production of and responsiveness to transforming growth factor
TRANSCRIPT

(CANCER RESEARCH 49, 6269-6274, November 15, 1989]
Production of and Responsiveness to Transforming Growth Factor-/? in Normal andOncogene-transformed Human Mammary Epithelial Cells1
Eva M. Valverius, Dorothy Walker-Jones, Susan E. Bates, Martha R. Stampfer, Robin Clark, Frank McCormick,Robert B. Dickson2, and Marc E. Lippman
Vincent Lombardi Cancer Research Center, Georgetown University Hospital, Washington DC 20007 ¡E.M. V.t M. E. L., R. B. D.J; Department of Zoology, HowardUniversity, Washington DC 20059 ¡D.W. J.J; Medicine Branch, National Cancer Institute, National Institutes of Health, Bethesda, Maryland 20892 fS. E. B.]; LawrenceBerkeley Laboratory, Berkeley, California 94720 [M. R. S.J; and Cetus Corporation, Emeryville, California 94608 [R. C., F. M.]
ABSTRACT
Transforming growth factors-/? (TGF/8) are a family of closely related,ubiquitously expressed growth factors with the common properties ofinduction of growth inhibition and expression of differentiation-relatedmarkers in epithelial cells. We investigated the role of TGF0, in growthregulation of normal human mammary epithelial cells and inbenzo(a)pyrene immortalized sublines further transformed by oncogenesin retroviral vectors. The normal cells were markedly growth inhibitedby TGF/9,, produced TGF0 in a latent form, and expressed TGF0receptors. In the immortalized cells, both TGF/?-induced growth inhibition and TGF/3 receptor binding were reduced. With the single oncogenesv-Ha-raî,\-mos, and SV40 T, growth sensitivity to TGF/?, increased, butTGF|8 production or TGF/3 receptor expression was not altered.Transformation to full malignancy by both SV40 T and v-Ha-ros led toescape from growth inhibition by TGF/? under anchorage-independent,but not anchorage-dependent, conditions without affecting TGF/? production or receptor characteristics. Thus, modulation of TGF/} growth responsiveness in these normal and oncogene transformed human mammaryepithelial cells apparently occurs at a level distal to TGF/? receptorbinding and is not solely correlated to expression of transforming oncogenes. Further, modulation of TGF/? production is not an indicator ofmalignant transformation in this system.
INTRODUCTION
The mechanisms by which normal and malignant humanmammary cell growth are regulated are incompletely understood. One difficulty is the lack of an appropriate in vitro modelsystem that would allow the comparison of malignant cells tothe normal epithelial cells from which they were derived.Growth regulation studies typically have employed establishedbreast cancer cell lines. We now have available a system ofHMEC,3 derived from histopathologically normal breast tissue
obtained from reduction mammoplasty, capable of rapid proliferation for up to 22 passages in a serum-free medium (MCDB170) before senescence (1). Following benzo(a)pyrene treatment (2), an immortalized clone, 184A1N4, was selected andsubsequently used as recipient for the oncogenes \-mos, SV40T,and v-Ha-ros using retroviral vectors (3). These transformedcell lines have previously been characterized and shown toexpress the appropriate proteins of the transduced oncogenes(3). Coexpression of SV40T and v-Ha-ras led to expression of
Received 1/5/89; revised 6/9/89, 8/16/89; accepted 8/21/89.The costs of publication of this article were defrayed in part by the payment
of page charges. This article must therefore be hereby marked advertisement inaccordance with 18 U.S.C. Section 1734 solely to indicate this fact.
1Supported by NIH Grant CA-24844, and the Office of Energy Research,
Office of Health and Environmental Research of the U. S. Department of Energy,(Contract DE-AC03-76SF00098).
1To whom requests for reprints should be addressed, at Vincent Lombardi
Cancer Research Center, Georgetown University Hospital, 3900 Reservoir RoadNW, Washington DC 20007.
3The abbreviations used are: HMEC, human mammary epithelial cells; TGF0,transforming growth factor-0; TGF«,transforming growth factor-«;EGF, epidermal growth factor; FBS, fetal bovine serum; BSA, bovine serum albumin;1MEM, Improved Modified Eagle's Medium; HEPES, 4-(2-hydroxyethyl)-l-
piperazineethanesulfonic acid; CM, conditioned medium; SDS, sodium dodecylsulphate; SSC, standard saline citrate (0.15 M sodium chloride: 0.015 M sodiumcitrate, pH 7.4).
the fully transformed and tumorigenic phenotype in the cells.Using fibroblasts as a model system to study growth regula
tion, it was initially observed that malignant transformation bya variety of oncogenic retroviruses led to the production ofgrowth promoting substances (4). These were later identified asTGFa and TGF/3. Subsequently, it was demonstrated that whileTGF/3 can stimulate the growth of cells of mesenchymal origin,such as fibroblasts (5, 6), it inhibits growth of epithelial cells.Observation of the growth inhibitory properties of TGF/3 led tothe proposal that transformation could result from either increased production of growth stimulatory factors or decreasedproduction of growth inhibitory factors, or altered responsiveness to either or both groups of growth regulatory substances(4).
We have previously proposed a possible role for TGF/3 as anantiestrogen-induced negative growth regulator for estrogenresponsive human breast cancer cell lines in vitro (7). Anotherrecent study has demonstrated that TGF/3 can also act as apotential autocrine growth inhibitor of estrogen receptor negative human breast cancer cells (8). It has also been shown thatnormal mouse mammary gland growth in vivo is reversiblyinhibited by TGF/3 (9). Further, we have reported that twoestrogen receptor negative, highly tumorigenic breast cancercell lines produce more TGF/3 than some estrogen receptorpositive, less malignant breast cancer cells (10). In the presentstudy, we examined the responsiveness of the normal andoncogene-transformed HMEC to TGF/3. We sought to determine whether a more malignant phenotype could be explainedon the basis of escape from the growth inhibitory actions ofTGF/3, and whether any differences in responsiveness would beaccompanied by alterations in the production of endogenousTGF/3 or the expression of TGF/3 cell surface receptors.
MATERIALS AND METHODS
Cells and Cell Culture. The normal HMEC from human reductionmammoplasty donor 184 were routinely cultured in the serum-freeMCDB 170 medium (University of California, San Francisco, CA)containing bovine pituitary extract (Collaborative Research, Bedford,MA), mouse EGF (Collaborative Research), and bovine insulin (Sigma,St. Louis, MO) as described (1) and used in experiments after 10-12passages in culture. The epithelial origin of these cells has been established using immunocytochemical markers and electron microscopy(11). The immortalized A1N4 cells were propagated in a simplifiedmedium supplemented with 0.5% FBS (GIBCO, Grand Island, NY),EGF, hydrocortisone (Sigma), and insulin, while all the oncogenetransformed cells were maintained in medium containing 10% FBS, aspreviously described (3). The oncogene-transformed cell lines werederived and propagated as mass populations rather than clonal isolates,and all experiments were performed with recently thawed cells in orderto avoid gradual selection of an atypical subpopulation. The cell lineshave been characterized previously (3). In brief, the A1N4-T and A1N4-M cell lines (expressing SV40T and \-mos, respectively) grow poorlyin soft agar and do not form tumors in nude mice. The A1N4-H andA1N4-MH cells (expressing v-Ha-roj alone or with \-mos) are weakly
6269
Research. on February 13, 2018. © 1989 American Association for Cancercancerres.aacrjournals.org Downloaded from

TGF0 IN HUMAN MAMMARY EPITHELIAL CELLS
tumorigenic but neither one clones well in soft agar. The AIN4-THcells (expressing both SV40T and v-Ha-ra.v) have a fully transformedphenotype, demonstrating extensive anchorage-independent growthand marked tumorigenicity in nude mice (3). Neither the normalparental 184 cells nor any of the immortalized oncogene transformedsublines have thus far been found to express the estrogen receptor.4
CC1-64 cells (provided by Dr. David Danielpour, Laboratory ofChemoprevention, NIH, Bethesda. MD), and A549 cells (obtained fromthe American Type Culture Collection, Rockville, MD) were maintained in IMEM (Biofluids, Rockville, MD) with 10% FBS.
TGF/i, and '"I-TGF/i, (50-100 nCi/Mg) for initial experiments were
provided by Dr. M. B. Sporn (Laboratory of Chemoprevention, NIH,Bethesda, MD), and subsequently obtained from R&D Systems (Minneapolis, MN). Recombinant human TGF«was supplied by Rik De-rynck (Genentech Inc., San Francisco, CA).
Monolayer Growth Assays. Cells in their respective culture mediumwere seeded in triplicates in 12-well cluster dishes (Costar, Cambridge,MA). Media were changed and growth factors added after an overnightincubation. At the end of the growth period, cells were trypsinized andcounted using a Coulter Particle Counter.
Anchorage-independent Growth Assays. Cells in a mixture of growthmedium with TGF« and/or TGF/3| and 0.36% Bacto-agar (Difco,Detroit, Ml) were plated in 35-mm dishes (Costar), incubated, andcounted as previously described (12).
Conditioned Media Collections. Cells were grown to 75-85% con-fluency in T-175 tissue culture flasks (Falcon) in their regular medium,then rinsed and given an 8-12-h wash in serum-free medium, andlimili) incubated for 48 h in serum-free medium. For the 184 cells,MCDB 170 medium was maintained minus bovine pituitary extract,EGF, and insulin. For the A1N4 cells, serum-free medium consisted ofIMEM with transferrin (2 mg/liter; Sigma). HEPES-buffer (20 ITIM;GIBCO), and trace elements (GIBCO), in addition to EGF, hydrocortisone, and insulin as in the regular medium. For all other cells, serum-free medium consisted of IMEM with transferrin, HEPES-buffer, andtrace elements. At the end of the conditioning period (when cells were85-95% confluent), media were collected, the protease inhibitors apro-tinin (0.2% v/v; Sigma), leupeptin (2 Mg/liter; Boehringer-Mannheim,Indianapolis, IN), and phenylmethylsulfonylfluoride (10~7 M; Sigma)
were added, and media were clarified of cellular debris by filtration(0.45-jim pore, Nalgene). CM were stored at -20°C until the time of
assay.Quantitation of TGF0 Activity in CM. At the time of assay, CM were
thawed, BSA (0.1%; Sigma) was added, and samples were used directlyfor quantification of bioactivity, or concentrated and dialyzed againstdistilled water on Centricon centrifugal devices (Amicon, Danvers, MA)for determination of TGF/J receptor-competing activity. In both assays,amounts of total TGF/3 activity were determined following transientacidification of the samples by treatment with HCI (45 ITIMfinalconcentration) for 5 min at 23°C,followed by neutralization with
NaOH and HEPES essentially as described (7). Parallel aliquots ofCM or media blanks received a mixture of HCI, NaOH, and HEPESto equal ionic strength.
TGF/j radioreceptor assays were performed using aliquots of dialyzedand concentrated CM and standard curves of porcine platelet TGFßiin competition with 50 pM '"I-TGF0, on confluent cultures of A549
human lung adenocarcinoma cells, as described (7). This procedure willallow quantitative determination of amounts of total TGF/i receptor-competing activity present in the CM, while the recovery of activeTGF/J after dialysis and concentration is unreliable. Quantitation ofTGF/3 activity was done by interpolation from the standard curves, andresults were normalized to DNA content ( 13) or cell numbers from theCM producing cells.
Inhibition of [3H]thymidine incorporation into CCI-64 mink lung
epithelial cells was tested as previously described (14) using aliquots ofunconcentrated, nondialyzed CM. Results were interpolated from parallel standard curves of porcine TGF/3,.
TGF/3 Receptor Binding Studies. Whole-cell monolayer binding assays were performed essentially as described (15) on subconfluent cellcultures. Saturation curves using 0.3-600 pM '"I-TGF/3, (in some cases
' E. M. Valverius. unpublished data.
up to 6 nM) with or without 100-fold excess unlabeled TGF/3i inDulbecco's modified Eagle's medium containing 0.1% BSA were incubated for 2 h at 23°C.Cells were then washed four times with ice-cold
buffer with 0.1% BSA, and lysed in 1% Triton X-100/20 ITIMHEPES/10% glycerol for l h at 37°C.Radioactivity in aliquots of lysates wasdetermined in an LKB gamma counter (efficiency for I25I79%), where
upon binding parameters were calculated using the program LIGAND(16) and Scatchard plots were generated. Binding parameters werenormalized to cell numbers determined from comparably treated parallel wells.
RNA Preparation and Northern Blot Analysis. Total cellular RNAwas isolated in guanidine isothiocyanate and separated in 1% agarose-6% formaldehyde gels as previously described (17). Gels found to havecomparably loaded and undegraded RNA samples upon ethidium bromide staining, were subjected to alkaline hydrolysis, neutralization, andNorthern transfer to nitrocellulose by capillary blot. Probes used werea human TGF/J, complementary' DNA insert probe encoding the mature
TGFfi, (XC1) (18) and a riboprobe from 36B4 complementary DNA(gift of P. Chambón, Strasbourg, France) subcloned into pGEM3, wasused as a control probe (19). The 36B4 probe, originally derived as anonestrogen regulated sequence in MCF-7 cells, has shown less variability for the 184 cells than the more commonly used fi-actin probe.
Hybridizations were performed for 18 h at 55'C in 50% formamide,5 X SSC, 5 x Denhardt's, 0.1% SDS, and 200 Mg/ml salmon sperm
DNA. Filters were then washed twice for 30 min at 2TC in 1 x SSC,0.1% SDS warmed to 68°C,and for l h at 68°Cin 0.1 x SSC, 0.1%
SDS. Autoradiography was performed with Kodak XAR-5 film at-70°C.
Statistical Analyses. For mitogenic assays, values are expressed asmeans with standard deviations. IC50values were calculated from Hillplots (20). Conditioned media data were calculated from interpolationsof best fitted standard curve using a simple curve-fitting analysis.Statistical significances were determined by ANOVA using the Statistical Analysis System computer program. The LIGAND computerprogram was used for calculating receptor binding parameters, andnonspecific binding was estimated together with the other bindingparameters. Generally, nonspecific binding did not exceed 5% of totallabeled ligand added for any receptor binding assays.
RESULTS
TGF/3 Effects on Cell Growth. In dose-response experiments,we found that the normal as well as the oncogene transformedHMEC were all growth inhibited to various degrees by TGFß,(Fig. 1, Table 1). The parental 184 cells, rapidly proliferatingat passages 10-12, were maximally inhibited to 75% of untreated control numbers after 5 days exposure to 100 pM (2.5ng/ml) TGF/3. While the majority of the 184 cells were growthinhibited and morphologically changed (Fig. 1 and data notshown; 21 ), some cells continued to actively proliferate throughout the treatment period. The TGF/3-induced decrease in cellnumbers was comparable in the 184, A1N4-T, A1N4-H, andA1N4-MH cells, although A1N4-T cell counts were significantly more reduced than the 184 HMEC (P < 0.05). A1N4and A1N4-TH were both significantly less growth inhibited inanchorage-dependent assays than all the other HMEC and didnot differ in sensitivity from each other. In both of these lines,a large proportion of the cells continued proliferating andremained morphologically unchanged. High or low serum content of the medium (10% or 0.5%) and the absence or presenceof EGF gave quantitatively and qualitatively comparable effectsof TGF/3 for the A1N4-T and A1N4-TH cells (Fig. 1 and datanot shown). The A l N4 cells showed the same diminished TGF/3responsiveness in both the MCDB 170 medium and in itsregular simplified 0.5% serum-containing medium (21).
Eight-day growth curves with various concentrations ofTGF/i, confirmed that close to maximal growth inhibition was
6270
Research. on February 13, 2018. © 1989 American Association for Cancercancerres.aacrjournals.org Downloaded from

TGFfi IN HUMAN MAMMARY EPITHELIAL CELLS
ooc2ou.oÕÎ<!rrÜJmz>"Z.
_lÃœJCJ100'90-80-70-60-50-40-30-20-1010090807060-50-40-30-20-101
nnIUU9080706050403020100l-ti^—-»....^^~°
"~~~§^^"^^0\'
\T•J....V....f.\
L «A1M\ •¿�—¿�AlNo\-0
o-184'
—¿�» t t t tlt| 1—¿�t t *•*++») 1—¿�t t t t Mt) ' t 1 t t Ml( 1—¿�t »»•¿�««*) 1 h t 1 t lltj
_.,^... . w, A"^^T' : -TV-,•*
' •¿�•¿�••T-;,s\
'-*. \\ '*. •¿�^\
*'.X\
'"'..'^.•.
VTX••¿� * 1 - -.•1
N. ' ,,** s •¿�-AINV\
X-v_.---v-MN
*.N
\v
'.A"'"*
A ""-
'T- T —¿�T'
1 t t t t H»| 1 1 t t *Mt| t ( t 1 tilt) t » t t-t-tt+1 1—¿�t * t tttff 1—¿�t |t«44t{V
"-ix-\r ^^\\" "•-«- - -«-TH\
\\L.\*-\""-'-.
l •¿�-H't-~-.*',--°-MH-o-—
l—¿� i •¿�,- -; ...i.
0.01 0.1 1.0 10 100 500
TGF0 CONCENTRATION (pM)Fig. 1. TGFfi dose titrations and anchorage-dependent growth. 5-10 x IO3
cells/well were seeded in 12-well cluster dishes. TGFfJ was added with freshmedium after an overnight incubation. 5 days later, cells were trypsinized andcounted. Results shown are averages ±SD of triplicate determinations from fourseparate experiments and expressed as percentage of control cell numbers. Somestandard deviations are smaller than the symbols used. Top, effect of immortalization upon cellular responsiveness; mÃdale,effect of single oncogenes; bottom,effect of combined oncogenes.
Table l TGFßgrowth effects on HMECCells were plated, treated, and counted as described in Fig. 1. IC50and maximal
percentage inhibition were determined from Hill plots of cell numbers fromtriplicate determination in four separate experiments for each cell and thenaverages ±SD calculated. Values for A1N4-M are from two experiments.
Cell184A1N4A1N4-TA1N4-MA1N4-HA1N4-MHA1N4-THIC50(PM)3.2
±0.22.6±0.81.2
±0.10.71.1
±0.81.5±1.42.9±2.0Maximal
inhibition(% ofcontrol)75.0
±0.144.3±10.986.0±1.763.078.3
±3.182.7±5.533.7±2.5TGF/3
concentration(PM)1001001001010100100
and morphological changes induced by TGF/3 were reversibleupon repeated washing (data not shown).
Under anchorage-independent conditions, TGF/3 could inhibit the cloning of A1N4-H and A1N4-MH, and counteractthe TGF«-induced cloning of A1N4-T (Fig. 3). The oppositeeffects on TGF« and TGF/3 on A1N4-T cloning could betitrated against each other such that one could cancel the effectof the other (data not shown). A1N4-M had such a low cloningefficiency (<0.5%) that effects of TGF/3 could not be accuratelycalculated. In contrast, A1N4-TH displayed a very high cloningefficiency (up to approximately 30%), and was refractory toeither TGF/3 or TGF«.
TGF/3 Cell Surface Receptor Expression. In order to investigate whether differences in the responsiveness to exogenousTGF/3 might correlate with altered TGF/3 receptor bindingcharacteristics, whole-cell binding assays were performed on allthe 184-derived cells. As shown in Table 2, all of the immortalized and oncogene transformed HMEC had 5,500 to 9,900receptor sites per cell with Kd values between 1.4 and 4.1 pM.There was no correlation between the presence of a particularoncogene and a specific receptor binding characteristic. Thenormal 184 cells displayed approximately 22,500 sites per cell,but the Kd was not significantly different from that found in theother HMEC. Scatchard plots from one representative assayare shown in Fig. 4.
Production of TGF/Î.To assess whether expression of themalignant phenotype or degree of responsiveness to exogenousTGF/3 might correspond to any qualitative or quantitative differences in endogenous TGF/J production, the amounts of activeand latent, i.e., acid-activated, TGFßsecreted into CM wereassayed. As shown in Table 3, all the 184-derived cells producedlow levels of radioreceptor-reactive and bioactive TGF/3 (corresponding to 1-5 pM before normalization to DNA content orcell numbers, respectively). The TGF/3 levels for the 184 cellswere difficult to measure accurately due to interference by theMCDB 170 medium in both assays; amounts of TGF/3 producedin the active form were negligible after subtraction of themedium background. CM from the A1N4 cells could only betested in the radioreceptor assay due to interference in thebioassay by the growth medium. Accurate determination ofamounts of TGF/3 secreted in the active form was difficult. Forthe radioreceptor assay, a significant proportion of active TGF/?was lost during the necessary preparation of the CM by dialysisand concentration. In the bioassay, amounts of active TGF/3may appear higher than expected due to partial activation oflatent TGF0 by the CC1-64 cells during the overnight incubation. For both assays, means were compared using analysis ofvariance (ANOVA) and Fischer's least significant difference
test, and were not found to differ significantly among the cells.Steady-state TGF/3, mRNA expression also failed to show
any significant differences among the immortalized and oncogene carrying cells (Fig. 5). In repeated hybridizations, the 184cells did not have consistently different levels of TGF/3 mRNAthan the other HMEC.
DISCUSSION
obtained for all the oncogene expressing cells with 10 pM (0.25ng/ml) TGF/i; increasing the TGF/3 concentration to 500 pMhad essentially no additional effect (Fig. 2). Cell doubling timeswere slowed by only 17% for the A1N4-TH and up to 67% forthe A1N4-T cells. For all the cells, both the growth inhibition
6271
The notion that the transformation of epithelial cells fromnormal to malignant growth patterns may involve escape fromnormal growth inhibitory mechanisms has been substantiatedin several in vitro model systems. For example, both v-Ha-rastransformed and chemically transformed rat liver epithelial cellswere found to be resistant to growth inhibition by TGF/3 (22,23). Normal human bronchial epithelial cells transformed by v-
Research. on February 13, 2018. © 1989 American Association for Cancercancerres.aacrjournals.org Downloaded from
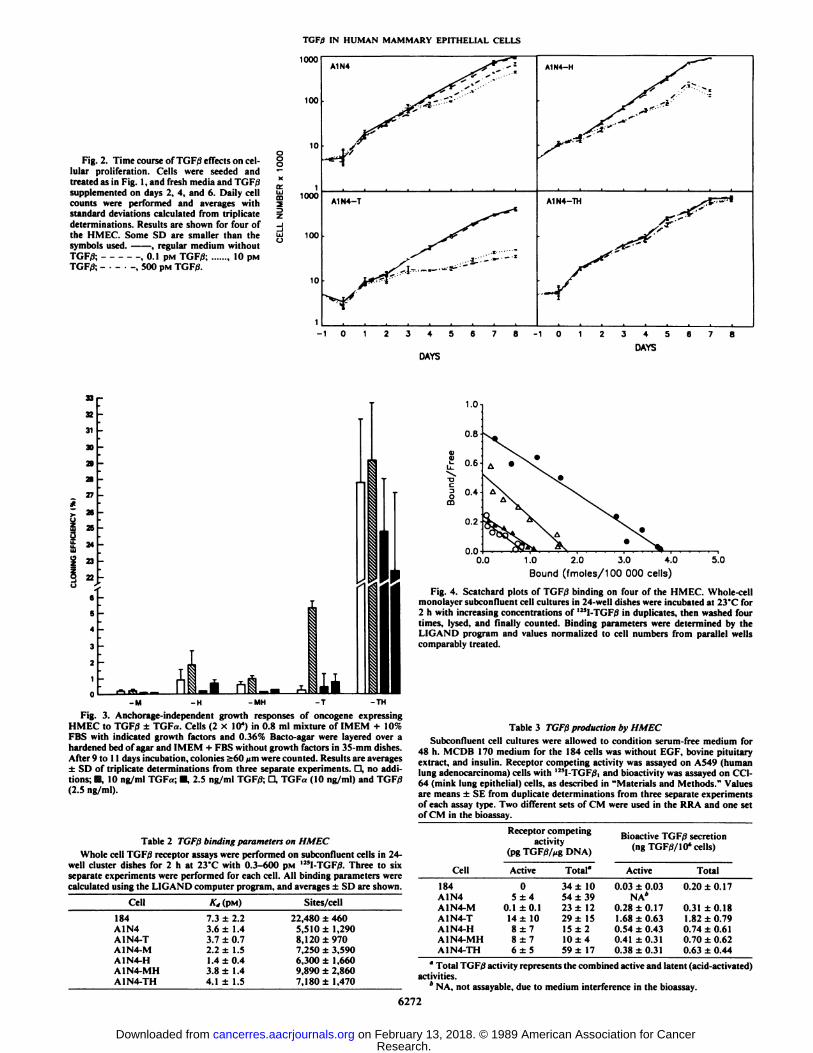
TGF0 IN HUMAN MAMMARY EPITHELIAL CELLS
1000
100
10
Fig. 2. Time course of TGF/3 effects on cellular proliferation. Cells were seeded andtreated as in Fig. I, and fresh media and K.I,;supplemented on days 2, 4, and 6. Daily cellcounts were performed and averages withstandard deviations calculated from triplicatedeterminations. Results are shown for four ofthe HMEC. Some SD are smaller than thesymbols used. , regular medium withoutTGF/3; ,0.1 pM TGF/3; 10 pinTGF/3; -•-•-, 500 PM TGFfi.
3=>
10
A1N4-T
345
DAYS
345
DAYS
£i0¡£CLONING
E33323130a2127*2423
22eC4321
o-----—
1:[•Õ-
,----~
_J_i
A^__¿Luni—*in^^^1$$Ii^^1s$1-M -H -MH -T -TH
Fig. 3. Anchorage-independent growth responses of oncogene expressingHMEC to TGFiJ ±TGFa. Cells (2 x 10") in 0.8 ml mixture of IMEM -I- 10%FBS with indicated growth factors and 0.36% Bacto-agar were layered over ahardened bed of agar and IMEM + FBS without growth factors in 35-mm dishes.After 9 to 11 days incubation, colonies >60 urn were counted. Results are averages±SD of triplicate determinations from three separate experiments. D, no additions; m, 10 ng/ml TGF«;•¿�,2.5 ng/ml TGF/3; D, TGFa (10 ng/ml) and TGF/3(2.5 ng/ml).
Table 2 TGFßbinding parameters on HMECWhole cell TGF/3 receptor assays were performed on subconfluent cells in 24-
well cluster dishes for 2 h at 23'C with 0.3-600 pM '"I-TGF0. Three to six
separate experiments were performed for each cell. All binding parameters werecalculated using the L1GAND computer program, and averages ±SD are shown.
Cell184A1N4A1N4-TA1N4-MA1N4-HA1N4-MHA1N4-TH*„(PM)7.3
±2.23.6±1.43.7±0.72.2±1.51.4
±0.43.8±1.44.1±1.5Sites/cell22,480
±4605,510±1,2908,
120±9707.250±3,5906.300±1,6609.890±2,8607,180±
1,470
0.0 1.0 2.0 3.0 4.0Bound (fmoles/100 000 cells)
Fig. 4. Scatchard plots of TGF/3 binding on four of the HMEC. Whole-cellmonolayer subconfluent cell cultures in 24-well dishes were incubated at 23'C for2 h with increasing concentrations of '"l-TGF/3 in duplicates, then washed four
times, lysed, and finally counted. Binding parameters were determined by theI l( .AM»program and values normalized to cell numbers from parallel wellscomparably treated.
Table 3 TGFßproduction by HMECSubconfluent cell cultures were allowed to condition serum-free medium for
48 h. MCDB 170 medium for the 184 cells was without EGF, bovine pituitaryextract, and insulin. Receptor competing activity was assayed on A549 (humanlung adenocarcinoma) cells with 125l-TGF/3,and bioactivity was assayed on CC1-64 (mink lung epithelial) cells, as described in "Materials and Methods." Values
are means ±SE from duplicate determinations from three separate experimentsof each assay type. Two different sets of CM were used in the RRA and one setof CM in the bioassay.
Receptor competingactivity
(pgTGF/3/xgDNA)
Bioactive TGF/3 secretion(ngTGFtf/10'cells)
Cell184A1N4A1N4-MA1N4-TA1N4-HA1N4-MHA1N4-THActive05±40.1
±0.114±108±78±76±5Total"34
±1054±3923±1229±1515±
210±459
±17Active0.03
±0.03NA*0.28
±0.171.68±0.630.54
±0.430.41±0.310.38
±0.31Total0.20
±0.170.31
±0.181.82±0.790.74±0.610.70
±0.620.63±0.44
" Total TGF/3 activity represents the combined active and latent (acid-activated)
activities.* NA, not assayable, due to medium interference in the bioassay.
6272
Research. on February 13, 2018. © 1989 American Association for Cancercancerres.aacrjournals.org Downloaded from

TGF)3 IN HUMAN MAMMARY EPITHELIAL CELLS
I I I I I•¿�<f -«fr •¿�*••**••<*•
<«•Z 2 2 2 Z00 *-*-*-*- i-
2.5--18S
36B4- * » » «»
Fig. 5. Steady state TGF0 mRNA expression in the HMEC. Northern analysisof 8 /ig total RNA extracted from subconfluent cultures of the HMEC. Theexpected 2.5-kilobase band (top) is apparent at comparable levels in all HMECwhen normalized for expression of the 36B4 mRNA (bottom).
Vii-ras were also resistant to TGF/3 inhibition (24). Neither theTGF|9 receptor status nor the levels of endogenous TGF/3production of the cells were analyzed in these reports.
The role of TGF/3 receptor expression in modulating cellularresponsiveness to TGF/3 is not unequivocally established. Intwo reports comparing normal human cell populations andcancer lines derived from the same organ, TGF/3 resistance inthe malignant cells was found to correlate with a loss of TGF/3receptors, specifically in retinoblastoma (25) and skin squamouscell carcinoma lines (26). However, in a third system, TGF/3resistant human bronchial carcinoma cell lines showed no significant change in TGF/3 receptor expression in comparison tonormal human bronchial epithelial cells (27). In contrast, otherreports have shown that neoplastic transformation does notalways lead to diminished TGF/3 responsiveness. No change inTGF/3 responsiveness (rat liver), or even growth stimulation byTGF/3 (human large cell lung carcinoma) have been demonstrated when malignant cells were compared to their normalcounterparts (28, 29).
To date, all estrogen receptor negative human breast cancercell lines studied have been found to be growth inhibited byTGF/3 and express TGF/3 receptors (7, 8, 30). In this paper, wehave extended these observations to a model system derivedfrom estrogen receptor negative, proliferating normal humanmammary epithelial cells and examined possible correlationsbetween responsiveness to exogenous TGF/3 and expression ofendogenous TGF/3 and TGF/3 cell surface receptors. We foundthat immortalization by the carcinogen benzo(a)pyrene wasassociated with attenuation of TGF/3 responsiveness in anchorage-dependent growth assays and diminished TGF/3 receptorexpression, while production of TGF/3 was essentially unchanged. In contrast, further transformation of the A1N4 cellsby the introduction of the single oncogenes v-mos, SV40 T, orv-Ha-ras apparently restored the cellular sensitivity (as deter
mined by reduction in cell numbers) to TGF/3. This enhancedresponsiveness occurred without further alterations in TGF/3receptor characteristics or TGF/3 production. Transformationto a fully malignant phenotype by the two oncogenes SV40 Tand v-Ha-ras completely desensitized the cells to TGF/3 underanchorage-independent conditions but only partially reducedtheir responsiveness under anchorage-dependent conditions.TGF/3 receptor expression and TGF/3 production were not
significantly altered in these fully transformed cells. Thus, thehypothesis that neoplastic transformation may involve escapefrom normal growth inhibitory pathways such as that of TGF/3is only supported by our observations of the reduced anchorage-independent TGF/3 responsiveness in the most fully transformed cells. The observation that cells of intermediate stagesof transformation may vary in their TGF/3 responsiveness issupported by a recent report on colon cancer cell lines of varyingdegrees of differentiation (31). It was found that one moderatelywell differentiated colon cancer cell line was unresponsive toTGF/3 whereas two other cell lines of the same classificationwere much more sensitive. Further, differential TGF/3 responsiveness in the colon cancer cell lines did not influence TGF/3receptor status (31). It may be that, just as transformation tofull malignancy appears to be a stepwise process, several consecutive events deregulating cell growth control are neededbefore effects on cell numbers can be measured. Clearly, theexpression of oncogenes in the human mammary epithelial cellscauses more profound perturbations in cell growth control thancan be explained by altered sensitivity to, receptors for, orproduction of TGF/3. These observations confirm the hypothesis that it is the cellular responsiveness to growth factors, ratherthan the nature of a particular growth factor or its level ofendogenous expression, that may correlate with the ability toexpress the transformed phenotype (32).
ACKNOWLEDGMENTS
We thank Drs. Lalage Wakefield and David Danielpour for helpfuldiscussions of methods for CM assays and for critical reading of themanuscript.
REFERENCES
1. Hammond, S. L., Harn, R. G., and Stampfer, M. R. Serum-free growth ofhuman mammary epithelial cells: rapid clonal growth in defined medium andextended serial passage with pituitary extract. Proc. Nati. Acad. Sci. USA81: 5435-5439, 1984.
2. Stampfer, M. R., and Bartley, J. C. Induction of transformation and continuous cell lines from normal human mammary epithelial cells after exposureto benzo-a-pyrene. Proc. Nati. Acad. Sci. USA, 82: 2394-2398, 1985.
3. Clark, R., Stampfer, M. R., Milley, R., O'Rourke, E., Walen, K. H., Kriegler,
M., Kopplin, J., and McCormick, F. Transformation of human mammaryepithelial cells by oncogenic retroviruses. Cancer Res., 48:4689-4694, 1988.
4. Sporn, M. B., and Roberts, A. B. Autocrine growth factors and cancer.Nature (Lond.), 313: 745-747, 1985.
5. Moses, H. L., Tucker, R. F., Leof, E. B., Coffey, R. J., Halper, J., andShipley, G. D. Type-/? transforming growth factor is a growth stimulator anda growth inhibitor. In: J. Feramisco, B. Ozanne, and C. Stiles (eds.), CancerCells, Vol. 3, pp. 65-71. Cold Spring Harbor, NY: Cold Spring HarborLaboratory', 1985.
6. Roberts, A. B., Anzano, M. A., Wakefield, L. M., Roche, N. S., Stern, D.F., and Sporn, M. B. Type ßtransforming growth factor: a bifunctionalregulator of cellular growth. Proc. Nati. Acad. Sci. USA, 82: 119-123, 1985.
7. Knabbe, C., Lippman, M. E., Wakefield, L. M., Flanders, K. C., Kasid, A.,Derynck, R., and Dickson, R. B. Evidence that transforming growth factor-3 is a hormonally regulated negative growth factor in human breast cancercells. Cell, 48: 417-428, 1987.
8. Arteaga, C. L., Tandon, A. K., Von Hoff, D. D., and Osborne, C. K.Transforming growth factor ß:potential autocrine growth inhibitor of estrogen receptor-negative human breast cancer cells. Cancer Res., 48: 3898-3904, 1988.
9. Silberstein, G. B., and Daniel, C. W. Reversible inhibition of mammary glandgrowth by transforming growth factor-/). Science (Wash. DC), 237:291-293,1987.
10. Bates, S. E., McManaway, M. E., Lippman, M. E., and Dickson, R. B.Characterization of estrogen responsive transforming activity in humanbreast cancer cell lines. Cancer Res., 46: 1707-1713, 1986.
11. Stampfer, M. R., and Bartley, J. C. Human mammary epithelial cells inculture: differentiation and transformation. In: M. E. Lippman and R. B.Dickson (eds.). Breast Cancer: Cellular and Molecular Biology, pp. 1-24.Boston: Kluwer Academic Publishers, 1988.
12. Valverius, E. M., Bates, S. E., Stampfer, M. R., Clark, R., McCormick, F.,Salomon, D. S., Lippman, M. E., and Dickson, R. B. Transforming growthfactor alpha production and EGF receptor expression in normal and onco-
6273
Research. on February 13, 2018. © 1989 American Association for Cancercancerres.aacrjournals.org Downloaded from

TGFßIN HUMAN MAMMARY EPITHELIAL CELLS
gene transformed human mammary epithelial cells. Mol. Endo., 3:203-214,1989.
13. Brunk, C. K., Jones, K. C., and James, T. W. Assay for nanogram quantitiesof DNA in cellular homogenates. Anal. Chem., 92:497-500, 1979.
14. Danielpour, D., Dart, L. L., Flanders, K. C., Roberts, A. B., and Sporn, M.B. Immunodetection and quantification of the two forms of transforminggrowth factor beta (TGF/3,, TGFfc) secreted by cells in culture. J. Cell.Physiol., 138:19-86, 1989.
15. Wakefield, L. M., Smith, D. M., Masui, T., Harris, C. C., and Sporn, M. B.Distribution and modulation of the cellular receptor for transforming growthfactor-beta. J. Cell. Biol., 105: 965-975, 1987.
16. Munson, P. J., and Rodbard, D. LIGAND: A versatile computerized approach for characterization of ligand-binding systems. Analyt. Biochem..107: 220-239, 1980.
17. Bates, S. E., Davidson, N. E., Valverius, E. M., Fréter,C. E., Dickson, R.B., Tarn, J. P., Kudlow, J. E., Lippman, M. E., and Salomon, D. S.Expression of transforming growth factor a and its messenger ribonucleicacid in human breast cancer: its regulation by estrogen and its possiblefunctional significance. Mol. Endo., 2: 543-555, 1988.
18. Derynck, R., Jarrett, J. A., Chen, F. Y., Eaton, D. H.. Bell, J. R.. Assoian.R. K., Roberts. A. B., Sporn, M. B., and Goeddel, D. V. Human transforminggrowth factor ßcomplementary DNA sequence and expression in normaland transformed cells. Nature (Lond.), 316: 701-705, 1985.
19. Masiakowski, P., Breathnach, R., Block, J., Gannon. F.. Krust, A., andChambón, P. Cloning of cDNA sequences of hormone-regulated genes fromthe MCF-7 human breast cancer cell line. Nucleic Acids Res., 10: 7895-7903, 1982.
20. Bennet, J. P., and Yamamura, H. I. Neurotransmitter, hormone, or drugreceptor binding methods. In: H. I. Yamamura, S. J. Enna, and M, J. Kuhar(eds.), Neurotransmitter Receptor Binding, Ed. 2, pp. 82-84. New York:Raven Press, 1985.
21. Hosobuchi, M. and Stampfer, M. R. Effects of transforming growth factorbeta on growth of human mammary epithelial cells in culture. In Vitro(Rockville), 25:705-713. 1989.
22. Houck, K. A., Strom, S. C., and Michalopoulos, G. Resistance to the growthinhibitory effect of transforming growth factor beta is induced by transfectionof an activated H-ras oncogene into rat liver epithelial cells (Abstract 254).
Proc. Am. Assoc. Cancer Res., 28: 64, 1987.23. McMahon, J. B., Richards, W. L., del Campo, A. A., Song, M-K. H., and
Thorgeirsson. S. S. Differential effects of transforming growth factor-f) onproliferation of normal and malignant rat liver epithelial cells in culture.Cancer Res., 46: 4665-4671, 1986.
24. Reddel, R. R., Ke, Y., Kaighn, M. E., Malan-Shibley, L., Lechner, J. F.,Rhim, J. S., and Harris, C. C. Human bronchial epithelial cells neoplasticallytransformed by v-Ki-ros: altered response to inducers of terminal squamousdifferentiation. Oncogene Res., in press, 1989.
25. Kimchi, A., Wang, X-F., Weinberg, R. A., Cheifetz, S.. and Massague, J.Absence of TGFfi receptors and growth inhibitory responses in retinoblas-toma cells. Science (Wash. DC), 240: 196-199, 1988.
26. Shipley, G. D., Pittelkow. M. R., Wille. J. J. Jr., Scott, R. E., and Moses,H. L. Reversible inhibition of normal human prokeratinocyte proliferationby type ßtransforming growth factor-growth inhibitor in serum-free medium.Cancer Res., 46: 2068-2071. 1986.
27. Masui, T., Wakefield, L. M., Lechner. J. F., La Veck, M. A., Sporn, M. B.,and Harris, C. C. Type ßtransforming growth factor is the primary differentiation-inducing serum factor for normal human bronchial epithelial cells.Proc. Nati. Acad. Sci. USA, 83: 2438-2442, 1986.
28. Wollenberg, G. K., Semple, E., Quinn. B. A., and Hayes, M. A. Inhibitionof proliferation of normal, preneoplastic, and neoplastic rat hepatocytes bytransforming growth factor-^. Cancer Res.. 47: 6595-6599, 1987.
29. Jetten, A. M., Shirley, J. E., and Stoner, G. Regulation of proliferation anddifferentiation of respiratory tract epithelial cells by TGF/3. Exp. Cell. Res.,767:539-549, 1986.
30. Fernandez-Pol, J. A., Klos, D. J., Hamilton, P. D., and Talkad, V. D.Modulation of epidermal growth factor receptor gene expression by transforming growth factor-rf in a human breast carcinoma cell line. Cancer Res.,47:4260-4265, 1987.
31. Hoosein, N. M., Mcknight, M. K., Levine, A. E., Mulder, K. M., Childress,K. E., Brattain, D. E., and Brattain, M. G. Differential sensitivity of subclasses of human colon carcinoma cell lines to the growth inhibitory effectsof transforming growth factor-rfl. Exp. Cell. Res., 181: 442-453, 1989.
32. Kaplan, P. L., and Ozanne, B. Cellular responsiveness to growth factorscorrelates with a cell's ability to express the transformed phenotype. Cell,33: 931-938. 1983.
6274
Research. on February 13, 2018. © 1989 American Association for Cancercancerres.aacrjournals.org Downloaded from

1989;49:6269-6274. Cancer Res Eva M. Valverius, Dorothy Walker-Jones, Susan E. Bates, et al. Mammary Epithelial Cells
in Normal and Oncogene-transformed HumanβFactor-Production of and Responsiveness to Transforming Growth
Updated version
http://cancerres.aacrjournals.org/content/49/22/6269
Access the most recent version of this article at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://cancerres.aacrjournals.org/content/49/22/6269To request permission to re-use all or part of this article, use this link
Research. on February 13, 2018. © 1989 American Association for Cancercancerres.aacrjournals.org Downloaded from