promoter-associated small double-stranded rna interacts ... · promoter to activate p21 expression....
TRANSCRIPT

Biochem. J. (2012) 447, 407–416 (Printed in Great Britain) doi:10.1042/BJ20120256 407
Promoter-associated small double-stranded RNA interacts withheterogeneous nuclear ribonucleoprotein A2/B1 to inducetranscriptional activationJia HU, Zhong CHEN1, Ding XIA, Jia WU, Hua XU and Zhang-Qun YE1
Department of Urology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430030, China
Several recent reports have demonstrated that small activ-ating dsRNA [double-stranded RNA; saRNA (small activatingdsRNA)] complementary to promoter regions can up-regulategene expression in mammalian cells, a phenomenon termedRNAa (RNA activation). However, the mechanism of RNAaremains obscure with regard to what is the target molecule forpromoter-targeted saRNA and what are the proteins involvedin this process. p21Waf1/Cip1 (p21) [CDKN1A (cyclin-dependentkinase inhibitor 1A)], an important tumour suppressor gene, isamong the genes that can be activated by RNAa in tumour cells.In the present study, we provide direct evidence that p21 promoter-targeted saRNA interact with its intended target on the p21promoter to activate p21 expression. This process is associated
with recruitment of RNA polymerase II and AGO2 (argonaute2) protein to the saRNA-target site. Additionally, we found thatseveral hnRNPs (heterogeneous nuclear ribonucleoproteins) (A1,A2/B1 and C1/C2) are associated with saRNA. Further studiesshow that hnRNPA2/B1 interacts with the saRNA in vivo andin vitro and is required for RNAa activity. These findings indicatethat RNAa results from specific targeting of promoters and revealsadditional mechanistic details of RNAa.
Key words: heterogeneous nuclear ribonucleoprotein A2/B1(hnRNPA2/B1), p21, promoter-associated, RNA activation(RNAa), small activating RNA (saRNA).
INTRODUCTION
RNAi (RNA interference) is a natural cellular process in whichsmall RNAs, such as siRNA (short interfering RNA) andmicroRNA target homologous mRNA sequences to degrade themRNA or inhibit their translation leading to post-transcriptionalgene silencing [1–3]. Previous studies have also found thatsmall RNAs complementary to gene promoters cause TGS(transcriptional gene silencing) by modulating epigenetic eventsassociated with the target promoter [4–8]. More recently,we and others reported that dsRNAs (double-stranded RNAs)complementary to target sequences within gene promoters caninduce gene expression [9–15], a phenomenon referred to asRNAa (RNA activation). These dsRNAs, termed saRNAs (smallactivating RNAs), are 21 nucleotide duplexes with a structureidentical to siRNA that targets mRNA sequences. RNAa requiresthe AGO2 (argonaute 2) protein to regulate RNAi, but possessesunique kinetics characterized by a delay in the start of geneactivation which subsequently lasts for over 10 days [16]. Theseobservations suggest that RNAa is a mechanism distinct fromRNAi. However, the molecular mechanism of RNAa remainselusive with regard to questions such as what is the target moleculeof saRNA and what proteins participate in this process.
hnRNPs [heterogeneous nuclear RNPs (ribonucleoproteins)]are families of RNA-binding proteins, which reside predomin-antly in the nucleus to form the core of the RNP complex thatassociates with nascent transcripts in eukaryotic cells [17]. Theprotein components of the RNPs participate in a number of cellularfunctions, including DNA maintenance and recombination,
transcription and processing of primary transcripts, and nuclearexport, subcellular localization, translation and stability of maturemRNA [18–20]. Previously hnRNPs have been shown to associatewith small regulatory RNAs and function in a wide range ofbiological processes. For example, a special RNP could transportsiRNAs into the nucleus in order to facilitate nuclear RNAiin Caenorhabditis elegans [21]. The K subfamily of hnRNPs(hnRNP-k) are involved in transcriptional regulation mediated bypromoter-targeted dsRNA or saRNA in human cells [22].
p21WAF1/CIP1 (p21) [CDKN1A (cyclin-dependent kinase inhibitor1A)] is a well-characterized cyclin-dependent kinase inhibitor thatbelongs to the Cip/Kip family of cyclin-dependent kinaseinhibitors and is a key regulator of the cell cycle as well ascell death, DNA repair, senescence, aging and as a barrier inreprogramming of iPSs (induced pluripotent stem cells) [23,24].Transcription regulation mediated by multiple transcriptionfactors (p53, Sp1/Sp3 and c-Myc) is considered to play a criticalrole in p21 expression and activity [25–27]. In addition, recentwork suggests that post-transcriptional control of p21 is equallyimportant. For instance, many ubiquitin ligases and proteinkinases regulate the stability and cellular localization of p21protein, thereby regulating its activity [28,29].
By targeting the p21 gene promoter region, we demonstratedthat a saRNA could induce cell-cycle arrest and apoptosis viaup-regulation of the expression of p21 in human prostate andbladder cancer cells [9,10]. In the present study, we performeddetailed analysis of RNAa using the saRNA and showed that thesaRNA could associate specifically with its intended target onthe promoter and interacts with hnRNPA2/B1, a subfamily of
Abbreviations used: AGO2, argonaute 2; ANXA7, annexin A7; ChIP, chromatin immunoprecipitation; DEPC, diethyl pyrocarbonate; dsRNA, double-stranded RNA; EST, expressed sequence tag; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; H3acK9, histone H3 acetylated at Lys9; hnRNP,heterogeneous nuclear ribonucleoprotein; IP, immunoprecipitation; LC-MS/MS, liquid chromatography tandem MS; ncRNA, non-coding RNA; PR,progesterone receptor; RACE, rapid amplification of cDNA end; RNAa, RNA activation; RNAi, RNA interference; RNA Pol II, RNA polymerase II; RNP,ribonuclear protein; RT, reverse transcription; qRT-PCR, quantitative RT-PCR; saRNA, small activating RNA; siRNA, short interfering RNA; TSS, transcriptionstart site; WB, Western blotting.
1 Correspondence may be addressed to either of these authors (email [email protected] or [email protected]).
c© The Authors Journal compilation c© 2012 Biochemical Society

408 J. Hu and others
hnRNPs. Knockdown of hnRNPA2/B1 could block the saRNA-mediated p21 induction. These findings reveal that as a coreprotein in RNAa process, hnRNPA2/B1 facilitates recognitionof the p21-specific promoter by the saRNA.
EXPERIMENTAL
dsRNA design and synthesis
dsRNAs were designed and synthesized as described previously[9]. A 21 nucleotide dsRNA targeting the p21 promoter atsequence position − 322 relative to the TSS (transcription startsite) (dsP21-322) was used to activate p21 expression [dsP21-322-S (sense), 5′-CCAACUCAUUCUCCAAGUAdTdT-3′ and dsP21-322-AS (antisense), 5′-UACUUGGAGAAUGAGUUGGdTdT-3′]. A 21 nucleotide dsRNA lacking significant homology to allknown human sequences (dsControl) was used as a non-specificcontrol [dsControl-S (sense), 5′-ACUACUGAGUGACAGUAG-AdTdT-3′ and dsControl-AS (antisense), 5′-UCUACUGUCA-CUCAGUAGUdTdT-3′]. The siRNAs sequences of hnRNPA2/B1were: sense, 5′-GGAACAGUUCCGUAAGCUCdTdT-3′ andantisense, 5′-GAGCUUACGGAACUGUUCCdTdT-3′. siCon(a scrambled dsRNA based on the mRNA sequence ofhnRNPA2/B1) was also synthesized and used as a negativecontrol. The synthetic dsRNAs were manufactured by Ribobio.
Plasmids and antibodies
pCI-FLAG-hnRNPA2/B1 plasmid (a gift from ProfessorChristopher Smith, University of Cambridge, Cambridge, U.K.)[31]. The sources of antibodies against the following proteinswere: monoclonal anti-AGO2 [ChIP (chromatin immunoprecip-itation) grade for IP (immunoprecipitation); catalogue numberab57113, Abcam], monoclonal anti-biotin (for IP; cataloguenumber sc-53179, Santa Cruz Biotechnology), monoclonal anti-hnRNPA2/B1 [for IP and WB (Western blotting); cataloguenumber sc-32316, Santa Cruz Biotechnology], normal mouseIgG (for IP; catalogue number 12-371, Millipore), monoclonalanti-(RNA Pol II) (RNA polymerase II) (for IP; cataloguenumber 05-623, Millipore), polyclonal anti-H3acK9 (histone H3acetylated at Lys9; for IP; catalogue number 07-352, Millipore),polyclonal anti-p21 (for WB; catalogue number ab7960, Abcam)and monoclonal anti-GAPDH (glyceraldehyde-3-phosphatedehydrogenase; for WB; catalogue number ab9484, Abcam).
Cell culture and transfection
PC-3 (A.T.C.C.) cells were maintained in RPMI 1640medium supplemented with 10% FBS (fetal bovine serum),2 mM L-glutamine, penicillin (100 units/ml) and streptomycin(100 μg/ml) in a humidified atmosphere of 5% CO2 maintainedat 37 ◦C. The day before transfection cells were plated insix-well plates or 150-mm culture dishes (Costar) withoutantibiotics at a density of 30–40%. Transfections of all RNAduplex oligonucleotides were carried out by using Entranster-R(Engreen) transfection reagent according to the manufacturer’srecommendations.
Nucleic acid extraction and protein expression
Total cellular RNAs were isolated with the TRIzol reagent(Invitrogen). Genomic DNA was isolated by using QIAamp DNABlood Maxi kit (Qiagen). The pCI-FLAG-hnRNPA2/B1 plasmidwas transcribed/translated hnRNPA2/B1 protein in vitro by
using TNT® T7 Quick Coupled Transcription/Translation system(Promega) according to the manufacturer’s recommendations.
RNA analysis
Expression of p21, hnRNPA2/B1 and non-coding transcriptsoverlapping the p21 promoter was evaluated by semi-quantitativeRT (reverse transcription)–PCR or qRT–PCR (quantitative RT–PCR). Each RNA sample was treated with RNase-free DNase I(Qiagen) to remove any potential DNA contamination. Samplesof 2 μg of total RNA were used for cDNA synthesis usingSuperScript reverse transcriptase (Invitrogen) and oligo(dT)primers or random primers. qRT–PCR was performed toquantify gene expression by using Platinum® SYBR® GreenqPCR SuperMix-UDG (Invitrogen) and an Stratagene Mx3000
TM
sequence detection system (Stratagene). Semi-quantitative RT–PCR was also used to evaluate gene transcript expression. Thespecific primer sequences used are listed in Supplementary TableS1 (at http://www.BiochemJ.org/bj/447/bj4470407add.htm).
5′ RACE (rapid amplification of cDNA ends)
5′ RACE was performed using the GeneRacer kit (Invitrogen).cDNA samples from PC-3 cells were prepared according to themanufacturer’s protocol. The 5′-end of cDNA was amplifiedthrough two nested PCR steps using Platinum Taq DNAPolymerase High Fidelity (Invitrogen). Appropriate primer sets(Supplementary Table S1) were used to maximize detection oftranscripts and reduce the likelihood of bias from any one primerset. After gel purification, the PCR products were cloned intoa pMD18T vector and sequenced (Invitrogen). We sequencedseveral clones from at least two independent experiments toconfirm the results.
ChIP assay
The ChIP assays were performed by using a ChIP assaykit (catalogue number 17-371, Millipore) according to themanufacturer’s instructions. A total of 3.5×106 cells were used forone separate immunoprecipitation. The RNase inhibitor RNasin(50 units/ml final) was added to each buffer used in this assayto avoid degradation of duplex RNAs. The following antibodieswere used for immunoprecipitations: anti-biotin, anti-AGO2, anti-hnRNPA2/B1, anti-(RNA Pol II), anti-H3m3K4 (histone H3trimethylated at Lys4), anti-H3acK9 and normal mouse IgG. Atotal of 5 μg of each of the appropriate antibody were used foreach ChIP. Immunoprecipitated DNA was reverse cross-linked,purified and analysed by real-time PCR or semi-quantitative PCR.The primers used are described in Supplementary Table S1.
Immunopurification, silver staining and MS
PC-3 cells were transfected with modified dsP21-322-S-3′Bioor dsControl-S-3′Bio. At 72 h after transfection, cellular lysateswere prepared by incubating the cells in 0.5% Nonidet P-40lysis buffer containing protease inhibitor cocktail (1 μl/ml final)and RNase inhibitor RNasin (50 units/ml final) in DEPC (diethylpyrocarbonate). Streptavidin affinity columns were prepared(Pierce) following the manufacturer’s suggestions. Cell lysateswere obtained from 5×108 cells, then added to the column andallowed to enter the resin bed. The bottom and top caps weresequentially replaced and incubated at room temperature (22 ◦C)for 2 h. After binding, the column was washed with the cold lysisbuffer. GdnHCl [guanidine hydrochloride; 8 M, pH 1.5 (Pierce)]was applied to the column to elute the biotinylated dsRNA–protein
c© The Authors Journal compilation c© 2012 Biochemical Society

Promoter-associated saRNA interacts with hnRNPA2/B1 409
Figure 1 Characterization of transcripts at the p21 promoter
(A) Schematic representation of the locations of PCR primers used for detecting the presence of transcripts throughout the p21 promoter. (B) Semi-quantitative RT–PCR detection of RNA at the p21promoter with ( + RT) or without (-RT) addition of reverse transcriptase. Genomic DNA (gDNA) was also amplified as controls for primer validation. Amplification of H20 was also included as anegative template control. (C) Agarose gel analysis of 5′ RACE products defining the 5′-termini of p21 mRNA in untreated and dsP21-322 transfected PC-3 cells. Total RNA used in RACE was treatedwith DNase before reverse transcription. Complete data including sequencing of amplified products are shown in Supplementary Figure S1 (at http://www.BiochemJ.org/bj/447/bj4470407add.htm).
complex following the manufacturer’s instructions. Fractions ofthe bed volume were collected and their pH immediately adjustedby adding 1 M Tris/HCl (pH 8.0). Then the eluted fractions wereresolved by SDS/PAGE (4–12% gel), silver stained (Pierce) andsubjected to LC-MS/MS (liquid chromatography tandem MS)sequencing and data analysis.
Biotin pull-down assay of duplex RNA–protein complexes
Biotinylated dsRNAs [200 μl (20 nM/l) dsP21-322-S-3′Bioand dsControl-S-3′Bio] were immobilized on 50 μl of theimmobilized Streptavidin slurry beads in 0.4 ml of biotinylatedpull-down binding buffer [25 mM Tris/HCl and 0.15 M NaCl(pH 7.0)] containing the RNase inhibitor RNasin (50 units/mlfinal concentration). After incubation for 1 h at 4 ◦C with rotation,the beads were briefly incubated and washed three times withbiotin blocking solution (2 mM biotin blocking buffer, pH 7.2,Pierce) containing the RNase inhibitor RNasin (50 units/ml final).Subsequently, the beads were washed three times with thebiotinylated pull-down binding buffer and resuspended in 0.4 mlof the biotinylated pull-down binding buffer before adding 5 μl ofin vitro transcribed/translated hnRNPA2/B1 for 2 h at 4 ◦C withrotation. The beads were then washed three times with 0.5 ml ofwash buffer (50 mM sodium acetate, pH 5.0). The bound proteinswere eluted by boiling in 25 μl of loading buffer and resolved onSDS/PAGE (12% gel). Immobilized Streptavidin and each bufferused in the biotin pull-down assay was consistent with Pull-DownBiotinylated Protein–Protein Interaction kit (Pierce). The RNaseinhibitor RNasin (50 units/ml final) was added to each buffer toavoid degradation of duplex RNAs.
Co-IP and WB
Cultured PC-3 cells were washed twice with ice-coldPBS containing Protease Inhibitor cocktail (1 μl/ml finalconcentration) and the RNase inhibitor RNasin (50 units/ml final).Then cellular lysates were prepared by incubating the cells in lysisbuffer [50 mM Tris/HCl (pH 7.5), 150 mM NaCl, 0.5% Nonidet
P40 and 2 mM EDTA) containing protease inhibitor cocktailand the RNase inhibitor in DEPC for 30 min at 4 ◦C, followedby centrifugation at 14000 g for 15 min at 4 ◦C. The proteinconcentration of the lysates was determined in the supernatantfraction by using the BCA (bicinchoninic acid) protein assaykit according to the manufacturer’s protocol (Pierce). For IP,500 μl of protein was incubated with 60 μl of immobilizedStreptavidin (Pierce) for 12 h at 4 ◦C with constant rotation;60 μl of 50% Protein A or G agarose beads were then addedand the incubation was continued for an additional 2 h at 4 ◦Cwith rotation. The beads were then washed five times usingthe lysis buffer. Between washes, the beads were collected bycentrifugation at 500 g for 1 min at 4 ◦C. The precipitated proteinswere eluted by resuspending the beads in 2× SDS/PAGE loadingbuffer and boiling for 5 min. The resultant materials from IP or celllysates were resolved by SDS/PAGE (10% gels) and transferredon to nitrocellulose membranes (Millipore). For WB, membraneswere incubated with appropriate antibodies for 1 h at roomtemperature (22 ◦C) or overnight at 4 ◦C followed by incubationwith a secondary antibody. Immunoreactive bands were visualizedusing WB Luminol reagent (Santa Cruz Biotechnology) accordingto the manufacturer’s instructions.
RESULTS
Characterization of the p21 TSS and promoter transcripts
Two models of action for RNAa have been proposed, with onestating that transcriptional activation is realized by targeting non-coding transcripts that overlap the target promoter [14,22], and theother suggesting that saRNA directly targets and binds promoterDNA. To determine whether there are non-coding transcriptsoriginating from, or overlapping with, the p21 promoter, weisolated total cellular RNA from PC-3 (prostate adenocarcinoma)cells which respond to RNAa of p21, and converted the resultingRNA into cDNA using random hexamer primers. We then detectedpotential p21 promoter transcripts by RT–PCR using severalprimer sets that cover different regions on the p21 promoter
c© The Authors Journal compilation c© 2012 Biochemical Society
410 J. Hu and others
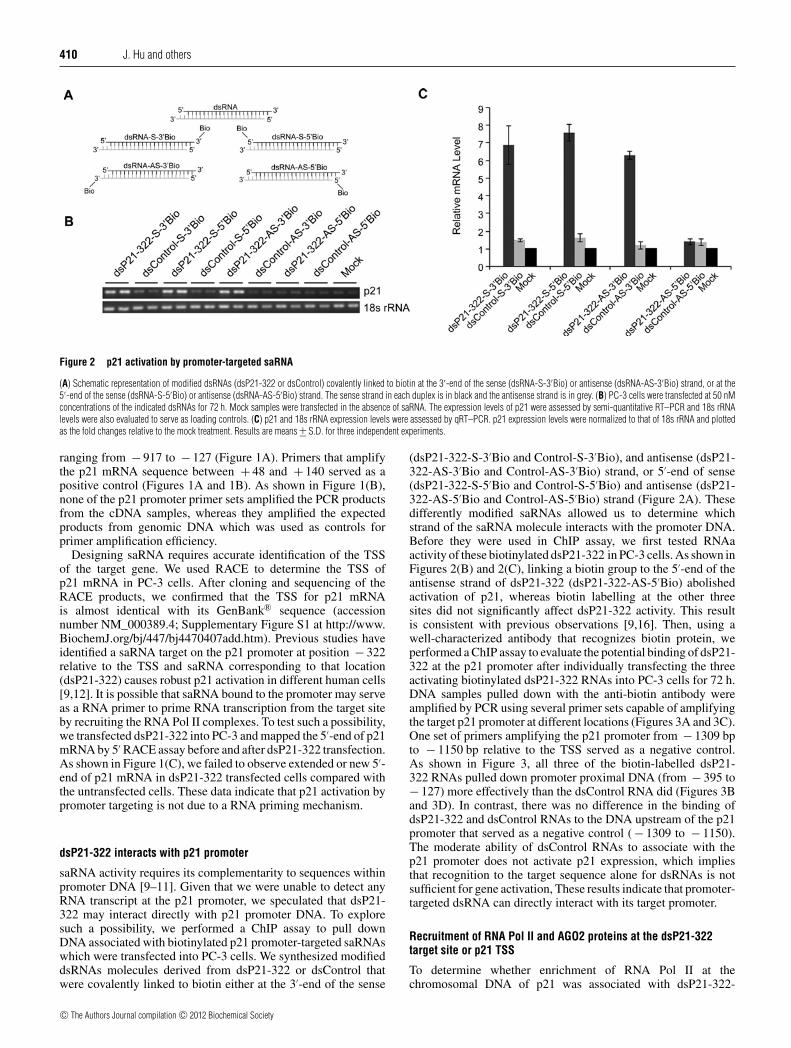
Figure 2 p21 activation by promoter-targeted saRNA
(A) Schematic representation of modified dsRNAs (dsP21-322 or dsControl) covalently linked to biotin at the 3′-end of the sense (dsRNA-S-3′Bio) or antisense (dsRNA-AS-3′Bio) strand, or at the5′-end of the sense (dsRNA-S-5′Bio) or antisense (dsRNA-AS-5′Bio) strand. The sense strand in each duplex is in black and the antisense strand is in grey. (B) PC-3 cells were transfected at 50 nMconcentrations of the indicated dsRNAs for 72 h. Mock samples were transfected in the absence of saRNA. The expression levels of p21 were assessed by semi-quantitative RT–PCR and 18s rRNAlevels were also evaluated to serve as loading controls. (C) p21 and 18s rRNA expression levels were assessed by qRT–PCR. p21 expression levels were normalized to that of 18s rRNA and plottedas the fold changes relative to the mock treatment. Results are means +− S.D. for three independent experiments.
ranging from − 917 to − 127 (Figure 1A). Primers that amplifythe p21 mRNA sequence between + 48 and + 140 served as apositive control (Figures 1A and 1B). As shown in Figure 1(B),none of the p21 promoter primer sets amplified the PCR productsfrom the cDNA samples, whereas they amplified the expectedproducts from genomic DNA which was used as controls forprimer amplification efficiency.
Designing saRNA requires accurate identification of the TSSof the target gene. We used RACE to determine the TSS ofp21 mRNA in PC-3 cells. After cloning and sequencing of theRACE products, we confirmed that the TSS for p21 mRNAis almost identical with its GenBank® sequence (accessionnumber NM_000389.4; Supplementary Figure S1 at http://www.BiochemJ.org/bj/447/bj4470407add.htm). Previous studies haveidentified a saRNA target on the p21 promoter at position − 322relative to the TSS and saRNA corresponding to that location(dsP21-322) causes robust p21 activation in different human cells[9,12]. It is possible that saRNA bound to the promoter may serveas a RNA primer to prime RNA transcription from the target siteby recruiting the RNA Pol II complexes. To test such a possibility,we transfected dsP21-322 into PC-3 and mapped the 5′-end of p21mRNA by 5′ RACE assay before and after dsP21-322 transfection.As shown in Figure 1(C), we failed to observe extended or new 5′-end of p21 mRNA in dsP21-322 transfected cells compared withthe untransfected cells. These data indicate that p21 activation bypromoter targeting is not due to a RNA priming mechanism.
dsP21-322 interacts with p21 promoter
saRNA activity requires its complementarity to sequences withinpromoter DNA [9–11]. Given that we were unable to detect anyRNA transcript at the p21 promoter, we speculated that dsP21-322 may interact directly with p21 promoter DNA. To exploresuch a possibility, we performed a ChIP assay to pull downDNA associated with biotinylated p21 promoter-targeted saRNAswhich were transfected into PC-3 cells. We synthesized modifieddsRNAs molecules derived from dsP21-322 or dsControl thatwere covalently linked to biotin either at the 3′-end of the sense
(dsP21-322-S-3′Bio and Control-S-3′Bio), and antisense (dsP21-322-AS-3′Bio and Control-AS-3′Bio) strand, or 5′-end of sense(dsP21-322-S-5′Bio and Control-S-5′Bio) and antisense (dsP21-322-AS-5′Bio and Control-AS-5′Bio) strand (Figure 2A). Thesedifferently modified saRNAs allowed us to determine whichstrand of the saRNA molecule interacts with the promoter DNA.Before they were used in ChIP assay, we first tested RNAaactivity of these biotinylated dsP21-322 in PC-3 cells. As shown inFigures 2(B) and 2(C), linking a biotin group to the 5′-end of theantisense strand of dsP21-322 (dsP21-322-AS-5′Bio) abolishedactivation of p21, whereas biotin labelling at the other threesites did not significantly affect dsP21-322 activity. This resultis consistent with previous observations [9,16]. Then, using awell-characterized antibody that recognizes biotin protein, weperformed a ChIP assay to evaluate the potential binding of dsP21-322 at the p21 promoter after individually transfecting the threeactivating biotinylated dsP21-322 RNAs into PC-3 cells for 72 h.DNA samples pulled down with the anti-biotin antibody wereamplified by PCR using several primer sets capable of amplifyingthe target p21 promoter at different locations (Figures 3A and 3C).One set of primers amplifying the p21 promoter from − 1309 bpto − 1150 bp relative to the TSS served as a negative control.As shown in Figure 3, all three of the biotin-labelled dsP21-322 RNAs pulled down promoter proximal DNA (from − 395 to− 127) more effectively than the dsControl RNA did (Figures 3Band 3D). In contrast, there was no difference in the binding ofdsP21-322 and dsControl RNAs to the DNA upstream of the p21promoter that served as a negative control ( − 1309 to − 1150).The moderate ability of dsControl RNAs to associate with thep21 promoter does not activate p21 expression, which impliesthat recognition to the target sequence alone for dsRNAs is notsufficient for gene activation, These results indicate that promoter-targeted dsRNA can directly interact with its target promoter.
Recruitment of RNA Pol II and AGO2 proteins at the dsP21-322target site or p21 TSS
To determine whether enrichment of RNA Pol II at thechromosomal DNA of p21 was associated with dsP21-322-
c© The Authors Journal compilation c© 2012 Biochemical Society

Promoter-associated saRNA interacts with hnRNPA2/B1 411
Figure 3 Specific binding of biotinylated dsP21-322 to the p21 promoter
Several primers sets capable of amplifying the target p21 promoter at different locations. (A) Primers used in ChIP to amplify p21 promoter regions ( − 395 bp to − 197 bp and − 1309 bp to− 1150 bp). (C) Primers used in ChIP to amplify p21 promoter regions ( − 190 bp to − 127 bp and − 1309 bp to − 1150 bp). (B and D) ChIP assays were performed by using an anti-biotinantibody to pull down DNA associated with biotinylated saRNA in PC-3 cells transfected at 50 nM concentrations of the indicated dsControl or activating dsP21-322 for 72 h. The resulted DNA wasamplified by qPCR using the above corresponding primer sets and normalized to the input levels. The input was the cellular lysates prior to treatment with antibody. IgG was used as a negativecontrol antibody. Results are means +− S.D. calculated from three independent transfection experiments with triplicate qPCR measurements for each. Statistical significance was tested using a pairedStudent’s t test.
induced gene expression, we performed ChIP assays usingan antibody specific against RNA Pol II and observed thatactivated dsP21-322 enhanced the association of RNA Pol II withthe dsP21-322 target site (Figure 4C) and p21 TSSs (Figure 4D).We also found RNA Pol II enrichment at the region upstreamfrom the dsP21-322 target site was increased after treatment withdsP21-322 (Figure 4A). These results indicate that up-regulationof p21 mediated by dsP21-322 occurs at the transcriptional level.
AGO2 has been shown to be involved in several models ofdsRNA-mediated gene activation as demonstrated by both loss-of-function study and ChIP assay [14,22,32,33]. To examine whetherintroduction of activating dsP21-322 could increase enrichmentof AGO2 protein at the p21 promoter, we also performeda ChIP assay using the anti-AGO2 antibody. Consistent withprevious reports [14,22,32,33], AGO2 enrichment was evidentat the dsP21-322 target site after transfection of dsP21-322. NoChIP product was observed after transfection with dsControl(Figures 4F and 4G). This data indicates that dsP21-322 recruitsAGO2 to its target site.
A loss of H3K27 trimethylation (H3K27me3), which is atranscription-suppressive chromatin mark, has observed at thep21 promoter after treatment with dsP21-322 [34]. To furtherdetermine the potential role of other histone modification
marks in the activation of p21 expression, we chose H3K9acetylation (H3acK9) which had been identified to be involvedin another model of RNAa [10]. However, no significantchanges were detected at the dsP21-322 targeted site or p21transcription start sites after transfecting dsP21-322 into PC-3 cell(Supplementary Figure S2 at http://www.BiochemJ.org/bj/447/bj4470407add.htm). This might reflect that H3acK9 is not adominant regulatory factor for the p21 gene.
Association of hnRNPs with dsP21-322 in vivo
We have demonstrated that dsP21-322 specifically binds to thep21 promoter. Characterizing the proteins involved in modulatinggene expression by promoter-bound dsP21-322 is essentialfor understanding the mechanism of RNAa. We employedaffinity purification and MS to identify proteins associatedwith dsP21-322. In these experiments, biotinylated dsP21-322 (dsP21-322-S-3′Bio) or dsControl (dsControl-S-3′Bio) weretransfected into PC-3 cells for 72 h. Cellular extracts were harves-ted and subjected to affinity purification using immobilizedStreptavidin-affinity columns. After exhaustively washing, thebound proteins were eluted from each column, resolved bySDS/PAGE electrophoresis and then visualized by silver staining.
c© The Authors Journal compilation c© 2012 Biochemical Society

412 J. Hu and others
Figure 4 Enrichment of RNA Poly II and AGO2 at dsP21-322-targeted promoters or the p21 TSS
(A) Schematic representation of locations of PCR primers used in the ChIP assay to amplify the p21 promoter or TSS. A ChIP assay was performed using an anti-(RNA Pol II) antibody to pull downthe p21 promoter upstream of the dsP21-322 target (B), dsP21-322-targeted promoters (C) or the p21 TSS associated with RNA Pol II (D). The resultant DNA was amplified by qPCR and normalizedto input levels. (E) Schematic representation of locations of PCR primers used in the ChIP assay to amplify the p21 promoter. (F) A ChIP assay was performed by using an anti-AGO2 antibody tomeasure the recruitment to dsP21-322-targeted promoter in PC-3 cells transfected with dsControl or activating dsP21-322. (G) Relative recruitment of AGO2 to the dsP21-322-targeted promoterwas determined by qPCR and normalized to the input levels. The input was the nuclear extract prior to treatment with antibody. IgG was used as a negative control antibody. Results are means +− S.D.calculated from three independent transfection experiments with triplicate qPCR measurements for each.
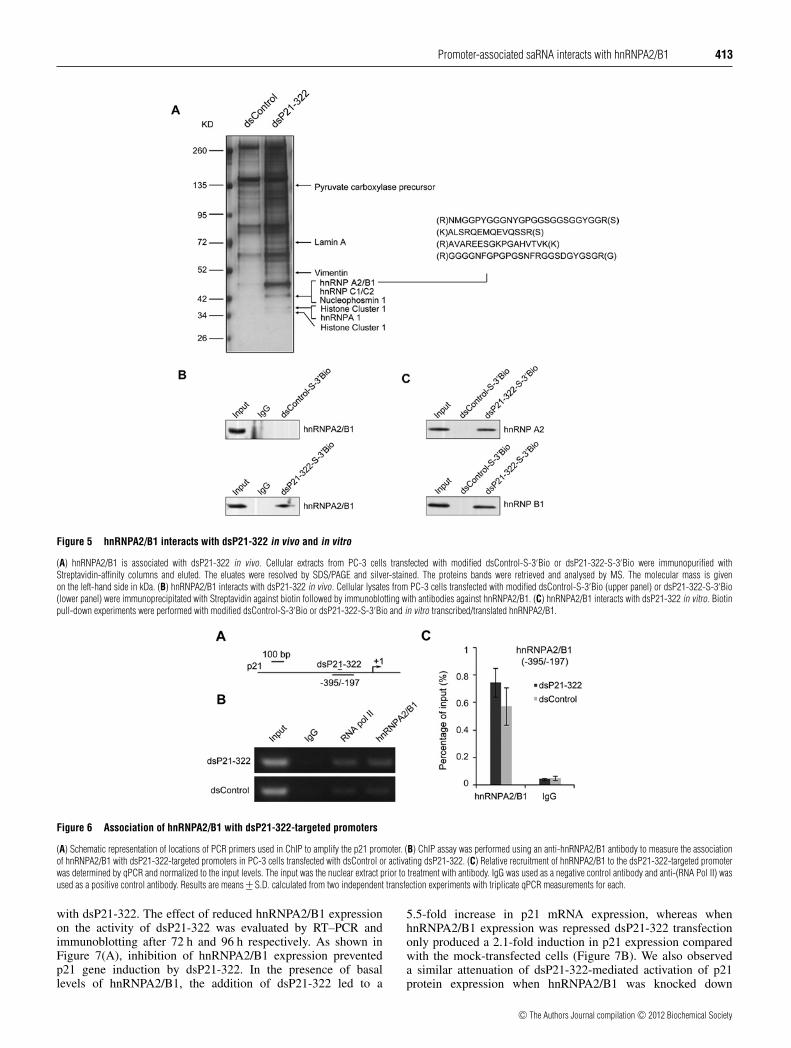
The protein bands on the gel were retrieved and analysed byLC-MS/MS. Compared with the dsControl (Figure 5A), ourresults revealed that dsP21-322 co-purified with several proteins,including pyruvate carboxylase precursor, lamin A, vimentin,nucleophosmin 1 and histone cluster 1 (Figure 5A). Of im-portance, three subfamilies of hnRNPs, including hnRNPA2/B1,hnRNPC1/C2 and hnRNPA1, were also identified in the dsP21-322-containing protein complex, suggesting that dsP21-322 isassociated with hnRNPs in vivo. Further details regarding theabove identified proteins are shown in Supplementary TableS2 (at http://www.BiochemJ.org/bj/447/bj4470407add.htm). Theidentification of four matching peptides from hnRNPA2/B1prompted us to study further the functional relevance of itsinteraction with dsP21-322.
Interactions between hnRNPA2/B1 and dsP21-322 in vivo andin vitro
In order to confirm the in vivo association between hnRNPA2/B1and dsP21-322, PC-3 cell lysates were prepared after treatmentwith biotinylated dsP21-322 (dsP21-322-S-3′Bio) or dsControl(dsControl-S-3′Bio) and co-IP assays were performed withimmobilized Streptavidin against biotin. The immunoprecipitateswere then analysed by immunoblotting with an anti-hnRNPA2/B1antibody. As shown in Figure 5(B), hnRNPA2/B1 was efficientlyco-immunoprecipitated with dsP21-322, whereas no band wasdetected with the dsControl. To confirm further a directinteraction between hnRNPA2/B1 and dsP21-322, biotin pull-down experiments were performed. We incubated in vitrotranscribed/translated hnRNPA2/B1 protein with biotinylateddsP21-322 (dsP21-322-S-3′Bio) or dsControl (dsControl-S-3′Bio). The binding of hnRNPA2/B1 to the dsRNAs was
collected through immobilized Streptavidin and analysed byimmunoblotting using an anti-hnRNPA2/B1 antibody. As shownin Figure 5(C) it was found that hnRNPA2/B1 could interact withthe dsP21-322 rather than the dsControl in vitro.
Association of hnRNPA2/B1 with the dsP21-322 target site on p21promoter
We have shown that dsP21-322 could bind to the p21 promoter andalso interact with hnRNPA2/B1, suggesting that hnRNPA2/B1probably also associates with the p21 promoter. To test sucha possibility, we performed a ChIP assay to pull downDNA associated with hnRNPA2/B1 using an anti-hnRNPA2/B1antibody in PC-3 cells transfected with dsP21-322. We thenamplified the DNA using primers that span the dsP21-322 targetsite (Figure 6A). As shown in Figure 6(B), the p21 promoterDNA was co-immunoprecipitated with hnRNPA2/B1, suggestingthat hnRNPA2/B1 could be associated with the p21 promoter andplay a role in p21 transcription. However, unlike our observationin RNA Pol II and AGO2, little significant enhancement ofhnRNPA2/B1 at the p21 target promoter was detected in dsP21-322-transfected cells compared with the dsControl-transfectedcells (Figure 6C). These data indicate that hnRNPA2/B1 binds todsP21-322 and localizes to the dsP21-322 target site.
hnRNP A2/B1 is required for dsP21-322-induced p21 expression
To test whether hnRNPA2/B1 is an transcriptional co-activatorand if its expression is required in RNAa, we performed aloss-of-function study of hnRNPA2/B1. We transfected PC-3cell with siRNAs targeting hnRNPA2/B1 mRNA in combination
c© The Authors Journal compilation c© 2012 Biochemical Society
Promoter-associated saRNA interacts with hnRNPA2/B1 413
Figure 5 hnRNPA2/B1 interacts with dsP21-322 in vivo and in vitro
(A) hnRNPA2/B1 is associated with dsP21-322 in vivo. Cellular extracts from PC-3 cells transfected with modified dsControl-S-3′Bio or dsP21-322-S-3′Bio were immunopurified withStreptavidin-affinity columns and eluted. The eluates were resolved by SDS/PAGE and silver-stained. The proteins bands were retrieved and analysed by MS. The molecular mass is givenon the left-hand side in kDa. (B) hnRNPA2/B1 interacts with dsP21-322 in vivo. Cellular lysates from PC-3 cells transfected with modified dsControl-S-3′Bio (upper panel) or dsP21-322-S-3′Bio(lower panel) were immunoprecipitated with Streptavidin against biotin followed by immunoblotting with antibodies against hnRNPA2/B1. (C) hnRNPA2/B1 interacts with dsP21-322 in vitro. Biotinpull-down experiments were performed with modified dsControl-S-3′Bio or dsP21-322-S-3′Bio and in vitro transcribed/translated hnRNPA2/B1.
Figure 6 Association of hnRNPA2/B1 with dsP21-322-targeted promoters
(A) Schematic representation of locations of PCR primers used in ChIP to amplify the p21 promoter. (B) ChIP assay was performed using an anti-hnRNPA2/B1 antibody to measure the associationof hnRNPA2/B1 with dsP21-322-targeted promoters in PC-3 cells transfected with dsControl or activating dsP21-322. (C) Relative recruitment of hnRNPA2/B1 to the dsP21-322-targeted promoterwas determined by qPCR and normalized to the input levels. The input was the nuclear extract prior to treatment with antibody. IgG was used as a negative control antibody and anti-(RNA Pol II) wasused as a positive control antibody. Results are means +− S.D. calculated from two independent transfection experiments with triplicate qPCR measurements for each.
with dsP21-322. The effect of reduced hnRNPA2/B1 expressionon the activity of dsP21-322 was evaluated by RT–PCR andimmunoblotting after 72 h and 96 h respectively. As shown inFigure 7(A), inhibition of hnRNPA2/B1 expression preventedp21 gene induction by dsP21-322. In the presence of basallevels of hnRNPA2/B1, the addition of dsP21-322 led to a
5.5-fold increase in p21 mRNA expression, whereas whenhnRNPA2/B1 expression was repressed dsP21-322 transfectiononly produced a 2.1-fold induction in p21 expression comparedwith the mock-transfected cells (Figure 7B). We also observeda similar attenuation of dsP21-322-mediated activation of p21protein expression when hnRNPA2/B1 was knocked down
c© The Authors Journal compilation c© 2012 Biochemical Society

414 J. Hu and others
Figure 7 hnRNPA2/B1 is required for dsP21-322-induced p21 gene expression
(A) PC-3 cells were transfected with 50 nM control siRNA (siCon), dsP21-322 or dsControl. Combination treatments of dsP21-322 and control siRNA or hnRNPA2/B1-specific siRNA (sihnRNPA2/B1)were performed with 50 nM of each RNA duplex. Mock samples were transfected in the absence of dsRNA. Control siRNA is scrambled dsRNA based on hnRNPA2/B1 siRNA. p21, hnRNPA2/B1and 18S rRNA expression levels were assessed by standard RT–PCR. 18S rRNA served as a loading control. (B) hnRNPA2/B1, p21 and 18S rRNA expression levels were assessed by qRT–PCR.Data were plotted as fold changes relative to the results from the mock cells. Results are means +− S.D for three independent experiments. (C) hnRNPA2/B1 and p21 protein expression levels wereconfirmed by immunoblot analysis. GAPDH levels were also detected and served as a loading control.
(Figure 7C). These results suggest that hnRNPA2/B1 is animportant component of the RNAa effector complex.
DISCUSSION
Targeting promoter sequences using small duplex RNA mediatedby AGO has the potential to influence transcription of targetgenes [9–11,35–37]. However the mechanism by which promoter-targeted dsRNA modulates transcription remains elusive. In thepresent study we showed that p21 promoter-targeted saRNAcan specifically recognize and interact with the p21 promoterand induce p21 expression. Such targeting causes increasedoccupancy of RNA Pol II and AGO2 protein at the dsP21-322target site which is close to the p21 TSS. We also identifiedhnRNPA2/B1 as a putative effector protein required for RNAa.
For traditional RNAi, only the antisense strand of the siRNAscan directly bind to mRNA and induce cleavage of their targettranscripts [30,38]. For the targeting of promoters it is unclearwhich strand guides RNAa activity. In the present study weshowed that either biotinylated sense strand or antisense strandof the dsP21-322 could interact with the p21 promoter andup-regulate p21 expression, suggesting a potentially importantmechanistic difference between the two processes in cells. Similarto the present study, it has been demonstrated that activation of thePR (progesterone receptor) and LDLR (low-density lipoproteinreceptor) genes by targeting promoter sequences with saRNAis facilitated through interacting with a non-coding antisensetranscript overlapping the promoter [14,22]. In these cases, thesense strand in the saRNA duplex triggers RNAa activity. Inanother report, in which PR expression was induced by targetingsequences beyond the 3′-termini of mRNA with saRNA [32],implying that RNAa activity is mediated by the antisense strandin this saRNA duplex. As such, both the sense and antisensestrand in the saRNA could serve as the guide and interact with thetarget.
RNA has long been known to hybridize to dsDNA [39] withhigh stability comparable with that of protein–DNA complexes[40]. It has been postulated that RNA hybridization with thepromoter could facilitate the binding of template strand by RNAPol II [41]. In the present study, specific promoters are recognizedby the sense or antisense strand of the dsRNAs with the help of anAGO protein, the interaction between them after hybridization
then acts as a recognition motif for directing RNA Pol II tothe corresponding targeted promoters to mediate transcriptionalregulation in human cells. A previous observation by Place etal. [11] indicated that the concurrent activation of E-cadherinand CSDC2 (cold shock domain containing protein C2) by miR-373 was specific for the microRNA target sites in both genepromoters and was associated with enrichment of RNA Pol II attargeted gene promoters [11]. Likewise, it has also been reportedthat signal-induced ncRNAs (non-coding RNAs) localized to thetranscriptional regulatory regions of the CCND1 (cyclin D1)promoter can recruit and modulate the activities of RNA-bindingco-regulators in response to DNA damage signals, causing gene-specific repression [42]. These observations suggest that smallRNAs recognition and interaction with gene promoters may be anatural and general mechanism for regulating gene transcription.
In contrast with the results of the present study, as mentionedabove, there have been previous reports that non-codingtranscripts serve as the molecular target of activating dsRNAsin human cells [14,22,32], though these models of RNAa havealso observed recruitment of RNA Pol II and AGO2 to genepromoters or TSSs. However, we did not detect any non-codingtranscripts that overlap the p21 promoter from − 1000 bp tothe TSS. Thus it is possible that saRNA interacts directlywith its DNA target without the need for a third ncRNA as adocking molecule. This viewpoint is further supported by recentobservations that chromatin-modifying complexes can be guidedto specific genomic sites by many ncRNAs in mammalian cells[43,44]. In addition studies from Morris and colleagues [34,45]reported that dsP21-322-mediated gene activation of p21 is not, intheory, the result of direct promoter targeting, but rather is an off-target effect of the dsP21-322 binding and cleaving an antisenseRNA [EST (expressed sequence tag) Bx332409] overlapping p21mRNA. However, Bx332409 is a rare EST which, according toNCBI Unigene database (http://www.ncbi.nlm.nih.gov/unigene/),is only expressed in human neuroblastoma. Li et al. [9] havealso identified that non-specific IFN (interferon) response is notinvolved in dsP21-322-induced transcriptional activation. OurChIP data demonstrated that saRNA could interact specificallywith the p21 promoter. However, the result still cannot excludethe possibility that small nascent promoter transcripts originatedfrom the saRNA target site serve as the target molecule of saRNA,even though our RACE and RT–PCR experiments failed to detectevidence for non-coding transcripts overlapping p21 promoter
c© The Authors Journal compilation c© 2012 Biochemical Society

Promoter-associated saRNA interacts with hnRNPA2/B1 415
(Figures 1B and 1C). Transcriptome studies have revealed thatover 70% of gene promoters are overlapped by ncRNA transcripts[46,47]. Therefore the role of ncRNA transcripts played in theprocess of dsP21-322-mediated gene expression requires furtherstudy.
A very interesting observation of the present study is thathnRNPA2/B1 plays an important role in saRNA-mediated geneactivation. The present (Figures 4C and 4D) and previousstudies [22,33] have found that AGO2 is a core effector proteinfor RNAa. Several lines of evidence from the present studysuggest that hnRNPA2/B1 is another core protein critical forRNAa. First, hnRNPA2/B1 is enriched at the p21 gene promoterand its expression is required for efficient gene activation.Secondly, hnRNPA2/B1 interacts with dsP21-322. As oneof the most abundant RNA-binding proteins, the interaction ofhnRNPA2/B1 with RNA has been well established with thebest-characterized hnRNPA2/B1-bound sequences being AU-richsequences [48,49]. dsP21-322 was designed to target an AT-rich region of the p21 promoter, which explains the finding thathnRNPA2/B1 prefer to bind dsP21-322 rather than the dsControl(Figure 5C). Furthermore, the interaction between dsP21-322and hnRNPA2/B1 facilitates the recognition of the p21-specificpromoter by dsP21-322.
Previous findings have shown that hnRNPA2/B1couldspecifically associate with multiple promoter sequences and actas a common transcriptional co-activator [50–52]. A recent reporthas suggested that hnRNPA2/B1 associated with the tumour-suppressor gene ANXA7 (annexin A7) promoter sequences andup-regulated ANXA7 expression in PC-3 cells [53]. These findingssupport our result that hnRNPA2/B1 associates with the p21promoter sequence and affects p21 transcriptional induction.
In the nematode, a special RNP is required to shuttle smallsiRNAs into nuclei in order to facilitate nuclear RNAi [21] andother researchers [54] have observed that small RNA activelymigrate into the nuclear compartment of living human cells.However, we have not yet known whether hnRNPA2/B1 arealso involved in trafficking dsP21-322 from the cytoplasm tothe nucleus, and what are the functions of additional proteinsassociated with dsP21-322, including other hnRNPs (A1 andC1/C2), NPM1 [nucleophosmin (nucleolar phosphoprotein B23,numatrin)] and histone cluster1.
In summary, we have demonstrated that the p21 promoter-targeting saRNA dsP21-322, in direct association withhnRNPA2/B1, enhances p21 expression through recognition andinteraction with the specific sequence within the p21 promoter.Our findings thus provide new insight toward the mechanism forsaRNAs-mediated gene activation and further support the viewthat RNAa is a distinct cellular mechanism by which small RNAparticipates in positive regulation of gene expression.
AUTHOR CONTRIBUTION
Jia Hu and Zhong Chen designed the experiments; Jia Hu, Ding Xia and Jia Wu performedthe experiments; Jia Hu, Zhong Chen and Hua Xu analysed the data; Jia Hu, Zhong Chenand Zhang-Qun Ye wrote the paper; and Zhong Chen obtained the funding.
ACKNOWLEDGEMENTS
We thank Dr Long-Cheng Li (University of California, San Francisco, San Francisco,CA, U.S.A.) for critical reading of the paper prior to submission and Feng Wang (PekingUniversity, Beijing, China) and Dr Fan Wang (Yale University, New Haven, CT, U.S.A.) forhelpful discussions. We also thank the Center of Biomedical Analysis (Tsinghua University,Beijing, China) for technical support and China Center for Type Culture Collection forprovision of the cell lines.
FUNDING
This work was supported by the National Natural Science Foundation of China [grantnumber 30873018].
REFERENCES
1 Czech, B. and Hannon, G. J. (2011) Small RNA sorting: matchmaking for Argonautes. Nat.Rev. Genet. 12, 19–31
2 Ghildiyal, M. and Zamore, P. D. (2009) Small silencing RNAs: an expanding universe.Nat. Rev. Genet. 10, 94–108
3 Krol, J., Loedige, I. and Filipowicz, W. (2010) The widespread regulation of microRNAbiogenesis, function and decay. Nat. Rev. Genet. 11, 597–610
4 Morris, K. V., Chan, S. W., Jacobsen, S. E. and Looney, D. J. (2004) Small interferingRNA-induced transcriptional gene silencing in human cells. Science 305, 1289–1292
5 Ting, A. H., Schuebel, K. E., Herman, J. G. and Baylin, S. B. (2005) Short double-strandedRNA induces transcriptional gene silencing in human cancer cells in the absence of DNAmethylation. Nat. Genet. 37, 906–910
6 Janowski, B. A., Huffman, K. E., Schwartz, J. C., Ram, R., Hardy, D., Shames, D. S.,Minna, J. D. and Corey, D. R. (2005) Inhibiting gene expression at transcription start sitesin chromosomal DNA with antigene RNAs. Nat. Chem. Biol. 1, 216–222
7 Napoli, S., Pastori, C., Magistri, M., Carbone, G. M. and Catapano, C. V. (2009)Promoter-specific transcriptional interference and c-myc gene silencing by siRNAs inhuman cells. EMBO J. 28, 1708–1719
8 Hawkins, P. G., Santoso, S., Adams, C., Anest, V. and Morris, K. V. (2009) Promotertargeted small RNAs induce long-term transcriptional gene silencing in human cells.Nucleic Acids Res. 37, 2984–2995
9 Li, L. C., Okino, S. T., Zhao, H., Pookot, D., Place, R. F., Urakami, S., Enokida, H. andDahiya, R. (2006) Small dsRNAs induce transcriptional activation in human cells. Proc.Natl. Acad. Sci. U.S.A. 103, 17337–17342
10 Janowski, B. A., Younger, S. T., Hardy, D. B., Ram, R., Huffman, K. E. and Corey, D. R.(2007) Activating gene expression in mammalian cells with promoter-targeted duplexRNAs. Nat. Chem. Biol. 3, 166–173
11 Place, R. F., Li, L. C., Pookot, D., Noonan, E. J. and Dahiya, R. (2008) MicroRNA-373induces expression of genes with complementary promoter sequences. Proc. Natl. Acad.Sci. U.S.A. 105, 1608–1613
12 Chen, Z., Place, R. F., Jia, Z. J., Pookot, D., Dahiya, R. and Li, L. C. (2008) Antitumoreffect of dsRNA-induced p21(WAF1/CIP1)gene activation in human bladder cancer cells. Mol.Cancer. Ther. 7, 698–703
13 Huang, V., Qin, Y., Wang, J., Wang, X., Place, R. F., Lin, G., Lue, T. F. and Li, L. C. (2010)RNAa is conserved in mammalian cells. PLoS ONE 5, e8848
14 Matsui, M., Sakurai, F., Elbashir, S., Foster, D. J., Manoharan, M. and Corey, D. R. (2010)Activation of LDL receptor expression by small RNAs complementary to a noncodingtranscript that overlaps the LDLR promoter. Chem. Biol. 17, 1344–1355
15 Wang, X., Wang, J., Huang, V., Place, R. F. and Li, L. C. (2012) Induction of NANOGexpression by targeting promoter sequence with small activating RNA antagonizesretinoic acid-induced differentiation. Biochem. J. 443, 821–828
16 Place, R. F., Noonan, E. J., Foldes-Papp, Z. and Li, L. C. (2010) Defining features andexploring chemical modifications to manipulate RNAa activity. Curr. Pharm. Biotechnol.11, 518–526
17 He, Y. and Smith, R. (2009) Nuclear functions of heterogeneous nuclearribonucleoproteins A/B. Cell. Mol. Life Sci. 66, 1239–1256
18 Krecic, A. M. and Swanson, M. S. (1999) hnRNP complexes: composition, structure, andfunction. Curr. Opin. Cell Biol. 11, 363–371
19 Dreyfuss, G., Kim, V. N. and Kataoka, N. (2002) Messenger-RNA-binding proteins and themessages they carry. Nat. Rev. Mol. Cell Biol. 3, 195–205
20 Glisovic, T., Bachorik, J. L., Yong, J. and Dreyfuss, G. (2008) RNA-binding proteins andpost-transcriptional gene regulation. FEBS Lett. 582, 1977–1986
21 Guang, S., Bochner, A. F., Pavelec, D. M., Burkhart, K. B., Harding, S., Lachowiec, J. andKennedy, S. (2008) An Argonaute transports siRNAs from the cytoplasm to the nucleus.Science 321, 537–541
22 Schwartz, J. C., Younger, S. T., Nguyen, N. B., Hardy, D. B., Monia, B. P., Corey, D. R. andJanowski, B. A. (2008) Antisense transcripts are targets for activating small RNAs. Nat.Struct. Mol. Biol. 15, 842–848
23 Abbas, T. and Dutta, A. (2009) p21 in cancer: intricate networks and multiple activities.Nat. Rev. Cancer 9, 400–414
24 Jung, Y. S., Qian, Y. and Chen, X. (2010) Examination of the expanding pathways for theregulation of p21 expression and activity. Cell. Signalling 22, 1003–1012
25 Chai, Y. L., Cui, J., Shao, N., Shyam, E., Reddy, P. and Rao, V. N. (1999) The secondBRCT domain of BRCA1 proteins interacts with p53 and stimulates transcription from thep21WAF1/CIP1promoter. Oncogene 18, 263–268
c© The Authors Journal compilation c© 2012 Biochemical Society

416 J. Hu and others
26 Gartel, A. L., Goufman, E., Najmabadi, F. and Tyner, A. L. (2000) Sp1 and Sp3 activatep21(WAF1/CIP1) gene transcription in the Caco-2 colon adenocarcinoma cell line. Oncogene19, 5182–5188
27 Gartel, A. L. and Radhakrishnan, S. K. (2005) Lost in transcription: p21 repression,mechanisms, and consequences. Cancer Res. 65, 3980–3985
28 Child, E. S. and Mann, D. J. (2006) The intricacies of p21 phosphorylation: protein/proteininteractions, subcellular localization and stability. Cell Cycle 5, 1313–1319
29 Kim, Y., Starostina, N. G. and Kipreos, E. T. (2008) The CRL4Cdt2 ubiquitin ligase targetsthe degradation of p21Cip1 to control replication licensing. Genes Dev. 22, 2507–2519
30 Meister, G., Landthaler, M., Patkaniowska, A., Dorsett, Y., Teng, G. and Tuschl, T. (2004)Human Argonaute2 mediates RNA cleavage targeted by miRNAs and siRNAs. Mol. Cell15, 185–197
31 McGlincy, N. J., Tan, L. Y., Paul, N., Zavolan, M., Lilley, K. S. and Smith, C. W. (2010)Expression proteomics of UPF1 knockdown in HeLa cells reveals autoregulation ofhnRNP A2/B1 mediated by alternative splicing resulting in nonsense-mediated mRNAdecay. BMC Genomics 11, 565
32 Yue, X., Schwartz, J. C., Chu, Y., Younger, S. T., Gagnon, K. T., Elbashir, S., Janowski,B. A. and Corey, D. R. (2010) Transcriptional regulation by small RNAs at sequencesdownstream from 3′ gene termini. Nat. Chem. Biol. 6, 621–629
33 Chu, Y., Yue, X., Younger, S. T., Janowski, B. A. and Corey, D. R. (2010) Involvement ofargonaute proteins in gene silencing and activation by RNAs complementary to anon-coding transcript at the progesterone receptor promoter. Nucleic Acids Res. 38,7736–7748
34 Morris, K. V., Santoso, S., Turner, A. M., Pastori, C. and Hawkins, P. G. (2008)Bidirectional transcription directs both transcriptional gene activation and suppression inhuman cells. PLoS Genet. 4, e1000258
35 Huang, V., Place, R. F., Portnoy, V., Wang, J., Qi, Z., Jia, Z., Yu, A., Shuman, M., Yu, J. andLi, L. C. (2012) Upregulation of Cyclin B1 by miRNA and its implications in cancer.Nucleic Acids Res. 40, 1695–1707
36 Kim, D. H., Saetrom, P., Snove, Jr, O. and Rossi, J. J. (2008) MicroRNA-directedtranscriptional gene silencing in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 105,16230–16235
37 Younger, S. T. and Corey, D. R. (2011) Transcriptional gene silencing in mammalian cellsby miRNA mimics that target gene promoters. Nucleic Acids Res. 39, 5682–5691
38 Siomi, H. and Siomi, M. C. (2009) On the road to reading the RNA-interference code.Nature 457, 396–404
39 Bekhor, I., Bonner, J. and Dahmus, G. K. (1969) Hybridization of chromosomal RNA tonative DNA. Proc. Natl. Acad. Sci. U.S.A. 62, 271–277
40 Roberts, R. W. and Crothers, D. M. (1992) Stability and properties of double and triplehelices: dramatic effects of RNA or DNA backbone composition. Science 258, 1463–1466
41 Frenster, J. H. (1976) Selective control of DNA helix openings during gene regulation.Cancer Res. 36, 3394–3398
42 Wang, X., Arai, S., Song, X., Reichart, D., Du, K., Pascual, G., Tempst, P., Rosenfeld,M. G., Glass, C. K. and Kurokawa, R. (2008) Induced ncRNAs allosterically modifyRNA-binding proteins in cis to inhibit transcription. Nature 454, 126–130
43 Gilbert, S. L., Pehrson, J. R. and Sharp, P. A. (2000) XIST RNA associates with specificregions of the inactive X chromatin. J. Biol. Chem. 275, 36491–36494
44 Tsai, M. C., Manor, O., Wan, Y., Mosammaparast, N., Wang, J. K., Lan, F., Shi, Y.,Segal, E. and Chang, H. Y. (2010) Long noncoding RNA as modular scaffold of histonemodification complexes. Science 329, 689–693
45 Morris, K. V. (2009) RNA-directed transcriptional gene silencing and activation in humancells. Oligonucleotides 19, 299–306
46 Gingeras, T. R. (2007) Origin of phenotypes: genes and transcripts. Genome Res. 17,682–690
47 Seila, A. C., Calabrese, J. M., Levine, S. S., Yeo, G. W., Rahl, P. B., Flynn, R. A., Young,R. A. and Sharp, P. A. (2008) Divergent transcription from active promoters. Science 322,1849–1851
48 Hamilton, B. J., Nagy, E., Malter, J. S., Arrick, B. A. and Rigby, W. F. (1993) Association ofheterogeneous nuclear ribonucleoprotein A1 and C proteins with reiterated AUUUAsequences. J. Biol. Chem. 268, 8881–8887
49 Kajita, Y., Nakayama, J., Aizawa, M. and Ishikawa, F. (1995) The UUAG-specific RNAbinding protein, heterogeneous nuclear ribonucleoprotein D0. Common modularstructure and binding properties of the 2xRBD-Gly family. J. Biol. Chem. 270,22167–22175
50 Thakur, S., Nakamura, T., Calin, G., Russo, A., Tamburrino, J. F., Shimizu, M., Baldassarre,G., Battista, S., Fusco, A., Wassell, R. P. et al. (2003) Regulation of BRCA1 transcriptionby specific single-stranded DNA binding factors. Mol. Cell. Biol. 23, 3774–3787
51 He, Y., Brown, M. A., Rothnagel, J. A., Saunders, N. A. and Smith, R. (2005) Roles ofheterogeneous nuclear ribonucleoproteins A and B in cell proliferation. J. Cell Sci. 118,3173–3183
52 Guha, M., Pan, H., Fang, J. K. and Avadhani, N. G. (2009) Heterogeneous nuclearribonucleoprotein A2 is a common transcriptional coactivator in the nuclear transcriptionresponse to mitochondrial respiratory stress. Mol. Biol. Cell 20, 4107–4119
53 Torosyan, Y., Dobi, A., Glasman, M., Mezhevaya, K., Naga, S., Huang, W., Paweletz, C.,Leighton, X., Pollard, H. B. and Srivastava, M. (2010) Role of multi-hnRNP nuclearcomplex in regulation of tumor suppressor ANXA7 in prostate cancer cells. Oncogene 29,2457–2466
54 Foldes-Papp, Z., Konig, K., Studier, H., Buckle, R., Breunig, H. G., Uchugonova, A. andKostner, G. M. (2009) Trafficking of mature miRNA-122 into the nucleus of live liver cells.Curr. Pharm. Biotechnol. 10, 569–578
Received 11 February 2012/26 July 2012; accepted 30 July 2012Published on the Internet 5 October 2012, doi:10.1042/BJ20120256
c© The Authors Journal compilation c© 2012 Biochemical Society

Biochem. J. (2012) 447, 407–416 (Printed in Great Britain) doi:10.1042/BJ20120256
SUPPLEMENTARY ONLINE DATAPromoter-associated small double-stranded RNA interacts withheterogeneous nuclear ribonucleoprotein A2/B1 to inducetranscriptional activationJia HU, Zhong CHEN1, Ding XIA, Jia WU, Hua XU and Zhang-Qun YE1
Department of Urology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430030, China
Figure S1 The TSS of the p21 gene
(A) p21 mRNA GenBank® sequence (accession number NM_000389.4). 5′ RACE PCR products(∼200 bp; Figure 1C from the main text) for p21 mRNA in PC-3 cells (B) and PC-3 cellstreated with activating dsP21-322 (C) were excised from the gel and subjected to cloning andsequencing. ATG, initiation codon.
Figure S2 Effect of adding dsP21-322 on acetylation of H3K9 (AcH3K9) atthe p21 promoter or TSS in PC-3 cells examined by ChIP
Relative effect of AcH3K9 was determined by qPCR and normalized to input levels. (A) ChIPassay of the p21 promoter using an anti-H3acK9 antibody after treatment with dsP21-322. (B)ChIP of the p21 TSS using an anti-H3acK9 antibody after treatment with dsP21-322. The inputwas the nuclear extract prior to treatment with antibody. IgG was use as a negative controlantibody. Results are means+−S.D. calculated from four independent transfection experimentswith triplicate qPCR measurements for each.
1 Correspondence may be addressed to either of these authors (email [email protected] or [email protected]).
c© The Authors Journal compilation c© 2012 Biochemical Society

J. Hu and others
Table S1 Primers used in the present study and their targets
The primers were used for either PCR, qPCR, ChIP or RACE. F, forward; R, reverse.
Name Sequence Assay used for Gene Targeted
p21-1309/-1150F 5′-GAGCAGCCTGAGATGTCAGTAATT-3′ ChIP p21 promoterp21-1309/-1150R 5′-TCCCCTGGACTTCACCTTTG-3′ ChIP p21 promoterp21-917/-822F 5′-CTTTGCTGCATGATCTGA-3′ PCR p21 promoterp21-917/-822R 5′-CTGTCTCCTGAATACTCCC-3′ PCR p21 promoterp21-666/-567F 5′-TCTCCAATTCCCTCCTTC-3′ PCR p21 promoterp21-666/-567R 5′-AGTCCCGTTTATTTCACA-3′ PCR p21 promoterp21-462/-341F 5′-TTGTGGGGCTTTTCTGGA-3′ PCR p21 promoterp21-462/-341R 5′-CTCTGGCAGGCAAGGATT-3′ PCR p21 promoterp21-395/-197F 5′-TAATGTCATCCTCCTGATCTTTTCA-3′ PCR and ChIP p21 promoterp21-395/-197R 5′-TCGCCTGCGTTGGTGC-3′ PCR and ChIP p21 promoterp21-190/-127F 5′-GGGGAGGAGGGAAGTGCC-3′ PCR and ChIP p21 promoterp21-190/-127R 5′-AGTTCCAGCAGGCCAGCC-3′ PCR and ChIP p21 promoterp21 + 48/ + 140F 5′-GCCGAAGTCAGTTCCTTGTGG-3′ PCR and ChIP p21 TSSp21 + 48/ + 140R 5′-GTTGTCTGCCGCCGCTCT-3′ PCR and ChIP p21 TSSGSP1 5′-TTCCATCGCTCACGGG-3′ RACE p21 mRNAGSP2 5′-CGCTGTCCACTGGGCCGAAGA-3′ RACE p21 mRNAGSP3 5′-GCCTTGCTGCCGCATGGGTTC-3′ RACE p21 mRNAp21F1 5′-TCTGTGAAATAAACGGGACT-3′ PCR p21 mRNAP21R1 5′-AGCACTGTTAGAATGGAGCC-3′ PCR p21 mRNAp21F2 5′-AGCAGCGGAACAAGGAGT-3′ qPCR p21 mRNAp21R2 5′-CGTTAGTGCCAGGAAAGACA-3′ qPCR p21 mRNAhnRNPA2/B1F1 5′-AACCAGCAACCTTCTAAC-3′ PCR hnRNPA2/B1 mRNAhnRNPA2/B1R1 5′-TACAATCCTTCCTCCACA-3′ PCR hnRNPA2/B1 mRNAhnRNPA2/B1F2 5′-TACTCAGGGCTTACTCTAT-3′ qPCR hnRNPA2/B1 mRNAhnRNPA2/B1R2 5′-ACTTCAGTGGTGGTCTTA-3′ qPCR hnRNPA2/B1 mRNA18s rRNA F 5′-GTAACCCGTTGAACCCCATT-3′ PCR and QPCR 18s rRNA mRNA18s rRNA R 5′-CCATCCAATCGGTAGTAGCG-3′ PCR and QPCR 18s rRNA mRNA
c© The Authors Journal compilation c© 2012 Biochemical Society

Promoter-associated saRNA interacts with hnRNPA2/B1
Table S2 Identified proteins that associated with dsP21-322 using LC-MS/MS
Protein name Coverage Peptide sequence
hnRNPA2/B1 24% (R)NMGGPYGGGNYGPGGSGGSGGYGGR(S)(K)ALSRQEMQEVQSSR(S)(R)AVAREESGKPGAHVTVK(K)(R)GGGGNFGPGPGSNFRGGSDGYGSGR(G)
hnRNPC1/C2 13% (R)MYSYPARVPPPPPIAR(A)(R)AAVAGEDGRMIAGQVLDINLAAEPKVNR(G)(R)MIAGQVLDINLAAEPK(V)
Nucleophosmin 1 24% (K)MSVQPTVSLGGFEITPPVVLR(L)(K)MQASIEKGGSLPKVEAK(F)(K)GPSSVEDIKAK(M)(K)SIRDTPAKNAQK(S)
Histone cluster 1, H1c 17% (K)ALAAAGYDVEKNNSR(I)(K)KPAAATVTKK(V)(R)KASGPPVSELITK(A)
Histone cluster 1, H1d 12% (R)KASGPPVSELITK(A)(K)ALAAAGYDVEKNNSR(I)(K)ASGPPVSELITK(A)
hnRNPA1 9% (R)SSGPYGGGGQYFAKPR(N)(R)NQGGYGGSSSSSSYGSGR(R)
Vimentin 57% (R)EMEENFAVEAANYQDTIGR(L)(R)KVESLQEEIAFLK(K)(R)DGQVINETSQHHDDLE( − )(K)TVETRDGQVINETSQHHDDLE( − )(R)TLLIKTVETR(D)(R)MFGGPGTASRPSSSR(S)
Lamin A 11% (R)TALINSTGEEVAMR(K)(R)SLETENAGLR(L)(R)NSNLVGAAHEELQQSR(I)(R)VAVEEVDEEGKFVR(L)(R)VAVEEVDEEGKFVRLR(N)
Received 11 February 2012/26 July 2012; accepted 30 July 2012Published on the Internet 5 October 2012, doi:10.1042/BJ20120256
c© The Authors Journal compilation c© 2012 Biochemical Society