prions first identified with “spongiform encephalopathies” characteristics of infection: –loss...
TRANSCRIPT

Prions
• First identified with “Spongiform encephalopathies”• Characteristics of infection:
– Loss of motor control– Dementia– Paralysis– Encephalitis– Widespread neuronal loss
• Ways of infection:– Infectious (including diet, after surgical
procedures, corneal transplants etc.)– Hereditary (autosomal and dominant)

vacuole
Source: UC Davis School of Veterinary Medicine
Brain Damage from Spongiform Encephalopathy

Transmissible spongiform encephalopathies
• Animals– Bovine spongiform encephalopathy (BSE)– Scrapie in sheep and goats– Transmissible mink encephalopathy– Chronic wasting disease of deer, elk
• Humans– Kuru– Creutzfeldt-Jacob disease (CJD)– Fatal familial insomnia (FFI)– Gerstmann-Straussler syndrome (GSS)
• TSEs are always fatal

Types of TSEs• Infectious
– e.g., kuru, BSE (mad cow disease), scrapie– Spread by
• consumption of infected material• Iatrogenic spread (organ transplant, esp. cornea)• transfusion
• Sporadic– 1-2 million infected worldwide, late in life– Evidence mounting that some sporadic TSE is really result of infection
• Familial– Due to autosomal dominant mutation of PrP– Inherited – at least 10-15% of total human TSE cases
• Each of these can be transmitted experimentally

Kuru• Identified by epidemiology in New Guinea base on
anthropological research by Robert and Louise Glasse in 1950’s
• 1% of the Fore tribe was afflicted; mostly women, some children, few adult males
• Symptoms: headache, joint pain, then 6-12 weeks later, difficulty walking, then death usually within 12 months, always within 2 years
• Disease was of recent origin: ~1910-1920• Epidemiological evidence led the Glasses to suggest that
endocannibalism was associated with disease• This hypothesis was not well accepted among medical
community

South Fore

Kuru• Australian government suppressed cannibalism among
North Fore in early 1950’s• South Fore were convinced to discontinue the practice in
1959• Incidence of kuru among North Fore ceased ~ 5 years
before South Fore; no child born since then has died of kuru• Carlton Gadjusek, a medical research scientist with NIH,
inoculated chimps with brain extracts of kuru victims; all chimps died after 50 months
• No unique antibodies were associated with disease, no virus particles or aberrant nucleic acids were identified
• Gadjusek got Nobel Prize; Glasses didn’t

Scrapie
• An animal model was needed to study TSEs• Scrapie disease of sheep had many similarities to
kuru in terms of symptomatology and etiology• Could be transmitted to hamsters and mice, kuru
could not• Scrapie was used as first good animal model TSE• 2 month incubation in rodents• Infectious agent purified 5000 fold
– Nuclease resistant– UV and heat resistant– Sensitive to protease (only at high levels) & protein
denaturants

Chemical Treatment: Concentration PSTV Scrapie(viroid) (prion)
NH2OH (hydroxylamine) 0.1-0.5mM + -Psoralen (binds NA) 10-500µg/ml + -Phenol Saturated - +SDS 1-10% - +Zn2+ 2mM + -Urea 3-8M - +Alkali pH 10 (-) +KSCN 1M - +Enzymatic Treatment: Concentration PSTV ScrapieRNAse A 0.1-100µg/ml + -DNAse 100µg/ml - -Proteinase K 100µg/ml - +Trypsin 100µg/ml - +
+ = inactivated; - = no change in infectivity

• Glasses research in1950’s and 60’s• In 1967 Tikvah Alper at Hammersmith Hospital
found “particles” responsible for transmittable spongioform encephalitis contained no nucleic acids. Following characterization of viroids in 1971, many pursued the viroid hypothesis for prions.
• In 1982 Stanley Prusiner at UCSF concluded no NA, first named “proteinaceous infectious particles that resist inactivation by procedures that modify nucleic acids” as PRIONS-received Nobel Prize in 1997.
• Carlton Gajdusek receive Nobel Prize in 1976
Major Contributors to the History of Prions

Bovine spongiform encephalopathy (BSE)“mad cow disease”
• In Britain in the 1970’s, hydrocarbon-solvent extraction of meat and bone meal (MBM) for cattle feed was abandoned
• In 1987, BSE emerged• In 1988, BSE became a “reportable” disease Epidemiology
suggested a prion disease, and MBM use was abandoned• BSE incubation period is ~5 years• Estimated that over 1,000,000 cattle were infected• In 1989, human consumption of bovine CNS tissue (thought
to have the highest prion concentration) banned based on fears of transmission to humans
• In 1996, a new type of CJD appeared in Britain and France; young patients (<40 years old) and different neuropathology

Evidence that BSE gave rise to vCJD in humans
• Disease was found in younger cohort• Course of vCJD disease was 14 months rather than 4-6
month for CJD, suggesting more distantly related source• Proteolytic degradation pattern suggests variant CJD (vCJD)
closer to BSE than other CJD strains• Mouse inoculations showed identical reactions with BSE and
vCJD, different from classical CJD; sporadic CJD and all scrapie variants also different from BSE and vCJD
• When transgenic mice expressing bovine PrPc gene were inoculated with vCJD or BSE, course of disease was identical and different from inoculations with CJD or scrapie

Time course of epidemic BSE in the UK 1986-2000, with dates of major precautionary interventions. Mammalian ban on meat and bone meal in March 1996 extended a 1994 ban for farmed food animal species to include all mammalian species. SBO = specified bovine offals (brain, spinal cord, thymus, tonsil, spleen, and intestines from cattle >6 months of age); MBM = meat and bone meal (protein residue produced by rendering).

BSE continues to spread to other areas, but has not become epidemic as it was in Great Britain. It is a major concern because finding it may result in quarantines against beef from the country in which it is found.
U.S. - 2003

Cost of Mad Cow Disease
• 3 BSE-infected cows identified in Canada in May, 2003• BSE identified in a cow, originally from Canada, in
Washington state in Dec., 2003; another in Texas in 2005• Embargoes against U.S. and Canadian beef brought
immediately by most importers• Loss to U.S. and Canadian beef industries so far due to
embargoes: approximately $10 billion• Canada and U.S. test only a small proportion (<1%) of
cattle; Europe and Japan test 100%• Practice of feeding cow remains, including blood meal, to
cattle still done in U.S. and Canada

THE FIGURES: THE U.S. STORY (EXPORTS)
Year
Total beef exports to Japan (in 1,000 USD)
Total beef exports to Canada
1998 1,296,265 242,802
1999 1,358,431 225,895
2000 1,449,734 245,003
2001 1,235,392 217,527
2002 831,489 217,690
Jan-May 2003
454,026 111,893
THE FIGURES: THE U.S. STORY (IMPORTS)
Year Total beef imports from Japan
Total beef imports from Canada
1997 678 603,022
1998 870 722,828
1999 1,435 918,940
2000 248 962,732
2001 0 1,083,866
2002 0 1,096,238
Source: Foreign Agriculture Service USDA (Figures in U.S. dollars)
After BSE was found in Japan in 2001, U.S stopped importing Japanese beef; Japanese consumption of beef also plunged
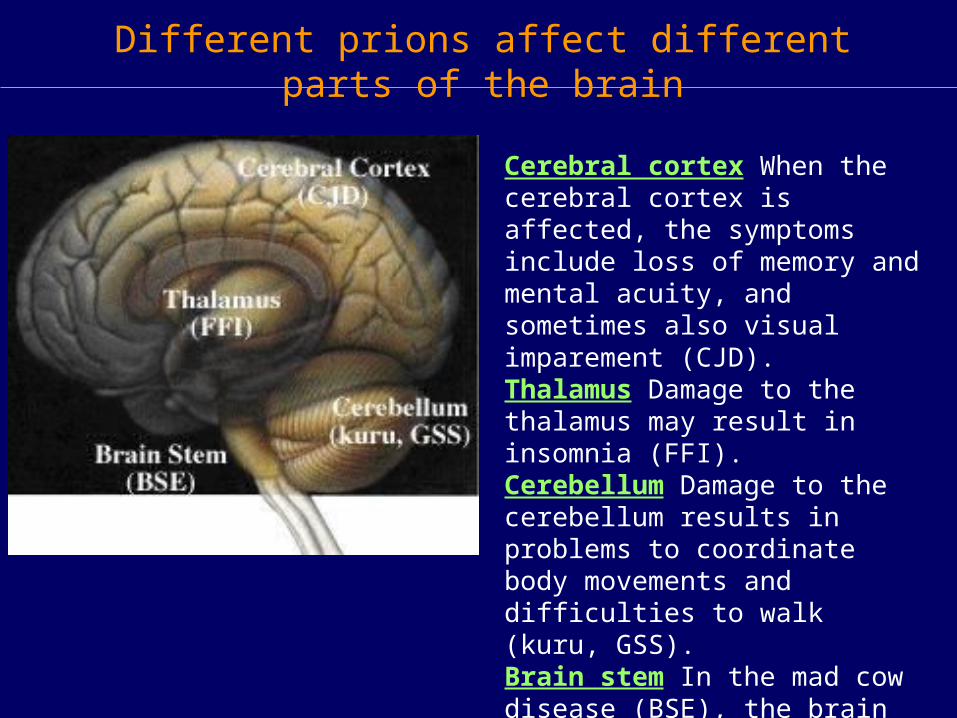
Different prions affect different parts of the brain
Cerebral cortex When the cerebral cortex is affected, the symptoms include loss of memory and mental acuity, and sometimes also visual imparement (CJD).Thalamus Damage to the thalamus may result in insomnia (FFI). Cerebellum Damage to the cerebellum results in problems to coordinate body movements and difficulties to walk (kuru, GSS). Brain stem In the mad cow disease (BSE), the brain stem is affected.

• Convert PrPc into PrSc• PrSc has identical primary structure but
different beta structures leading to resistance of protease cleavage.
• Brain tissue collects PrSc causing too much protein accumulation.
• Distinguished by nerve cell death causing large vacuoles and plaques in brain tissue
Effect of prions on neural tissue

How do prions function?
PrPSc
PrPc
The prion protein exists in two forms. The normal, innocuous protein (PrPc) can change its shape to a harmful, disease-causing form (PrPSc). The conversion from PrPc to PrPSc then proceeds via a chain-reaction. When enough PrPSc proteins have been made they form long filamentous aggregates that gradually damage neuronal tissue. The harmful PrPSc form is very resistant to high temperatures, UV-irradiation and strong degradative enzymes.

Prion biology
• For a prion (PrPSc) to infect a host, the host must have a recognizable cellular form (PrPc) of that prion
• Generally, the closer the phylogenetic relationship between the donor host and the recipient, the greater the chance for infection, and the more rapidly symptoms occur
• Level of accumulation of prion does not necessarily correspond to level of disease
• Mice in which PrPc copy is knocked out have altered sleep/wake cycles and circadian rhythm

Species barrier
• Infectous dose between species is usually higher than between animals of the same species (possibly a million fold), but it is sometimes the same (e.g. between scrapie doses for mink)
• When a species has been infected with a TSE of a different species it can then go on to infect a range of animals that the original species could not, and with a different dose.
• When a species has been infected, it can infect additional animals of the same species with much lower doses of agent.
• The histopathology of the disease in an animal infected from another species is not the same as if it had been infected from one of the same species.
• The incubation period of an animal infected from another species is much longer than that of an animal from one of the same species.

Criteria for prion demonstration
• Transmissible and associated with phenotype• Reversible curability – from “cured” individual,
phenomenon can arise again because the same event may reoccur in the same genotype
• Overproduction of normal protein increases frequency of prion formation – more normal molecules will be converted to prion form
• Phenotype relationship of prion and mutation of the normal gene for its protein in the host

Sequence of prion protein

Cellular trafficking of PrPC and PrPSc. PrPC (yellow dots) follows the secretory pathway of the cell through the endoplasmic reticulum (ER) and the Golgi. Mature PrPC is inserted via its GPI anchor into plasma membrane lipid rafts. The conversion of PrPC to PrPSc (orange ovals) occurs either on the cell surface or, following endocytosis, in a cellular compartment such as the endosome. PrPSc formed at the surface and released into the extracellular space may cause the plaques seen in TSE diseases such as human vCJD. The diffuse PrPSc deposits and neuronal vacuolation common to many sheep scrapie strains may be due to PrPSc formation in endocytic compartments or to endocytosed surface PrPSc accumulating inside the cell. Misfolded PrPC (squiggle) accumulating in the cytosol may also trigger PrPSc formation. (Inset) Structure of PrPC showing the GPI anchor, the glycan chains, the copper-binding octapeptide repeats, and the regions where the αhelices and loop structure of PrPC (red, blue) may be converted to the βsheets of PrPSc. ERAD, endoplasmic reticulum associated degradation.
Cellular trafficking of PrPC and PrPSc

Advances in prion control• BSE-resistant cattle
• Bovine PrPc gene cloned, modified by site-directed mutagenesis to produce BSE-resistance form
• Cattle were transformed with modified form of the gene, targeted to replace natural PrPc gene
• Transgenic animals homozygous for mutant gene express mutant copy and are resistant to BSE, but do not show altered sleep/wake cycles as seen in knockout mice
• Depleting neuronal PrPc in prion infection prevents disease and reverses spongiosis• Using transgenic mice, first demonstration that prion
infection and pathology can be reversed by ceasing expression of endogenous PrPc copy

Prions of yeast and fungi• Yeast and filamentous fungi make great experimental
tools because they are eukaryotes that normally grow as haploids with small genome sizes and powerful genetics
• Prions in yeast first identified by Wickner as non-Mendelian elements associated with nitrogen metabolism [URE3], then as a component of a suppressor tRNA activity [PSI].
• The first prion in filamentous fungi was identified in association with heterokaryon (vegetative) incompatibility in the ascomycete Podospora anserina – This is the only prion identified to date that is not associated
with a diseased state

Identity of alleles at the het-s locus is required for hyphae of different Podospora colonies to fuse. However, an encounter of het-s and het-S colonies will only result in the lethal reaction that comprises the incompatibility reaction if the Het-s protein is in its prion form (called [Het-s]).
From Wickner, 1999, J.Biol. Chem. 274: 555
The [Het-s] prion results in heterokaryon incompatibility in the filamentous fungus Podospora anserina.

Structural model of the SHaPrPC molecule. The model depict relative sizes of and locations of the Asn-linked oligosaccharides relative to the published structure of SHaPrP fragments inferred from NMR spectroscopy. SHaPrPC is shown attached to the plasma membrane by its GPI anchor to indicate how the range of movement of the N-terminal half of the molecule might be constrained in vivo. The putative protein X binding sites are indicated with an ‘X’ with lines pointing to the discontinuous epitope on helices C and B with which it interacts (Kaneko et al., 1997). Adapted from DeArmond et al. (DeArmond et al., 1999).