photochemical synthesis of 1-aminonorbornanes via strain ...€¦ · this report describes a new...
TRANSCRIPT

1
Photochemical Synthesis of 1-Aminonorbornanes via Strain-Driven, Formal [3+2] Cycloadditions
Daryl Staveness; Taylor M. Sodano; Corey R.J. Stephenson*
University of Michigan, Department of Chemistry, Ann Arbor, MI 48109, USA.
Abstract
This report describes a new route toward 1-aminonorbornanes via strain-driven, visible light-mediated formal [3+2] cycloadditions
with aminocyclopropanes. Lewis acidic salts (LiBF4, ZnCl2) were found to facilitate the oxidation of a variety of amine-containing
heterocycles, and consequently, these additives led to improved conversion and isolated yields. This operationally-simple method
tolerates a variety of functional handles (e.g. alcohols, protected amines), can generate optically-pure products through
diastereoselective variations, and affords entry to unique chemical space through the diversity of accessible substitution patterns.
Providing flexible access to 1-aminonorbornanes is expected to benefit modern drug design and development efforts seeking to
incorporate more sp3-rich motifs into lead scaffods. Further, gram-scale operation proceeds smoothly in continuous flow, suggesting
that this chemistry can be readily translated beyond the academic- or discovery-scales.
Introduction Development of this new, flexible route toward 1-aminonorbornanes, saturated building blocks favorably aligned with the now
popular Fsp3 metric,1 arose from a desire to leverage a fundamental property of amine oxidation chemistry: the dramatic weakening
of -C-C bonds upon generating amine radical cations. Extensive work by Whitten,2 Mariano,3 and others defined many fundamental
aspects and even the synthetic utility4 of these reactive intermediates, but these efforts were all but exclusively focused on heterolysis
of -C-R bonds (see Figure 1a). Even modern implementation of amine radical cations5 (largely driven by the revival and subsequent
exponential growth of photoredox catalysis6) has relied heavily on heterolytic decomposition pathways, primarily leveraging the
analogously weakened (and more acidic) -C-H bonds to generate -amino radicals, a potential precursor to iminium species.6,7
Homolytic decomposition pathways offer an intriguing counterpart, however, as -C-C bond homolysis generates reactive
intermediates (namely, an iminium and a carbon-centered radical) that are both of value and are fundamentally distinct from the -
amino radicals available through heterolysis.8 Interestingly, the relative lack of implementation of this method belies the simplicity
through which it can be achieved; our recent catharanthine fragmentation efforts9 demonstrate that as little as 4.2 kcal/mol of strain
energy in the natural product are enough to drive -scission upon oxidation to the amine radical cation.
Aminocyclopropanes (aminoCPs) clearly offer more strain energy,10 but despite the analogy to the well-studied cyclopropylcarbinyl
radical,11 only a handful of reports have employed oxidized aminoCPs. The -iminium radical intermediate that arises via homolytic
decomposition (see Figure 1b) has been shown to be a competent coupling partner with olefins12 or alkynes12d,13 (toward cyclopentane
or cyclopentene derivatives), with molecular oxygen (toward endoperoxides14 or ketone/amide derivatives15,16), and with
dicyanoanthracene.17 Unfortunately, most of these reports rely on harsh oxidants in (super)stoichiometric amounts. Zheng and co-
workers offer a departure from this limitation, recently demonstrating that anilinocyclopropanes12c,12d,13 are viable substrates within
photoredox catalysis. Our discovery that Lewis acidic salts decrease the oxidation potential of a number of amine heterocycles (vide
infra) provided an opportunity to greatly expand the diversity of amine functionality that can reliably participate within visible light
photocatalysis, including aminoCP derivatives. Further, it was envisioned that an aminoCP with an appropriately-tethered olefin
would afford bridged bicyclic structures, such as 1-aminoNBs, through homolytic C-C bond fragmentation of the oxidized aminoCP
and serial radical cyclizations (see Figure 1c). As 1-aminoNBs are well-suited to modern, sp3-centric drug design principles,1 these
motifs seemed to be ideal targets through which to demonstrate the broader utility of our strain-driven, homolytic C-C bond
fragmentation efforts.

2
Figure 1. a) Summation of previously studied pathways for heterolytic decomposition of amine radical cations. b) Accessing
homolytic decomposition pathways. c) Generic representation of visible light-mediated approach to 1-aminoNBs as driven by
homolytic fragmentation of oxidized aminocyclopropanes.
Interestingly, while 1-aminoNBs are uncommon in modern times,18 these structures played a critical role in the history of organic
chemistry, mostly appearing in classical reports19 seeking to define the basic principles of SN1 and SN2 reactivity.20 This includes
the landmark manuscript from Bartlett and Knox19b in which bridgehead-substituted 1-apocamphanes (7,7-dimethylnorbornanes)
were used as negative controls for SN2 reactivity, thereby providing proof for the Walden inversion mechanism.21 Perhaps
unsurprisingly, these classic approaches generally employed commercially-available terpenes as starting materials (e.g. ketopinic
acid, camphor), thereby limiting the diversity of accessible substitution patterns; de novo approaches have proven similarly
inflexible. This supply issue has largely precluded biological evaluation of 1-aminoNB-based scaffolds,22 yet these substructures
aptly address the need for new, saturated building blocks in drug discovery and can also be envisioned to serve as aniline
bioisosteres,23 complementing recent work with amine-substituted [1.1.1]bicyclopentanes24 and cubanes.25 The work detailed herein
seeks to enable the integration of 1-aminoNBs into the medicinal chemistry toolbox by providing more efficient access via
environmentally-benign methods. The following describes the optimization, scope, scalability, and performance in continuous flow
of this formal [3+2] cycloaddition toward 1-aminoNBs.
Initial optimization was performed with 7,7-dimethyl aminoCP 1a. N,N-Dialkylamine-based aminoCPs were targeted due to their
ease of access via Kulinkovich cyclopropanation26 of the corresponding amides (see Supporting Information). Heteroleptic Ir(III)
photocatalyst [Ir(dF[CF3]ppy)2(dtbbpy)](PF6)27 (3) in degassed acetonitrile was identified as the best starting point, affording 63%
of 1-aminoNB 2a after 12 hrs of irradiation (see Figure 2a, entry 1). It was found that simple Lewis acidic salts improved production
even at sub-stoichiometric loadings (20 mol%). LiBF4 and ZnCl2 proved to be the most effective, yielding 79% and 84% of 1-
aminoNB 2a, respectively (entries 2-3). The addition of ZnCl2 even enabled partial conversion to product when employing the less
oxidizing photocatalyst Ru(bpy)3Cl2∙6H2O (entries 10-11; for 4: Ru(II)*/Ru(I) Ered = +0.77 V vs. SCE6a; for 3: Ir(III)*/Ir(II) Ered =
+0.89 V vs. SCE27). Other Lewis acids provided little enhancement (NaBF4; entry 4) or facilitated degradation pathways (Me2AlCl,
BF3∙OEt2, MgBr2∙OEt2; entries 5-7), while redox non-innocent metal salts inhibited reactivity (CuBr2, NiCl2; entries 8 and 9); of
note, ZnCl2 is not necessarily redox-innocent under these conditions given the efforts of Bernhard and co-workers,28 though only the
most inefficient substrates afforded a grey precipitate that could indicate reduction to Zn(0) (see Supporting Information). Of note,
the reaction did not proceed to any extent in the absence of photocatalyst, without light, or under purely thermal conditions (entries
12-14)
Importantly, this formal [3+2] cycloaddition methodology was readily translated to continuous flow processing (compare entry 2
and entry 15; 79% and 78%, respectively), including efforts on gram-scale (entry 16). ZnCl2 results were similar (81% in flow), but
the modest heterogeneity of these reaction mixtures brought concerns of clogging. Importantly, demonstrating viability in continuous
flow aligns with the steadily increasing industrial investment in flow photochemistry.6c

3
Figure 2. Optimization of the formal [3+2] cycloaddition methodology; [a]Recov. SM = recovered starting material, approximate
values indicate estimated values from impure recovered material, [b]Reaction run in absence of light, [c]Reaction run in absence of
light at 80 °C, [d]Reaction run in continuous flow (detailed descriptions of flow and batch setups are provided in the Supporting
Information), [e]Yield obtained from processing 1.0 g of aminoCP 1a.
Differential pulse voltammetry (DPV) data suggests that the Lewis acidic salts are eliciting an effect on reaction outcome through
modulation of amine oxidation potential. Morpholines are unusually difficult to oxidize relative to other N,N-dialkylamines (for N-
methylmorpholine (NMM): Eox = +1.28 V vs. SCE, for N-methylpiperidine (NMP): Eox = +1.06 V vs. SCE; see Figure 3a), thus
prior morpholine oxidation efforts in the field have necessitated the use of an Ir(ppy)3/O2-based oxidative quenching cycle29
(operating via the Ir(IV) species) or the development of new Ir(III) photocatalysts with excited state oxidizing power that vastly
exceeds that of photocatalyst 3.30 Alternatively, this work demonstrates that morpholine oxidation is enabled by the addition of
readily-available Lewis acidic salts (for NMM: Eox = -170 mV; of note, ZnCl2 generally produced moderately heterogeneous
mixtures, thus LiBF4 was employed for all mechanistic studies). The effect on aminoCP 1a was less pronounced (Eox = -40 mV),
but this seemingly small change could still prove impactful given that the Nernst equation defines 59 mV shifts as equivalent to a
full order of magnitude adjustment in equilibrium processes at room temperature. Of note, the discrepancy in oxidation potential
between aminoCP 1a and NMM (260 mV lower) can be rationalized by the facile post-oxidation fragmentation pathway, as single-
electron transfer processes with amines are highly influenced by post-oxidation reactivity.3 The impact of the Lewis acidic salts was
not limited to morpholines, facilitating conversion to product for a variety of nitrogen heterocycles (i.e. piperidine, piperazine,
azetidine, pyrrolidine (1-aminoNBs 2d-2g); see Figure 3b; see Supporting Information for additional DPV data).
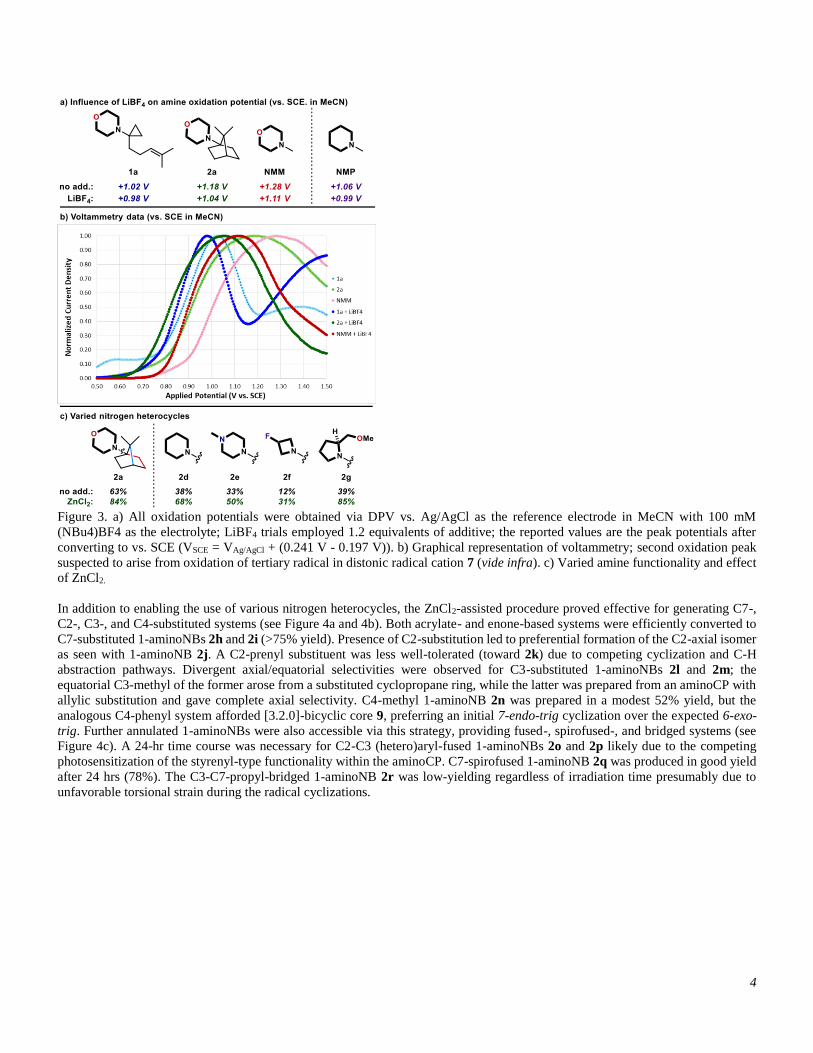
4
Figure 3. a) All oxidation potentials were obtained via DPV vs. Ag/AgCl as the reference electrode in MeCN with 100 mM
(NBu4)BF4 as the electrolyte; LiBF4 trials employed 1.2 equivalents of additive; the reported values are the peak potentials after
converting to vs. SCE (VSCE = VAg/AgCl + (0.241 V - 0.197 V)). b) Graphical representation of voltammetry; second oxidation peak
suspected to arise from oxidation of tertiary radical in distonic radical cation 7 (vide infra). c) Varied amine functionality and effect
of ZnCl2.
In addition to enabling the use of various nitrogen heterocycles, the ZnCl2-assisted procedure proved effective for generating C7-,
C2-, C3-, and C4-substituted systems (see Figure 4a and 4b). Both acrylate- and enone-based systems were efficiently converted to
C7-substituted 1-aminoNBs 2h and 2i (>75% yield). Presence of C2-substitution led to preferential formation of the C2-axial isomer
as seen with 1-aminoNB 2j. A C2-prenyl substituent was less well-tolerated (toward 2k) due to competing cyclization and C-H
abstraction pathways. Divergent axial/equatorial selectivities were observed for C3-substituted 1-aminoNBs 2l and 2m; the
equatorial C3-methyl of the former arose from a substituted cyclopropane ring, while the latter was prepared from an aminoCP with
allylic substitution and gave complete axial selectivity. C4-methyl 1-aminoNB 2n was prepared in a modest 52% yield, but the
analogous C4-phenyl system afforded [3.2.0]-bicyclic core 9, preferring an initial 7-endo-trig cyclization over the expected 6-exo-
trig. Further annulated 1-aminoNBs were also accessible via this strategy, providing fused-, spirofused-, and bridged systems (see
Figure 4c). A 24-hr time course was necessary for C2-C3 (hetero)aryl-fused 1-aminoNBs 2o and 2p likely due to the competing
photosensitization of the styrenyl-type functionality within the aminoCP. C7-spirofused 1-aminoNB 2q was produced in good yield
after 24 hrs (78%). The C3-C7-propyl-bridged 1-aminoNB 2r was low-yielding regardless of irradiation time presumably due to
unfavorable torsional strain during the radical cyclizations.

5
Figure 4. a) Generic depiction of aminoCPs converted to 1-aminoNBs. b) Evaluation of scope; [a]84% yield obtained with the trans-
C3-Me aminoCP, the cis-C3-Me aminoCP yielded 81% 1-aminoNB 2l also in 2.3:1 dr, [b]78% corresponds to a 3.3:1 mix of the
3R,4R and 3S,4S isomers (both axial) from a 4:1 mix of the anti:syn aminoCPs. c) Production of bridged- and fused-1-aminoNBs; [c]Reagents and conditions: 3 (2 mol%), ZnCl2 (20 mol%), MeCN, rt, 24 hrs, blue LEDs; [d]3 (5 mol%), MeCN, rt, 72 hrs, blue LEDs.
Importantly, optically-pure 1-aminoNBs are also readily-accessible through this formal [3+2] methodology. Controlling the absolute
configuration at C7 was achieved via aminoCPs of form 10 (see Figure 5a), in which the diastereoselectivity (7S-epimer 11 vs. 7R-
epimer 12) positively correlated to the accessibility of the C8-oxygen lone pairs (-OH > -OBn > -OAc > -OTBS). It is speculated
that the oxygenation can engage in an electrostatic interaction with the iminium early in the 5-exo-trig transition state when
progressing toward the 7S,8R isomer (11), and this donation lowers this barrier relative to the 5-exo-trig toward the 7R,8R isomer
(12). To our knowledge, no prior reports detail this means of diastereoselectivity in which -hydroxy functionality dictates the
relative configuration obtained in radical additions to carbonyl equivalents. Additionally, C3--aminoethyl aminoCPs (13; Figure
5b) cleanly afford a single isomer (reinforcing the C3-axial selectivity alluded to above), necessarily providing control over the
configuration of both C1 and C4. Significantly, these two series of aminoCPs also serve to demonstrate the functional group tolerance
of this strategy, proceeding in good to high yields while incorporating alcohol, ether, acyloxy, amide, and carbamate functionality.

6
Figure 5. a) Controlling absolute configuration at C7 via lactate-derived aminoCPs 10; [a]Reagents and conditions: 3 (2 mol%), ZnCl2
(20 mol%) [unless otherwise noted], MeCN, rt, 12 hrs, blue LEDs; [b]n.d. = no detectable trace of corresponding isomer (TLC,
GCMS, 1H NMR). b) Controlling absolute configuration at C1 and C4 via from lactate-derived aminoCPs 13.
Mechanistically, these intramolecular formal [3+2] cycloadditions are suspected to initiate via reductive quenching of the excited
state photocatalyst with the aminoCP substrate (see Figure 6), followed by homolytic decomposition of the amine radical cation (5)
to the -iminium radical (6). 6-Exo-trig radical cyclization would generate distonic radical cation (7), requiring 5-exo-trig cyclization
to forge the norbornane core18a and reduction of radical cation 8 to provide the product. Reduction via a radical chain mechanism
(path b) is favored given the kinetic challenges of the closed catalytic cycle (path a) and the DPV data (vide supra) indicating an
energetically-favorable electron transfer between radical cation 8 and aminoCP 1. Additionally, Chen, Zheng, and co-workers have
recently provided evidence for propagative pathways for anilinocyclopropane-based formal [3+2] cycloadditions.31 For the system
presented herein, quantum yield measurements32 do not directly indicate a highly propagative system ( = 0.34 for aminoCP 1a to
1-aminoNB 2a), and the high quenching efficiency and presence of facile post-oxidation fragmentation (thus minimizing back-
electron transfer) eliminate two common explanations for artificially low quantum yields.33 Instead, it is suspected that the 5-exo-
trig cyclization is sufficiently slow relative to the electron transfer processes that it renders the measured quantum yield <1 due to
photons lost during the lifetime of distonic radical cation 7. Defining the role of these kinetic idiosyncrasies could have ramifications
for a variety of photochemical transformations beyond this methodology.
Figure 6. Mechanism. [a]See Supporting Information for further details on all photophysical data.
This report details the most efficient and flexible access to 1-aminoNBs to date in an effort to provide the medicinal chemistry
community with valuable new saturated building blocks via environmentally-benign methods. This visible light-mediated formal
[3+2] cycloaddition tolerates a wide variety of substitution patterns and functional handles for further diversification, and generates

7
optically-pure 1-aminoNBs via diastereoselective variations. Operation in continuous flow, including gram-scale preparations,
suggests that this chemistry can translate beyond discovery scale. Future investigations will seek to demonstrate the value of these
substructures for generating bioactive leads.
AUTHOR INFORMATION
Corresponding Author
Notes
The authors declare no competing financial interests.
ACKNOWLEDGMENT
The authors acknowledge the financial support for this research from the NIH NIGMS (R01-GM096129), NSF (CHE-1565782), the
Camille Dreyfus Teacher-Scholar Award Program, and the University of Michigan. DS was supported by a Postdoctoral Fellowship,
PF-16-236-01 - CDD, from the American Cancer Society.
REFERENCES
(1) a) Lovering, F.; Bikker, J.; Humblet, C. J. Med. Chem. 2009, 52, 6752-6756; b) Lovering, F. Med. Chem. Commun. 2013, 4, 515-
519; c) Meanwell, N. Chem. Res. Toxicol. 2016, 29, 564-616.
(2) a) DeLaive, P.; Foreman, T.; Giannotti, C.; Whitten, D. J. Am. Chem. Soc. 1980, 102, 5627-5631; b) Gaillard, E.; Whitten, D.
Acc. Chem. Res. 1996, 29, 292-297.
(3) Yoon, U.; Su, Z.; Mariano, P. in CRC Handbook of Organic Photochemistry and Photobiology, 2nd ed. (Eds.: Horspool, W.;
Lenci, F.), 2004, CRC Press.
(4) Su, Z.; Mariano, P.; Falvey, D.; Yoon, U.; Oh, S. J. Am. Chem. Soc. 1998, 120, 10676-10686.
(5) For recent reviews of our work in the field, see: a) Beatty, J.; Stephenson, C. Acc. Chem. Res. 2015, 48, 1474-1484; b) Staveness,
D.; Bosque, I.; Stephenson, C. Acc. Chem. Res. 2016, 49, 2295-2306.
(6) For selected recent reviews, see: a) Douglas, J.; Nguyen, J.; Cole, K.; Stephenson, C. Aldrichim. Acta 2014, 47, 15-25; b) Shaw,
M.; Twilton, J.; MacMillan, D. J. Org. Chem. 2016, 81, 6898-6926; c) Douglas, J.; Sevrin, M.; Stephenson, C. Org. Process Res.
Dev. 2016, 20, 1134-1147.
-desilylation has proven valuable for pioneering works in asymmetric photocatalysis, e.g.: Ruiz Espelt, L.;
McPherson, I.; Wiensch, E.; Yoon, T. J. Am. Chem. Soc. 2015, 137, 2452-2455.
(8) Hu, J.; Wang, J.; Nguyen, T.; Zheng, N. Beilstein J. Org. Chem. 2013, 9, 1977-2001.
(9) Beatty, J.; Stephenson, C. J. Am. Chem. Soc. 2014, 136, 10270-10273.
(10) For a recent comprehensive review of aminocyclopropane reactivity, see: Rassadin, V.; Six, Y. Tetrahedron 2016, 72, 4701-
4757.
(11) Hancock, A.; Tanko, J. in Encyclopedia of Radicals in Chemistry, Biology and Materials (Eds.: Chatgilialoglu, C.; Studer, A.),
2012, John Wiley & Sons, Inc.
(12) a) Ha, J.; Lee, J.; Blackstock, S.; Cha, J. J. Org. Chem. 1998, 63, 8510-8514; b) Takemoto, Y.; Yamagata, S.; Furuse, S.; Hayase,
H.; Echigo, T.; Iwata, C. Chem. Commun. 1998, 651-652; c) Maity, S.; Zhu, M.; Shinabery, R.; Zheng, N. Angew. Chem. Int. Ed.
2012, 51, 222-224; d) Nguyen, T.; Morris, S.; Zheng, N. Adv. Synth. Catal. 2014, 356, 2831-2837.
(13) Nguyen, T.; Maity, S.; Zheng, N. Beilstein J. Org. Chem. 2014, 10, 975-980.
(14) a) Wimalasena, K.; Wickman, H.; Mahindaratne, M. Eur. J. Org. Chem. 2001, 3811-3817; b) Madelaine, C.; Six, Y.; Buriez,
O. Angew. Chem. Int. Ed. 2007, 46, 8046-8049; c) Madelaine, C.; Buriez, O.; Crouse, B.; Florent, I.; Grellier, P.; Retailleau, P.; Six,
Y. Org. Biomol. Chem. 2010, 8, 5591-5601.
(15) a) Itoh, T.; Kaneda, K.; Teranishi, S. Tet. Lett. 1975, 32, 2801-2804; b) Lee, H.; Sung, M.; Blackstock, S.; Cha, J. J. Am. Chem.
Soc. 2001, 123, 11322-11324; c) Blackburn, A.; Bowles, D.; Curran, T.; Kim, H. Synthetic Communications 2012, 42, 1855-1863.
(16) Toward enones via photochemical oxidation with dicyanobenzene: Lee, J.; Sun, J.; Blackstock, S.; Cha, J. J. Am. Chem. Soc.
1997, 119, 10241-10242.
(17) Wang, Y.; Luttrull, D.; Dinnocenzo, J.; Goodman, J.; Farid, S.; Gould, I. Photochem. Photobiol. Sci. 2003, 2, 1169-1176.
(18) To our knowledge, the following lists all known reports post-1980 in which a 1-aminoNB has been prepared: a) Della, E.; Knill,
A. Aust. J. Chem. 1994, 47, 1833-1841; b) Martinez, A.; Vilar, E.; Fraile, A.; Cerero, S.; Herrero, M.; Ruiz, P.; Subramanian, L.;
Gancedo, A. J. Med. Chem. 1995, 38, 4474-4477; c) Braslau, R.; Kuhn, H.; Burrill II, L.; Lanham, K.; Stenland, C. Tetrahedron
Lett. 1996, 37, 7933-7936; d) Buser, S.; Vasella, A. Helv. Chim. Acta 2005, 88, 3151-3173; e) Dejmek, M.; Hrebabecky, H.; Sala,
M.; Dracinsky, M.; Prochazkova, E.; Leyssen, P.; Neyts, J.; Balzarini, J.; Nencka, R. Bioorg. Med. Chem. 2014, 22, 2974-2983; f)
Aissa, C.; Ho, K.; Tetlow, D.; Pin-No, M. Angew. Chem. Int. Ed. 2014, 53, 4209-4212.

8
(19) a) Houben, J.; Pfankuch, E. Justus Liebigs Ann. Chem. 1931, 489, 193-223; b) Bartlett, P.; Knox, L. J. Am. Chem. Soc. 1939,
61, 3184-3192; c) Wilt, J.; Parsons, C.; Schneider, C.; Schultenover, D.; Wagner, W. J. Org. Chem. 1968, 33, 694-708; d) Beak, P.;
Harris, B. J. Am. Chem. Soc. 1974, 96, 6363-6372; e) Kropp, P.; Poindexter, G.; Pienta, N.; Hamilton, D. J. Am. Chem. Soc. 1976,
98, 8135-8144.
(20) Applequist, D.; Roberts, J. Chem. Rev. 1954, 54, 1065-1089.
(21) a) Walden, P. Ber. Dtsch. Chem. Ges. 1896, 29, 133-138; b) Kenyon, J.; Phillips, H. Trans. Faraday Soc. 1930, 26, 451-458;
c) Olson, A.; Long, F. J. Am. Chem. Soc. 1934, 56, 1294-1299.
(22) For examples of saturated ring systems that have impacted the clinic, see: Stockdale, T.; Williams, C. Chem. Soc. Rev. 2015,
44, 7737-7763.
(23) For a review highlighting common structural alerts in drug development, including anilines, see: Kalgutkar, A.; Dalvie, D.
Annu. Rev. Pharmacol. Toxicol. 2015, 55, 35-54.
(24) For a recent lead reference, see: Stepan, A.; Subramanyam, C.; Efremov, I.; Dutra, J.; O’Sullivan, T.; DiRico, K.; McDonald,
S.; Won, A.; Dorff, P.; Nolan, C.; Becker, S.; Pustilnik, L.; Riddell, D.; Kauffman, G.; Kormos, B.; Zhang, L.; Lu, Y.; Capetta, S.;
Green, M.; Karki, K.; Sibley, E.; Atchison, K.; Hallgren, A.; Oborski, C.; Robshaw, A.; Sneed, B.; O’Donnell, C. J. Med. Chem.
2012, 55, 3414-3424.
(25) For a recent lead reference, see: Chalmers, B.; Xing, H.; Houston, S.; Clark, C.; Ghassabian, S.; Kuo, A.; Cao, B.; Reitsma, A.;
Murray, C.; Stok, J.; Boyle, G.; Pierce, C.; Littler, S.; Winkler, D.; Bernhardt, P.; Pasay, C.; De Voss, J.; McCarthy, J.; Parson, P.;
Walter, G.; Smith, M.; Cooper, H.; Nilsson, S.; Tsanaktsidis, J.; Savage, P.; Williams, C. Angew. Chem. Int. Ed. 2016, 55, 3580-
3585.
(26) a) Cha, J.; Kulinkovich, O. in Organic Reactions, Vol. 77 (Ed.: Denmark, S.), 2012, John Wiley & Sons, Inc.; b) The
cyclopropanation of amides is commonly referred to as the Kulinkovich-de Meijere reaction: Chaplinski, V.; de Meijere, A. Angew.
Chem. Int. Ed. 1996, 35, 413-414.
(27) Lowry, M.; Goldsmith, J.; Slinker, J.; Rohl, R.; Pascal, R.; Malliaras, G.; Bernhard, S. Chem. Mater. 2005, 17, 5712-5719.
(28) Brooks, A.; Basore, K.; Bernhard, S. Inorg. Chem. 2013, 52, 5794-5800.
(29) Douglas, J.; Cole, K.; Stephenson, C. J. Org. Chem. 2014, 79, 11631-11643.
(30) Musacchio, A.; Lainhart, B.; Zhang, X.; Naguib, S.; Sherwood, T.; Knowles, R. Science 2017, 355, 727-730.
(31) Cai, Y.; Wang, J.; Zhang, Y.; Li, Z.; Hu, D.; Zheng, N.; Chen, H. J. Am. Chem. Soc. 2017, ASAP, DOI: 10.1021/jacs.7b06319.
(32) Cismesia, M.; Yoon, T. Chem. Sci. 2015, 6, 5426-5434.
(33) For a recent review on mechanistic investigations as applied to photoredox catalysis, see: Arias-Rotondo, D.; McCusker, J.
Chem. Soc. Rev. 2016, 45, 5803-5820.