phylogenetic reconstruction. types of data used in phylogenetic inference: distance-based methods:...
Post on 20-Dec-2015
226 views
TRANSCRIPT

Phylogenetic reconstruction

Types of data used in phylogenetic inference:
Distance-based methods: Transform the sequence data into pairwise distances, and use the matrix during tree building.
A B C D E Species A ---- 0.20 0.50 0.45 0.40 Species B 0.23 ---- 0.40 0.55 0.50 Species C 0.87 0.59 ---- 0.15 0.40 Species D 0.73 1.12 0.17 ---- 0.25 Species E 0.59 0.89 0.61 0.31 ----
Character-based methods: Use the aligned characters directly. Taxa Characters
Species A ATGGCTATTCTTATAGTACGSpecies B ATCGCTAGTCTTATATTACASpecies C TTCACTAGACCTGTGGTCCASpecies D TTGACCAGACCTGTGGTCCGSpecies E TTGACCAGTTCTCTAGTTCG
Example 1: Uncorrected“p” distance(=observed percentsequence difference)
Example 2: Kimura 2-parameter distance(estimate of the true number of substitutions between taxa)

Tree-Building Methods
• Distance• UPGMA, NJ, FM, ME
• Character• Maximum Parsimony• Maximum Likelihood

Distance Methods
• Measure distance (dissimilarity)
• Methods• UPGMA (Unweighted Pair Group
Method with Arithmetic Mean)• NJ (Neighbor joining)• ME (Minimal Evolution)

UPGMA: Distance measure
ijd
Clustering: All leaves are assigned to a cluster, which then are iteratively merged according to their distance.
The distance between two clusters i and j can be defined as:
Number of mismatches in gap free alignment

2jlil
kl
ddd
jik CCC (1)
(2)
UPGMA: ReplacingNode k replaces nodes i and j with their union:
The new distances between the new node k and all other clusters l are computed according to:

UPGMA: Algorithm
• Initialization:• Assign each sequence i to its own cluster Ci .• Define one leaf of T for each sequence, and place at height zero.
• Iteration • Determine the two clusters i, j for which di,j is minimal.• Define a new cluster k by Ck = Ci U Cj, and define dkl for all l by (2).• Define a node k with daughter nodes i and j, and place it at height
di,j/2.• Add k to the current clusters and remove i.
• Termination• When only two clusters i, j remain, place the root at height di,j/2.

UPGMA example: Step 1 Alignment -> distance
A B C D E F G
A -
B 63 -
C 94 79 -
D 111 96 47 -
E 67 20 83 100 -
F 23 58 89 106 62 -
G 107 92 43 16 96 102 -
Example: observed percent sequence differenceDistance:
Distance matrix:

GD
Step 2: distance -> cladeA B C D E F G
A -
B 63 -
C 94 79 -
D 111 96 47 -
E 67 23 83 100 -
F 20 58 89 106 62 -
G 107 92 43 16 96 102 -

A B C E F DG
A -
B 63 -
C 94 79 -
E 67 23 83 -
F 20 58 89 62 -
DG 109 94 45 98 104 -
GD
Step 3: merge D and G
A B C D E F G
A -
B 63 -
C 94 79 -
D 111 96 47 -
E 67 23 83 100 -
F 20 58 89 106 62 -
G 107 92 43 16 96 102 -
2jlil
kl
ddd

A B C E F DG
A -
B 63 -
C 94 79 -
E 67 23 83 -
F 20 58 89 62 -
DG 109 94 45 98 104 -
GD A F
Step 4

AF B C E DG
AF -
B 61 -
C 92 79 -
E 65 23 62 -
DG 107 94 45 98 -
A F
Step 5
GD
A B C E F DG
A -
B 63 -
C 94 79 -
E 67 23 83 -
F 20 58 89 62 -
DG 109 94 45 98 104 -

AF B C E DG
AF -
B 61 -
C 92 79 -
E 65 23 62 -
DG 107 94 45 98 -
A F
Step 6
GD B E
AF BE C DG
AF -
BE 63 -
C 92 71 -
DG 107 96 45 -
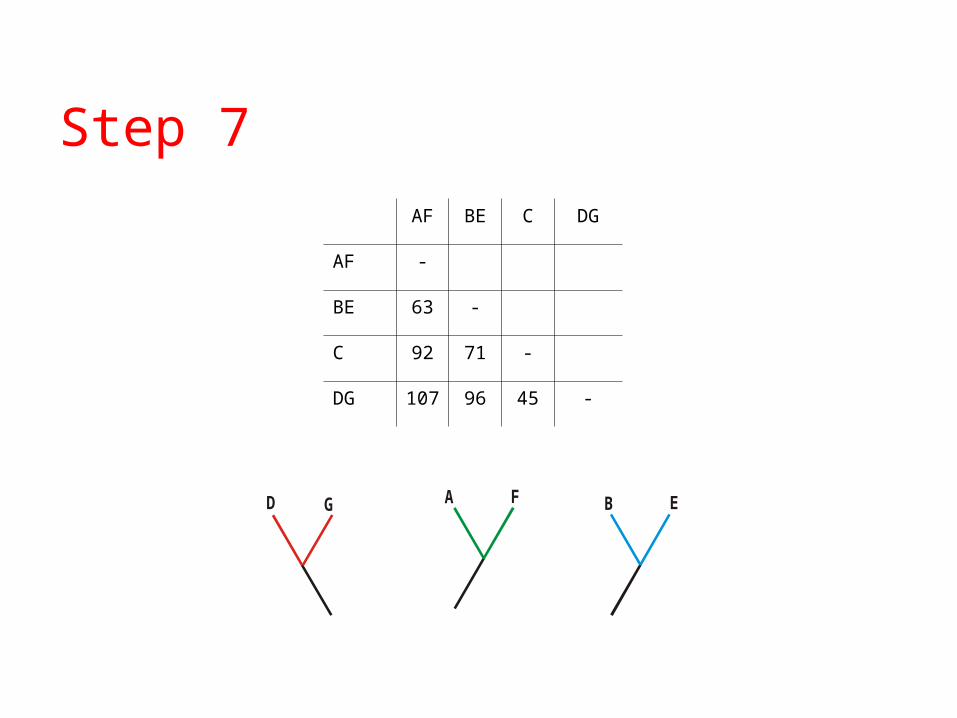
Step 7
A FGD B E
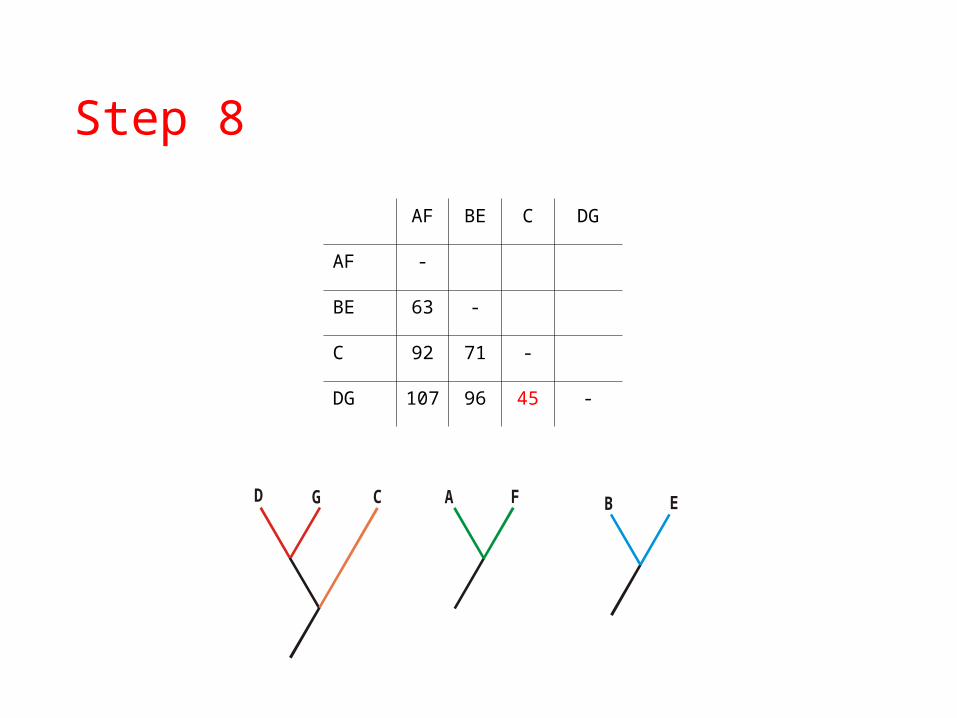
AF BE C DG
AF -
BE 63 -
C 92 71 -
DG 107 96 45 -
GD C
Step 8
A F B E

GD C
Step 9
A F B E
AF BE CDG
AF -
BE 63 -
CDG 102 88 -

GD C
Step 10
AF BE CDG
AF -
BE 63 -
CDG 102 88 -
B EA F

Root
B E
AFBE CDG
AFBE -
CDG 94 -
GD C A F
UPGMA: distance -> phylogeny
B E GD CA F

Additivity
a
b
ec
d
A
B
C
D
eddedcbadddd CDABBCADBDAC 22
Neighbors
Fourpoint condition
BDACCDAB dddd
BCADCDAB dddd

Neighbor-joining (N-J)
• Starts with a star-like tree
• Neighbors are sequentially merged, if they minimize the total length of branches
A
BC
D
EF
A
B
C
DEF
ididdNQ 21)2( 1212Find 1 and 2 that give minimum Q

Clustering methods (UPGMA & N-J)
Optimality criterion: NONE. The algorithm itself builds‘the’ tree.
UPGMA: rootet treeN-J: unrootet tree

Minimum evolution (ME) methods
Optimality criterion: The tree with the shortest overall tree lengthis chosen as the best tree.

Character Methods
• Maximum Parsimony• minimal changes to produce data
• Maximum Likelihood

Parsimony methods:Optimality criterion: The ‘most-parsimonious’ tree is the one thatrequires the fewest number of evolutionary events (e.g., nucleotidesubstitutions, amino acid replacements) to explain the sequences.
• Occam’s razor: “Of two competing theories or explanations, all other things being equal, the simpler one is to be preferred.” (William of Occam, 1280-1347)

Parsimony
1 CCCAGG2 CCCAAG3 CCCAAA4 CCCAAC
Comparison # changes
1-2 1
1-3 2
1-4 2
2-3 1
2-4 1
3-4 1
1,2 can be sister taxaAND
3,4 can be sister taxa
Infer ancestor of 1,2 and 3,4

Parsimony
CCCAGGCCCAAG->
CCCAAGCCCAAA->
CCCAAACCCAAA->
CCCAAC
3 changes

Calculate # changes | tree
acgtatgga acgggtgca aacggtgga aactgtgca
: c
: c
: a
: a
: c
: c
: a
: a
: c
: a
: a
: c
Total tree length: 7 Total tree length: ? Total tree length: ?

Calculate # changes | tree
acgtatgga acgggtgca aacggtgga aactgtgca
: c
: c
: a
: a
: c
: c
: a
: a
: c
: a
: a
: c
Total tree length: 7 Total tree length: 8 Total tree length: 8

Maximum likelihood (ML) methodsOptimality criterion: ML methods evaluate phylogenetic hypothesesin terms of the probability that a proposed model of the evolutionaryprocess and the proposed unrooted tree would give rise to theobserved data. The tree found to have the highest ML value isconsidered to be the preferred tree.

Using modelsObserved differences
Actual changes
A G
C T
Q
3 3 3 3
Example: Jukes-Cantor
pij 1
4
1
4e 4t
pij 1
4
3
4e 4t
, if i=j
, if i≠j
pt : proportion of different nucleotides

Maximum likelihood
• Given two trees, the one with the higher likelihood, i.e. the one with the higher conditional probability of observing the data, is the better
LH P(D | H ) P(D | T ,)

-55.0
-54.5
-54.0
-53.5
-53.0
-52.5
-52.0
-51.5
-51.0
-50.5
0 0.02 0.04 0.06 0.08 0.1
30 nucleotides from -globin genes of two primates on a one-edge tree
* *Gorilla GAAGTCCTTGAGAAATAAACTGCACACTGGOrangutan GGACTCCTTGAGAAATAAACTGCACACTGG
There are two differences and 28 similarities
28
42
4 3116
11
16
1
tt eeL
t
lnL
t= 0.02327lnL= -51.133956
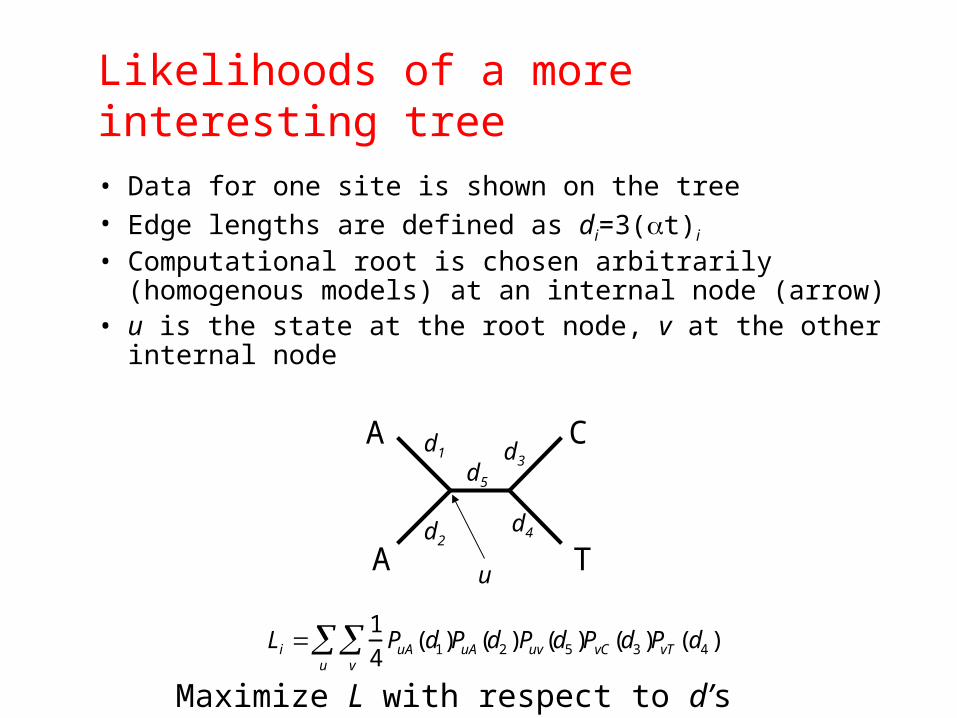
Likelihoods of a more interesting tree
• Data for one site is shown on the tree• Edge lengths are defined as di=3(t)i
• Computational root is chosen arbitrarily (homogenous models) at an internal node (arrow)
• u is the state at the root node, v at the other internal node
Li 1
4PuA(d1)
v
u PuA(d2 )Puv(d5 )PvC (d3 )PvT (d4 )
A
A
C
T
d1 d3
d4d2
d5
u
Maximize L with respect to d’s

Confidence assesment
• Bootstrap

1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20Aus C G A C G G T G G T C T A T A C A C G ABeus C G G C G G T G A T C T A T G C A C G GCeus T G G C G G C G T C T C A T A C A A T ADeus T A A C G A T G A C C C G A C T A T T G
Original data set with n characters.
2 3 13 8 3 19 14 6 20 20 7 1 9 11 17 10 6 14 8 16Aus G A A G A G T G A A T C G C A T G T G CBeus G G A G G G T G G G T C A C A T G T G CCeus G G A G G T T G A A C T T T A C G T G CDeus A A G G A T A A G G T T A C A C A A G T
Draw n characters randomly with re-placement. Repeat m times.
m pseudo-replicates, each with n characters.
Aus
Beus
Ceus
Deus
Original analysis, e.g. MP, ML, NJ.
Aus
Beus
Ceus
Deus
75%
Evaluate the results from the m analyses.
Aus
Beus
Ceus
Deus
Aus
Beus
Ceus
Deus
Aus
Beus
Ceus
Deus
Aus
Beus
Ceus
Deus
Aus
Beus
Ceus
Deus
Aus
Beus
Ceus
Deus
Repeat original analysis on each of the pseudo-replicate data sets.
Bootstrap

Pros and cons of some methods• Pair-wise, algorithmic approach
+ Fast
+ Models can be used when transforming to distances
- Information is lost when transforming to pair-wise distances
- One will get a tree, but no measure of goodness to compare with other hypotheses
• Parsimony+ Philosophically appealing
- Models can not be used
- Can be computationally slow
• Maximum likelihood+ Model based
- Model based
- Computationally veeeeery slow

Computation
• For large data sets (many taxa) exact solutions for any method employing an optimality criterion (parsimony, maximum likelihood, minimum evolution) are not possible
• => Use Neghbor-Joining

What can go wrong?
• Reality• A tree may be a poor model of the real
history• Information has been lost by subsequent
evolutionary changes• “Species” vs. “gene” trees

Canis MusGadus
What is wrong with this tree?
100
100
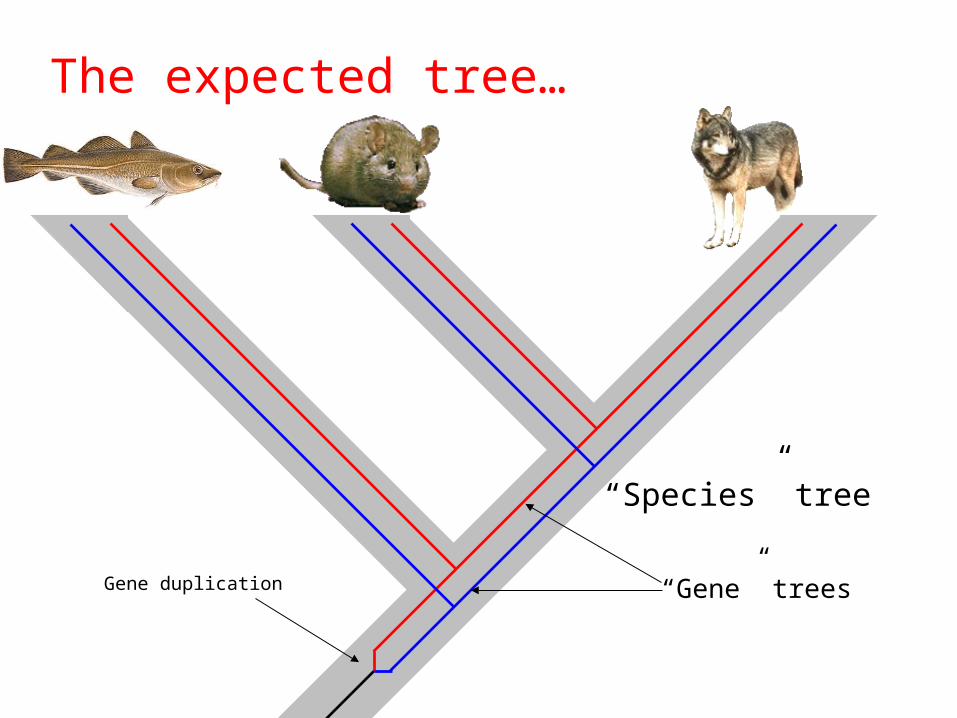
Gene duplication
“Species” tree
“Gene” trees
The expected tree…

Canis Mus Gadus Gadus Mus Canis
Two copies (paralogs) present in the genomes
Paralogous
Orthologous Orthologous

Canis Gadus Mus
What we have studied…

HIV Genome Diversity
• Error prone (RT) replication
• High rate of replication• 1010 virions/day
• In vivo selection pressure

HIV tree
Recombinants!
ENV
GAG
AIDS 1996, 10:S13

To conclude–
• Trash in, trash out : Alignment crucial• Try several methods, for consistency• Beware of paraloges• Choos outgroup wisly: related sequence of
more ancient origin than ”ingroup”• If recombinations possible: each site has its
own tree.