce organ toxicity. Example: 0.25 ng of TCDD(dioxin) can increase the susceptibility of mice to influenza virus.

• Heavy metals exert their toxic effects by combining with one or more reactive groups (ligands) essential for normal physiological functions. Heavy-metal antagonists (chelating agents) are designed specifically to compete with these groups for the metals and thereby prevent or reverse toxic effects and enhance the excretion of metals.
• Heavy metals, particularly those in the transition series, may react in the body with ligands containing oxygen (-OH, -COO-, - OPO3H-, >CO), sulfur (-SH,-S-S-), and nitrogen (-NH2 and >NH). The resulting metal complex (or coordination compound) is formed by a coordinate bond in which both electrons are contributed by the ligand.
• The stability of chelates varies with the metal and the ligand atoms. For example, lead and mercury have greater affinities for sulfur and nitrogen than for oxygen ligands; calcium, however, has a greater affinity for oxygen than for sulfur and nitrogen. These differences in affinity serve as the basis of selectivity of action of a chelating agent in the body.

• The effectiveness of a chelating agent for the treatment of poisoning by a heavy metal depends on several factors, including the relative affinity of the chelator for the heavy metal as compared with essential body metals, the distribution of the chelator in the body as compared with the distribution of the metal, and the capacity of the chelator to mobilize the metal from the body once chelated.
GI absorption of lead varies with age; adults absorb approximately 10% of ingested lead, whereas children absorb up to 40%.
• Iron deficiency also enhances intestinal absorption of lead apparently because in the absence of iron, the divalent metal transporter (DMT) can readily transport lead in place of iron. Absorption of inhaled lead varies with the form (vapor versus particle) as well as with concentration. Approximately 90% of inhaled lead particles from ambient air are absorbed

CaNa2EDTA is initiated at a dose of 30 to 50 mg/kg per day in two divided doses either by deep intramuscular injection or slow intravenous infusion for up to 5 consecutive days. • The first dose of CaNa2EDTA should be delayed until 4 hours after the
first dose of dimercaprol. Colic may disappear within 2 hours; paresthesia and tremor cease after 4 or 5 days.Urinary elimination of lead usually is greatest during the initial infusion.
Dimercaprol is given intramuscularly at a dose of 4 mg/kg every 4 hours for 48 hours, The combination of dimercaprol and CaNa2 EDTA is more effective than is either chelator alone.
Penicillamine is effective orally and may be included in the regimen at a dosage of 250 mg given four times daily for 5 daysSuccimer. Succimer is the first orally active lead chelator available for children, with a safety and efficacy profile that surpasses that of D-penicillamine. Succimer usually is given every 8 hours (10 mg/kg) for 5 days and then every 12 hours for an additional 2 weeks.

General Principles. In any chelation regimen, the blood lead concentration should be reassessed 2 weeks after the regimen has been completed; an additional course of therapy may be indicated if blood lead concentrations rebound. Treatment of organic lead poisoning is symptomatic. Chelation therapy will promote excretion of the inorganic lead produced from the metabolism of organic lead, but the increase is not dramatic.
Short-term exposure to the vapor of elemental mercury may produce symptoms within several hours, including weakness, chills, metallic taste, nausea, vomiting, diarrhea, dyspnea, cough, and a feeling of tightness in the chest. Pulmonary toxicity may progress to an interstitial pneumonitis with severe compromise of respiratory function.
Chronic exposure to mercury vapor produces a more insidious form of toxicity that is dominated by neurological effects . The syndrome, termed the asthenic vegetative syndrome, consists of neurasthenic symptoms in addition to three or more of the following findings: goiter, increased uptake of radioiodine by the thyroid, tachycardia, labile pulse, gingivitis, dermographia, and increased mercury in the urine .
With continued exposure to mercury vapor, tremor becomes noticeable, and psychological changes consist of depression, irritability, excessive shyness, insomnia, reduced self-confidence, emotional instability, forgetfulness, confusion, impatience, and vasomotor disturbances (such as excessive perspiration and uncontrolled blushing, which together are referred to as erethism). Common features of intoxication from mercury vapor are severe salivation and gingivitis.

The short-chain organic mercurials, especially methylmercury, are the most difficult forms of mercury to mobilize from the body presumably because of their poor reactivity with chelating agents.
Dimercaprol is contraindicated in methylmercury poisoning because it increases brain concentrations of methylmercury in experimental animals. Although penicillamine facilitates the removal of methylmercury from the body, it is not clinically efficacious, and large doses (2 g/day) are needed
Clinical experience with various treatments for methylmercury poisoning in Iraq indicates that penicillamine, N-acetylpenicillamine, and an oral nonabsorbable thiol resin all can reduce blood concentrations of mercury; however, clinical improvement was not clearly related to reduction of the body burden of methylmercury .
With long-term use, penicillamine induces several cutaneous lesions, including urticaria, macular or papular reactions, pemphigoid lesions, lupus erythematosus, dermatomyositis, adverse effects on collagen, and other less serious reactions, such as dryness and scaling. Cross-reactivity with penicillin may be responsible for some episodes of urticarial or maculopapular reactions with generalized edema, pruritus, and fever that occur in as many as one-third of patients taking penicillamine

• Arsine gas, generated by electrolytic or metallic reduction of arsenic in nonferrous metal products, is a rare cause of industrial intoxication. A few hours after exposure, headache, anorexia, vomiting, paresthesia, abdominal pain, chills, hemoglobinuria, bilirubinemia, and anuria occur. The classic arsine triad of hemolysis, abdominal pain, and hematuria is noteworthy. Jaundice appears after 24 hours.
• Deferoxamine is isolated as the iron chelate from Streptomyces pilosus and is treated chemically to obtain the metal-free ligand. Deferoxamine has the desirable properties of a remarkably high affinity for ferric iron (Ka = 1031) coupled with a very low affinity for calcium (Ka = 102).
• Studies in vitro have shown that it removes iron from hemosiderin and ferritin and, to a lesser extent, from transferrin. Iron in hemoglobin or cytochromes is not removed by deferoxamine.
• Deferoxamine is metabolized principally by plasma enzymes, but the pathways have not yet been defined. The drug also is excreted readily in the urine.
Deferoxamine causes a number of allergic reactions, including pruritus, wheals, rash, and anaphylaxis. Other adverse effects include dysuria, abdominal discomfort, diarrhea, fever, leg cramps, and tachycardia. Occasional cases of cataract formation have been reported.

Principles in management of Poisoned PatientNecessary measures to prevent further deterioration of the patient; 1. Stabilization of the patient,2. Diagnosis of the poison,3. Prevention and treatment of poisoning,4. Administration of antidotes (specific antidotes or using the antidote cocktail),5. Continuing care.
1. Stabilization of the patient (ABCDEs measures)A. Evaluation of Airway obstruction Causes (Mucosal swelling, Secretions, Posterior displacement of the tongue and Foreign
bodies). Signs and Symptoms (Dyspnea, Dysphoria, Air hunger, Cyanosis, Diaphoresis and Tachypnea). Measures (Clearing the airway, use of nose-pharyngeal tube, Intubation or Crico-
thyroidotomy).
B. Evaluation of Breathing (by ventilation and oxygenation) Causes (Respiratory depressant drugs, Pneumonia, Pulmonary edema, Lung abscess,
Pulmonary emboli, Bronchospasm from numerous environmental & occupational sources and Tetanus).
Signs and Symptoms (Tachypnea, Cyanosis, Hypoventilation and altered mental state).Evaluated by measuring of blood gases (PaCO2, PaO2), Chest X-ray, or Tidal volume. Measures (Assisted ventilation and supplemental O2 delivered by nasal catheters and
cannulae).

Principles in management of Poisoned Patient (cont.)
C. Evaluation of (C) Circulation
Signs and Symptoms of inadequate tissue perfusion is shock (Depressed
consciousness, Decreased blood pressure, Peripheral vasoconstriction, Metabolic
acidosis and Oliguria)
Treatment (Position change, Vasopressors as Dopamine and Norepinephrine, and
Fluids).
D. Evaluation of Depression (D) or Excitation (E)
Depression is evaluated by (measuring the pupillary size, pupillary light reflex,
motor responses to pain, and /or spontaneous eye movements).
Treatment of depressed patient (coma cocktail: Glucose, Thiamine & Naloxone )
Excitation is manifested as seizures.
Treatment of generalized seizures secondary to toxins (Diazepam, Phenytoin,
Phenobarbital, General anaesthesia, Enhancement of drug elimination by
Hemodialysis).
Treatment of violent patient (Benzodiazepines with Haloperidol and stabilization
of blood glucose level).

2. The diagnosis of poisons
Once the patient has been stabilized, the potential poison has to be identified. The
diagnosis of poisoning involves the following;
1. History given by the patient himself or relatives.
2. Physical examination of the patient.
3. Laboratory investigations.
I. History
Adults (Conscious or unconscious patients).
Children (presence of traces, disintegrated tablets, abnormal behaviour or GIT
disturbances).
II. Physical examination of the patient (Blood pressure, pulse, respiration, temperature,
eyes, mouth, skin, abdomen, nervous system).
III. Laboratory investigations (Toxicant extraction from Urine, Blood, Hair, Meconium,
Saliva or Sweat samples, screening by TLC, GLC, Enzyme-mediated immunoassay
techniques.
Principles in management of Poisoned Patient (cont.)

3. Prevention and treatment of poisoningA- Non ingested poison
1. Inhalation exposures Immediate, cautious removal of the patient from the hazardous environment. Administration of 100% humidified O2, assisted ventilation, and bronchodilators. Observe for edema of the respiratory tract and later non-cardiogenic pulmonary edema. Arterial blood gas assays, chest examination, and blood tests for the criminal substance
(e.g., cyanide) should be performed. Treatment should not await laboratory results. 2. Dermal exposures Attendees should wear protective gear (gloves, gown, shoe covers). Remove the patient’s contaminated clothes, contact lenses, and jewelry immediately. Gently rinse and wash the skin with copious amounts of water for at least 30 minutes. Do not use forceful flushing in a shower. Use slightly cold water and soap of oily substances. Toxic substances such as organophosphorous compounds, metal compounds, phenol, may
penetrate the intact skin and must be handled with proper protective equipment.3. Ocular exposures Ocular decontamination consists of at least 15 minutes of immediate irrigation of eyes with
normal saline or water. Alkaline or acid irrigating solutions should be avoided. Continue to irrigate the eye for as long as the pH is abnormal. Alkaline corneal burns are requiring ophthalmic consultation.
Principles in management of Poisoned Patient (cont.)

B- Ingested poison1- Dilution of the poison with water .2- Prevention of further absorption of poison. Induction of Emesis (Syrup of ipecac and Apomorphine). Gastric lavage (noso-gastric or an oro-gastric tube). Adsorption by activated charcoal (exceptions poisonings with heavy metals). Cathartics (hyper-osmotic saline, bulk-forming stimulant, and lubricant laxatives).
3. Enhancement of elimination of absorbed poison. A. Forced diuresis (mannitol or furosemide) and pH alteration (NaHCO3).B. Extracorporeal techniques: Peritoneal dialysis (Diffusion of toxins from mesenteric capillaries across the peritoneal
membrane into dialysate dwelling in the peritoneal cavity). Hemodialysis (Two catheters are inserted into the patient’s femoral vein. Blood is pumped
from one catheter through the dialysis unit (a cellophane bag) and returned through the other catheter.
Hemofiltration (Similar to hemodialysis, except that the blood is pumped through a hemifilter, where waste products and water are removed by hydrostatic pressure. Replacement fluid is added and the blood is returned to the patient).
Hemoperfusion (The blood is withdrawn from the patient and passed directly over the adsorbing material contained in sterile columns to remove toxic materials).
Plasmapheresis and Plasma exchange (separation of cellular blood components from plasma then cells are resuspended in fresh frozen plasma and reinfused again).
Exchange transfusion (removal of the patient’s blood, replacement with fresh whole blood).
Plasma perfusion (combination of plasmapheresis and hemoperfusion).
Principles in management of Poisoned Patient (cont.)

Teratogenesis
Teratogenesis is a prenatal toxicity characterized by structural or functional defects in the developing embryo or fetus. It also includes intrauterine growth retardation, death of the embryo or fetus, and transplacental carcinogenesis.
Stages of intrauterine human development: pre-implantation and post-implantation stages (0 8 weeks), teratogens may produce
abortion, no effect at all, an anatomic defect (teratogenesis), or a metabolic or functional defect that may not be detected until later in life.
fetal development (9 weeks birth), influence neurologic development, growth, physiologic and biochemical functioning, mental development, and reproduction or death of the fetus.
Teratology education:
Anencephalic newborn Cleft lip and palate Microtia
Congenital abnormalities (birth defects) comprise > 1/5 of all fatalities among newborns/infants. Of these, the largest portion consists of cardiac abnormalities followed by lung abnormalities
and chromosomal aberrations.
Mercury (Hg) is a toxic metal that can exist as a pure element or in a variety of inorganic and organic forms and can cause immune, sensory, neurological, motor, and behavioral dysfunctions similar to traits defining or associated with autism.Mercury compounds (thimerosal) are used as preservatives in nasal solutions, opthalmic drugs,vaccines etc which has generated controversy on the possible toxicity.

Mutations: Examples Gene Mutations
The inheritance of monogenetic diseases occurs in accordance with Mendel's laws. In this, one
distinguishes among dominant and recessive genes. 1. Autosomal dominant inheritance
D = dominant trait. Dd = children with trait "Dominant" means that having a mutation in just one of the two copies of a particular gene is
all it takes for a person to have a trait. Examples of such diseases are:1. Marfan's syndrome (abnormalities in connective tissues) due to mutations in the fibrillin-1
gene on 15q21.1.2. Aniridia (incomplete formation of the iris).3. von Recklinghausen's disease (Neurofibromatosis: changes in skin coloring and the growth
of tumors along nerves in the skin, brain, and other parts of the body).
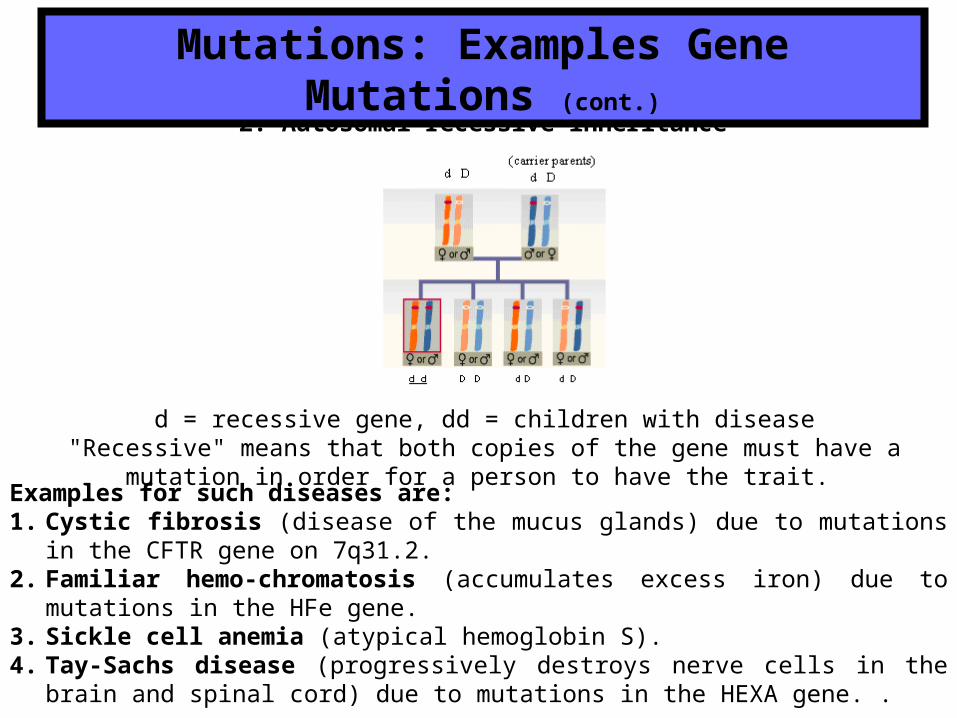
2. Autosomal recessive inheritance
Mutations: Examples Gene Mutations (cont.)
d = recessive gene, dd = children with disease"Recessive" means that both copies of the gene must have a mutation in order for a person to
have the trait.
Examples for such diseases are: 1. Cystic fibrosis (disease of the mucus glands) due to mutations in the CFTR gene on 7q31.2.2. Familiar hemo-chromatosis (accumulates excess iron) due to mutations in the HFe gene.3. Sickle cell anemia (atypical hemoglobin S).4. Tay-Sachs disease (progressively destroys nerve cells in the brain and spinal cord) due to
mutations in the HEXA gene. .

Mutations: Examples Gene Mutations (cont.)
3. X chromosomal inheritance
All male descendents are ill. Among the female descendents there are healthy and carriers. Both are phenotypically healthy.
Examples for X chromosome-linked recessive diseases are: 1. Duchenne's muscular dystrophy due to mutations in the gene DMD, in Xp21.2. Hemophilia A and B (deficiencies of clotting factor VIII and factor IX, respectively).3. Red-green blindness.

Mutations: Structural Chromosome Aberrations2. Structural Chromosome Aberrations
Structural Chromosome Aberrations are changes in chromosome structure and involve gross alteration of the genetic material and are detected by light microscopy.
Structural aberrations are the result of chromosomal breaks that occur during cell division. Here deletion, rings and duplication (unstable) lead to an abnormal phenotype, while inversion, insertion as well as translocation (stable) can be balanced. This means that the carrier of this structural aberration can escape notice phenotypically, because the entire genetic material is present.
Examples of structural chromosome aberrations are1. Cri du chat syndrome (cry of the cat) due to the deletion of part of the short arm of
chromosome 5. 2. Wolf-Hirshchhorn syndrome (mental and growth retardation) is due to the partial deletion
of part of the short arm of chromosome 4. 3. CATCH 22 syndrome (cardiac, abnomal faces, thymic hypoplasia, cleft palate, hypocalcemia)
is due to the deletion of the q11.13 region on chromosome 22.

Mutations (cont.)
3. Numerical Chromosome Aberrations These are changes in the number of chromosomes in the genome. When mutations change the number of whole chromosome sets present, polyploid cells
result. When mutations change parts of chromosome sets, aneuploid cells result. The normal diploid genome is euploid, and contains a complete set of chromosomes from
each parent, e.g. 2n = 46 for humans. Thus, 45 or 47 chromosomes would be described as aneuploid, whereas cells with 69 chromosomes would be described as triploid (3n).
Aneuploid nomenclature for autosomes in diploid organisms: The aneuploid condition 2n-1 is called monosomic (meaning “one chromosome loss”), 2n+1 is called trisomic, and 2n-2 (where the -2 represents homologs) is nullisomic.
Aneuploid nomenclature for sex-chromosome in diploid organisms: The symbolism simply lists the copies of each sex chromosome, such as XXY, XYY, XXX, or XO.
Mechanisms of Aneuploidy Induction Non-disjunction of chromosomes at anaphase or Chromosome loss during cell division.

Mutations: Numerical Chromosome Aberrations (cont.)
Aneuploidy due to non-disjunction in autosomes during spermatogenesis
Patau's syndrome (47, Y-13-13: 90% lethality in the first year of life). Edwards's syndrome (47, Y-18-18: 90% lethality in the first year of life).
Down’s syndrome (47, Y-21-21)

Mutations: Numerical Chromosome Aberrations (cont.)
Examples of Aneuploidy due to non-disjunction in sex chromosomes during spermatogenesis
Klinefelter’s syndrome(47, X-X-Y)
Turner's syndrome(45, X-O)

DefinitionsPHARMACOVIGILANCE
Adverse Drug Reaction (ADR)
An adverse drug reaction (ADR) is any untoward medical occurrence in a patient administered a pharmaceutical product, which is suspected to have a causal relationship with this treatment.
Spontaneous reports from consumers and healthcare professionals should be regarded as
suspected ADRs.
Lack of Efficacy (Lack of Drug Effect)
Failure to produce the expected pharmacological action for an approved indication.
Example: A patient received an oral contraceptive and became
pregnant.

?Central questions
What information should be reported ?
How is the information reported ?
To whom should I report ?
What information should be reported ?
Any information
on an ADR or lack of efficacy connected with the use of a product.

What information should be reported ?
Any information
on ADRs occurring
– in the course of the use of a drug – from drug overdose whether accidental or intentional– from drug abuse / misuse / non-approved use– from drug withdrawal– in the infant of a nursing mother– possibly as a result of exposure of the mother or the fetus during
pregnancy.– even if no ADR has been observed,
– from drug overdose whether accidental or intentional– from drug abuse / misuse / non-approved use– from drug administration during pregnancy.

What information should be reported ?
results in death is life-threatening requires inpatient hospitalization or prolongation of existing
hospitalization results in persistent or significant disability/incapacity is a congenital anomaly/birth defect is an important medical event.
Serious ADRsAny ADR occurring at any dose which fulfills one of the following criteria:
Minimum information required for a case:• An identifiable patient• An identifiable reporter• A suspect drug• A suspect ADR• “Identifiable”Patient/reporter does not need to be identified at time of report but is identifiable
if some effort is taken.

What information should be reported ?
Product Technical Complaints
Please also report any information regarding the product quality of a product you become aware of.
Examples are wrong product (label and contents are different products) correct product but wrong strength faulty packaging, e.g. wrong or missing batch number or
expiry date.

How is the information reported ?Adverse Events
Document any Adverse Event on the
ADR Short Report Form.
Pregnancy
Inform about any exposure to a product during pregnancy.
ADR Short Report Form
• Who has experienced the event ?
• What event has the patient experienced ?
• Which drugs were involved ?
• Who has reported the event ?
?

How is the information reported ?
ADR Short Report Form
20 June 2004

THERAPEUTIC DRUG MONITORING• Why are there so many ADRs? Here are just a few of the many reasons.• First, more drugs – and many more combinations of drugs are given chronically – are
being used to treat patients than ever before. To exemplify this point, 66% of all patient visits to physicians result in prescriptions, and visits to specialists result in 2.3 prescriptions per visit.1
• Secondly, 3.42 billion prescriptions were filled in the year 2006.2 That is approximately 11 prescriptions for every person in the United States.
• A survey of 36,901 Medicare patients obtained in 2003 (before Medicare had begun Part D, which provided a prescription drug benefit) gives a snapshot of the extent of prescription drug use by seniors in the U.S. It also demonstrates the complexity of prescribing to this population because of their use of multiple physicians, pharmacies and sources for their medicines. Overall, 5% of seniors with coverage purchased their medicines from Canada or Mexico, compared to 10.5% of those without a prescription benefit.3
• Finally, the rate of ADRs increases exponentially after a patient is on four or more medications.4.Efforts to reduce unnecessary prescribing are important, but for many patients, the number of medications cannot always be reduced without losing benefit. That is why it is important to understand the basis for drug interactions. This will allow us to make the most appropriate choices in prescribing and avoiding preventable ADRs.

Therapeutic Drug Monitoring• Relates concentrations of drug in blood to response• Blood concentrations surrogate for the concentration
at the site of action• Has been established on the principle that the
concentration correlates better than the dose with the drug effect
• Is important when – the dose cannot be titrated against response eg INR,
cholesterol– the drug is being used to prevent infrequent occurrences -
eg epilepsy

Conditions that must be met• Blood concentrations can be accurately reliably
and economically measured• There is sufficient inter-individual variation in drug
handling to warrant individualisation of dose• There is a clear relationship between
concentration and beneficial and/or adverse effects, particularly if there is a narrow therapeutic index
• The effects are due to the parent drug and not its metabolites

Purpose of TDM• To confirm ‘effective’ concentrations• To investigate unexpected lack of efficacy• To check compliance• To avoid or anticipate toxic concentrations• Before increasing to unusually large doses• Limited role in toxicology - drug screen

Pharmacokinetic Considerations• Is the aim to provide constant concentrations? - eg
anticonvulsants• Is the aim to achieve transient high concentrations
without toxicity? - eg gentamicin • Are drug concentrations likely to vary greatly
between individuals on the same dose? - eg phenytoin
• Remember it takes around 5 half-lives to reach steady state

Practical considerations• Can the lab actually measure the drug?• What sample is needed?• What is the right timing?• Is there an accepted ‘therapeutic range’
– MEC - threshold concentration above which efficacy is expected in most patients with the disorder
– MTC - upper concentration above which the rate and severity of adverse effects become unacceptable

Methodological Difficulties in establishing ‘Therapeutic Range’
• Good data relating concentration to effect are seldom available
• Ideally it would require trials where participants were randomised to different plasma concentrations with follow-up and accurate and unbiased measurement of the outcomes
• See diagram of therapeutic range

TDM - examples• Lithium - used for bipolar disorder• Toxic - neurological, cardiac, renal• Narrow therapeutic range:
– 0.8 - 1.2 mmol/L acutely– 0.5 - 0.75 mmol/L for maintenance– Chronic concentrations of 3.0 are potentially
lethal• Renal clearance of Li can be affected by
diuretics and NSAIDs

Anticonvulsants
• Variable dose dependant kinetics• Most metabolised through cytochrome P450 system• Concentration-related CNS toxicity can be partly
avoided by TDM• However severe skin rashes, liver and marrow
toxicity cannot be predicted or avoided• With phenytoin small dose increases can produce
disproportionate rises in blood levels and toxicity• Sometimes free (unbound) concentrations need to
be measured - eg hypoalbuminaemia, pregnancy

Digoxin• Has variable bioavailability• Has variable clearance (by kidney) - remember the
elderly• Drug interactions are fairly common• Relationship between concentration and effect is not
constant - concentrations soon after dosing are difficult to interpret. Range is approx 1 to 2 nmol/L
• Patients may become more ‘sensitive’ to a given concentration - eg hypokalalaemia, hypothyroidism
• In atrial fibrillation titrate against the ventricular rate• Concentrations should be measure at least 6-8 hours
after the last dose

Cyclosporin
• Used as immunosuppressant in transplant rejection
• Low therapeutic index and toxicity (kidney) is severe
• Interactions are common - eg calcium channel antagonists
• Plasma range 50-300 mg/L

Theophylline
• Declining use in asthma• Very narrow therapeutic index: 55 - 110
umol/L (should be lower)• At the high end toxicity is common• Toxicity is severe - GI, neuro, cardiac• Interactions are common - erythromycin,
cyclosporin, cimetidine, smoking

Gentamicin
• Practice is changing - trend to once/daily dosing
• Toxicity relates to trough concentrations, particularly with prolonged therapy
• Desirable range:– peak 6 - 10 mg/L– trough 1-2 mg/L

Toxicity Testing in Lab Animals Exposure:• Acute: Usually a single dose within 24 hours.• Subacute: repeated exposure to a chemical for one month or less.• Subchronic: 1-3 months• Chronic: > 3 months
How are toxicants metabolized? Summary:• By a wide array of enzymes: Convert lypophilic to hydrophilic.• Phase-one reactions in which a polar reactive group is introduced into the xenobiotic molecule.Example: cytochrome P450s.• Phase-two reactions in which the enzymes conjugate the xenobiotic to sugars, aminoacids etc,forming water-soluble products that are readily excreted. Sometimes the metabolites are more toxic than the parent molecule.
• TOXICITY TESTS IN ANIMALS • Testing Lab animals• Commonly used:– Rats, mice, dogs, monkeys etc• Safety in experimental animals does not necessarily indicate the same in humans.– Example: Thalidomide a human teratogen shows toxicity at doses as low as 0.5-1 mg/kg and has little or no effect in mice or rats at doses as high as 4000mg/kg.

Acute toxicity tests: Skin & eye studies (Dermal)
Such testing may provide information on the adverse effects resulting
from a dermal application of a single dose of a test substance. The acute dermal test also provides the initial toxicity data for
regulatory purposes, labelling, classification & subsequent subchronic
& chronic dermal toxicity studies. Comparison of acute toxicity by the oral & dermal route may provide
evidence of the relative penetration of a test material. Draize test
1. It is a simple and generalized test developed to study eye irritation in
rabbits.
2. It is used as the animal test to identify human eye irritants.
3. The Draize test can adequately identify most of the moderate to
severe human eye irritants

Acute toxicity tests: Pyramidal single dose test Large number of dogs ≈ 100 are given a single daily X dose of a
compound under test. At the end of the day, the number of dogs which died & those
which survived is observed. The procedure is continued, till all dogs die, then plotting is done.
It helps in studying the mechanism of drug toxicity. It can be used to determine any pathological changes by
examination of the animal after death. The effect of the drug on all body organs can be examined. Clinical chemical tests can be performed on living animals
(hematology, and detection of different biotransformation and
excretion product (metabolites), and determination of t½ of the
compound).

Prolonged toxicity studies
They predict any cumulative effect of the drug.
Compound under test is given daily in 3 dose levels for 2 – 4 weeks (Subacute), for
90 days (Subchronic) or more than 90 days (Chronic).
Animals are observed for different parameters: physiological, clinical and chemical
tests, behaviour, CNS & autonomic profiles.
At the end of the test, animals are subjected to the following tests & then are killed.
1. Hematological studies: hemoglobin, RBCs, WBCs, platelets.
2. Clinical chemistry studies: serum creatinine, ALT, AST.
3. Histopathological studies: for different organs (spinal cord, heart, kidney).
Life – Span Toxicity Test
1. The same previous procedures are applied but treatment with chemicals starts after
weaning of offsprings (litters).
2. Administration of the chemical is continued till death of animals.
3. When animals die spontaneously, the same previous parameters are determined.

Specific Toxicity Studies: Reproductive toxicity (toxicity on male or female reproductive system) Toxic effects may cause: Decreased libido and impotence, Infertility, Interrupted
pregnancy (abortion, fetal death, or premature delivery), Infant death or childhood morbidity, Altered sex ratio and multiple births, Chromosome abnormalities and Childhood cancer.
Developmental Toxicity (toxicity on developing embryo or fetus) Embryolethality (Failure to conceive, spontaneous abortion), Embryotoxicity (Growth retardation or delayed growth of specific organ systems),
Teratogenicity (Irreversible conditions that leave permanent birth defects in live offspring).
Carcinogenic studies Carcinogenicity is a complex multistage process of abnormal cell growth and
differentiation which can lead to cancer. The initial neoplastic transformation results from the mutation of the cellular genes that control normal cell functions. Mutation may lead to abnormal cell growth. It may involve loss of suppresser genes that usually restrict abnormal cell growth. Many other factors are involved (e.g., growth
factors, immune suppression, and hormones).

Exposure and Toxic Responses
Exposure classes (toxicants in food, air, water, and soil as well as
toxicants characteristic of domestic and occupational settings).
Toxic effects may be systemic or local at the site of exposure.
The target organs that are affected may vary depending on dosage and
route of exposure.
Sings and symptoms are the effects produced by the action of a
particular poison on the physiological function of the body.
Certain general symptoms suggested the possibility of a number of
poisons;
1. Sudden death (aconitine, cyanide and barium compounds).
2. Eyes (ergot, morphine, pilocarpine, atropine and cocaine).
3. Breath (acetic acid, ammonia, phenol, ether and iodine).
4. Mouth (atropine, pilocarpine and ammonia).

5. Skin (atropine, pilocarpine, strong acids and alkalies, cyanosis
produced by aniline, acetanilide).
6. GIT (metals, ergot and food poisons).
7. Cardiovascular system (quinidine, digitalis, ephedrine and reserpine).
8. Liver (carbone tetrachloride and chloroform).
9. Kidney (phenol and sulphonamides).
10. Nerves (peripheral neuritis due to antimony and arsenic).
11. Skeletal muscle (curare and flaxedil).
12. Blood changes (anaemia by benzene, haemolysis due to saponins,
leukopenia by benzene).
13. CNS (strychnine, picrotoxin, barbiturates, ether, alcohol).
Exposure and Toxic Responses (cont.)

Factors That Influence Toxicity
There are numerous factors which may modify the patient's responses to the toxic agent. For examples;
Physicochemical composition of the toxicant (solids, liquids, particle size, pH),
Dose and concentration (aspirin tablets, dilution),
Routes of administration (inhalation > IV > IP > SC > IM > ID > oral > topical), Metabolism of the toxic agent (more polar or more toxic),
State of health (person with severe hepatic or renal disease, hypertension, head injuries),
Age and maturity (gray-baby syndrome, reduced pharmacokinetics in elderly people),
Nutritional state and dietary factors (stomach contents, types of food, proteins),
Pharmacogenetics or idiosyncrasy (succinyl-choline, aspirin, fava beans),
Sex (erythromycin, lipid sol., larger BV and tissue mass in men which dilute the chemical),
Environmental (temperature, occupation (liver-metab), living conditions), Chemical interactions (the increase or decrease of toxicity by simultaneous or
consecutive exposure to another one).

Evaluation of Safety of Chemicals and drugs
Sources of information on safety1. Experimental studies2. Controlled clinical studies3. Epidemiological studies
Experimental studies Goals of toxicity studies. Adv. & disadv. of toxicity tests using experimental animals. Characteristics of ideal animal species. Examples. Strains of rats 1. Specific pathogen free animals, 2. Germ free animals, 3. Dirty animals, 4. Rats for specific purposes.
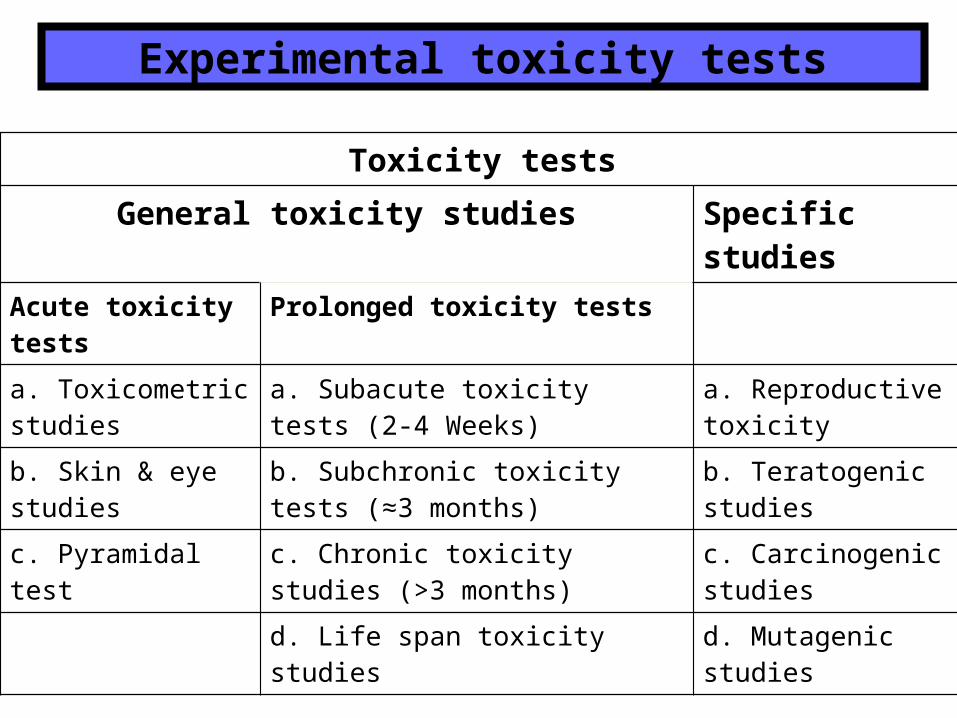
Toxicity tests
General toxicity studies Specific studies
Acute toxicity tests
Prolonged toxicity tests
a. Toxicometric studies
a. Subacute toxicity tests (2-4 Weeks)
a. Reproductive toxicity
b. Skin & eye studies
b. Subchronic toxicity tests (≈3 months)
b. Teratogenic studies
c. Pyramidal test c. Chronic toxicity studies (>3 months)
c. Carcinogenic studies
d. Life span toxicity studies d. Mutagenic studies
Experimental toxicity tests

LD50: Median lethal dose (the dose that causes 50% mortality in a population).LC50: Median lethal concentration (inhaled drugs). LD0: Represents the dose at which no individuals are expected to die. This is just below the threshold for lethality. LD10: Refers to the dose at which 10% of the individuals will die. EDs: Effective Doses that are used to indicate the effectiveness or harmful effect
(paralysis) of substances. TI: The Therapeutic Index (is used to compare the therapeutically effective dose to
the toxic dose = LD50 / ED50).
Acute toxicity tests: Toxicometric studies

Adverse effects Type A (augmented)
• Are induced by:same pharmacological mechanisms as the therapeutic effects– By increase of the therapeutic or other pharmacological effect
of the drug• Is directly dose-dependent (or plasma concentration dependent)
– It is mostly associated with inappropriate dosage schedule (inappropriately high dose and/or short dosing interval)
– It can arise from changes in drug pharmacokinetics (e.g., impaired drug elimination or plasma protein binding)
• As a result of the pathology (kidney, liver failure and hypoalbuminemia)
• As a result of aging (e.g. Lower renal elimination in elderly)– It can arise from changes in drug pharmacodynamics
• Predisposition due to the concomitant pathology– pay appropriate attention on CONTRAINDICATIONS
• Or patient non-compliance (e.g. failure to follow all instructions)
• Are well predictable with respect to both – their clinical manifestation and probability of onset
• Type A is the most frequent type of adverse effects (76%)• They have relatively less dangerous outcomes with lower rate of
mortality

• Examples:– Anticoagulants (e.g., wafarin, heparin) – bleeding– Antihypertensives (e.g.. α1-antagonists) – hypotension– Antidiabetics (e.g. insulin) - hypoglycemia– 1-blockers (e.g. metoprolol)
• Symptomatic heart failure inpatients with previous systolic dysfunction
• Bronchoconstriction in patients with COPD– Antiepileptics blocking Na+ channel (e.g., phenytoin) –
neurological symptoms - vertigo, ataxia, confusion
• Intervention – dose reduction in most cases, use of antagonist in serious circumstances
• Prevention: dose titration, adverse effects monitoring, pharmacotherapy monitoring (PK and PD principle)
Adverse effectsType A (augmented)

• Develop on the basis of:Immunological reaction on a drug (allergy)– Genetic predisposition (idiosyncratic reactions)
• Have no direct relationship to– the dose of the drug– The pharmacological mechanism of drug action
• Are generally unexpected and therefore unpredictable• They appear with much lower frequency (0,1-0,01%)• Have more serious clinical outcomes with higher overall
mortality• Intervention: instant drug withdrawal, symptomatic treatment
– pharmacological approach in allergy: antihistamines, adrenalin (epinephrine) , glucocorticoids …
• Prevention: troublesome, the risks can be reduced by dutiful drug-related anamnesis, by avoiding certain drugs with known significant risk of B-type reactions– Allergy: dermatological testing, in vitro testing (mixed
outcomes), desensitization– Idiosyncratic reactions: genotyping, phenotyping
Adverse effects Type B (bizzare)

Adverse effects - Type BAllergic reactions• Based on immunological mechanism • They require previous exposition before actual manifestation • Molecular weight of most drugs is low (Mr<1000) which is NOT
enough for direct immunogenicity– Exception: peptides and proteins of non-human origin – Immunogenicity can be acquired– By binding of LMW drug (as a hapten) on the
macromolecular carrier• Covalent bond is usually needed• Carrier is usually protein – e.g.. Plasma proteins
(albumin) or proteins on the cell surface• E.g. penicillin is covalently bound to albumin
– LMW drug (prohapten) is metabolized to the reactive metabolite, which acts as a hapten and is bound to the carrier
• E.g., sulfamethoxazole– LMW drug interacts with receptors of immunity systems
• Direct binding to T-cell receptors (TCR), enhanced by MHC system

• Route of administration impact – Higher probability of both occurrence and increased severity
after parenteral (injectional) administration !); – mind the effectiveness of antigen presenting process– Relatively high probability after application on the skin
• Significantly lower probability after p.o. administration• Not only a active substance can be responsible for allergic
reactions– excipients – antimicrobial agents, preservants
- E.g., parabens - must be listed in the Summary of Product
Characteristics (SPC!)- In the case of known allergy to common excipients the generic prescription should be avoided
- Drug decomposition products, impurities etc.: they are under control of the national authorities (FDA…)
- appropriate storage, use and expiration should be followed
Adverse effects - Type B Allergic reactions

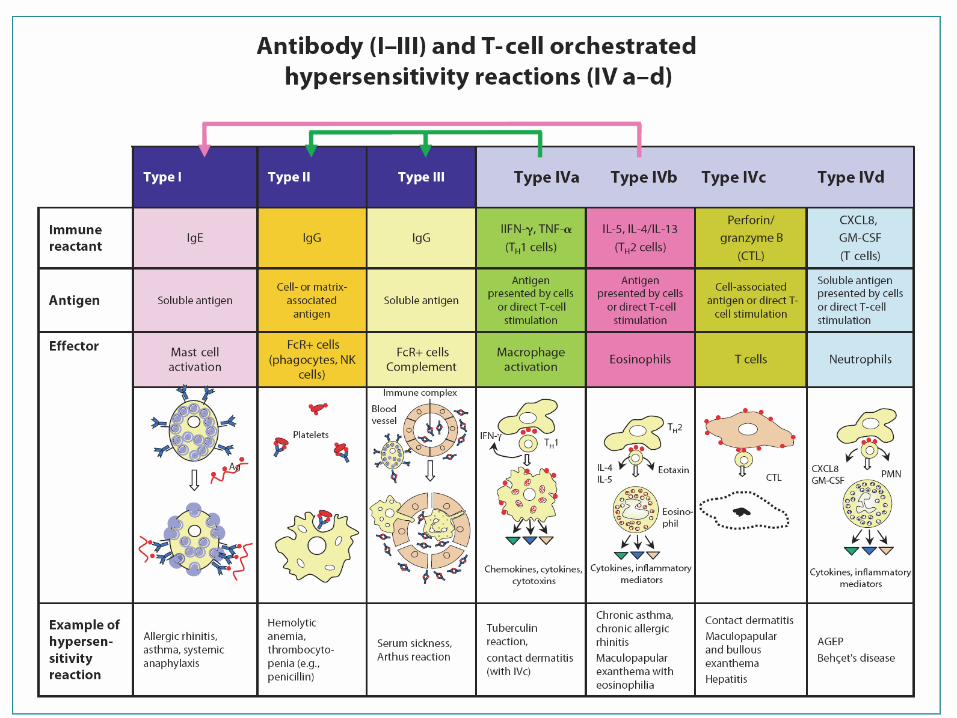
• They are divided according to the prevailing immunological mechanism into 4 groups (Gell-Coombs classification system):
– TYPE I (IgE-mediated, immediate reactions)– TYPE II (cytotoxic reactions)– TYPE III (immunocomplex reactions)– TYPE IV (delayed, cell-mediated reactions)
• Newer classification:– Taking into account T-cell subtypes (Th1/Th2, Cytotox. T-
cells), specificity of the cytokine signaling and different effectors (monocytes, eaosinophils, CD8 T-cells, neutrophils)
– TYP IV – a, b ,c, d
Adverse effects - Type B Allergic reactions - classification

• Sensitisation phase– Immunogenic complex (drug-carrier) induces production
of specific IgE antibodies– IgE ab is bound on the cell surface of mast cells and
basophiles via high affinity receptors• Allergic reaction triggering
– After re-exposition, the drug+carrier is directly bound on the IgE
– Cross-linking of the IgE– Degranulation of the mast cells = release of histamine,
leukotriens, prostaglandins → inflammatory reaction! • Rubor, calor, dolor a tumor
• Clinical manifestation: urticaria, itching, nose/eye hyperemia and secretion, soft-tissue swelling, bronchospasm, anaphylactic reaction
• Time window: after previous sensitization the onset is very rapid one (seconds to minutes)
• Examples: penicilins, cephalosporins, quinolones, macrolides, streptokinase, thiazides, salicylates and skeletal muscle relaxants, local anesthetics
Allergic reactions – TYPE I IgE-mediated

• Hemolytic anemia– Associated with cephalosporins, penicilins, quinidine,
levodopa, methyldopa, some NSAIDs– Symptoms: like in other anemia + jaundice, dark urine– Lab. picture: erythrocytopenia, reticulocytosis and
billirubin (unconjugated); hemoglobin a hemosiderin in urine
• Thrombocytopenia– Associated with heparin (up to 5% patients), quinine
quinidine, sulfonamides and biologicals (-mabs, e.g., bevacizumab)
– Symptoms: petechial bleeding to the skin and mucosa, GIT and urogenital tract bleeding
– Reversibility: in usually in 3-5 days
Allergic reactions – TYPE II Cytotoxic

Allergic reactions – TYPE III Immunocomplex reactions
• Drug-carrier or drug as a chimeric protein induces production of IgG antibodies
• Formation of IgG-drug(carrier) complexes
• Normally these complexes are cleared by the RES with only some decrease in the clinical response
• In some circumstances (huge amount of complexes, deficient decomposition system) it results to development of symptomatic reaction
• Time window: 1-3 weeks after exposition• Epidemiology: 1-3:100 000 patients

– Clinical manifestation: vasculitis and/or serum sickness,• Urticaria, dermatol. affections, pruritus, fever, arthritis/arthralgia,
glomerulonephritis, lyfmadenopathy
– Serum sickness first described after passive immunization with animal serum
• Within 4-10 day the abs were produced and formed complexes with antigenic proteins.
• These complexes were deposited in postcapillary venules and attracted neutrophils
• Development of inflammation with release of proteolytic enzymes destructing vessel and surrounding tissue
– Drugs: chimeric abs (e.g., infliximab) or cephalosporins (cefaclor, cefalexin), amoxicillin, sulfamethoxazole/trimethoprim, NSAIDs, amiodaron
Allergic reactions – TYPE III Immunocomplex reactions

• Cellular reaction mediated by T-cells General principle: drug-carrier complex is presented by APC to T-cells with their following clonal proliferation
• After re-exposition the drug gets into contact with T-cells with release of specific cytokines and inflammatory mediators which activate the target cells
• Clinical manifestation: mostly drug-related contact dermatitis (rash) in many forms + pruritus, tuberculin reaction, maculopapular exanthema or e.g. allergic hepatitis
• Drugs: aminoglycosides, penicillins…..Time window: 2-8 days
Allergic reactions – TYPE IV Delayed, cell-mediated reaction

Pseudoalergic reactions• Are NOT immune reactions • The are induced by direct activation of mast cells or by displacing histamine from
granules
• IgE are NOT increased• Are as frequent as true type I reactions (IgE-mediated)• Clinical manifestation is very close or even indistinguishable from type I reactions
– Mostly less severe (erythema, urticaria)– Onset can be slower then in true type I– May require higher doses– Anaphylactoid forms can occur
• Drugs: NSAIDs, vancomycin, opiates, radiocontrast agents
• Severe adverse effects:Uncommon, but explainable extensions of known pharmacologic effects– Unexpected, may not be recognized until a drug has been marketed for
years, sometimes unexplainable (Thalidomide)• Often represent immunological reactions
– Urticaria, angioedema, – Lupus-like, serum sickness, cell mediated allergies– Severest form --> Anaphylactic shock!

• CLINICAL TRIALSPHASE I
• Studies carried out in healthy volunteers• Carried out by pharmaceutical companies or major hospitals• In some cases patients with the disease in question may be enrolled
(cancer chemotherapy)• Initially small doses (as little as one fiftieth of intended dose)• Toxicity evaluated with routine hematology and biochemical
monitoring of liver and renal function• Dose is escalated until pharmacologic effect is observed or toxicity
occurs• Used to study the disposition, metabolism and main pathways of
elimination of the new drug in humans• Identify the most suitable dose and route of administration for
further clinical studies• Use of isotope-labeled (usually beta-emitting) compounds to
investigate pharmacokinetics and metabolism

PHASE II
Pharmacology of the new drug is determined in patients with the intended clinical condition
• Principal aim is to define relationship between dose and pharmacological and/or therapeutic response in humans
• During phase II some evidence of beneficial effect may emerge
• Address subjective element in human illness (placebo effect)
• Additional studies:• Special populations (elderly, etc.)• Tests for potential interactions with other drugs• Optimum dosage established for use in phase III trials

Phase III• Main clinical trial
– Drug is compared to placebo, or if this would be unethical (effective treatment for the disease in question already exists), an established drug in use for this disease
– Comparison to other established treatments– Addition to established treatment with placebo control
• Random placebo-controlled studies– Randomization of patient population– Sometimes there is double-blinding of the study– Between patient population studies
• Separate patient population arms• Requires greater number of patients
– Within patient population studies (“crossover”)• Alternate treatment with new drug and standard therapy or
placebo => each patients gets both treatments sequentially
• Takes longer

Measurements of adverse effects and possible benefit made at regular intervals.Attention to detecting likely occurring side effects (type A reactions), and unpredictable, rarer complications (type B reactions)Majority of type B reactions may not be seen until post marketing because during the Phase III trial usually only 2000-3000 people will take the drug, usually for short periods.Type B reactions typically occur in one in 1000 to 10,000 patients
PHASE IV• Postmarketing Surveillance
– Ongoing monitoring of drug safety under actual conditions of use in large numbers of patients. (Pharmacovigilance)
– Physician and pharmacist reporting of adverse drug events– No fixed duration– Picks up adverse events occurring in less than one in 1000
subjects

Drug Development
• The drug development phase is significantly more expensive in terms of time and money than either lead discovery or drug design and many drugs will fall during the wayside.
• On average, for every 10000 structures synthesized during drug design, 500 will reach animal testing, 10 will reach phase I clinical trials and only 1 will reach the market place.
• The average overall development cost of a new drug was
recently estimated as $ 800 million or $ 444 million.
91

Three main issues are involved in drug development
1. The drug has to be tested to ensure that it is not only safe and effective, but can be administered in a suitable fashion. This involves preclinical and clinical trials covering toxicity, drug metabolism, stability, formulation, and pharmacological tests.
2. There are the various patenting and legal issues.
3. The drug has to be synthesized in ever-increasing quantities for testing and eventual manufacture (this is known as chemical and process development).
92

• Toxicity tests are carried out in vivo on drug candidates to assess acute and chronic toxicity. During animal studies, blood and urine samples are taken for analysis.
• Individual organ are analyzed for tissue damage or abnormalities. • Toxicity testing is important in defining what the initial dose level should
be for phase I clinical trials. • Drug metabolism studies are carried out on animals and human to
identify drug metabolites. The drug candidate is labeled with an isotope in order to aid the detection of metabolites.
• Pharmacology testes are carried out to determine a drug's mechanism of action and to determine whether it acts at targets other than the intended one.
• Formulation studies aim to develop a preparation of the drug which can be administered during clinical trials and beyond.
• The drug must remain stable in the preparation under variety of environmental conditions.
93

Clinical trials involve four phases.• In phase I healthy volunteers are normally used to evaluate the
drug's safety, its pharmacokinetics, and the dose levels that can safely be administered.
• Phase II studies are carried out on patients to assess whether
the drug is effective, to give further information on the most effective dosing regime and to identify side effects.
• Phase III studies are carried out on larger numbers of patients
to ensure that results are statistically sound, and to detect less common side effects.
• Phase IV studies are ongoing and monitor the long-term use of
the drug in specific patients, as well as the occurrence rare side effects.
94

• Patent are taken out as soon as a useful drug has been identified. They cover a structural class of compounds rather than a single structure.
• A significant period of the patent is lost as a result of the time taken to get a drug to the market place.
• Patents can cover structures, their medicinal uses, and their method of synthesis.
• Regulatory bodies are responsible for approving the
start of clinical trials and the licensing of new drugs for the market place.
95

• Drugs that show promise in a field which devoid of a current therapy may be fast tracked.
• Special incentives are given to companies to develop orphan drug-drug that are effective in rare diseases.
• Pharmaceutical companies are required to abide by professional codes of practice known as good laboratory practice, good manufacturing practice, and good clinical practice.
• Chemical development involves the development of
a synthetic route which is suitable for large scale synthesis of a drug.
96

• The priorities in chemical development are to develop a synthetic route which is straightforward, safe, cheap, and efficient, has the minimum number of synthetic steps, and provide a consistency good yield of high-quality product that meets predetermined purity specifications.
• An early priority in chemical development is to define the purity specifications of the drug and to devise a purification procedure which will satisfy these requirements.
• Process development aims to develop a production process which is safe, efficient, economic, environmentally friendly, and produces product of a consistent yield and quality to satisfy purity specification.
• Drugs derived from natural sources are usually produced by harvesting the natural source or through semi-synthetic methods.
97

/ 20 98
“Rational use of drugs requires that patientsreceive medications appropriate to theirclinical needs, in doses that meet their ownindividual requirements, for an adequateperiod of time and at the lowest costs to thecommunity”
ESSENTIAL DRUGSRational prescription

/ 20 99
• Introduction of Essential drug list limits the use of non-essential drugs.
• Provided details of pharmacokinetics help the physician in selecting right kind of drug and dosage form.
RATIONALIZATION OF PRESCRIPTION PRACTISES

/ 20 100
Prescription of Rational drugs requires:• Accurate diagnosis.• Selection of best drug from the available.• Prescribing adequate drug for a sufficient
length of time.• Choosing the most suitable drug, weighing of
effectiveness, safety, and availability and cost.
RATIONALIZATION OF PRESCRIPTION PRACTISES

/ 20 101
Most of the illness respond to simple, inexpensive drugs,
Physician should avoid :• Use of expensive drugs.• Use of drugs in nonspecific conditions (e.g.,
use of vitamins).• Use of not required forms (e.g. injection in
place of capsules, syrup in place of tablets)
RATIONALIZATION OF PRESCRIPTION PRACTISES

/ 20 102
Most of the illness respond to simple, inexpensive drugs, even of improve with no therapy at all.
Physician should avoid :• Multiple drug prescription (bullet treatment)
even if it is considered in the best of the patient in a given situation.
RATIONALIZATION OF PRESCRIPTION PRACTISES

/ 20 103
The Concept of p-drug list
• There is a need for evidence based, rational prescription
• Each GP has his/her own context with different needs and priorities
• Scientists suggested a method where each doctor prepares a list of essential drugs for different conditions
• P-drug concept has been propagated by the WHO Action Program on Essential Drugs world wide
Kawakami J, Mimura Y, Adachi I, Takeguchi N. [Application of personal drug (P-drug) seminar to clinical pharmacy education in the graduate school of pharmaceutical sciences]. Yakugaku Zasshi. 2002 Oct;122(10):819-29

/ 20 104
• Step 1: Define the patient’s problem
• Step 2: Specify the therapeutic objective
• Step 3: Verify the suitability of your P-treatment
• Step 4: Start the treatment
• Step 5: Give information, instructions and warnings
• Step 6: Monitor (and stop?) treatment
http://p-drug.umin.ac.jp/34th-gakkai/slide/DrSunami/SunamiP-drug.PPT
The process of rational treatment

/ 20 105
• Step i : Define the diagnosis
• Step ii : Specify the therapeutic objective
• Step iii : Make an inventory of effective groups of drugs
• Step iv : Choose an effective group according to criteria
• Step v : Choose a P-drug
• Step iv: Choose an effective group according to criteria
Selecting a P-drug

/ 20 106
• Imagine you go shopping for a shirt. What
would be your criteria to select a product?
– Efficacy
– Safety
– Suitability
– Cost
/ 20 107
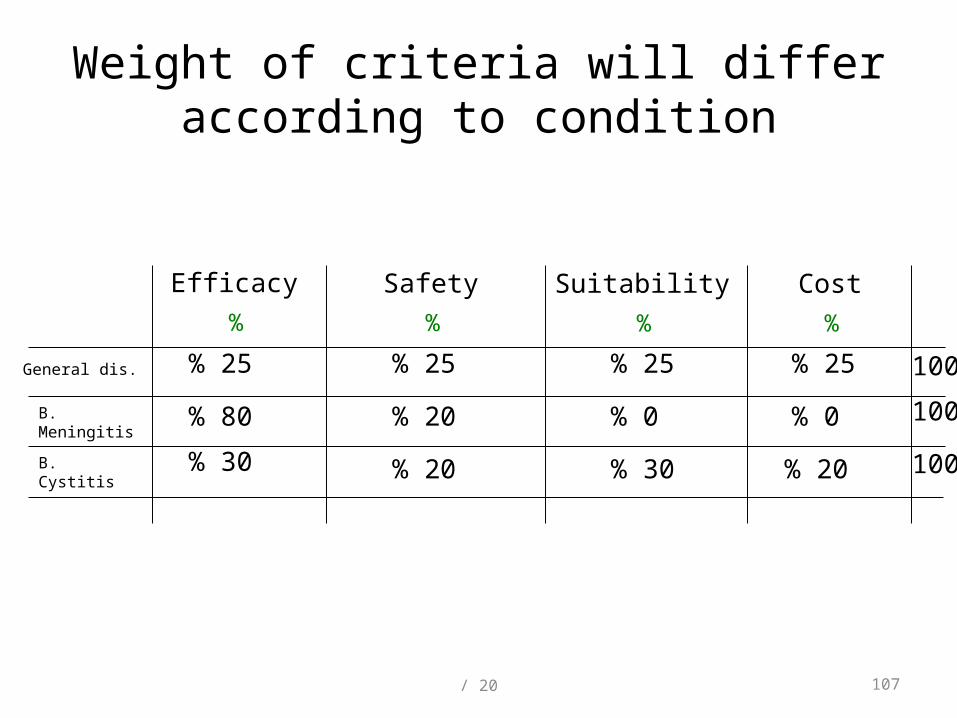
Weight of criteria will differ according to condition
Efficacy Safety Suitability Cost
B. Meningitis
B. Cystitis
% % % %
% 25 % 25 % 25 % 25General dis.
% 80 % 0% 0% 20
% 30 % 20 % 30 % 20
100
100
100

/ 20 108
P-drug example: angina pectoris
• Step i : Define the diagnosis– 60 y male. Since one month repeated attacks of
constricting chest pain starting with exercise and relieving with rest
– Diagnosis: stable angina pectoris due to partial coronary occlusion and myocardial ischemia.

/ 20 109
P-drug example: angina pectoris
• Step ii : Specify the therapeutic objective– To prevent pain coming with effort– To decrease the oxygen need of myocardium– To increase the perfusion of myocardium

/ 20 110
P-drug example: angina pectoris
• Step iii : Make an inventory of effective groups of drugs (look for the evidence)
Preload Contractility Rate Afterload
Nitrates + + - - ++
Beta-blockers + ++ ++ ++
Ca channel blockers + ++ ++ ++

/ 20 111
P-drug example: angina pectoris• Step iii : Make an inventory of effective groups of drugs
(look for the evidence)Efficacy (pharmacodynamics) Safety (Side effects) Suitability (Contraind.)
Nitrates
Peripheral vasodilatation Headache, nitrate intoxication
Hypotension, SIIP, anemia, Sildenafil (Viagra) usage
Beta-blockers
Decrease in heart contractility and rate
Hypotension, bradiarrythmia, impotence
Asthma, raynould, decompansated heart failure, DM, severe bradicardia
Calcium channel blockers
Coronary and peripheral vasodilatation, decrease in heart rate and contractility
Hypotension, dizziness, bradicardia, Heart f.
Hypotension, congestive heart failure, AV block
/ 20 112
P-drug example: angina pectoris
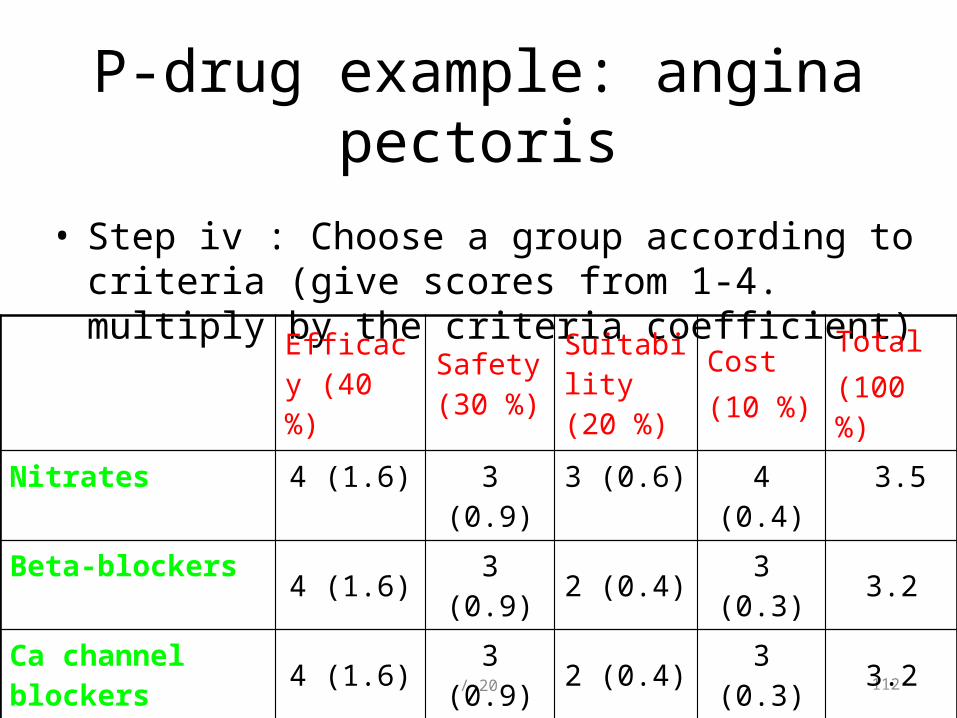
• Step iv : Choose a group according to criteria (give scores from 1-4. multiply by the criteria coefficient)
Efficacy (40 %)
Safety (30 %)
Suitability (20 %)
Cost (10 %)
Total (100 %)
Nitrates 4 (1.6) 3 (0.9) 3 (0.6) 4 (0.4) 3.5
Beta-blockers 4 (1.6) 3 (0.9) 2 (0.4) 3 (0.3) 3.2
Ca channel blockers 4 (1.6) 3 (0.9) 2 (0.4) 3 (0.3) 3.2

/ 20 113
P-drug example: angina pectoris
• Step v : Choose a P-drug (choose your brand name from the medicine available in the market)
Efficacy (40 %)
Safety (30 %)
Suitability (20 %)
Cost (10 %)
Total (100 %)
Isosorbid dinitrate tablets sublingual 5 mg
4 (1.6) 3 (0.9) 3 (0.6) 4 (0.4) 3.5
Glyceril trinitrate spray 4 (1.6) 3 (0.9) 4 (0.8) 2 (0.2) 3.5
Isosorbid dinitrate tablets 10 mg
3 (1.2) 3 (0.9) 3 (0.6) 3 (0.3) 3.0
Isosorbid mononitrate tablets
3(1.2) 3 (0.9) 3 (0.6) 3 (0.3) 3.0

/ 20 114
P-drug form for this exercise
• Indication: stable angina pectoris• P-drug 1 (first choice)
Name: Isosorbid dinitrateDose available: 5mg, 50 tablets package Use: One tablet sublingually when pain arisesDuration: Until next control visit
• P-drug 2 (second choice)Name: Glyceril trinitrateDose available: …Use: …Duration: …

Seven steps to get a new medicine onthe WHO Model List of Essential Drugs 1. Identification of public-health need for a medicine2. Development of the medicine; phase I - II - III trials3. Regulatory approval in a number of countries
> Effective and safe medicine on the market4. More experience under different field circumstances; post-marketing surveillance5. Price indication for public sector use6. Review by WHO disease programme; define comparative effectiveness and safety in real-life
situations, comparative cost-effectiveness and public health relevance> Medicine included in WHO treatment guideline
7. Submission to WHO Expert Committee on Essential Drugs> Medicine included in WHO Model List

• With the development of new drugs, the accrual of new information on marketed agents, the findings emerging from clinical trials, ongoing regulatory decisions, and the updated guidelines for disease management, the information base available to guide drug therapy is in a state of constant and brisk evolution.
• Among the available sources are textbooks of pharmacology and therapeutics, medical journals, published guidelines for treatment of specific diseases, analytical evaluations of drugs, drug compendia, professional seminars and meetings, and advertising. Developing a strategy to extract objective and unbiased data is required for the practice of rational therapeutics.
• As is the case with all continuing medical education, patient-centered acquisition of the relevant information is a centerpiece of such a strategy. This requires access to the information in the practice setting and increasingly is facilitated by electronic availability of information resources.

• Facile online access to the primary medical literature that forms the basis for therapeutic decisions is available via PubMed, a search and retrieval system of the National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov/entrez/query.fcgi).
• Depending on their aim and scope, textbooks of pharmacology may provide, in varying proportions, basic pharmacological principles, critical appraisal of useful categories of therapeutic agents, and detailed descriptions of individual drugs or prototypes that serve as standards of reference for assessing new drugs.
• In addition, pharmacodynamics and pathophysiology are correlated. Therapeutics is considered in virtually all textbooks of medicine, but often superficially.
• A source of information often used by physicians is the Physicians' Desk Reference (PDR), available in printed form and online (http://www.pdr.net).

• No comparative data on efficacy, safety, or cost are included. The information is identical to that contained in drug package inserts, which are largely based on the results of phase 3 testing; its value includes designation of the FDA-approved indications for use of a drug and summaries of pharmacokinetics, contraindications, adverse reactions, and recommended doses; some of this information is not available in the scientific literature. Many commonly used drugs that are no longer patent-protected are not covered by the PDR.
Several unbiased sources of information on the clinical uses of drugs provide balanced and comparative data. All recognize that the clinician's legitimate use of a drug in a particular patient is not limited by FDA-approved labeling in the package insert. The Medical Letter (http://www.medletter.com) provides objective summaries in a biweekly newsletter of scientific reports and consultants' evaluations of the safety, efficacy, and rationale for use of drugs.

• The "Drug Therapy" section of the New England Journal of Medicine provides timely evaluations of specific drugs and areas of therapeutics. The United States Pharmacopeia Dispensing Information (USPDI) comes in two volumes. One, Drug Information for the Health Care Professional, consists of drug monographs that contain clinically significant information aimed at minimizing the risks and enhancing the benefits of drugs.
• Monographs are developed by USP staff and are reviewed by advisory panels and other reviewers. Drug Facts and Comparisons also is organized by pharmacological classes and is updated monthly (online at http://www.factsandcomparisons.com).
• Information in monographs is presented in a standard format and incorporates FDA-approved information, which is supplemented with current data obtained from the biomedical literature. A useful feature is the "Cost Index," an index of the average wholesale price for equivalent quantities of similar or identical drugs.

• Particularly helpful in obtaining a comprehensive overview of therapy for specific diseases are the guidelines published by a number of professional organizations. These can be accessed by judicious use of a search engine; for example, a search for heart failure guidelines on Google will yield the Web address for the American College of Cardiology/American Heart Association Guidelines for the Evaluation and Management of Chronic Heart Failure in the Adult.
Industry promotion-in the form of direct-mail brochures, journal advertising, displays, professional courtesies, or the detail person or pharmaceutical representative-is intended to be persuasive rather than educational. The pharmaceutical industry cannot, should not, and indeed does not purport to be responsible for the education of physicians in the use of drugs.
More than 1500 medical journals are published regularly in the United States.

• However, of the two to three dozen medical publications with circulations in excess of 70,000 copies, the great majority are sent to physicians free of charge and paid for by the industry.
• In addition, special supplements of some peer-reviewed journals are entirely supported by a single drug manufacturer whose product is featured prominently and described favorably.
• Objective journals that are not supported by drug manufacturers include Clinical Pharmacology and Therapeutics, which is devoted to original articles that evaluate the actions and effects of drugs in human beings, and Drugs, which publishes timely reviews of individual drugs and drug classes.
• The New England Journal of Medicine, Annals of Internal Medicine, Journal of the American Medical Association, Archives of Internal Medicine, British Medical Journal, Lancet, and Postgraduate Medicine offer timely therapeutic reports and reviews

• Sources of drug information and drug nomenclature(brand vs genetic or proprietary vs no-proprietary)
The existence of many names for each drug, even when the names are reduced to a minimum, has led to a lamentable and confusing situation in drug nomenclature. In addition to its formal chemical name, a new drug is usually assigned a code name by the pharmaceutical manufacturer.
If the drug appears promising and the manufacturer wishes to place it on the market, a U.S. adopted name (USAN) is selected by the USAN Council, which is jointly sponsored by the American Medical Association, the American Pharmaceutical Association, and the United States Pharmacopeial Convention, Inc.
This nonproprietary name often is referred to as the generic name. If the drug is eventually admitted to the United States Pharmacopeia (see below), the USAN becomes the official name. However, the nonproprietary and official names of an older drug may differ. Subsequently, the drug also will be assigned a proprietary name, or trademark, by the manufacturer.

• If more than one company markets the drug, then it may have several proprietary names. If mixtures of the drug with other agents are marketed, each such mixture also may have a separate proprietary name.
There is increasing worldwide adoption of the same nonproprietary name for each therapeutic substance. For newer drugs, the USAN is usually adopted for the nonproprietary name in other countries, but this is not true for older drugs. International agreement on drug names is mediated through the World Health Organization and the pertinent health agencies of the cooperating countries.
• One area of continued confusion and ambiguity is the designation of the stereochemical composition in the name of a drug. The nonproprietary names usually give no indication of the drug's stereochemistry except for a few drugs such as levodopa and dextroamphetamine. Even the chemical names cited by the USAN Council often are ambiguous.

• Physicians and other medical scientists frequently are ignorant about drug stereoisomerism and are likely to remain so until the system of nonproprietary nomenclature incorporates stereoisomeric information . This issue becomes especially important when a drug's different diastereomers produce different pharmacologic effects, as is the case with labetalol, for instance .
• The nonproprietary or official name of a drug should be used whenever possible. The use of the nonproprietary name is clearly less confusing when the drug is available under multiple proprietary names and when the nonproprietary name more readily identifies the drug with its pharmacological class.
• The facile argument for the proprietary name is that it is frequently more easily pronounced and remembered as a result of advertising. Not all proprietary names for drugs are included because the number of proprietary names for a single drug may be large and because proprietary names differ from country to country.

• The Drug Price Competition and Patent Term Restoration Act of 1984 allows more generic versions of brand-name drugs to be approved for marketing. When the physician prescribes drugs, the question arises as to whether the nonproprietary name or a proprietary name should be employed.
• A pharmacist may substitute a preparation that is equivalent unless the physician indicates "no substitution" or specifies the manufacturer on the prescription.
• In view of the preceding discussion on the individualization of drug therapy, it is understandable why a physician who has carefully adjusted the dose of a drug to a patient's individual requirements for chronic therapy may be reluctant to surrender control over the source of the drug that the patient receives

• In the past, there was concern that prescribing by nonproprietary name could result in the patient's receiving a preparation of inferior quality or uncertain bioavailability; indeed, such therapeutic failures owing to decreased bioavailability have been reported .
• To address this issue, the FDA has established standards for bioavailability and compiled information about the interchangeability of drug products, which are published annually (Approved Drug Products with Therapeutic Equivalence Evaluations).
• Because of potential cost savings to the individual patient and simplification of communication about drugs, nonproprietary names should be used when prescribing except for drugs with a low therapeutic index and known differences in bioavailability among marketed products