prenatal diagnosis of loeys ... - thieme-connect.de · 75 % have de-novo mutations (c. van...
TRANSCRIPT

Title Page 391
Ultraschall in Med 2014; 35
Prenatal Diagnosis of Loeys–Dietz Syndrome
Introduction ▼
Loeys–Dietz is an autosomal dominant aortic aneurysm syndro-me with widespread systemic involvement. The disease is cha-racterized by the triad of arterial tortuosity and aneurysms, hy-pertelorism, and bifid uvula or cleft palate and is caused by he-terozygous mutations in the genes encoding transforming growth factor β receptors 1 and 2 (TGFBR1 and TGFBR2, respectively) (B.L. Loeys et al. Engl J Med 2006; 355: 788–798; B.L. Loeys et al. Nat Genet 2005; 37: 275–281; C. Van Hemelrijk et al. Curr Opin Car-diol 2010; 25: 546–551).
Aortic aneurysm is a common finding in abnormal connective tissue diseases such as Marfan syndrome, Ehlers-Danlos syndro-me, and LDS (P. Pomianowski et al. Ann Cardiothorac Surg 2013; 2: 271–279). Cardiovascular disease in LDS is more widespread and aggressive than in Marfan syndrome, leading to earlier sur-gery with a smaller aortic diameter (C. Van Hemelrijk et al. Curr Opin Cardiol 2010; 25: 546–551).
LDS is a recently discovered syndrome and should be suspected in fetuses and children with an aortic aneurysm. Only 25 % of newly diagnosed patients have an affected parent while the other 75 % have De-novo mutations (C. Van Hemelrijk et al. Curr Opin Cardiol 2010; 25: 546–551). Approximately 75 % of LDS patients presenting with typical facial dysmorphic features (cleft palate, craniosynostosis or hypertelorism) are designated LDS type 1. LDS type 2 was used to describe the part of the syndrome wit-hout major craniofacial features, but with cutaneous manifesta-tions (C. Van Hemelrijk et al. Curr Opin Cardiol 2010; 25: 546–551).
Case description ▼
A couple was referred for genetic counseling in their third preg-nancy. The couple were of Jewish origin (mixed Ashkenazi and non-Ashkenazi). Their first daughter (15 years old now) was born with dilatation of the aortic root, cleft palate, wide anterior fon-tanel and club feet. At the age of three months, she was diagnosed clinically with Marfan syndrome, but no further investigation was done. At the age of 12, she had to undergo an operation due to severe aortic aneurysm. Four years later, a healthy son was born (second pregnancy). In the third pregnancy in 2010, a nor-mal nuchal translucency of 1.9 mm was detected and amniocen-tesis was performed due to advanced maternal age, revealing a normal karyotype (46XY). At 21 weeks of gestation, the mother was referred to fetal echocardiography due to the diseased daughter’s history. Dilatation of the aortic root was diagnosed in the fetus. An anomaly scan revealed a wide aortic root with tortuous aorta, club foot, cleft palate and wide anterior fontanel (q Fig. 1). At 25 weeks of gestation after approval from the hos-pital committee, the pregnancy was terminated. The combinati-on of the shape and wideness of the aorta and the family history raised the suspicion of Loeys-Dietz syndrome (LDS, OMIM ID #609192).
Pränatale Diagnostik des Loeys-Dietz-Syndroms
Einleitung ▼
Das Loeys-Dietz-Syndrom (LDS) ist ein autosomal-dominant ver-erbtes Aortenaneurysmen-Syndrom mit ausgeprägter systemi-scher Beteiligung. Die Krankheit wird durch den Trias Gefäß-schlängelung und Aneurysmen, Hypertelorismus und Uvula bifi-da oder Gaumenspalte charakterisiert. Sie wird durch heterozy-gote Mutationen der Gene für die Transforming-Growth-Factor-ß-Rezeptoren 1 und 2 (TGFBR1 und TGFBR2) hervorgerufen (B.L. Loeys et al. Engl J Med 2006; 355: 788–798; B.L. Loeys et al. Nat Genet 2005; 37: 275–281; C. Van Hemelrijk et al. Curr Opin Car-diol 2010; 25: 546–551).
Das Aortenaneurysma ist ein häufiger Befund bei Erkrankungen mit abnormalem Bindegewebe, wie zum Beispiel beim Marfan-Syndrom, dem Ehlers-Danlos-Syndrom und LDS (P. Pomianowski et al. Ann Cardiothorac Surg 2013; 2: 271–279). Die kardiovas-kuläre Erkrankung bei LDS ist noch ausgeprägter und aggressiver als beim Marfan-Syndrom und führt zu früheren Operationen bei geringeren Aortendurchmessern (C. Van Hemelrijk et al. Curr Opin Cardiol 2010; 25: 546–551).
LDS ist ein kürzlich entdecktes Syndrom und sollte bei allen Fe-ten mit Aortenaneurysmen in Betracht gezogen werden. Nur 25 % der neu diagnostizierten Patienten haben ein betroffenes Eltern-teil, während die anderen 75 % auf Neumutationen zurückzufüh-ren sind (C. Van Hemelrijk et al. Curr Opin Cardiol 2010; 25: 546–551). Etwa 75 % der LDS-Patienten zeigen typische Dismorphien des Gesichts (Gaumenspalte, Kraniosynostose oder Hyperteloris-mus) und werden dem LDS Typ1 zugerechnet. Der Typ 2 be-schreibt gewöhnlich den Anteil der Patienten, die keine wesent-lichen kraniofazialen Merkmale aufweisen, dafür jedoch kutane Manifestationen (C. Van Hemelrijk et al. Curr Opin Cardiol 2010; 25: 546–551).
Fallbeschreibung ▼
Ein Paar jüdischer Herkunft (halb aschkenasisch und nicht asch-kenasisch) besuchte in der 3. Schwangerschaft auf Empfehlung eine genetische Beratungsstelle. Ihre erstgeborene Tochter (jetzt 15 Jahre alt) wurde mit einer Dilatation der Aortenwurzel, einer Gaumenspalte, einer großen vorderen Fontanelle und Klumpfü-ßen geboren. Im Alter von 3 Monaten wurde bei ihr klinisch ein Marfan-Syndrom diagnostiziert, aber es erfolgte keine weiter-führende Untersuchung. Im Alter von 12 Jahren musste sie sich aufgrund eines schweren Aortenaneurysmas einer Operation un-terziehen. 4 Jahre später (2. Schwangerschaft) wurde ein gesun-der Sohn geboren. In der 3. Schwangerschaft 2010 wurde eine normale fetale Nackentransparenz von 1,9 mm bestimmt und eine Amniozentese wegen Altersindikation durchgeführt, die ei-nen normalen Karyotyp ergab (46XY). In der 21. Schwanger-schaftswoche (SSW) wurde bei der Mutter aufgrund der Anam-nese der kranken Tochter eine fetale Echokardiografie veranlasst. Beim Feten wurde eine Dilatation der Aortenwurzel diagnosti-ziert. Eine Untersuchung auf Anomalien zeigte eine weite Aor-
Thi
s do
cum
ent w
as d
ownl
oade
d fo
r pe
rson
al u
se o
nly.
Una
utho
rized
dis
trib
utio
n is
str
ictly
pro
hibi
ted.
Title Page392
Ultraschall in Med 2014; 35
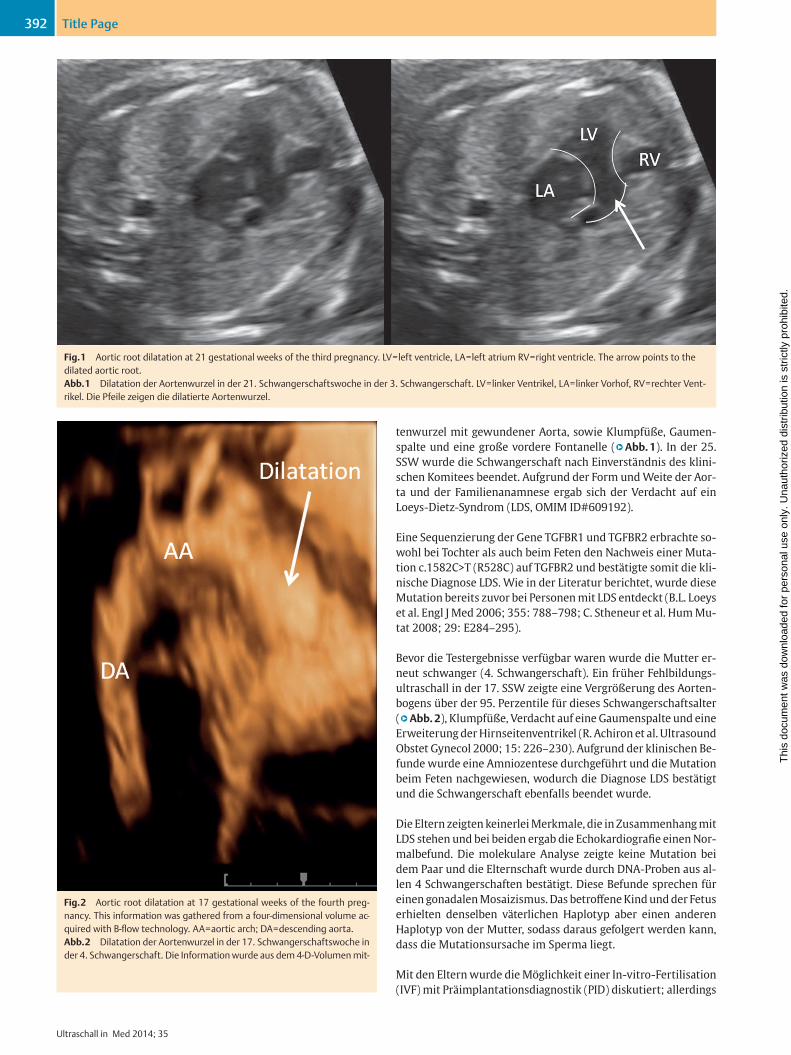
tenwurzel mit gewundener Aorta, sowie Klumpfüße, Gaumen-spalte und eine große vordere Fontanelle (q Abb. 1). In der 25. SSW wurde die Schwangerschaft nach Einverständnis des klini-schen Komitees beendet. Aufgrund der Form und Weite der Aor-ta und der Familienanamnese ergab sich der Verdacht auf ein Loeys-Dietz-Syndrom (LDS, OMIM ID#609192).
Eine Sequenzierung der Gene TGFBR1 und TGFBR2 erbrachte so-wohl bei Tochter als auch beim Feten den Nachweis einer Muta-tion c.1582C>T (R528C) auf TGFBR2 und bestätigte somit die kli-nische Diagnose LDS. Wie in der Literatur berichtet, wurde diese Mutation bereits zuvor bei Personen mit LDS entdeckt (B.L. Loeys et al. Engl J Med 2006; 355: 788–798; C. Stheneur et al. Hum Mu-tat 2008; 29: E284–295).
Bevor die Testergebnisse verfügbar waren wurde die Mutter er-neut schwanger (4. Schwangerschaft). Ein früher Fehlbildungs-ultraschall in der 17. SSW zeigte eine Vergrößerung des Aorten-bogens über der 95. Perzentile für dieses Schwangerschaftsalter (q Abb. 2), Klumpfüße, Verdacht auf eine Gaumenspalte und eine Erweiterung der Hirnseitenventrikel (R. Achiron et al. Ultrasound Obstet Gynecol 2000; 15: 226–230). Aufgrund der klinischen Be-funde wurde eine Amniozentese durchgeführt und die Mutation beim Feten nachgewiesen, wodurch die Diagnose LDS bestätigt und die Schwangerschaft ebenfalls beendet wurde.
Die Eltern zeigten keinerlei Merkmale, die in Zusammenhang mit LDS stehen und bei beiden ergab die Echokardiografie einen Nor-malbefund. Die molekulare Analyse zeigte keine Mutation bei dem Paar und die Elternschaft wurde durch DNA-Proben aus al-len 4 Schwangerschaften bestätigt. Diese Befunde sprechen für einen gonadalen Mosaizismus. Das betroffene Kind und der Fetus erhielten denselben väterlichen Haplotyp aber einen anderen Haplotyp von der Mutter, sodass daraus gefolgert werden kann, dass die Mutationsursache im Sperma liegt.
Mit den Eltern wurde die Möglichkeit einer In-vitro-Fertilisation (IVF) mit Präimplantationsdiagnostik (PID) diskutiert; allerdings
Fig. 1 Aortic root dilatation at 21 gestational weeks of the third pregnancy. LV = left ventricle, LA = left atrium RV = right ventricle. The arrow points to the dilated aortic root.Abb. 1 Dilatation der Aortenwurzel in der 21. Schwangerschaftswoche in der 3. Schwangerschaft. LV = linker Ventrikel, LA = linker Vorhof, RV = rechter Vent-rikel. Die Pfeile zeigen die dilatierte Aortenwurzel.
Fig. 2 Aortic root dilatation at 17 gestational weeks of the fourth preg-nancy. This information was gathered from a four-dimensional volume ac-quired with B-flow technology. AA = aortic arch; DA = descending aorta.Abb. 2 Dilatation der Aortenwurzel in der 17. Schwangerschaftswoche in der 4. Schwangerschaft. Die Information wurde aus dem 4-D-Volumen mit-
Thi
s do
cum
ent w
as d
ownl
oade
d fo
r pe
rson
al u
se o
nly.
Una
utho
rized
dis
trib
utio
n is
str
ictly
pro
hibi
ted.

Title Page 393
Ultraschall in Med 2014; 35
Sequencing of the genes TGFBR1 and TGFBR2 was performed, and the mutation c.1582C>T (R528C) in TGFBR2 was identified in the daughter and the affected fetus, confirming the clinical diagnosis of LDS. As reported in the literature, this mutation has been pre-viously identified in individuals with LDS .(B.L. Loeys et al. Engl J Med 2006; 355: 788–798; C. Stheneur et al. Hum Mutat 2008; 29: E284–295).
Before the test results arrived, the mother became pregnant again (fourth pregnancy). An early anomaly scan at 17 weeks of gesta-tion revealed enlargement of the aortic arch of above the 95th percentile for the gestational age (q Fig. 2), club feet, suspected cleft palate and dilatation of the lateral ventricles of the brain (R. Achiron et al. Ultrasound Obstet Gynecol 2000; 15: 226–230). Due to the clinical findings, amniocentesis was performed, and the mutation was identified in the fetus, confirming the diagno-sis of LDS, and thus, the pregnancy was again terminated.
The parents had never shown any physical features consistent with LDS, and echocardiography was normal in both. Molecular analysis had not revealed the mutation in this couple, while pa-renthood was confirmed on DNA samples from all four pregnan-cies. These findings are compatible with gonadal mosaicism. The affected child and fetus received the same haplotype from the father, but a different haplotype from the mother, leading to the conclusion that the origin of the mutation was in the sperm.
The possibility of in-vitro fertilization (IVF) with pregestational diagnosis (PGD) procedure was discussed with the parents, but the mother conceived spontaneously (fifth pregnancy). An early
wurde die Mutter spontan schwanger (5. Schwangerschaft). Der Fehlbildungsultraschall in SSW 12 + d 1 zeigte keine Anomalien. Die Chorionzottenbiopsie zeigte einen normalen Karyotyp und die Sequenzierung wies ein Wildtyp-Allel nach.
Diskussion ▼
Nach Sichtung der medizinischen Fachliteratur zeigte sich, dass unser Fall der erste war, der pränatal diagnostiziert wurde. Der Fetus zeigte typische Ultraschallbefunde: eine weite Aortenwur-zel mit gewundener Form, Klumpfüße, Gaumenspalte und eine große vordere Fontanelle. Aufgrund des Geschwisterkindes mit ähnlichen Merkmalen und der sonografischen Befunde ergab sich die Verdachtsdiagnose LDS, die durch die genetische Unter-suchung bestätigte wurde. Es brauchte mehr als 2 weitere Schwangerschaften bis die Eltern einer vollständigen geneti-schen Untersuchung der gesamten Familie zustimmten. Beim 1. Kind wurde der Verdacht auf ein Marfan-Syndrom gestellt, da LDS zu diesem Zeitpunkt noch nicht beschrieben war und die korrekte Diagnose erst nach Feststellung der Erkrankung im Fe-ten in der 4. Schwangerschaft erfolgte.
Die Datenlage zur pränatalen Diagnostik des LDS ist dürftig. In der Literatur wurde nur 1 Fall eines Feten der 19. SSW mit Aor-tenaneurysmas beschrieben (V. Viassolo et al. Prenat Diagn 2006; 26: 1081–1083). Der Karyotyp war normal und in der 33. SSW wurden Klumpfüße beobachtet. Nach der Geburt wurde eine weiche Gaumenspalte entdeckt und die molekulare Analyse zeig-te eine Neumutation in TGFBR2.
Thi
s do
cum
ent w
as d
ownl
oade
d fo
r pe
rson
al u
se o
nly.
Una
utho
rized
dis
trib
utio
n is
str
ictly
pro
hibi
ted.

Title Page394
Ultraschall in Med 2014; 35
anomaly scan at 12w1 d showed no abnormality. Chorionic vill-ous sampling revealed a normal karyotype and sequencing re-vealed the wild type allele.
Discussion ▼
After a search of the medical literature, our case turned out to be the first diagnosed prenatally. The fetus presented with typical ultrasound findings of a wide aortic root with torturous shape, club foot, cleft palate and wide anterior fontanel. The combinati-on of a previous child with similar signs together with the ultra-sound findings raised the suspicion of LDS, and the genetic inves-tigation confirmed the diagnosis. It took the parents two more pregnancies before they agreed to complete genetic testing of the family. The first child was suspected to have Marfan syndrome, since LDS still had not been described at that point, and the cor-rect diagnosis was made only after diagnosing the fetus during the fourth pregnancy.
Data concerning the prenatal diagnosis of LDS is scarce. Only one case has been reported in the literature of an aortic root aneu-rysm in a 19-week fetus (V. Viassolo et al. Prenat Diagn 2006; 26: 1081–1083). The karyotype was normal, and at 33 weeks of ge-station, club feet were observed. After birth, a cleft soft palate was found, and molecular analysis revealed a de-novo mutation in TGFBR 2.
The severe prognosis of LDS results mainly from the high poten-tial for rupture of aneurysms in childhood, a devastating compli-cation that occurs with a smaller diameter of the artery compared to Marfan syndrome (C. Van Hemelirjk et al. Curr Opin Cardiol 2010; 25: 546–551). Aortic aneurysm is a life-threatening condi-tion that can deteriorate to dissection and death. Therefore, ac-curate diagnosis of this syndrome is extremely important for the whole family. Routine follow-up with echocardiography and sur-gery for repair of the aneurysm can be life-saving.
Prenatal diagnosis of LDS is feasible as was demonstrated in this family. Aortic root dilatation is a constant feature of LDS and can be detected by looking at the outflow tracts during ultrasound scanning. If an enlarged diameter of the aorta is suspected, fetal echocardiography by an expert cardiologist and detailed anoma-ly scan are recommended. Genetic counseling should raise the possibility of connective tissue disorder, such as Marfan syndro-me or LDS. In LDS, meticulous prenatal ultrasound examination can detect additional findings, including aneurysmatic ductus ar-teriosus, hypertelorism, cleft palate, retrognathia, craniosynosto-sis, postaxial polydactyly, camptodactyly, club feet, Chiari mal-formation and hydrocephalus. These types of features can diffe-rentiate between Marfan syndrome and LDS.
In conclusion, awareness of this syndrome should be expanded to include the possibility of its prenatal diagnosis. The finding of aortic dilatation and aneurysm in fetuses is a major sign that should prompt genetic counseling due to the possibility of a con-nective tissue disorder. The specific shape of the dilated aorta and the diagnosis of additional dysmorphic features serve as the car-dinal signs of LDS. Molecular testing for TGFBR1 and TGFBR2 should lead to the final diagnosis. This will play a major role in identifying additional affected family members and the need for prenatal diagnosis in future pregnancies.
Die schlechte Prognose bei LDS ist vor allem auf das hohe Risiko für einen Riss des Aneurysmas in der Kindheit zurückzuführen, eine verheerende Komplikation, die kleinere Arteriendurchmes-ser betrifft als beim Marfan-Syndrom (C. Van Hemelirjk et al. Curr Opin Cardiol 2010; 25: 546–551). Ein Aortenaneurysma ist eine lebensbedrohliche Erkrankung, die sich verschlechtern und zu Sektion und Tod führen kann. Daher ist eine genaue Diagnose-stellung bei diesem Syndrom äußerst bedeutsam für die ganze Familie. Ein Routine-Follow-Up mit Echokardiografie und Opera-tion des Aneurysmas kann lebensrettend sein.
Die pränatale Diagnostik des LDS ist – wie bei dieser Familie ge-zeigt – machbar. Die Dilatation der Aortenwurzel ist ein konstan-tes Merkmal des LDS und kann in der Ultraschall-Untersuchung nachgewiesen werden, indem man die Ausflusskanäle beobach-tet. Wenn der Verdacht auf einen vergrößerten Aortendurchmes-ser besteht, wird empfohlen eine fetale Echokardiografie bei ei-nen erfahrenen Kardiologen und einen Feinultraschall zum Aus-schluss von Fehlbildungen durchführen zu lassen. Die genetische Beratung sollte eine mögliche Bindegewebskrankheit wie das Marfan-Syndrom oder LDS ansprechen. Bei LDS kann die sorgfäl-tige pränatale Sonografie zusätzliche Merkmale erkennen, ein-schließlich eines aneurysmatischen Ductus arteriosus, Hyper-telorismus, Gaumenspalte, Retrognathie, Kraniosynostose, postaxiale Polydaktylie, Kamptodaktylie, Klumpfüße, Chiari-Fehlbildung und Hydrozephalus. Diese Merkmale dienen der Dif-ferenzierung zwischen Marfan-Syndrom und LDS.
Abschließend sei angemerkt, dass der Bekanntheitsgrad dieses Syndroms noch zunehmen sollte, auch in Bezug auf die Möglich-keit der pränatalen Diagnostik.
Der Befund einer Aortendilatation und eines Aneurysmas im Fe-ten ist das Hauptmerkmal und sollte eine schnelle genetische Be-ratung zur Folge haben, da eine mögliche Bindegewebskrankheit vorliegen kann. Die besondere Form der dilatierten Aorta und die Diagnose zusätzlicher dysmorpher Merkmale sind die Haupt-marker eines LDS. Mit der molekularen Untersuchung von TGFBR1 und TGFBR2 kann die endgültige Diagnose gestellt wer-den. Das ist insbesondere wichtig, um zusätzliche betroffene Fa-milienmitglieder zu entdecken und da bei weiteren Schwanger-schaften eine pränatale Diagnostik indiziert ist.
L. Gindes, M. Berkenstadt, H. Reznik-Wolf, E. Pras,R. Achiron, Tel-Aviv, Ramat Gan
Thi
s do
cum
ent w
as d
ownl
oade
d fo
r pe
rson
al u
se o
nly.
Una
utho
rized
dis
trib
utio
n is
str
ictly
pro
hibi
ted.