platelets: thrombotic thrombocytopenic purpura
TRANSCRIPT

Hematology 2002 315
Platelets: Thrombotic Thrombocytopenic Purpura
James N. George, J. Evan Sadler, and Bernhard Lämmle
Abnormalities of plasma von Willebrand factor(VWF) have been recognized to be associated withthrombotic thrombocytopenic purpura (TTP) forover 20 years. Patients with chronic, relapsing TTPhave VWF multimers that are larger than normal,similar in size to those secreted by cultured endo-thelial cells. Recent observations have documentedthat a deficiency of a VWF-cleaving protease(termed ADAMTS13) may be responsible for thepresence of these unusually large VWF multimers.Multiple mutations of the ADAMTS13 gene canresult in ADAMTS13 deficiency and cause congeni-tal TTP; autoantibodies neutralizing ADAMTS13protease activity have been associated with ac-quired TTP.
In Section I, Dr. Evan Sadler reviews the struc-ture, biosynthesis, and function of the ADAMTS13protease. He describes the mutations that havebeen identified in congenital TTP and describes therelationship of ADAMTS13 deficiency to the devel-opment of both congenital and acquired TTP. Dr.Sadler postulates that the development of TTP maybe favored by conditions that combine increasedVWF secretion, such as during the later stages ofpregnancy, and decreased ADAMTS13 activity.
In Section II, Dr. Bernhard Lämmle describesthe assay methods for determining ADAMTS13activity. Understanding the complexity of thesemethods is essential for understanding the diffi-culty of assay performance and the interpretationof assay data. Dr. Lämmle describes his extensiveexperience measuring ADAMTS13 activity inpatients with TTP as well as patients with acutethrombocytopenia and severe illnesses not diag-
nosed as TTP. His data suggest that a severedeficiency of ADAMTS13 activity (< 5%) is a spe-cific feature of TTP. However, he emphasizes that,although severe ADAMTS13 deficiency may bespecific for TTP, it may not be sensitive enough toidentify all patients who may be appropriatelydiagnosed as TTP and who may respond to plasmaexchange treatment.
In Section III, Dr. James George describes theevaluation and management of patients withclinically suspected TTP, as well as adults who maybe described as having hemolytic-uremic syn-drome (HUS). Dr. George presents a classificationof TTP and HUS in children and adults. Appropriateevaluation and management are related to theclinical setting in which the diagnosis is consid-ered. A clinical approach is described for patientsin whom the diagnosis of TTP or HUS is considered(1) following bone marrow transplantation, (2)during pregnancy or the postpartum period, (3) inassociation with drugs which may cause TTP eitherby an acute immune-mediated toxicity or a dose-related toxicity, (4) following a prodrome of bloodydiarrhea, (5) in patients with autoimmune disorders,and (6) in patients with no apparent associatedcondition who may be considered to have idio-pathic TTP. Patients with idiopathic TTP appear tohave the greatest frequency of ADAMTS13deficiency and appear to be at greatest risk for aprolonged clinical course and subsequent relapse.Management with plasma exchange has a high riskof complications. Indications for additional immu-nosuppressive therapy are described.
I. ADAMTS13, VON WILLEBRAND FACTOR,AND THE PATHOPHYSIOLOGY OF
THROMBOTIC THROMBOCYTOPENIC PURPURA
J. Evan Sadler, MD, PhD*
Thrombotic thrombocytopenic purpura (TTP) is char-acterized by microangiopathic hemolytic anemia andthrombocytopenia, often accompanied by fever, renalfailure, and neurological deficits. Platelet-rich microvas-
cular thrombi appear to be responsible for the renal andcerebral lesions, and often damage other organ systemsas well. If untreated, TTP is almost always fatal. How-ever, intensive plasma exchange therapy has reduced themortality to approximately 25%.1,2
* Departments of Medicine and Biochemistry and MolecularBiophysics, and Howard Hughes Medical Institute, WashingtonUniversity School of Medicine, 660 South Euclid Avenue, Box8022, St Louis, MO 63110.

316 American Society of Hematology
The pathophysiology of TTP has been mysteriousuntil recently, although several clues have implicated aplasma protein defect. For example, from the first de-scription of the disease,3 transfusions of blood wereknown to cause remissions in occasional patients, andthe striking efficacy of plasma therapy suggests that aplasma protein deficiency or a circulating toxic factor isresponsible. Twenty years ago, Moake et al found thatsome patients with chronic, relapsing TTP had plasmavon Willebrand factor (VWF) multimers that were largerthan normal, similar in size to those secreted by cul-tured endothelial cells. They proposed that “unusuallylarge” VWF multimers (ULVWF) persisted after secre-tion in vivo because the patients lacked a protease or adisulfide reductase activity that reduces the size of VWFmultimers and suggested that ULVWF could cause TTPby promoting uncontrolled platelet agglutination, throm-bosis, and ischemia. Thus, the efficacy of plasma ex-change could be due to the replacement of the missing“depolymerase” or the removal of an inhibitor.4
During the past decade, the proposed role of ULVWFin TTP has received considerable further support, start-ing with the independent discovery by Furlan et al5,6 andTsai and Lian7 that most patients with TTP are deficientin a plasma metalloprotease that cleaves a specific pep-tide bond in the VWF subunit,8,9 thereby decreasing thesize of VWF multimers. Congenital TTP is associatedwith the constitutional absence of the protease, whereas
adults with acquired TTP usually have IgG autoantibod-ies that inhibit protease activity. Last year, this proteasewas purified to homogeneity and partially sequenced,10,11
which showed that it belongs to the ADAMTS family ofmetalloproteases.10 The corresponding gene and cDNAwere cloned quickly, and the VWF cleaving proteasewas designated ADAMTS13.12,13 In a remarkable con-vergence of distinct experimental strategies, theADAMTS13 gene was identified simultaneously by thepositional cloning of mutations in families affected byautosomal recessive inherited TTP.14 These discoverieshave provided a new molecular focus for investigationsof TTP.
Structure of ADAMTS13ADAMTS13 is one of 19 ADAMTS proteases describedto date (http://www.gene.ucl.ac.uk/nomenclature/genefamily/adamts.html). The family name “ADAMTS”is an acronym for “a disintegrin-like and metalloproteasewith thrombospondin type 1 repeats,”15,16 andADAMTS13 has all of the structural motifs that definethe family (Figure 1). The ADAMTS13 precursor con-sists of 1427 amino acid residues and contains a signalpeptide, a relatively short propeptide, a metalloproteasedomain of the reprolysin/adamalysin type, a disintegrindomain, a thrombospondin type 1 repeat (TSP1), a char-acteristic Cys-rich and spacer domain, and 7 additionalTSP1 repeats. ADAMTS13 is the most divergent mem-
Figure 1. Structure of ADAMTS13 and mutations in congenital thrombotic thrombocytopenic purpura.
The ADAMTS13 gene (A) contains 29 exons in ~ 37 kb on chromosome 9q34. Dashed lines show the relationship of exons to theADAMTS13 protein (B). Structural domains include signal peptide (S), propeptide (P), metalloprotease, disintegrin domain (Dis),thrombospondin 1 repeats (numbered 1-8), cysteine-rich domain (Cys), spacer domain, and CUB domains. Mutations in patients withinherited TTP are shown (C).
Reproduced with permission from Zheng X, Majerus EM, Sadler JE. ADAMTS13 and TTP. Curr Opin Hematol. 2002;9:389-394.

Hematology 2002 317
ber of the family and, unlike any other ADAMTS pro-tease, ADAMTS13 also has 2 CUB domains at its C-ter-minus. CUB domains are named for complement compo-nents C1r/C1s, urinary epidermal growth factor, and bonemorphogenetic protein-1 (a synonym for procollagen C-proteinase), each of which has 1 or more CUB domains.17
The protease domain is a typical reprolysin-like oradamalysin-like metalloprotease, with a conserved ac-tive site sequence of HEXXHXXGXXHD in which theHis residues coordinate a catalytic Zn2+ ion. Molecularmodeling based on the structure of adamalysin II sug-gests that several other residues coordinate a structuralCa2+ ion and predicts the locations of 3 intrachain disul-fide bonds.12 Chelation of either Zn2+ or Ca2+ inhibits theprotease,8,9 which is consistent with the proposed pres-ence of both metal ions in the metalloprotease domain.The functions of the various other domains are not es-tablished, although comparisons with related ADAMTSproteases suggest they may interact with other proteinsor glycosaminoglycans. Disintegrin domains are foundin many snake venom proteins and are so named be-cause they can bind integrins and disrupt cellular inter-actions. However, the disintegrin domains of ADAMTSproteases are not known to have this activity, and otherdomains may be better candidates for intermolecularinteractions. The Cys-rich domain of ADAMTS13 doescontain an Arg-Gly-Asp sequence that might bind cer-tain integrins.12,14 An Arg-Gly-Asp sequence is also presentin the Cys-rich domain of ADAMTS2, but not in otherfamily members.18 The potential integrin-binding activityof these Arg-Gly-Asp sequences has not been tested.
Several ADAMTS domains have been implicatedin binding to other macromolecules. RecombinantADAMTS1 binds to extracellular matrix, and binding isinhibited by heparin, suggesting that the ligand is a gly-cosaminoglycan. Matrix binding appears to depend onthe ADAMTS1 spacer region and certain TSP1 repeats.19
The TSP1 repeats in platelet thrombospondin interactwith a number of ligands, including fibrinogen, CD36,and several glycosaminoglycans.20,21
The CUB domains of ADAMTS13 are unique withinthe ADAMTS family but are common in the closely re-lated astacin-like family and can determine substratespecificity. For example, procollagen C-proteinase is ametalloprotease with a C-terminal extension that con-tains 5 CUB domains. This protease removes the C-ter-minal propeptide from certain procollagens, and at least3 CUB domains are required to recognize the procollagensubstrate.22 Furthermore, procollagen cleavage is en-hanced by a cofactor protein, POLCE, that also contains2 essential CUB domains.23
At the present early stage of the field, one can onlyspeculate about the purpose of TSP1, spacer, and CUB
domains in ADAMTS13. These domains are completelyconserved among ADAMTS13 from human, mouse, andthe Japanese puffer fish (J.E. Sadler, unpublished data),suggesting they are likely to be important for function.Based on analogies developed in the preceding para-graphs, these domains are potential sites for binding tosubstrates, cofactors, cell surfaces, or the extracellularmatrix.
Biosynthesis and CatabolismBy Northern blotting, full-length 4.6-kb ADAMTS13mRNA is found only in the liver, suggesting that the liveris the principal source of plasma ADAMTS13.12-14 Al-ternatively spliced mRNA forms occur in many tis-sues,12-14,18 but whether they give rise to functional pro-tein is unknown.
ADAMTS13 undergoes extensive posttranslationalprocessing, involving proteolysis as well as glycosyl-ation. ADAMTS13 appears to be synthesized as a zy-mogen that is activated by propeptide cleavage. The pro-peptide ends in a typical furin-like recognition sequence,Arg-Gln-Arg-Arg, suggesting that activation could oc-cur intracellularly. The propeptide has been cleaved fromADAMTS13 that is purified from plasma.10,11 Therefore,some ADAMTS13 circulates as an active enzyme, butthe existence of a significant pool of ADAMTS13 zy-mogen has not been excluded. The plasma protein isextensively glycosylated, which is consistent with thepresence of 10 potential N-glycosylation sites through-out the protein and 7 potential O-glycosylation sites, onefor each TSP1 repeat except the fourth.12 Glycosylationprobably accounts for much of the discrepancy betweenthe apparent mass of purified ADAMTS13 on gel elec-trophoresis (190 kDa) and the calculated mass for theADAMTS13 polypeptide (145 kDa).
The plasma concentration of ADAMTS13 is notknown precisely but is estimated to be 1 µg/mL.11 Itshalf-life in the circulation is approximately 2 to 3 days,24
and this exceptional stability allows patients with con-genital deficiency to be treated with plasma infusionsevery 2 to 3 weeks to prevent the recurrence of throm-botic microangiopathy.25,26
EnzymologyThe activity of ADAMTS13 depends on both Zn2+ andCa2+; the required calcium ion can be replaced by Ba2+
or Sr2+ but not by Mg2+.8 The enzyme is active betweenpH 7 and pH 11, with optimal activity at pH 8, and ac-tivity is increased substantially at an ionic strength muchlower than that of plasma.8 Aside from metal chelators,none of the usual protease inhibitors inhibitADAMTS13.8,9 The resistance of ADAMTS13 to allplasma protease inhibitors is relatively unusual but is

318 American Society of Hematology
consistent with its long circulatory half-life.The only known substrate for ADAMTS13 is VWF,
which is cleaved between Tyr1605-Met1606 (Tyr842-Met843
in mature subunit numbering) in the second of the 3 con-secutive A domains in the center of the VWF subunit.8,9
This cleavage produces fragments of 176 kDa and 140kDa that are found in normal plasma VWF, suggestingthat ADAMTS13 may be responsible for most of theproteolytic degradation of VWF subunits in vivo.27 Therate of VWF cleavage by ADAMTS13 is increased mark-edly by mild denaturation with low concentrations of urea8
or guanidine hydrochloride,9 or by fluid shear stress.9
Increased susceptibility of VWF to digestion per-sists after the removal of denaturants or shear stress,9
suggesting that these treatments unfold the VWF sub-strate and expose the cleavage site. This conclusion issupported by studies of mutant VWF. The common vonWillebrand disease (VWD) type 2A mutation Arg1597Gln(Arg834Gln in mature subunit numbering) is withinVWF domain A2, only 8 residues from the ADAMTS13-sensitive Tyr-Met bond. The mutation does not impairmultimer assembly but increases the sensitivity of VWFto proteolytic degradation in the circulation, and recom-binant VWF with this mutation is digested readily byADAMTS13 in the absence of shear stress or denatur-ants.28 Therefore, a conformational change in VWF thatpromotes proteolytic cleavage may be localized to theA2 domain.28 For many VWD type 2A mutations, cleav-age of the mutant subunits by ADAMTS13 probablycauses the deficit in large multimers, which in turn causesa hemostatic defect.
Mutations in Congenital TTPSoon after the development of an assay for plasma VWFcleaving protease activity, severe deficiency was foundto be associated with autosomal recessive inheritedTTP,5,29 or Upshaw-Schulman syndrome.30,31 Last year,genome-wide linkage analysis in 4 affected familiesmapped the responsible locus to chromosome 9q34 andshowed that it encodes ADAMTS13.14 The ADAMTS13gene spans 37 kb and contains 29 exons. Among 15 af-fected ADAMTS13 alleles, 12 different mutations wereidentified (Figure 1): 9 were single amino acid substitu-tions at residues conserved between human and mouse,2 were frameshift mutations, and 1 was a splice site mu-tation. No patient had obvious null mutations on bothalleles, suggesting that total ADAMTS13 deficiencycould be lethal and, in fact, ADAMTS13 activity levelswere extremely low but detectable (2%-7% of normal)in all patients. This landmark study demonstrates con-clusively that ADAMTS13 deficiency causes inheritedTTP, rather than being a secondary consequence of an-other molecular defect.14
ADAMTS13 and the Mechanism ofThrombotic Microangiopathy
With the benefit of recent knowledge concerningADAMTS13, the ULVWF model of TTP4 can be adaptedreadily to account for microvascular thrombosis, the mostdangerous aspect of TTP (Figure 2, see Color Figures,page 520). Platelets in flowing blood adhere transientlyto exposed, immobilized VWF. Transient adhesion is fol-lowed by the engagement of other adhesive and signal-ing receptors, which causes platelet activation, immobi-lization, and spreading. This platelet surface can recruitmore VWF and more platelets by the same mechanism.Under the influence of fluid shear stress, the VWF isrecognized by ADAMTS13, which cleaves VWFmultimers, releases the platelets, and limits the growthof the thrombus. Without ADAMTS13, this feedback in-hibitory mechanism fails, and microvascular thrombi con-tinue to grow, causing tissue ischemia and infarction.
This model accounts for several details of the clini-cal picture of TTP and the behavior of VWF. DecreasedADAMTS13 activity would be expected to cause accu-mulation of ULVWF, as is observed in patients with in-herited TTP due to ADAMTS13 deficiency.5 The ad-ministration of desmopressin releases ULVWF into thecirculation and might be expected to exacerbate TTP,and this phenomenon has been reported.32 When sub-jected to fluid shear stress, the platelets in blood aggre-gate in a process that depends on VWF, and the thresh-old for shear-induced platelet aggregation is shifted tolower values of shear stress when ADAMTS13 activityis decreased.33,34 Certain inherited disorders are associ-ated with increased VWF-platelet binding. In VWD type2B, mutant VWF binds platelet GPIb with increased af-finity.35,36 In platelet-type pseudo-VWD, mutant GPIbαbinds VWF with increased affinity.37 Both of these con-ditions are characterized by bleeding rather than throm-bosis, possibly because ADAMTS13 cleaves VWF inany growing platelet aggregate and prevents microvas-cular thrombosis.
The ULVWF model is consistent with several bio-chemical and clinical observations but raises many im-portant questions. For example, according to the model,platelet thrombi form on immobilized VWF that is ex-posed to the blood, but the origin and properties of thisVWF are not fully understood. When the vasculature isdisrupted, VWF bound to connective tissue mediatesplatelet adhesion. However, it seems unlikely that enoughdeendothelialized vessels could be diffusely distributedto account for the disseminated microvascular thrombo-sis caused by ADAMTS13 deficiency. A more plausiblesource of exposed VWF may be the ULVWF multimerssecreted by a relatively intact endothelium. In the ab-sence of ADAMTS13, long strings of secreted ULVWF

Hematology 2002 319
remain bound to endothelial cells and become decoratedwith adherent platelets. In the presence of ADAMTS13,these VWF strings are cleaved rapidly, and the plateletsare released.38 Thus, TTP may be favored by conditionsthat combine increased VWF secretion and decreasedADAMTS13 activity. Such a “two-hit” model could ex-plain the substantial variation in the age at which pa-tients with inherited TTP develop symptoms. Deficiencyof ADAMTS13 may set the stage, so that thromboticmicroangiopathy supervenes after a triggering event in-jures or activates microvascular endothelial cells andcauses the secretion of ULVWF.39 For example, the in-crease in VWF secretion during the third trimester of preg-nancy may contribute to the tendency of TTP, whether in-herited or acquired, to present during late pregnancy.39,40
Other Potential Factors inThrombotic Microangiopathy
The developing conceptual framework involving VWFand ADAMTS13 not only has wonderful explanatorypower but also highlights issues that need further study.Perhaps the most relevant for clinical practice is the ob-servation that some patients with thrombotic micro-angiopathy have normal ADAMTS13 activity, and oth-ers have ADAMTS13 deficiency but no demonstrableinhibitor.41,42 These cases suggest there may be other le-sions in the ADAMTS13 pathway, or mechanisms inde-pendent of ADAMTS13, that can cause TTP. If suffi-ciently common, alternative pathophysiologic mecha-nisms may reduce the utility of ADAMTS13 assays forthe diagnosis and management of TTP. Suitable clinicaltrials are needed to address this important point.
Additional factors in the ADAMTS13 pathway havenot been identified, but recent case reports suggest thata search for them may be appropriate. Two unrelatedfamilies have been described in which children born ofa consanguineous marriage developed TTP that was char-acterized by ULVWF multimers and a good clinical re-sponse to prophylactic plasma transfusions; however,these patients had normal endogenous ADAMTS13 ac-tivity.43,44 Such anomalous cases would be consistent witha mutation in an unknown ADAMTS13 cofactor, or po-tentially with a mutation in ADAMTS13 that affectedfunction in vivo but not in vitro.
Cleavage by ADAMTS13 is not the only mecha-nism by which VWF multimers can be decreased in size.Many other proteases can cleave VWF, and cleavage ofVWF subunits at several sites has been observed in vivo.45
Plasma thrombospondin-1 recently was shown to havedisulfide reductase activity toward VWF, with the abil-ity to reduce intersubunit disulfide bonds and decreaseVWF multimer size.46 It is not known whether deficien-cies in these processes could cause TTP, or whether they
may sometimes complement ADAMTS13 deficiency andprevent TTP. One possibly critical parameter may bewhether shear stress can increase the efficiency of VWFmultimer scission by other proteases or by thrombo-spondin-1. If not, these mechanisms may be unable toinhibit platelet thrombus growth.
A study of TTP patients with normal ADAMTS13levels has uncovered an unexpected potential risk fac-tor. The current model for the pathogenesis of TTP em-phasizes the proposed role of VWF in the developmentof platelet-rich thrombi at sites of high fluid shear stressand is consistent with the histopathologic demonstrationthat TTP lesions are rich in VWF and poor in fibrin.47 Incontrast, risk factors for venous thrombosis, with lesionscharacteristically rich in fibrin and poor in platelets,would not usually be thought of as risk factors for TTP.However, among 11 Caucasian patients with TTP andnormal ADAMTS13 levels, 4 (36%) were heterozygousfor factor V Leiden compared with 6 (3%) of 186 con-trol subjects (P < 0.001).42 The possible involvement offactor V Leiden suggests that some mechanisms ofthrombotic microangiopathy may involve fibrin deposi-tion, which is not a prominent feature of TTP.
II. THE ROLE OF ADAMTS13 IN THE
EVALUATION AND MANAGEMENT OF PATIENTS WITH
THROMBOTIC THROMBOCYTOPENIC PURPURA
Bernhard Lämmle, MD*
Many hypotheses concerning the pathogenesis of TTPhave been proposed (for review, see Moake and Chow,1
Ruggenenti and Remuzzi,2 and Furlan and Lämmle3).One of them, originally put forward in 1982 by Moakeet al,4 has attracted much interest in the past few years.These researchers observed unusually large multimersof von Willebrand factor (ULVWF) in the plasma of sev-eral patients with chronic relapsing TTP and suggestedthat these extremely adhesive VWF multimers were di-rectly responsible for in vivo platelet clumping in themicrocirculation leading to ischemic neurologic and re-nal dysfunction. ULVWF multimers, present during re-mission, disappeared from plasma during acute relapses,presumably by consumption during the platelet clump-ing process. Moake et al4 suggested that their presencein plasma might be due to an excessive release from en-
* Central Hematology Laboratory, University Hospital,Inselspital, CH 3010 Bern, Switzerland
Dedication: I dedicate this paper to my beloved son Gregor, athird-year medical student, who died on July 12, 2002, at theage of 24 years.

320 American Society of Hematology
dothelial cells and/or an impaired degradation by a then-hypothetical VWF “depolymerase.”
In 1996, our group5 and Tsai6 independently isolatedand partially characterized a novel protease from hu-man plasma that specifically cleaved VWF at Tyr842-Met843,5 the peptide bond known to be cleaved in vivo.7
Shortly thereafter, 4 patients, including 2 brothers, withchronic relapsing TTP and ULVWF in their plasma werefound to completely lack the VWF-cleaving proteaseactivity.8 Infusion of fresh frozen plasma (FFP) into the2 brothers led to a quantitative recovery of VWF-cleav-ing protease activity and disappearance of the ULVWFmultimers. The half-life of the protease in plasma wasestimated to be 2-4 days.9
Another patient with severe sporadic TTP came toour attention whose plasma lacked any VWF-cleavingprotease activity due to an autoantibody inhibiting itsactivity.10 Two large retrospective (multicenter) studieson a large series of patients diagnosed with TTP11,12 orhemolytic uremic syndrome (HUS)11 revealed that mostpatients with acute TTP had a severely depressed VWF-cleaving protease activity (less than 5% of the activityin normal human plasma [NHP]), most often due to in-hibiting IgG autoantibodies,11,12 and that those patientsdiagnosed with HUS had normal (higher than 50%) orsubnormal (26-50%) protease activity.11
The VWF-cleaving protease has been further puri-fied from plasma,13,14 and the N-terminal amino acid se-quence allowed its characterization as a novel memberof the ADAMTS family of metalloproteases14 and eluci-dation of its cDNA and gene structure.15-17 The approachof Levy et al17 using genome-wide positional cloning inpatients with hereditary TTP and their family membersled to the identification of the ADAMTS13 gene on chro-mosome 9q34 and of several mutations of this gene aspresumably being responsible for severely decreasedVWF-cleaving protease activity and disease in doublyheterozygous or homozygous carriers of mutated alleles.This latter observation17 strongly supported a causal link-age of severely deficient ADAMTS13 activity with thehereditary form of TTP.8
In this overview, I will (1) describe the methods tomeasure ADAMTS13 activity, (2) report on our experi-ence with hereditary ADAMTS13 deficiency in patientswith constitutional TTP (the Upshaw-Schulman syn-drome), (3) assess the specificity and sensitivity of se-vere ADAMTS13 deficiency for the diagnosis of TTP,and (4) estimate the value of ADAMTS13 activity mea-surement for diagnostic purposes and therapeutic decisions.
Assays of ADAMTS13 Activity and ofADAMTS13 Inhibiting Autoantibodies
Several assays have been developed for measuringADAMTS13 activity (for review, see Furlan andLämmle18). Our original assay5,8 involves activation ofthe diluted patient plasma with 10 mM BaCl
2, admix-
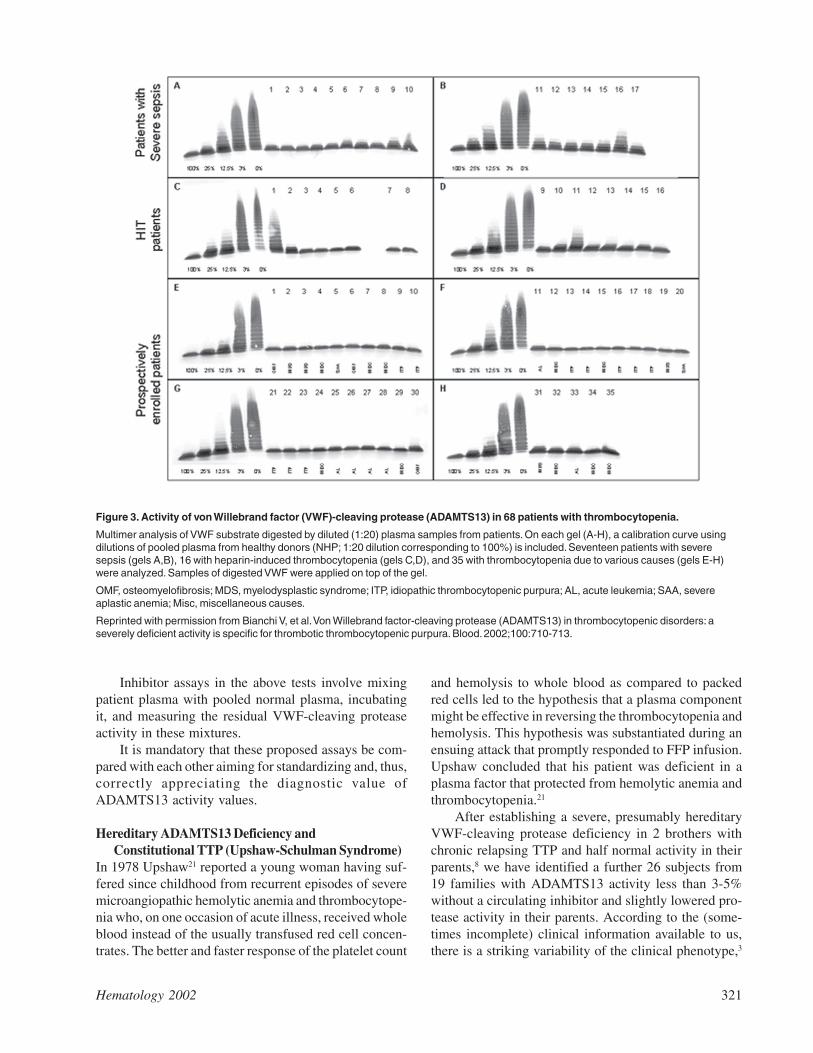
ture of purified protease-free VWF substrate, digestionof VWF during 16-18 hours on a hydrophilic filter mem-brane lying on top of a buffer consisting of 1.5 M urea, 5mM Tris, pH 8.0, followed by sodium dodecyl sulphate(SDS) -1.4% agarose gel electrophoresis and immuno-blotting of the degraded multimers. The assay takes 4working days to be completed, is reproducible, and isvery sensitive, allowing one to discriminate 1-3% of pro-tease activity from 0% by comparing the VWFmultimeric pattern produced by the sample with that ofa standard curve obtained with serial dilutions of poolednormal plasma defined to contain 100% of ADAMTS13activity (Figure 3).
The method reported by Tsai and Lian6,12 measuresthe generation of disulfide-linked dimers of C-terminal(amino acids 843-2050) and/or N-terminal (amino ac-ids 1-842) proteolytic fragments of the VWF subunit af-ter incubation of guanidinium-treated VWF substratewith diluted plasma in the presence and absence ofEDTA, followed by unreduced SDS polyacrylamide gelelectrophoresis and immunodetection with 125I-labeledanti-VWF antibodies and autoradiography. The differ-ence of the intensity of the 350 kD band representingthe dimer of C-terminal VWF fragments between thereaction mixture with and without EDTA reflects theprotease activity in the test sample.
In order to make the measurement of VWF-cleav-ing protease activity more generally available to routinelaboratories, we set out to simplify the assay by measur-ing the residual collagen binding of the VWF substrateafter its degradation by BaCl
2 activated protease in pa-
tient plasma in the presence of 1.5 M urea and low ionicstrength conditions.19 Despite its advantages of simplic-ity (ELISA technique using microtiter plates) and speed(completion within a few hours), the assay is less sensitiveand less precise in the low activity range, and we havesometimes encountered problems with reproducibility.
A very elegant assay was reported by Obert et al,20
in which BaCl2 activated protease in patient plasma di-
gests recombinant VWF substrate. Protease-digestedVWF is assayed using a 2-site immune radiometric as-say (IRMA) with a capturing monoclonal antibody di-rected against the C-terminal part of the VWF subunitand a mixture of radiolabeled monoclonal antibodiestoward the N-terminal part of VWF for detection. Theapparent loss of VWF antigen is directly related toADAMTS13 activity.
Hematology 2002 321
Inhibitor assays in the above tests involve mixingpatient plasma with pooled normal plasma, incubatingit, and measuring the residual VWF-cleaving proteaseactivity in these mixtures.
It is mandatory that these proposed assays be com-pared with each other aiming for standardizing and, thus,correctly appreciating the diagnostic value ofADAMTS13 activity values.
Hereditary ADAMTS13 Deficiency andConstitutional TTP (Upshaw-Schulman Syndrome)
In 1978 Upshaw21 reported a young woman having suf-fered since childhood from recurrent episodes of severemicroangiopathic hemolytic anemia and thrombocytope-nia who, on one occasion of acute illness, received wholeblood instead of the usually transfused red cell concen-trates. The better and faster response of the platelet count
and hemolysis to whole blood as compared to packedred cells led to the hypothesis that a plasma componentmight be effective in reversing the thrombocytopenia andhemolysis. This hypothesis was substantiated during anensuing attack that promptly responded to FFP infusion.Upshaw concluded that his patient was deficient in aplasma factor that protected from hemolytic anemia andthrombocytopenia.21
After establishing a severe, presumably hereditaryVWF-cleaving protease deficiency in 2 brothers withchronic relapsing TTP and half normal activity in theirparents,8 we have identified a further 26 subjects from19 families with ADAMTS13 activity less than 3-5%without a circulating inhibitor and slightly lowered pro-tease activity in their parents. According to the (some-times incomplete) clinical information available to us,there is a striking variability of the clinical phenotype,3
Figure 3. Activity of von Willebrand factor (VWF)-cleaving protease (ADAMTS13) in 68 patients with thrombocytopenia.
Multimer analysis of VWF substrate digested by diluted (1:20) plasma samples from patients. On each gel (A-H), a calibration curve usingdilutions of pooled plasma from healthy donors (NHP; 1:20 dilution corresponding to 100%) is included. Seventeen patients with severesepsis (gels A,B), 16 with heparin-induced thrombocytopenia (gels C,D), and 35 with thrombocytopenia due to various causes (gels E-H)were analyzed. Samples of digested VWF were applied on top of the gel.
OMF, osteomyelofibrosis; MDS, myelodysplastic syndrome; ITP, idiopathic thrombocytopenic purpura; AL, acute leukemia; SAA, severeaplastic anemia; Misc, miscellaneous causes.
Reprinted with permission from Bianchi V, et al. Von Willebrand factor-cleaving protease (ADAMTS13) in thrombocytopenic disorders: aseverely deficient activity is specific for thrombotic thrombocytopenic purpura. Blood. 2002;100:710-713.
322 American Society of Hematology
an observation also made by other investigators.22 Abouthalf of the patients identified in our laboratory3 had theirfirst TTP attack between the neonatal period and an ageof 5 years. Diagnosis was often delayed; in one childconstitutional TTP was recognized only after severalhospital admissions when severe ischemic brain lesionswere detected by magnetic resonance imaging;23 and sev-eral siblings of affected patients had died. Very recently,we encountered the case of an 8-year-old boy who diedafter several bouts of acute illness since childhood; thediagnosis of TTP was made only at autopsy, and VWF-cleaving protease activity in his premortem serum wasless than 3%, both parents showing about 50% of activ-ity. Pediatricians should be aware of the existence ofconstitutional TTP, given that effective treatment is avail-able (see below).
The other half of patients diagnosed by our labora-tory became clinically symptomatic only in adulthood,and 2 subjects are still asymptomatic at an age older than35 years.3 Most patients, both those with early and thosewith late onset of TTP, showed often multiple relapses
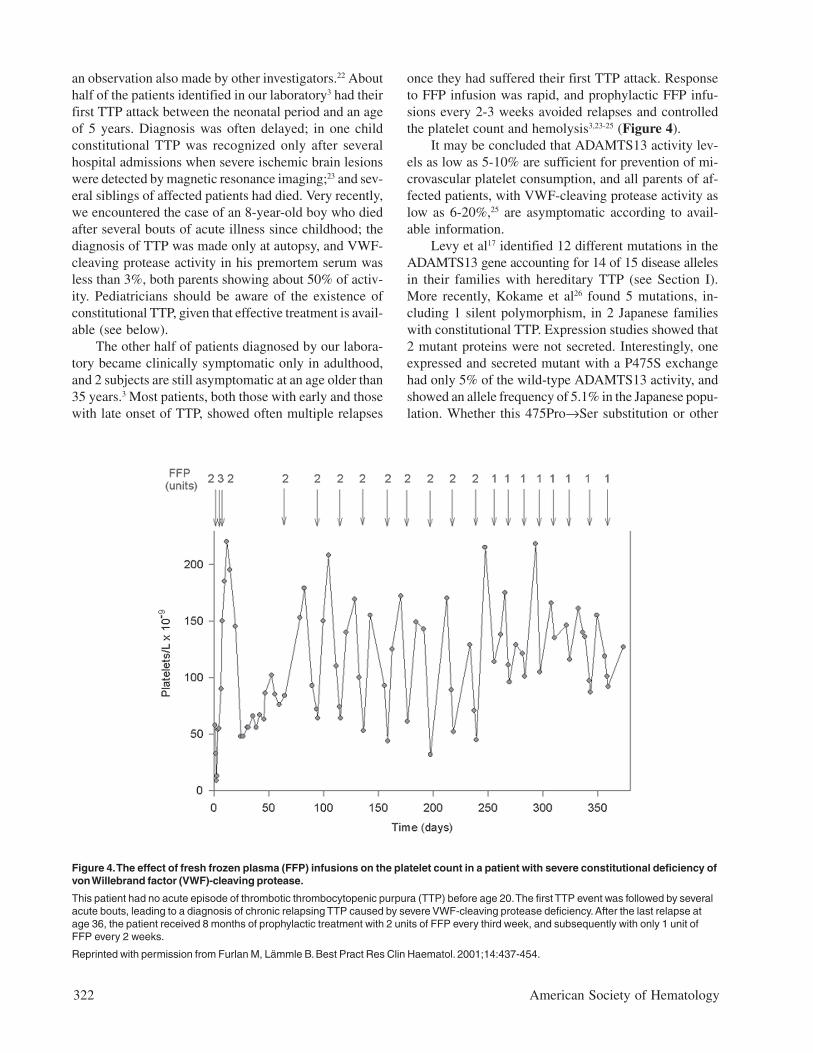
once they had suffered their first TTP attack. Responseto FFP infusion was rapid, and prophylactic FFP infu-sions every 2-3 weeks avoided relapses and controlledthe platelet count and hemolysis3,23-25 (Figure 4).
It may be concluded that ADAMTS13 activity lev-els as low as 5-10% are sufficient for prevention of mi-crovascular platelet consumption, and all parents of af-fected patients, with VWF-cleaving protease activity aslow as 6-20%,25 are asymptomatic according to avail-able information.
Levy et al17 identified 12 different mutations in theADAMTS13 gene accounting for 14 of 15 disease allelesin their families with hereditary TTP (see Section I).More recently, Kokame et al26 found 5 mutations, in-cluding 1 silent polymorphism, in 2 Japanese familieswith constitutional TTP. Expression studies showed that2 mutant proteins were not secreted. Interestingly, oneexpressed and secreted mutant with a P475S exchangehad only 5% of the wild-type ADAMTS13 activity, andshowed an allele frequency of 5.1% in the Japanese popu-lation. Whether this 475Pro→Ser substitution or other
Figure 4. The effect of fresh frozen plasma (FFP) infusions on the platelet count in a patient with severe constitutional deficiency ofvon Willebrand factor (VWF)-cleaving protease.
This patient had no acute episode of thrombotic thrombocytopenic purpura (TTP) before age 20. The first TTP event was followed by severalacute bouts, leading to a diagnosis of chronic relapsing TTP caused by severe VWF-cleaving protease deficiency. After the last relapse atage 36, the patient received 8 months of prophylactic treatment with 2 units of FFP every third week, and subsequently with only 1 unit ofFFP every 2 weeks.
Reprinted with permission from Furlan M, Lämmle B. Best Pract Res Clin Haematol. 2001;14:437-454.

Hematology 2002 323
high-frequency alleles with a similar effect explain therather wide normal range of about 50-150%, also inhealthy Caucasian subjects, remains to be investigated.
Specificity and Sensitivity ofADAMTS13 Deficiency for TTP
The specificity of VWF-cleaving protease deficiency forTTP has been recently challenged.27-29 Loof et al27 re-ported decreased VWF-cleaving protease activity (36%± 24% [mean ± SD] as compared to the activity of nor-mal pooled plasma) in 14 patients with disseminated in-travascular coagulation (DIC). Moore et al28 found mod-erately or severely decreased VWF-cleaving proteaseactivity in several patients with thrombocytopenic dis-orders different from TTP and even in some healthy con-trols. Moreover, Mannucci et al29 reported decreasedADAMTS13 activity in newborns; during the second andthird trimesters of pregnancy; in liver cirrhosis, uremia,and various acute inflammatory disorders; and in thepostoperative period. They concluded that VWF-cleav-ing protease deficiency was not a specific beacon of TTP.It should be noted, however, that most subjects with lowVWF-cleaving protease activity described by these au-thors29 had activity levels of some 20-50% and none hadvalues less than 10%.
We performed a formal study on 68 patients withthrombocytopenia not due to TTP or HUS, including 17with severe sepsis or septic shock, 16 with heparin-in-duced thrombocytopenia, and 35 with thrombocytope-nia caused by various hematologic conditions, such asidiopathic (immune) thrombocytopenic purpura, idio-pathic osteomyelofibrosis, myelodysplastic syndrome,acute leukemia, severe aplastic anemia, and miscella-neous disorders.30 Twelve of the 68 patients had a VWF-cleaving protease activity ≤ 30% of NHP, but none hada value < 10% (Figure 3). The median ADAMTS13 ac-tivity level in the 17 patients with severe sepsis was 40%(range 15-80%), being in agreement with the slightly ormoderately decreased values reported in patients withinflammatory conditions.29 The ADAMTS13 values inour series of thrombocytopenic patients30 contrast sharplywith our data in TTP patients.11 Our experience is basedon the analysis of more than 1500 plasma samples pro-vided by clinicians from more than 100 hospitals in some25 countries. As of June 2002, we have identified > 130patients with an ADAMTS13 activity < 5% of NHP.According to available clinical information, all had aclinical picture consistent with TTP with the exceptionof 3 cases: 2 subjects were brothers of affected siblingsfrom 2 families with adult-onset hereditary TTP, bothstill asymptomatic at age > 35 years.3 In addition, onechild with < 5% ADAMTS13 activity, caused by a tran-sient autoantibody, belonged to a series of 29 children
with Escherichia coli 0157:H7-associated D+ HUS.31
Therefore, in contrast to a recent editorial on thedebate,32 severe deficiency of VWF-cleaving proteaseactivity (< 5% of the activity in NHP) is a specific fea-ture for a thrombotic microangiopathy commonly diag-nosed as TTP.12,30
This statement is supported not only by the above-mentioned study on thrombocytopenic patients,30 but alsoby data on 74 hospitalized or healthy controls12 and 120previously investigated healthy subjects,11 all showingADAMTS13 activity levels of at least 45% of NHP.
Even though a severely deficient VWF-cleaving pro-tease activity is specific for TTP, the sensitivity of thislaboratory finding for the diagnosis of TTP remains ques-tionable. In retrospective large-scale studies, Furlan etal11 found 26 of 30 patients (86%) with TTP to lack anymeasurable protease activity, and Tsai and Lian12 re-ported a severe VWF-cleaving protease deficiency in37 of 37 patients (100%) with acute TTP. A recent ret-rospective study from Japan33 on 27 patients with throm-botic microangiopathy showed a VWF-cleaving protease< 3% due to a circulating inhibitor in 12 of 18 patients(66%) with TTP, the remaining 6 patients having activ-ity levels of 6-28%, whereas the 9 patients diagnosedwith HUS had values between 28-70%. These resultslargely confirmed previous studies from Europe.11,34 Inthis latter prospective multicenter study on 111 patientswith thrombotic microangiopathies, Veyradier et al34
found a severe ADAMTS13 deficiency in 47/66 patients(71%) being diagnosed as TTP, whereas most patientspresenting with HUS had normal or subnormal proteaseactivity.
Thus, with sensitivities of 66-100%, it is evident thatmany but not all patients being diagnosed with acute TTPhave severe ADAMTS13 deficiency. This suggests thatother pathogenetic factors1-3 may lead to a clinical con-dition indistinguishable from that seen with severe ac-quired or hereditary ADAMTS13 deficiency.
Patients with bone marrow transplantation–associ-ated thrombotic microangiopathy had normal35 and 4 pa-tients with disseminated neoplasia-associated thromboticmicroangiopathy had subnormal or normal ADAMTS13activity.36 In contrast, ticlopidine-induced37 and possi-bly clopidogrel-induced thrombotic microangiopathy38
were reported to be associated with severe, autoantibody-mediated ADAMTS13 deficiency, as was the thromboticmicroangiopathy in one patient with acquired immuno-deficiency syndrome.39

324 American Society of Hematology
Value of Measuring ADAMTS13 Activity andIts Inhibitors for the Diagnosis of andTreatment Decisions in TTP
Diagnosis of acute TTP is often both difficult and ur-gent.40 It may be difficult because not all patients withTTP will show the complete pentad of diagnostic crite-ria,40,41 and clinical diagnosis may have to be based onthrombocytopenia and microangiopathic (schistocytic)hemolysis that are not explained by another condition,even in the absence of ischemic organ dysfunction40 (seeSection III). Moreover, the diagnosis is urgent becausemortality in untreated patients may be as high as 80-90%40,41 and plasma exchange with replacement by FFPhas reduced the mortality to about 20%.40,42 Recognitionof a large proportion of acute idiopathic TTP patients asdisplaying an autoantibody-mediated severe VWF-cleav-ing protease deficiency may explain the empirically es-tablished effectiveness of plasma exchange with FFPreplacement that removes the inhibiting autoantibody andsubstitutes the lacking ADAMTS13 activity. Still, I be-lieve that based on the present evidence it would not bejustified or appropriate to withhold plasma exchange orFFP infusion in a patient with a bona fide clinical diag-nosis of TTP showing normal ADAMTS13 activity.
The retrospective study by Mori et al33 is notewor-thy. Ten of the 12 TTP patients with a severe acquiredADAMTS13 deficiency survived the acute episode,whereas 4 of 6 patients with moderate protease defi-ciency (~25% activity) died. These results may suggestthat treatment with plasma exchange is not the optimaltherapy for a thrombotic microangiopathy that is not dueto severe acquired VWF-cleaving protease deficiency.However, in the absence of knowledge on the underly-ing pathophysiology, and hence specific therapy, stan-dard treatment with plasma exchange still seems man-datory until improved therapeutic measures based on theunderlying pathogenesis become available.
There is an indication that corticosteroids may haveefficacy in TTP.43 Even though no controlled study isavailable, the demonstration of autoantibodies abolish-ing VWF-cleaving protease activity in many cases ofTTP would certainly support their use. Splenectomy hasbeen performed empirically in desperate cases of plasmarefractory or frequently relapsing TTP44,45 and we haveprovided evidence that its efficacy may be related to theelimination of the autoantibody-producing B cells.10 Es-tablishment of a high-titer autoantibody inhibitingADAMTS13 activity would certainly facilitate the deci-sion to proceed to splenectomy, which may entail con-siderable risk in a patient with ongoing active TTP.
The relapse rate in patients surviving an initial TTPattack is rather high.40 The observation of disappearancewith remission and reappearance of an autoantibody in-
hibiting ADAMTS13 heralding a TTP relapse within afew weeks in our initially studied patient with acquiredTTP10 may suggest that assaying ADAMTS13 activity andits inhibitor could be useful for estimating the relapse riskin patients having survived an initial bout of TTP.
At present, probably the best clinical indication formeasuring VWF-cleaving protease and its inhibitor is todistinguish between acquired and hereditary ADAMTS13deficiency. A severely deficient VWF-cleaving proteaseactivity in a symptomatic patient, a lack of circulatinginhibitor, and mildly decreased activity levels in the par-ents prove hereditary TTP. Treatment of acute attacksusing simple FFP infusion instead of performing plasmaexchange is in order for these rare patients,21-25 and pro-phylactic FFP infusions are warranted to prevent other-wise frequent relapses3,23-25 (Figure 4).
Measuring ADAMTS13 activity in other clinicalconditions, such as sepsis, heparin-induced thrombocy-topenia, various inflammatory disorders, disseminatedneoplasia, and other conditions, is not routinely indicatedbut may—if performed in the setting of appropriate clini-cal studies—further our understanding of this novelmember of the ADAMTS family of metalloproteases.
Conclusions• The rather complex assays reported for the measure-
ment of ADAMTS13 activity need comparison andstandardization in the frame of multisite studies.Severely diminished activity values (< 3-5%) mustbe distinguished from moderately (10-25%) orslightly (25-50%) decreased values, the former be-ing a specific beacon of TTP.
• Hereditary TTP is often, if not always, caused bysevere ADAMTS13 functional deficiency, and vari-ous causal mutations of the ADAMTS13 gene havebecome known. Some childhood-onset cases ofUpshaw-Schulman syndrome are probably not di-agnosed during the life time because pediatriciansare not always familiar with this rare but potentiallyfatal disease for which clearly effective treatment isavailable. On the other hand, a sizable proportionof severely ADAMTS13-deficient subjects will notbecome symptomatic until adulthood. It is unclearat present whether this phenotypic variability is dueto different genetic defects or variable residualADAMTS13 activity levels below the detectionthreshold of current assays, or whether other con-stitutional factors or circumstantial triggering events,such as endothelial activation or damage, may benecessary to provoke acute TTP.
• Sporadic idiopathic TTP is often caused by autoan-tibodies inhibiting ADAMTS13 activity, but other

Hematology 2002 325
pathogenetic mechanisms may lead to a clinicallyindistinguishable thrombotic microangiopathy.
• A diagnosis of acute sporadic TTP without under-lying severe acquired VWF-cleaving protease defi-ciency should not—at present—preclude the patientfrom being subjected to plasma exchange therapy.However, the distinction between hereditary and ac-quired ADAMTS13 deficiency is of major importancebecause patient management will be different.
III. EVALUATION AND MANAGEMENT OF PATIENTS
WITH CLINICALLY SUSPECTED
THROMBOTIC THROMBOCYTOPENIC PURPURA OR
HEMOLYTIC-UREMIC SYNDROME
James N. George, MD*
The diagnosis of TTP is appropriately suspected in apatient who has acute and severe thrombocytopenia andmicroangiopathic hemolytic anemia, without anotherexplanation.1-3 But these are obviously non-specific cri-teria; there is no “gold standard” for diagnosis. TTP canbe associated with a deficiency of ADAMTS13 (seeSection I) and a severe deficiency of ADAMTS13 (<5% activity) may be specific for TTP (see Section II).However, severe ADAMTS13 deficiency does not de-fine TTP and is not a definitive diagnostic test. The sen-sitivity of severe ADAMTS13 deficiency remains un-known: patients with only moderate ADAMTS13 defi-ciency or even normal activity can have presenting fea-tures and clinical courses, including response to plasmaexchange treatment, identical to patients with severedeficiency. The difficulty of diagnosis is emphasized byobservations that among patients diagnosed with TTPand who have severe ADAMTS13 deficiency, approxi-mately half have normal renal function and approxi-mately one-fourth have no neurologic abnormalities.These observations are consistent with the current diag-nostic criteria of only thrombocytopenia and micro-angiopathic hemolytic anemia.1-3
The frequent uncertainty about the diagnosis of TTPmakes management decisions difficult. The most impor-tant initial decision is whether or not to begin plasmaexchange, a treatment with documented efficacy1 but alsowith a high risk for major complications (discussed fullybelow and in ref. 4).
This, therefore, is the clinical dilemma: uncertaindiagnosis of a disorder that can be rapidly fatal, and avail-
ability of an effective treatment which has major risks.Table 1 describes the clinical situations in which pa-tients with suspected TTP are encountered. Each of theseclinical categories has distinct diagnostic and manage-ment issues; they are the basis for a physician’s evalua-tion and for this discussion. The distinction of adult syn-dromes as either TTP or HUS is not important for theinitial management decision regarding plasma exchange.Patients with acute renal failure, a defining criterion forHUS, may respond as well to plasma exchange as pa-tients with normal renal function. Therefore all adultsyndromes are described in this discussion simply as TTP,even if acute renal failure is present.
* Hematology-Oncology Section, Department of Medicine,University of Oklahoma Health Sciences Center, P.O. Box26901, Oklahoma City, OK 73190
Table 1. Thrombotic thrombocytopenic purpura (TTP) andhemolytic-uremic syndrome (HUS): clinical presentations andassociated conditions.
Children
• “Typical” HUS(prodrome of bloody diarrhea caused by an enterohemorrhagicE. coli strain; predominant acute renal failure)
• “Atypical” HUS(predominant acute renal failure, no diarrhea prodrome)
• TTP(syndromes with no or minimal renal insufficiency are uncommonin children)
Adults
• Bone marrow transplantation(in most patients, the etiology of the acute disorder diagnosed asTTP is actually sepsis and/or acute GVHD)
• Pregnancy(75% occur peripartum/postpartum; distinction from preeclamp-sia, eclampsia, and HELLP syndromes may be impossible)
• Drug-associated
o Acute, immune-mediated toxicity(quinine most common; also ticlopidine, clopidogrel)
o Insidious, dose-related toxicity(mitomycin C, alpha-interferon, cyclosporine, tacrolimus, otherchemotherapeutic and immunosuppressive agents)
• Bloody diarrhea prodrome(as in typical HUS of children, usually caused byenterohemorrhagic E. coli; however in adults, may or may not beassociated with renal failure)
• Autoimmune disorders(patients with systemic lupus erythematosus, anti-phospholipidantibody syndrome, scleroderma, and polyarteritis nodosa canhave signs suggesting consideration of TTP. In some patients,pathologic lesions and response to plasma exchange treatmentare consistent with TTP as an additional diagnosis)
• Alternative disorders that may mimic TTP(sepsis [e.g., meningococcus, cytomegalovirus, aspergillosis,rocky mountain spotted fever], disseminated malignancy,malignant hypertension)
• Idiopathic, “typical” TTP(may have a prolonged clinical course with multiple exacerba-tions and subsequent relapses)

326 American Society of Hematology
Childhood Syndromes“Typical HUS” is a frequent diagnostic term used forchildren, usually less than 5 years old, who present witha prodrome of bloody diarrhea caused by an enterohemor-rhagic strain of E. coli, usually E. coli O157:H7.5-7 E. coliO157:H7 and related organisms produce Shiga toxin,which causes the diarrhea prodrome and further causesacute HUS in 5-10% of infected children.8 Acute renalfailure is the principal abnormality; the associated throm-bocytopenia and microangiopathic hemolytic anemia arenot critical problems; conventional management is onlysupportive care for the renal failure; mortality in largecase series is 3-5%.5-7 Because mortality is not great,because plasma infusion has no clearly documented ef-ficacy,9,10 and because plasma exchange catheter inser-tion may have greater risks in small children, plasmaexchange treatment is rarely considered. However ifchildren with HUS have an “atypical” presentation (i.e.,without a diarrhea prodrome), mortality may be greater,spontaneous remissions are less certain, and plasma ex-change may be considered.11 Rarely, children maypresent with minimal or no renal insufficiency, compa-rable to “typical TTP” in adults. These children, likeadults, do not spontaneously resolve and plasma ex-change treatment is required.
Adult Syndromes
Bone marrow transplantationAlthough there are many case series describingTTP and/or HUS following bone marrow trans-plantation (BMT), whether this is a valid diagno-sis is uncertain. Uncertainty is emphasized by theextreme variation of both the reported incidenceof TTP following BMT (2-76% following alloge-neic BMT, 0-27% following autologous BMT) andits mortality (0-93%).12 Inconsistency of diagno-sis and clinical outcomes is inevitable in these pa-tients who have multiple, severe transplant-relatedcomplications which share many clinical featureswith TTP. Diagnosis of TTP following allogeneicBMT is further complicated by dose-dependentrenal and neurotoxicities of the GVHD prophy-laxis/treatment medications, cyclosporine andFK506 (see Table 1), which can mimic TTP orperhaps may even cause a TTP-like syndrome.
Table 2 describes the experience with patientswho had clinically suspected TTP following BMTat the University of Oklahoma.13 These patientshad greater risks for transplant-related complica-tions as well as a greater frequency of complica-tions. In the 17 patients who were treated withplasma exchange for suspected TTP, sepsis and/
or acute GVHD could have caused the signs suggestingthe diagnosis of TTP. Although plasma exchange forpossible TTP may seem appropriate for a patient withcritical deterioration of uncertain cause, the outcomesare poor. None of these patients responded to plasmaexchange and only 1 of 17 patients currently survives.Therefore our practice for patients with clinically sus-pected TTP following BMT is to intensify efforts to di-agnose and treat GVHD and sepsis, and to delay a deci-sion for plasma exchange. This practice is distinct fromthe urgency for initiating plasma exchange in other pa-tients with clinically suspected TTP.
Pregnancy/postpartumClinical features suggesting a diagnosis of TTP in preg-nant or postpartum women are described in Table 3. Thedistinction of TTP from pregnancy-related complica-tions, pre-eclampsia/eclampsia/HELLP syndrome, maybe impossible.14-16 However the association of TTP withpregnancy is clear. In large published case series of TTP,approximately 70% of patients are women; in 12-25%of all women, TTP is diagnosed during pregnancy orpostpartum, with 75% of episodes occurring around thetime of delivery.14 The association of pregnancy withTTP is further documented by five reports of familialTTP in which sisters had their initial episode at the endof their first pregnancy.14,17,18
Table 2. Clinical features of patients with suspected thromboticthrombocytopenic purpura following allogeneic bone marrowtransplantation.*
Patients with Patients inClinically Whom TTP Was
Suspected TTP Not Suspected(n = 17) (n = 245) P
Risk factors for BMT-related complications
Severe primary disease 29% 13% 0.08
>1 transplant 18% 9% 0.20
Unrelated donor 71% 39% 0.02
HLA mismatch 35% 16% 0.04
Occurrence of BMT-related complications
Acute GVHD (grade III-IV) 47% 13% <0.01
Bacterial sepsis 82% 57% 0.04
Viral sepsis 65% 16% <0.01
Fungal sepsis 65% 28% <0.01
* Adapted from reference 13. These data describe the experience withallogeneic BMT at the University of Oklahoma, 1989-1998. No patients hadclinically suspected TTP following autologous stem cell transplants. Clinicallysuspected TTP was defined by treatment with plasma exchange. TTP is usedas a comprehensive diagnostic term, including syndromes with acute renalfailure that may be described as HUS.

Hematology 2002 327
The decision to initiate plasma exchange in a womanwith suspected TTP who is pregnant or postpartum isoften difficult. The thrombocytopenia and microangio-pathic hemolytic anemia associated with pre-eclampsia/HELLP syndrome can be severe, and may only occurfollowing delivery.19-21 Seizures, defining eclampsia, of-ten first occur after delivery.22 By definition, these syn-dromes spontaneously resolve within several days fol-lowing delivery, though case reports describe more pro-longed courses. The decision for plasma exchange isbased on an estimate of the chance for spontaneous reso-lution versus the risk of progressive multi-organ failureand death (Table 3). If the woman is not acutely ill, ob-servation for several days after delivery may allow timefor spontaneous resolution to begin;23 women with moreacute and severe multi-organ failure require promptplasma exchange.
The risk for recurrent TTP with a subsequent preg-nancy is a difficult issue. Subsequent pregnancies havea risk for recurrence of TTP. However even in womenwith prolonged, severe, and relapsing TTP following anearlier pregnancy, a subsequent pregnancy can be un-complicated with delivery of a healthy infant (see “TTP,the patient’s perspective: Christy’s story,” http://moon.ouhsc.edu/jgeorge).
Drug associationDrug-associated syndromes appear to be of two types:acute, immune-mediated syndromes caused by drug-dependent antibodies and insidious, dose-related toxiceffects.24 In our experience, quinine is by far the mostcommon cause of immune-mediated TTP.25 This is notdescribed in some discussions of drug-induced TTP26
because quinine-associated syndromes are often consid-ered to be HUS, rather than TTP. However the clinicalfeatures of quinine-associated syndromes emphasize theinability to distinguish TTP from HUS. Although acuterenal failure is a common manifestation, some patientshave no renal insufficiency. Even more striking, one re-ported patient had acute renal failure with her first epi-sode and no renal insufficiency with two subsequentquinine-induced episodes.25 The frequency of neurologicabnormalities and the severity of thrombocytopenia andanemia are not different in patients with quinine-associ-ated TTP than in other patients with TTP; the serum LDHincrease is greater, emphasizing the severity of systemicischemia.25 The severe systemic ischemia may explainthe extremely rapid rise of serum creatinine in patientswith acute renal failure, far greater than the assumedmaximum daily increase of 0.5-1.0 mg/dL per day, de-rived from observations on anephric patients. The char-acteristic presenting features of quinine-associated TTPare the sudden onset of nausea, vomiting, diarrhea (oc-casionally bloody), fever, and chills occurring severalhours after quinine ingestion.25 Sepsis is a common ini-tial, incorrect diagnosis.27 Patients may have severe men-tal status abnormalities, even coma.25 In addition to thesigns of TTP, leukopenia, disseminated intravascular co-agulation, and liver function abnormalities may occur,all of which can be caused by quinine sensitivity.28-30
These syndromes are caused by quinine-dependent an-tibodies to epitopes on multiple cell types.28 Plasma ex-change treatment appears to be effective.
Quinine is also one of the most common drugs thatcan cause isolated thrombocytopenia.31 In patients withisolated thrombocytopenia, the absence of anemia andany other systemic symptoms and signs exclude consid-eration of TTP. However, one important case report de-scribes a patient with quinine-induced isolated thromb-ocytopenia who developed TTP following a subsequentexposure to quinine.32 This observation is consistent withthe development of only quinine-dependent antiplateletantibodies causing the initial episode and the subsequentdevelopment of quinine-dependent antibodies to mul-tiple tissues causing the systematic disorder of TTP.32
Ticlopidine24,33 and clopidogrel24,34 have also beenreported to be associated with TTP and HUS. In someof these patients, a deficiency of ADAMTS13 and thepresence of inhibitors of ADAMTS13 activity have been
Table 3. Clinical features suggesting a diagnosis of thromboticthrombocytopenic purpura (TTP) and consideration for plasmaexchange treatment in pregnant/postpartum women.
Hematologic abnormalities
• Severe thrombocytopenia and microangiopathic hemolyticanemia that continue to worsen > 3 days after delivery
• Normal coagulation parameters, or resolving DIC
Neurologic parameters
• Severe mental status abnormalities
• Focal abnormalities (e.g., aphasia or focal motor deficits)
• Seizures not associated with severe hypertension, oroccurring > 3 days after delivery
Renal abnormalities
• Oliguric acute renal failure
Systemic abnormalities
• Resolving hypertension
• Progressive abnormality of serum lactate dehydrogenase(LDH) associated with resolving abnormalities of liver functiontests (serum alanine aminotransferase [ALT] and aspartateaminotransferase [AST])
Adapted from reference 14. These abnormalities may all be due tosevere preeclampsia/eclampsia/HELLP syndrome, and may allresolve spontaneously following delivery with only continuedobservation and supportive care. However, the clinical featuresdescribed here suggest progressive disease and are appropriateindications for plasma exchange treatment, for presumptive TTP.

328 American Society of Hematology
observed.34,35 However, drug-dependence of the antibod-ies to ADAMTS13 has not been documented, and there-fore how an acute, apparently immune-mediated drugreaction may be associated with these autoantibodies isunknown. In patients with ticlopidine-associated TTP,the ticlopidine had been taken for less than 2 weeks in15% of patients and less than one month in 80% of pa-tients.33 In the largest report on clopidogrel-associatedTTP, the onset of TTP was 3-14 days after beginningclopidogrel in 10 of 11 patients. However since 2 of the11 reported patients had recurrences of TTP without re-exposure to clopidogrel,34 the initial drug association mayhave been coincidental.
In contrast to the acute onset of immune-mediateddrug-associated TTP, the onset of syndromes, often de-scribed as HUS, following mitomycin C, cyclosporine,and related immunosuppressive agents is insidious andmay only become manifest after the drug has been dis-continued. These toxic syndromes have the same renalpathology as TTP and HUS,24 but hematologic manifes-tations may be minimal. The efficacy of plasma exchangeis uncertain. In some patients with cyclosporine ortacrolimus-associated TTP, simple discontinuation ordose adjustment of the drug is sufficient to reverse theprocess.
Prodrome of bloody diarrheaSimilar to the typical HUS of young children,enterohemorrhagic strains of E. coli can also cause acuteTTP or HUS in adults.36 Although these syndromes maybe similar to HUS in children, in some adults no renalinsufficiency occurs,36-38 supporting the use of a com-prehensive diagnostic term, TTP-HUS, or simply TTP.Although children with typical HUS are not treated withplasma exchange, mortality is high in adults and plasmaexchange may be efficacious.37 Bloody diarrhea may alsooccur in patients who have severe ADAMTS13 defi-ciency, without a documented enteric infection.
Association with autoimmune disordersIn some patients with severe multi-organ failure causedby an established diagnosis of systemic lupus erythema-tosus, antiphospholipid antibody syndrome, scleroderma,or polyarteritis nodosa, the clinical and pathologic fea-tures may be indistinguishable from TTP.39-41 In thesecritically ill patients, a trial of plasma exchange treat-ment for presumed TTP may be appropriate, in additionto intensive immunosuppressive treatment.
Alternative disorders that may mimic TTPSince the diagnostic criteria for TTP are not specific andsince there is urgency to begin plasma exchange, an al-ternative explanation for acute multi-organ failure, such
as sepsis,42 disseminated malignancy,43,44 or malignanthypertension,45 may become apparent after plasma ex-change is begun. This experience suggests that somepatients who die with apparently refractory TTP and whodo not have an autopsy could have had a disorder otherthan TTP. Although it is often stated that HIV-positivepatients are at greater risk for developing TTP,46 thereare no epidemiologic data to support a causal associa-tion of HIV infection and TTP. The abnormalities sug-gesting a diagnosis of TTP in HIV-positive patients mayonly be features of opportunistic infections.47 Therefore,similar to the management of patients following BMTdescribed above, it is appropriate to carefully consideralternative diagnoses, specifically infectious etiologies,before initiating plasma exchange treatment.
Idiopathic TTPWhen all of the preceding disorders are excluded, pa-tients are considered to have idiopathic TTP, or “typicalTTP,” presumably caused by an acquired autoantibodyto ADAMTS13. However, even patients within the idio-pathic category are heterogeneous. Among the subsetof patients within the idiopathic category who have asevere deficiency of ADAMTS13, some may have a rap-idly fatal course with multi-organ failure while othersmay have no neurologic or renal manifestations and re-quire only few plasma exchange treatments to achieve adurable remission.
ManagementOnce the diagnosis of TTP is made (or seriously consid-ered), urgent plasma exchange is indicated. If plasmaexchange is unavailable, as in a distant rural site, initialtreatment with plasma infusion and glucocorticoids isappropriate until the patient is moved to a facility withplasma exchange ability. Since plasma exchange isclearly superior to plasma infusion,1 the patient shouldbe moved as soon as possible.
Once the decision for plasma exchange is made, ourtreatment plan is to exchange one plasma volume oncedaily.3 In patients with severe disease who do not re-spond within the first several days, or in patients whoimprove and then exacerbate while on daily plasma ex-change, twice daily plasma exchange may be more ef-fective. Whole FFP and the cryosupernatant fraction ofplasma appear to be equally efficacious.48 Glucocorti-coids are frequently used, and their potential efficacy issupported by observations that autoantibodies toADAMTS13 may be the etiology in some patients. How-ever, many patients respond promptly and completelywithout glucocorticoid treatment,1 even patients whohave severe ADAMTS13 deficiency with an associatedinhibitor. Therefore, reasonable practice in patients who

Hematology 2002 329
are not critically ill may be to observe the response toplasma exchange alone, adding glucocorticoids if theresponse is not prompt or complete, or if an exacerba-tion occurs when plasma exchange frequency is dimin-ished.3 Rarely are patients truly refractory to plasmaexchange. Continued plasma exchange will achieve re-sponses and ultimately remissions in most every patient.The benefit of additional immunosuppressive therapy orof splenectomy is uncertain.3 There are case reports andsmall series of selected patients describing efficacy ofsplenectomy, vincristine, cyclophosphamide, rituximab,and other immunosuppressive agents, but the unpredict-able clinical course of TTP makes interpretation of thesereports uncertain.
Complications of Plasma ExchangeAlthough plasma exchange is often considered to be asafe procedure, the risks are great in patients treated forTTP, perhaps because of the severity of illness and therequirement for prolonged treatment in many patients(Table 4).4 Deaths, or cardiac arrest with near-death, maybe caused by complications of central venous catheterinsertion, such as hemorrhage, pneumothorax, or peri-cardial tamponade due to cardiac perforation by the cen-tral venous catheter guide wire. Allergic reactions toplasma can cause severe hypotension and hypoxia. Sep-sis related to the central venous catheter may be fatal.
An unappreciated complication of plasma exchangeis unintentional plateletpheresis, causing persistentthrombocytopenia which may be interpreted as continu-ing active TTP and may result in inappropriate treat-ment.49 In our studies, the Fresenius AS 104 apheresisinstrument caused greater platelet losses than eitherCOBE Spectra or Haemonetics LN-9000 instruments,but platelet loss occurred with all 3 instruments.49 Inap-propriate apheresis instrument settings that acceleratethe procedure by removing plasma closer to the centri-fuged cell layer can also cause unintentional platelet-pheresis.
Long-Term Clinical OutcomesThe risk for relapse is related to the clinical categoryand presumably related to the presence of autoantibod-ies causing ADAMTS13 deficiency. Among patients withno renal failure who have severe ADAMTS13 deficiency,the risk for relapse may be greater than 50% over thefirst 5 years following recovery. Most relapses occurwithin the first year of the initial episode, although re-lapses after more than 10 years of complete remissionmay occur. Patients may have multiple relapses, althoughthe occurrence of only a single relapse may be morecommon. Mortality is minimal with recurrence of TTP,because the patient responded to plasma exchange pre-viously and because there is no delay in the diagnosisand therefore less delay in the initiation of treatment.
The incidental observation of asymptomatic throm-bocytopenia during follow-up of a patient who has re-covered from TTP creates a difficult issue. Withoutknowledge of a patient’s history, TTP would never besuspected, yet asymptomatic thrombocytopenia may bethe first sign of an acute relapse. In some patients, inter-mittent thrombocytopenia has been observed for severalmonths prior to their first episode of TTP. Some descrip-tions suggest the occurrence of ITP and TTP in the samepatient at different times.50 This may be analogous tothe report of quinine-induced thrombocytopenia followedon the next occasion of quinine exposure by TTP, as thespectrum of drug-dependent antibodies broadens fromonly antiplatelet antibodies to antibodies reacting withmultiple cell types in multiple organs.32
Many patients describe incomplete recovery ofphysical and cognitive abilities even though all objec-tive parameters suggest complete remission. These ob-servations suggest that even after complete hematologicand renal function recovery, the effects of profound sys-temic ischemia may continue for months or years. For-tunately, continued observations suggest improvementin most patients.51
Table 4. Complications of plasma exchange treatment forthrombotic thrombocytopenic purpura (TTP).*
Percentage ofPatients with
Complications Major Complications
Central venous catheter-related
Insertion procedure 4%
Sepsis 15%
Thrombosis 10%
Plasma-related
Allergic 4%
Infection 0
Instrument-related
Unintentional plateletpheresis Unknown
* Adapted from references 4 and 49. Major catheter-relatedcomplications included pneumothorax or hemothorax caused by theinsertion procedure; sepsis requiring systemic antimicrobialtreatment; and thrombosis requiring line replacement or anticoagu-lant treatment. Major plasma-related complications includedhypoxemia or hypotension requiring more than oxygen administra-tion or volume replacement and allergic reactions requiring morethan Benadryl and hydrocortisone treatment. No transfusion-transmitted infections were observed. Unintentional plateletpheresismay cause persistent thrombocytopenia, that may be mistaken forcontinued activity of TTP.

330 American Society of Hematology
REFERENCES
I. ADAMTS13, Von Willebrand Factor,and the Pathophysiology ofThrombotic Thrombocytopenic Purpura
1. Amorosi EL, Ultmann JE. Thrombotic thrombocytopenicpurpura: report of 16 cases and review of the literature.Medicine. 1966;45:139-159.
2. Rock GA, Shumak KH, Buskard NA, et al. Comparison ofplasma exchange with plasma infusion in the treatment ofthrombotic thrombocytopenic purpura. Canadian ApheresisStudy Group. N Engl J Med. 1991;325:393-397.
3. Moschcowitz E. An acute febrile pleiochromic anemia withhyaline thrombosis of the terminal arterioles and capillaries.Arch Intern Med. 1925;36:89-93.
4. Moake JL, Rudy CK, Troll JH, et al. Unusually large plasmafactor VIII: von Willebrand factor multimers in chronicrelapsing thrombotic thrombocytopenic purpura. N Engl J Med.1982;307:1432-1435.
5. Furlan M, Robles R, Solenthaler M, Wassmer M, Sandoz P,Lämmle B. Deficient activity of von Willebrand factor-cleavingprotease in chronic relapsing thrombotic thrombocytopenicpurpura. Blood. 1997;89:3097-3103.
6. Furlan M, Robles R, Solenthaler M, Lämmle B. Acquireddeficiency of von Willebrand factor-cleaving protease in apatient with thrombotic thrombocytopenic purpura. Blood.1998;91:2839-2846.
7. Tsai HM, Lian EC. Antibodies to von Willebrand factor-cleaving protease in acute thrombotic thrombocytopenicpurpura. N Engl J Med. 1998;339:1585-1594.
8. Furlan M, Robles R, Lämmle B. Partial purification andcharacterization of a protease from human plasma cleaving vonWillebrand factor to fragments produced by in vivo proteolysis.Blood. 1996;87:4223-4234.
9. Tsai H-M. Physiologic cleavage of von Willebrand factor by aplasma protease is dependent on its conformation and requirescalcium ion. Blood. 1996;87:4235-4244.
10. Fujikawa K, Suzuki H, McMullen B, Chung D. Purification ofhuman von Willebrand factor-cleaving protease and itsidentification as a new member of the metalloproteinase family.Blood. 2001;98:1662-1666.
11. Gerritsen HE, Robles R, Lämmle B, Furlan M. Partial aminoacid sequence of purified von Willebrand factor-cleavingprotease. Blood. 2001;98:1654-1661.
12. Zheng X, Chung D, Takayama TK, Majerus EM, Sadler JE,Fujikawa K. Structure of von Willebrand factor-cleavingprotease (ADAMTS13), a metalloprotease involved inthrombotic thrombocytopenic purpura. J Biol Chem.2001;276:41059-41063.
13. Soejima K, Mimura N, Hirashima M, et al. A novel humanmetalloprotease synthesized in the liver and secreted into theblood: possibly, the von Willebrand factor-cleaving protease? JBiochem. 2001;130:475-480.
14. Levy GG, Nichols WC, Lian EC, et al. Mutations in a memberof the ADAMTS gene family cause thrombotic thrombocy-topenic purpura. Nature. 2001;413:488-494.
15. Hurskainen TL, Hirohata S, Seldin MF, Apte SS. ADAM-TS5,ADAM-TS6, and ADAM-TS7, novel members of a new familyof zinc metalloproteases: general features and genomicdistribution of the ADAM-TS family. J Biol Chem.1999;274:25555-25563.
16. Kaushal GP, Shah SV. The new kids on the block: ADAMTSs,potentially multifunctional metalloproteinases of the ADAMfamily. J Clin Invest. 2000;105:1335-1337.
17. Bork P, Beckmann G. The CUB domain: a widespread modulein developmentally regulated proteins. J Mol Biol.1993;231:539-545.
18. Cal S, Obaya AJ, Llamazares M, Garabaya C, Quesada V,Lopez-Otin C. Cloning, expression analysis, and structuralcharacterization of seven novel human ADAMTSs, a family ofmetalloproteinases with disintegrin and thrombospondin-1domains. Gene. 2002;283:49-62.
19. Kuno K, Matsushima K. ADAMTS-1 protein anchors at theextracellular matrix through the thrombospondin type I motifsand its spacing region. J Biol Chem. 1998;273:13912-13917.
20. Adams JC. Thrombospondins: multifunctional regulators of cellinteractions. Annu Rev Cell Dev Biol. 2001;17:25-51.
21. Panetti TS, Kudryk BJ, Mosher DF. Interaction of recombinantprocollagen and properdin modules of thrombospondin-1 withheparin and fibrinogen/fibrin. J Biol Chem. 1999;274:430-437.
22. Sieron AL, Tretiakova A, Jameson BA, et al. Structure andfunction of procollagen C-proteinase (mTolloid) domainsdetermined by protease digestion, circular dichroism, bindingto procollagen type I, and computer modeling. Biochemistry.2000;39:3231-3239.
23. Moschcovich L, Bernocco S, Font B, et al. Folding and activityof recombinant human procollagen C-proteinase enhancer. EurJ Biochem. 2001;268:2991-2996.
24. Furlan M, Robles R, Morselli B, Sandoz P, Lämmle B.Recovery and half-life of von Willebrand factor-cleavingprotease after plasma therapy in patients with thromboticthrombocytopenic purpura. Thromb Haemost. 1999;81:8-13.
25. Sasahara Y, Kumaki S, Ohashi Y, et al. Deficient activity of vonWillebrand factor-cleaving protease in patients with Upshaw-Schulman syndrome. Int J Hematol. 2001;74:109-114.
26. Kinoshita S, Yoshioka A, Park YD, et al. Upshaw-Schulmansyndrome revisited: a concept of congenital thromboticthrombocytopenic purpura. Int J Hematol. 2001;74:101-108.
27. Dent JA, Berkowitz SD, Ware J, Kasper CK, Ruggeri ZM.Identification of a cleavage site directing the immunochemicaldetection of molecular abnormalities in type IIA vonWillebrand factor. Proc Natl Acad Sci U S A. 1990;87:6306-6310.
28. Tsai HM, Sussman II, Ginsburg D, Lankhof H, Sixma JJ, NagelRL. Proteolytic cleavage of recombinant type 2A vonWillebrand factor mutants R834W and R834Q: inhibition bydoxycycline and by monoclonal antibody VP-1. Blood.1997;89:1954-1962.
29. Furlan M, Robles R, Galbusera M, et al. von Willebrand factor-cleaving protease in thrombotic thrombocytopenic purpura andthe hemolytic-uremic syndrome. N Engl J Med.1998;339:1578-1584.
30. Schulman I, Pierce M, Lukens A, Currimbhoy Z. Studies onthrombopoiesis, I: a factor in normal human plasma requiredfor platelet production; chronic thrombocytopenia due to itsdeficiency. Blood. 1960;16:943-957.
31. Upshaw JD Jr. Congenital deficiency of a factor in normalplasma that reverses microangiopathic hemolysis and thromb-ocytopenia. N Engl J Med. 1978;298:1350-1352.
32. Hara T, Kitano A, Kajiwara T, Kondo T, Sakai K, Hamasaki Y.Factor VIII concentrate-responsive thrombocytopenia,hemolytic anemia, and nephropathy: evidence that factorVIII:von Willebrand factor is involved in its pathogenesis. Am JPediatr Hematol Oncol. 1986;8:324-328.
33. Yagi H, Konno M, Kinoshita S, et al. Plasma of patients withUpshaw-Schulman syndrome, a congenital deficiency of vonWillebrand factor-cleaving protease activity, enhances theaggregation of normal platelets under high shear stress. Br JHaematol. 2001;115:991-997.

Hematology 2002 331
34. Ajzenberg N, Denis CV, Veyradier A, Girma JP, Meyer D,Baruch D. Complete defect in vWF-cleaving protease activityassociated with increased shear-induced platelet aggregation inthrombotic microangiopathy. Thromb Haemost. 2002;87:808-811.
35. Randi AM, Rabinowitz I, Mancuso DJ, Mannucci PM, SadlerJE. Molecular basis of von Willebrand disease type IIB:candidate mutations cluster in one disulfide loop betweenproposed platelet glycoprotein Ib binding sequences. J ClinInvest. 1991;87:1220-1226.
36. Cooney KA, Nichols WC, Bruck ME, et al. The moleculardefect in type IIB von Willebrand disease: identification of fourpotential missense mutations within the putative GpIb bindingdomain. J Clin Invest. 1991;87:1227-1233.
37. Miller JL, Cunningham D, Lyle VA, Finch CN. Mutation in thegene encoding the a chain of platelet glycoprotein Ib inplatelet-type von Willebrand disease. Proc Natl Acad Sci U SA. 1991;88:4761-4765.
38. Dong JF, Moake JL, Nolasco L, et al. ADAMTS-13 rapidlycleaves newly secreted ultra-large von Willebrand factormultimers on the endothelial surface under flowing conditions.Blood. 2002: prepublished onine July 25, 2002; DOI2010.1182/blood-2002-2005-1401l.
39. Furlan M, Lämmle B. Aetiology and pathogenesis of throm-botic thrombocytopenic purpura and haemolytic uraemicsyndrome: the role of von Willebrand factor-cleaving protease.Best Pract Res Clin Haematol. 2001;14:437-454.
40. Bell WR, Braine HG, Ness PM, Kickler TS. Improved survivalin thrombotic thrombocytopenic purpura-hemolytic uremicsyndrome: clinical experience in 108 patients. N Engl J Med.1991;325:398-403.
41. Veyradier A, Obert B, Houllier A, Meyer D, Girma JP. Specificvon Willebrand factor-cleaving protease in thromboticmicroangiopathies: a study of 111 cases. Blood. 2001;98:1765-1772.
42. Raife TJ, Lentz SR, Atkinson BS, Vesely SK, Hessner MJ.Factor V Leiden: a genetic risk factor for thromboticmicroangiopathy in patients with normal von Willebrand factor-cleaving protease activity. Blood. 2002;99:437-442.
43. Daghistani D, Jimenez JJ, Moake JL, Ledford MR, Yunis AA.Familial infantile thrombotic thrombocytopenic purpura. JPediatr Hematol Oncol. 1996;18:171-174.
44. Savasan S, Taub JW, Buck S, Botterill M, Furlan M,Ravindranath Y. Congenital microangiopathic hemolyticanemia and thrombocytopenia with unusually large vonWillebrand factor multimers and von Willebrand factor-cleaving protease. J Pediatr Hematol Oncol. 2001;23:364-367.
45. Galbusera M, Noris M, Rossi C, et al. Increased fragmentationof von Willebrand factor, due to abnormal cleavage of thesubunit, parallels disease activity in recurrent hemolytic uremicsyndrome and thrombotic thrombocytopenic purpura anddiscloses predisposition in families. Blood. 1999;94:610-620.
46. Xie L, Chesterman CN, Hogg PJ. Control of von Willebrandfactor multimer size by thrombospondin-1. J Exp Med.2001;193:1341-1349.
47. Asada Y, Sumiyoshi A, Hayashi T, Suzumiya J, Kaketani K.Immunohistochemistry of vascular lesion in thromboticthrombocytopenic purpura, with special reference to factor VIIIrelated antigen. Thromb Res. 1985;38:469-479.
II. The Role of ADAMTS13 in the Evaluation andManagement of Patients with ThromboticThrombocytopenic Purpura
1. Moake JL, Chow TW. Thrombotic thrombocytopenic purpura:understanding a disease no longer rare. Am J Med Sci.1998;316:105-119.
2. Ruggenenti P, Remuzzi G. The pathophysiology and manage-ment of thrombotic thrombocytopenic purpura. Eur J Haematol.1996;56:191-207.
3. Furlan M, Lämmle B. Aetiology and pathogenesis of throm-botic thrombocytopenic purpura and haemolytic uraemicsyndrome: the role of von Willebrand factor-cleaving protease.Best Pract Res Clin Haematol. 2001;14:437-454.
4. Moake JL, Rudy CK, Troll JH, et al. Unusually large plasmafactor VIII: von Willebrand factor multimers in chronicrelapsing thrombotic thrombocytopenic purpura. N Engl J Med.1982;307:1432-1435.
5. Furlan M, Robles R, Lämmle B. Partial purification andcharacterization of a protease from human plasma cleaving vonWillebrand factor to fragments produced by in vivo proteolysis.Blood. 1996;87:4223-4234.
6. Tsai HM. Physiologic cleavage of von Willebrand factor by aplasma protease is dependent on its conformation and requirescalcium ion. Blood. 1996;87:4235-4244.
7. Dent JA, Berkowitz SD, Ware J, Kasper CK, Ruggeri ZM.Identification of a cleavage site directing the immunochemicaldetection of molecular abnormalities in type IIA vonWillebrand factor. Proc Natl Acad Sci U S A. 1990;87:6306-6310.
8. Furlan M, Robles R, Solenthaler M, Wassmer M, Sandoz P,Lämmle B. Deficient activity of von Willebrand factor-cleavingprotease in chronic relapsing thrombotic thrombocytopenicpurpura. Blood. 1997;89:3097-3103.
9. Furlan M, Robles R, Morselli B, Sandoz P, Lämmle B.Recovery and half-life of von Willebrand factor-cleavingprotease after plasma therapy in patients with thromboticthrombocytopenic purpura. Thromb Haemost. 1999;81:8-13.
10. Furlan M, Robles R, Solenthaler M, Lämmle B. Acquireddeficiency of von Willebrand factor-cleaving protease in apatient with thrombotic thrombocytopenic purpura. Blood.1998;91:2839-2846.
11. Furlan M, Robles R, Galbusera M, et al. Von Willebrand factor-cleaving protease in thrombotic thrombocytopenic purpura andthe hemolytic-uremic syndrome. N Engl J Med.1998;339:1578-1584.
12. Tsai HM, Lian ECY. Antibodies to von Willebrand factor-cleaving protease in acute thrombotic thrombocytopenicpurpura. N Engl J Med. 1998;339:1585-1594.
13. Gerritsen HE, Robles R, Lämmle B, Furlan M. Partial aminoacid sequence of purified von Willebrand factor-cleavingprotease. Blood. 2001;98:1654-1661.
14. Fujikawa K, Suzuki H, McMullen B, Chung D. Purification ofhuman von Willebrand factor-cleaving protease and itsidentification as a new member of the metalloproteinase family.Blood. 2001;98:1662-1666.
15. Zheng X, Chung D, Takayama TK, Majerus EM, Sadler JE,Fujikawa K. Structure of von Willebrand factor-cleavingprotease (ADAMTS13), a metalloprotease involved inthrombotic thrombocytopenic purpura. J Biol Chem.2001;276:41059-41063.
16. Soejima K, Mimura N, Hirashima M, et al. A novel humanmetalloprotease synthesized in the liver and secreted into theblood: possibly, the von Willebrand factor-cleaving protease? JBiochem (Tokyo). 2001;130:475-480.

332 American Society of Hematology
17. Levy GG, Nichols WC, Lian EC, et al. Mutations in a memberof the ADAMTS gene family cause thrombotic thrombocy-topenic purpura. Nature. 2001;413:488-494.
18. Furlan M, Lämmle B. Assays of von Willebrand factor-cleavingprotease: a test for diagnosis of familial and acquired throm-botic thrombocytopenic purpura. Sem Thromb Hemost.2002;28:167-172.
19. Gerritsen HE, Turecek PL, Schwarz HP, Lämmle B, Furlan M.Assay of von Willebrand factor (vWF)-cleaving protease basedon decreased collagen binding affinity of degraded vWF: a toolfor the diagnosis of thrombotic thrombocytopenic purpura(TTP). Thromb Haemost. 1999;82:1386-1389.
20. Obert B, Tout H, Veyradier A, Fressinaud E, Meyer D, GirmaJP. Estimation of the von Willebrand factor-cleaving protease inplasma using monoclonal antibodies to vWF. Thromb Haemost.1999;82:1382-1385.
21. Upshaw JD. Congenital deficiency of a factor in normal plasmathat reverses microangiopathic hemolysis and thrombocytope-nia. N Engl J Med. 1978;298:1350-1352.
22. Allford SL, Harrison P, Lawrie AS, Liesner R, Mackie IJ,Machin SJ. Von Willebrand factor-cleaving protease activity incongenital thrombotic thrombocytopenic purpura. Br JHaematol. 2000;111:1215-1222.
23. Häberle J, Kehrel B, Ritter J, Jürgens H, Lämmle B, Furlan M.New strategies in diagnosis and treatment of thromboticthrombocytopenic purpura: case report and review. Eur JPediatr. 1999;158:883-887.
24. Barbot J, Costa E, Guerra M, et al. Ten years of prophylactictreatment with fresh-frozen plasma in a child with chronicrelapsing thrombotic thrombocytopenic purpura as a result of acongenital deficiency of von Willebrand factor-cleavingprotease. Br J Haematol. 2001;113:649-651.
25. Kinoshita S, Yoshioka A, Park YD, et al. Upshaw-Schulmansyndrome revisited: a concept of congenital thromboticthrombocytopenic purpura. Int J Hematol. 2001;74:101-108.
26. Kokame K, Matsumoto M, Soejima K, et al. Mutations andcommon polymorphisms in ADAMTS13 gene responsible forvon Willebrand factor-cleaving protease activity. Proc NatlAcad Sci U S A. 2002;99:11902-11907.
27. Loof AH, van Vliet HHDM, Kappers-Klunne MC. Low activityof von Willebrand factor-cleaving protease is not restricted topatients suffering from thrombotic thrombocytopenic purpura.Br J Haematol. 2001;112:1087-1088.
28. Moore JC, Hayward CPM, Warkentin TE, Kelton JG. Decreasedvon Willebrand factor protease activity associated withthrombocytopenic disorders. Blood. 2001;98:1842-1846.
29. Mannucci PM, Canciani MT, Forza I, Lussana F, Lattuada A,Rossi E. Changes in health and disease of the metalloproteasethat cleaves von Willebrand factor. Blood. 2001;98:2730-2735.
30. Bianchi V, Robles R, Alberio L, Furlan M, Lämmle B. VonWillebrand factor-cleaving protease (ADAMTS13) in thromb-ocytopenic disorders: a severely deficient activity is specific forthrombotic thrombocytopenic purpura. Blood. 2002;100:710-713.
31. Hunt BJ, Lämmle B, Nevard CH, Haycock GB, Furlan M. VonWillebrand factor-cleaving protease in childhood diarrhoea-associated haemolytic uraemic syndrome. Thromb Haemost.2001;85:975-978.
32. Kelton JG. Thrombotic thrombocytopenic purpura andhemolytic uremic syndrome: will recent insight into pathogen-esis translate into better treatment? Transfusion. 2002;42:388-392.
33. Mori Y, Wada H, Gabazza EC, et al. Predicting response toplasma exchange in patients with thrombotic thrombocytopenicpurpura with measurement of vWF-cleaving protease activity.
Transfusion. 2002;42:572-580.34. Veyradier A, Obert B, Houllier A, Meyer D, Girma JP. Specific
von Willebrand factor-cleaving protease in thromboticmicroangiopathies: a study of 111 cases. Blood. 2001;98:1765-1772.
35. Van der Plas RM, Schiphorst ME, Huizinga EG, et al. VonWillebrand factor proteolysis is deficient in classic, but not inbone marrow transplantation-associated, thrombotic thromb-ocytopenic purpura. Blood. 1999;93:3798-3802.
36. Fontana S, Gerritsen HE, Kremer Hovinga J, Furlan M, LämmleB. Microangiopathic haemolytic anaemia in metastasizingmalignant tumours is not associated with a severe deficiency ofthe von Willebrand factor-cleaving protease. Br J Haematol.2001;113:100-102.
37. Tsai HM, Rice L, Sarode R, Chow TW, Moake JL. Antibodyinhibitors to von Willebrand factor metalloproteinase andincreased binding of von Willebrand factor to platelets inticlopidine-associated thrombotic thrombocytopenic purpura.Ann Intern Med. 2000;132:794-799.
38. Bennett CL, Connors JM, Carwile JM, et al. Thromboticthrombocytopenic purpura associated with clopidogrel. N EnglJ Med. 2000;342:1773-1777.
39. Sahud MA, Claster S, Liu L, Ero M, Harris K, Furlan M. VonWillebrand factor-cleaving protease inhibitor in a patient withhuman immunodeficiency syndrome-associated thromboticthrombocytopenic purpura. Br J Haematol. 2002;116:909-911.
40. George JN. How I treat patients with thrombotic thrombocy-topenic purpura-hemolytic uremic syndrome. Blood.2000;96:1223-1229.
41. Moake JL. Thrombotic thrombocytopenic purpura: Thesystemic clumping “plague.” Annu Rev Med. 2002;53:75-88.
42. Rock GA, Shumak KH, Buskard NA, et al. Comparison ofplasma exchange with plasma infusion in the treatment ofthrombotic thrombocytopenic purpura. Canadian ApheresisStudy Group. N Engl J Med. 1991;325:393-397.
43. Bell WR, Braine HG, Ness PM, Kickler TS. Improved survivalin thrombotic thrombocytopenic purpura-hemolytic uremicsyndrome. N Engl J Med. 1991;325:398-403.
44. Veltman GAM, Brand A, Leeksma OC, ten Bosch GJA, vanKrieken JHJM, Briet E. The role of splenectomy in thetreatment of relapsing thrombotic thrombocytopenic purpura.Ann Hematol. 1995;70:231-236.
45. Mant MJ, Turner AR, Bruce D, Ritchie C, Larratt LM.Splenectomy during partial remission in thrombotic thrombocy-topenic purpura with prolonged plasma exchange dependency.Am J Hematol. 1999;62:56-57.
III. Evaluation and Management of Patients withClinically Suspected Thrombotic ThrombocytopenicPurpura or Hemolytic-Uremic Syndrome
1. Rock GA, Shumak KH, Buskard NA, et al. Comparison ofplasma exchange with plasma infusion in the treatment ofthrombotic thrombocytopenic purpura. N Engl J Med.1991;325:393-97.
2. Thompson CE, Damon LE, Ries CA, Linker CA. Thromboticmicroangiopathies in the 1980s: Clinical features, response totreatment, and the impact of the human immunodeficiency virusepidemic. Blood. 1992;80:1890-1895.
3. George JN. How I treat patients with thrombotic thrombocy-topenic purpura-hemolytic uremic syndrome. Blood.2000;96:1223-29.
4. Rizvi MA, Vesely SK, George JN, et al. Complications ofplasma exchange in 71 consecutive patients treated forclinically suspected thrombotic thrombocytopenic purpura-

Hematology 2002 333
hemolytic uremic syndrome. Transfusion. 2000;40:896-901.5. Martin DL, MacDonald KL, White KE, Soler JT, Osterholm
MT. The epidemiology and clinical aspects of the hemolyticuremic syndrome in Minnesota. N Engl J Med. 1990;323:1161-67.
6. Milford DV, Taylor CM, Guttridge B, Hall SM, Rowe B,Kleanthous H. Haemolytic uraemic syndromes in the BritishIsles 1985-8: association with Verocytotoxin producingEscherichia coli. Part 1: clinical and epidemiological aspects.Arch Dis Child. 1990;65:716-21.
7. Rowe PC, Orrbine E, Wells GA, McLaine PN. Epidemiology ofhemolytic-uremic syndrome in Canadian children from 1986 to1988. J Pediat. 1991;119:218-24.
8. Besser RE, Griffin PM, Slutsker L. Escherichia coli O157:H7gastroenteritis and the hemolytic uremic syndrome: Anemerging infectious disease. Annu Rev Med. 1999;50:355-67.
9. Rizzoni G, Claris-Appiani A, Edefonti A, et al. Plasma infusionfor hemolytic uremic syndrome in children: results of amulticenter controlled trial. J Pediat. 1988;112:284-90.
10. Loriat C, Sonsino E, Hinglais N, Jais JP, Landais P, FermanianJ. Treatment of childnood haemolytic uraemic syndrome withplasma. A multicenter randomized controlled trial. PediatrNephrol. 1988;2:279-85.
11. Fitzpatrick MM, Walters MDS, Trompeter RS, Dillon MJ,Barratt TM. Atypical (non-diarrhea-associated) hemolytic-uremic syndrome in childhood. J Pediatr. 1993;122:532-37.
12. Pettitt AR, Clark RE. Thrombotic microangiopathy followingbone marrow transplantation. Bone Marrow Transplant.1994;14:495-504.
13. Roy V, Rizvi MA, Vesely SK, George JN. Thromboticthrombocytopenic purpura-like syndromes following bonemarrow transplantation: an analysis of associated conditions andclinical outcomes. Bone Marrow Transplant. 2001;27:641-6.
14. McMinn JR, George JN. Evaluation of women with clinicallysuspected thrombotic thrombocytopenic purpura-hemolyticuremic syndrome during pregnancy. J Clin Apheresis.2001;16:202-9.
15. McCrae KR, Samuels P, Schreiber AD. Pregnancy-associatedthrombocytopenia: pathogenesis and management. Blood.1992;80:2697-714.
16. McCrae KR, Bussel JB, Mannucci PM, Cines DB. Platelets: anupdate on diagnosis and management of thrombocytopenicdisorders. In: Schecter GP, Broudy VC, Williams ME, eds.Hematology. Washington, D.C.: The American Society ofHematology; 2001: 282-305.
17. Fuchs WE, George JN, Dotin LN, Sears DA. Thromboticthrombocytopenic purpura. Occurrence two years apart duringlate pregnancy in two sisters. JAMA. 1976;235:2126.
18. Furlan M, Lämmle B. Aetiology and pathogenesis of throm-botic thrombocytopenic purpura and haemolytic uraemicsyndrome: the role of von Willebrand factor-cleaving protease.Best Pract Res Clin Haematol. 2001;14:437-54.
19. Martin JN, Blake PG, Perry KG, McCaul JF, Hess LW, MartinRW. The natural history of HELLP syndrome: patterns ofdisease progression and regression. Am J Obstet Gynecol.1991;164:1500-1513.
20. Chandran R, Serra-Serra V, Redman CWG. Spontaneousresolution of pre-eclampsia-related thrombocytopenia. Br JObstet Gynaecol. 1992;99:887-90.
21. Sibai BM, Ramadan MK, Usta I, Salama M, Mercer BM,Friedman SA. Maternal morbidity and mortality in 442pregnancies with hemolysis, elevated liver enzymes, and lowplatelets (HELLP syndrome). Am J Obstet Gynecol.1993;169:1000-1006.
22. Katz VL, Farmer R, Kuller JA. Preeclampsia into eclampsia:
Toward a new paradigm. Amer J Obstet Gynecol.2000;182:1389-94.
23. Katz VL, Thorp JM, Jr., Rozas L, Bowes WA, Jr. The naturalhistory of thrombocytopenia associated with preeclampsia. AmJ Obstet Gynecol. 1990;163:1142-43.
24. Medina PJ, Sipols JM, George JN. Drug-associated thromboticthrombocytopenic purpura-hemolytic uremic syndrome. CurrOpin Hematol. 2001;8:286-93.
25. Kojouri K, Vesely S, George JN. Quinine-associated thromboticthrombocytopenic purpura-hemolytic uremic syndrome:frequency, clinical features, and long-term outcomes. AnnIntern Med. 2001;135:1047-51.
26. Moake JL. Thrombotic thrombocytopenic purpura. In: KitchensCS, Alving BA, Kessler CM, eds. Consultative Hemostasis andThrombosis. Philadelphia: W.B. Saunders Co.; 2002: 343-54.
27. Schattner A. Quinine hypersensitivity simulating sepsis. Am JMed. 1998;104:488-90.
28. Stroncek DF, Vercellotti GM, Hammerschmidt DE, Christie DJ,Shankar RA, Jacob HS. Characterization of multiple quinine-dependent antibodies in a patient with episodic hemolyticuremic syndrome and immune agranulocytosis. Blood.1992;80:241-48.
29. Kedia RK, Wright AJ. Quinine-mediated disseminatedintravascular coagulation. Postgrad Med J. 1999;75:429-30.
30. Farver DK, Lavin MN. Quinine-induced hepatotoxicity. AnnPharmacother. 1999;33:32-34.
31. George JN, Raskob GE, Shah SR, et al. Drug-induced thromb-ocytopenia: A systematic review of published case reports. AnnInt Med. 1998;129:886-90.
32. Glynne P, Salama A, Chaudry A, Swirsky D, Lightstone L.Quinine-induced immune thrombocytopenic purpura followedby hemolytic uremic syndrome. Am J Kidney Dis.1999;33:133-37.
33. Bennett CL, Weinberg PD, Rozenberg-Ben-Dror K, Yarnold PR,Kwaan HC, Green D. Thrombotic thrombocytopenic purpuraassociated with ticlopidine - A review of 60 cases. Ann IntMed. 1998;128:541-44.
34. Bennett CL, Connors JM, Carwile JM, et al. Thromboticthrombocytopenic purpura associated with clopidogrel. N EnglJ Med. 2000;342:1773-77.
35. Tsai HM, Rice L, Sarode R, Chow TW, Moake JL. Antibodyinhibitors to von Willebrand factor metalloproteinase andincreased binding of von Willebrand factor to platelets inticlopidine-associated thrombotic thrombocytopenic purpura.Ann Intern Med. 2000;132:794-99.
36. Dundas S, Todd WTA, Stewart AI, Murdoch PS, ChaudhuriAKR, Hutchinson SJ. The central Scotland Escherichia coliO157:H7 outbreak: Risk factors for the hemolytic uremicsyndrome and death among hospitalized patients. Clin InfectDis. 2001;33:923-31.
37. Dundas S, Murphy J, Soutar RL, et al. Effectiveness oftherapeutic plasma exchange in the 1996 Lanarkshire Escheri-chia coli O157:H7 outbreak. Lancet. 1999;354:1327.
38. Kovacs MJ, Roddy J, Grégoire S, Cameron W, Eidus L, DrouinJ. Thrombotic thrombocytopenic purpura following hemor-rhagic colitis due to Escherichia coli O157:H7. Am J Med.1990;88:177-79.
39. Devinsky O, Petito CK, Alonso DR. Clinical and neuropatho-logical findings in systemic lupus erythematosus: the role ofvasculitis, heart emboli, and thrombotic thrombocytopenicpurpura. Ann Neurol. 1988;23:380-384.
40. Asherson RA, Cervera R, Piette JC, et al. Catastrophicantiphospholipid syndrome. Clinical and laboratory features of50 patients. Medicine. 1998;77:195-207.
41. Laszik Z, Silva F. Hemolytic-uremic syndrome, thrombotic

334 American Society of Hematology
thrombocytopenia purpura, and systemic sclerosis (systemicscleroderma). In: Jennett JC, Olson JL, Schwartz MM, SilvaFG, eds. Heptinstall’s Pathology of the Kidney. 5 ed. Philadel-phia: Lippincott-Raven; 1998:1003-57.
42. Marty AM, Dumler JS, Imes G, Brusman HP, Smrkovski LL,Frisman DM. Ehrlichiosis mimicking thrombotic thrombocy-topenic purpura. Case report and pathological correlation. HumPathol. 1995;26:920-925.
43. Antman KH, Skarin AT, Mayer RJ, Hargreaves HK, CanellosGP. Microangiopathic hemolytic anemia and cancer: A review.Medicine. 1979;58:377-84.
44. Lin Y-C, Chang H-K, Sun C-F, Shih L-Y. Microangiopathichemolytic anemia as an initial presentation of metastatic cancerof unknown primary origin. South Med J. 1995;88:683-87.
45. Brain MC, Dacie JV, Hourihane OB. Microangiopathichemolytic anemia: the possible role of vascular lesions inpathogenesis. Br J Haematol. 1962;8:358.
46. Sahud MA, Claster S, Liu L, Ero M, Harris K, Furlan M. vonWillebrand factor-cleaving protease inhibitor in a patient withhuman immunodeficiency syndrome-associated thrombotic
thrombocytopenic purpura. Br J Haematol. 2002;116:909-11.47. Moore RD. Schistocytosis and a thrombotic microangiopathy-
like syndrome in hospitalized HIV-infected patients. Am JHematol. 1999;60:116-20.
48. Zeigler ZR, Shadduck RK, Gryn JF, et al. Cryoprecipitate-poorplasma does not improve early response in primary adultthrombotic thrombocytopenic purpura (TTP). J Clin Apheresis.2001;16:19-22.
49. Perdue JJ, Chandler L, Vesely S, et al. Unintentional plateletremoval by plasmapheresis. J Clin Apheresis. 2001;16:55-60.
50. Baron BW, Martin MS, Sucharetza BS, Jeon H-R, Baron JM.Four patients with both thrombotic thrombocytopenic purpuraand autoimmune thrombocytopenic purpura: The concept of amixed immune thrombocytopenia syndrome and indications forplasma exchange. J Clin Apheresis. 2001;16:179-85.
51. Perdue JJ, Vesely SK, George JN, Clark KR, Terrell DR.Residual effects of thrombotic thrombocytopenic purpura-hemolytic uremic syndrome (TTP-HUS) on health-relatedquality of life (QOL): limitations in vitality and daily activitiesamong survivors. Blood. 2001;98:62b-3b.