plant pathology in the post-genomics era
TRANSCRIPT

Plant pathology in the post-genomics era
Sophien Kamoun
http:KamounLab.net @KamounLab

PhytophthoraIrish potato famine pathogen Greek for plant destroyer fungus-like oomycete

Crop losses due to fungal/oomycete diseases Fisher et al. Nature 2012
potato blight
rice blast
wheat rusts
corn smut
soybean rust
0 400 800 1200 1600
million people could be fed by lost food

Crop losses due to fungal/oomycete diseases Fisher et al. Nature 2012
rice blast
wheat rusts
corn smut
potato blight
soybean rust
0 400 800 1200 1600
million people could be fed by lost food

Filamentous pathogens
• Fungi and oomycetes
• Highly adaptable - can rapidly overcome plant resistance
• Mixed modes of reproduction (asexual and sexual)
• Large population sizes

Why the misery?
The secret of these pathogens’ success is their ability to rapidly adapt to resistant plant varieties
How did they keep on adapting to ensure their uninterrupted survival over evolutionary time?

Pathogenomics - an emerging field of plant pathology
The genome sequence of the rice blastfungus Magnaporthe griseaRalph A. Dean1, Nicholas J. Talbot2, Daniel J. Ebbole3, Mark L. Farman4, Thomas K. Mitchell1, Marc J. Orbach5, Michael Thon3,Resham Kulkarni1*, Jin-Rong Xu6, Huaqin Pan1, Nick D. Read7, Yong-Hwan Lee8, Ignazio Carbone1, Doug Brown1, Yeon Yee Oh1,Nicole Donofrio1, Jun Seop Jeong1, DarrenM. Soanes2, Slavica Djonovic3, Elena Kolomiets3, Cathryn Rehmeyer4, Weixi Li4, Michael Harding5,Soonok Kim8, Marc-Henri Lebrun9, Heidi Bohnert9, Sean Coughlan10, Jonathan Butler11, Sarah Calvo11, Li-Jun Ma11, Robert Nicol11,Seth Purcell11, Chad Nusbaum11, James E. Galagan11 & Bruce W. Birren11
1Center for Integrated Fungal Research, North Carolina State University, Raleigh, North Carolina 27695, USA2School of Biological and Chemical Sciences, University of Exeter, Washington Singer Laboratories, Exeter EX4 4QG, UK3Department of Plant Pathology and Microbiology, Texas A&M University, College Station, Texas 77843, USA4Department of Plant Pathology, University of Kentucky, Lexington, Kentucky 40546, USA5Department of Plant Pathology, University of Arizona, Tucson, Arizona 85721, USA6Department of Botany and Plant Pathology, Purdue University, West Lafayette, Indiana 47907, USA7Institute of Cell and Molecular Biology, University of Edinburgh, Edinburgh EH9 3JH, UK8School of Agricultural Biotechnology, Seoul National University, Seoul 151-742, Korea9FRE2579 CNRS-Bayer, Bayer Cropscience, 69263 Lyon Cedex 09, France10Agilent Technologies, Wilmington, Delaware 19808, USA11Broad Institute of MIT and Harvard, Cambridge, Massachusetts 02141, USA
* Present address: RTI International, Research Triangle Park, North Carolina 27709, USA
...........................................................................................................................................................................................................................
Magnaporthe grisea is the most destructive pathogen of rice worldwide and the principal model organism for elucidating themolecular basis of fungal disease of plants. Here, we report the draft sequence of the M. grisea genome. Analysis of the gene setprovides an insight into the adaptations required by a fungus to cause disease. The genome encodes a large and diverse set ofsecreted proteins, including those defined by unusual carbohydrate-binding domains. This fungus also possesses an expandedfamily of G-protein-coupled receptors, several new virulence-associated genes and large suites of enzymes involved in secondarymetabolism. Consistent with a role in fungal pathogenesis, the expression of several of these genes is upregulated during the earlystages of infection-related development. The M. grisea genome has been subject to invasion and proliferation of activetransposable elements, reflecting the clonal nature of this fungus imposed by widespread rice cultivation.
Outbreaks of rice blast disease are a serious and recurrent problemin all rice-growing regions of the world, and the disease is extremelydifficult to control1,2. Rice blast, caused by the fungusMagnaporthegrisea, is therefore a significant economic and humanitarianproblem. It is estimated that each year enough rice is destroyed byrice blast disease to feed 60 million people3. The life cycle of the riceblast fungus is shown in Fig. 1. Infections occur when fungalspores land and attach themselves to leaves using a special adhesivereleased from the tip of each spore4. The germinating spore developsan appressorium—a specialized infection cell—which generatesenormous turgor pressure (up to 8MPa) that ruptures the leafcuticle, allowing invasion of the underlying leaf tissue5,6. Subsequentcolonization of the leaf produces disease lesions from which thefungus sporulates and spreads to new plants. When rice blast infectsyoung rice seedlings, whole plants often die, whereas spread of thedisease to the stems, nodes or panicle of older plants results in nearlytotal loss of the rice grain2. Different host-limited forms ofM. griseaalso infect a broad range of grass species including wheat, barley andmillet. Recent reports have shown that the fungus has the capacity toinfect plant roots7.Here we present our preliminary analysis of the draft genome
sequence of M. grisea, which has emerged as a model system forunderstanding plant–microbe interactions because of both itseconomic significance and genetic tractability1,2.
Acquisition of the M. grisea genome sequenceThe genome of a rice pathogenic strain of M. grisea, 70-15, wassequenced through a whole-genome shotgun approach. In all,greater than sevenfold sequence coverage was produced, and a
summary of the principal genome sequence data is provided inTable 1 and Supplementary Table S1. The draft genome sequenceconsists of 2,273 sequence contigs longer than 2 kilobases (kb),ordered and orientated within 159 scaffolds. The total length of allsequence contigs is 38.8 megabases (Mb), and the total length of thescaffolds, including estimated sizes for the gaps, is 40.3Mb. Thegenome assembly has high sequence accuracy—96% of the baseshave quality scores of greater than 40—and long-range continuity,with 50% of all bases residing in scaffolds longer than 1.6Mb.
Reconstruction of the M. grisea genome was aided by theavailability of genome maps8,9 (Supplementary Methods S1).Thirty-three scaffolds, representing 32.8Mb or 85% of the draftassembly, were ordered on the genetic map and assigned to each ofthe seven chromosomes by virtue of containing an anchored geneticmarker. In addition, 19 scaffolds (65% of genome assembly)contained more than one marker and could thus be oriented onthe map. The ends of chromosomes were identified by the telomererepeat motif (TTAGGG)n. Thirteen telomeric sequences wereplaced at the ends of scaffolds, of which six could be placed at theends of chromosomes, whereas the remainder were associated withunanchored scaffolds (Supplementary Table S2). Genome coveragewas estimated by aligning 28,682 M. grisea expressed sequence tags(ESTs), representing genes expressed during a range of develop-mental stages and environmental conditions10,11. Approximately94% of the ESTs were aligned to the genome assembly, despitemany of these ESTs being from different strains.
The gene content of a plant pathogenic fungusWithin the M. grisea genome, 11,109 genes were predicted with
articles
NATURE | VOL 434 | 21 APRIL 2005 | www.nature.com/nature980© 2005 Nature Publishing Group
2005!
2006!
intrinsic light scattering in bR) will help establishthe exact nature of the field interaction. The mainconclusion is that the experiment is clearly in theweak field limit and the laser field is not stronglyperturbing the underlying thermally populatedmodes, but rather inducing their interference inthe excited-state surface. Isomerization yieldswerealso compared with and without phase control(Fig. 6C), keeping spectral amplitudes constant(Fig. 6D; spectral profiles were confirmed usinga tunable monochromator with 0.2-nm spec-tral resolution). The data show a clear phasedependence indicative of coherent control.
Pure amplitude modulation alters the temporalprofile of the pulse (fig. S5); therefore, removingthe phase modulation affects the isomerizationyield by only 5 to 7%. The phase sensitivity ofthe control efficiency further illustrates the co-herent nature of the state preparation.
The temporal profiles of the shaped optimal andanti-optimal pulses and the observed degree of theisomerization yield control are consistent with theknown fast electronic dephasing of bR (10, 38).The largest field amplitudes are confined to ap-proximately 300-fs widths to yield 20% control. Inthe case of transform-limited pulses, all the vibra-tional levels within the excitation bandwidth areexcited in phase and there is a fast decoherence inthe initial electronic polarization (38). However,with the phase-selective restricted bandwidths inthe shaped pulses, there is an opportunity to ma-nipulate different vibrational stateswithmuch long-er coherence times than the electronic polarization.The resultant constructive and destructive interfer-ence effects involving vibrational modes displacedalong the reaction coordinate offer the possibility ofcontrolling isomerization. Experimental obser-vations presented here show that the wave proper-ties ofmatter can play a role in biological processes,to the point that they can even be manipulated.
References and Notes1. M. Shapiro, P. Brumer, Rep. Prog. Phys. 66, 859 (2003).2. S. H. Shi, H. Rabitz, J. Chem. Phys. 92, 364 (1990).3. W. T. Pollard, S. Y. Lee, R. A. Mathies, J. Chem. Phys. 92,
4012 (1990).4. Q. Wang, R. W. Schoenlein, L. A. Peteanu, R. A. Mathies,
C. V. Shank, Science 266, 422 (1994).5. K. C. Hasson, F. Gai, P. A. Anfinrud, Proc. Natl. Acad. Sci.
U.S.A. 93, 15124 (1996).6. T. Ye et al., J. Phys. Chem. B 103, 5122 (1999).7. R. Gonzalez-Luque et al., Proc. Natl. Acad. Sci. U.S.A. 97,
9379 (2000).8. T. Kobayashi, T. Saito, H. Ohtani, Nature 414, 531 (2001).9. J. Herbst, K. Heyne, R. Diller, Science 297, 822 (2002).
10. J. T. M. Kennis et al., J. Phys. Chem. B 106, 6067 (2002).11. S. Hayashi, E. Tajkhorshid, K. Schulten, Biophys. J. 85,
1440 (2003).12. S. C. Flores, V. S. Batista, J. Phys. Chem. B 108, 6745 (2004).13. Y. Ohtsuki, K. Ohara, M. Abe, K. Nakagami, Y. Fujimura,
Chem. Phys. Lett. 369, 525 (2003).14. G. Vogt, G. Krampert, P. Niklaus, P. Nuernberger,
G. Gerber, Phys. Rev. Lett. 94, 068305 (2005).15. K. Hoki, P. Brumer, Phys. Rev. Lett. 95, 168305 (2005).16. J. L. Herek, W. Wohlleben, R. J. Cogdell, D. Zeidler,
M. Motzkus, Nature 417, 533 (2002).17. J. Tittor, D. Oesterhelt, FEBS Lett. 263, 269 (1990).18. S. L. Logunov, M. A. El-Sayed, J. Phys. Chem. B 101, 6629
(1997).19. H. J. Polland et al., Biophys. J. 49, 651 (1986).
20. F. Gai, K. C. Hasson, J. C. McDonald, P. A. Anfinrud,Science 279, 1886 (1998).
21. See supporting data on Science Online.22. V. I. Prokhorenko, A. M. Nagy, R. J. D. Miller, J. Chem.
Phys. 122, 184502 (2005).23. A. K. Dioumaev, V. V. Savransky, N. V. Tkachenko,
V. I. Chukharev, J. Photochem. Photobiol. B Biol. 3, 397(1989).
24. G. Schneider, R. Diller, M. Stockburger, Chem. Phys. 131,17 (1989).
25. A. Xie, Biophys. J. 58, 1127 (1990).26. S. P. Balashov, E. S. Imasheva, R. Govindjee, T. G. Ebrey,
Photochem. Photobiol. 54, 955 (1991).27. J. Paye, IEEE J. Quant. Electron. 28, 2262 (1992).28. H. L. Fragnito, J.-Y. Bigot, P. C. Becker, C. V. Shank, Chem.
Phys. Lett. 160, 101 (1989).29. A. B. Myers, R. A. Harris, R. A. Mathies, J. Chem. Phys. 79,
603 (1983).30. B. X. Hou, N. Friedman, M. Ottolenghi, M. Sheves,
S. Ruhman, Chem. Phys. Lett. 381, 549 (2003).31. R. Morita, M. Yamashita, A. Suguro, H. Shigekawa, Opt.
Commun. 197, 73 (2001).32. K. Blum, Density Matrix Theory and Applications (Plenum,
New York, 1981).33. D. Gelman, R. Kosloff, J. Chem. Phys. 123, 234506 (2005).34. J. Hauer, H. Skenderovic, K.-L. Kompa, M. Motzkus, Chem.
Phys. Lett. 421, 523 (2006).35. D. Oesterhelt, W. Stoeckenius, in Methods in Enzymology,
vol. 31 of Biomembranes (Academic Press, New York,1974), pp. 667–678.
36. The saturation energy is related to the absorption crosssection s as Es 0 1/strans (for a negligibly smallcontribution of the excited-state emission), and s is
related to the extinction coefficient e as s 0 [log(10)/NA]e 0 3.86 ! 10j21e, where NA is Avogadro’s number.
37. The fraction of excited molecules can be estimated as aratio between the number of absorbed photons np 0Eexc ! pd20/4 and the number of retinal molecules Nm 0C ! V in an excited volume V0 ; l0 ! pd20/4, where theconcentration is C 0 2.303A0/strans, A0 is OD at 565 nm(0.9), l0 0 0.04 cm is the path length in the cell used,and d0 0 0.015 cm is the beam diameter in the sample.This gives a fraction of excited molecules of 0.0236 (thatis, 1 out of 42.3 molecules will be excited during theexcitation pulse at a given fluence). The fraction ofdouble-excited molecules can be estimated from thePoisson distribution f(k) 0 ejl(l)k/k!, where l 0 0.0236,and k is the number of occurrences (k 0 1 for thesingle excitation, k 0 2 for the double excitation, etc.).Thus, the fraction of double-excited moleculesf(2)/f(1) 0 l/2; that is, 1.18%.
38. V. F. Kamalov, T. M. Masciangioli, M. A. El-Sayed, J. Phys.Chem. 100, 2762 (1996).
39. K. Edman et al., Nature 401, 822 (1999).40. This work was supported by the National Sciences and
Engineering Research Council of Canada. The authorsthank J.T.M. Kennis, Vrije Universiteit Amsterdam, forhelpful discussions of preliminary results.
Supporting Online Materialwww.sciencemag.org/cgi/content/full/313/5791/1257/DC1Materials and MethodsFigs. S1 to S5References
1 June 2006; accepted 10 August 200610.1126/science.1130747
Phytophthora Genome SequencesUncover Evolutionary Origins andMechanisms of PathogenesisBrett M. Tyler,1* Sucheta Tripathy,1 Xuemin Zhang,1 Paramvir Dehal,2,3 Rays H. Y. Jiang,1,4
Andrea Aerts,2,3 Felipe D. Arredondo,1 Laura Baxter,5 Douda Bensasson,2,3,6 Jim L. Beynon,5
Jarrod Chapman,2,3,7 Cynthia M. B. Damasceno,8 Anne E. Dorrance,9 Daolong Dou,1
Allan W. Dickerman,1 Inna L. Dubchak,2,3 Matteo Garbelotto,10 Mark Gijzen,11
Stuart G. Gordon,9 Francine Govers,4 Niklaus J. Grunwald,12 Wayne Huang,2,14
Kelly L. Ivors,10,15 Richard W. Jones,16 Sophien Kamoun,9 Konstantinos Krampis,1
Kurt H. Lamour,17 Mi-Kyung Lee,18 W. Hayes McDonald,19 Monica Medina,20
Harold J. G. Meijer,4 Eric K. Nordberg,1 Donald J. Maclean,21 Manuel D. Ospina-Giraldo,22
Paul F. Morris,23 Vipaporn Phuntumart,23 Nicholas H. Putnam,2,3 Sam Rash,2,13
Jocelyn K. C. Rose,24 Yasuko Sakihama,25 Asaf A. Salamov,2,3 Alon Savidor,17
Chantel F. Scheuring,18 Brian M. Smith,1 Bruno W. S. Sobral,1 Astrid Terry,2,13
Trudy A. Torto-Alalibo,1 Joe Win,9 Zhanyou Xu,18 Hongbin Zhang,18 Igor V. Grigoriev,2,3
Daniel S. Rokhsar,2,7 Jeffrey L. Boore2,3,26,27
Draft genome sequences have been determined for the soybean pathogen Phytophthora sojae andthe sudden oak death pathogen Phytophthora ramorum. Oomycetes such as these Phytophthoraspecies share the kingdom Stramenopila with photosynthetic algae such as diatoms, and thepresence of many Phytophthora genes of probable phototroph origin supports a photosyntheticancestry for the stramenopiles. Comparison of the two species’ genomes reveals a rapid expansionand diversification of many protein families associated with plant infection such as hydrolases, ABCtransporters, protein toxins, proteinase inhibitors, and, in particular, a superfamily of 700 proteinswith similarity to known oomycete avirulence genes.
Phytophthora plant pathogens attack awide range of agriculturally and orna-mentally important plants (1). Late blight
of potato caused by Phytophthora infestans re-sulted in the Irish potato famine in the 19th cen-
tury, and P. sojae costs the soybean industrymillions of dollars each year. In California andOregon, a newly emerged Phytophthora species,P. ramorum, is responsible for a disease calledsudden oak death (2) that affects not only the live
RESEARCH ARTICLES
www.sciencemag.org SCIENCE VOL 313 1 SEPTEMBER 2006 1261
LETTERS
Genome sequence and analysis of the Irish potatofamine pathogen Phytophthora infestansBrian J. Haas1*, Sophien Kamoun2,3*, Michael C. Zody1,4, Rays H. Y. Jiang1,5, Robert E. Handsaker1, Liliana M. Cano2,Manfred Grabherr1, Chinnappa D. Kodira1{, Sylvain Raffaele2, Trudy Torto-Alalibo3{, Tolga O. Bozkurt2,Audrey M. V. Ah-Fong6, Lucia Alvarado1, Vicky L. Anderson7, Miles R. Armstrong8, Anna Avrova8, Laura Baxter9,Jim Beynon9, Petra C. Boevink8, Stephanie R. Bollmann10, Jorunn I. B. Bos3, Vincent Bulone11, Guohong Cai12, Cahid Cakir3,James C. Carrington13, Megan Chawner14, Lucio Conti15, Stefano Costanzo16, Richard Ewan15, Noah Fahlgren13,Michael A. Fischbach17, Johanna Fugelstad11, Eleanor M. Gilroy8, Sante Gnerre1, Pamela J. Green18,Laura J. Grenville-Briggs7, John Griffith14, Niklaus J. Grunwald10, Karolyn Horn14, Neil R. Horner7, Chia-Hui Hu19,Edgar Huitema3, Dong-Hoon Jeong18, Alexandra M. E. Jones2, Jonathan D. G. Jones2, Richard W. Jones20,Elinor K. Karlsson1, Sridhara G. Kunjeti21, Kurt Lamour22, Zhenyu Liu3, LiJun Ma1, Daniel MacLean2, Marcus C. Chibucos23,Hayes McDonald24, Jessica McWalters14, Harold J. G. Meijer5, William Morgan25, Paul F. Morris26, Carol A. Munro27,Keith O’Neill1{, Manuel Ospina-Giraldo14, Andres Pinzon28, Leighton Pritchard8, Bernard Ramsahoye29, Qinghu Ren30,Silvia Restrepo28, Sourav Roy6, Ari Sadanandom15, Alon Savidor31, Sebastian Schornack2, David C. Schwartz32,Ulrike D. Schumann7, Ben Schwessinger2, Lauren Seyer14, Ted Sharpe1, Cristina Silvar2, Jing Song3, David J. Studholme2,Sean Sykes1, Marco Thines2,33, Peter J. I. van de Vondervoort5, Vipaporn Phuntumart26, Stephan Wawra7, Rob Weide5,Joe Win2, Carolyn Young3, Shiguo Zhou32, William Fry12, Blake C. Meyers18, Pieter van West7, Jean Ristaino19,Francine Govers5, Paul R. J. Birch34, Stephen C. Whisson8, Howard S. Judelson6 & Chad Nusbaum1
Phytophthora infestans is the most destructive pathogen of potatoand a model organism for the oomycetes, a distinct lineage offungus-like eukaryotes that are related to organisms such as brownalgae and diatoms. As the agent of the Irish potato famine in themid-nineteenth century, P. infestans has had a tremendous effect onhuman history, resulting in famine and population displacement1.To this day, it affects world agriculture by causing the most destruc-tive disease of potato, the fourth largest food crop and a criticalalternative to the major cereal crops for feeding the world’s popu-lation1. Current annual worldwide potato crop losses due to lateblight are conservatively estimated at $6.7 billion2. Managementof this devastating pathogen is challenged by its remarkable speedof adaptation to control strategies such as genetically resistant cul-tivars3,4. Here we report the sequence of the P. infestans genome,
which at 240 megabases (Mb) is by far the largest and most com-plex genome sequenced so far in the chromalveolates. Its expansionresults from a proliferation of repetitive DNA accounting for 74%of the genome. Comparison with two other Phytophthora genomesshowed rapid turnover and extensive expansion of specific familiesof secreted disease effector proteins, including many genes that areinduced during infection or are predicted to have activities that alterhost physiology. These fast-evolving effector genes are localized tohighly dynamic and expanded regions of the P. infestans genome.This probably plays a crucial part in the rapid adaptability of thepathogen to host plants and underpins its evolutionary potential.
The size of the P. infestans genome is estimated by optical map andother methods at 240 Mb (Supplementary Information). It is several-fold larger than those of the related Phytophthora species P. sojae
*These authors contributed equally to this work.
1Broad Institute of MIT and Harvard, Cambridge, Massachusetts 02141, USA. 2The Sainsbury Laboratory, Norwich NR4 7UH, UK. 3Department of Plant Pathology, The Ohio StateUniversity, Ohio Agricultural Research and Development Center, Wooster, Ohio 44691, USA. 4Department of Medical Biochemistry and Microbiology, Uppsala University, Box 597,Uppsala SE-751 24, Sweden. 5Laboratory of Phytopathology, Wageningen University, 1-6708 PB, Wageningen, The Netherlands. 6Department of Plant Pathology and Microbiology,University of California, Riverside, California 92521, USA. 7University of Aberdeen, Aberdeen Oomycete Laboratory, College of Life Sciences and Medicine, Institute of MedicalSciences, Foresterhill, Aberdeen AB25 2ZD, UK. 8Plant Pathology Programme, Scottish Crop Research Institute, Invergowrie, Dundee DD2 5DA, UK. 9University of Warwick,Wellesbourne, Warwick CV35 9EF, UK. 10Horticultural Crops Research Laboratory, USDA Agricultural Research Service, Corvallis, Oregon 97330, USA. 11Royal Institute of Technology(KTH), School of Biotechnology, AlbaNova University Centre, Stockholm SE-10691, Sweden. 12Department of Plant Pathology and Plant-Microbe Biology, Cornell University, Ithaca,New York 14853, USA. 13Center for Genome Research and Biocomputing and Department of Botany and Plant Pathology, Oregon State University, Corvallis, Oregon 97331, USA.14Biology Department, Lafayette College, Easton, Pennsylvania 18042, USA. 15Plant Molecular Sciences Faculty of Biomedical and Life Sciences, Bower Building, University of Glasgow,Glasgow G12 8QQ, UK. 16USDA-ARS, Dale Bumpers National Rice Research Center, Stuttgart, Arkansas 72160, USA. 17Department of Molecular Biology, Massachusetts GeneralHospital, Boston, Massachsetts 02114, USA. 18Delaware Biotechnology Institute, University of Delaware, Newark, Delaware 19711, USA. 19Department of Plant Pathology, NorthCarolina State University, Raleigh, North Carolina 27695, USA. 20USDA-ARS, Beltsville, Maryland 20705, USA. 21Department of Plant and Soil Sciences, University of Delaware,Newark, Delaware 19716, USA. 22Department of Entomology and Plant Pathology, University of Tennessee, Knoxville, Tennessee 37996, USA. 23Institute for Genome Sciences,University of Maryland School of Medicine, Baltimore, Maryland 21201, USA. 24Department of Biochemistry, Vanderbilt University School of Medicine, Nashville, Tennessee 37232,USA. 25The College of Wooster, Department of Biology, Wooster, Ohio 44691, USA. 26Department of Biological Sciences, Bowling Green State University, Bowling Green, Ohio 43403,USA. 27University of Aberdeen, School of Medical Sciences, College of Life Sciences and Medicine, Institute of Medical Sciences, Foresterhill, Aberdeen AB25 2ZD, UK. 28Mycologyand Phytopathology Laboratory, Los Andes University, Bogota, Colombia. 29Institute of Genetics and Molecular Medicine, University of Edinburgh, Cancer Research Centre, WesternGeneral Hospital, Edinburgh EH4 2XU, UK. 30J. Craig Venter Institute, Rockville, Maryland 20850, USA. 31Department of Plant Sciences, Tel Aviv University, Tel Aviv 69978, Israel.32Department of Chemistry, Laboratory of Genetics, Laboratory for Molecular and Computational Genomics, University of Wisconsin Biotechnology Center, University of Wisconsin-Madison, Madison Wisconsin 53706, USA. 33University of Hohenheim, Institute of Botany 210, D-70593 Stuttgart, Germany. 34Division of Plant Science, College of Life Sciences,University of Dundee (at SCRI), Invergowrie, Dundee DD2 5DA, UK. {Present addresses: 454 Life Sciences, Branford, Connecticut 06405, USA (C.D.K.); Virginia BioinformaticsInstitute, Virginia Polytechnic and State University, Blacksburg, Virginia 24061, USA (T.T.-A.); Biomedical Diagnostics Institute, Dublin City University, Dublin 9, Ireland (K.O.).
Vol 461 | 17 September 2009 | doi:10.1038/nature08358
393 Macmillan Publishers Limited. All rights reserved©2009
2009
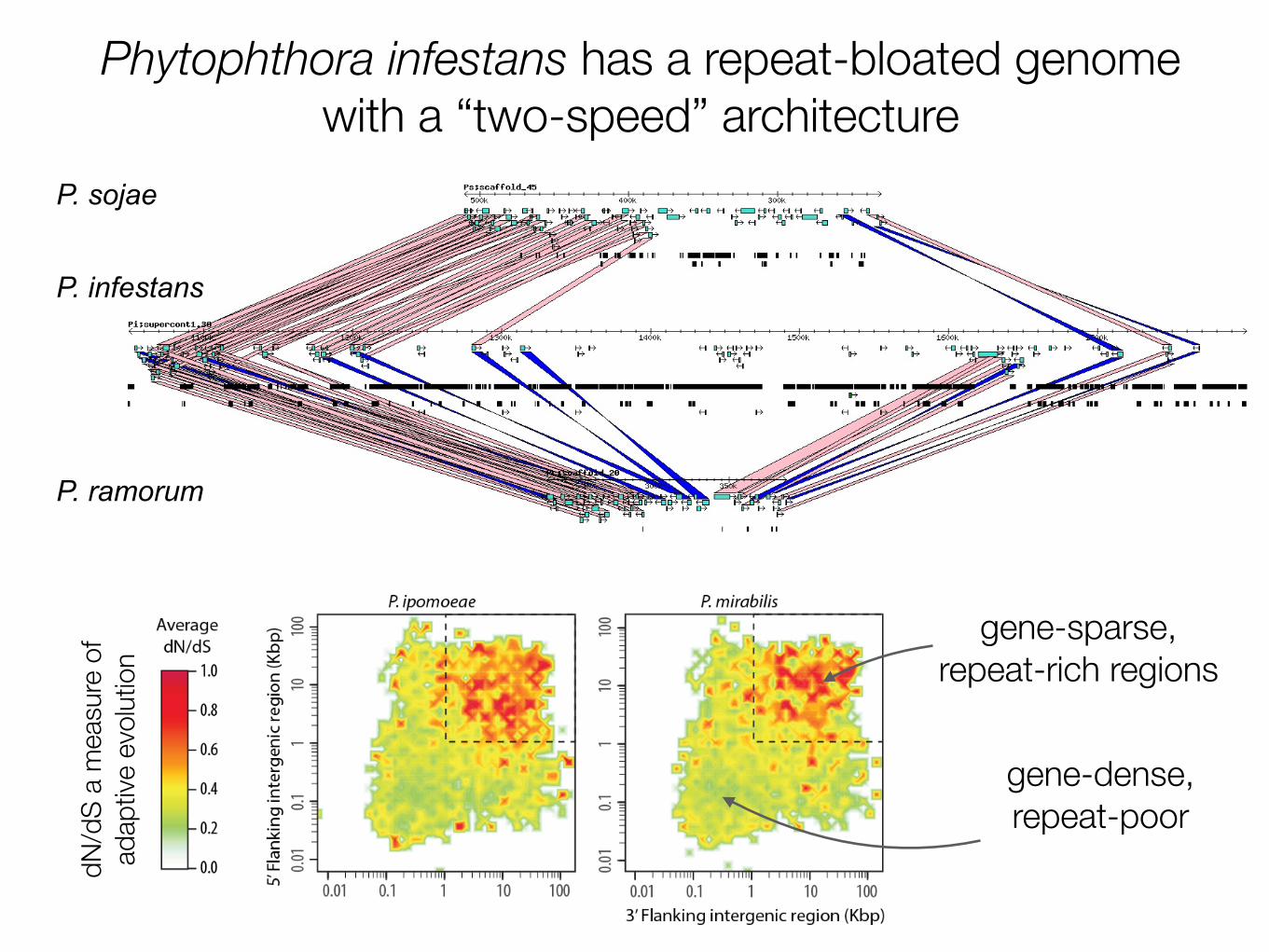
Phytophthora infestans has a repeat-bloated genome with a “two-speed” architecture
P. infestans
P. sojae
P. ramorum
P. sojae
P. infestans
P. ramorum
gene-sparse, repeat-rich regions
gene-dense, repeat-poor
dN/d
S a
mea
sure
of
adap
tive
evol
utio
n
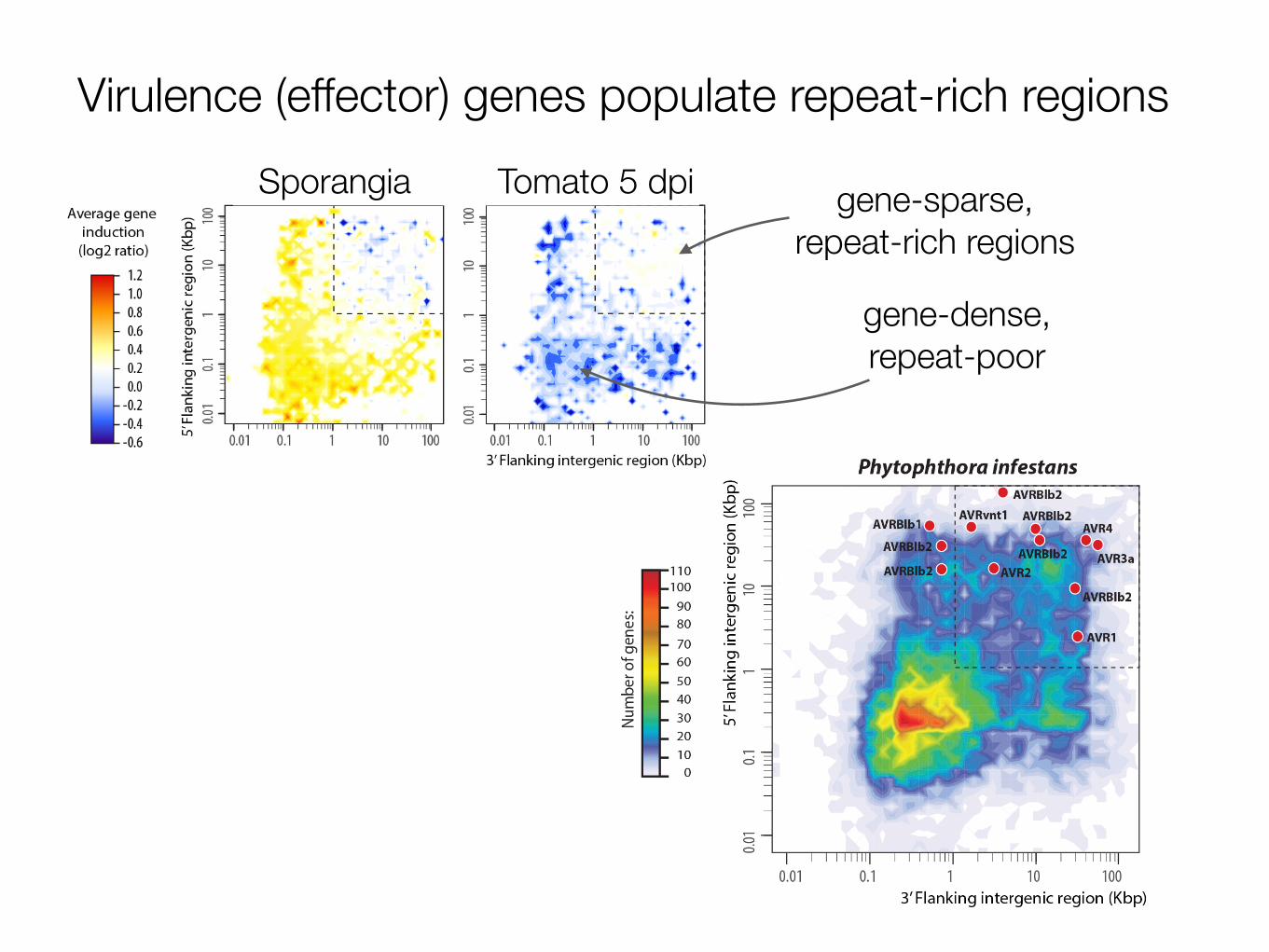
Virulence (effector) genes populate repeat-rich regions Sporangia Tomato 5 dpi gene-sparse,
repeat-rich regions
gene-dense, repeat-poor

Convergence towards a “two-speed” genome architecture in independent pathogen lineages

Genome architecture underpins ability to rapidly adapt to resistant plant genotypes.
• Structural genome variation – increased genome instability and structural variation, deletions, duplications etc.
• Horizontal gene/chromosome transfer – mobile effectors
• Increased local mutagenesis – RIP mutation leakage
• Epigenetics – heterochromatin leakage?
• …more to be discovered
How?

New way of doing business Pathogen and host genomes Integrated conceptual model
increased numbers of mobile elements across diverse families ascompared to P. sojae and P. ramorum, with ,5 times as many LTRretrotransposons and ,10 times as many helitrons (Supplemen-tary Fig. 7).
Consistent with a model of repeat-driven expansion of the P. infestansgenome, the vast majority of repeat elements in the genome are highlysimilar to their consensus sequences, indicating a high rate of recenttransposon activity (Supplementary Fig. 8). In addition, we haveobserved and experimentally confirmed examples of recently activeelements (Supplementary Figs 9–11).
Phytophthora species, like many pathogens, secrete effectorproteins that alter host physiology and facilitate colonization. Thegenome of P. infestans revealed large complex families of effectorgenes encoding secreted proteins that are implicated in patho-genesis10. These fall into two broad categories: apoplastic effectorsthat accumulate in the plant intercellular space (apoplast) and cyto-plasmic effectors that are translocated directly into the plant cell by aspecialized infection structure called the haustorium11. Apoplasticeffectors include secreted hydrolytic enzymes such as proteases,lipases and glycosylases that probably degrade plant tissue; enzymeinhibitors to protect against host defence enzymes; and necrotizingtoxins such as the Nep1-like proteins (NLPs) and PcF-like smallcysteine-rich proteins (SCRs) (Supplementary Table 6).
As in the other Phytophthora species5, candidate effector genes arenumerous and typically expanded compared to non-pathogenic rela-tives (Supplementary Table 6). Most notable among these are theRXLR and Crinkler (CRN) cytoplasmic effectors, described later.
The archetypal oomycete cytoplasmic effectors are the secretedand host-translocated RXLR proteins12. All oomycete avirulencegenes (encoding products recognized by plant hosts and resultingin host immunity) discovered so far encode RXLR effectors, modularsecreted proteins containing the amino-terminal motif Arg-X-Leu-Arg (in which X represents any amino acid) that defines a domainrequired for delivery inside plant cells11, followed by diverse, rapidlyevolving carboxy-terminal effector domains13,14. Several of these Ctermini have been shown to exhibit virulence activities as host celldeath suppressors15,16. We exploited the known motifs and otherconserved sequence features to predict 563 RXLR genes in theP. infestans genome (Supplementary Tables 6, 7 and Supplemen-tary Information). RXLR genes are notably expanded in P. infestans,with ,60% more predicted than in P. sojae and P. ramorum (Sup-plementary Tables 6 and 7). We observed that 70 of these are rapidlydiversifying (Supplementary Table 8). Approximately half ofP. infestans RXLRs are lineage-specific, largely accounting for theexpanded repertoire (Supplementary Figs 12 and 13). In contrastto the core proteome, RXLR genes show evidence of high rates ofturnover with only 16 of the 563 genes with 1:1:1 orthology relation-ships (Supplementary Table 2) and many (88) putative RXLR
pseudogenes (Supplementary Table 9). This high turnover inPhytophthora is probably driven by arms-race co-evolution with hostplants5,13,14,17.
RXLR effectors show extensive sequence diversity. Markov cluster-ing (TribeMCL18) yields one large family (P. infestans: 85, P. ramorum:75, P. sojae: 53) and 150 smaller families (Supplementary Fig. 14). Thelargest family shares a repetitive C-terminal domain structure(Supplementary Figs 15 and 16). Most families have distinct sequencehomologies (Supplementary Fig. 14) and patterns of shared domains(Supplementary Fig. 17) with greater diversity than expected if allRXLR effectors were monophyletic.
In contrast to the core proteome, RXLR effector genes typicallyoccupy a genomic environment that is gene sparse and repeat-rich(Fig. 2g and Supplementary Figs 18 and 19). The mobile elementscontributing to the dynamic nature of these repetitive regions mayenable recombination events resulting in the higher rates of gene gainand gene loss observed for these effectors.
CRN cytoplasmic effectors were originally identified from P. infestanstranscripts encoding putative secreted peptides that elicit necrosisin planta, a characteristic of plant innate immunity19. Since their dis-covery, little had been learned about the CRN effector family. Analysisof the P. infestans genome sequence revealed an enormous family of 196CRN genes of unexpected complexity and diversity (SupplementaryTable 10), that is heavily expanded in P. infestans relative to P. sojae(100 CRNs) and P. ramorum (19 CRNs) (Supplementary Table 6). LikeRXLRs, CRNs are modular proteins. CRNs are defined by a highlyconserved N-terminal ,50-amino-acid LFLAK domain (Supplemen-tary Fig. 20) and an adjacent diversified DWL domain (Fig. 3a, b). Most(60%) possess a predicted signal peptide. Those lacking predicted signalpeptides are typically found in CRN families containing members withsecretion signals (Supplementary Table 10). CRN C-terminal regionsexhibit a wide variety of domain structures, with 36 conserved domainsand a further eight unique C termini identified among the 315Phytophthora CRN proteins (Supplementary Table 11). We observedevidence of recombination between different clades as a mechanismdriving CRN diversity (Supplementary Figs 21–23).
We explored the ability of diverse CRNs to perturb host cellularprocesses. In assays for necrosis in planta (Supplementary Infor-mation), deletion mutants of the previously described CRN2 secretedprotein19 defined a C-terminal 234 amino-acid region (positions173–407, domain DXZ) that is sufficient to induce cell death whenexpressed inside plant cells (Supplementary Fig. 24). Assays withrepresentative P. infestans CRN genes identified four other distinctC termini that also trigger cell death inside plant cells (Fig. 3c). Theseinclude the newly defined DC domain (P. infestans: 18 genes and 49pseudogenes (y)) and the D2 (14 and 43y) and DBF (2 and 1y)domains, which have similarity to protein kinases (SupplementaryTable 11). These results indicate that the CRN protein domains
P. ramorum (65 Mb) scaffold_51
100,000 200,000
P. sojae (95 Mb)scaffold_16 500,000600,000700,000800,000
P. infestans (240 Mb)scaffold1.16
1.5 Mb 1.6 Mb 1.7 Mb 1.8 Mb 1.9 Mb 2 Mb 2.1 Mb 2.2 Mb 2.3 Mb
Figure 1 | Repeat-driven genome expansion in Phytophthora infestans.Conserved gene order across three homologous Phytophthora scaffolds.Genome expansion is evident in regions of conserved gene order, a
consequence of repeat expansion in intergenic regions. Genes are shown asturquoise boxes, repeats as black boxes. Collinear orthologous gene pairs areconnected by pink (direct) or blue (inverted) bands.
NATURE | Vol 461 | 17 September 2009 LETTERS
395 Macmillan Publishers Limited. All rights reserved©2009
NLR$%triggered%immunity%
effectors%
bacterium%
fungus%
oomycete%
haustorium%
NB$LRR%immune%receptors%plant%cell%
pathogen)associated.molecular.pa3erns.(PAMPs).
Pa3ern.recogni9on.receptors.(PRRs).

New way of doing business Discovery pipelines Effectors and immune receptors
understanding of oomycete-plant interactions, and ulti-mately of plant processes that are perturbed by these pathogens. Oomycete pathosystems are an excellent case
study illustrating how plant pathogen genomes can be a remarkable resource for basic and applied plant biology (Figure 1).
Plant parasitic oomycete genome structureCurrently, draft genome sequences are available for ten oomycete species, nine of which are plant pathogens [3,9-16] (Figure 2). One striking feature of these oomycete genomes is the considerable variability in size, ranging from 37 Mb for the biotrophic plant pathogen Albugo laibachii (an obligate parasite of the model plant Arabidopsis thaliana), to 240 Mb for the hemibiotrophic P. infestans (which parasitizes tomato and potato). Th ese observed diff erences in genome size are largely due to the proliferation of transposable elements and repetitive DNA, which in P. infestans account for 74% of the genome content [3]. At 100 Mb, the downy mildew Hyaloperono-spora arabidopsidis, an obligate parasite of A. thaliana, also has a relatively large genome size, a recurrent trend in biotrophic oomycete and fungal pathogens [12,15,17]. Repeat regions in these expanded genomes tend to be unstable; they may promote genome duplication and shuffl ing, increased rates of mutagenesis and gene silencing [17,18].
Th e genomes of Phytophthora species have a peculiar bipartite architecture. Th ey show a characteristic struc-ture comprising blocks of conserved gene order, con-taining approximately 90% of core ortholog genes, separated by regions in which gene order is not conserved
(a) Sequencing of the plant pathogen genome
(b) Computational prediction of pathogen effectors
(c) Library of effector clones
(d) In planta expression of effectors
(e) Insights into plant processes
TC C A ATC A GG TT CGA
TC C A A TC A GGTT CGA
TC C A A TC A GGTT CGA
(i) (ii)
(iii) (iv)
Figure 1. High-throughput pipeline for using plant pathogen eff ectors to unravel plant processes. (a) Genome sequences are currently available for a variety of plant pathogens. (b) This allows the computational prediction of candidate eff ector genes using the knowledge gained from characterized eff ector proteins. In the case of Phytophthora pathogens, prediction is facilitated by the presence of eff ector genes in gene-sparse, repeat-rich regions of the genomes, and by the modular structure of eff ectors. (c) Cloning of these eff ector genes and (d) expression of candidate eff ectors in planta using Agrobacterium-mediated transient expression systems such as agroinfi ltration (left) and wound inoculation (right). (e) These help us understand diverse plant processes. For instance, (i) identifi cation of host proteins that interact with pathogen eff ectors can give insights into the plant pathways targeted and perturbed during the infection process. (ii) In addition, eff ectors can be used as molecular probes to study the structural changes that occur during plant infection at a subcellular level. For instance, eff ectors can be fused to fl uorescent proteins to assess their localization in planta. (iii) The suppression of plant immune responses such as the production of reactive oxygen species (ROS) can also be studied. Blue line shows induction of ROS by fl agellin peptide in the absence of eff ector proteins. This ROS burst is reduced (red) by the expression of an oomycete eff ector. (iv) The activation of host immunity by eff ector proteins helps the dissection of plant susceptibility and resistance mechanisms. For example, the hypersensitive response (brown spots on leaves) against eff ectors transiently exp ressed in planta can be used to identify immune receptors (R genes) of high value for plant breeding.
Pais et al. Genome Biology 2013, 14:211 http://genomebiology.com/2013/14/6/211
Page 2 of 10
Supplemental Table 3 online). However, none of these additionalhomologs induced the HR on Rpi-blb2-expressing leaves.
TheAvrblb2 Family IsHighly Variable and underDiversifyingSelection in P. infestans
Weelected to study theAvrblb2 family inmore detail because theforthcoming release of potato cultivars carrying Rpi-blb2 wouldbenefit from a better understanding of the targeted effector. Tomine further sequence polymorphisms of Avrblb2 in P. infestans,we used the strategy that we previously applied for the smallCys-rich protein SCR74 (Liu et al., 2005). We performed PCRamplifications with genomic DNA from six diverse P. infestansisolates, 88069, 90128, IPO-0, IPO-428, IPO-566, andUS980008(Table 2; see Supplemental Table 2 online). Direct sequencing ofamplicons obtained from genomic DNA of the six isolatesresulted in mixed sequences, indicating that the primers ampli-fied multiple alleles or paralogs of Avrblb2. Therefore, we clonedthe amplicons and generated high-quality sequences (phredQ>20, phred software; CodonCode) of the inserts of 85 differentclones. In addition, we included seven Avrblb2 paralogoussequences from the genome sequence of strain P. infestansT30-4 (Haas et al., 2009).
A total of 24 different nucleotide sequences, encoding 19predicted amino acid sequences, could be identified for Avrblb2(Figure 6A, Table 2; see Supplemental Data Set 2 online).Polymorphisms were detected in 24 of the 279 examined nucle-otides. None of the Avrblb2 sequences contained prematurestop codons or frameshift mutations. Multiple alignments of the24 predicted AVRblb2 amino acid sequences revealed a highlypolymorphic family (Figure 6A). A total of 14 polymorphic aminoacid sites were identified, 10 of which localize to the C-terminaldomain (after the RSLR motif).
To determine the selection pressures underlying sequence diver-sification in the AVRblb2 family, we calculated the rates of non-synonymous (dN) and synonymous (dS) mutations across the 24sequences. We found that dN was greater than dS (v = dN/dS > 1) in121 of 276 pairwise comparisons (see Supplemental Figure 3 andSupplemental Data Set 3 online). In the C-terminal (after RSLR)
Figure 5. Functional Identification of Avrblb1 and Avrblb2.
(A) Wound inoculation screening of the pGR106-PexRD library on N.
benthamiana leaves expressing the S. bulbocastanum R genes Rpi-blb1
(left panel) and Rpi-blb2 (right panel). The two HR-inducing PexRD6/IpiO
clones (PexRD641-3/IpiO1-K143N and PexRD641-10/IpiO2) and two of the
positive PexRD39 and PexRD40 clones (PexRD39169-6 and PexRD40170-1)
are shown. Additional PexRD clones that yielded negative responses
are also shown. All tested clones are labeled RD# for the corresponding
PexRD clone number. The negative and positive controls were A.
tumefaciens strains carrying pGR106-DGFP (dGFP) and pGR106-
PiNPP1 (NPP1), respectively.
(B) to (D) Confirmation of Avrblb cloning using agroinfiltration. Agro-
infiltration of the positive A. tumefaciens pGR106 strains carrying Avrblb1
(PexRD641-3/IpiO1-K143N and PexRD641-10/IpiO2, top and bottom right
panels, respectively) and Avrblb2 (PexRD39 and PexRD40, top and
bottom panels, respectively) was performed in N. benthamiana corre-
sponding to control plants (B) or leaves expressing Rpi-blb1 (C) or Rpi-blb2 (D). A. tumefaciens strain carrying pGR106-DGFP (dGFP) was used
as a negative control (top and bottom left panels of leaves). Coinfiltration
was performed with A. tumefaciens solutions mixed in 1:2 ratio (Avr:R
gene). Hypersensitive cell death was observed starting at 4 DAI, and the
photograph was taken at 7 DAI. The experiment was repeated three
times with similar results.
RXLR Effector Activities 2935
Now there is a new, strong weapon in the battle
against late blight (Phytophthora infestans),
which has been the scourge of potato ultivation
for centuries – Fortuna, the high performance
potato variety with in built special, natural resis-
tance! Late blight is the most common fungal
disease which affects potatoes. It is estimated
that late blight is responsible for around 20% of
potato harvest failures around the globe each
year, which represents 14 million tons with a
value of €2.3 billion. Fighting this disease costs
potato farmers a lot of time and money.
Until now, it was only possible to protect against
this fungal disease using fungicides. But nature
has had a solution at its fi ngertips for thousands
of years. In the high valleys of Mexico, a wild
potato was discovered which is completely
resistant to late blight. Breeders have been
trying for more than 50 years to integrate this
resistance into an agronomically useful potato
variety. They have not been successful, because
undesired negative characteristics of the wild
potato, such as low yield, were always transfer-
red with the resistance gene.
Fortuna, the potato variety for your peace of mind.
2 | 3
BASF Plant Science has made a major breakthrough. The natural potato mechanism which protects against late blight in the wild potato has been
introduced into the leading potato variety used for making fries, whose proven, outstanding characteristics are valued by farmers and processors
alike. The result of this targeted biotechnological process is a high-performance potato variety called Fortuna, which completely protects itself
against late blight, caused by the pathogen Phytophthora infestans. Fortuna will be available to you from 2014, once the approval process is complete.
-20%
It is estimated that late blight is responsible for 20% of potato harvest failures around the globe each year. With its natural resistance, Fortuna guarantees outstanding quality and high yield.
A new weapon against Phytophthora –
... for peace of mind in potato cultivation
UNDER DEVELOPMENT FOR YOUR CONVENIENCE

Field pathogenomics - rapid assessment of pathogen diversity from field samples
2. Purify and multiply field isolates
3. Infect wheat lines and score infection type
4.0
3.0
2.0
1.00.0
Susceptible
Resistant
Phenotype
2. Sequence genes with latest technology
3. Assess pathogen genotypic diversity
4. Determine wheat variety
Traditional Pathology
Field Pathogenomics
1. Receive sample from the field

COMMENTARY Open Access
Crowdsourcing genomic analyses of ash and ashdieback – power to the peopleDan MacLean1*, Kentaro Yoshida1, Anne Edwards2, Lisa Crossman3, Bernardo Clavijo3, Matt Clark3,David Swarbreck3, Matthew Bashton4, Patrick Chapman5, Mark Gijzen5, Mario Caccamo3, Allan Downie2,Sophien Kamoun1 and Diane GO Saunders1
Abstract
Ash dieback is a devastating fungal disease of ash trees that has swept across Europe and recently reached the UK.This emergent pathogen has received little study in the past and its effect threatens to overwhelm the ashpopulation. In response to this we have produced some initial genomics datasets and taken the unusual step ofreleasing them to the scientific community for analysis without first performing our own. In this manner we hopeto ‘crowdsource’ analyses and bring the expertise of the community to bear on this problem as quickly as possible.Our data has been released through our website at oadb.tsl.ac.uk and a public GitHub repository.
Keywords: Crowdsource, Genomics, Ash dieback, Open source, Altmetrics
Main textoadb.tsl.ac.uk: A new resource for the crowdsourcing ofgenomic analyses on ash and ash diebackAsh dieback is a devastating disease of ash trees causedby the aggressive fungal pathogen Chalara fraxinea.This fungus emerged in the early 1990s in Poland andhas since spread west across Europe reaching native for-ests in the UK late last year. The emergence of Chalarain the UK caused public outcry where up to 90% of themore than 80 million ash trees are thought to be underthreat. The disease, which is a newcomer to Britain, wasfirst reported in the natural environment in October2012 and has since been recorded in native woodlandthroughout the UK. There is no known treatment forash dieback, current control measures include burninginfected trees to try and prevent spread [1] and theimplications for the UK environment and the economyremain stark.To kick start genomic analyses of the pathogen and
host, we took the unconventional step of rapidly gener-ating and releasing genomic sequence data. We releasedthe data through our new ash and ash dieback website,oadb.tsl.ac.uk, which we launched in December 2012.Speed is essential in responses to rapidly appearing and
threatening diseases and with this initiative we aim tomake it possible for experts from around the world toaccess the data and analyse it immediately, speeding upthe process of discovery. We hope that by providing dataas soon as possible we will stimulate crowdsourcing andopen community engagement to tackle this devastatingpathogen.
The transcriptomics and genomics data we have releasedso farWe have generated and released Illumina sequence dataof both the transcriptome and genome of Chalara andthe transcriptome of infected and uninfected ash trees.We took the unusual first step of directly sequencing the“interaction transcriptome” [2] of a lesion dissected froman infected ash twig collected in the field. This enabledus to respond quickly, generating useful informationwithout time-consuming standard laboratory culturing;the shortest route from the wood to the sequencer tothe computer.The Chalara transcriptome data, generated at The
Sainsbury Laboratory (TSL, Norwich, UK) was derivedfrom two infected ash samples collected at Ash-wellthorpe Lower Wood, near Norwich; the location ofthe first confirmed case of ash dieback in the wild in theUK. Here we extracted RNA from branches of twoinfected ash trees, prepared cDNA libraries from each
* Correspondence: [email protected] Sainsbury Laboratory, Norwich Research Park, Norwich NR4 7UH, UKFull list of author information is available at the end of the article
© 2013 MacLean et al.; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the CreativeCommons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, andreproduction in any medium, provided the original work is properly cited.
MacLean et al. GigaScience 2013, 2:2http://www.gigasciencejournal.com/content/2/1/2

0
1000
2000
3000
4000
All puzzles
05
10152025
0 5 10 15 20 25 30 35 40 45 50 55 60 65 70 75 80 85 90 95 100
High scoring user %, who has aligned different to software
puzz
le c
ount
0
1000
2000
3000
4000SNP puzzles
0
200
400
600
INDEL puzzles
0
5
10
15
0 5 10 15 20 25 35 40 45 50 55 60 65 70 75 80 85 90 95 100
0
5
10
15
0 10 15 25 30 35 40 50 60 65 70 75 80 85 90 100
High scoring user %, who has aligned different to software
puzz
le c
ount
% readalignmentsdifferent
05101520253035404550556065707580859095100
INDEL(n=10348)
SNP(n=47486)
ALL(n=57834)
0% 25% 50% 75% 100% 0% 25% 50% 75% 100%
Identity Score
Player Better Both Equal Software Better
a b
c

• Genomics-enabled gene mapping and cloning
• From Marker Assisted Selection (MAS) to Genomic Selection (GS)
• Next-generation crop (disease resistance) breeding
©20
12 N
atur
e A
mer
ica,
Inc.
All
righ
ts r
eser
ved.
NATURE BIOTECHNOLOGY ADVANCE ONLINE PUBLICATION 1
A RT I C L E S
The world population is predicted to reach 9 billion within the next 40 years, requiring a 70–100% increase in food production relative to current levels1. It is a major challenge to ensure sustainable food production without further expanding farmland and damaging the environment, in the midst of adverse conditions such as rapid climatic changes. Crop breeding is important for improving yield and toler-ance to existing and emerging biotic and abiotic stresses2. However, current breeding approaches are mostly inefficient, and have not incorporated the findings of the genomics revolution3.
Most crop traits relevant to agronomic improvement are con-trolled by several loci, including quantitative trait loci (QTL), that when disrupted lead to minor phenotypic effects4. To enable plant breeding by marker-assisted selection, it is important to identify the locus or chromosome region harboring each gene contributing to an improved trait. However, compared with genes with major effects that determine discrete characteristics, allelic substitutions at agronomic trait loci lead to only subtle changes in phenotype. Consequently, cloning genes that control agronomic traits is not straightforward.
Here we describe MutMap, a method of rapid gene isolation using a cross of the mutant to wild-type parental line, and apply it to a large population of mutant lines of an elite Japanese rice cultivar. We used MutMap to localize genomic positions of rice genes controlling agronomically important traits including semidwarfism. As mutant plants and associated molecular markers can be made available to plant breeders, this approach could markedly accelerate crop breed-ing and genetics.
RESULTSMutMap methodWe explain the principle of MutMap (Fig. 1) using the example of rice. We first use a mutagen (for example, ethyl methanesulfonate) to mutagenize a rice cultivar (X) that has a reference genome sequence. Mutagenized plants of this first mutant generation (M1) are self- pollinated and brought to the second (M2) or more advanced genera-tions to make the mutated gene homozygous. Through observation of phenotypes in the M2 lines or later generations, we identify recessive mutants with altered agronomically important traits such as plant height, tiller number and grain number per spike. Once the mutant is identified, it is crossed with the wild-type plant of cultivar X, the same cultivar used for mutagenesis. The resulting first filial generation (F1) plant is self-pollinated, and the second generation (F2) progeny (>100) are grown in the field for scoring the phenotype. Because these F2 progeny are derived from a cross between the mutant and its parental wild-type plant, the number of segregating loci responsible for the phenotypic change is minimal, in most cases one, and thus segrega-tion of phenotypes can be unequivocally observed even if the pheno-typic difference is small. All the nucleotide changes incorporated into the mutant by mutagenesis are detected as single-nucleotide polymor-phisms (SNPs) and insertion-deletions (indels) between mutant and wild type. Among the F2 progeny, the majority of SNPs will segregate in a 1:1 mutant/wild type ratio. However, the SNP responsible for the change of phenotype is homozygous in the progeny showing the mutant phenotype. If we collect DNA samples from recessive mutant F2 progeny and bulk sequence them with substantial genomic coverage
Genome sequencing reveals agronomically important loci in rice using MutMapAkira Abe1,2,7, Shunichi Kosugi3,7, Kentaro Yoshida3, Satoshi Natsume3, Hiroki Takagi2,3, Hiroyuki Kanzaki3, Hideo Matsumura3,4, Kakoto Yoshida3, Chikako Mitsuoka3, Muluneh Tamiru3, Hideki Innan5, Liliana Cano6, Sophien Kamoun6 & Ryohei Terauchi3
The majority of agronomic traits are controlled by multiple genes that cause minor phenotypic effects, making the identification of these genes difficult. Here we introduce MutMap, a method based on whole-genome resequencing of pooled DNA from a segregating population of plants that show a useful phenotype. In MutMap, a mutant is crossed directly to the original wild-type line and then selfed, allowing unequivocal segregation in second filial generation (F2) progeny of subtle phenotypic differences. This approach is particularly amenable to crop species because it minimizes the number of genetic crosses (n = 1 or 0) and mutant F2 progeny that are required. We applied MutMap to seven mutants of a Japanese elite rice cultivar and identified the unique genomic positions most probable to harbor mutations causing pale green leaves and semidwarfism, an agronomically relevant trait. These results show that MutMap can accelerate the genetic improvement of rice and other crop plants.
1Iwate Agricultural Research Center, Kitakami, Japan. 2United Graduate School of Agricultural Sciences, Iwate University, Morioka, Japan. 3Iwate Biotechnology Research Center, Kitakami, Japan. 4Gene Research Center, Shinshu University, Ueda, Japan. 5Graduate University for Advanced Studies, Hayama, Japan. 6The Sainsbury Laboratory, Norwich Research Park, Norwich, UK. 7These authors contributed equally to this work. Correspondence should be addressed to R.T. ([email protected]).
Received 28 July 2011; accepted 14 December 2011; published online 22 January 2012; doi:10.1038/nbt.2095
©20
12 N
atur
e A
mer
ica,
Inc.
All
righ
ts r
eser
ved.
NATURE BIOTECHNOLOGY ADVANCE ONLINE PUBLICATION 1
A RT I C L E S
The world population is predicted to reach 9 billion within the next 40 years, requiring a 70–100% increase in food production relative to current levels1. It is a major challenge to ensure sustainable food production without further expanding farmland and damaging the environment, in the midst of adverse conditions such as rapid climatic changes. Crop breeding is important for improving yield and toler-ance to existing and emerging biotic and abiotic stresses2. However, current breeding approaches are mostly inefficient, and have not incorporated the findings of the genomics revolution3.
Most crop traits relevant to agronomic improvement are con-trolled by several loci, including quantitative trait loci (QTL), that when disrupted lead to minor phenotypic effects4. To enable plant breeding by marker-assisted selection, it is important to identify the locus or chromosome region harboring each gene contributing to an improved trait. However, compared with genes with major effects that determine discrete characteristics, allelic substitutions at agronomic trait loci lead to only subtle changes in phenotype. Consequently, cloning genes that control agronomic traits is not straightforward.
Here we describe MutMap, a method of rapid gene isolation using a cross of the mutant to wild-type parental line, and apply it to a large population of mutant lines of an elite Japanese rice cultivar. We used MutMap to localize genomic positions of rice genes controlling agronomically important traits including semidwarfism. As mutant plants and associated molecular markers can be made available to plant breeders, this approach could markedly accelerate crop breed-ing and genetics.
RESULTSMutMap methodWe explain the principle of MutMap (Fig. 1) using the example of rice. We first use a mutagen (for example, ethyl methanesulfonate) to mutagenize a rice cultivar (X) that has a reference genome sequence. Mutagenized plants of this first mutant generation (M1) are self- pollinated and brought to the second (M2) or more advanced genera-tions to make the mutated gene homozygous. Through observation of phenotypes in the M2 lines or later generations, we identify recessive mutants with altered agronomically important traits such as plant height, tiller number and grain number per spike. Once the mutant is identified, it is crossed with the wild-type plant of cultivar X, the same cultivar used for mutagenesis. The resulting first filial generation (F1) plant is self-pollinated, and the second generation (F2) progeny (>100) are grown in the field for scoring the phenotype. Because these F2 progeny are derived from a cross between the mutant and its parental wild-type plant, the number of segregating loci responsible for the phenotypic change is minimal, in most cases one, and thus segrega-tion of phenotypes can be unequivocally observed even if the pheno-typic difference is small. All the nucleotide changes incorporated into the mutant by mutagenesis are detected as single-nucleotide polymor-phisms (SNPs) and insertion-deletions (indels) between mutant and wild type. Among the F2 progeny, the majority of SNPs will segregate in a 1:1 mutant/wild type ratio. However, the SNP responsible for the change of phenotype is homozygous in the progeny showing the mutant phenotype. If we collect DNA samples from recessive mutant F2 progeny and bulk sequence them with substantial genomic coverage
Genome sequencing reveals agronomically important loci in rice using MutMapAkira Abe1,2,7, Shunichi Kosugi3,7, Kentaro Yoshida3, Satoshi Natsume3, Hiroki Takagi2,3, Hiroyuki Kanzaki3, Hideo Matsumura3,4, Kakoto Yoshida3, Chikako Mitsuoka3, Muluneh Tamiru3, Hideki Innan5, Liliana Cano6, Sophien Kamoun6 & Ryohei Terauchi3
The majority of agronomic traits are controlled by multiple genes that cause minor phenotypic effects, making the identification of these genes difficult. Here we introduce MutMap, a method based on whole-genome resequencing of pooled DNA from a segregating population of plants that show a useful phenotype. In MutMap, a mutant is crossed directly to the original wild-type line and then selfed, allowing unequivocal segregation in second filial generation (F2) progeny of subtle phenotypic differences. This approach is particularly amenable to crop species because it minimizes the number of genetic crosses (n = 1 or 0) and mutant F2 progeny that are required. We applied MutMap to seven mutants of a Japanese elite rice cultivar and identified the unique genomic positions most probable to harbor mutations causing pale green leaves and semidwarfism, an agronomically relevant trait. These results show that MutMap can accelerate the genetic improvement of rice and other crop plants.
1Iwate Agricultural Research Center, Kitakami, Japan. 2United Graduate School of Agricultural Sciences, Iwate University, Morioka, Japan. 3Iwate Biotechnology Research Center, Kitakami, Japan. 4Gene Research Center, Shinshu University, Ueda, Japan. 5Graduate University for Advanced Studies, Hayama, Japan. 6The Sainsbury Laboratory, Norwich Research Park, Norwich, UK. 7These authors contributed equally to this work. Correspondence should be addressed to R.T. ([email protected]).
Received 28 July 2011; accepted 14 December 2011; published online 22 January 2012; doi:10.1038/nbt.2095

©20
12 N
atur
e A
mer
ica,
Inc.
All
righ
ts r
eser
ved.
2 ADVANCE ONLINE PUBLICATION NATURE BIOTECHNOLOGY
A RT I C L E S
(>10× coverage), we expect to have 50% mutant and 50% wild-type sequence reads for SNPs that are unlinked to the SNP responsible for the mutant phenotype. However, the causal SNP and closely linked SNPs should show 100% mutant and 0% wild-type reads. SNPs loosely linked to the causal mutation should have >50% mutant and <50% wild-type reads. If we define the SNP index as the ratio between the number of reads of a mutant SNP and the total number of reads corresponding to the SNP, we expect that this index would equal 1 near the causal gene and 0.5 for the unlinked loci. SNP indices can be scanned across the genome to find the region with a SNP index of 1, harboring the gene responsible for the mutant phenotype.
The rate of false positives can be assessed because allelic segrega-tion follows a binomial distribution with a probability parameter of 0.5 (probability of mutant SNP equals 0.5) at a SNP with no link-age to the causal SNP. If the sample size (read depth of the site) is 10, the probability of having a SNP index of 1 is P = (0.5)10 = 10−3. Therefore, in a data set with a known number of genotyped SNPs (L),
the expected number of clusters of SNPs with SNP index of 1 ( k) would be approximately pkL = 10−3kL. In our case, the maximum estimate of L is 2,225 (Supplementary Table 1), and the probability of observing a cluster of more than four consecutive SNPs with SNP index of 1 would be 2.3 × 10−9. Statistical considerations of how the number of F2 progeny to be bulked and the average coverage (depth) of genome sequencing affect the false-positive rate, and how misclassi-fication of phenotypes between mutant and wild type affects true positives, are in Supplementary Data.
MutMap applied to pale green leaf mutantsWe have maintained 12,000 ethyl methanesulfonate–mutagenized rice lines of third and fourth mutant generations (M3–M4) with a back-ground of Hitomebore, an elite cultivar of Northern Japan5. Whole-genome resequencing of five independent mutants indicated that each line harbors 1,499 469 (mean s.d.; range 960–2,225) SNPs that are different from wild type (Supplementary Table 1). Using our mutant stock, we set out to isolate genes of agronomic importance. As a proof-of-principle experiment, we applied MutMap to two mutants showing pale-green leaf phenotypes with slightly lower chlorophyll concentrations compared with wild type (Hit1917-pl1 and Hit0813-pl2; Fig. 2a).
We crossed these mutants to the Hitomebore wild type in 2009, and obtained F1 progeny. F1 plants were self-pollinated, and >200 F2 progeny were obtained for each cross. For both mutants we observed segregation between wild-type and mutant phenotypes in field-grown
EMS mutagenesis
Wild-typeparental line
Mutant
Selfing
F2 progeny
Sequence the bulked DNA
F2 progeny showing mutant phenotype
Sequence(reference)
SNP mapping
WTSeq.
SNPs not linked tothe phenotype
SNP index = 3/7 = 0.43 SNP index = 7/7 = 1
SNPs linked tothe phenotype
Mapping ofSNPs
X
F1
Figure 1 Simplified scheme for application of MutMap to rice. A rice cultivar with a reference genome sequence is mutagenized by ethyl methanesulfonate (EMS). The mutant generated, in this case a semidwarf phenotype, is crossed to the wild-type plant of the same cultivar used for the mutagenesis. The resulting F1 is self-pollinated to obtain F2 progeny segregating for the mutant and wild-type phenotypes. Crossing of the mutant to the wild-type parental line ensures detection of phenotypic differences at the F2 generation between the mutant and wild type. DNA of F2 displaying the mutant phenotype are bulked and subjected to whole-genome sequencing followed by alignment to the reference sequence. SNPs with sequence reads composed only of mutant sequences (SNP index of 1; see text) are closely linked to the causal SNP for the mutant phenotype.
a
b
SN
P in
dex
1.0
0.9
0.8
0.7
0.6
0.5
0.4
0.3
0.20 10 20 Mb
Hit1917-pl1_Chr.10 Hit0813-pl2_Chr.1
0 10 20 30 40 Mb
1.0
0.9
0.8
0.7
0.6
0.5
0.4
0.3
0.2
SP
AD
val
ue
50
WT Hit1917-pl1
Hit0813-pl2
403020100
Figure 2 Identification of genomic regions harboring causal mutations for two pale green leaf mutants, Hit1917-pl1 and Hit0813-pl2, using MutMap. (a) Leaf color and SPAD (stability of soil plant analytical development) values (an estimate of chlorophyll content) of wild-type (WT) Hitomebore and two mutants. Error bars, s.d. (b) SNP index plots for two leaf color mutants (Hit1917-pl1 and Hit0813-pl2) showing chromosomes 10 and 1, respectively. Red regression lines were obtained by averaging SNP indices from a moving window of five consecutive SNPs and shifting the window one SNP at a time. The x-axis value of each averaged SNP index was set at a midpoint between the first and fifth SNP.
Semi-dwarf
Salt tolerant
Next-Gen crop breeding - MutMap

Kaijin!
• Salt tolerant rice variety “Kaijin” bred in just two years
• Seawater contaminated paddies/groundwater after 2011 tsunami
• MutMap revealed the causal gene
• Kaijin differs from parental variety Hitomebore by only 201 SNPs
mutant�WT�
WT�
mutant�
Kai
jin�
WT�
mutant�
Kai
jin�
Control� Treated with 0.375% NaCl�

www.sciencemag.org SCIENCE VOL 341 23 AUGUST 2013 833
BACTERIA MAY NOT ELICIT MUCH SYMPA-
thy from us eukaryotes, but they, too, can get
sick. That’s potentially a big problem for the
dairy industry, which often depends on bac-
teria such as Streptococcus thermophilus to
make yogurts and cheeses. S. thermophilus
breaks down the milk sugar lactose into tangy
lactic acid. But certain viruses—bacterio-
phages, or simply phages—can debilitate the
bacterium, wreaking havoc on the quality or
quantity of the food it helps produce.
In 2007, scientists from Danisco, a
Copenhagen-based food ingredient com-
pany now owned by DuPont, found a way to
boost the phage defenses of this workhouse
microbe. They exposed the bacterium to
a phage and showed that this essentially
vaccinated it against that virus (Science,
23 March 2007, p. 1650). The trick has
enabled DuPont to create heartier bacterial
strains for food production. It also revealed
something fundamental: Bacteria have a
kind of adaptive immune system, which
enables them to fi ght off repeated attacks
by specifi c phages.
That immune system has suddenly
become important for more than food scien-
tists and microbiologists, because of a valu-
able feature: It takes aim at specific DNA
sequences. In January, four research teams
reported harnessing the system, called
CRISPR for peculiar features in the DNA of
bacteria that deploy it, to target the destruc-
tion of specifi c genes in human cells. And in
the following 8 months, various groups have
used it to delete, add, activate, or suppress tar-
geted genes in human cells, mice, rats, zebra-
fi sh, bacteria, fruit fl ies, yeast, nematodes,
and crops, demonstrating broad utility for the
The CRISPR CrazeA bacterial immune system yields a potentially
revolutionary genome-editing technique
CR
ED
IT: E
YE
OF
SC
IEN
CE
/SC
IEN
CE
SO
UR
CE
NEWSFOCUS
Fighting invasion. When
viruses (green) attack
bacteria, the bacteria
respond with DNA-targeting
defenses that biologists
have learned to exploit
for genetic engineering.
Published by AAAS
on
Augu
st 2
3, 2
013
ww
w.s
cien
cem
ag.o
rgD
ownl
oade
d fro
m
www.sciencemag.org SCIENCE VOL 341 23 AUGUST 2013 833
BACTERIA MAY NOT ELICIT MUCH SYMPA-
thy from us eukaryotes, but they, too, can get
sick. That’s potentially a big problem for the
dairy industry, which often depends on bac-
teria such as Streptococcus thermophilus to
make yogurts and cheeses. S. thermophilus
breaks down the milk sugar lactose into tangy
lactic acid. But certain viruses—bacterio-
phages, or simply phages—can debilitate the
bacterium, wreaking havoc on the quality or
quantity of the food it helps produce.
In 2007, scientists from Danisco, a
Copenhagen-based food ingredient com-
pany now owned by DuPont, found a way to
boost the phage defenses of this workhouse
microbe. They exposed the bacterium to
a phage and showed that this essentially
vaccinated it against that virus (Science,
23 March 2007, p. 1650). The trick has
enabled DuPont to create heartier bacterial
strains for food production. It also revealed
something fundamental: Bacteria have a
kind of adaptive immune system, which
enables them to fi ght off repeated attacks
by specifi c phages.
That immune system has suddenly
become important for more than food scien-
tists and microbiologists, because of a valu-
able feature: It takes aim at specific DNA
sequences. In January, four research teams
reported harnessing the system, called
CRISPR for peculiar features in the DNA of
bacteria that deploy it, to target the destruc-
tion of specifi c genes in human cells. And in
the following 8 months, various groups have
used it to delete, add, activate, or suppress tar-
geted genes in human cells, mice, rats, zebra-
fi sh, bacteria, fruit fl ies, yeast, nematodes,
and crops, demonstrating broad utility for the
The CRISPR CrazeA bacterial immune system yields a potentially
revolutionary genome-editing technique
CR
ED
IT: E
YE
OF
SC
IEN
CE
/SC
IEN
CE
SO
UR
CE
NEWSFOCUS
Fighting invasion. When
viruses (green) attack
bacteria, the bacteria
respond with DNA-targeting
defenses that biologists
have learned to exploit
for genetic engineering.
Published by AAAS
on
Augu
st 2
3, 2
013
ww
w.s
cien
cem
ag.o
rgD
ownl
oade
d fro
m
infects plant scientists

REVIEW Open Access
Plant genome editing made easy: targetedmutagenesis in model and crop plants using theCRISPR/Cas systemKhaoula Belhaj†, Angela Chaparro-Garcia†, Sophien Kamoun* and Vladimir Nekrasov*
Abstract
Targeted genome engineering (also known as genome editing) has emerged as an alternative to classical plantbreeding and transgenic (GMO) methods to improve crop plants. Until recently, available tools for introducingsite-specific double strand DNA breaks were restricted to zinc finger nucleases (ZFNs) and TAL effector nucleases(TALENs). However, these technologies have not been widely adopted by the plant research community due tocomplicated design and laborious assembly of specific DNA binding proteins for each target gene. Recently, aneasier method has emerged based on the bacterial type II CRISPR (clustered regularly interspaced short palindromicrepeats)/Cas (CRISPR-associated) immune system. The CRISPR/Cas system allows targeted cleavage of genomic DNAguided by a customizable small noncoding RNA, resulting in gene modifications by both non-homologous endjoining (NHEJ) and homology-directed repair (HDR) mechanisms. In this review we summarize and discuss recentapplications of the CRISPR/Cas technology in plants.
Keywords: CRISPR, Cas9, Plant, Genome editing, Genome engineering, Targeted mutagenesis
IntroductionTargeted genome engineering has emerged as an alter-native to classical plant breeding and transgenic (GMO)methods to improve crop plants and ensure sustainablefood production. However, until recently the availablemethods have proven cumbersome. Both zinc fingernucleases (ZFNs) and TAL effector nucleases (TALENs)can be used to mutagenize genomes at specific loci, butthese systems require two different DNA binding proteinsflanking a sequence of interest, each with a C-terminalFokI nuclease module. As a result these methods have notbeen widely adopted by the plant research community.Earlier this year, a new method based on the bacterialCRISPR (clustered regularly interspaced short palindromicrepeats)/Cas (CRISPR-associated) type II prokaryoticadaptive immune system [1] has emerged as an alter-native method for genome engineering. The ability toreprogram CRISPR/Cas endonuclease specificity usingcustomizable small noncoding RNAs has set the stagefor novel genome editing applications [2-8]. The system is
based on the Cas9 nuclease and an engineered singleguide RNA (sgRNA) that specifies a targeted nucleic acidsequence. Given that only a single RNA is required to gen-erate target specificity, the CRISPR/Cas system promisesto be more easily applicable to genome engineering thanZFNs and TALENs.Recently, eight reports describing the first applications
of the Cas9/sgRNA system to plants have been published[9-16]. In this review, we summarise the methods andfindings described in these publications and provide anoutlook for the application of the CRISPR/Cas system as agenome engineering tool in plants.
Plant genome editing using the CRISPR/Cas systemThe application of the bacterial CRISPR/Cas system toplants is very recent. In the August 2013 issue of NatureBiotechnology three short reports described the first ap-plications of the Cas9/sgRNA system to plant genomeengineering [9-11]. Shortly after, five more reportsfollowed [12-16]. The papers mainly focused on testingthe CRISPR/Cas technology using transient expressionassays (Table 1 and Figure 1), such as protoplast trans-formation and in planta expression using Agrobacterium
* Correspondence: [email protected]; [email protected]†Equal contributorsThe Sainsbury Laboratory, Norwich Research Park, Norwich, UK
PLANT METHODS
© 2013 Belhaj et al.; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the CreativeCommons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, andreproduction in any medium, provided the original work is properly cited. The Creative Commons Public Domain Dedicationwaiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwisestated.
Belhaj et al. Plant Methods 2013, 9:39http://www.plantmethods.com/content/9/1/39
