phosphorus chernical shift and indirect spin-spin coupling...
TRANSCRIPT

Phosphorus Chernical Shift and Indirect 31P-3LP Spin-Spin Coupling Teasors in Compounds Containhg P-P Bonds: A Solid-State '*P NMR
and Theoretical Study
by
Myrlene Gee
Submitted in partial hilfiilrnent of the requirements for the degree of D w o r of Philosophy
Dalhousie University Halifax, Nova Scotia
lune, 2001
@ Copyright by Myrlene Gee, 2001

National Libmy Bibliothèque nationale du Canada
Acquisitions and Acquisitions et Bibliographie Services services bibliographiques
The author has granted a non- exclusive licence allowing the National Library of Canada to reproduce, loan, distri'bute or seii copies of this thesis in microforrn, paper or electronic formats.
The author retains ownership of the copyright in this thesis. Neither the
L'auteur a accordé une licence non exclusive pefmettant à la Bibliothèque nationale du Canada de reproduire, prêter, distribuer ou vendre des copies de cette thèse sous la forme de microfiche/film, de reproduction sur papier ou sur format électronique .
L'auteur conserve la propriété du droit d'auteur qui protège cette thèse.
thesis nor substantial extracts fiom it Ni la thèse ni des extraits substantiels may be p ~ t e d or othenwise de ceiie-ci ne doivent être imprimés reproduced without the author's ou autrement reproduits sans son permission. autorisation.

For M m und Dad
7 went to the wu& because I wished to live deliberately, tofront only the essential facts of life. and see if1 could not learn whm it had to teuch, and not, when I came to die, discover that I had not lived. "
"1 leji the wmds for as g w d a reason as I weni there. Perhaps it seemed to me that I had several more lives to live, and could not spre any more time for thor one. "
Front Waùien. by Henry David nioreau

Table of Contents Page
............................................................................. Table of Figures vïii
.............................................................................. List of Tables.. xi
Abstract ....................................................................................... xii
...................................................... List of Abbreviations aml Symbols xiii
Chapter 1 : Introduction and Scope
................................................................... 1 . 1 Introduction 1
................................................................. 1.2TtiesisOutline 5
Chapter 2: Background Theory
2.1 NMR Interactions
........................................................... 2.1.1 Overview
............................................... 2.1.2 Zeeman Interaction
.................................... 2.1 .3 Radio Frequency interaction
2.1.4 Nuclear Magnetic Shieldhg and Chernical Shift ............ ................................................. 2.1.5 Dipolar Coupling
2.1.6 J Coupling ......................................................... 2.2 NMR Spectra Arising from Isolated Spin Pairs
......................................................... 2.2.1 Overview
2.2.2 The Dipolar Chernical Shi& Method ......................... .................................... 2.2.3 2D Spin-Echo Spectroscopy
2.3 Ab Initio Calculations of NMR Parameters
.......................... 2.3.1 Nuclear Magnetic Shielding Tensors
................................. 2.3.2 Indirect Spinspin (5) Tensors
Chapter 3 : Phosphocus Chernid Shift Teosors for Tetr amethy ldiphosphine Disulphide: A Single-Crystal P NMR, Dipolar-Chernical Shift NMR and A b Initio Molecular Orbital Study
.................................................................. 3.1 Introduction.. 43

................................ 3.2 Experimental and Computational Details 46
3.3 Results and Discussion
..................... 3.3.1Phosphonis31NMRofaSingleCrystal 51
..... 3.3.2 Phospho~s-3 1 NMR of Crystalline Powder Samples 60
... 3.3.3 Ab Initio Calculation of Phosphocus Shielding Tensors 65
3.3.5 Trends in Phosphorus Chernical Shifts for .................................... Al@ ldiphosphine Disulfides 67
Chapter 4: Characterization of Phosphonis Chernical Shift Tensors in a Phosphole Tetramer: A Combiwd Experimental NMR and Theoretical Study
................................................................... 4.1 introduction 70
................................ 4.2 Experimental and Computational Deuils 71
4.3 Results and Discussion
4.3.1 Experimental Determination of Phosphorus Chernical Shift Tensors ......................... .... ......................... 74
4.3.2 Ab Initio Calculations of Phosphorus Shielding Tensors . . 84
4.3.3 Trends in Phosphonis Chemical Shifts for Phospholes .... 87
................................................................. 4.4 Conclusions 90
Chapter 5: lJelP. "P) Coupling Tensors . Conformational Shidies in Mode1 Systems and Structural Characterization of [Ph, P.PPhJ[GaClJ
................................................................... 5.1 Introduction 91
................................ 5.2 Experimental and Computational Details 94
...................................................... 5.3 Results and Discussion 97
5 .3.1 Phosphorus-3 1 NMR Spectra of [Ph, P.PPhJ [GaClJ ...... 97
5.3.2 Dependence of 1J(31P. "P) on conformation: HLP.PH, vs . ................................. ................... HIP-PH2+ .... 105

vii
.......................... ...............*.....-................ Chapter 6: Conclusions .. 117
Chapter 7: Future Research Directions
................................................................... 7.1 Introduction 120
..................................................... 7.2 [Ph2(CI)P-PPhJ [ChCd 120
7.3 Ab Inirio Calculation of Phosphorus Chemicai Shift Tepsors ........ 122
7.5 NMR Spectra Arising from Three Coupled Homonuclear Spin-% ................................................... Nuclei in the Solid State 127
Appendix 1: Performing the 2D Spin-Echo NMR Experiment on the CMX ....................................... 2ûû at the University of Alberta 129
............................... Appendix 2: Handling Air-Sensitive NMR Samples 131
.................................................................................... References 133

List of Figures
Page
Figure 1.1 Structures of the compounds studied this thesis 3
Figure 2.1 Zeeman ewrgy as a function of B, 10
Figure 2.2 Angles d e m g the orientation of $, rK, and the PAS of the nuclear magnetic shielding tensor 13
Figure 2.3 Typical Iine shapes for an isolated spin in a powder crystailine 16 sample
Figure 2.4 Calculated NMR spectra when the Zeeman and dipolar interactions are present 19
Figure 2.5 Calculated NMR spectra illustrating the effect of dipolar and J couplhg 27
Figure 2.6 Calculated NMR spectra iilustrating the effect of dipolar coupling and anisotropic nuctear magnetic shielding 28
Fipre 2.7 Euler angles relating the PAS of the nuclear magnetic shielding tensor to the molecular frame of reference 29
Figure 2.8 The 2D spin-echo pulse sequence 3 1
Figure 3.1 Structure of tetramethyldiphosphine disulfde, TMPS 45
Fi- 3.2 Phosphorus-3 1 NMR specua of a single crystal of TMPS 52
Figure 3.3 Example of a 31P NMR spectrum of the TMPS single crystal 53
Figure 3.4 Phosphorus chernical shift as a huiction of crystal rotation for TMPS 55
Figure 3.5 Dipolar splitthg as a function of crystal rotation angle TMPS 55
Figure 3.6 Orientation of the phosphorus chemical shift tensor for TMPS 58
Figure 3.7 Experimentai and calculated "P NMR spectra of stationary powder samples of TMPS 61
viii

Figure 3.8
F i r e 3.9
F i r e 4.1
Figure 4.2
Figure 4.3
Figure 4.4
Figure 4.5
Figure 4.6
Fipre 4.7
Figure 4.8
Figure 5. 1
Fipre 5.2
Figure 5.3
Figure 5.4
Figure 5.5
Experimental and calculated 3ïP NMR spectra of MAS samples of TMPS
Experimental and calculated Fi projections of the 2D spin-echo "P NMR spectnim of TMPS
Structure of the phosphole tetramer and the mode1 system used for ab inirio calculations
Experimentai and calculated "P NMR spectra of stationary samples of the phosphole tetramer
Experimental and calculaied 31P NMR spectra of MAS samples of the phosphole tetramer at 4.7 T
Experimental and caiculated NMR spectra of MAS samples of the phosphole t e m e r at 9.4 T
The calculated and experimental Fi projections of the 2D spin- echo 3LP NMR spectrum of the phosphole tetramer
The relative orientation of the two phosphorus chemical shifk teosors in the phosphole tetramer
The phosphorus chemical shifi tensor orientations for the phosphoie tetramer determinecl b y ub initio calculat ions
Structures of compound 5, 6, and 7 in table 4.2
Structures of the molecules discussed in chapter 5
Experimental and calculated 31P NMR spectra of MAS samples of [Ph3P-PPhJ[GaCb] at 4.7 T
Experimentai and calculated 'lP NMR spectra of MAS samples of [Ph,P-PPhJ[GaC4] at 9.4 T
Experirnental and calculated FI projections of the 2D spin-echo 31P NMR spectru~~~ of [Ph,P-PPhJ[GaClJ at 4.7 T
Experirnental and calculated 31P NMR spectra of stationary wwder sam~les of IPhtP-PPh711GaCL1

Figure 5.6
Figure 5.7
Figure 5.8
F g u e 5.9
Figure 7.1
Figure 7.2
Calculated orientation of the phosphorus chernical shifi tenson for (CH,),P-P(CH,),+
Plot of the total energy for H2P-PH2 a huiction of 4, The dependence of 1Je1P,31P) in H,P-PH, on +pp
The depenâence of 1Je1P,31P) in H,P-PH2+ on @,+
Phosphonis-3 1 NMR spectra of solid [Ph,(Cl)P-PPhJ [GaCh] (MAS)
Structure of a phosphonis-containing homonuclear three-spin s ystem
Figure A2.1 A Varian-Chemagnetics rotor

List of Tables
Page
Table 3.1 Selected bond lengths and bond angles for TMPS 47
Table 3.2 Direction cosiaes orienting the crystal axes of a single crystal of TMPS to the NMR cube fiame 48
Table 3.3 Linear least-squares coefficients for the phosphocus chemical shifi and spin-spin coupling interactions in TMPS as functions of crystal rotation 56
Table 3.4 Principal components and orientations of the phosphonis chemical shift and dipolar coupling tensors relative to the crystal axes (a*bc) for TMPS 57
Table 3.5 Spin-spin coupling data for TMPS 57
Table 3.6 The isotropic chemical shift, principal components, Q, and K of the phosphorus chernical shift tensors for TMPS 64
Table 3.7 Cornparison of 8, and C-P-C bond angle for &P(S)], 68
Table 4.1 Experimental and calculateci phosphorus chemical shift tensor principal components for the phosphole tetramer 78
Table 4.2 Phosphorus chernical shift tensor principal components for some phospholes 89
Table S. 1 Phosphorus chernical shift tensor principal components and spin-spin coupling parameters for [Ph,P-PPhJ[GaClJ 101
Table 5.2 1J(31P,31P) for H2P-PH, as a function of @,, 110
Table 5.3 1J(3iP,31P) for H,P-PH,+ as a function of @,,+ 112
Table 5.4 Geometry dependence of 'le tP,31P), for H,P-PH, 114

The combination of solid-state NMR and theoretid calculations to characterize
NMR parameters is applied to compounds containiog P-P bonds where phosphorus
participates in a variety of bonding modes. In particular, phosphorus chemical shifi
tensors and one-bond indirect spin-spin or J-coupling temors, 1J("P,31P), have been
investigated experimentally by NMR and complemented by first-prhciples calculations.
The phosphorus chernical shift and spin-spin coupling tensors for
tetramethy ldiphosphine disul fide are characterized by analysis of 31P NMR spectra
obtained at 4.7 T for a single crystal. These results were compared to those obtained
ftom spectra aquired at 4.7 T and 9.4 T for powder samples. The data obtained from
both methods are in exceilent agreement. It was aiso found that the upper limit on the
anisotropy in 1J(31P,nP), hl. is approximately 450 Hz.
The phosphorus chemical shifi tensors for a phosphole tetramer were
characterized by analysis of "P NMR spectra aquired at 4.7 T and 9.4 T for powder
samples . The experimental NMR results are supplemented by f i s t principles
calculations of the chemical shift tensor. The calculatioos are useful for proposing
chemical shift tensor orientations.
The final example presented here is an investigation of the
phosphinophosphonium sali, ph,P-PPhJ[GaClJ. The structure is not available fiom X-
ray diffraction studies, thus characterization by other means, such as solid-state NMR,
is important. To determine the P-P boad length from the effective dipolar coupling
constant, R,, AJ needs to be estimated, in this case by ab initio calculations on a mode1
system, H,P-PH,+. Combination of the calculated AJ value and the measured Re, yields
a P-P bond length of 2.25 A. Further calculations of 1J(31P,31P) for H,P-PH, offers an
opportunity to iavestigate the dependence of this parameter on conformation. First-
principles calculations allows one to map out the detailed orientation dependence as well
as the potential energy surface.

Lis# of Abbreviatbns and Symbols
10)
B
b
Bo
Bn
Brf
B,
Bi,
CASSCF
CC-pVTZ
CSGT
CP '
D
DFT
DSO
E(B, M)
LIE
ground electronic state
a magnetic field vector
applied magnetic field vector
magnitude of Bo
applied radio frequency field vector
magnitude of B,
amplitude of B,
induced magnetic field vector
complete active space selfconsistent field
correlation-consistent polarization valence triple-zeta
coatinuous set of gauge transformations
cross polarization
dipolar coupling tensor
density hinctiooal theory
diamagnetic spin-orbit
the energy of a molecule
the ewrgy separation between the + Yz and -95 nuclear spin States of I in
the presence of Bo
Fermi contact
finite field
xiii

xiv
ûee induction decay
finite perturbation
Fourier tram form
gauge-iiifluding atomic orbitals
Planck's constant divided by 2%
Hamiltonian describing the chernid shift interaction for I
Hamiltonian describing the dipolar couphg between I and S
Hamiltonian describing the DSO contribution to J
Hamiltonian describing the FC contribution to J
Hamiltonian descr ibing the indirect spin-spin coupling (J-coupling )
between I and S
Hamiltonian describing the PSO contribution to J
Hamiltonian describing the interaction of I with 8,
Hamiltonian describing the SD contribution to J
Hamiltonian describing the Zeeman interaction
Hartree Fock
nuclear spin angular momentun operators
ith component of 1 or S
nuclei with spin of 'h
individuai gauge for localized orbitals
intemediate neglect of differential overlap

LORG
indirect spin-spin coupling tensor
tensor describing the anisotropic part of J
the DSO contribution to J
the FC contribution to J
the FC x SD contribution to J
the PSO contribution to J
the SD contribution to J
J coupling over n bonds between 1 and S (Hz)
principal components of J (Hz)
a component of J (Hz)
component of an axially symmetric J which is perpendicular to the bond
component of an axially symmetric J which is pardlel to the bond
isotropic J coupling (Hz)
anisotropy in J (Hz)
reduced J-coupling tensor (N A-I
Boltzmann constant
angular momentum operator for the &th electron (N denotes with respect
to a nucleus)
localized orbital local origin
the set of nuclear magnetic moment vectors in a molecule
the nuclear magnetic moment vector or nucleus I

xvi
me
ml
MAS
Mes'
MCLR
MCSCF
MP
I n)
An1
N
NMR
o. d.
Pi
PAS
PSO
6 s
ris
=k* rkN
%D
9 n
lm
stt,), ~ ( l r )
efectron mass
z-compownt of the nuclear angular momentum of 1
magic angle splliaing
(2,4,6-tri-t-butyi) phenyl
multiconfipratiod liwar respoose
multiconfiprational sel €-consistent field
Msller-Plesset
nth excited electronic state
population difference between the + H and -95 nuclear spin States of I
total number of I nuclei in a sample
nuclear magne tic resomce
outer diameter
intensity of the vi transition for a homonuclear spin pair
principal axis system
paramagnetic spin-or bit
vector between spin I and S of magnitude r,
distance between I and S
position vector for the kth eeleciroa (N denotes with respect to a nucleus)
the dipolar coupling constant (kHz)
the effective dipolar coupling constant (kHz)
rotational resonance
FDs for the 2D spin-echo NMR experiment

xvii
SD
SO
SOS-CI
SOPPA
STO
T
tl
t2
TBPS
TEPS
TMPS
spindipolar
spin-orbit
sum-over-states configuration interaction
second-order polarization propagator approximation
Slater-type orbital
temperature
evolution time in 2D spin-echo experiment
acquisition tirne in 2D spin-echo experiment
tetrabutyldiphosphine disulfide
teuaethyldiphosphine disulfide
te tramethy ldiphosphine disul fide
phase angle for Bd
polar angle defining the position of q, with respect to the PAS of (3
Euler angles
difference between the values of the Euler angle a for chernical shift
teosors of a spin pair
rnagwtogyric ratio of nucleus I (rad T1s")
chernical shifi tensor (ppm)
principal compownts of 8 (ppm)
a component of 6 (ppm)

xviii
isotropic chernid shifi (pprn)
the angle between B, anci r,
asymmetry in J
polar angles defining the position of $ with respect to the PAS of (J
tip angle
skew
the set of parameters that dehe the wavefunction for a molecule
permeability of vacuum
frequency corresponding to the ith (i = 1,2. 3 or 4) transition for a
homonuclear spin pair
Vii resoaance frequency for a sample
V,f resonance frequency for a reference compound
V rf Frequency of an applied radio frequency field
VKN MAS spinning frapuency
VO Larmor frequency
(3 nuclear magnetic shielding tensor
a,, , or, q3 principal cornponents of (f (ppm)
d diamagnetic contribution to a (ppm)
paramagnetic contribution to a (pprn)
uii a component of U (ppm)
0, isotropie ouclear rnagnetic shielduig (ppm)

xix
nuclear magnetic shielding for a ceference cornpourd (ppm)
duratioa of radio f r q u e ~ ~ y irradiation
dihedrai angle for (CH,),P-P(CH,), +
dihedral angle for H2P-PH,
dihedral angle for H3P-PH,+
angle describing the relative orientation of adjacent phosphole rings in the
phosphole tevamer
rotation angle of a single crystal in a goniorneter about its X, Y or Z axis
span m m )

Acknowledgments
1 would like to thaiilr my Ph. D. supervisor. Rod Wasylishen, for his patience
and support. For his constant encouragement anâ guidance, 1 am grateful. The
members of the solid-state NMR group at Dalhousie University and at the University of
Alberta are acknowledged for al1 their help. In particular, 1 would like to thank Klaus
Eichele for help with the spectrometers and suggestions on many projects. 1 would also
like to thank Dave Bryce, Guy Bernard, Shelley Forgeron. Rob Schurko. Mike
Lumsden, and Scott Kroeker for their interest in my research projects. Special thanks
to Dave and Guy for proofreading this thesis.
Neil Bdord and Paul Ragogna at Daihousie University and Edgar Ocando-
Mavarez at IVIC are acknowledged for samples and suggestions. 1 am grateful to Gang
Wu at Queen's University for suggestions on the phosphole tetramer project. Sian T.
Cameron at Dalhousie University is acknowledged for his contribution to the phosphole
tetramer project as well. I am gratefùl to Jim Britten at McMaster University for his
contribution to the TMPS project. 1 would also like CO thank Brian Millier at Dalhousie
University for his help.
I acknowledge the hancial support of the Natural Sciences and Engineering
Research Council (NSERC) of Canada. the Izaak Walton Killam Trust, and the Walter
C. Sumner Foundation for postgraduate scholarships. Al1 NMR spectra were acquired
at the Atlantic Regional Magwtic Resonance Centre (ARMRC) which is also funded by
NSERC.
Starting my Ph. D. degree at Dalhousie University and finishing at the
XX

xxi
University of Alberta means that 1 have twice the number of friends and colleagues to
thank cumpared to the average Ph. D. candidate. To everyow 1 met at both places.
thank you. F W y . 1 would like to th& my family for al1 their support and
encouragement.

Chapter 1: Introduction and Seope
1.1 Introduction
Nuclear magnetic resonance (NMR) spectroscopy is one of the most powerful
tools avaiiable for characterizing molecular structure and dynamics in chernistry.
However, it is important to recognize ihat some fundamental questions regarding the
relationship between NMR parameters anâ structure remain. An NMR spectnim is
characterized by a number of parameters, for example the chemical shift and the indirect
spin-spin coupling (or J-coupling) constant. The majority of the NMR literature
concentrates on the measurement of the isotropic values of these parameters and the
application of various empirical niles and correlations to exuact information on
structure and dynamics. For example, one of the most important relationships between
an isotropic NMR parameter and molecular structure is the Karplus relationship, which
describes how the indirect spin-spin coupling over three bonds depends on the dihedral
angle.' However, the chernicd shift and J couplhg are tensor propertie~;~~ hence, it is
essential to characterize the anisotropic nature of these interactions to fully understand
the origins of observed trends. For example, considering only the isotropic chemical
shift leads to the observation of trends which often de@ simple explaaations in terms of
local structure, particularly in the case of phosphorus chemicai shifts."
The determination of the chernical shift tensor, ôoth its principal components and
the orientation of its principal anis system (PAS) with respect to the moletule, is
particularly valuabte due to its close relationship with the local structure and electronic
1

2
proprties of the rnole~ule.~ Nuclear magnetic resooance spectroscopy of solids is ideal
for investigating the tensor nature of these parameters since the interactions are not
usually averaged as in the case of solutioa NMR stuciies . The field of solid-state NMR
is curreatly enjoying an expanding popularity, thanks in part to a number of
experimental advances which have led to applications of this technique to a range of
research problerns, from protein structure to materials science. Recent advances in
computatiod chemistry, particularly with regards to ob initio calculation of NMR
para me ter^,^^*^*' bave resulted in the use of calculated results as a complement to
experimental data.
Ideally, NMR studies using single crystals are desirable since, in principle. the
chernical shift tensor as well as dipolar and J-coupling tensors may be determined
unambiguously. Unfortunately, large single crystals are rarely available; hence, powder
samples must be used. If the system of interest contains an isolated spin pair, some
orientation information can be extracted from NMR studies (see section 2.2).
One objective of my Ph.D. research was to use solid-state I1P NMR combineci
with first-principles calculatioos to characterize the NMR parameters for systems
containhg P-P bonds. The three examples illustrated in figure 1.1 have been selected
for discussion in ihis thesis. For one of these systems, tetramethyldiphosphine
disulfide, 1, abbreviated as TMPS, a singlecrystai "P NMR investigation was carried
out. For another compound, a phosphole tetramer, 2, the NMR parameters were
characterized experimentally using a crystalline powder sample and by theoretical
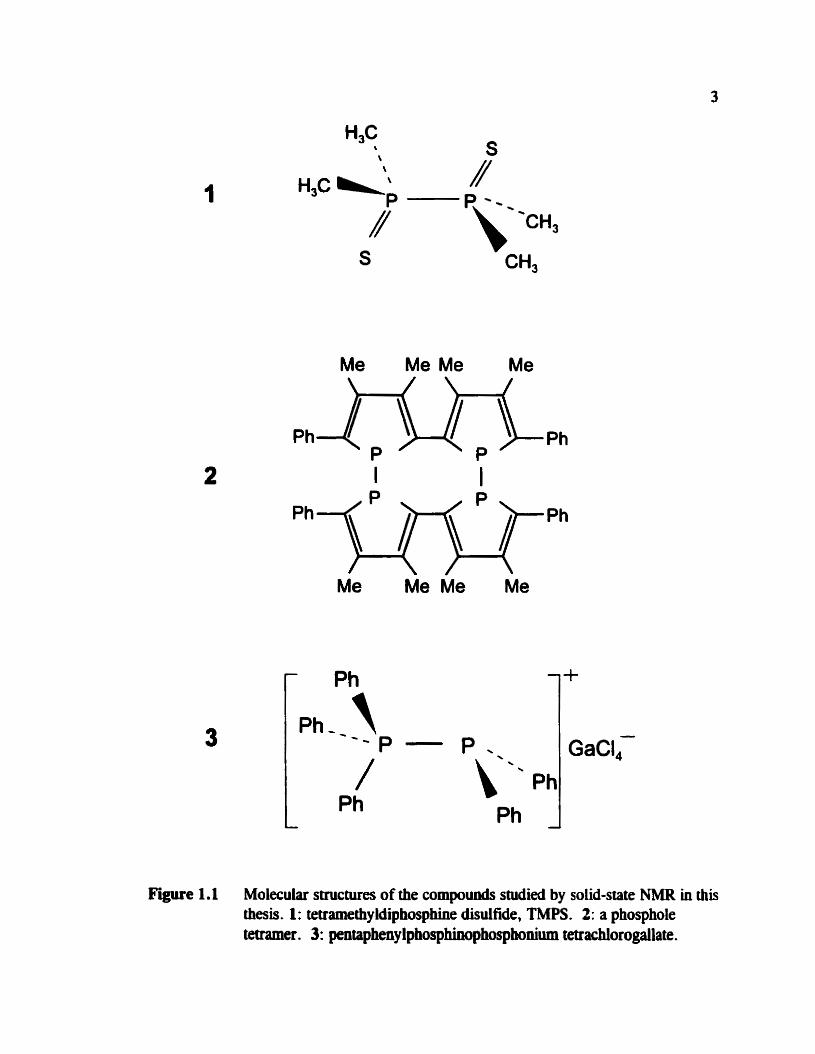
Figure 1.1 Molecular ssllctures of the compoumls studied by solid-state NMR in this thesis. 1 : tetramethy ldiphosphhe disulfide, TMPS . 2: a phosphole tetraxner. 3: pentapheny lphosphinopho~honium tetrachlorogallate.

4
calculations. In addition, the X-ray crystai structure was redetermined in the course of
this study. For a third compound, pentaphenylphosphinophosphonium
tetrachlorogallate, [Ph,P-PPhJ[GaCb], 3, the X-ray structure was unavailable aad NMR
investigations of solid samples have provided valuable structural information.
There are three themes running through the examples presented in this thesis.
The fist theme concems the orientation of the phosphorus chemical shift teasor as a
reflection of molecular structure. It has been shown that for compounds such as
tetraethy ldiphosphine disuifide, (CH3CH3,P(S)P(S)(CHZCH3)Z , abbreviated as TEPS.
the most shielded component of the phosphonis chemical shift tensor is duected dong
the P = S bond. 1°*11 This observation has also been made in (CH3),PS. l2 Investigation of
the phosphonis chemical shift tensor orientations in TMPS may aid in establishing this
observation as a general phenornenon. On a related note, one assumption about
chemical shift tensor orientations is that the direction of greatest shielding corresponds
to the direction of greatest electron density." Studies of phosphonis shielding in a
phosphole tetramer and a phosphinophosphonium cation, where there are formal lone
pairs on the phosphorus nuclei, affords an opportunity to investigate these trends
m e r .
A second theme concems the indirect spin-spin couplhg tensor between
phosphonis nuclei that are bonded to each other, 1J(31P,31P), and in particular its
aaisotropy, Aï. The investigation of hl in TMPS is relevant here, as is explained in
fiirther detail in section 1.2. Ab initio dculations of 1J(31P,3'P) in mode1 compounds,
H,P-PH, and H3P-PH,+, are potentially usehl for exploring the dependence of

1Je1P,31P) on conformatioa.
The third theme concems the value of ob initio calculations of NMR parameters
as a cornpiement to experimental &ta. The accuracy of calculated chernid shift tensor
orientations is explored in the chapter on TMPS (chapter 3) and calculations of J on a
mode1 system, H,P-PH,+, are applied to the determination of the dipolar coupling
constant, R,, between the phosphorus nuclei in [Ph,P-PPhJ[GaClJ, and hence estimate
the P-P bond length (chapter 5). The contents of each chapter are discussed in more
detail in the following section.
1.2 Thesis Outline
Following a chapter detailing the relevant background theory (chapter 2). the
work on TMPS is describeci in chapter 3. There have been a few NMR studies of
alkyldiphosphine disulfides in the literature. Many solution studies stem from an
interest in the relationship between conformation and indirect spin-spin coupling
between phosphorus nuclei that are directly bonded to each other, 1J~1P,xP).14*15
TEPS1o*ll and tetrabutyldiphosphine disulfidei6 (TBPS) have beeo testing grounds for the
determination of anisotrop y in indirect spin-spin coupling teasors. Ear 1 y sing leîry stal
NMR studies reporteci large anisotropies for 1J(3 LP,31 P) in TEPS and TBPS of 2.2 kHz1
and 1.9 kHz,'6 respectively. The phosphorus chemicai shift and spin-spin coupling
tenson for TMPS (figure 1 .l, structure 1) are characterized by a single-crystal jlP
NMR study and by an iodependent anaiysis of jlP NMR spectra obtained from
crystaliine powdered samples combined with ub inirio calculations. As so much

6
information is available about this system. it also serves as a benchmark for evaluating
experimentai NMR methods as well as ub initio approaches for the characterization of
NMR parameters. Since accurate P-P bond lengths are required for a reliable
determination of AJ, the X-ray crystal structure was redetermined. Data available in the
literature for related compounds provide an opportunity to consider some trends in
phosphonis magnetic shielding.
Chapter 4 summarizes the results for a phosphole tetramer, figure 1.1, structure
2. This compound contains two phosphonis spin pairs, each one consisting of two
three-coordinate phosphorus nuclei directîy bonded to each other ; characterization of
phosphorus chernical shift tensors and spin-spin coupling parameters in such
environments are relatively rare.13*17*18*1920 Ab initio calculations prove to be an
extremely valuable addition to the experimental "P NMR data for a complete
characterization of the two phosphorus chernical shift tensors since single crystals large
enough for an NMR investigation were unavailable for this compound. It was found
that the relative orientation of the two chernical shift tensors reflects the local structure.
Inconsistencies between the NMR data and the published X-ray crystal structurefi
prompted a redetermiaation of the structure.
The structural characterization of pentapheny lphosphinophosphonium
tetrachlorogailate, [Ph,P-PPhJ [GaCL] (figure 1.1, structure 3), by solid-state NMR is
detailed in chapter 5. Phosphinophosphonium saltsa represent a new class of
compounds and are thus important to characterize. X-ray crystal structures are not
avaiiable in the Literanire; hence, characterization by other means is vital. For the

7
example presented in this chapter, ~h3P-PF%J[GaQ]. some structural information is
avaiiable from analysis of "P NMR spectra of solid samples combined with ab initio
caiculation of 1Jc1P,31P). Trends in 1Jc1P,31P) are explored by ab initio caiculatioas on
mode1 systems, H2P-PH2 and H3P-PH2+. in particular. the effect of the conformation is
considered.
Finally, extensions of the research projects describeci here are proposeci in
chapter 7 of this thesis.

Chapter 2: Background Theory
2.1 NMR Interactions
2.1.1 O v e ~ e w
The systems of interest in this thesis consist of homonuclear spin pairs, IS,
where 1 and S refer to spin-% nuclei. To d y z e the NMR spectra for such a spin pair,
the following Hamihonian needs to be considered:
where Nz(l) and MZ(S) are the Harniltonian operators that describe the interaction of 1
and S respectively with the applied external magnetic field, $, termed the Zeeman
interaction. WC) and W S ) describe the interaction of each spin with an applied radio
frequency field, B,. and G ( S ) are the nuclear magnetic shielding (or chernical
shift) Hamiltonians, %,(I,S) describes the dipolar interaction between I and S, and
H,(I,S) is the indirect spin-spin or J-couplhg interaction between the two nuclei. Hz
and 9& are under the control of the spectroscopist, hence they are termeà exzenuzl
Hamiltonians; the remaining ternis are dependent on the Panire of the system anci are
hence referred to as intemal Hamiltonians.

2.1.2 The Zeeman Interaction
The huidamental phenornenon upon which NMR is based involves the interaction
of the nuclear magnetic moment with B, as described by the following Hamiltonian:
where y, is the magnetogyric ratio (in rad T' s'l) of nucleus I and 1 is the nuclear spin
angular momenturn operator. For NMR studies of spin-% nuclei, this is the largest of
the interactions described in equation 2.1. The spin states for I are described by the
quantum number m, which gives the z-component of angular momentum. For a spin-%
nucleus, ml = + '/i or -%. In the presence of Bo, these two states are separated by an
energy, Al?, that is dependent on the magnitude of the applied magnetic field, Bo, as
illustratecl in figure 2.1. The corresponding frequency ,
is called the Larmor frequency.
2.1.3 Radio-Frequency Interaction
To obtain an NMR spectrum, it is necessary to induce transitions between the
allowed nuclear spin states. This is achieved by applying electromagnetic radiation at or
near the Larmor frequency d usually in a direction that is perpendicular to $.

Figure 2.1 The Zeeman enetgy as a fuoction of the magnitude of the applied magnetic field, Bo, for a spin-% nucleus with y, > 0.
Typically , vo fdls in the radio frequency region of the electromagnetic spectrum. The
Hamihonian describing the interaction between I and an applied radio frequency field,
B,, is identical to the Zeeman Harnilionian (equation 2.2) except that the magnetic field
is not static. The magnitude of Bd in a direction perpendicular to B, is B,:
where B, is the amplitude of the applied radio frequency field, v, is its frequency (at or
near vo), and a is its phase. Under the influence of B , the net magnebtion resulting

11
from the nuclear magnetic moments are tipped away from their equiiibrium positions by
an angle 0, = y,B,r, where r, is the duration of the radio fiequency irradiation.
NMR is an inherently insensitive technique, as AE is much less than the thermal
ewrgy, kBT. As a result, the population difference between the m, = + 'A and m, = -!h
States, An,, is very small. Under the high-temperature approximation,
where N, is the total nurnber of nuclei, I , in the sample. Typically , An, / N, is on the
order of 1 O-5 ?
2.1.4 Nuclear Magneâic Shieldhg and Chernieal SWt
The nuclear rnagnetic shielding interaction, described by a second-rank tensor,
0, arises from the interaction of electrons around the nucleus with B, to produce a local
induced rnagnetic field at the nucleus, Bi,:
The auclear rnagnetic shielding Hamiltonkm is:
&-i3c,(~ = y,1*0*B~

in general, O can be expressed as the sum of symmetric and antisymmetric parts
The symmetric tensor, which has compownts such that oi,=oji, influences the line shape
in NMR of solids, whereas the antisymmetric part does not to tint order.*l in
principle, the antisymmetric part can play a role in the relaxation of the nu cl eu^.^ In
this thesis, the symbol O refers exclusively to the symmetric put:
in which there are six independent components. It rnay be transformeci, as with any
symmetric second-rank tensor, to a frarne of reference known as the principal mis
system (PAS), in which the tensor is diagonal:
where a,, , a,, and a, are the principal compownts (in units of ppm) of the nuclear
magwtic shielding tensor. The PAS is related to the molecular frame of reference by
Euler angles, a, P. and y ." There are still six parameters to determine (three principal
components and three Euler angles) to fdly characterize the tensor. In the soiid state,

Figure 2.2 Illustration of the angles which define the orientation of the applied magnetic field, &, and the dipolar vector, rû, with respect to each other and to the principal axis system of the nuclear magnetic shielding tensor for I .
the observed frequency is dependent on the orientation of the crystallite with respect to
B, if the shielding is anisotropic; thus equation 2.3 must be modified to include both the
Zeeman and nuclear magnetic shielding interactions :
i - (a,, sin20 cos2 4 + a, sin29 sin2 4 + a,, cos20) 1 where 0 and 4 are polar angles definhg the position of B, in the PAS of the shielding
tensor, as shown in figure 2.2.

By convention, a,, a a, s a,,, therefore a, corresponds to the direction of
greatest shieldirig. Experimentally, the chemicai shift teasor, 6, is measured rather than
the nuclear shielding tensor, the difference king thai (3 is referenced to the bare nucleus
while 6 is referenced to a prllnary standard:
V.. - oü = u 'Ef 106
h m ) "Ri
where Vii and v, refer to the resonance frequencies of the sample and the reference
respectively. The chemical shift and nuclear magnetic shielding are related as follows:
Since 1 - o, = 1 .O, equation 2.12 simplifies to:
The largest component of the chemical shift teosor, &,,, corresponds to the srnailest
component of the nuclear magnetic shielding tensor, a, ,, in accordance with equation
2.13. Hence 6,, 2 42 2 &33. The isotropie nuclear magnetic shieldiag is '1, the trace of
(3:

with an analogous expression for the isotropic chemid shift, 6,. One can also define
two parameters, the span, Q, and the skew, K. which describe the breadth and sbape of
the powder pattern:27
Span = Q = a,,- a,, = o,*- il3,
3 ( 0 ~ - 0 , ) 3(6,-hu0) skew = n = - -
Q Q
Altematively, the anisotropy and asymrnetry are ofien used; however they have k e n
defined in various ways by different au th or^?^ For a powder sample, in which al1
possible orientations of crystallites are preseat, the NMR spectrum of an isolateci spin
cm have one of several line shapes as shown in figure 2.3, depending on the nature of
the chernical shift teosor. In one case, 6, , = 6, = 6 , (isotropic), in the second case
6, , = 6n or 6 , = b3 (axially symmetric) and in the third case none of the components
are equivalent (non-axially symmetric) .
2.1.5 DipIar Coupüng
Dipolar or direct spin-spin coupling is an interaction between auclear magnetic
moments separated by a vector r, where the subscript indicates nuclei I and S
re~pectively.~ '~ This interaction is anaiogous to the classical interaction between two
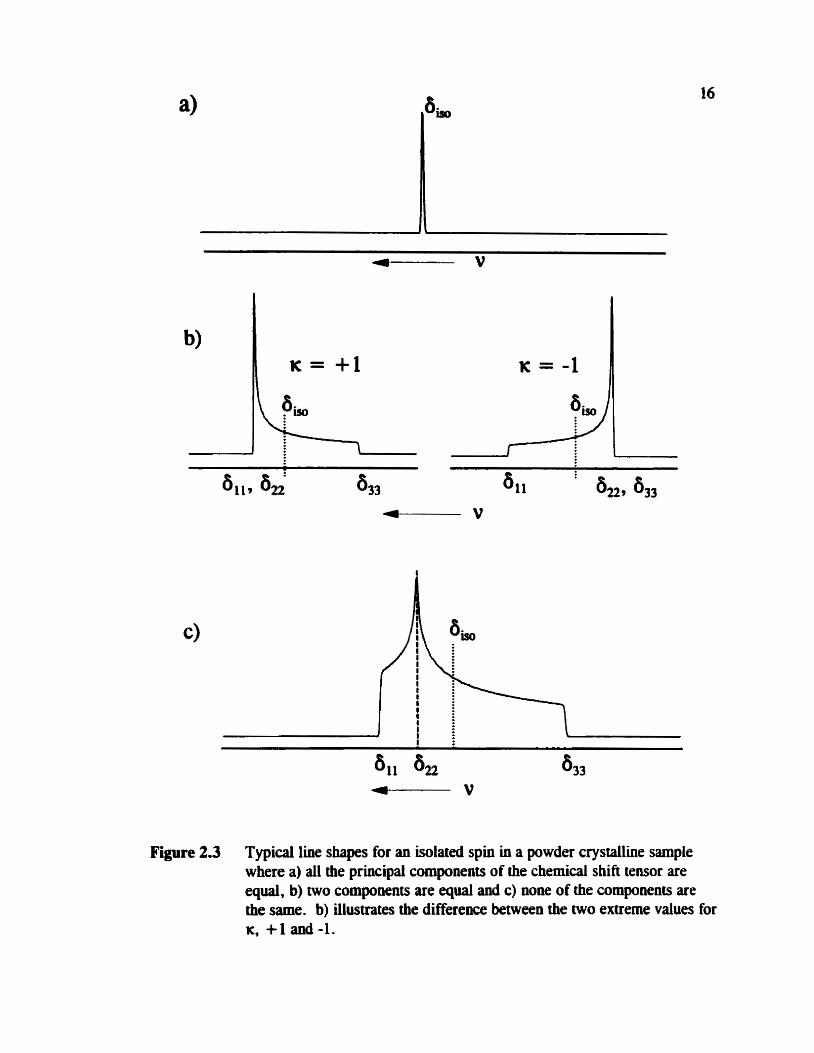
Figure 2.3 Typical line shapes for an isolated spin in a powder crystalline sample where a) al1 the principal components of the chernical shift tensor are equal, b) two compownts are equal and c) ww of the components are the same. b) illustrates the difference between the two extreme values for K, +l and-1.

bar magnets.
The dipolar H d t o n i a n is:
where S is the nuclear spin angular momentum operator for nucleus S and D is the
dipolar coupling tensor which is diagonal in its PAS:
if I and S are not magneticaily equivalent, e.g . , a heteronuclear spin pair and:
if I and S are magnetically equivalent, e.g., a homonuclear spin pair. Different types of
homonuclear spin pairs are discussed in further detail in section 2.2. Udike 0, D is
traceless; hence, dipolar interactions are not observed d k t l y in NMR studies of
isotropie fluids. The unique axis of D is dong r, uniess there is anisotropic motion.

The dipolar coupling constant, RD,, in frequency wits (Hz) is defined as follows:
where CI, is the permeability of vacuum and kW-') refers to the vibrationai average of the
cubed intemuclear distauce. Since RDD depends only on (r,-3). the remaining factors
king constants, the dipolar coupling constant is a valuable parameter for structurai
determination by NMR of solids, as will be discussed in greater detail in section 2.1.6
and chapter 5 . The specvum for I coupled to a spin-% nucleus, S, in the absence of any
other interaction except the Zeeman interaction. consists of a doublet centred at the
Larmor frequency of I with a splitting given by RD, (3 cos2 C - 1) if I aod S are not
magnetically equivalent (figure 2.4(a)) and by '1, RD, (3 cos2 C - 1) if I and S are
rnagnetically equivaient (figure 2.4(b)), where C is the angle between Bo and r,. In a
powdered crystalline sample, al1 possible values of C are present, hence a characteristic
powder pattern with a Pake doublet3' is observed. Figure 2.4(c) shows the Pake doublet
for a heteronuclear spin pair while the specuum arising fiom a pair of magnetically
equivalent nuclei is shown in figure 2.4(d). The two subspectra, shown by dotted lines,
illustrate the fact that the dipolar line shape arises from two transitions conesponding to
%,- two spin States of S.

w - . - . - - - - - = - - - - - - - - - = - ~ - = - - - - - - -
1800 1400 1OOO 600 200 -200 -600 -1000 -1400 -1800
Hz Figure 2.4 Calculated NMR spectra for the I nucleus of a spin pair where 1 and S are
a) not magnetidly equivalent aod b) where they are magnetidy equivalent. Bo and r, are perpendicular in this case, i .e., F =!JO0. The corresponding line shapes for powder crystalline sample, where al1 values of C are present, are shown in c) for noomagneticaiiy equivalence and d) for magnetic equivalence. The subspectra shown by doned lines in c) correspond to the two spin States of S. These spectra were calculateci using RD, = 700 Hz.

2.1.6 J Coupüng
Another interaction between two nuclei that may be measured in NMR
experiments is the indirect spin-spin or J-mupling interaction. This is an interaction
whereby a nucleus pemirbs the surrounding electrons. This perturbation is
subsequently transmitted to a second nucleus through the electron network of the
molecule. The streogth of the coupling depends on the nature of the electron network,
hence J coupling is an extremely useful parameter for characterizhg systems where
nuclei are separated by one or more covalent The Hamiltonian for J couphg
between two nuclei is:29S0
where J is a second-rank tensor. Like the nuclear magnetic shielding tensor, J can be
represented by a symmetric and antisymmetric part;' however the antisymmetric part
does not perturb the NMR line shape to first order? In its PAS, the symmetric part of
J is given by :

The isotropie J coupling is:
While J coupling is measured experimentally in units of Hz. cornparison of indirect
spin-spin couplings for different spin pairs is facilitated by using the reduced coupling
teosor, K. which is independent of the magnetogyric ratios of the coupled nuclei:
where K is in units of N A" m-3 or equivalently in TL J? The ensuing defultions also
apply to K.
The features of J may be described by the anisotropy, hl, and the asymmetry,
:2935."
and
where the p ~ c i p a l compownts are assigned accordhg to 1 J, - J., 1 r 1 JI, - J,1 2

22
1 J - J . If J is axially symmetric, then hl = J, - J , where J, is the component dong
the intemuclear axis while J , is perpendicular to it. The indirect spin-spin couplhg
Hamiltonian (equation (2.21)) can be expressed in terms involving only J , and an
anisotropic term which is identical in form to the dipolar Hamiltonian (equation
2. 17):"j2
where
Consequently, it is impossible to experimentally distinguish between the dipolar
coupling and anisotropy in J. Experimentally, one measures an effective dipolar
coupling constant:
In practice, RD, can be calculated fiom equation 2.20 if the intemuclear distance is
known. If RDD differs sipnificantly from the experimental value of R,, then one can
estimate AJ. R& is available from solid-state NMR experiments or, for simple diatomic

molecules, from hyperfine constants measured in molecular beam or high-resolution
microwave spectroûcopy experiment~.~' Since RH and RD, c m have the same or
opposite signs, two possible values of hl result €rom equation 2.29, one of which is
usualiy discardeci since it is much larger than J.,. For coupling involviag lighter nuclei,
hl is often assumed to be negligible; for heavier nuclei, hl can be quite large.33*u For
example, in thallium fluoride, A J ~ T 1 , l ~ = -13.3 * 0.7 While reliable
experimental determinations of A J are scarce,m-3739flA1-42 theoretical calculations of J
otfer an avenue for examiniag the relationship between indirect spin-spin coupling and
structure. Calculations of I from fust pruiciples will be discussed in section 2.3.2.
2.2 NMR Spectra Arising from isolatcd Spin Pairs
2.2.1 Overview
For a system consisting of an isolated spin where the chemical shift is the only
internai interaction present, analysis of NMR spectra arishg from single crystals an, in
principle, yield the orientation of the chemical shift tensor with respect to the molecule.
Unfortunately, single crystals of sufficient size for NMR studies are rarely available.
NMR spectra of powder sampies yield the three principal components of the chemical
shifi tensor; however there is no means of determinhg the tensor orientation relative to
the molecular frame of reference unless the chernical shift tensor is axially symmeuic.
If a dipolar interaction is also present, as in the case of systems containhg isolated spin
pairs, then the spectra of powder samples become sensitive to the relative orientation of
the chernical shîft tensors with respect to r, which of course is in the mlecular frame

of reference .43
Systems where an isolated spin pair is present have been extensively
investigated in the solid state."" As mentioned previously, if two nuclei have the same
magnetogyric ratio, the system is referred to as a homonuclear spin pair; otherwise they
form a heteronuclear spin pair. For a homonuclear spin pair, a further distinction can
be made between magnetic equivdence where the two nuclei are relaied by a centre of
inversion and crystallographic equivalence where the two nuclei are related typically by
a C, axis or a &or plane.2 in the case of magnetic quivalence, the two spins have
identical chetnical shift tensor components and orientations. For nuclei that are
crystallographically equivalent but magnetically non-equivalent, the chemicai shift
tensor components are the same but their orientations differ. The most general case is
observed when the two nuclei are neither magnetically nor crystallographically
equivalent. Spin pairs are often classified as A,, AB, or AX spin systerns. If the two
nuciei are magneticall y equivalent, the y have identical resonance frequencies for any
orientation of the spin pair in $, hence they form an A, spin system. On the other
hand, if the difference between their resomce frequencies is much greater than the
magnitude of the spin-spin couplings (dipolar and indirect), then the spin system is
designateci as an AX spin system. The AB spin system lies between these two extremes.
It should be wted that such a iabelling system is not entirely suitable in solid-state NMR
since the difference in the resonance frequencies as well as the magnitudes of the spin-
spin couplings are orientation dependent. It is possible to have a spin pair that is A, at
one crystallite orientation but AB or AX at another.

In general, homouuclear spin pairs give rise to four iramitions in the solid state
hteWities, pi: lOM45.4W7
where v, and v, depend on v,, a,, , a,, and a,,, and the orientation of PAS of the
shielding tensor with respect to $, given by the polar angles 0 and (figure 2.2). for
each nucleus, accordhg to equation 2.10. The terms A, B, and D are:
1
D = [ (v, - v,)' + BZ]'
For an AX spin system, (v, - v a w B; hence, D = v, - v,. Four peaks are observed, two

26
in the region of the I nucleus and two in the region of the S nucleus. In both cases, the
splitting of the I and S peaks is ) J , - &(3 cos2C - 1) 1 . For an A, spin system, v, = v,,
hence D = B, thus the intemities of v, and v, are zero and the remaining two peaks are
split by 1 J , - ' J , ReH (3 cos2 C - 1)l. An interesthg situation a r k s in NMR spectra of
solids containhg isolated AB spin systems. It is possible for the two outer peaks to be
more intense than the hvo inner peaks." An example of this is presented in chapter 3.
Such spectra are not observed for isotropie fiuids. For a powder crystalline sarnple, the
addition of J coupling modifies the splittiags shown in figure 2.4, as illustrated in figure
2.5. The observed l k shape is dependent on the relative signs of Re, and J,.
2.2.2 The Dipoiar-Chemical Shift Methoà
The combination of anisotropic chernid shifi and dipolar coupling interactions
results in a specirum where each component of the chernical shift tensor is split
according to (in units of freq~ency):~~
for an AX spin pair, where a and P are the polar angles defining the position of r, with
respect to the PAS of in figure 2.2. The expected line shape is illustrated in figure
2.6(a). Although one cannot masure the sign of Av,, the sum of the three splittings

Figure 2.5 Calculatecl NMR spectra for the A nucleus of an AX spin pair illustrating the effect of dipolar and J coupling . For a) J , = +200 Hz, while J , = O Hz for b). J , = -200 Hz for c). Al1 spectra were calculated ushg &fi = 700 Hz and in the absence of anisotropic magnetic shielding.
mut be zero since the dipolar coupling tensor is traceless. One of the subspectra
shown in figure 2.4(c) is stretched out while the other is compressed, as shown in
figures 2.6(b) and (c). For an AB spin system, the overlap of the subspectra due to both
nuclei leads to complex h e shapes, examples of whifh will be discussed in this thesis.
As i.: evident from equations 2.37 to 2.39, the spectnun is sensitive to the
relative orientation of the chernid shift teosor and r,, which is coincident with the
unique component of the dipolar couplhg tensor. Thus sorne information regardhg the
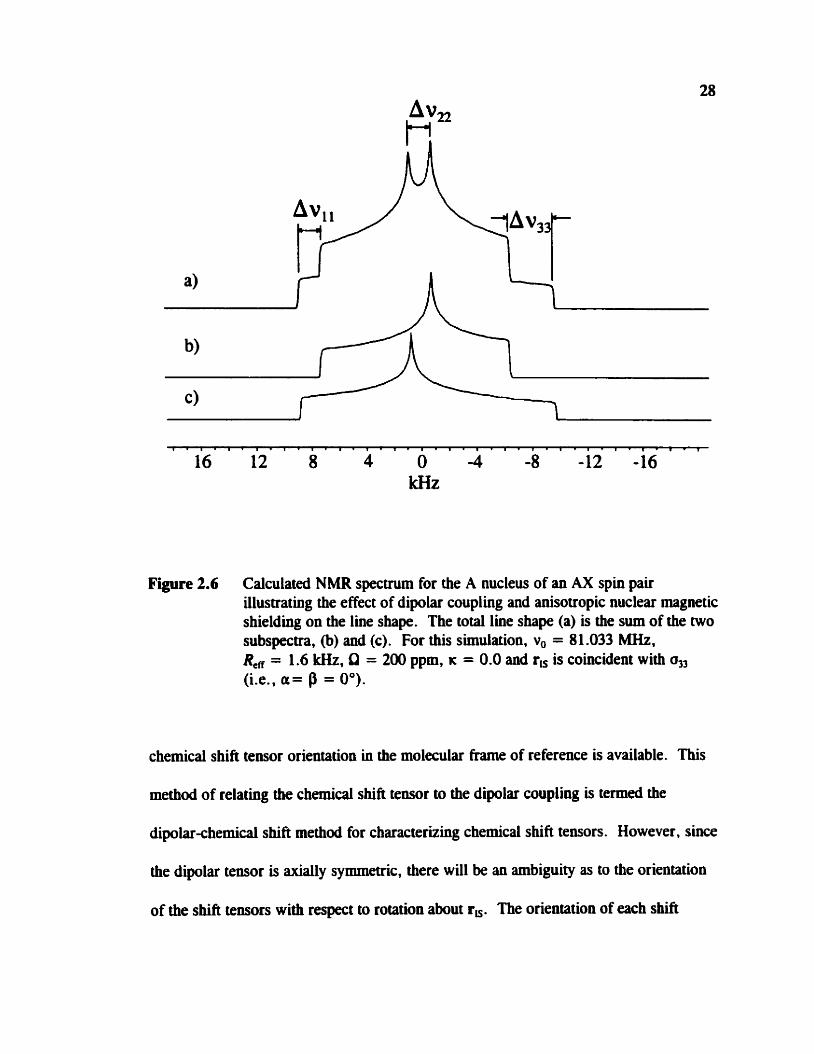
Figure 2.6 Calculated NMR spectrum for the A nucleus of an AX spin pair illustrating the e ffect of dipolar coupling and anisotropic nuclear magnetic s hielding on the line shape . The to ta1 line shape (a) is the sum of the two subspectra, (b) and (c). For this simulation, v, = 81 .O33 MHz, R, = 1.6 kHz, Q = 2 0 ppm, K = 0.0 and r, is coincident with (Le., a= P = O").
chemicaf shift tensor orientation in the molecular frame of ceference is available. This
method of relating the chernical shift tensor to the dipolar coupling is temed the
dipolar-chernical shifi method for characterizing chernical shift tensors. However, since
the dipolar tensor is axially symmetric, there will be an ambiguity as to the orientation
of the shift tensors with respect to rotation about r,. The orientation of each shin

Figure 2.7 Euler angles relating the PAS of the nuclear magnetic shielding tensor to the molecular ftame of reference for nucleus I of a spin pair, IS, where aZ0Q is the original position of a,.
tensor relative to re can be described by a set of Euler angles, a , P. and y,26 as
illustrateci in figure 2.7. The angle P positions r, relative to o, while y determines
whether r, is closer to a,, or a=. The angle a describes the rotation of the chernid
shift tensors about r,; since the dipolar tensor is axiaiiy symmetric, a carmot be
measured relative to an axis fixed in the molecular frame of refereace, hence its value is
arbitrary. However, the difference between the values of a for the two chernical shift
teusors (which wiU be denoted by ha in this thesis) may be determined. Aa defmes the
angle between the projections of the a,, coniponents for I and S on to the same plane. It

should be notai that the angles above are dEerent from those defiaed in figure 2.2.
The analysis of spectra depenàent on as many as six chemical shifi tensor
components, two sets of Euler angles and two spin-spin coupling parameters may seem
difficult if not impossible; however, computer pro gram^^^^-" are available for
caiculating spectra based on the Hamiltonian presented in section 2.1. allow ing a
cornparison with the experimental results. The dipolar coupling constant, RD,, cm be
estimated using bond lengths from diffraction structures if avaiiable. From magic angle
spinning (MAS) NMR spectra, J.,, 6,, and, in some cases. the principal components of
V2 for I and S may be detennined. As well, techniques like 2D spin-echo spectroscopy
are available for homonuclear spin pairs to separate the effect of dipolar and J coupling
from the chemical shift interaction (see section 2.2.3). Acquirhg spectra at two or
more applied magnetic fields is essential as the various spin interactions described in
section 2.1 scale differently with B,,. In practice, al1 the NMR spectra are simulated
until one obtains a set of NMR parameters which gives a satisfactory reproduction of al1
experirnental spectra. In addition, reliable ab Niitio calculations can provide uiformation
concerning the orientations of chernical shift tensors and spin-spin coupling tensors with
respect to the molecular frarne of reference. Ab initio calculations of these two NMR
parameters are discussed in section 2.3.
2.2.3 2D Spin-Echo NMR Spectroscopy
The 2D spin-echo NMR experiment is extremely useful in the characterization of
spin-spin coupihg parameters in systems consisting of isolated homonuclear spin pairs.

CP- spin-echo - i acquisition
contact
Figure 2.8 The 2D spinecho pulse sequence which consists of a CP sequence followeâ by a spin-echo pulse and then the acquisition of an FID.
The spin-spin coupling interactions are separated fiom the chernical shift, allowing for
an independent measurement of R,, and determination of the relative sigm of R, and
I,. The basic 2D pulse seqwnce consists of a preparation period, an evolution time of
duration t , followed by an acquisition tirne, t2.53 The evolution t h e is incremented to
produce a 2D &ta set. The 2D spin-echo pulse sequence,I3 shown in figure 2.8,
consists of the standard cross polarizationM (CP) experiment, under the Hartmann-Hahn
match condition," for the preparation period followed by the evolution time during
which the x spin-echo pulse is applied. The spin-echo pulse has the effect of refocusing
inhomogeneities due to nuclear magnetic shielding, leaving the rnagnetization to evolve

32
exclusively under the influence of spin-spin coupling. In the resulting 2D spectnim, the
Fi projection contains the spin-spin coupling data while the R projection is the normal
ID powder pattern of a stationary sample. Nalcai and McDowellnM have derived
expressions for the fkee induction decays (FïDs). s(tJ and s(ta, of the 2D spin-echo
NMR expriment:
where cos 26 = (v, - v,)ID. sin 26 = BID and the V i s (i = I to 4) are given by
equations 2.30 to 2.33. For an A, or AX spin system in the absence of J coupling , the
dipolar splittings at the 'bonis' of the Pake doublet in the Fl projection will be R, or
R, respectively; however, the d y s i s of 2D spin-echo spectra is complicated by the
possibility of the spin system king A,, AB or AX at different orientations, as
mentioned previously (section 2.2.1). If J , is also present then the splitting in the R
projection is modifieci, sunilar to the case illustrated in figure 2.5. A centre artifact is
ofien observed which has been attributed to imperfect rf pulses.13 Fortunately , the Fl
projection can stüi be caiculated and compareci to the experimental results to extract the
coupling constants. Twodimemional spin-echo spectroscopy has b e n used extensively

to investigate homonuclear spin pairs in the solid, especially in molecules containing
P-P bonds, for example, Mes'P = PMeso (Mes' =(2,4,6-tri-t-butyl) phenyl),17
Wes'NP-PPh3] [S03CF3] 7 Ph2PPPh2, l3 the pymphosphate anion in Ni4P2O7- 10H20,
and Lawesson's reagent. (C,J14ûCH3PS&.59 Specific details regarding performance of
this experiment on the CMX Infinity 200 spectrometer at the University of Alberta are
given in Appedix 1.
2.3 Ab Initio Calculations of NMR Parameters
2.3.1 Nuclear Magnetic Shielding Teiwrs
The theoretical basis for understanding nuclear magnetic properties in ternis of
modem quantum chemistry was established by Ramsey in a series of papers published
between 1950 and 1953.461*62 A commentary on the significauce of these papers was
publis hed in 2ûûû.63 In this section, Ramsey ' s formulation for uuclear magnetic
shielding and modern approaches to the calculaiion of this parameter wül be discussed.
Rarnsey's perturbation approach separates nuclear magnetic shielding into a
diamagnetic, d, and paramagnetic, d , contribution:"
This partitioning is analogous to the separation of magnetic susceptibility into
diamagnetic and paramagnetic parts? For shieldiag, the lull expressions for ad and op
are:60*65

where r, is the position vector for electron k and L, is its angular momentum operator,
both with respect to a chosen origia. The position vector and angular momentum
operator of electron k with respect to the nucleus of interest are denoted by r, and k,
respectively. One Unportant conclusion from the above equations is that d depends
only on the ground electronic state, IO), hence it is a fust-order property and easiiy
caiculated by ab initio methods. However, op is a second-order property , involving
mixing between ground and excited electronic States, 1 d, and is thus more challenging
to calculate.
Nuclear magnetic shielding is very sensitive to the molecular electroaic
structure, hence very accurate electronic wavefunctioos are required for ab initio
calculations at any level of t h e ~ r y . ~ ~ - ~ " - " The development and implementation of
methods to overcorne the gauge origin problem (vide infia) bas lead to hirther progress
in calculations of shielding tensors. Ab initio calculation of nuclear magnetic shielding
tensors may be usefui to resolve the ambiguity involved in assigning tensor orientations
obtained from a diplatchernical shift analysis (section 2 -2.2) The present statu
of theoreticai methods has been swnmarized in a number of ment r e v i e ~ s ~ * ~ * ~ * ~ - ~ ~ * ~ ~ and

wiii be discussed ody briefly in this thesis.
Rather than ushg Ramsey's formulation for nuclear magnetic shieldiag directly,
most modern ab initio computational approaches use an quivalent expression for the
shielding for I in terms of a second derivative of the total electronic energy, E, which is
dependent on a magnetic field, B, and the set of al1 nuclear magnetic moments, M, for a
mole~ule:~
where M, is the nuclear magnetic moment of I . The calculation of a thus reduces to
evaluating derivatives of molecular electronic energies. Methods in which the
derivatives are evaluated numerically are termed f ~ t e perturbation (FP) or fmite field
(FF). In general, though. quantum chernical programs evaluate the derivatives
analytically , which is more computationaily efficient than numerical analys is . In an
analytical evaluation. it is necessary to determine how the wavefunction respoods to the
perturbation arising from the presence of B andor M. Below, equation 2.45 is stated
more explicitly to show the dependence on the wavefûnction:
where 1 is the set of parameters that define the wavefunction. For a given molecule, it
is necessary to determine the response of the wavefunction with respect to the magnetic

field. via the factor allaB, to calculate the shielding for al1 the nuclei.
With the introduction of a magnetic field, a problem arises due to the need to
describe B using a vector potential.Md-mJ1*R It was recognized early on that calculated
nuclear magnetic sbielding varied dependhg on where one placed the origin of the
vector potential, tenned the gauge origin problem. While the choice of gauge does not
affect the results of an exact quantum mechanical treabnent, this type of calculatioa is
generaily irnpractical; hence, the gauge origin wül be a factor in the quality of the
results when one uses approximate wavehinctions constnicted from standard basis sets
included in quanhim chemistry programs. Several methods are used to overcome this
difficulty , including GIAOn and IGLO ." The GiAO (gauge-including atomic orb itals)
method places the gauge origin for the atomic orbitals at the nucleus on which the
orbitais are centred. The GXAOs are also referred to as London orbitaka The IGLO
(individual gauge for localized orbitals) method uses the origin of the molecular orbital
as the gauge origin. Other meihods include LORG (localized orbital local origin
rneth~d)'~ and CSGT (continuous set of gauge transformations rneth~d).'~
The Hartree-Fock (HF) approach, a common model for shielding calculations,
describes the electronic wavehinction by a single electronic configuration and the
influence of the other electrons is introduced as an effective, static field. This
approximation obviously does not account for the effect arising from the motion of
electrons. Electron correlation is the term used to describe the difference between the
HF model anci an exact description of the system. The neglect of elecaon correlation
can cause severe erron in the calculated nuclear magnetic shielding for systems with

37
multiple bonds or formal lone Ab initio methods which allow for electron
correlation, such as Meller-Plesset (MP) perturbation theoryn or multiconfigurational
selfconsistent field (MCSCF) theory,78-79 ofien give better results that HF in such
situations. ql though not strictly an ub initio method, density frinctional theory (DFT)
has aiso been applied to the calculation of nuclear magnetic ~hielding.~*" Recently,
relativistic effects have been considered in conjunction with DFT.= While the Gaussian
suite of pro gram^"*^ hcludes DFT methods, the currently available functionals do not
include a dependence on the magnetic field. hence it is not an improvement on HF
methods in this case?
To assess the quaiity of an ub initio calculation of nuclear magnetic shielding, it
is necessary to compare calculated results with experiment. However, it is the chemical
shift, 8, and wt the nuclear magnetic shielding, o. which is measured experimentally .
Conversion from one sale to the other requires an absolute shielding scale? If the
shielding for the nucleus of interest is known for a reference compound, then
measurement of the chemical shift relative to this compound can be converted to
shielding according to equation 2.13. Absolute shieMing scales have been established
for a nurnber of elements. including carbon," ~ x y g e n , ~ fluorine," and pbosphorus.~
Recently, we proposed a revised absolute shielding d e for chlo~ine.~'
W ith advances in computational techniques and faster cornputers, the quality of
the caîculations is certain to improve. Signifiant progress has been made in the last
few years and ab initio calculations of nuclear magnetic shielding are becoming
integrated into the rnethodology for aaalyzing NMR ~pectra ."*~*~

2.3.2 Indirect SpinSpin Coupiing (4 Teiisors
For reasons outlined below, reliable ab initio calculations of J couplhg are
challenging. Thus, reports have been relatively scarce compared to calculations of
nuclear magnetic shielding tensors. However, the introduction of some new
computational approaches has prompted interest in the ab initio dculation of J. In
addition. the cecent observation of J couplhg across hydrogen bonds95 has resulted in a
number of papers focussing on the caiculation of J coupling in such s y ~ t e m s . % ~ * ~ ~ The
current state of J-coupling calculations has been recently re~iewed.~~"
In Ramsey's thcory of J c ~ u p l i n g , ~ ~ ~ ' ~ several mechanisms for the interaction
between the auclei and electrons are identified. One rnechanism invoives the coupling
between the nuclear spin and the orbital motions of the electron, temed the spin-orbit
(SO) mechanism. A formai distinction is usually made between a diamagnetic (DSO)
and paramagnetic (PSO) contribution. Another mefhanism is the dipolar interaction
between the nuclear and electron magnetic moments, called the spindipolor (SD)
mechanism. The Femi contact (FC) mechanism describes the interaction between a
nucleus and elecuons that have a tinite probability of being at the nucleus of interest.
The J-coupling Harniitonian may be expressed in terms of a contribution fkom
each mechanism:
Secondsrder perturbation theory is used to solve the above Hamiltonian for expressions

describing each contribution to J:
J = J , + J ~ m - ~ S ~ + 'SD-SD + J ~ ~ - ~ ~ + 'FC-SD
where Jwo involves only the ground electronic states of the molecule. J,, has
contributions involving ground and excited singlet electronic states, while the
contributions involvhg SD and FC (J,,, J,,, and J,d depend on ground and
excited triplet states. The full expressions for each tenn are given el~ewhere.~~*l~I It is
important to note that J,, is completely isotropie; hence. it contributes only to I,.
The FC-SD cross term. J,,. is completely anisotropic; hence, it contributes only to
hl. Ramsey's formalism for J coupling does not account for relativistic effects. An
extension of Ramsey's theory to include relativistic effects was developed by Pyykk6.l"
As with calculations of nuclear magnetic shielding , modem ab inifio programs
calculate J as a second derivative of E, in this case with respect to the nuclear magnetic
moments of the coupled nuclei, M, and Ms. Here, it is given in terms of the reduceà
coupling tensor, K(I,S):8*1*
K(M) =
As with nuclear magnetic shielding, the derivatives can be evaluated
numerically , lw*lm*lq or anaîytically . Equation 2.49 may be rewritten as follows to show
explicitly the dependence of the wavefiinction on M:

For each K(I,S) it is necessary to determine the response of the wavefunction with
respect to the magnetic moment of one of the nuclei, via the factor dA/dM,.
Determination of the full set of Jcoupling tensors for a molecule is thus considered
computatiody more expensive than calculations of nuclear magnetic shieldiag, where
al laB need only be determined once to obtain all the shielding tensors.
A nurnber of different models have been used for the calculation of J coupling .
While HF methods have been applied, this approach usually fails because it cannot
accurately describe the contribution from triplet States to the FC and SD mechanisms,
termed the 'triplet iastabiiity" pr~blem.'-~~ This can lead to calculations of Jcoupling
constants with significantly different magnitudes comparecl to experimentim or even the
wrong sign. It has been generally recognized that molecules containing multiple bonds
andfor electron lone pairs should not be investigated using the HF appr~ach.'*~~ More
success has ken obtained with the MCSCF approach. Calculations of I coupling using
MCSCF theory with analytical evaluation of the derivatives in equation 2.49 (tenned
multiconfiguratiooal linear response (MCLR) theory) were fust reported in 1992.'"
The agreement between calculated and experimental results are generally very
good,n*lOg*L1O although this rneihod is still limited to small molecuies. A coupled cluster
approach has also been used in the calculation of J coupiing . l 1 1 ~ 1 1 2 ~ 1 l3 Polarization
propagator methods with acronyms such as SOPPA (second-order polarization

propagator approximation) and CCDPPA (coupled cluster doubles polarization
propagator approximation) focus on the perturbation rather than the
wavehinction.114~115*116*117 Density fiuictional theory has also been applied; the first
attempt was by Fukui in 1976.118 The combination of D R methoâs with a completely
andfical evaluation of the derivatives in equation 2.49 was reported recently . la Some
recent examples of I coupling where relativistic effects are accounted for include results
for MH, (M=C. Si, Ge, Sn, Pb) and Pb(CH,),H.119 A relativistic correction to just the
FC term within the DFT formalism has been d i s c ~ s s e d . ~ ~ Examples of systems where
relativistic corrections have been applied to DFT calculation of J couplhg have ken
reponed. 12l.lZl23
The choice of buis set is crucial in calculations of J coupling. In particular,
accurate representation of the electronic structure near the nucleus is necessary. The
convergence behaviour of correlationconsistent basis sets (denoted cc-pVXZ, where
X=2 to 6) indicaies that the good results are obtained when core-valence s-type orbitals
(cc-pCVXZ) are included, and the best results are obtained with the cc-pVXZ-sun bais
set, where the s-type tùnctions are decontracted and n tight s-hinctions are added.
Unfortunately, these basis sets are not generally available for al1 elements.
If reports of reliable first-principles calculations of J-couplhg constants are
scarce, then reports of calculations of J are rarer still. As discussed previously (section
2.1.6). experimental measurement of l is difficult. However, it is imperative to
characterize the tensor nature of this interaction to understand its relationship to
molecular and electronic structure. Some examples of ab initio calculations of J include

42
work on J(X,193 (X = lH , 13C, l9F)lU and J(*F ,%i). lm Extensive calculations of J in
ditomic molecules established the importance of spin-spin coupling mechanisms other
than FC."*lU The symmetry properties of JPC1,l9F) and J(IgF,l9F) for CFj and
J(19F,170) for OF2 were explored in a subsequent publication. l l0 It has been
demonstrated that the inclusion of relativistic considerations tends to increase AJ. '26

Chapter 3: Phosphorus Chernical Shift Tensors for Tetramet hy ldiphmphine DiSulphide: A Single-Crystal "P NMR, Dipolar-Chernid Shift NMR and Ab Initia Molecular Orbital Study
3.1 Introduction
Alkyldiphosphine disulfides have been of some interest to NMR spectroscopists
due to the relationship between the indirect 31P-31P spin-spin coupling and structure.
The investigation of diphosphine disulfides with various alkyl groups bonded to the
phosphorus nuclei allows one to examine the effect of geometry differences such as the
P-P bond iength or the C-P-C bond angle on the phosphorus chemical shift and spin-
spin coupling temors. Solution NMR studies have been performed, l4 and a few
alkyldiphosphine disulfides have k e n investigated by NMR in the solid state, for
example TEPS (tetraethy ldiphosphine disul fide)lO*" and TBPS (teuabuty ldiphosphine
disulfide).16 Initiai reports indicated that anisotropy in 1J(31P,3'P) for both TEPS and
TBPS was substantial, 2.2 kHz1' and 1.9 kHz, respectively . However, subsequent
work on TEPS indicated that the upper Iimit is 462 Hz.l0
There are no extensive ub inirio studies of phosphorus rnagnetic shielding teosors
in the licerature for these compounds and ab initio calculations of J in molecules of this
size are impractical at the present the. Some experimental and ub in- shielding
investigations of related compounds, the dithiadiphosphetaws, [RSP(S)S], (R = H,
CH,, CH2CH3, Ph, cyclo-C,H,,, CH,Ph, or 4-methylphenyl), and
dithioxophosphoranes, RPS, (R = H, CH,, phenyl, or 2,6dimethylphenyl) have ken

reported.ln For A&P,O,, a signifiant anisotropy in 1Jc'P.3'P) has been reported,
W = 800 f 80 Hz.'=
In this chapter, the characterization of phosphocus chernid shift and spin-spin
coupling tensors for TMPS, 1, by "P singlecrystal NMR is described. The structure is
shown again in figure 3.1 (a) for convenience. These results are compared with lhose
obtaîned from an independent aaalysis of "P NMR specua of crystalline powder
samples. In addition, the quality of ab initio calculations of phosphocus magnetic
shieldiog tensors is evaluated by comparison with the experimental results. Fioally, our
analysis ieads to an upper limit on the anisotropy of 1J(31P,31P) for TMPS.
Cornparison of results obtained from combining NMR studies of crystalline
powder samples with ab initio calculations us results from a single-crystal NMR study
allows one to evaluate the various methods for characterizing chernical shift and spin-
spin coupling tensors. 19*'2g ln principle. the latter experiments provide the principal
components and orientations of the chernical sbiH tensors. as well as dipolar and indirect
spin-spin coupling teasors. Recent advances in hardware and software have made the
single-crystal NMR experiment more efficient.lM This NMR investigation of TMPS is
ideal for such a comparison of data obtained from single-crystal NMR studies vs data
from NMR studies of crystaliiae powder samples and ub initio shielding caiculations. It
is straightforward to grow a large single crystal of TMPS and the molecule is
sufficiently smail, allowing for ub initio caiculations with acceptably large basis sets and
various levels of theory .
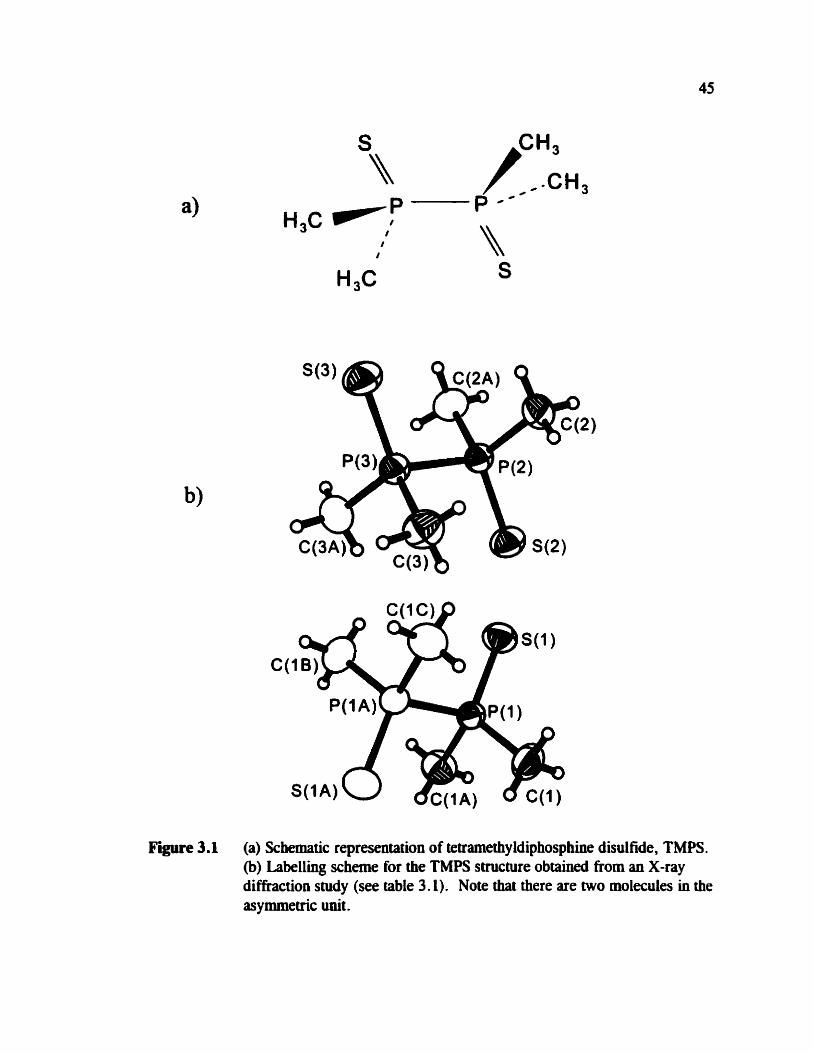
Figure 3.1 (a) Schernatic representation of tetramethy ldiphosphine disul fide, TMPS . (b) Labelling scheme for the TMPS structure obtained h m an X-ray diffraction study (see table 3.1). Note that there are two molecules in the asymmetric unit.

3.2 Experimetltal and Computational DdPils
A sample of TMPS was obtained ffom Johnson Matthey Electronics.
X-ray Dam CoLIection Md Processing: For reasons diiussed below, the X-ray
structure of TMPS was redetermined by J. F. Britten at McMaster University. A single
crystal of dimensions 0.02 mm x 0.20 mm x 0.45 mm, fiom a sample of TMPS
recrystallized in CHFI,, was mounted on a glass fiber. X-ray diffraction experiments
were carrieci out on a Siemens P4RA diMactometer (Mo Ka, A = 0.710 69 A, graphite
monochrornator) at room temperature, using the o and 4 scan technique with a CCD
area detector. The maximum 28 value was 55.0". The parameters for the monoclinic
ce11 were o = 18.860(6) A , b = 10.693(6) A, c = 7.021(4) A, and P = 94.608(3)",
with Z = 6. Wiîh formula weight of 186.20 glmol, the calculated density is 1.3 1 1
g/cm3. Of the 5 143 reflectiotis collecteci, 1686 were unique. The data were corrected
for Lorentz and polarization effects and for absorption using an empirical model. 13'
The structure was refmed in C21m,1x where al1 atoms with the exception of
hydrogen atoms were refiwd ushg anisotropic temperature factors. Hydrogen atoms in
their observed positions were refined isotropically. The final cycle of full-manix least-
squares refmement, using al1 unique reflections ( I > 3o(f)) with 101 variable parameters,
converged with unweighted and weighted agreement factors of R = 0.0327 and
R, = 0.0706, respectively. On the final difference Fourier map, the maximum and
minimum peaks correspondeci to 0.384 and -0.260 electrons k3. AU calculations were
performed with SHELXTL. 13' Structural parameters are given in table 3.1 and the atom
labelling scheme is show in figure 3.1 (b) .
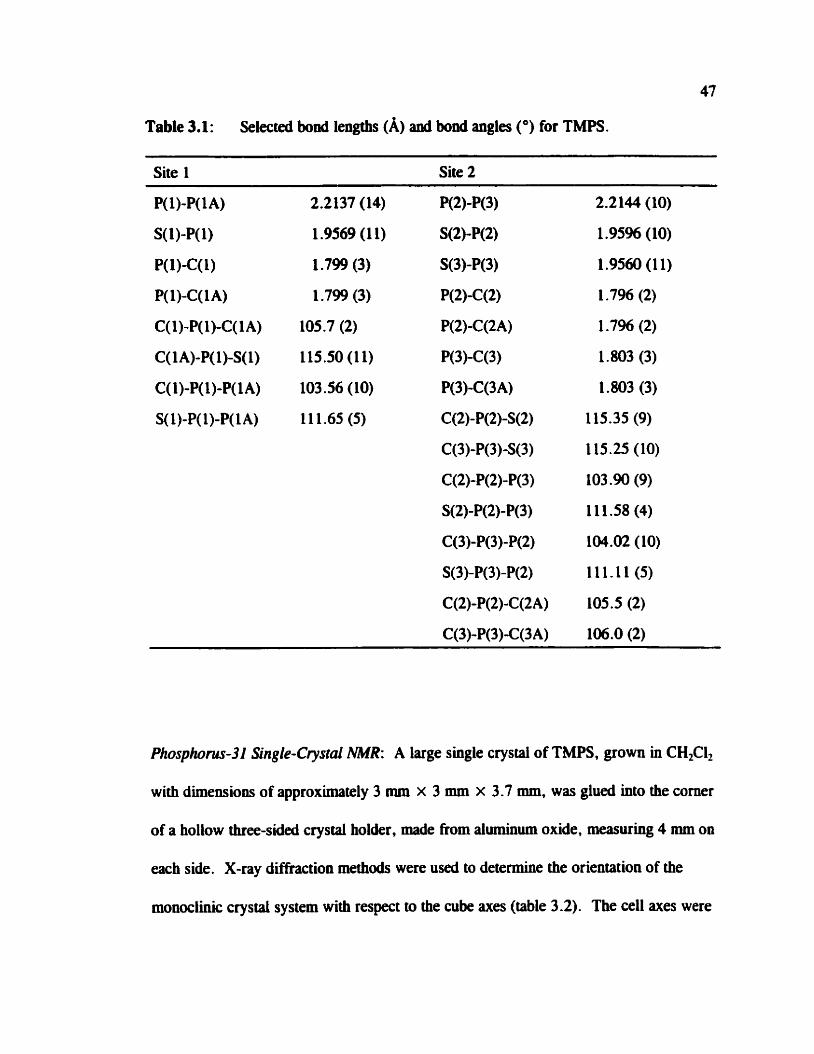
47
Table 3.1: Selected bond lengtbs (A) anâ bond angles (O) for TMPS.
Site 1 Site 2
Phosphom-31 Single-Cwal AMR: A large single crystal of TMPS, grown in CHzCiz
with dimensions of approximately 3 mm x 3 mm x 3 -7 mm, was glued into the corner
of a hollow three-sided crystal holder, made from aluminum oxide, measuring 4 mm on
each side. X-ray diffraction methods were used to determine the orientation of the
rnonociinic crystai system with respect to the cube axes (table 3.2). The ce11 axes were

48
onhogoaaiized using Rollett's convention. * The rotation marrix. RJy)It&P)lQ(a),'U
relates the orthogoaalized crystal system and the cube frame of refereace. For our
sample, a = 120.33O, P = 3.M0, and y = 242.72'.
Table 3.2: Direction cosines to orient the monoclinic crystal axes (a, b, c) with respect to the orthogonal NMR cube fiame ( X , Y, Z) as determineci by X- ray diffraction.
X Y z
Phosphorus-3 1 NMR data fiom the single crystai were obtained on a Bruker
MSL 200 spectrometer (4.7 T. correspondhg to a 31P NMR frequency of 81.03 MHz),
using an automated single-crystal goniorneter probe manufacturecl by Doty Scientific.
Rotations were performed about each of the X, Y, andZ axes of the cube, from O" to
180" in 9" increments. Al1 31P NMR spectra were aquired using C p with high power
proton decoupling. A IH xI2 pulse width of 3.1 p. contact tirne of 5.0 ms, acquisition
time of 41 ms, and a recycle delay of 6 s were used. For each spectnun the sweep
width was 50 kHz; 64 transients were adequate to obtain a good signal-to-noise ratio.
For the FID, 3072 K of zero pohts were added to give a total of 40% K data points

49
before Fourier transformation with 50 Hz of gaussian line broadening . AU spectra are
referenccd to 85% H,PO,(q). The peaks in each spectrum were fitted with a gaussian
hinction to obtain the frequencies of the peak maxima. The "P single-crystal NMR data
for each site were analyzed by linear least squares fit to:lM
where f; is the NMR parameter of interest and Ji tracks the rotation angle of the crystal
in the goniorneter about the im mis (i = X, Y, 2). For the 31P chernical shift data of
each site, the position at the centre of each doublet was ploned. In the analysis of the
31P-31P dipolar couplhg interaction, the splitting between the doublets was plotted for
both sites. Phase angles of -2" for the X rotation, -3 for the Y rotation and -6" for the
Z rotation were introduced in order to compensate for errors in the initial goniorneter
positions. The standard analysis of single-crystal NMR data is described e l~ewhere . l~*~~
Phosphom-32 MUR of Powder Soniples: Phosphorus-3 1 NMR spectra of stationary or
MAS powdered samples were aquued on a Bruker MSL 200, a Chernagnetics CMX
Infinity 200 (4.7 T for both, corresponding to a frequency of 81.03 MHz for "P), or a
Bniker AMX 400 spectrometer (9.4 T, corresponding to a frequency of 161.90 MHz - for "P). C F and high power proton decoupling were used for all experirnents.
Double-air bearing MAS probes were used throughout. Samples were packed hto 7
mm (MSL) , 7 -5 mm (Infinity) and 4 mm ( A m 0.d. zirconium oxide rotors. Typical
parameters were lH ~ 1 2 pulse widths of 4 ps, contact times of 1 ms, and recycle delays

of 4 s for MAS and 8 s for statioaary samples. The sweep width was 62.5 lcHz with
typical acquisition iimes of 41 ms. The FlDs were zero fiiied by 40% K points to a
total of 8192 K points before Fourier transformation. AU spectra are referenced to 85 %
H,PO,(q) by using solid NH,H2Pû4 which bas an isotropie phosphorus chernical shift
of 0.8 ppm from 85 96 HjPû4(aq).
Phosphorus-3 1 NMR spectra of stationary samples were simulated using
WSOLIDS,49 a program developed in this laboratory which incorporates the PûWDER
routine of Alderman et al. 13' NMR spectra of MAS samples were caiculated using
NMRLAB,M which uses the Monte Car10 method to sample crystal orientations for
powder averag hg. In Our calculations 10,000 cry stallite orientations were used for the
powder averaging.
The 2D spin-echo NMR spectnun was acquired on the CMX Infuiity 200, using
a standard spin-echo pulse sequence with the phase cycling of Rance and Byrd. The
experimental parameters were similar to those used for the 1D experiments. To obtain a
2D data set, 64 increments of C, ( s e figure 2.8) were sufficient. A sweep width of 12.5
kHz in the FI dimension was used. The data size for the 2D spectnun was 512 x 128
after zero filling. Gaussian Line broadening of 100 Hz was applied to both dimensions,
then the data were pracessed in magnitude mode. The FI projection of the 2D spectrum
was calculated using SpinE~hoI~~ which uses equatioos 2.40 and 2.41.
Comp~ctc~lio~l Detaik: Ab initio calculations were perfonned using the Gaussian 98
suite of programsM running on an IBM Ml6000 cornputer. The phosphorus nuclear
magnetic shielding was caiculated using the atomic coordinates from the X-ray structure

51
deterrnined in this work. Calculations were performed using the GIA0 methodn at the
HF level of theory as well as with DFT. For the DFT calculations, the k k e three
parameter functi~nal'~ ushg the Lee, Yang, and Parr correlation functio~all*~ (B3LYP)
was used. To compare calculated results with experimental results, the calculated
phosphorus nuclear magnetic shielding was converted to chemicai shift using the
absolute shielding of 328.35 ppm for the phosphorus nucleus in the reference, 85%
w w 0 q )
3.3 Results and Discussion
3.3.1 Phosphorus-31 NMR of a Sigle Crystal
The "P NMR spectra obtained from a single crystal of TMPS are shown in
figure 3.2. The X-ray structure of TMPS indicates the presence of six molecules in the
unit cell. In two of the six molecules, the two phosphorus nuclei are related by
inversion symmetry, hence they are crystallographically and magnetically equivalent,
Le., they form an A, spin p a p (site 1). In the singleîrystal NMR study , site 1 gives
rise to a doublet where the splitting is the 3LP-31P effective spin-spin coupling at that
particular orientation of the crystal in the applied magnetic field, B,,. Since 1 l J(31P,31P) 1
measured in solution is less than 20 Hz,142 the splitthg is dominated by Ren. The
remaining four molecules (site 2) in the unit ce11 of TMPS possess &or planes that
include the S-P-P-S plane; however the phosphonis nuclei are not related by a centre of
inversion; hence, they are not cry stallographidy equivalent .

52 x-rotation y-rotation z- rotation
Figure 3.2 Phosphorus-3 1 NMR spectra of a single crystal of TMPS for rotations of the crystal holder about its X, Y, and Z axes, acquired at 4.7 T .
In most of the NMR spectra of the TMPS single crystal (figure 3.2), four peaks are
evident. The two of lesser intensity are assigned to site 1 and the more intense set is
assigned to site 2 since the two sites are present in a 1:2 ratio. It is not obvious in
figure 3.2 that site 2 is an AB spin system, since the expected four transitions (section
2.2.1) are not immediately apparent. In fact, very few of the rotations produce NMR
spectra where there is any evidence of an AB quartet; one example is sbown in figure
3.3. Since there are kufficient data to analyze site 2 as an AB spin system, it bas been
analyzed as an A, spin system. The plots of the chernical shift and the dipolar splining

Figure 3.3 Example of a 3'P NMR spectcum of the TMPS single crystal (from the rotation about the Y axis of the crystal holder, at 36" €tom the initial position). The asterisks indicate the four peaks attributed to site 2.

54
as a function of crystal rotation are shown in figures 3.4 and 3 3, respectively . The
coefficients of the linear least squares fit to equation 3.1 for the chemical shift tensors
are given in table 3.3. The components of the chemical shift tensors, b,, , an and a,,
and theu respective direction cosines relative to the orthogoaalhed crystal frame of
reference are given in table 3.4. As is evident in this table, the principal components of
the phosphorus chemical shift tensors for sites 1 and 2 are virtually identical. The
orientation of the tensor is illustrated in figure 3.6(a) for site 1. The principal
component of the phosphorus chemical shift tensor which corresponds to the direction of
greatest shielding , 6,,, is closest to the P-S bond, while 6, , is approximately
perpendicular to the plane containhg the S-P-P-S bonds. with a P-P-6,, angle of 83".
The angle between 4, and the P-P bond is 124" for site 1. The chemical shift tensor
orientation at the other phosphocus nucleus of site 1 is shply an inversion of the one
shown in figure 3.6(a). For site 2, the phosphorus chemical shift tensor is oriented in a
similar fashion with respect to the molecule, with a,, at 2.5" fkom the P-S bond and the
P-P6, , angle of 100". The presence of a mirror plane in site 2 requires that the P-P-6,,
angle be exactly 90" ; however, the magnitude of b,, and 6= are similar , thus it is
difficult to determine theu orientations indepeodently . Io
The principal components of the dipolar coupling tensors are given in table 3.4
with their respective direction cosines. The spin-spin couplhg data for TMPS are also
summarized in table 3 .S. As mentioned in section 2.1.5, the dipolar coupling tensor in
its principal axis system is axiaüy symmetric and traceless, with the unique axis dong
the intemuclear vector. The non-zero trace and very slight non-axial symmetry of our

rotation angle / degrees
Figure 3.4 Phosphorus chernical shifi as a fuoction of rotation angle, Ji, for sites 1 and 2. The curves represent the linear least-squares best fit to the experimentai data, shown as O for site 1 and as O for site 2.
-$O00 --
. 1 . , . , . , ~ . 1 . ~ . [ . ~ 1 . 1 . 1 S , . ~
O 2 0 4 0 6 0 0 0 0 2 0 4 0 6 0 8 0 0 2 0 4 0 6 0 8 0
rotation angle / degrees
Figure 3.5 Dipolar spliuiog as a hinction of rotation angle, $, for sites 1 and 2. The curves represent the iinear least squares best fit to the experimental data, shown as O for site 1 anâ as O for site 2.
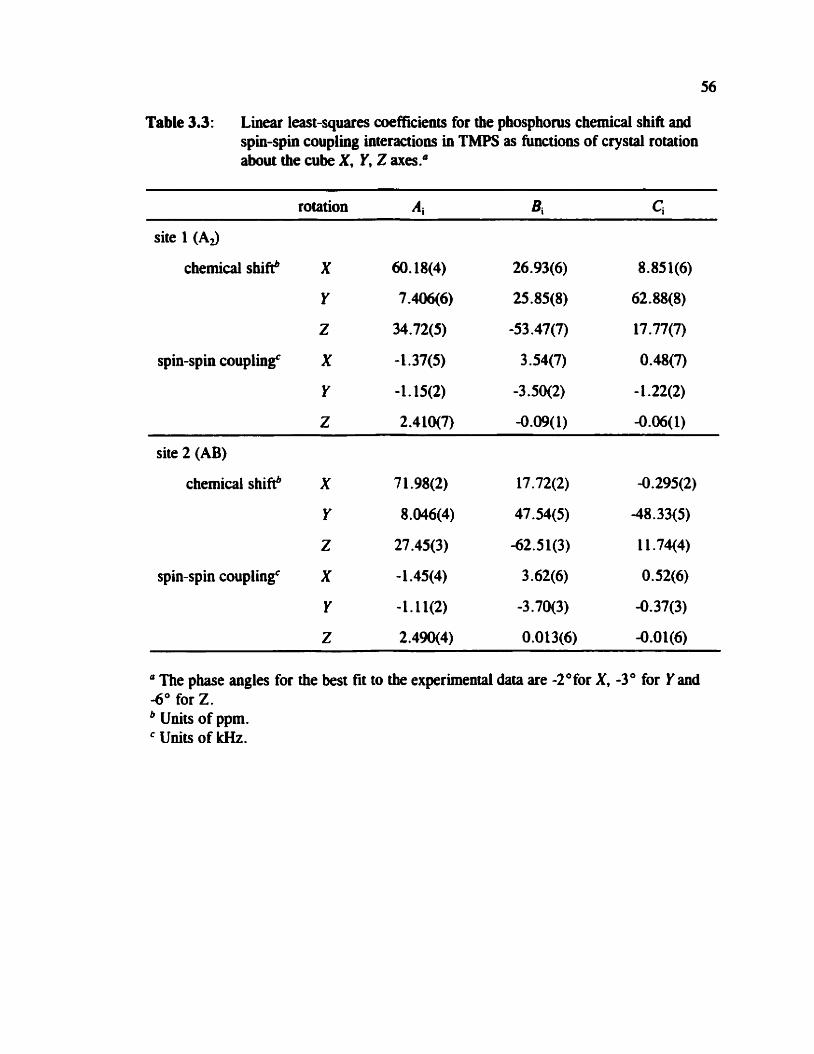
Table 3.3: Linear least-squares coefficients for the phosphorus cheraical shift and spin-spin coupling interactions in TMPS as functioas of crystal rotation about the cube X, Y, Z axes."
rotation Ai si ci - - -
site 1 (A3
chernical shifeb X 60.18(4) 26.93(6) 8.851(6)
Y 7.406(6) 25.85(8) 62.88(8)
Z 34.72(5) -53.47(7) 17.77(7)
spin-spin couplh@ X -1.37(5) 3.54(7) 0.48(7)
Y -1.15(2) -3.50(2) - 1.22(2)
Z 2.4 lO(7) -0.09(1) -0.06(1)
site 2 (AB)
chernical shift! X 7 1.98(2) 17 .72(2) 4. 295(2)
Y 8.046(4) 47.54(5) -48.33(5)
Z 27.45(3) -62.51(3) 1 1.74(4)
spin-spin couplingc X - 1 .45(4) 3.62(6) O. 52(6)
Y -1.1 l(2) -3.70(3) -Q.37(3)
Z 2.490(4) 0.013(6) -0 .O 1(6)
The phase angles for the best fit to the experimental data are -2Ofor X, -3 O for Y and -6" for 2.
Units of ppm. ' Units of kHz.

Table 3.4: Principal cornponents and orientations (direction cosines) of the phosphorus chernical shift and dipolar coupling tensors relative to the orthogonalized crystal axes (a*bc) for TMPS.
a* b c
site 1 (A3 61, / P P ~ 90.6 -0.1417 0.9836 O. 1 140
/ P P ~ 74.9 -0.5171 -0.1717 0.8385
633 / P P ~ -63.2 0.8441 0 .O599 0.5328
DI 2.538 O .9805 4.1405 0.1373
DdkHz 2.365 O. 1430 0.9897 -0.008I
h3/kHz -5 .O08 -0.1348 0.02753 0.9905
site 2 (AB)
51 I / P P ~ 9 1.8 -0.1120 0.9795 -0.1621
W P P ~ 74.4 0.3955 0.1964 0.897 1
W P P ~ -58.8 0.9106 0.0435 -0.41 10
D, ,/kHz 2.566 0.9916 -0.1273 0.0237
D&Hz 2.358 O. 1280 0.9946 4.0239
D3311rH~ -4.994 -0.0204 0.0268 O. 9994
Table 3.5: Spin-spin coupling data for TMPS.
Method &/kHz ReRIWz RDDIlrHz (JhIIHz W l H z - --
single crysral site 1 (Ad -0.035 1.669" 1.818 18.7b 447
site 2(AB) 4.023 1 -665" 1.816 - 453
2D spin echo - 1.6W - - -
a Estimateci experirnentai error is f 0-050 kHz. From reference 142. ' Experimentai error is f 0.080 kHz.

Figure 3.6 Orientation of the phospliocus chernical shift tensor for TMPS as determinet! by (a) a single-crystal NMR study and (b) ab initio (HF) calculations. The orientations are shown for site 1. The plane of the page coatains the S-P-P-S moiety. In both cases, 6 , , is approximateiy perpendicular to the plane of the page.
dipolar coupling tensors is likely a consequence of experimental erron in the
single-crystal NMR experiment. In addition, anisotropic molecular motion may
c~nt r ibu te .~~*l~~ The experimental values of Rcn are 1.669 f 0.050 lcHz for site 1 and
1.665 * 0.050 Wz for site 2, detennined by averaging Rar obtained from each of the
diagonal components of D (table 3.4). The rotation plot for the dipolar splining (figure
3 -5) indicates that the unique axis of the dipolar teasor, whicb is dong the P-P bond, is
very close to the 2-axis of the cube, since the dipolar splitting for both sites is

59
essentially invariant to rotation of the crystal about that axis. This impiies that r, anci
B,, are approximately perpendicular (c =go0) during the rotation about the Z axis; hence,
a splitting of '1, Rd is observed.
From RD, calculated ushg the P-P bond leagths reporteci in the X-ray crystal
stnicture'" and Re, determined experimentally, one can estimate hl as describeci in
section 2. 1.6.29 The most recently published crystal structure of TMPSIU reports a
signifiant and unexplained difference in the P-P bond lengths of sites 1 and 2,
r = 2.245(6) A and r = 2.16 l(4) A respectively , correspoading to RD, = 1.74 & 0.04
kHz and, using equation 2.29, hl = 0.3 & 0.05 kHz for site 1, while RD, = 1.95 * 0.03 kHz and AJ = 0.9 * 0.05 kHz for site 2. This is clearly suspicious, prompting
our redetermination of the X-ray crystal structure. From our investigation, the general
features of the X-ray structure are similar to those previously reporteci; however the
difference in the geomevy between the two sites is much less dramatic. The P-P bond
lengths from Our X-ray data are 2.2137(14) A and 2.2144(10) j\ for site I and site 2
respectively, resulting in more reasonable values of R, (table 3.5). For site 2, the
major stmctural difference between the two (CH,),PS fragments is the C-P-C bond
angle (106.0(2)" vs 105.5(2)"; see table 3.1). Using our values of RD, for both sites,
the upper limit on AJ(31P,NP) is approximately 450 Hz. in addition, there may also be a
contribution h m librationai motion of the rnolecule, which results in a smaller
observed value for the dipolar coupling constant. The reduction in the dipolar coupling
constant has been estîmated to be between 3 and 5 Sb .45(a)*14S*'Jb

3.3.2 Wospho~s-31 NMR Spectra of Crystalliae Powder Spmpks
The "P NMR spectra of powder sarnples of TMPS were analyzed independently
of the single-crystal work. Before aoalyzhg the 31P NMR spectra of statioaary (figure
3.7) and MAS (figure 3.8) crystalline powder samples of TMPS. the effective dipolar
coupling constant can be measured independently using the 2D spin-echo e~perirnent.~'
From simulations of the Fi projection (figure 3.9) of the 2D spin-echo NMR spectnun,
Rd = 1.69 f 0.08 kHz, in good agreement with the single crystal data. in the
simulations, it is assumed that 11J("P,31P)I = 18.7 Hz which is the value measured in a
previous solution NMR study . 14'
The NMR spectra of the statioaary samples (figure 3.7) are simulated using the
value of Reff from the 2D spin-echo NMR spectrum to obtain the chemical shift tensor
principal components given in table 3.6 and tensor orientation information discussed
below. The principal components of the phosphorus chemical shift tensors as obtained
from the single-crystal NMR study (table 3.4) are reproduced in table 3.6 for
cornparison. The NMR spectra of MAS samples, spinniag at various kequencies, v,,
(figure 3.8) were calculatecl ushg the same parameters as obtained from the stationary
samples, with the exception of the isotropic phosphom chemical shifts for sites 1 and
2. More accurate isotropic chemical shifis are available from the "P NMR spectra of
MAS samples. For site 1, 6 , = 34.9 ppm while for site 2, two peaks are evident at
37.4 ppm and 37.0 ppm (figure 3.8(a), inset). The 31P NMR spectra of MAS samples
c m be successfidly simulated based on this Uiformation and the data obtained from the

Figure 3.7 Experimental and calculated 31P NMR spectra of stationary powder samples of TMPS, acquired at (a) 4.7 T (500 transients, 20 Hz gaussian Liw broadening) d @) 9.4 T (7010 transients, 25 Hz gaussian line broadening). The same experimental NMR parameters were used to simulate the observed spectra at both fields.

Fgw 3.8 Experimental and calculateû "P MAS NMR spectra of TMPS aquired at (a) 9.4 T with v, = 4.00 kHz (172 transients, 10 Hz gaussian line broadening). The inset shows details of the isotropic region. The bottom spectrum (b) was obtained at 4.7 T with v, = 4.00 lcHz (64 transients, 10 Hz gaussian line broadening). The asterïsks hdicate the isotropic

v 1 . w m.
4 2 O -2 -4 k H z
Figure 3.9 Experimentai and calculateci F1 projection of the 2D spin-echo "P NMR spectm aquired at 4.7 T. The large centre peak (mincated in this figure) is an experimental artifact.
simulation of the NMR spectra of statiooary samples. Since the single-crystal NMR
spectra exhïbited very littie AB character, the subtle difference between the non-
quivalent phosphorus environments in site 2 was not apparent from that analysis.
While one cm, in principle, fit the "P NMR spectra to obtain al1 the chernical shift and
coupling information, these spectra (figure 3.8) are essentially featureless, heace it was
necessary to also analyze the NMR spectra of stationary samples, acquired at different
applied magnetic fields (figure 3.7). In addition, one cannot apply the Henfeld-Berger
appr~achl'~ to the spinning sideband manifold of the MAS specua due to the presence of
strong dipolar c~upling.'~
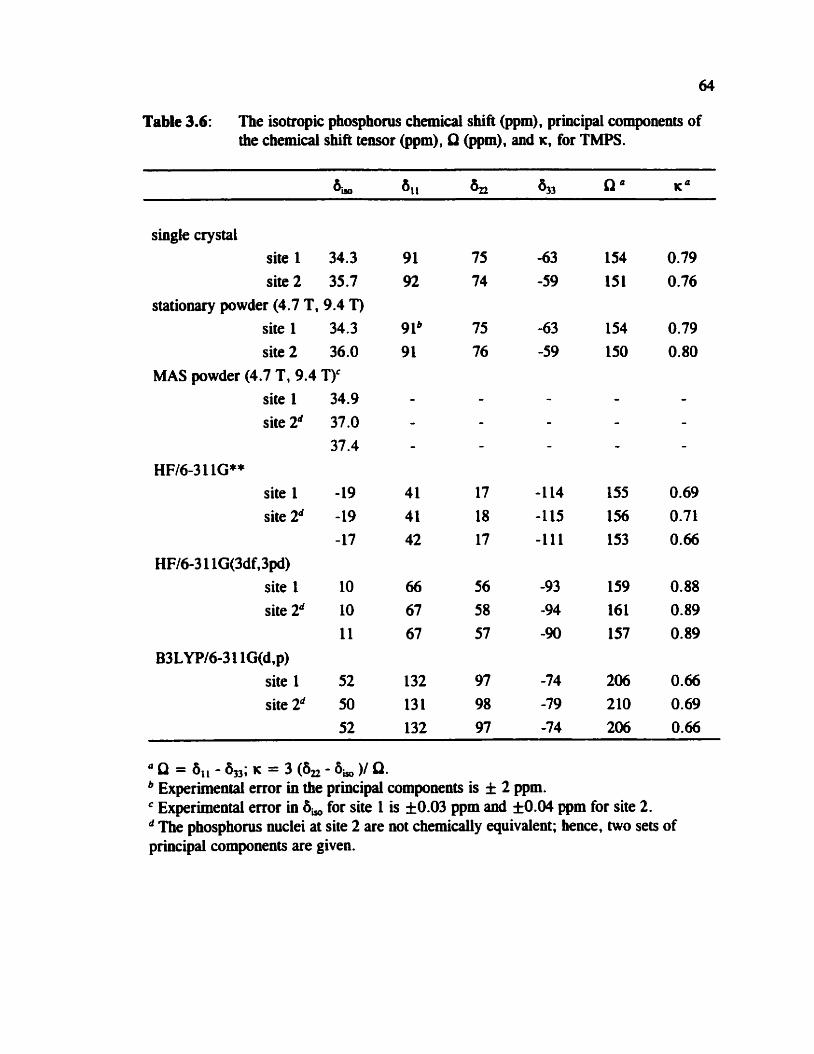
Table 3.6: The isotropie phosphow chemicai shift (ppm), principal componenis of the chernical shift tensot (ppm), Q (ppm), and K. for TMPS.
single crystal site 1 34.3 site2 35.7
stationary powder (4.7 T, 9.4 T) site 1 34.3 site 2 36.0
MAS powder (4.7 T, 9.4 TT site 1 34.9 site 2d 37.0
37.4 HF16-3 1 1G4*
site 1 -19 site 2d -19
-17 HF16-3 1 lG(3df,3pd)
site 1 10 site 2d 10
11 B3LYP16-3 1 lG(d,p)
site 1 52 site 2d 50
52
u n = a,, - 5n; K = 3 - 5, y Q. Experirnental error in the principal components is f 2 ppm. Experirnental error in 6, for site 1 is f 0.03 ppm and f 0.04 ppm for site 2. The phosphocus nuclei at site 2 are not chemically quivalent; hence, two sets of
principal components are given.

In addition to obtaining the principal components of the phosphorus chernical
shift tensors, some tensor orientation information is available from the anaiysis of NMR
spectra of powders containing isolated spin pairs such as those in TMPS. As describeci
in section 2.2.2 the orientations are commoniy described in t e m of Euler angles, a, P
and y .26 The 31P NMR spectra of stationary samples (figure 3.7) were successfully
simulated using A a = 0' for both site 1 and 2, indicating that the &, components for
adjacent phosphorus nuclei lie in the same plane. The angle between 6 , and the P-P
bond, P, is 63" * 3' for site 1 and 65" f 3" for site 2, equally welldescribed by the
supplement angles, 1 17' and 1 lSO, respectively . The orientation of the principal
components of the phosphorus shielding tensor obtained from a dipolar-chernical shift
analysis is clearly consistent with the single-crystal NMR results.
3.3.3 Ab Inilto Caicuiation of Phosphorus Shielding Tensors
The calculated principal compownts of the phosphorus shielding tensors for
TMPS are given in table 3.6 and the orientation for one phosphorus nucleus in site 1 is
shown in figure 3.6(b). The calculated phosphorus chernical shift tensor orientations for
site 2 are similar. Using the 6 3 1 1G** bais set, the HF results for 6 , are about
53 ppm too shielded for both sites; however, the line shapes, as described by 0 and K,
are remarkably accurate. Increasing the size of the basis set to 6-3 11 + +G(3df,3pd)
improves the calculated value of 6,; however, it is stiU too shielded by about 24 ppm
for both sites. The values from D R calculatioos are deshielded by only 16 ppm
compared to experimental resuits, but Q is overestimated. Differences between the

66
isotropie chemical shift measured in the solid state compared with results from ub initio
caiculations are expected since the calcuiatiom are performed on an isolated molecule.
The intermolecular effects may be substantiai. For example, PH&) is more shielded by
28 ppm compared to PH3(1)90 and P4(g) is more sbielded by 93.0 ppm compared to
P4(f).'4g This is a general trend that nuclei in the gas phase are invariably shielded
compareâ to the condeased phase. lM The ub initio meth& reproduce phosphocus
shielding tensor orientations quite well (figure 3 A).
3.3.4 Cornparison of Results
The phosphorus chemicai shift and spin-spin coupling tensors for TMPS obtained
by analysis of 31P NMR spectra of crystalline powder samples are in excellent
agreement with the single-crystal NMR study. The value of R, measured by both
single-crystal NMR and the 2D spin-echo experiment compared to RDD calculated from
P-P bond lengths places an upper limit hl (about 450 Hz), substantially smaller than
originally estimated for similar compounds.~~~ The phosphorus chemical shift tensors
obtained from both single-crystal and powder methods are virniaily identical. In
addition, apalysis of 31P NMR spectra of MAS samples at 9.4 T provide additional
details regarding the chemically noa-equivalent phosphorus nuclei of site 2. From fhe
analysis of the NMR spectra of stationary powder samples, the principal components of
the phosphorus chernical shift tensors are determineci. The orientations of the most
sbielded components relative to the P-P bond as well as to each other are obtained. For
both site 1 and 2, the direction of greatest shieldhg is in the same plane and are closest

67
to the P-S bond. This is consistent with the orientation in other systems coutainhg P=S
bonds.Lo*11*12 Unfortunately, the orientation of the teasor in the molecular frame of
reference can not be detennined by powder NMR methods. The hi11 tensor orientation
is available experimentally from the single-crystal NMR study (figure 3.6(a)). The
orientation information obtaiwd from the powder methods are consistent with the
single-crystal resuits.
The ub inirio rnethods employed in this work produce line shapes that are in
good agreement with experimental results. in addition, the calculated nuclear magnetic
shielding tensor orientations are aimost identical to those obtairied from the single-
crystal NMR study. The invariance of the nuclear rnagaetic shielding tensor orientation
with basis set suggests that calculations at the HF level of theory with moderate-sized
basis sets are adequate for obtaining the shielding tensor orientation. This is
particularly usehl for large systems, where high-level caiculations are very difficult due
to limi ted computational resources .
3.3.5 Trends in Phosphorus Chernieal Shifts for Akyldiphospbiw DisuIfides
It has been suggested that the isotropie chemical shift at the phosphonis nucleus
is directly related to the C-P-C bond angle in alLyldiphosphine disulfides." With the
characterization of the phosphonis chemical shifi tensor in TMPS, this correlation can
be examinec! more rigorously. Table 3.7 lists the phosphorus chernical shift data and
the C-P-C bond angle for [R,P(S)12 where R = CH, ( t h work), CHFH3, 'O n-propyl, 15'
or n-butyl. l6 It is difficult to draw any detinite conclusion based on the first three

compods in the series, partly due to the expetunenml uncertainty in the C-P-C bond
angle; however, it appears that the isotropic shielding trend does not follow the bond
angle, i.e., increasing bond angle does not correlate directiy with an increase or
decrease in the isotropic chernical shift. Attempts have also been made to explain the
difference between the phosphorus chernid shift for R=n-propyl and R = n-butyl by
an effect sirnilu to the y -effect in 13C M. I l b Trends in the principal components of
the phosphorus shift tensors should be considered, as has been done, for example, in the
case of the dithiadiphosphetanes, [RSP(S)S], (R = alkyi or For the
aikyldiphosphine disulfides, the lirnited data available on the phosphonis chemical shifi
tensors (table 3.7) indicate that the intermediate component, &, changes the most (by
about 23 ppm) upon going fkom R = CH, to R = CHFH,. The smallest component,
6, , , changes by about 17 ppm and q3 by about 7 ppm. By contrast, al1 the principal
components are similar for R = CH2CH, compared to R = n-butyl. Unfortunately, no
data on the phosphocus chemical shift tensor are available for R = n-propyl.
Table 3.7: Cornparison of 6, and C-P-C bond angle for [R,P(S)],.
bYO / ppm 8 , ,1 ppm & 1 ppm 833 1 ppm C-P-C ! deg
CH, (site 1)" 34.1 91 75 -63 105.7(2)
n-propyl ' 45.9 - - - 1 07.8(2)
n-butyl 49.5 104.7 97.5 -53 -7 - (multiple sites)d 49.7 106.3 % -53.1
a This thes is. Reference 10. NMR &ta fiom reference 15, X-ray crystal structure from ref 151. NMR &ta from reference 16, m crystai structure available.

3.4 Conclusions
The characterization of the phosphorus chexnical shift anâ spin-spin coupling
tensors for TMPS presented in this paper serves as a benchmark for the evaluation of
experimental NMR powder methods as well as the reliability of ab inirio methods. The
excellent agreement between the calculated and experimental phosphorus chernical sbift
tensor orientations is particularly encourzighg . For TMPS, as for TEPS, l0 AJ(31P,3LP) is
less than 500 Hz. A number of aikyldiphosphiw disulfides have been characterized by
NMR and attempts have been made to account for trends in the isotropie phosphorus
chemical shifts; however, it is c1ear that the entire phosphorus chemical shift teusor
must be considered. In addition, the structural data available in the literature for
alkyldiphosphhe disulfides are limited, hence correlations between a particular
structural feature in this class of compounds aod the phosphom chemical shih are
tenuous at bes t .

Chapter 4: Cbaracterizltion of Phosphorus Chemicai S M Tenson in a Phosphole Tetramer: A Combined Experimentai NMR and Theoreticai Study
4.1 introduction
The phosphole tetramer, 2 (figure 4. l), was first synthesized in 1982lS and the
X-ray crystai structure was reported s~bsequently.~~ Solid-state NMR investigations of
similar systems where two three-coordinate phosphorus nuclei are directly bonded are
relatively rare.i3*17*19a In the case of the phosphole tetramer, analysis of the "P NMR
spectra acquired with cross polarhtion and MAS revealed a difference in the isotmpic
phosphorus chernical shifis of 1.7 ppm with 1J(31P,31P)I, = -362 Hz.153*Ls Here, we use
the presence of phosphonis spin pairs to characterue the phosphorus chernical shift
tensors via the dipolar-chernical shift NMR method (section 2.2). In contrat to the
earlier 31P NMR investigations of MAS sarnples,lB-lY NMR spectra of stationary
samples obtaiaed at different applied magnetic fields are hilly analyzed. It is also
demonstrated that analysis of the "P 2D spin-echo NMR spectnun of the phosphole
tetramer is invaluable for refiniog the coupling parameters. Ab inifio calculaiions
complement the experimental data and suggest orientations of the phosphonis chernical
shift tensors in the molecular frame of reference. Inconsistencies between the NMR
data (vide infa) and the original X-ray data2' prompred a redetermhtion of the crystal
structure.
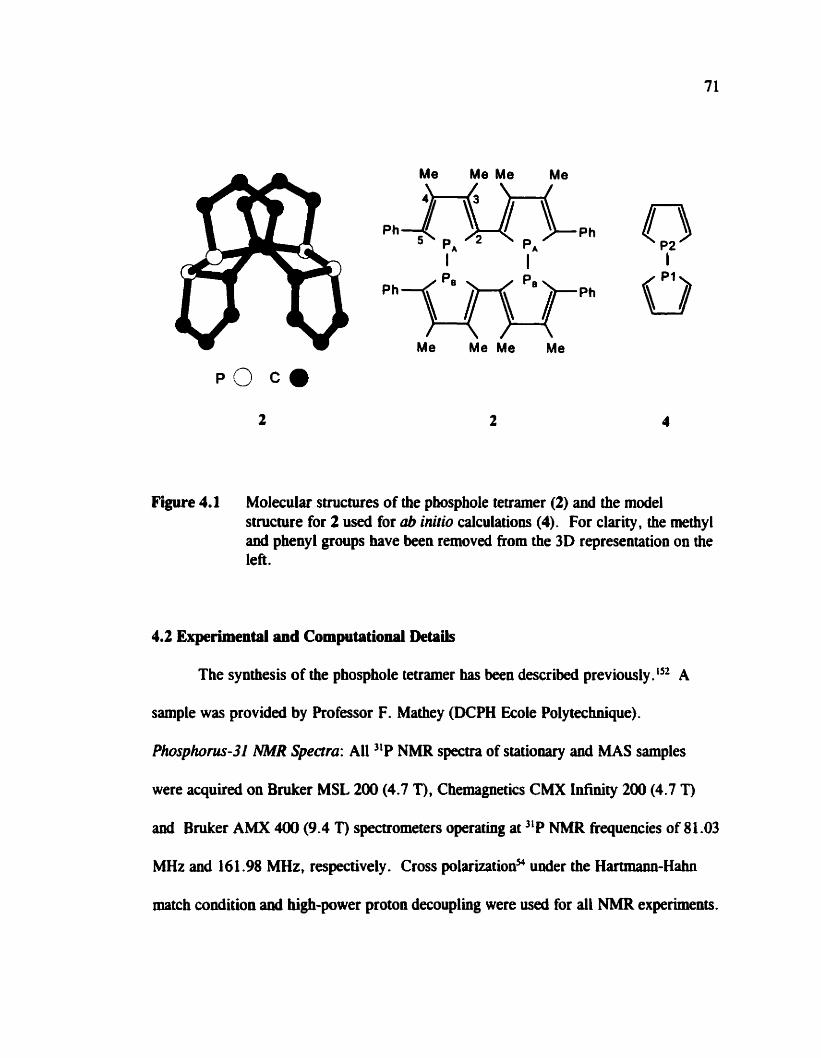
Figure 4.1 Molecular structures of the phosphole teuamer (2) and the mode1 structure for 2 used for ub initio calculations (4). For clarity, the methyl and phenyl groups have ben removed f'om the 3D representation on the left .
4 3 Experimentai and Computatiod DetPitF
The synthesis of the phosphole tetramer has been describeû previ~usly.'~~ A
sarnple was provided by Professor F. Mathey (DCPH Ecole Polytechnique).
Phosphorus-31 W R S p e m : Al1 )'P NMR spectra of stationary and MAS sarnples
were acquired on Bniker MSL 200 (4.7 T), Chemagnetics CMX Infinity 200 (4.7 T)
anci Bruker AMX 400 (9.4 T) spectrometers operating at NMR frequencies of 81.03
MHz and 16 1.98 MHz, respectively . Cross polarizations under the Hartmann-Hahn
match condition and hi&-power proton decouplhg were us& for d l NMR experiments.

72
At the lower field, double air beariag MAS probes were useâ with the sample packed in
7 mm O .d. (for the MSL) and 7.5 mm O .d. (for the C m zirconium dioxide rotors.
For the stationary sample, a lH ni2 pulse of 3.6 ps, contact time of 5 ms, acquisition
tirne of 43 ms, and a recycle delay of 5 s were used. A sweep width of 24 kHz was
used and 100 Hz of gaussian line broadening was applied before Fourier transformation.
For spectra acquired with MAS at 1.50 or 2.00 kHz, simüar pulse widths were used.
The acquisition time was 20 ms and the sweep width was 40 kHz. A total of 40% data
points were acquired. Before Fourier transformation, 40% zero points were added and
10 Hz of gaussian line broadening was applied.
A double air bearing MAS probe was used at the higher tield as well, with the
sample packed in a zirconium dioxide rotor (4 mm 0.d.). For both MAS and statîonary
saniples, a IH x/2 pulse of 3.6 ps with a contact t h e of 3 ms and a recycle delay of 10
s were used. For the stationary sample, an acquisition time of 8 ms was used. The
sweep width was 65 kHz and 1024 points were acquired. The data were zero filled by
1024 points and 100 Hz of gaussian line broadening was applied. With MAS at 2.00 or
4.00 kHz the sweep width was 100 kHz and acquisition time was 30 ms. In total, 4096
points were aquireû and zero filleci by 40% points. Gaussian line broadening of 20 Hz
was applied before Fourier transformation. Al1 IIP NMR spectra are referenced to the
primary standard, 85% HIPO,(aq) by setting the jlP isotropie peak of solid JY&H,PO,
to +O. 8 ppm. Spectra of stationary samples at both fields were sirnulateci using
WSOLIDS.4g Spectra of MAS samples were calculated using NMRLAB." In our
simulations, 10,000 cry stallite orientations were used.

73
2D Spin-Echo 31P NMR Specttum: The 2D spinecho NMR specmim was acquired at
4.7 T (Bniker MSL 200) with a standard spinecho pulse sequence using the phase
cycling of Rance and Byrd. 13' A lH x12 pulse of 5.5 ps was used with a contact time of
5.5 ms and a recycle delay of 10 S. For each t , increment, 64 transients were acquired
for a total of 64 increments. Gaussian line broadenings of 200 Hz and 100 Hz were
applied to the FI and FL projections respectively. The experimental and calculated 2D
NMR spectra were processecl in magnitude mode. The Fi projection was calculated
using SpinEcho. lS9
X-Ray Data Collection and Processing: The X-ray difiaction study was carried out by
T. Stanley Carneron at Dalhousie University. A crystaî of approximate dimensions 0.10
x 0.10 x 0.10 mm, recrystallized from chlorobenzene, was mounted on a glas fibre.
A Rigaku AFCSR diffiactometer was used for al1 measurements. Calculations were
performed us h g texsan. lS5 The crys tals have ce11 dimensions a = 1 1 .523 (4) A, b =
23.083 (1) A. c = 15.115 (1) A, a = 89.99 (l)", P = 92.71 (1)" and y = 89.99 (1)".
The merging R, for the reflection data, merged on the assumption that the ce11 is
monoclinic, is 3 .O448 for 244 quivalent reflections with I > 50(1). The reflections for
which h = 2n + 1 are weak in intensity and are gewrally diffuse. The strong
reflections thus belong to a ce11 where a = 5.762 (1) A. Both cells have systernatic
absences consistent with a space group Pt,lc. The molecule for the structure in the
larger ce11 refmes to R = 4.796. The cefinement used unit weights.
Cbmputatio~i Details: Theoretical caiculations were perfomed with the Gaussian 94
suite of programsm running on aa iBM RISC16000 cornputer. Using RHF tûeory and

74
the GIA0 methoci," the phosphorus nuclear magnetic shielding tensors were determined
for the mode1 geometry, 4 (figure 4. l), of the phosphole tetramer baseâ on the X-ray
crystallography results for half of the molecule with the methyl and phenyl groups
replaced by hydrogen atoms. A partial geometry optimization using HF theory with the
6-31 1G basis set on d l the atoms was canied out. The P-P bond length, the dihedral
angle between the rings, and the C-H bond lengths were fixed at the X-ray structure
values for the optimization. Ab inirio calculations of nuclear magnetic shielding were
perfomed using the 6-3 1 1 + +G(d,p) bais set on phosphonis and wighbouring nuclei,
while the 6-3 11G basis set was used on the remaining atoms to keep computation times
within reasonable limits . IM To compare with experirnental results , the calculated
phosphorus shielding is converted to chernical shift by usiog the absolute shielding of
328.35 ppm for the phosphorus reference, 85 96 H3Pû,(q).90
4.3 Results and Discussion
4.3.1 Experimental Deteradnath of Phosphorus Chemicai Shift Terisors
Tbe observed and calculated 31P NMR spectra of stationary sarnples of the
tetramer are show in figure 4.2, while NMR spectra of MAS samples are shown in
figures 4.3 and 4.4. Best-fit parameters are given in table 4.1. In the text to follow , a
brief account of how these parameters were obtained is presented.
The "P NMR spectra of the phosphole tetramer with MAS indicate the presence
of a unique phosphonis spin pair in the crystalLn** and, as indicated in table 4.1, the
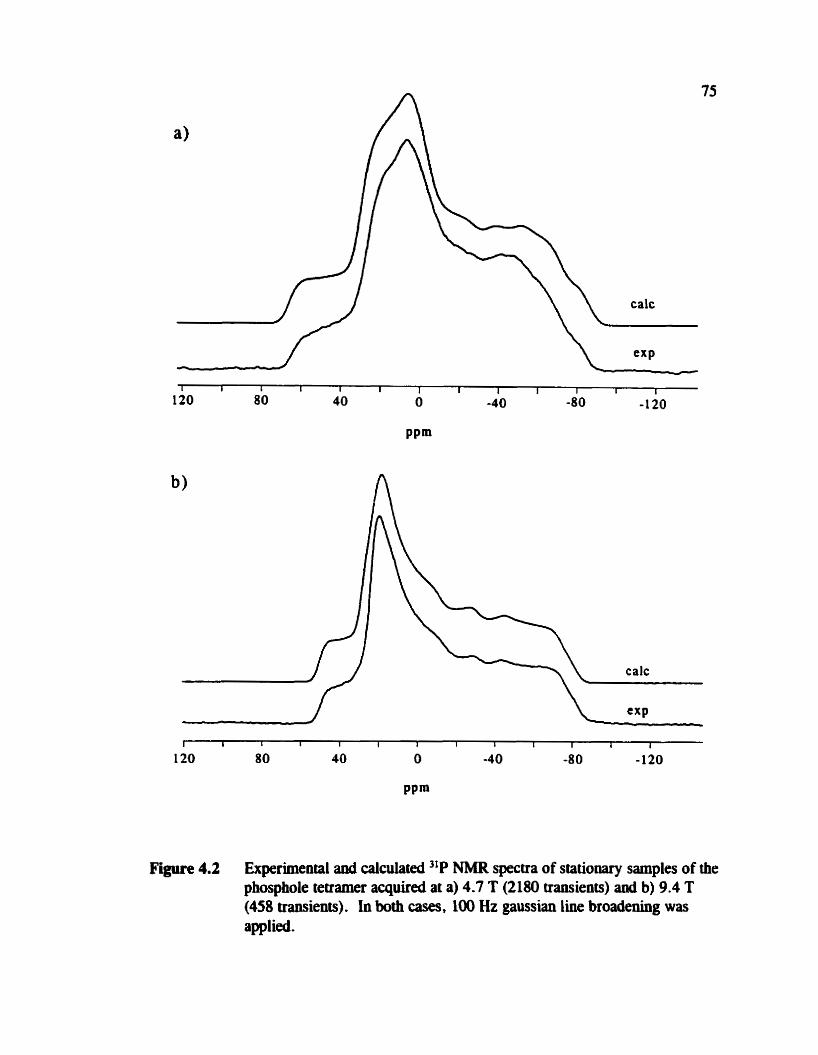
Figure 4.2 Experimental anà calculated "P NMR spectra of stationary samples of the phosphole tetramer acquired at a) 4.7 T (2 180 transients) and b) 9.4 T (458 transients). in both cases, 100 Hz gaussian line broadeaiag was applied .

calc
calc J V L
Figure 4.3 Experimental and calculated 31P NMR spectra of crystalline powder sarnples of the phosphole tetramer spinning at the magic angle, acquired at 4.7 T for v, of a) 2 kHz or b) at 1.5 Wz. For each specmim 256 mients were aquired and 10 Hz of gaussian line broadening was applied. The asterisic indicates O,.

Figure 4.4 Experimental and calculated 31P NMR spectra of crystailine powder samples of the phosphole tetramer spioniog at the rnagic angle, acquired at 9.4 T for v, of a) 4 kHz or b) 2 kHz. For each spectnim 512 transients were aquired ad 20 Hz of gaussian line broadening was applied. The asteris k indicates 6,.

Table 4.1: Experimentai and caiculated isotropic phosphom chernical shift, principal componentsa of the phosphorus chemical shift iensoa, 0, and K for the phosphole tetramer.
Site
a All chemical shifts and Q are given in ppm. Esthteci error on the principal components is f 5 ppm. GIAOs were used with the 6-31 1 + +G(d,p) basis set on phosphorus and adjacent
atoms for structure 4. The 6-3 1 1G basis set was used on the remaining atoms.
NMR spectra have been aaalyzed accordingly. However, in the prevîously reporteâ
crystal structure of the phosphole tetrame8' there are two molecules in the asymmetric
unit related by a pseudo mirror plane, hence four unique P-P bond lengths were
reported; r = 2.198(6), 2.191(6), 2.201(6) and 2.175(6) A.21 Given the reported X-ray
structure, one would expect to see potentially 8 different phosphorus sites in the jlP
MAS NMR spectrum; simüarly up to 16 different methyl carbon sites could be
anticipated in the 13C MAS NMR specmim. Expehentally, we observe only two
unique "P sites at both 4.7 and 9.4 T. Carbon-13 NMR spectra of MAS samples
revealed two I3C sites at both fields (6 , = 15.5 I: 0.1 ppm and 17.1 * 0.1 ppm).
This apparent disagreement between our NMR results and the reported X-ray data

prompted a redetermination of the X-ray data. Repeated attempts were made to grow
suitable single crystals; however, the best crystals obtaiwd were less than ideal for
X-ray diffraction. The R factor for our X-ray &ta is 0.047, compared to the R factor of
O -089 reporteci previously .'' The space group obtained from the new X-ray data is f2,lc with the asyrnmetric
unit consisting of one molecule, rather than a space group of Pr with two molecules in
the asyrnmetric unit as had been previously r e p ~ n e d , ~ ~ hence there are four
crystailographicaily non-equivalent phosphorus sites and potentiaily eight non-equivalent
methyl groups. In both cases. the unit ce11 contains four molecules. The rnolecular
structure obtained fiom our X-ray data is essentially the same as the reported
structure;21 however, our results indicate that the molecule contains a near perfect C,
axis which relates the two phosphom spin pairs (figure 4.1, structure on the left). This
also reduces the number of different phosphom sites fiom four to two and the number
of different methyl carbons dom to four. Given that the isotropie jLP chernical shifts of
the two phosphorus sites only differ by 1.7 ppm,'53*Ls it is not surprishg that the four
methyl carbons in the C, position (figure 4.1, middle structure) are isochronous;
similarly for those in position 4.
Within the unit ceil the two phosphorus spin pairs related by the near perféct C,
axis have very similar environments though the two atorns of each pair are in different
envirooments. From the difhise reflections for which h = 2n + 1, it would appear that
there are stacking enors in the a direction. The molecules are packed in layers
approximately perpendicular to the a direction; however, there are too few reflections

80
for which h = 2n + 1 (51 out of a total of 766) to test this suggestion. Using equation
2.20, and an average P-P bond length of 2.192(9) A ftom the new X-ray crystal
structure, RDD = 1.873 kHz with an estimated error of f 0.05 kHz. based on the error
in the X-ray &ta. Also, with four phosphorus nuclei in each molecule. each ''P-"P
spin pair is not truly isolated since there WU be muaial dipolar coupling. From the
internuclear distances between phosphonis nuclei that are not directly bonded to each
other, the values of RD, are 233 Hz (r = 4.392 A) and 169 Hz (r = 4.882 A), hence
the NMR spectral features are largely detennined by the much larger dipolar coupling
between phosphorus nuclei that are directly bonded to each other rather than the
phosphorus nuclei fonning the other spin pairs in the rnolecule.
The value of Re, and the relative signs of R, and 11(31P,"P), are obtained from
the 2D spin-echo experiment. Figure 4.5 shows the experimentai and calculated FI
projections of the 2D spin-echo NMR spectrum of the phosphoie tetramer. Using
J , = -362 Hz,"~*~" the best value of R, is 1.80 * 0.05 kHz (figure 4.5(a)), which is
in good agreement with the value calculated fiom the crystal structure. The difference,
Reg - RDD = 73 f 50 Hz, is very srnall and could arise ftom vibrational averaging and
possibly A.J. The stnailer dipolar couplings between the phosphorus nuclei that are not
directly bonded to each other wwt likely contribute to line broadening; however, it is
possible they couid slightly modify the value of Rew The semitivity of the Fl projection
to R, c m be seen in figure 4.5(b), where the agreement between the experimental
spectnim and one calculated using %, = RDD = 1.873 kHz is not as good. As well, the

F i 4.5 The calculated (dashed line) and experimental (solid line) R projections of the 2D spinecho 31P NMR spectnm of the phosphole tetramer, acquired at 4.7 T. The best fit is obtained for a) Rcff = 1-80 kHz, J , = -362 Hz. If & = RDD = 1.873 kHz and J , = -352 Hz, the projection shown in b) is obtained. If Refi = 1.80 lrHz and J , = + 362 Hz the splitting between the 'honis' is increased, as shown in d).

82
poor fit in the simulation using 1Jc1P,3'P), = + 362 Hz with Rer = 1.80 Wz (figure
4.5(c)) indicates that R, and l JC1P,3 'P), must have opposite sigm. In fact, for
1J(31P,xP), to be positive. Re* must be less than 1.20 Wz to obtain a satisfactory fit of
the two most intense peaks. which seems unreasonable in light of previous investigations
1JC1P,31P). 'O A negative J-coupling constant is consistent with I N W (intermediate
wglect of differential overlap) calculations of 1J(3'P, IIP). For example, the calculated
1Je1P,31P) in (CH3)2P-P(CH1)2 is -254 HZ for a dihedral angle between the forma1 low
pairs of 74" which is similar to the dihedral angle for the phosphole tetramer, 78".
The dependence of J on conformation is discussed in more detail in chapter 5.
The spin-spin coupling data (1J(3'P,NP), = -362 Hz a d R, = 1.80 Hz) were
used to simulate the experimental 31P NMR spectra of a stationary sample shown in
figure 4.2 to obtain the principal components of the chemical shifi tensor given in table
4.1 and tensor orientation information discussed below . The experimental error in the
principal components of the two chemical shift tensors is estimatecl to be * 5 ppm. As
a result, a,, , oz, and &,, are virtually identical for the two phosphorus chernical shift
tensors.
The NMR spectra of MAS samples, sbown in figure 4.3 and 4.4. demonstrate
that the quality of the simulations using these new parameters is comparable to
previously published w ~ r k . ' ~ + ' ~ Although the agreement between the calculated and
experimental NMR spectra is good at both fields for stationary and MAS samples, there
are some subtle differences between observed and calculated NMR spectra. These
differences probably arise because neither of the two phosphonis spio pairs in the

Figure 4.6 The relative orientation of the two phosphorus chemical shift tensors in the phosphole tetramer, determined from simulation of the "P NMR spectra acquired at 4.7 T aad 9.4 T. For PA, the Euler angles are EL = 0°, P = 78O and y = 167". For PB, u = 82", P = 78" and
cule is strictly isolated (Le., these spin pairs interact with one another).
As detailed in section 2.2, it is possible to obtain some chemical shift tensor
orientation information from the NMR spectra of systems containing spin pairs. For the
phosphole tetramer, the NMR line shape is panicularly sensitive to Aa, which is the
dihedral angle between the &, components of the two phosphorus chernical shift tensors.
Best-fit calculated NMR spectra were obtained with Aa = 82 f 3 O - For both
phosphorus chemical shift tensors, the &, componeat is tipped away fiom the
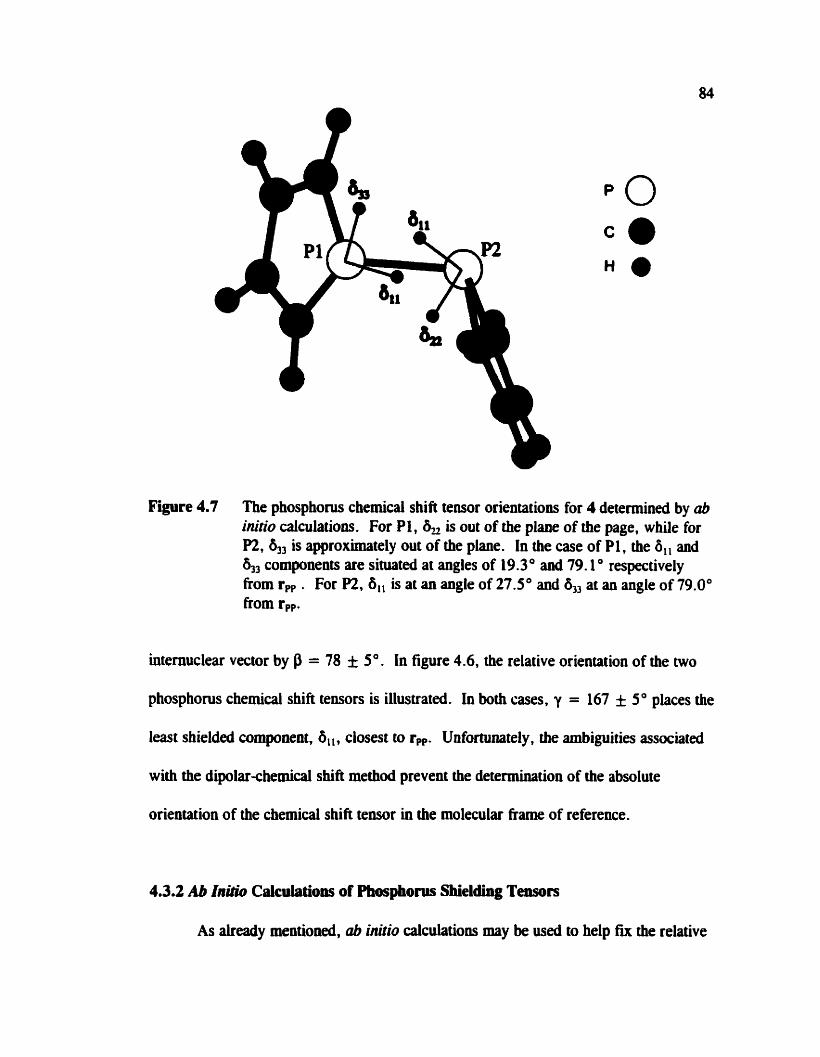
Figure 4.7 The phosphorus chernical shift tensor orientations for 4 determined by ub inirio calculations. For PI, b, is out of the plme of the page, while for PZ, &,, is approximately out of the plane. In the case of P 1. the a,, and ti3, components are situated at angles of 19.3 O and 79.1 O respectively from rpp . FOC PZ, 4, is at an angle of 27.5" and 8, at an angle of 79.0" from q,.
internuclear vector by P = 78 * 5'. In figure 4.6, the relative orientation of the two
phosphorus chemical shift tensors is illustrated. In both cases, y = 167 & 5 places the
least shielded component, 6 , , , closes t to r,. Unfortunately , the ambipities associated
with the dipolarchernical shift method prevent the determination of the absolute
orientation of the chernical shift tensor in the molecular fiame of teference.
4.3.2 Ab ln& Caiculations of Pûosphonis Shielding Tensors
As aiready mentioned, ab initio calculations may be used to help fix the relative

85
orientations (figure 4.6) in the moleculai axis system (figure 4.7). Ab initio calculation
results for the phosphonis nuclear magnetic shielding teasors in the mode1 system, 4,
for the phosphole tetramer are given in table 4.1. The two phosphorus sites are labelled
as Pl and PZ rather than PA and PB (figure 4.1) since it is not known how the two sites
in the NMR spectra should be assigned to the molecule. In a partial optimization of the
geometry, the P-C bond lengths and the ring bond angles were allowed to change,
resulting in changes that were largely within the errors stated in the crystal structure.'l
The calculated tensor orientations, depicted in figure 4.7. are similar for both Pl
and P2 with respect to the local geometry about each phosphocus nucleus. The &,,
components are closest to the P-P bond at angles of 19.3" and 27.5' for Pl and P2,
respectively while the 4, component for P l f o m an angle of 79.1 O with the
internuclear vector. and the correspondiiig angle for P2 is 79.0". The calculated
orientations of &,, and 4, for both phosphonis nuclei are consistent with the
exper imental observations. In addition, the dihedral angle between the 4, principal
components is W.$", in good agreement with the value of 82" determiad by analysis
of "P NMR spectra. These ab initio calculaiions indicate thai the 6, components are
not aligned in the general direction of the formal lone pairs, as had been previously
sugge~ted.~~*~" In fact, 4, is closest to the direction of the fornial lone pairs. For the
phosphole tetramer, the 633 components are in the plane of the respective phosphole
rings and approximately perpendicular to the P-P bond.
With respect to the magnitudes of the principal components. the experimental
and calculated results show s idar treids (table 4.1). The small calculated isotropie 3'P

chetnical shift difference, 4 ppm, between the two sites is consistent with experiment.
The calculations also predict that 6,, and b, are similar, while 6, is quite distinct, as
reflected in the skews of about 0.5, comparable to the skews of 0.7 observed
experimentally. However, the calculated isotropic cherniai shifts are too srnall by
about 43 ppm and the values of are significantly larger than the experimental results.
Some reasons for this discrepancy between calculated and experimental results
have been discussed in the previous chapter (section 3.3.3). In addition, the presence of
formal lone pairs in the phosphole teaamer makes ab initio calculations more
chailenging. Inclusion of electron correlation has been shown to be important for
obtaining good quantitative agreement between calculated and experimental shielding
tensors in systems with formal lone pairs a d multiple b ~ o d s . ~ ~ * ~ * " The hprtance of
lone pairs in detemiining the nuclear magnetic shielding in phospholes has been
demonstrated in a series of ub initia calculations on phospholes and the phospholide
ion.Isa A recent investigation of some model phosphorus systems, for example PH3,
inâicates that the hclusion of electron correlation in ob initio calculations yields
improved results for isotropic shielding . '" The use of a model geometry consisting of
half of the phosphole tetramer may also contribute to this discrepancy. Given these
factors, quantitative agreement between the experimentai and ab inifio values is not
expected, and seemingly good calculateci results may be due to the fortuitous
canceilation of errors. The primary goal with respect to ab initio calculations of nuclear
magnetic shielding teosors is to obtain the teasor orientation in the molecular fiame of
reference. The results for TMPS (chapter 3) and previous re~ul ts~ '*~*~@ suggest that ab

Nlitio methoch repruduce well the expehental tensor orientation.
The relative orientation of the 8, wmponents of the two phosphows shielding
tensors in the phosphole tetramer appears to be related to the relative orientation of the
adjacent phosphole rings (see figure 4.7). in the crystal structure. the rings are
arrangeci such that the ring planes are approximately parallel to each other and twisted
about the bridging P-P bond (figure 4.1. structure on the left). The twist angle between
adjacent phosphole rings, @,, ranges between 102°-1050, which can be equally well
described by the supplement angle range of 75"-78". This agrees weU with Aa which is
82 O detedned experimentally and 87.8 " from ab initio calculations . This structure-
teasor relationship can be m e r demonstrated by ab inirio calculation of the
phosphorus tensor orientations at various values of 4,. Further calculations on Our
mode1 system using 6, = O", 70". 110" and 180' indicate that the 4, components are
orienteci relative to the local geometry in a similar marner as show in figure 4.7, with
Aa reflecting a, (Aa = 0°, 60.5", 109.5" and 180° for the correspondhg listed
above).
4.3.3 T r e d in Phosphorus Cbemicai Sbilts for Phospholes
There has been some discussion in the literature over the aromaticity of
phospb~les,'~~ recently revived by the synthesis of some planar phospholes. 162
Aromaticity in phosphocus heterocycles was recentiy reviewed.la While 31P solution
NMR has been used routinely to characterize this important class of compounds, there
are few comprehensive NMR investigations of the chernical shift tensors. The

phosphorus chetnid shift teosors of several phosphole derivatives have also been
characterwd recently by solid-state NMR? Compatison of the phosphocus cherniai
shift teasors in phospholes (table 4.2 with structures given in figure 4.8). reveals that
6, in the phosphole tetramer is smaller compared to the other phospholes. For ali
phospholes investigated by 31P solid-state NMR thus fa-. the spans of the phosphorus
chemical shift tensors are relatively small cornpared with systems where the phosphorus
nucleus is known to be participahg in a delocalized electron ftamework. For example,
the span of the phosphorus chemical shift tensors in Mes0P=PMes' is 1239 ppm17 and
Q = 779 ppm in benzo-1,3,2dithiaphospholium aluminurn ch1oride.l" The iarge spans
in these systems have been amibuted to the piesence of low-lying excited States with the
proper symmetry to mix with the highest occupied molecular orbitals (equation 2.44).'"

Table 4.2: Phosphorus chemicai shift tensor principal compents" for some phospholesb.
5-pheny ldibenzophosp hole -17.3 12 -19 -45 57 O 163 (two sites) (5) 164
cis-2,Lû- 75.3(PS3 129 122 -25 154 0.9 168 dimethyl[l,2,3]benzothia-di-
phospholo[2.3b][l,2.3] 53.7(PCJ 190 25 -54 244 O beozothiad iphosphole (7)
a Al1 chernical shifts and Q are given in ppm. Structures are shown in figure 4.8.
F ' i 4.8 Molecular structwes of compounds 5.6, and 7 in table 4.2.

4.4 Conclusions
The analysis of 31P NMR spectra of a stationary and MAS samples of the
phosphole tetramer indicates that the principal compownts of the two phosphorus
chernid shift tensors are virtually identical. Regarding the phosphorus chemical shiH
tensor orientation, the &,, components of bth tenson are closest to r,, while the q3
components are tilted away at an angle of 78". in addition to king twisted relative to
each other by an angle of 82". This latter angle is thought to reflect the relative
orientation of the phosphole rings, which are related by a dihedral angle ranging from
75" to 78 O . Ab initio results support the experimental conclusions and iedicate that the
8, components are not aligned with the formal lone pairs on the phosphorus nuclei.
The characterization of the phosphorus chemical shifi tensors in the phosphole tetramer
is greatly aided by the 2D spinaho experiment as well as by employîng ub initio
calculations to suggest an orientation of the tensor in the molecular hame.

Cbapter 5: 'JPP,"P) Coupüng Tellsors - Conformational Studies in Mode1 Systems and Structural Cbaracterizatioa of [WQ-PPhJ[GaCW
5.1 Introduction
Phosphinophosphonium cations are representative of a new class of
cornpound~;~ however , the lack of X-ray diffraction dataa means that alternative
methods such as NMR are vital for characterizhg these compounds in the solid state.
in addition, 1J(31P,xP) is not observed in solution NMR for some of these compo~ods,~~
most likely due to interrnolecular exchange. One can apply the dipolar-chernical shift
method (section 2.2.2) to characterize the phosphorus chernical shift tensors as well as
the 3iP,3LP spin-spin coupling interactions. From the dipolar coupling constant, the P-P
bond length may be estimateci according to equation 2.20. However, as discussed in
section 2.1.6, the Harniltonians describing the dipolar interaction and the contribution
from the anisotropy in J are identical in form; hence, they are inseparable
experimentally . For this reason, accurate measurement of intemuclear distances via RDD
requires some knowledge of AJ.
Comprehensive understanding of how trends in J reflet molecular and electronic
structure is still lacking, primarily a consequence of the difficulty in characterizhg the
complete tensor experimentally. For one-bond J-coupling involvhg phosphorus nuclei,
' Since the preparation of this chapter, the X-ray crystal smctwes of [Pû,P-PPùJ[GaCiJ and [Ph,P-PPhJ[SO,CF,] have been obtained (N. Buiford, T. S W y Cameron, P. J. Ragogna, E. Ocmddo- Mavarez, M. Gee, Robert McDonald, and R. E. Wasylishen, J. Am. Chem. Soc. î23,7947 (2001)).
91

92
reports in the literature indicates that hl may be s ~ b s t a n t i a l ; ~ ~ * ~ ~ ~ however, as is evident
from the work on TMPS (chapter 3) anâ the phosphole tetramer (chapter 4). it is
difficult to distinguish the effect of hl from the consequences of vibrational averaging.
A possible avenue for determinhg the contribution of hl to Re, is via <ib initio
calculations. Details of ab iriitio methods for calculating J have been discussed in
section 2.3.2. In addition, first-p~ciples calculaiions of 1J(31P,31P) allow one to
investigate the infiuence of formal electron lone pairs as well as determine rhe
dependence on conformation of the low pairs with respect to rotation about the P-P
bond. While variable temperature NMR studies in solution bave provided some
insight,'' many conformations are not experimentally accessible.
For a representative phosphinophosphonium sait, [Ph,P-PPhJ[GaCi,J, compound
3 (figure S.l(a)), the calculation of 1J(3'P,3'P) has been used to obtain an estimate of
RD,, from the experimentally measured value of Re, (see equation 2.29). From RD, the
P-P bond length may be estimated according to equation 2.20. H,P-PH,+ is used as a
model system for Ph3P-PPh,+ since ab initio computational methods currently avaiîable
are limited to small molecules for J-coupling calculations. To further examine the
relationship between molecular structure and trends in J, calculations on H2P-PH2 have
also been performed. In addition, the phosphorus chernical shift tensors have ben
characterized and these experimental results are complemented by ab inifio calculation
of phosphorus shielding tensors in a model compound, (CH3,P-P(CH,),+.

Structures of the molecules discussed in tbis chapter; (a) [Ph3P-PPhJ [ W h ] , indicahg the labelling convention for the two phosphorus nuclei; (b) (CH,)3P-P(CH,)2+ and a Newman projection defining &&p; (c) H2P-PHI and a Newman projection defining epp; (d) H3P-PH2+ and a Newman projection denning, @,+ .

5.2 Experimental and Computationai Deta&
SMiple Preprdon: A sample of IW,P-PPhJ[GaClJ was provided by Dr. Edgar
Ocando-Mavarez and Professor Neü Burford. AU samples for NMR work were
prepared in an inert atmosphere as this compound is air sensitive. Details and
suggestions for handling air-sensitive NMR samples are presented in Appendix 2.
NMR S p e m : Phosphorus-3 1 NMR spectra were acquired for stationary or MAS
samples of powdered crystalline [W3P-PPhJ [GaCl,] at applied magnetic fields of 4.7 T
(Chemagnetics CMX 200 spectrometer) and 9.4 T (Bruker AMX 400 spectrometer).
The standard CP experimentY with high-power proton decouplhg was used in both
wes. For experiments at 4.7 T, the sample was packed into a 4 mm (0.d.) zirconium
oxide rotor and a proton 90" pulse of 3.0 ps, contact time of 5 .O ms, and recycle delay
of between 5 and 8 s were used. Typical sweep widths were 50 kHz with acquisition
tirnes of 41 ms. Before Fourier transformation, 2048 points of zero filling was applied.
Experiments at 9.4 T were perfomied on a sample packed into a 2.5 mm (0.d.)
zirconium oxide rotor. Parameters similar to those for experiments at 4.7 T were used.
Al1 31P NMR spectra are referenced to the prirnary standard, 85% H,PO,(aq). by setting
the isotropie peak in the "P NMR spectnun of solid bM4H2P04 to +O. 8 ppm. Gaussian
line broadening of between 20 and 40 Hz for the specûa of MAS samples and between
100 and 200 Hz for speura of stationary samples was applied.
The 2D spin-echo experimeat was perfonned at 4.7 T using parameters identical
to the ID experiments. For each r , iofrement, 224 transients were acquired, for a total
of 64 increments. The sweep width in the E! dimension was 28.6 kHz and the

95
acquisition time was 12 m. In the FI dimension, the sweep width was 12.5 kHz.
Gaussian line broadening of 200 Hz in F1 and 100 Hz in F2 was applied, followed by
processing in magnitude mode. The expimental &ta were zero tilled to 512 points x
512 points.
The spectra of MAS samples were sirnulateci using the prograrn NMRLAB."
whiie the specua of statiooary samples were simulateci using WSOLIDS.49 The F1
projection of the 2D spinccho spectnun was calculated using SpuiEcho. n9
C u m p ~ u t i u ~ l Details: Calculation of nuclear magnetic shielding tensors for
(CH,),P-P(CH3)2+ was carrieci out at the HF level using the 6-3 11G** basis set on the
phosphorus and carbon atom and 3-21G on the hydrogen atoms. The structure was
first optimized at the HF level using the 6-3 1 1G** basis set on al1 atoms, resulting in
(see figure 5.1@) for atom labelling) P(1)-P(2) = 2.219 A, P(2)-C = 1.848 A,
P(1)-C(1) = 1.816 A, P(1)-C(1)' = 1.814 A, C(1)-P(1)-C(1)' = 108.3".
C(1)'-P(1)-C(1)' = lO8.2", C-P(2)-C = 1û2. lo , C(1)'-P(1)-P(2) = 108.0" and
@MePP = 180°. +Mep is the dihedral angle between the plane containing the P(1)-P(2)
bond and dso bisecting the C-P(2)-C bond angle and the plane containing the P(1)-P(2)
and P(1)-C(1) bonds. Al1 C-H distances were 1.08 À and H-C-H bond angles were
lO9.5 O . Al1 caiculations were performed with Gaussian 98" using an iBM R S i 6 0
workstation.
Indirect 31P,3iP spin-spin coupling tensors for H,P-PH, and H,P-PH,' were
calculated using the MCSCF approach with complete active space self-consistent field
(CASSCF) wa~efunctions~~*~~ as implemented in DALTON, an ub initio electronic

structure pr~gram,'~l instaîled on an IBM RSi6000 workstation. For CASSCF
wavefunctions, it is necessary to choose active aad inactive space cornbhtions. The
active space consists of the molecular orbitais into which electrons may be placed to
produce configurations that are used to construct the electronic wavehinctions. It
usually consists of a number of occupied and virnial molecular orbitals. The inactive
space consists of the molecular orbitals which remain doubly occupied for al1 electron
configurations. The selection of active and inactive spaces was made based on MP2
n a ~ a l molecular orbital occupation numbers and energies.In The inactiveiactive space
used are given in the following format: for each molecular orbital symmetry , the
number of ioactive orbitals and active orbitals is given. For example, 101012131
indicates that there are four different molecular orbital symmetries. For the first
symmetry type, 1 molecular orbital is inactive aed 2 are active, O inactive and 1 active
for the second symmetry, 1 inactive anâ 3 active for the third symmetry. and O inactive
and I active for the fourth syrnmetry. Calculations of J for H,P-PH, and H,P-PH,+
were perforrned with cc-pVTZ (correlationconsistent polarkation valence triple-zeta)
basis setdn on al1 atoms.
An experimental ge~metryl'~ was used for H,P-PH, with the exception of @,,
which was varieâ between O' and MO0 in 30" increments, keeping al1 other bond
lengths and bond angles fïxed. 4, is the dihedral angle between the planes which both
contain the P-P bond but each one bisects one of the H-P-H bond angles (see figure
Sl (c ) ) The bond lengths and angles are as follows: P-P = 2.2191 A, P-H = 1.4155
A, H-P-H = 92.0". The experimental value of a,, is 74.0". For H2P-PH2, the

97
inactive/active space used to calculate 'Je1P,31P) as a fiiaction of ePp (table 5.2)
consisteci of 55/54. which corresponds to a total of two unoccupied orbitals in the active
space. one per molecular orbital symmetry. For sndying the effect of other geometry
factors (table 5.4). an inactive/active space consisting of 55/43, which does not include
any uwccupied orbitals. was used.
For HIP-PH2+, the geometry was optimized at the HF level with the
6-3 11 + +G(3df,3pd) basis set on al1 atoms. The optimized structure is as foilows (see
figure 5.l(d) for atom labelling): P(1)-P(2) = 2.209 A. P(1)-H(1) = 1.387 A, P(1)-
H(1)' = 1.386 A, P(2)-H = 1.403 A, H(1)-P(1)-P(2) = 116.6". H(1)'-P(1)-P(2) =
110.2". H-P(2)-P(1) = 92.4". H(1)'-P(1)-H(1)' = 106.3". md H-P(2)-H = %.SO.
Similar to the calculations for H2P-PH2, the angle a,+ was varied between O0 and
180". a,,+ is the dihedral angle between the plane containing the P(1)-P(2) boad and
also bisecting the H-P(2)-H bond angle and the plane containing the P(1)-P(2) and
P(1)-H(1) bonds. MCSCF calculations for the dependence of 'J(xP,3'P) on g,,, for
H,P-PH,' (table 5.3) were performed using an inactiveJactive space 10/8, i.e., one
uwccupied orbital in the active space.
5.3 R d t s and Discussion
5.3.1 Phosphoriis-31 NMR Sptxtra of [Wp-PPûJ[GaCiJ
The ''P NMR spectra for a crystalline powder sample of [Ph,P-PPhJ[GaCh] are
shown in figures 5 -2 (obtained at 4.7 T) and 5.3 (obtained at 9.4 T) for various MAS
rates. From the spectra aquired at the higher MAS rates, the isotropie chernical shifts

expt
Figure 5.2 Experimental and calculated 31P NMR spectra of MAS samples of [Ph,P-PPhJ [GaCi,] , aquired at 4.7 T. The spioning rates are (a) 2.17 kHz (128 transients), (b) 2.72 kHz (256 traasieats), and (c) 5.02 lrHz (256 transients). in each case. 40 Hz of gaussian line broadening was applied. The asterisk aad dagger mark the isotropie region for P(l) and P(2) respectively .

P . . . . . i r . . . . . . . . a
60 40 20 O -20 -40 -6û -80 ppm
C . . . . . . . i . i . . . . . l
60 40 20 O -20 -40 -60 -80 ppm
Figure 5.3 Experhnental and calculated 3'P NMR spectra of MAS samples of [Ph3P-PPhJ[GaCi,,], aquired at 9.4 T. The spioning rates are (a) 2.65 kHz, (b) 5.38 kHz, aiid (c) 7.03 kHz. In each case, 512 transients were acquired and 20 Hz of gaussian liw broadenhg was applied. The asterisk and dagger mark the isotropie rcgioa for P(1) and P(2) respectively . A signai possibly due to a decomposition product is indicated by $.

100
for each phosphorus nucleus and the magnitude of 1Je'P,31P), may be estimatecl and
refined in subsequent simulations of dl the NMR specba. The data are summarized in
table 5.1. The assigrnent of the phosphorus chemical shifts to sites in the molecule is
made on the basis of hown trends and on mults of ob iniiio calculation of phosphorus
nuclear magnetic shielding in a mode1 system. (CH,XP-P(CH3),+ (vide infa).
Calculations for nuclear rnagnetic shielding in Ph,P-PPh2+ would entaii a the-
consuming geometry optimization since an experhental geometry is not available.
For this compound, [Ph,P-PPhJ [GaCî,] , the phosphorus cbemical shifts obtauied
from a sample dissolved in CH,CI, compared to the solid state differ by less than 8
ppm. The impurity peak in figure 5.3 is attributed to a decomposition product. it is not
observed in the spectra at 4.7 T (figure 5.2), probably due to differences in the
effeftiveness of the rotors at excluding air fiom the sample.
The difference between the isotropic chemical shifts of the two coupled
phosphorus nuclei is 33.2 ppm, which is 2.61 Wz or 5.38 kHz at an applied magnetic
field of 4.7 T or 9.4 T respectively. These conditions are ideal for the rotational
resoaance (RR) e~periment,'~~ whereby the dipolar coupling interaction is restored for a
homonuclear spin pair when the difference between the isotropic chemical shifts is nv,
where n is typically 1 to 3. Indeed, the "P NMR spectra shown in figures 5.2@) and
5.3(a).(b) were aquired under conditions of RR. While Rer may have been meamred
from Rit by generating Zeeman magwtization exchange curves, for this compound it
was simpler to use the results of the 2D spinecho experiment, presented below, to
ob<ain Re@

Table 5.1: Phosphorus chernical shift t ewr principal components and spin-spin coupiing parameters for [Ph3P-PP~LJ[G~C~].
solution: a
4 m 1 PPm LJ("P.3'P) I Hz
solid state and ab initio data:
6 , / ppm '
4 1 1 PPm
b,/ PPm
4 3 1 PPm
Q 1 P P ~ K
1J,(31P,31P) 1 Hz
Ren I kHz
expt . ab initio expt. ab initio
15.4 -40 -17.8 -79
36 -22 23 3
23 -43 -8 -73
-14 -56 -68 - 167
50 34 91 170
0 A8 0.28 0.32 O. 10
-323 * 2
1.70 * 0.05
a E. Ocando, N. Burford, unpublished results. The solvent is CHFI,. Ab initio data are for the optimized (CH3,P-P(CH,h+ structure. Calculations are
carried out at the HF level using 6-3 1 1G** basis set on phosphocus and carbon atoms while the 3-21G basis set was used on al1 the hydrogen atoms.
Experimental error on &,, is * 0.1 ppm, obtained from specaa of MAS samples. Experimental error on the principal components is f 2 ppm, obtained from spectra of
stationary samples .

102
The FI projection fiom the 2D spin-echo 31P NMR specinun is show in figure
5.4 dong with simulations using opposite signs (figure 5.4(a)) aid the same signs for
l J(31P,31P)I, and R& (figure 5.4(b)). From these simulations, Rem = 1.70 * 0.05 kHz
and 1Jc1P,31P), is clearly negative relative to R,. Using the data from 31P NMR
spectra of MAS samples and the spin-spin foupling data from the 2D spin-echo
spectnun, the 31P NMR spectra obtained at two applied magwtic fields for a stationary
sample may be analyzeâ. The best simuiatioas are shown in figure 5.5 and the principal
components of the two phosphorus chemicai shifi teasors as well as spin-spin couplhg
parameten which give the best overall fit to al1 the experimental spectra are given in
table 5.1. The two features of note in the phosphorus nuclear magnetic shielding are
that one of the phosphorus nuclei is more shielded than the other and its chemical shift
tensor has a larger span. Based on kwwn trends in phosphonis shielding, that
phosphines are in gewral more shielded than phosphonium the specua may
be assigned to the phosphorus nuclei as in table 5.1. in addition, calculated phosphonis
chemical shift tensors in (CH,),P-P(CH&+ indicate that the phosphine centre, P(2). is
more shielded and has a significantly larger span (table 5.1).
Regarding shielding tensor orientations in the molecular frame of reference, the
position of the 4, components relative to the P-P bond, given by P, and the orientations
relative to each other, given by Au, have been determined experimentally. For P(L) and
P(2), P = 81 * 2" and 78 * 2" respectively and A a = 25". The third Euler angle, y,
is 2 * 5" for P(1) and 78 f 2" for P(2). Note that y for P(l) bas a larger error than y
for P(2). The angle y detemines whether o1, or b, is closest to r,,. The spectrum is

expt calc
expt
calc
Figure 5.4 Experimental and caiculated F I projections of the 2D sph-echo jLP NMR specmun of ph,P-PPhJ[GaC&] acquired at 4.7 T. The peak rnarked by an asterisk is an experimentai artifact. Simulations are shown with Ra = 1.70 kHz and (a) 1J(31P,31P)m = -323 Hz and (b) 1Je1P,31P), = + 323 Hz.
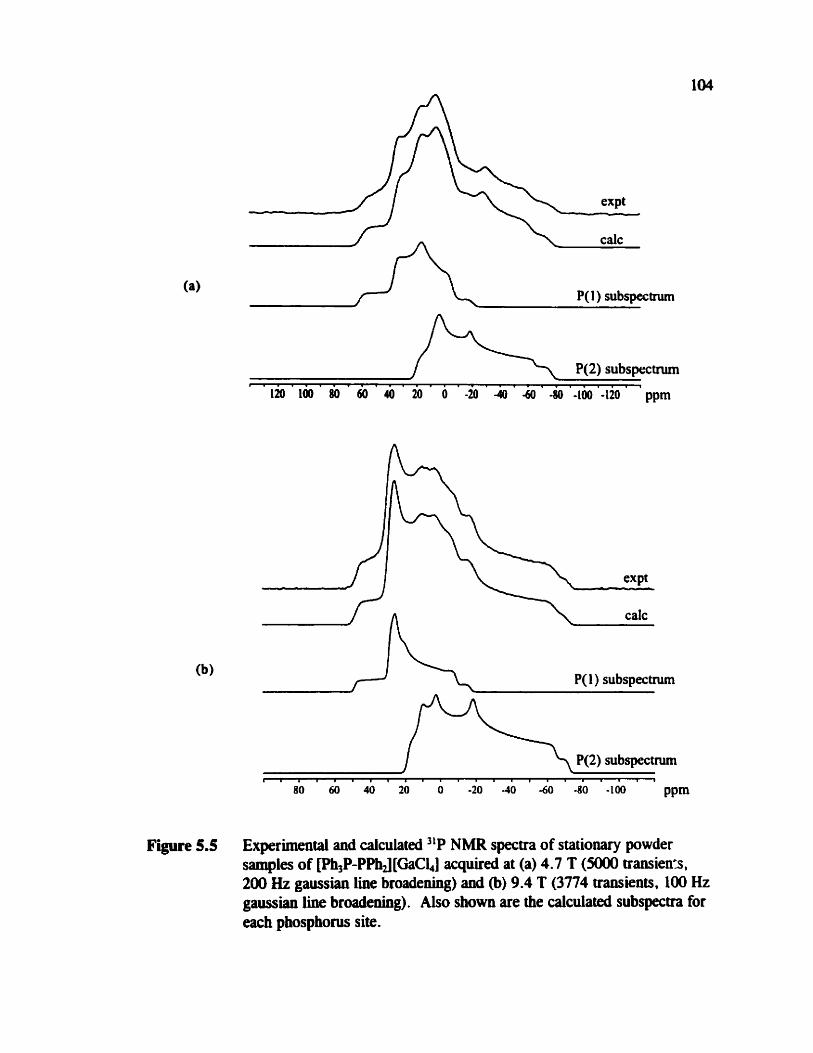
P(1) subspectnun
~ . . . . . . . . . r . . r . . . . r i . . . . lm 100 NI M) 40 20 O -20 4 UI -80 -100 '-120 ' ' ppm
expt
calc
P( 1 ) subspecüum
P(2) subspectnim t i i i i r i r r i i i i i i i m i i . i i l
80 60 40 20 O -20 -40 -60 -80 -100 ppm
Figure 5.5 Experimeatal and calculated "P NMR spectra of stationary powder sampks of [W,P-PPhJ[GaCLJ aquked at (a) 4.7 T ( 5 0 transien%, 200 Hz gaussian line broadening) and (b) 9.4 T (3774 traasients, lûû Hz gaussian liw broadening). Aiso shown are the calcuiated subspectra for each phosphorus site.

relatively insensitive to y for P(1) since the difference between 6,i and 6, for P(l) is
much smaller than for P(2); hence the position of 6,, and &a is w t as easily defined in
the case of P(1). The calculated orientation of the q3 component for P(1) (figure 5.6(a))
is in agreement with experiment; however, this is not the case for P(2) where the 6,,
component is calculated to be 34" away from the P-P bond (figure 5.6(b)). This is
significantl y different €tom the experimentai result , P = 78 O . in addition, the
calculations predict that the 6, components for both tensors are aligned, i.e., Ba = O",
whereas the experimental results predict an offset, Aa = 25 * 2". While it may be
tempthg to further compare the experimental and calculated phosphorus shielding tensor
orientations, it should be noted that calculations are performed on an optimized
geometry of a mode1 cation. in contcast to the situation for TMPS (chapter 3) and the
phosphole tetramer (chapter 4), much less information is available about the
experimental structure of [Ph,P-PPhJ [GaCL].
5.3.2 Dependeuce of lJ('lP,'lp) on codocm8tion: H2P-PH2 vs. H,P-PH,+
If one wishes to compare experimental and cakulated results, as well as use the
calculated J coupling to obtain structural information in these types of systerns,
knowledge of how J depends on the conformation is essential. H2P-PH2 and H,PPH,+
are prototypical molecules for such an investigation. Indirect experimental evidence that
'Je1P,"P), depends on conformation was first reporteci in 1970 in a variable
temperature NMR study of P2F41n and in 197 1 for 1,2dimethyl- 1.2-diphenyl
biphosphine. Since then, chis phenomewn has been observeci in a number of other

Figure 5.6 Calculated orientation of the phosphorus chernical shift tensors for (a) P(1) and (b) P(2) in (CH,)3P-P(CH3k+. In both cases, the il, compownt is perpendicular to the plane of the page. The hydrogen atom have been ornitteci for clarity .

s y s t e n ~ s - ~ ~ e ~ ~ ~ The general trend for biphosphiaes appears to be as fouows: a mal1
positive 1J(31P,31P), is observed for the conformation where the formai lone pairs are
tram with respect to each other (+, = 180°) whiie 1Je1P,31P)b is large and wgative
for the cis conformation (4, = in addition, biphosphines are not fixed in a
particular conformation, hence the observed value of 1J(31P,31P), must be averaged over
several conformations. The calculated potential energy curve for rotation of the PH2
fragment about the P-P bond in H,P-PH, indicates rhat the cis and t r m arrangements of
the formai electron lone pairs about the P-P bond have ewrgies of 11.3 and 3.3 Id mol"
respectively relative to the energy minimum at @, = 77" (note that RT = 2.45 W mol-'
at T = 295 K). Our calculations at the HF level of theory using the cc-pVTZ basis
set on al1 atoms give results that are in qualitative agreement with reference 181 and
indicate that the energy for rotation about the P-P bond is relatively Bat for
conformations between @pp = 60' and +,, = 180" (figure 5.7).
Sbown in figure 5.8 is the dependence of 1J(31P,31P) on 4, in H2P-PH,. The
data are given in table 5.2. The experimental value for 'J(31P,xP), is -108.2 * 0.2
Hz. '" While it is apparent that the quantitative agreement is no< very good, it is evident
ihat 1J(3 l P,31P) is highly dependent on the dihedral angle. nie FC mechanism is largely
responsible for the dihedral angle depenâence of L/(f1P,3iP)),, although the SD
mechanism makes a non-negligiôle contribution to the magnitude. An early molecular
orbital SCF study also found a similar dependence of 1J(3'P,31P), on 4pp.1(D Our results
are compared to results fkom otber c a l ~ u l a t i o n s ~ ~ ~ ~ ~ in table 5.2. it is intetesthg to

Figure 5.7 Plot of the totaî energy for H,P-PH, as a fùnction of Al1 ewrgies are relative to aPp = 180'.
note that the calculated 1J(31P,31P), results differ significantly with respect to the
magnitude of the FC contribution; however , the trends are the same. The anisotropy in
J is calculated to be significantly larger than LJ(31P,31P), and it is also very dependent
on @,, for H,P-PH, as illustrated in figure 5.8(b).
in contrast to H2P-PH2, lJCIP:lP) for H3P-PH,+ is relatively dependent of
4,+, defined in figure S.l(d). Figure 5.9 illustrates the dependence of J in H,P-PH,'
on a,, , with the &ta given in table 5 -3. This result is teasonable, since rotation of the
PH, group does not result in a signifiant change in the overall structure as îs the case
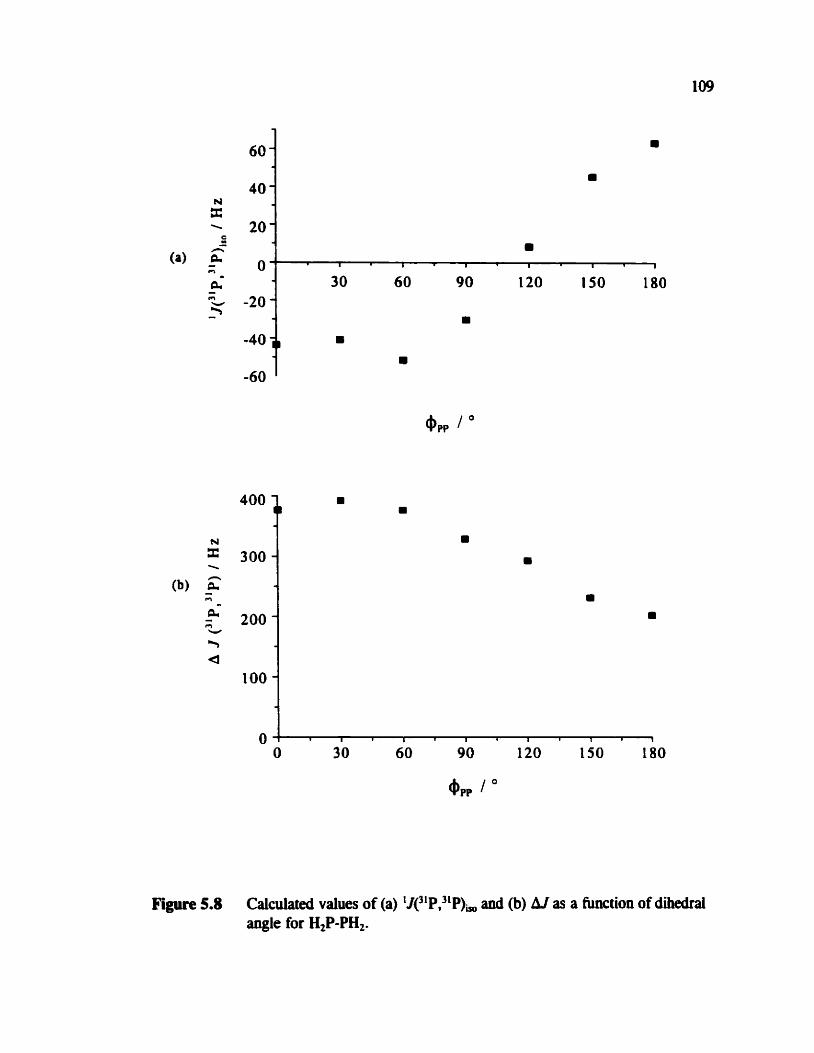
Figure5.8 Calculatedvalws~f(a)'J~'P,~'P),and(b)hlasahurtionofdihedral angle for H2P-PH2.
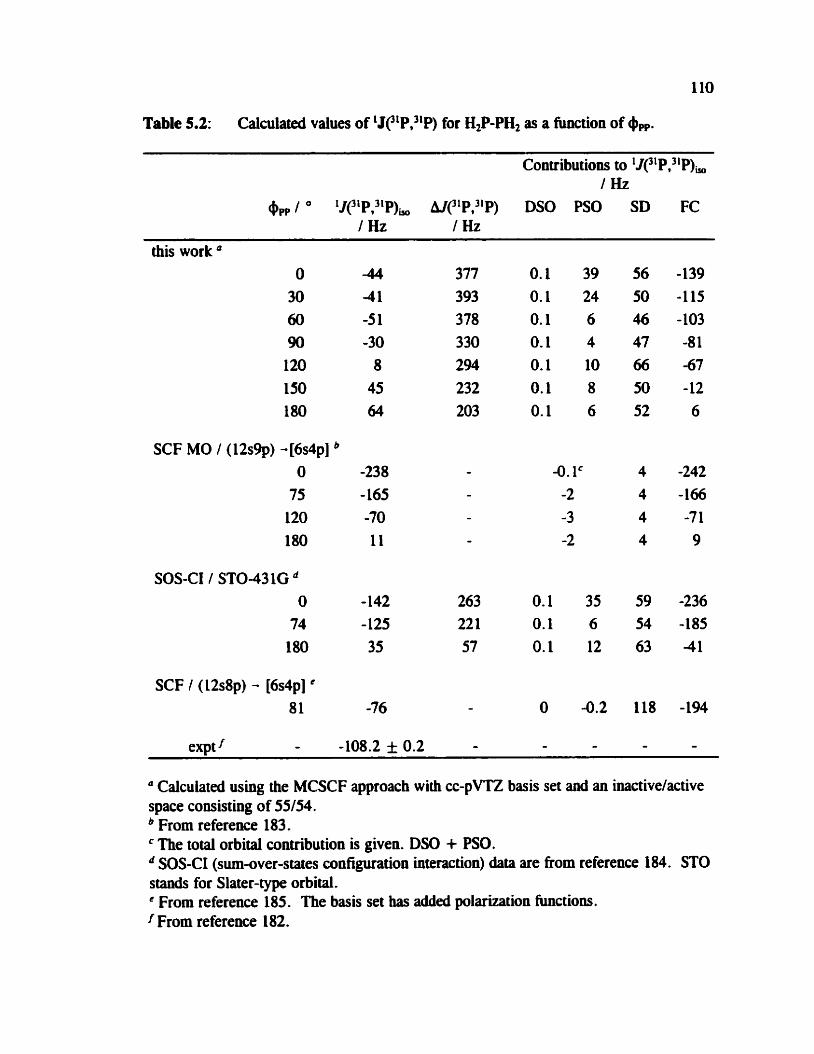
Table 5.2: Calculated values of lJCIP,"P) for H2P-PH2 as a m i o n of 6,.
Contributions to 1Jc1P,31P), / Hz
aPP 1 O 1J(31P,31P)to AJ(31P,31P) DSO PSO SD FC /Hz /Hz
this work O 30 60 90
120 150 180
SCF MO 1 (12s9p) -[6s4p] O
75 120 180
SOS-CI / STO-431G O
74 180
SCF 1 ( 1 2 ~ 8 ~ ) - [6s4p] ' 81
" Calculated using the MCSCF approach with cc-pVTZ bais set and an inactivelactive space consisting of 55/54.
From reference 183. ' The total orbital contribution is given. DSO + PSO. SOS-CI (sum-over-states configuration interaction) &ta are from reference 184. STO
stands for Slater-type orbital. ' From reference 185. The bais set has added polarization functions.
From reference 182.

Figure 5.9 Calculateci values of (a) 11(3'P,3iP), and @) AJ as a function of dihedral angle for H,P-PH,+. The scale is the same as in figure 5.8 to emphasize the difterence benveen the two molecules.

112
Table 5.3: Calcufared values of LJe1P,31P) for H3P-PH2+ as a function of @,+.a
Contributions to LJC1P,31P), /Hz
@pp+ 1 O 'J(3LPT3iP)y. Hz h1(31P,31P) / Hz DSO PSO SD FC
Calculations performed at the MCSCF levei with the cc-pVTZ basis set using an inactive/active space of 1018.
when one of the PH, groups in H2P-PH, is rotated with respect to the P-P bond. For
H3P-PH2+, the magnitude of 1J(31P,31P), is much larger than in H,P-PH,, with the
dominant contribution fiom the FC mechanism. For H3P-PH,+, the anisotropy in J is
smaller relative to 1J(NP,31P), in coatrast to the situation for H2P-PH,. The trend in
J(31P ,"P) for H,P-PH2 vs. H3P-PH2+ bas also been reportai in semi-empirical
calculations of J(15N, "N), for the aoalogous nitrogencontaining compounds . l M As
weîl, it was noted that the calculated valw of 1J(13C,"N), in methylamiw is virtuaily
independent of rotation of the MI, fragment about the C-N bond.'"
The replacement of one of the loue pairs in H2P-PH, with a bonding pair (CO a
hydrogen atom) in H3P-PH2+ r d t s in a decrease in iJ('1P,3'P), as well as a decrease
in the anisotropy in J. In t e m of reduced coupling constants (see epuation 2.24),

113
iK(P,P), for H,P-PH, vs. H W H 2 + with 6, = +,+ = O0 are -22 x IOi9 NA-%nm3 and
-99 x IOi9 NA%-3 respectively. This is contrary to the generaily observed lone pair
effect, where an increase in IK, is observai when a lone pair is replaced by a
For example. 'K(N,C), for CH3CH2cH,NH2 is 12.7 x lOI9 N A k 3 vs.
14.4 x IOi9 NA-k3 for CH3CH&H2NH,+, la and for P(CH,), vs. P(CH3),+, 'K(P,C),
= -1 1.1 x 1019 NA-%m3 and 45.3 x IOs9 NA-%rr3 respectively . Two exceptions are
(CH,),P-P(CH3), vs. (CH,)2P-P(S)(CH3)2, with IK(P,P), = 91 x lOI9 NA%-' and
'K(P,P), = -1 14 x IOL9 NA-%r3 respectively,ln and Ph2P-PPh, ('K(P,P), = -101 x
IOL9 NA-%r3)I3 vs. Ph,P-PPh2+ (IK(P,P), = -164 x IOt9 NA%-), this work). It should
be noted that lK(P.P), for ?h,P-PPh2 is lilcely very dependent on the lone pair
conformation and hence changes in the dihedral angle rnay change the trend with respect
to Ph3P-PPh2+.
Another factor that must be considered is a change in structure besides rotation
about the P-P bond. It has been claimed that 'J~'P,"P), for RR'P-PRR' is sensitive CO
the R-FR' bond angle (where R, R' are any one of the following: H, an alkyl group, or
phenyl).lW To investigate the influence of geoinetry, iJ(31P,3'P), was calculated for
H2P-PH, geometries where one bond length or angle was changed to match the
corresponding parameter in the geornetry used for H3P-PH,' (table 5.4). in this
manner, the effect of changing each bond length or angle may be determineci. The
calculated iJ(31P.HP), values for H,P-PH, using two different active spaces are given in
the f ~ s t two liws of table 5.4. This illustrates that using a larger active space (first
line) does not significantly change the calculated cwpling constant compared to the

114
results obtained using a smaller active space (second line). The wxt four lhes present
the calculaied 1J(31P,3LP), values obtaioed when a given bonâ length or angle in
H2P-PH2 is changed to match the correspondhg boad length or angle in H3P-PH2+. As
is evident from the &ta in table 5.4, none of the geometry differences between H,P-PH,
anci H,P-PH,' can account for the difference in 1J(31P,NP), for these two molecules.
Unforninately, m e r MCSCF caiculations on systems of this size are oot possible with
available computatiooal resources .
Table 5.4: Geometry dependence of 1J(31P,31P), for H2P-PH,."
geometry alteration 1J(31P.xP), I Hz -- -
H,P-PH2 experimental geometry (&, = O") a
H,P-PH2 experimental geomerry (#,, = O")
P-P in H,P-PH, cbaoged to 2.2192 A H-P-H in H,P-PH2 changed to 95"
H-P-H in H2P-PH2 changed to 106.37O
H-P in H,P-PH2 changed to 1.386 A H3P-PH2+ (+pp+ = 0")
a From table 5.2. MCSCF calculations using the cc-pVTZ basici set on al1 atoms and an inactivelactive
space of 55/43. From table 5.3.

115
5.3.3 Estimate of r, in ~@PPhJ[GaCW
As was stated in the introduction (section 5. l), to obtain an estimate of the P-P
bond length in ph3P-PPhJ[OaCi,], it is necessary to have an estimate of the anisotropy
in the J coupling, shce it is experirnentally inseparable fiom RD,. With a value for AJ,
one can obtah RDD from & according to equation 2.29. As mentioned in section 2.1.6,
reliable measurements of hl are scarce. An estimate of hl may be obtained tiom ab
initio calculations; however cornputer resources lirnit the size of the molecule one can
use for J calculations. In this case, H,P-PH,+ is the smallest possible mode1 compound
for Ph3P-PPh2+. As the results for H,P-PH,+ indicate that AJ is vimially independent of
app+ (figure 5.9). one may use the average AJ over al1 conformers of H,P-PH,+,
121 Hz. Using this value in equatioa 2.29. a value of 1.74 I0.05 l d z is obtained for
RD,, hence r, = 2.25 f 0.03 A in [Ph3P-PPhJ[GaCiJ, accordhg to equation 2.20. If
one assumes that R, = RD,, then r,, = 2.26 f 0.03 A which is within experimental
error of the fust estimate. In this case, A J is relatively small, hence a reliable estimate
of the vibratioaally averaged P-P bond length may be obtained for R,,. However, the
ob initio calculations of J for H2P-PH, indicate that one m o t necessarily assume that
AJ for d l one-bond P-P J-coupling teosors will be small.
5.4 Conclusions
The phosphorus chernical shift and 31P-31P dipolar coupling tensors in
Ph3P-PPhJ[GaCW have been characterimi by solid-state NMR. By combining the
experimentai results with ab initio dculations of 'J(3'P,"P) for H,P-PH,+, a reliable

Il6
estimate of the P-P bond le- in [Pb3P-PPhJ[GaClJ has been obtained, 2.25 * 0.03 A. It should be mted that vibrationai averaging and possible rockhg motion of the
P-P bond have been ignored. in addition, the dependence of indirect spin-spin couplhg
on lone pair conformation has been investigated for H,P-PH, and H,P-PH,+ using the
MCSCF approach. It was found that 1J(3LP,nP) for H2P-PH, is very depenâent on
conformation, in contrast to H,P-PH,+.

Cbapter 6: Conclusions
The phosphorus chemical shift and spin-spin coupling tensors for tbtee different
compounds containing P-P bonds have been characterized by solid-state "P NMR. The
combination of experimental methods and ab inirio calculations has proven to be very
usehl for explorhg the tensor nature of each interaction.
For the first example, TMPS (chapter 3), the phosphorus NMR parameters were
characterized by NMR studies on both a single crystal and a powder crystailine sample.
There are two molecules in the asymmetric unit; however the difference between the
two sites is very subtle, as evident in a redetenninatioa of the X-ray crystal structure.
This difference is also clearly evident in the )lP NMR spectra, from which two unique
phosphorus chemical shift tensors have been characterized. The tensor orientation is
such that the direction of greatest shielding, a,,, lies dong the P=S bond. The
experimental and calculated orientation of the phosphonis chemical shift tensors are in
excellent agreement. It was also shown that the anisotropy in the nP,31P indirect spin-
spin coupling for TMPS is less than 500 Hz, contrary to earlier reports for similar
cornpo~nds.~*~~~ This compound serves as a benchmark for testing the reliability of ub
initio calculations of nuclear magnetic shieldiag tensors.
The second system described in this thesis was the phosphole tetramer. Since a
large single crystal was not available, the phosphorus NMR parameters for this
compound were characterizeû by analysis of "P NMR spectra of powdered sarnples

118
(chapter 4). In this case, ab initio calculations of chernical shift tensor orientaiions
proved to be invahiable. It was found that il, is closest to the P-P bond for both
phosphorus chernical shift tensors. The b33 component is situated in the same plane as
the phosphole ring. The relative orientation of the 6, components for the phosphorus
nuclei reflects the local geometry of the phosphole rings.
The third compound described here, [Ph3P-PPhJ [GaCl.,] , was prepared in the
Burford lab. The solid-state NMR investigation of this compowid provided the P,P
bond length (chapter 5). Ab initio caiculations of 1J(31P,31P) on mode1 systems provided
insight into the dependence of J on molecular structure. For H,P-PH,, 1J(31P,31P) is
very dependent on the rotation of the PH, fragment about the P-P bond while it is n a l y
independent of rotation in H,P-PH2+.
From the results of these three projects some general conclusions rnay be drawn.
It is evident from the results for the phosphole tetramer and for [Ph,P-PPhJ[GaCl,] that
one cannot assume that the direction of greatest shielding corresponds to the generaily
perceived direction of greatest electron density, Le., dong the formal lone pairs.
Furthemore, knowledge of the anisotropy in 1J(31P,31P) is important for obtaining an
estimate of the bond length from ReR. The investigation of hl in TMPS M e r
establishes the observation that AJ(3iP,31P) is small in diphosphine disulfides, contrary
to earlier rep~rts.l'*'~ On the other W, first-priociple calculations indicate signifiant
anisotropies for LJ(31P,3LP) in HIP-PH2 and H,P-PH,+. In each of the projeds discussed
in this thesis, ub initio calculations of NMR parameters play a significant role. The
reliabiiity of d~ initio calculations for characterizhg phosphorus chernical shift tensor

orientations is established by compafison with results fiom a single-crystal "P NMR
study of TMPS. For the phosphole tetramer, it is possible to fU the chemicai shift
tensor orientation in the molecula. frame of reference by cornparhg calculated tensor
orientations with the orientation information available from the analysis of spectra
obtained from powder crystalline samples . First-principles calculations of 'Je 1P,31 P)
were aecessary to establish tbat hJC1P,31P) mkes a very small contribution to RM,
hence a reliable estimate of r, could be obtained for m3P-PPhJ[GaClJ.
In the past few years, the value of ab initio caiculations as a complement to
experimental results in the field of NMR has been recognized. The continuous
improvement and availability of computational methods and resources have undoubtedly
contributed to extending the range of systems which may be studid. While ub initio
calculations are yielding reliable qualitative results for nuclear magnetic shielding
tensors, much more work needs to be dom io reach the same level of confidence with J
coupling. in this respect, it is essential that the results from first-principle calculations
be compared with reliable experimental data. Some specific suggestions are given in the
next chapter. At the same the , it is important for NMR spectroscopists to use more
quantum chemistry methods and keep up to date with the latest developments Ui this
field.

Chapter 7: Future Research Directions
7.1 Introduction
Several extensions of the research presented in this thesis are worthy of
consideration. Suggestions include an investigation of phosphom chernical shift teasors
in other phosphinophosphoaium cations (section 7.2) as well as further 1 initio
calculations (section 7.3). While the calculations of J appears to be promising, much
more effort neeâs to be expeaded in this area. Ab inirio calculations are presently
limited to relatively small systems. The recent success of DFT approaches at
calculating J for numerous spin pairs are most encouraging . 12'g'u*1U Obviously , the
potential of this approach for calculating 1J(3iP,31P) should be investigated. Section 7.4
presents some preliminary resuits and suggestions for hirther theoretical studies of
indirect spin-spin coupling for systems containhg phosphorus spin pairs. All the spin
systems discussed in this thesis involve two coupled spin-% nuclei. An example of a
system consisting of three coupled spin-% nuclei is outlined in section 7.5.
7.2 [W,(Cî)P-PPbJ[GaCiJ
The precursor to [Ph,P-PPhJ [GaCb] (chapter 5), [Ph2(Cl)P(1)-P(2)PhJ [GaCl.,] ,
has also been isolated.l* In this case, the coupling of P(1) to a chlorine nucleus
cornplkates the appearance of the spectra. Chlorhe hm two naniraliy occurring
isotopes; 3sCl: spinin3/,, N.A. = 75.53 % and )'Cl: ~ p i o - ~ l ~ , N.A. = 24.47 1.
Phosphotus-31 NMR spectra of MAS samples are shown in figure 7.1, obtained at
120
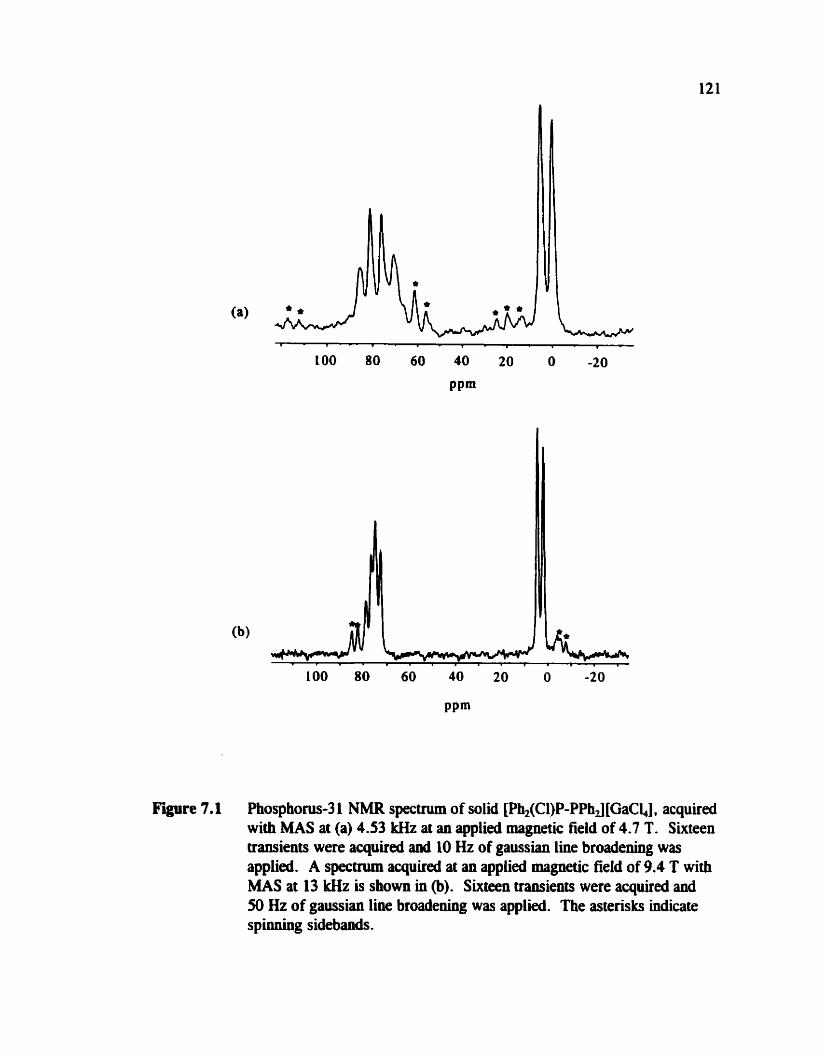
. - , . . - , . . - , . , . LOO 80 60 40 20 0 -20
LOO 80 60 40 20 O -20
PPm
Figure 7.1 Phosphocus-3 1 NMR spectnun of solid [pb,(Cl)P-PPhJ[GaCî,,], with MAS at (a) 4.53 kHz at an applied magnetic field of 4.7 T.
acquired Sixteen
transients were acquired and 10 Hz of gaussian line broadening was applied. A specrnim aquired at an applied magnetic field of 9.4 T with MAS at 13 lrHz is s h o w in (b). Sixteen iransients were acquired and 50 Hz of gaussian line broadening was applied. The asterisks indicate spioning sidebands.

applieâ magnetic fields of 4.7 T and 9.4 T. The adysis of the spectra arising from a
spin-% nucleus couplai to a quadrupolar nucleus has been presented in the
literat~re;'~'- '~ however, many of the simulations are suspect, e.g., reference 192.
Carbon-13 coupld to 3sa7Cl bas been investigated in chloroketosulfones. L93 It is
important that experiments be carrieci out at several applied magnetic fields, as is
evident in re ference 193. The line shape for [PhZ(Cl)P-PPhJ [GaCî.,] is further
cornplicated by the coupling of P(1) to the phosphine centre. Initial simulation of the
MAS spectra proved to be unsuccessful. Reliminary 31P NMR studies of
[Ph2(Cl)P-PPhJ[GaCW by CP experiments indicate that the proton relaxation times are
long; recycle delays in excess of 2 minutes were needed to avoid saturation. Funher
experiments on this compound are in progress.
7.3 Ab Inirio Calculatioiis of Phosphorus Chernid Shift Tenson
The results from ub initio calculations of phosphonis chemical shift tensors
presented in this thesis indicate that qualitative agreement with expriment may be
expected. However, there appears to be a systematic discrepancy between the
experirnental and calculated results. For al1 three compounâs presented in this thesis,
the values of 6, calcdatd ai the HF level are more shielded by about 50 ppm. As
mentioned in section 3.3.3, changes in the phosphonis chernicd shift on the order of
teas of ppm have been observed upon changing from the gas to liquid state. Clearly,
the influence of intennolecular effefts on the phosphonis chemical shift weds to be
considered if one is to obtain quantitatively reliable results. Investigations dong these

123
lines on mode1 systems, such as PH3 or P,, for which the gas to liquid shifts are known
(see section 3.3.3), are recornrnended.
7.4 Characterization of 'J(31P:1P)
While 1J(31P,31P), is large and negative in H2P-PH2,Ln a large positive value,
+720 Hz, is observed in molecules of the type H,(CH,),,PPF5,lw and a value of +766
Hz was reported for the potassium sait of F02PPû2F+. lW Successful calculation of
'J(%P,"P), for these molecules which, together with H,P-PH,, span the range of
observed lJ("P?'P), values, would be of great value. A survey of MW results (FC
contribution only ), which includes the above-mentioned molecules, has ken reported ."
In addition, a large wgative values of 1J(31P,31P),, approximately 480 Hz, have been
observed for a series of dipho~pholes.~% Given the success of MCSCF37*1Q*110 and DFT
r n e t h ~ d s ~ ~ ~ * ~ ~ * ~ ~ for calculating J coupling, application of these theoretical approaches to
the above systems would be of interest. For the larger molecules, DFT may be the only
possibility with cunentiy available cornputer resources. initial results are promising;
however, systematic studies to reproduce experimental trends are warrant&.
There are a number of other small phosphonis-containing molecules besides
H,P-PH, for which experimental data are avaiiable." One system of fiindamental
importance is diphosphene, P,H,, as well as the analogous nitrogen compound, diazene,
N,H,. Results for J from calculations using the MCSCF approach with the cc-pVTZ
bais set for the tronr isomen of these two compounds are given in table 7.1. For
cornparison, data from an early ub initio studylW using smaller basis sets are also given.

Contributions to 'JJHz
'J(X,X)f iz hl(X,X)Rlz DSO PSO DS FC trm-N,H, O< = 15N)
CAS1 -249 -220 O -115 -16 -118 CASII -25 74 O 10 1 -36 SOS CI -17 - O -15 0.2 -2
diazenes * 10 to 20 tram-P,H, (X = 31P)
CASüI -1985 1076 0.2 -531 -1069 -385 CASIV -348 - 1463 O -386 49 -12 SOS CI -552 - O -498 9 -76
diphosphenes' & 5 10 to 670
Contributions to 'K, / IOi9 N A - k 3
lKO(,X)J ,I(X,X)/ DSO PSO DS FC 10i9 N A - k 3 1019 NA-%r3
trm-N2H2 (X = N) CAS1 -20 17 -1782 O -932 -130 -956 CAS11 -203 598 -0.3 79 12 -293 SOS CI -138 - 0.1 -122 2 - 16 diazenes & 80 to 160
tram-P,H, (X = P)
CASm -1006 174 0.1 -269 -542 -195
CASIV -176 -742 0.1 -1% 25 -6 SOS CI -280 - O -262 5 -39
diphosphenes' * 260 to 340
a Calculations p e r f o d at the MCSCF level with cc-pVTZ basis set. From reference 197. The geometq for tram N2H2 was not given. From reference 201. From reference 197. using the ST016-3 lGf* basis set. The geometry used for truns
P,H, is as foîlows: P-P = 2.034 A, P-H = 1.44 A, and P-P-H = 95.6". From reference 204.

The reduced coupling tensor, K (equation 2-24), is also presented in table 7.1.
The molecular geometries used are as foliows. For trm-P2H2, the P-P bond
length = 2.00 A, the P-H bond length = 1.40 A aad the P-P-H angle = 108". based on
typical values for diphosphenes. 198-199 For tm-N,H,, the N-N bond length = 1.247 A,
the N-H bond length = 1 -029 A, and the N-N-H bond angle = 106.3 O .m
The smallest active spaces used in both cases, 10101 1120 (denoted by CASI) for N2H,
and 414113 120 (CASIII) for P2H, do not include virtual molecular orbitals, and thus
give the same results as an HF ~alculation.'~ The larger active spaces, 10101223 1
(CAS@ for N2H, and 414114231 (CASiV) for P2H2, include a virhial orbitai for each
molecular orbital symmetry. As is evident for N,H,, the magnitude of 'J(lSN * NI,
calculated with CASII is in reasonable agreement with experirnental values that are
typicai of diazenes?' Unfortunately , the sign of this coupling is not known; however,
early semi-empuical calculations predicted that 'J(15N, I J N ) is negative except possibly in
hydrazine type c o m p o u n d ~ . ~ ~ ~ ~ The MCSCF results discussed above and early ab
initio resultsL* for N,H, in table 7.1 are in agreement with this prediction.
For P2H2, calculations using the larger active space, CASIV, predict a value of
-348 Hz for 1J(3tP,3'P), and a large negative hl of -1463 Hz. Unfortunately, no
experimental data are available for P2H2; however typical values of ' J(31P,3 'P), for
diphosphenes range between 5 10 and 670 Hz in magnitude, depending on the
substituent~.~~ The sign of the coupling is not known, but it bas been s u g g e ~ t e d ' ~ ~
that it is negative on the bais of early ab initio dcuiatiom which predicted a negative
value,tn 1J(31P,31P)im = -552 Hz, due to the large negative contribution from the orbital

126
mechanism. The type of basis set used for the early ab initio calculations,
ST016-3 le**, is known to be unreliable for indirect spin-spin coupling ~alculations.~
Semi-ernpiricai approaches predict a large positive value, +830 Hz when ody the FC
mechanism is conside~ed.'~
Also given in table 7.1 are the contributions of each cnechanism to lJh and 'Kb.
For P2H2, the magnitude of lJCIP,NP), is dominated by the PSO contribution; however,
it not definitive which mechanism dominates 'J(15N,1"N in NN,H2. Cornparison of the
contributions obtained using an active space with no virtual orbitals vs the results using
one that does include virtual orbitals illustrates the triplet iastability problem discussed
in section 2.3.2 where the contributions that depend on the triplet States are known to be
poorly calculated if virtual orbitals are not included in the MCSCF space. While
calculations qualitatively predict the trend in ' J , for P2H, vs N2H2, cornparison of the
values of 'Km is more telling of problems with the ub initio results. In this case, the
calculated results are clearly wrong ~~rnpated to the experimental results, i.e., using the
larger active space, the magnitude of [Km for N2H2 is larger than that for P2H2,
contrary to the experimental trend. For these systems, hirther work is necessary ,
possibly ushg restricted active s p a c e ~ . " * ~ ~ ~ ~ Even for a molecule as small as P2H2,
extensive study of the dependence of the calculated J on the active space is not possible
with currently available computational resources. It should be noted that systems such
as N,H, and P2HL, where there are multiple bonds and formal electron lone pairs,
present a difficult computatiorial chaUenge.'

127
7.5 NMR Spcdra Arising from Tbnc Coupied Homonuclear Spin-% Nuclei in the
Solid State
A logical extension to the investigations of spin pairs presented in this thesis is to
systems of multiple coupled spins. Analysis of spectra arising fkom multiple spin
systems are of interest in biomolecules where multiple labelling allows for more
information to be ext ra~ ted .~ The theory for the analysis of spectra arising fiom three
coupled heteronuclear spin-% nuclei has been presented in the literature, for the
AA'X,m ABXVm and AXZm cases. The iterative fitting of MAS spectra arising fmm
an ABX spin system to obtain chernical shift tensors has been reported.*1° In principle,
the ambiguity regarding the orientation of the chemicai shik tensor with respect to the
molecular frame of reference, which is an inherent limitation of the dipolar-chernical
shift method (section 2.2.2), is elhinateci in the analysis of spectra for three coupled
spins. For example, it was demoostrated for doubly 13C labelled L-alanine that the
carbon chemical shift tensors obtained from an analysis of powder samples211 agreed
well with results from a single-crystal NMR study.'12 In this case, the presence of both
the 13C-i3C, and L3Ca-14N spin pair was exploited to fm the carbon chemical SM tensors
relative to the molecule.
The above discussion pertains to multiple heteronuclear spin systems. A
homonuclear four spin system, the fully 'C labeUed monoarnmonium salt of maleic
acid, has been investigated by solid-state An example of a homonuclear three
spin system is iiiustrated in figure 7.2, where a linear arrangement of three phosphorus

Figure 7.2 Structure of a phosphorus-containing homonuclear three-spin system.
nuclei is pre~ent.~" A linear arrangement of phosphorus nuclei will simplify the
analysis of the spectra. An investigation of the phosphorus chernical shift tensors and
spin-spin coupling parameters in such a unique molecule would be a valuable
contribution to an understanding of the relationship between structure and NMR
parameters. A program available for simulating solid-state NMR spectra, SIMPSONVs'
is capable of calculating spectra arisiag from multiple coupled spins with the inclusion
of chernical shift, dipolar aod spin-spin coupling interactions and may be useful for this
project .

Appendix 1: Performing the 2D Sph-Echo NMR Expriment on the CMX Infinity 200 at the University of Alberta
The 2D spin-echo NMR spectra presented in this thesis were obtained ushg a
Bruker MSL 2 0 sptxtrometer (figure 4.5) or a Varian Chemagnetics CMX Infinity 2ûû
(figures 3.9 and 5.4). For the benefit of future users, the details of the procedure used
to obtain 2D spin-echo NMR spectra using the CMX Infhty 200 and to process the
data to a fonn that can be used for simulation with the program SpinEcholY> are
presented here. The following description pertains to the pulse program show in figure
2.8. The phase cycling of Rance and Byrd was used.IM
After setting up the parameters for a standard 1D NMR expriment using CP,
the x pulse on the X chamel needs to be caiibrated. This must be doue using the sarne
X power settings as used for CP. The important 2D parameters are dw2, tl - evolve,
and a12. The dwell tirne for the second dimension, dw2, should be set so that the sweep
width in the Fi projection (sweep width = lldw2) is larger than the largest splitting
expected, i.e., larger than 2& for a spin pair where the two nuclei are aot magnetically
equivalent anci 3R, for the case of magnetic equivalence (see figure 2.4). The
parameter t l - evolve corresponds to the time between the CP part of the pulse program
and the x pulse as well as the time following the x pulse aod before acquisition. In the
version of the 2D spin-echo pulse sequence used on the CMX mnity 200, the
parameter is calculated based on the value of a12, which is the number of times
129

130
t1 - evolve is incrernented, and dw2. The maximum value of tl-evolve is dwPai2.
UsuaUy a12 = 64 experiments is acceptable.
Once the data are collected, they are pracessed using Spinsight software.21s It is
recommeeded that zero filling and gaussian line broadeniog be applied in both
dimensions. To obtah a symmetric 2D NMR specmun, it is necessary to process in
magnitude mode for both dimensions. To ease simulation of the Fl projection with
S p i n E ~ h o , ~ ~ ~ it is usehl to bave the Ne in a format that is usable with WSOLIDS49 or
other processing software available for PCs. At the t h e of preparation of this thesis,
there is no direct method for doing this. uistead, a print out of the R projection is
scannai and a program called SPECMAKI? which is part of the WSOLIDS49 software
package, is used to convert the scanned image to a data file. The print out of the Fi
projection is obtained by executing a macro with Spinsight, called ldshowgroj . Note
that in the display, Spinsight uses the wrong value of the dwell t h e to calculate the
sa le (dw instead of dw2). This is corrected by changing the value of dw to the value
used for dw2 in the acquisition panel before the FI projection is printed out. If the 2D
spectnun is p ~ t e d out, the scale for the fl projection is correct. While it is not
necessary to convert the spectrum data in this way, simulation of the specaa and
preparation for publication is easier.

This appendix is intended to provide some details with regards to handling air-
sensitive sarnples for NMR studies. Some generai strategies will first be presented,
followed by some comments pertalliing to specific rotor designs.
For samples that are extremely air-sensitive, sealed Pyrex MAS rotor inserts
may be ~sed .~" The sample is heat sealed into the inserts or an epoxy sealant may be
used. Heat sealing is generally more difficult given the srnail size of the inserts and the
need to obtain as symmetric a seal as possible so as not to harnper MAS. If the sample
is packed directly into the MAS rotors, an additional precautionary masure to avoid
contact with air is to seal the rotor itself into a glass tube and break it open once the
NMR experirnent is set up.
The rotors used to obtain the NMR spectra presented in this thesis are
manufactured by either Bniker or Varian-Chemagnetics. It has been the experience of
the solid-state NMR group at Dalhousie University and University of Alberta that the
Bruker rotors are gewrally sufficiently air-tight and no special precautions are needed
beyond ensuring b t the rotor caps are tight fitting. For the VarianChemagnetics
rotors, there are a number of strategies for dealing with air-sensitive smples, for
example the use of glas inserts as discussed above is a cornmon solution. Lnserts are
commerciaiiy available for various Varian-Chemagmtics rotors. The rotor illustrated in
figure A2.1 was used to obtain NMR spectra of &-sensitive sarnples on the Varian-
Chemagnetics spectrometer. The ody modification to the commercial design is an
131

Figure A2.1 An illustration of a Varian-Chemagnetics rotor. (a) drive tip. (b) spacer . (c) zirconjum o d e sleeve. (d) epoxy seal. (e) end cap, with a hole for variable temperature experiments .
epoxy seal applied to the inner surface of the end cap, which is manufactureci with a
vent hole for variable temperature experiments. The drive tips usually fit very tightly,
hence there is no difficulty with excluding air at the other end of the rotor.

References
D. L. Pavia, G. M. Lampman, and G . S. Kriz. htroduction to Spectroscopy, 1 Ed. Saunders College Riblishing, Fort Worth, Texas. 19%.
U. Haeberlen. In Advances in Magnetic Resonance. Edired by J . S . Waugh. Acadernic Press, New York. 1976, Supplement 1, p. 1.
J. B. Robert and L. Wiesenfeld, Physics Repons 86, 363 (1982).
K. R. Dixon. In Multinuclear NMR. Edired by J . Mason. Plenum Press, New York. 1987, p. 369.
C. J. Jameson, AMU. Rm. Phys. Chem. 47, 135 (19%).
H. Fukui, Prog. Nucl. Magn. Reson. Spectrosc. 31.3 17 (1997).
A. C. de Dios, Prog. Nucl. Mugn. Reson. Spectrosc. 29, 229 (l996).
T. Helgaker, M. Jasniiiski, and K. Ruud, Chem. Rev. 99, 293 (1999).
C. J. Jameson. In Modeling NMR Chernical Shifts: Gaining Insights into Structure and Envuonment. Edired 6y J. C . Facelli and A. C. de Dios. American Chernical Society, Washington, K. 1999, p. 1.
K. Eichele, G. Wu, R. E. Wasylishen, and J. F. Britten, J. Phys. Chem. 99, 1030 (1995).
P. N. Tuniojian and J. S. Waugh, J. Chem. Phys. 76, 1223 (1982).
J. B. Robert and L. Wiesenfeld, Mol. Phys. 44, 319 (1981).
T. Nakai and C. A. McDowell, J. Am. Chem. Soc. 116, 6373 (1994).
(a) S. Aime, R. K. Hams. E. M. McVicker, and M. Fild, J. C h . Soc., Dalton T r m . 2144 (1976). (b) F. Sarürahya, Y. Sarikahya, 1. Topaloglu, and 1. O. Sentiirk, Chimica Acta Turcica 25, 35 (1997).
R. K. Harris, L. H. Merwin, and G. Hiigele, J. Chem. Soc., Farodoy Trans. 1. 83, 1055 (1987).
P. N. Tuaiajian aod J. S. Waugh, J. Magn. Reson. 49, 155 (1982).

K. W. Zilm, G. G . Webb, A. H. Cowley, M. Pakulski, and A. Orendt, J. Am. Chem. Sac. 110,2032 (1988).
R. Chaiionet, C. A. McDoweîi, M. Yoshifuji, K. Toyota, and J. A. Tosseiî, J. Mugn. Reson. A 104,258 (1993).
K. Eichek, G. C. Ossenkamp, R. E. Wasylishen, and T. S. Cameron, Inorg. Chem. 38, 639 (1999).
G. M. Bernard, G . Wu, M. D. Lumsden, R. E. Wasylishen, N. Maigrot, C. Charrier, and F. Mathey , J. Phys. Chem. A 103, 10L9 (WW).
J. Fischer, A. Mitschler, F. Mathey, anâ F. Mercier, J. Chem. Soc. D a h n T r m . 841 (1983).
(a) F. S. Shagvaleev, T. V. Zykova, R. 1. Tarasova, T. Sh. Sitdikova, and V. V. Moskva, J. Gen. Chen. USSR 60, 1585 (1990). (b) N. Burford, T. S. Cameron, J. A. C. Clybunie, K. Eichele, K. N. Robertson, S. Sereda, R. E. Wasylishen, and W. A. Whitla, Inorg. Chem. 35, 5460 (19%). (c) N. Burford, T. S. Cameron, D. J. LeBlanc, P. Losier, S. Sereda, and G. Wu, Organometallics 16, 4712 (1997). (d) N. Burford and D. J . LeBlanc, Inorg. Chem. 38,2248 (1999).
R. K. Harris. Nuclear Magnetic Resooaoce Spectroscopy . John Wiley and Sons, inc., New York. 1986, p. 10.
F. A. L. Anet and D. J. O'Leary, Concepts in Magn. Reson. 3, 193 (1991).
F. A. L. Anet and D. J. O'Leary, Concepts in Magn. Reson. 4, 35 (1992).
K. Schmidt-Rohr and H. W. Spiess. Multidimensional Solid State NMR and Polymers. Academic Press, London. 1994, p. 444.
J. Masoa, Solid Srare Nucl. Magn. Reson. 2,285 (19%).
(a) C . J. Jarneson, Solid Stae Nucl. Magn. R e m . 11,265 (1998). (b) R. K. Harris, Solid State Nucl. Magn. Reson. 10, 177 (1998).
R. E. Wasylishen. In Encyclopedia of Nuclear Magnetic Resoaance. Edited by D. M. Grant and R. K. Harris. Wiley hc., Chichester, UK. 1996, p. 1685.
C . I. lameson and J. Mason. In Multinuclear NMR. Edited by J . Mason. Plenum Press, New York. 1987, p. 3.

G. E. Pake, J. *m. Phys. 16,327 (1948).
C. J. Jarneson. In Multinuclear NMR. Edired by J . Mason. Plenum Ress, New York, 1987, p. 89.
A. D. Buckingham, P. Pyykko, J. B. Robert, and L. Wieseofeld, Mol. Phys. 46, 177 (1982).
W. T . Raynes, Magm Reson. Chem. 30,686 (1992).
A. D. Buckingham and 1. Love, J. Magn. Reson. 2, 338 (1970).
H. W. Spiess, hM.R Basic Princ. Prog. 15, 55 (1978).
D. L. Bryce and R. E. Wasylishen. J. Am. Chem. Soc. 122, 3197 (2000).
R. von Boeckh, G. Griff, and R. Ley, 2. Phys. 179,285 (1%4).
R. E. Wasylishen, K. C. Wright, K. Eichele, and T. S. Cameron, Inorg. Chem. 33,407 (1994).
M. D. Lumsden, R. E. Wasylishen. and J. F. Britten, J. Phys. Chem. 99, 166M (1995).
M. D. Lumsden, K. Eichele, R. E. Wasylishen, T. S. Cameron, and J. F. Britten, J. Am. Chem. Soc. 116, 11 129 (1994).
(a) W. P. Power, M. D. Lumsden, and R. E. Wasylishen, J. Am. Chem. Soc. 113, 8257 (1991). (b) W. P. Power and R. E. Wasylishen, Inorg. Chem. 31, 2176 (1992).
(a) D. L. VanderHart and H. S. Gutowsky, J. Chem. Phys. 49, 261 (1%8). (b) M. Linder, A. Hohener, and R. R. Ernst, J. Chem. Phys. 73,4959 (1980). (c) R. E. Wasylishen, R. D. Curtis, K. Eichele. M. D. Lumsden. G. H. Pemer, W. P. Power, and G. Wu. In Nuclear Magnetic Shieldings and Molecular Structure. Edited &y J . A. TosseIl. Kluwer Academic Publishers, Dordrecht. 1993, p. 297.
W. P. Power anci R. E. Wasylishen, Ann. Rep. NMR Spectrusc. 23, 1 (1991).
(a) K. W. Zilm and D. M. Grant, J. Am. Chem. Soc. 103. 2913 (198 1). @) G . Wu and R. E. Wasylishen, M d Sfate Nucl. Magn. Reson. 4-47 (1995).

H. van Wiiiigen, R. G. Griffin, and R. A. Haberkom, J. Chem. Pliys. 67,5855 (1977).
(a) R. D. Curt'i, J. W. Hiibom, G. Wu, M. D. Lumsden, R. E. Wasylishen, and l. A. Pincock, J. Phys. Chem. 97, 1856 (1993). (b) M. D. Lumsden, G. Wu, R. E. Wasylishen, and R. D. Curtis, J. Am. Chem. Soc. 115,2825 (1993).
K. Eichele a d R. E. Wasylishen, J. Mugn. Reson. A 106, 46 (1994).
K. Eichele and R. E. Wasylishen, WSOLIDS, 1994 and 2000.
B. Sun and R. G. Griffin, unpublished results.
M. Bak, J. T. Rasmussen, and N. C. Niclsen, J. Magn. Reson. 147. 2% (2000).
(a) A. Kubo and C. A. McDowell, J. Chem. Phys. 92, 7156 (1990). (b) E. Klaus a d A. Sebald, Angew. Chem. Iht. Ed. 34,667 (1995). (c) S. Dusold, E. Klaus, A. Sebald, M. Bak, and N. C. Nielsen, J. Am. Chem. Soc. 119,7121 (1997)
R. R. Ernst, G. Boâenhausen, and A. Wokaun. Principles of Nuclear Magnetic Resonance in One and Two Dimensions. Clarendon Press, Oxford. 1987.
A. Pines, M. G. Gibby, and J. S. Waugh, J. Chem. Phys. 59, 569 (1973).
S. R. Hartmann and E. L. Hahn, Phys. Rev. 128,2042 (1%2).
T. Nakai and C. A. McDowell, Chem. Phys. Leu. 217,234 (1994).
K. Eichele, R. E. Wasylishen, R. W. Schurko, N. Burford. and W. A. Whitla, Gm. J. Chem. 74, 2372 (1996).
S. Dusold, J. Kümmerlen, and A. Sebald. J. Phys. Chem. A 101,5895 (1997).
T. Nakai and C. A. McDowell, Solid State Nucl. Magn. Reson. 4, 163 (1995).
(a) N. F. Ramsey, Phys. Rev. 77,567 (1950). (b) N. F. Ramsey, Phys. Rev. 78, 699 (1950). (c) N. F. Ramsey, Phys. Rev. 83, 540 (1951). (d) N. F. Ramsey, Phys. Rev. 86, 243 (1952).
N. F . Ramsey, Phys. Rev. 91, 303 (1953).
N. F. Ramsey . Molecular Beams. Oxford University Press, London. 1956.
P. Pyykk6, nieor. Chem. Acc. 103,214 (2000).

J. H. Van Vleck. The Theory of Electric and Magnetic Susceptibilities. Oxford University Press, London. 1932.
C. J. Jameson and J. Mason. In Muttinuclear NMR. Edited liy J. Mason. Plenum Press, New York. 1987, p. 51.
D. B. Chesnut, Ann. Rep. NMR Spearosc. 29.71 (1994).
R. W. Schurko and R. E. Wasylishen, J. Phys. Chem. A 104, 3410 (2000).
D. L. Bryce and R. E. Wasylishen, J. Phys. Chem. A 104,7700 (2000).
U. Fleischer, C. van Wüllen, and W. Kutzelnigg . In Encyclopedia of Computational Chemistry. Edited by P. von R. Schleyer. John Wiley and Sons, Chichester. 1998, p. 1927.
W. Kutzelnigg, U. Fleischer , a d M. Schindler, NMR Basic Princ. Prog. 23, 163 (1990).
S. 1. Chan and T. P. Das, J. a e m . Phys. 37, 1527 (1962).
C. W. Kern and W. N. Lipscomb, J. Chem. Phys. 37, 260 (1%2).
(a) R. Ditchfield, Mol. Phys. 27, 789 (1974). (b) K. Wolinski, J. F. Hinton, and P. Pulay, J. Am. Chm. Soc. 112, 825 1 (1990). (c) G. Rauhut, S. Puyear, K. Wolinski, and P. Pulay, I. Phys. Chem. 100, 6310 (19%).
M. Schindler and W. Kutzelnigg, J. Chem. Phys. 76, 19 19 (1982).
A. E. Hansen and T. D. Bouman, J. Chem. Phys. 82, 5035 (1985).
T. A. Keith and R. F. W. Bader, Chem. Phys. Laa. 194, 1 (1992).
(a) J. Gauss, Chem. Phys. Lm. 191,614 (1992). @) J. Gauss, Chem. Phys. L m . 229, 198 (1994).
B. O. Roos, Adv. Chen. Phys. 69,399 (1987).
K. Ruud, T. Helgaker, R. Kobayashi, P. Jmgensen, K. L. Bak, and H. J. Aa. Jensen, J. Chem. Phys. 100,8178 (1994).
(a) M. Biihl, M. Kaupp, O. L. M a l b , and V. G. Malkin, J. Cornp. Chem. 20 91 (1999). (b) V. G. Maikin, O. L. MalLina, M. E. Casida, a d D. R. Salahub, J. Am. Chem. Soc. 116,5898 (1994). (c) V. G. Malkin, O. L. Maikina, and D. R. Salahub, Chem. Phys. Lm 204.87 (1993). (d) V. G. MalLin, O. L.

Mafkina, and D. R. Salahub, &m. Phys. Le#. 2-04. 80 (1993).
G. Scbreckenbach. R. M. Dichon, Y. Ruiz-Morales. anci T. Ziegler. In Chemical Applications of Density Funaional Theory . Edited by B. B. Laird, R. B. Ross, and T. Ziegler. American Chemical Society, Washington. 19%. p. 328.
(a) G. Schreckenbach, S. K. Wolff, a d T. Ziegler, J. Phys. Chem. A 184, 8244 (2W). (b) S. K. Wolff, T. Ziegler, E. van Lenthe, ami E. J. Baerends, J. Chem. Phys. 110,7689 (1999).
Gaussian 94, Revision 8.2. M. J. Frisch, G. W. Trucks, H. B. Schlegel, P. M. W. Gill, B. G. Johnson, M. A. Robb, J. R. Cheeseman, T. Keith, G. A. Petersson, J. A. Montgomery, K. Raghavachari, M. A. Al-Laham, V. G. Zaknewski, J. V. Ortiz, J. B. Foresman, J. Cioslowski, B. B. Stefanov, A. Nanayakkara, M. Challacombe. C. Y. Peng, P. Y. Ayala. W. Chen. M. W. Wong, J. L. Andres, E. S. Replogle, R. Gomperts, R. L. Martin, D. J. Fox. J. S. Binkley, D. J. Defrees, J. Baker. J. P. Stewart, M. Head-Gordon, C. Gonzalez, ancî J. A. Pople, Gaussian, hc., Pittsburgh, PA, 1995.
Gaussian 98, Revision A.4, M. J. Frisch, G. W. Trucks, H. B. Schiegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, J. A. Montgomery, Jr., R. E. Stramÿinn, J. C. Burant, S. Dapprich, J. M. Millam, A. D. Daniets, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi, R. Carnmi, B. Mennuai, C. Pomelli, C. Adamo, S. Clifford, I. Ochterski, G. A. Petersson, P. Y. Ayala, Q. Cui, K. Morokuma. D. K. Malick. A. D. Rabuck, K. Raghavachari, J. B. Foresmao. J. Cioslowski, J. V. Ortiz, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskon, 1. Komaromi, R. Gomperts, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, C. Gonzalez, M. Challacombe, P. Gill, B. Johiison, W. Chen, M. W. Wong, J. L. Andres, C. G o d e z , M. Head-Gordon, E. S. Replogle. aod J. A. Pople, Gaussian, ïnc., Pittsburgh PA, 1998.
E. Frisch and M. J. Frisch. Gaussian 98 User's Reference, 2"d Ed. Gaussian hc., Pittsburgh PA. 1999, p. 126.
C.J. Jameson. In Encyclopedia of Nuclear Magnetic Resonance. Edited by D. M. Grant anâ R. K. Harris. Wiley Inc. Chichester, UK. 19%, p. 1273.
A. K. Jarneson and C. J. Jameson, Chm. Phys. Len. 134, 461 (1987).
R. E. Wasylishen, S. Mooibrœk, aad J. B. Macdonald, J. Chem. Phys. 81, 1057 (1984).

C. J. Jameson, A. K. Jarneson, aod P. M. Bumll, J. Chem. Phys. 73,6013 (1980).
C. J. lamson, A. C. de Dios, and A. K. Jameson, Chem. Phys. Let?. 167,575 (1gm.
M. Gee, R. E. Wasylishen, and A. Laaksouen, J. Phys. Chem. A 103, 10805 (1999) =
J. E. Rich, M. N. Mando, and A. C. de Dios, J. Phys. Chem. A 104,5837 (20OQ
B. Celda, C. Biamonti, M. J. Arneau, R. Tejero, and G. T. Montelione, J. Biomol. NMR 5, 161 (1995).
D. D. Laws, A. C. de Dios, d E. Oldfield, J. Biomol. NMR 3,607 (1993).
(a) A. J. Dingley and S. Grzesiek, J. Am. C h . Soc. 120, 8293 (l998). (b) A. J. Dingley, J. E. Masse, R. D. Peterson, M. Barfield, J. Feigon, and S. Grzesiek, J. Am. Chem. Soc. 121, 6019 (1999). (c) F. Cordier and S. Grzesiek, J. Am. Chem. Soc. 121. 1601 (1999). (d) G. Cornilescu, J.-S. Hu, and A. Bax, J. Am. Chem. Soc. 121,2949 (1999). (f) F. Cordier, M. Rogowski, S. Grzesiek, and A. Bax, J. Magn. Reson. 1 4 , 510 (1999).
C. Scheurer and R. Briischweiier, J. Am. Chem Soc. 121, 8661 (1999).
D. L. Bryce and R. E. Wasylishen, J. Biomol. NMX 19, 371 (2001).
H. Beuedict, 1. G. Shenderovich, O. L. Malkina, V. G. Malkin, G. S. Denisov, N. S. Golubev, and H.-H. Limbach, J. Am. Chem. Soc. 122. 1979 (2000).
H. Fukui, Prog. Nucl. Magn. Reson. 35,267 (1999).
N . F. Ramsey . In -Experimental Nuclear Physics . Edited by E . Segr&. John Wiley and Sons, New York. 1953, vol. 1, part III, p. 358.
J. Kowalewski and A. laaksouen. In Theoretical Models of Chernical Bonding. Edited by 2. B. Maksic. Springer, Berlin. 1991, part 3, p. 387.
P. PyykE, Chem. Pliys. 22,289 (1977).
V. Sychrovsw, J. Grâfenstein, and D. Cremer, J. Chem. Phys. 113, 3530

1. Carmichael, J. Phys. Chen. 97, 1789 (1993).
O. L. Malkina, D. R. Salahub, and V. G. MalLin, J. Chem. Phys. 105, 8793 (19%)
V. G. MalLin, O. L. Malkioa, and D. R. Salahub, Chem. Phys. Lan. 221.91 (1994) -
M. F. Guest, V. R. Saunders, and R. E. Overill, Mol. Phys. 35, 427 (1978).
O. Vahtras, H. Agren, P. Jergensen, J. J. Aa. Jensen, S. B. Padkjzr, and T. Helgaker, J. Chem. Phys. 96, 6120 (1992).
(a) A. Barszczewicz, T. Helgaker, M. Jasnidski, P. Jmgensen, and K. Ruud, J. Magn. R e m . A 114, 212 (1995). (b) A. Barszczewicz, T. Helgaker, M. JaszuBski, P. Jergensen, and K. Ruud, J. Chem. Phys. 101, 6822 (1994). (c) J. Kaski, P. Lantto, J. Vaara, and I. Jokisaari, J. Am. Chem. Soc. 120, 3993 (1998). (d) J. Kaski, P. Lantto, T. T. Raniala, 1. Schroderus, J. Vaara, and J. Jokisaari, J. Phys. mem. A 103, %69 (1999).
D. L. Bryce and R. E. Wasyiishen, J. Am. Chem. Soc. 122, 11236 (2000).
J. F. Stanton and R. J. Bartien, J. Chem. Phys. 98,7029 (1993).
S. A. Perera, H. Sekino, and R. J. Bartlett, I. Chem. Pliys. 101, 2186 (1994).
S. A. Perera, R. J. Bartien, and P. von R. Schleyer, J. Am. Chem. Soc. 117, 8476 (1995).
J. Oddershede, P. Jergensen, and D. L. Yeager, Cornput. Phys. Rep. 33,2 (1984).
J. Oddershede. In Methods in Computatioiial Molecuiar Physics. Edited &y S. Wilson and G. H. F. Diercksen. Plenum Press, New York, 1992, p. 303.
T. Enevoldsen, J. Oddershede, and S. P. A. Sauer, Theor. Chem. Acc. 100,275 ( 19%).
S. A. Perera and R. J. Bartien, J. Am. a e m . Soc. 122, 1231 (2000).
H. Fukui, J. Chem. Phys. 65, 844 (1976).
T. Enevoldsen, L. Vischer, T. Saue, H. J. Aa. Jensen, and J. Oddershede. J. Chem. Phvs. 112.3493 (2000).

3. Khandogin and T. Ziegler, J. Phys. Qirm. A 104, 1 13 (2ûûû).
J. Autschbach and T. Ziegler, J. Am. C'hm. Soc., 123, 3341 (2001).
J. Autschbach and T. Ziegler, J. Chem. Phys. l U , 936 ( 2 0 ) .
J. Autschbach and T. Ziegler, J. Chen. PM. 113, 9410 (2000).
T. Helgaker, M. Jasnihski, K. Ruud, aiid A. Gorska, nieor. Chem. Acc. 99, 175 (1998).
J. Vaara, J. Kaski, and J. Jokisaari, J. Phys. C h . A 103, 5675 (1999).
A. Viste, M. Hotokka, L. LaaiCsonen, and P. Pyykk6, Chem. Phys. 72,225 (1982).
(a) K. Krüger, G. Grossmann, U . Fleischer, R. Franke, and W. Kutzelnigg , Mogn. Reson. Chem. 32, 5% (1994). (b) G. Ohms, U. Fleischer, and V. Kaiser, J. G e m . Soc., Dalron Tram. 1297 (1995).
A.-R. G r b e r , R. Peter, and E. Fechner, 2. Chem. 18, 109 (1978).
(a) R. J. Iuliucci, C. G. Phung, J. C. Facelli, and D. M. Grant, J. Am. Chem. Soc. 118. 4880 (1996). (ô) R. J. Iuliucci, C. G. Phung, J. C. Facelli, and D. M. Grant, J. Am. Chem. Soc. 12û, 9305 (1998). (c) R. W. Schurko, R. E. Wasylishen, and H. Foerster, J. Phys. Qlom. A 102,9750 (1998). (d) D. L. Bryce and R. E. Wasylishen, J. Phys. Chem. A 103,7364 (1999).
(a) T. Vosegaard, P. Daugaard, E. Haid, and H. J. Jakobsen, J. MW. Reson. 142, 379 ( 2 0 ) . (b) T. Vosegaard, J. Skibsted, and H. J. Jakobsen, J. Phys. Chem. A 103.9144 (1999). (c) T. Vosegaard, E. Hald, V. Langer, H. J. Skov, P. Daugaard, H. Bildsae, and H. I. Jakobsen, J. Magn. Reson. 135, 126 (1998).
(a) SADABS. G. M. Sheldrick, and Bruker AXS, Inc., Madison, Wisconsin, 53719 USA. (ô) R. H. Blessing, Acta Crysrallogr. A51, 33 (1995).
S. N. Dutta, and M. M . Woolfson, Acta. Cpal logr . 14, 178 (1%1).
SHELXTL (5.10) program library (G. Sheldrick, Siemens XRD, Madison Wisconsin).
J. S. Rollen, Computing Methods in Crystallography . Pergamon Press, New York. 1965.

E. Prince. Mathmaticai Techniques in Crystailography and Material Science. Springer-Ver lag , Berlin. 1994.
(a) M. A. Kemedy and P. D. Ellis, Concepts in Magn. Reson. 1, 35 (1989). (b) M. A. Kennedy and P. D. Ellis, Concepts in Mogn. Reson. 1, 109 (1989). (c) K. Eichele, J. C. C. Chan, R. E. Wasylishen, aad J. F. Britten, J. Phys. Chem. A 101,5423 (1997).
D. W. Aldemian, M. S. Solum, and D. M. Grant, J. Chem. Phys. 84,3717 (1986).
M. Rance and R. A. Byrd, J. Magn. Reson. 52, 221 (1983).
SpinEcho, K. Eichele and R. E. Wasylishen, unpublished.
A. D. Becke. J. Chem. Phys. 98, 5648 (1993).
C. Lee, W. Yang, anâ R. G. Parr, Phys. Rev. B. 37,785 (1988).
R. K. Harris and R. G. Hayter, Can. I. C h . 42,2282 (1964).
(a) J. M. Mfllar, A. M. T'hayer, D. B. Zax. and A. P k s , J. Am. Chem. Soc. 108, 5113 (1986). (b) T. Nakai, J. Ashida, and T. Terao, Mol. Phys. 67, 839 (1989).
J. D. Lee and G. W. Goodacre, Acra. CrystaZlogr. B27, 302 (1971).
M. G. Munowitz and R. G. Griffin, J. Chenz. Phys. 76,2848 (1982).
Y. Ishii, T. Terao, and S. Hayashi, I. Chem. Phys. 107, 2760 (1997).
J. Henfeld and A. E. Berger, J. Chem. Phys. 73,6021 (1980).
R. E. Wasylishen, W. P. Power, G. H. P e ~ e r , and R. D. Curtis, Can. 3. Chem. 67, 1219 (1989).
G. Heckmann and E. Fluck, Mol. Phys. 23, 175 (1972).
C. 1. Jarneson, Buü. Magn. Reson. 3, 3 (1980).
R. Sillanpaa. O. Ergin, and S. Çelebi, Am C'alfogr. C49, 767 (1993).
F. Mathey, F. Mercier, F. Nief, J. Fischer, aod A. Mitschier, J. Am. Ckm. Soc. 104,2077 (1982).

143
G. Wu and R. E. Wasylishen, J. Chem. Phys. 99, 6321 (1993).
G. Wu. B. Sun, R. E. Wasylishen, and R. G. Gnnin, J. Magn. Reson. 124,366 ( 1997)
teXsan for Windows version 1-05: Crystai Structure Analysis Package, Molecular Structure Corporation (1997-8).
(a) D. B. Chesnut and K. D. Moore, J. C o q . Chem. 10,648 (1989). (b) D. B. Chesnut, B. E. Rusiloski, K. D. Moore, and D. A. Egolf, J. C o q . Chem. 14, 1364 (1993).
S. Dumgthai and G. A. Webb, Orgarùc Magn. Reson. 21, 199 (1983).
D. B. Chesnut and L. D. Quin, J. Am. Chem. Soc. 116, %38 (1994).
D. B. Chesnut and E. F. C. Byrd, H e t e r ~ m Chem. 7, 307 (19%).
G. M. Bernard, G. Wu, and R. E. Wasylishen, J. Phys. Chem. A 102, 3 184 (1998).
(a) F. Mathey, Chem. Rev. 88,429 (1988). (b) F. Mathey, Coordination Chem. Rev. 137, 1 (1994).
(a) L. Nyulhzi, 3. Phys. Chem. 100, 6194 (1996). (b) A. Dransfeld, L. Nyuihzi, anâ P. v. R. Schleyer, Iiwrg. Chem. 37,4413 (1998). (c) F. G. N. Cloke, P. B. Hitchcock, P. Humble, I. F. Nixon, L. Nyulhszi, E. Niecke, and V. Thelen, Angew. Chem. IM. Ed. 37, 1083 (1998).
L. Nyulâszi, Chem. Rev., in press, 2001.
K. Eichele, R. E. Wasylishen, J. M. Kessler, L. Solujic, and J. H. Nelson, Inorg. Chem. 35, 3904 (19%).
R. D. Curtis, B. W. Royan, R. E. Wasylishen, M. D. Lumsden, and N. Bdord, Inorg. &m. 31,3386 (1992).
J. B. Gmtzner . In Recent Advances in Organic NMR Spectroscopy . Edited ûy 3. B. Lambert a d R. Rittrier. Noreii Press, Landisville, NJ. 1987, chapter 2, p. 17.
G. H. Penner and R. E. Wasylishen, Clm. J. Chem. 67, 1909 (1989).
G. Wu, R. E. Wasylishen, W. P. Power, anâ G. Baccolini, Cnn. J. Chem. 70,

N. Burford, E. Ocando-Mavara, unpubüshed.
W. Haubenreisser, U. Sternberg, and A.R. Grimmer, Mol. Phys. 9, 15 1 (1987).
"DALTON, an ob initio electronic structure program", written by T. Helgaker, H.J. À. Jensen, P. Jargemea, J. Oka, K. Ruud, H. Agren, T. Andersen, K.L. Bak, V. Bakken, O. Christiansen, P. Dahle, E.K. Dalskov, T. Euevoldsen, B. Fernandez, H. Heiberg, H. Hettema, D. Jonsson, S. Kirpekar, R. Kobayashi, H. Koch, K.V. Mikkelseo, P. N o m , M.J. Packer, T. Saue, P.R. Taylor, and O. Vahtras; Release 1 .O (1997).
(a) H. J. Aa. Jensen, P. Jergensen, H. Àgren, and J. Olsen, J . Chem. Phys. BS, 3834 (1988). (b) J. Guilleme and J. S. Fabian, J. Chem. Phys. 109, 8168 (1998).
T. H. Dunning, Jr., J. Chem. Phys. 90, 1007 (1989).
J. R. Durig, L. A. Carreira, and J. D. Odorn, J. Am. Chem. Soc. 96,2688 (1974).
(a) E. R. Aadrew, A. Bradbury, R. G. Eades, and V. T. Wym, Phys. Lm. 4, 99 (1963). (b) D. P. Raleigh, M. H. Levitt, and R. G. Griffin, Chem. Phys. Len. 146, 71 (1988). (c) D. L. Bryce and R. E. Wasylishen. In Encyclopedia of Spectroscopy and Spectrometry. Edited by J. C. Lindon, G. E. Tranter, and J. L. Holmes. Academic Press, San Diego. 2000, p. 2 136.
(a) G. A. Olah and C. W. McFarland, J. Org. Cliem. 34, 1832 (1%9). (b) B. E. Mann, J. Chem. Soc., Perkin Trans. 2.30 (1972). (c) S. O. Grim, E. F. Davidoff, and T. J. Marks. 2. Nmuforsch. 26b, 184 (1971). (d) G. P. Schiemenz, Phosphorus 3, 125 (1973).
R. W. Rudolph and R. A. Newmark, J. Am. Chem. Soc. 92, 1 195 (1970).
H. C. E. McFarlane and W. McFarlane, J. Chem. Soc., Chem. Commun. 1589 (1971).
W. McFarIane, N. H. Rees, L. Constanza, M. Patel, anci 1. J. Colquhoun, J. Chem. Soc., Dalton Tram. 4453 (2000).
C. I. Jarneson. In Phosphorus-31 NMR Spectroscopy in Stereochemical Analysis. Edired by J. G. Verkaâe and L. D. Quin. VCH Publishers, Inc., Deerfield Beach. Fla. 1987. D. 205.

U. C. Singh, P. K. Basu, and C. N. R. Rao, J. Mol. Stnîct. 87, 125 (1982).
R. M. Lyaden-Bell, J. Chem. Six., Farad@ Tron. 57,888 (1961).
J. P. Albrand, H. Faucher, D. Gagnaire, and J. B. Robert, Chem. Phys. Lm. 38, 521 (1976).
V. Galasso, J. C h m . Phys. 80,365 (1984).
D. Chakraborty and P. Chandra, J. Mol. Struct. 434,75 (1998).
J. M. Schulman, J. Ruggio, and T. J. Venanzi, J. Am. Chem. Soc. 99,2045 (1977).
V. M. S. Gil and W. von Philipsbom, Magn. Reson. Chem. 27,409 (1989).
S. Berger and J. D. Roberts, J. Am. Chem. Soc. 96, 6757 (1974).
W. McFarlane, Proc. R. Soc. Lordon, Ser. A. 306, 185 (1968).
H. C. E. McFarlane and W. McFarlane, J. Chem. Soc., Chem. Commun. 582 (1975).
(a) D. L. VanderHart, H. S. Gutowsky, and T. C. Farrar, J. Am. Chem. Soc. %9, 5056 (1%7). (b) H. W. Spiess, U. Haeberlen, and H. Zimmermann, J. Magn. Reson. 25, 55 (1%7). (c) M. M. Maricq and J. S. Waugh, J. Chem. Phys. 70, 3300 (1979). (d) E. Menger and W. S. Veeman, I. Magn. Reson. 46,257 (1982). (e) J. G. Hexem, M. H. Frey, anci S. J. Opella, J. Chem. Phys. 77, 3847 (1982). (f) J. Bohm, D. Fenzke, and H. Pfeifer, J. Magn. Reson. 55, 197 (1983). (g) D. L. Sastry, A. Naito, and C. A. McDowell, Chem. Phys. Lm. 146, 422 (1988). (h) A. C. Olivieri and R. K. Harris, Prog. Nucl. Magn. Reson. Spectrosc. 24,435 ( l m ) .
N. Zumbulyadis, P. M. Henrichs, and R. H. Young, J. Chem. Phys. 75, 1603 (198 1).
K. Eichele, R. E. Wasylishen, J. S. Grossert, and A. C. Oüvieri, J. Phys. Chem. 99, 101 10 (1995).
C. W. Schultz and R. W. Rudolph, J. Am. Chem. Soc. 93, 1898 (1971).
H. Falius and M. Murray, J. Magn. Reson. 10, 127 (1973).

F. Breitsameter, K. Polborn, and A. Schmidpeter, Eur. J. Inorg. Chem. 1907 (1998).
V. Galasso, (Tiem. Phys. 83,407 (1984).
A. H. Cowley and N. C. Norman, Prog. Inorg. Chem. 34, 1 (1986).
D. G. Gilheany . In The Chemistry of Organophosphonis Cornpounds. Edired by F. R. Hartley. John Wiley and Sons, Chichester. 1990, p. 43.
J. Demaison. F. Hegeluad, and H. Burger, J. Mol. Strun. 413,447 (1997).
M. Witanowski, L. Stefaniak, and G. A. Webb, Ann. Rep. W R Spectrosc. l lb , 1 (1981).
T. Khin and G. A. Webb, J. Mugn. Reson. 33, 159 (1979).
M . Witanowski, L. Stefaniak, and G. A. Webb, Am. Rep. MMR Spectrosc. l lb , 140 (1981).
H .-P. Schrikiel and A. Schmidpeter, Phosphorus, Suiphur, und Silicon 129, 69 ( l m *
P.-A. Malmqvist, A. Rendell. and B. O. Roos, J. Phys. Chem. 94,5477 (1990).
(a) Y. Ishii and R. Tycko, J. Am. Chem. Soc. 122, 1443 (2000). (b) F. M. Marassi, A. Ramarnoorthy, and S. J. Opella, Nd. Aca. Sci. U.S.A. 94, 8551 (1997). (c) C. H. Wu, A. Ramamoorthy, and S. J. Opella, J. Magn. Reson. A, 109, 270 (1994). (d) A. Ramamaorthy, C. H. Wu, and S. J. Opella, J. Magn. Reson. B , 107, 88 (1995).
G. Wu and R. E. Wasylishen, Mol. Phys. 83, 539 (1994).
R. Challoner and C. A. McDowell, J. Magn. Reson. 98, 123 (1992).
H. Bai and R. K. Harris, J. Magn. Reson. 96.24 (1992).
S. Dusold and A. Sebald, Mol. Phys. 95, 1237 (1998).
M. Bak and N. C. Nielsen, J. Chern. Phys. 106,7587 (1997).
A. Naito, S. Ganapathy, K. Akasaka, and C. A. McDowell, I. Chem. Phys. 74, 3190 (1981).
S. Dusold. H. Maisel. and A. Sebald, J. Mam. Reson. 141, 78 (1999).

214. H. Luo, R. McDonald, and R. G. Cavell, Angew. Chem. Int. Ed. 37, 1098 ( 1998).
215. Spinsight 4.1, Varian Associates, Inc., 1998.
216. SPECMAKE, a program by K. Eichele, 1998.
217. P. J. Giarnmatteo, W. W. H e h t h , F. G. Ticehurst, and P. W. Cope, J. Magn. Reson. 71, 147 (1987).