protein folding i and ii -...
TRANSCRIPT

Protein Folding I and II
Sepideh Khorasanizadeh September 2008September 2008
Material adapted from text books and journal articles
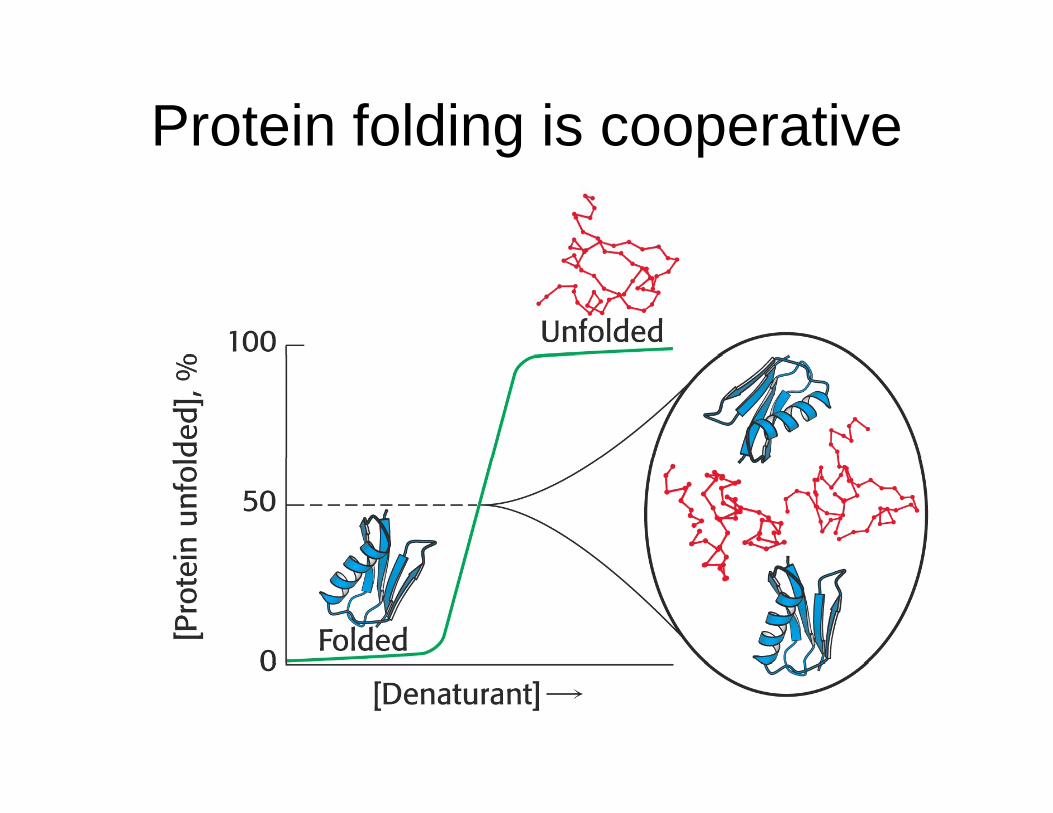
Protein folding is cooperativeg p
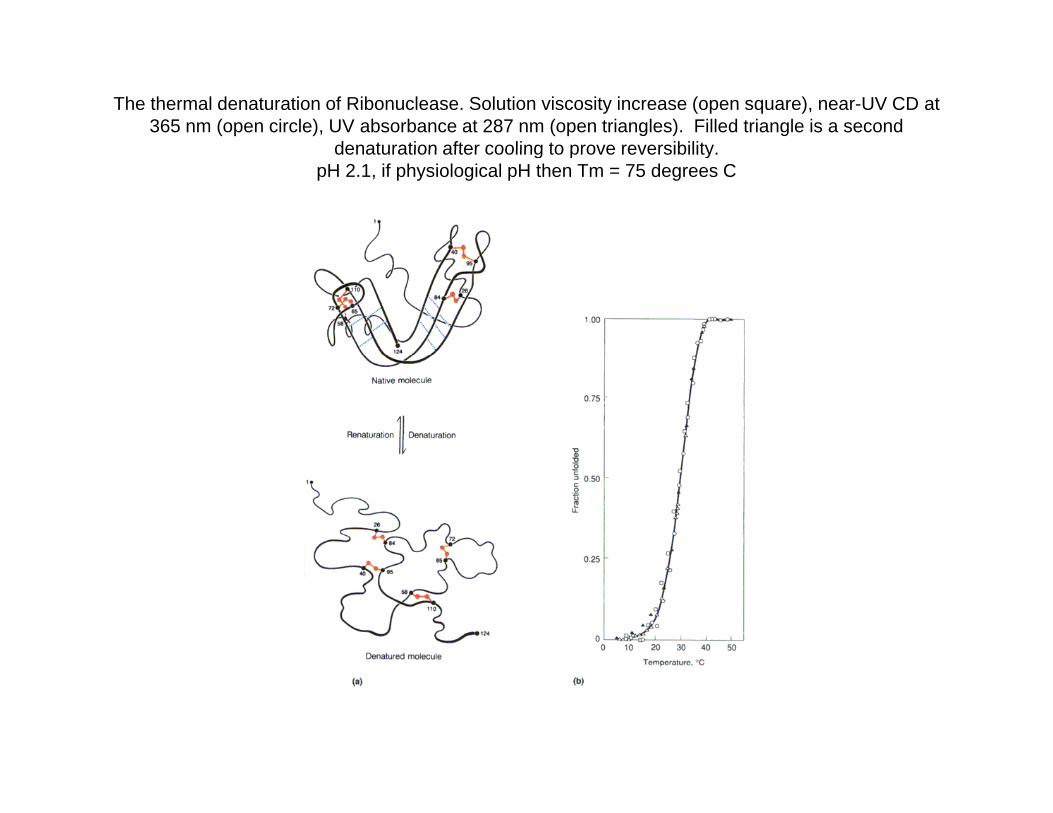
The thermal denaturation of Ribonuclease. Solution viscosity increase (open square), near-UV CD at 365 nm (open circle), UV absorbance at 287 nm (open triangles). Filled triangle is a second
denaturation after cooling to prove reversibilitydenaturation after cooling to prove reversibility.pH 2.1, if physiological pH then Tm = 75 degrees C

• Amino acid sequence determines secondary and tertiary structure.
• The decrease in conformational entropy when a protein folds disfavors folding this is compensated in part by energy stabilization through internal noncovalent bonding.
• Details of H-bonding in a typical protein is shown.
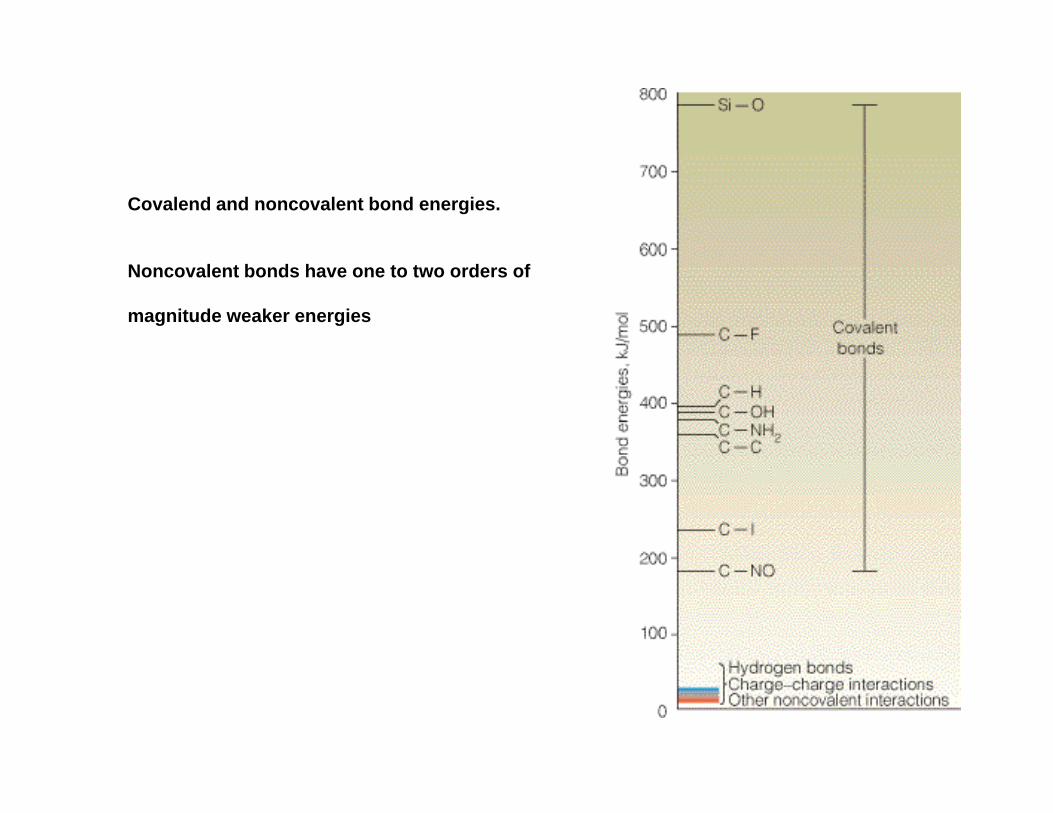
Covalend and noncovalent bond energies.
Noncovalent bonds have one to two orders of
magnitude weaker energies
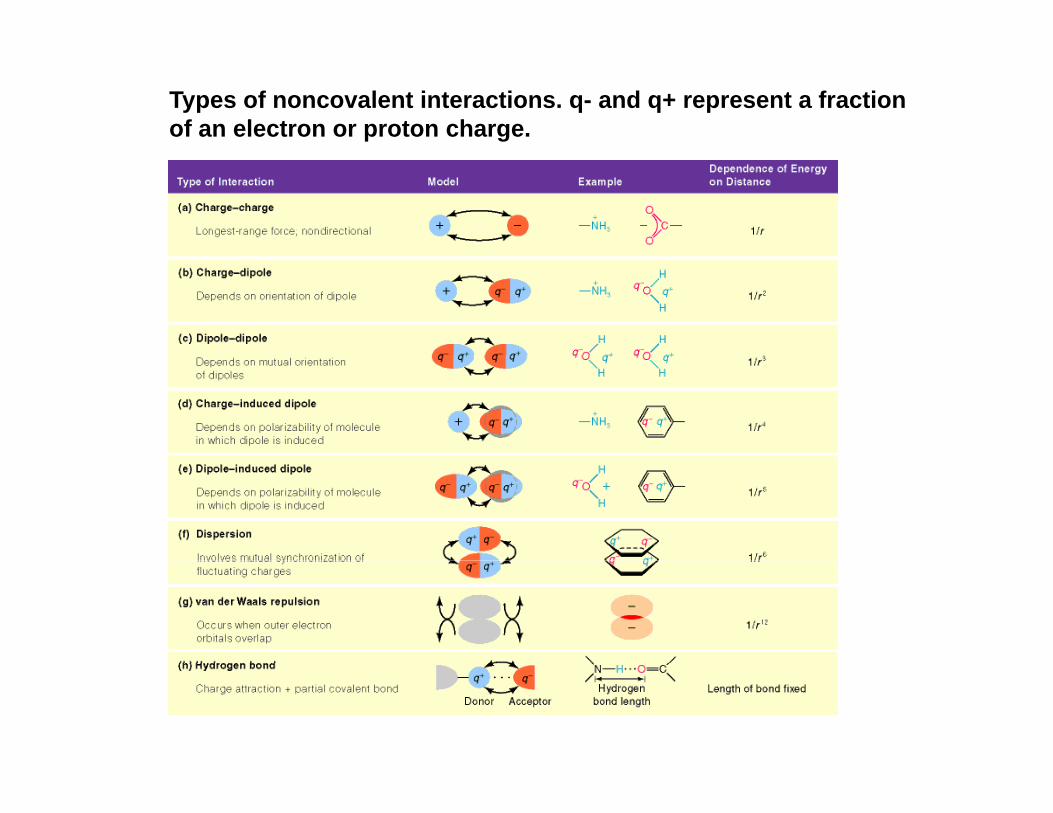
Types of noncovalent interactions. q- and q+ represent a fraction of an electron or proton charge.
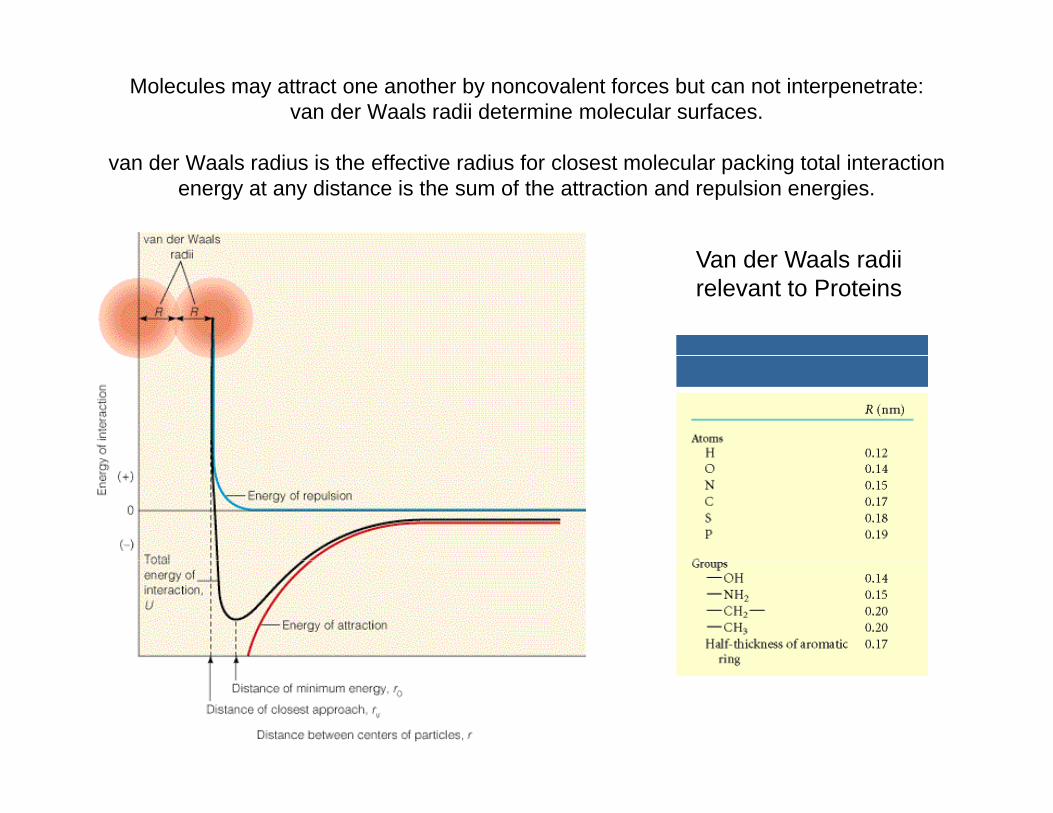
Molecules may attract one another by noncovalent forces but can not interpenetrate: van der Waals radii determine molecular surfaces.
van der Waals radius is the effective radius for closest molecular packing total interaction energy at any distance is the sum of the attraction and repulsion energies.
V d W l diiVan der Waals radii relevant to Proteins
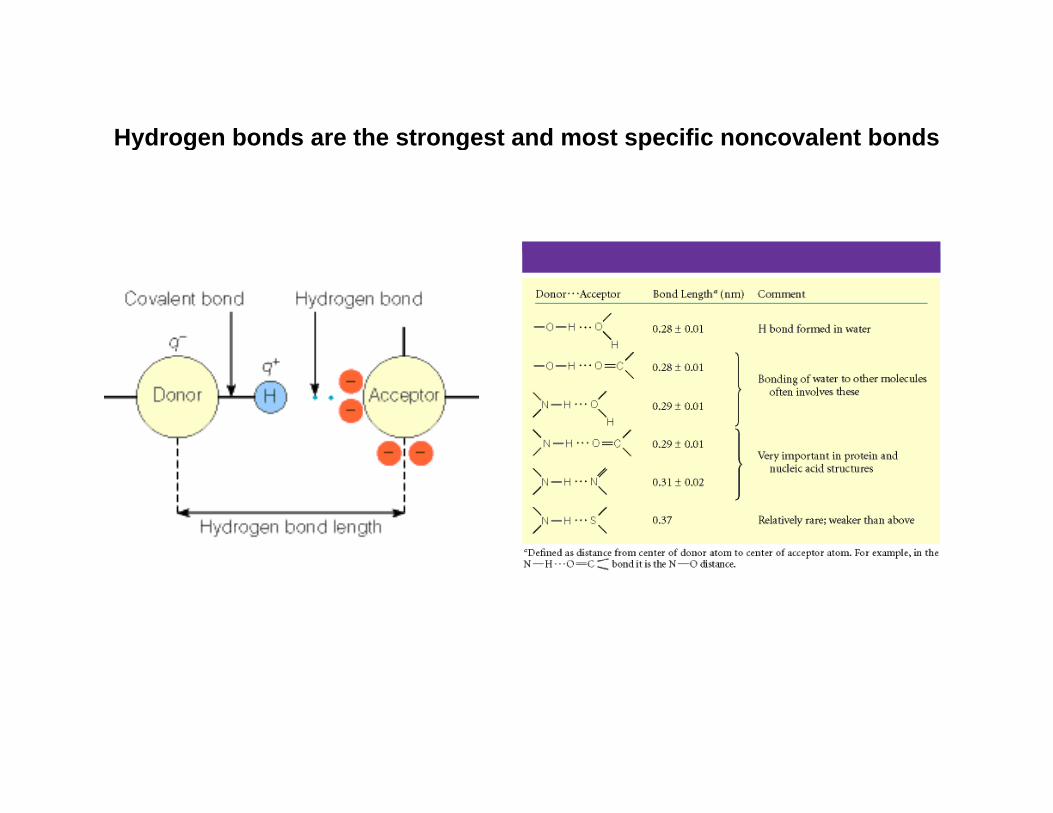
Hydrogen bonds are the strongest and most specific noncovalent bondsy g g

The internal energy of a system includes all forms of energy that can be exchanged via simple physical process and chemical reactions.
First Law of Thermodynamics: The internal energy can change only by the exchange of heat or k ith th di Th h t l d i ti t t t i l t thwork with the surrounding. The heat evolved in a reaction at constant pressure is equal to the
enthalpy, ΔH. The enthalpy change in a reaction is the energy change of most interest to biochemists.
Reversible processes occur always near a state of equilibrium; irreversible processes drive toward equilibrium.q
Entropy is a measure of the randomness or disorder in a system.
Second Law of Thermodynamics: The entropy of a system will tend to increase to a maximum value.
The free energy change for a process atconstant pressure is:
ΔG = ΔH T ΔSΔG = ΔH – T ΔS
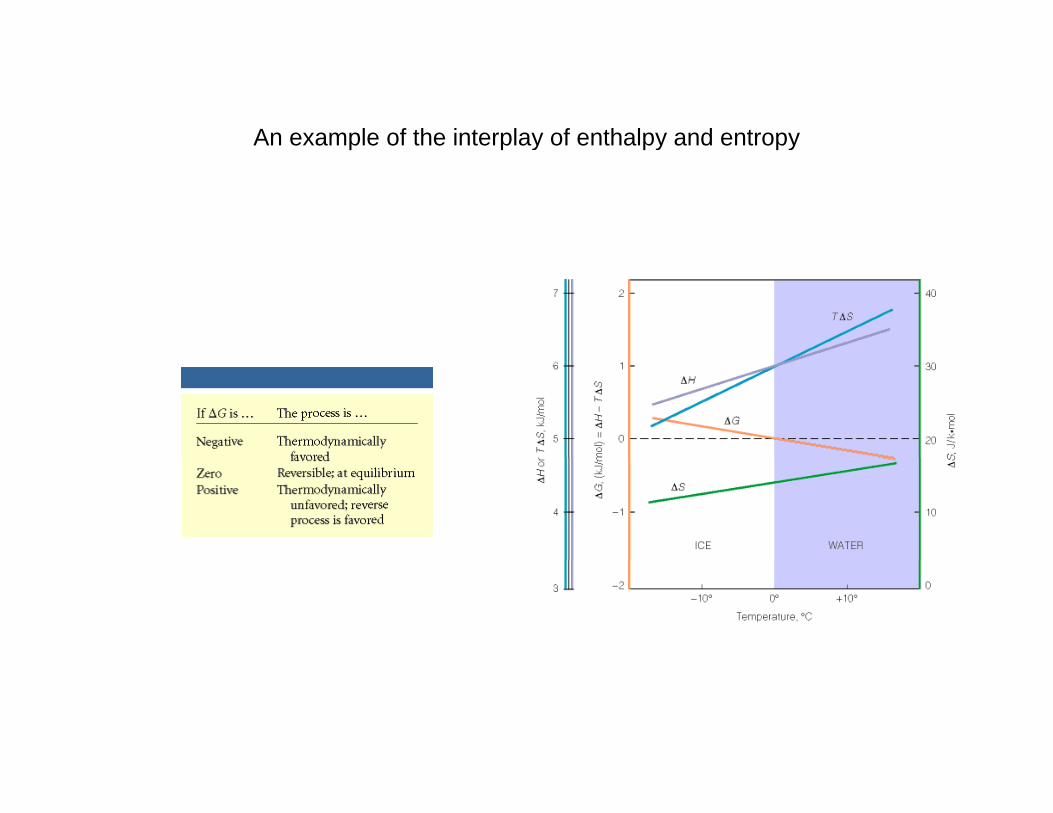
An example of the interplay of enthalpy and entropyy y y

The signs of ΔH and ΔS determine the effect of temperature on processes or reactionsprocesses or reactions
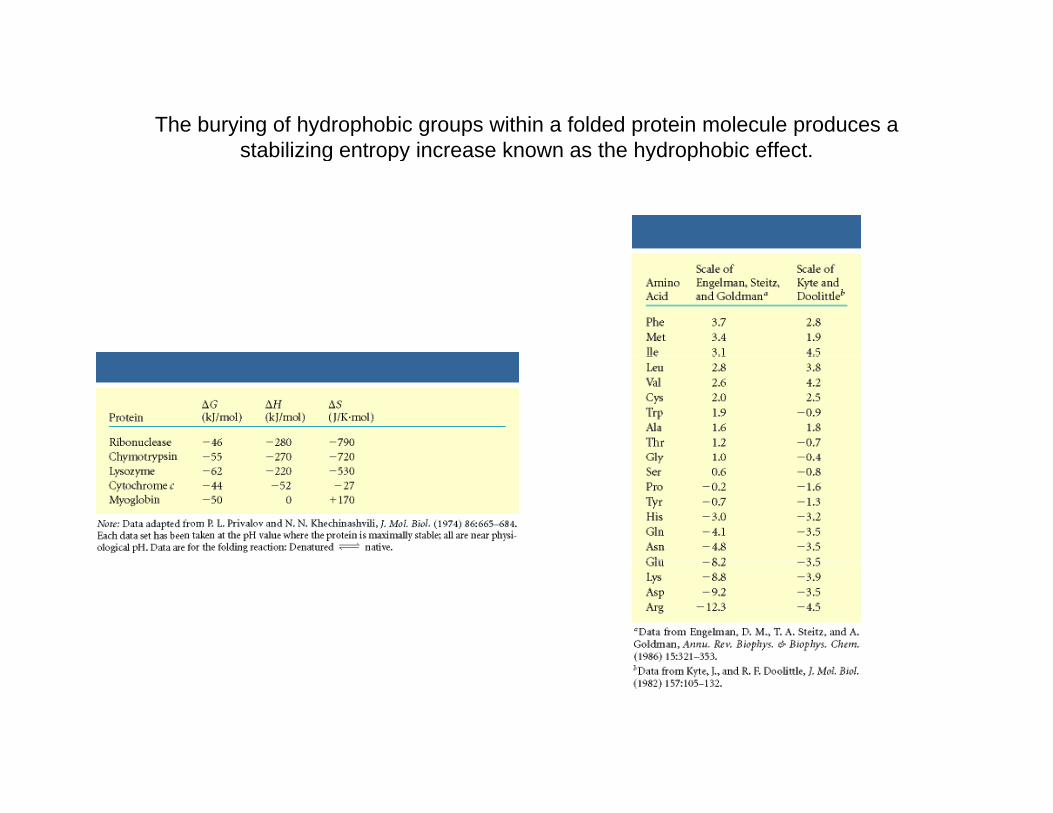
The burying of hydrophobic groups within a folded protein molecule produces a stabilizing entropy increase known as the hydrophobic effectstabilizing entropy increase known as the hydrophobic effect.
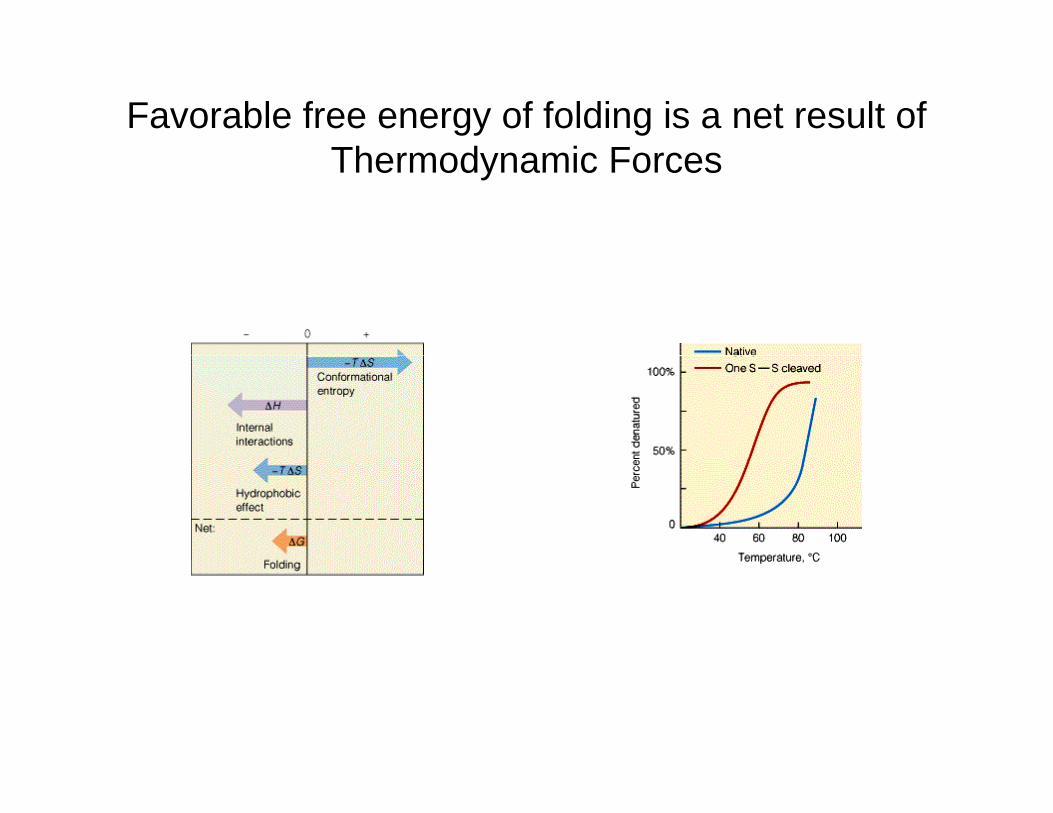
Favorable free energy of folding is a net result of Th d i FThermodynamic Forces
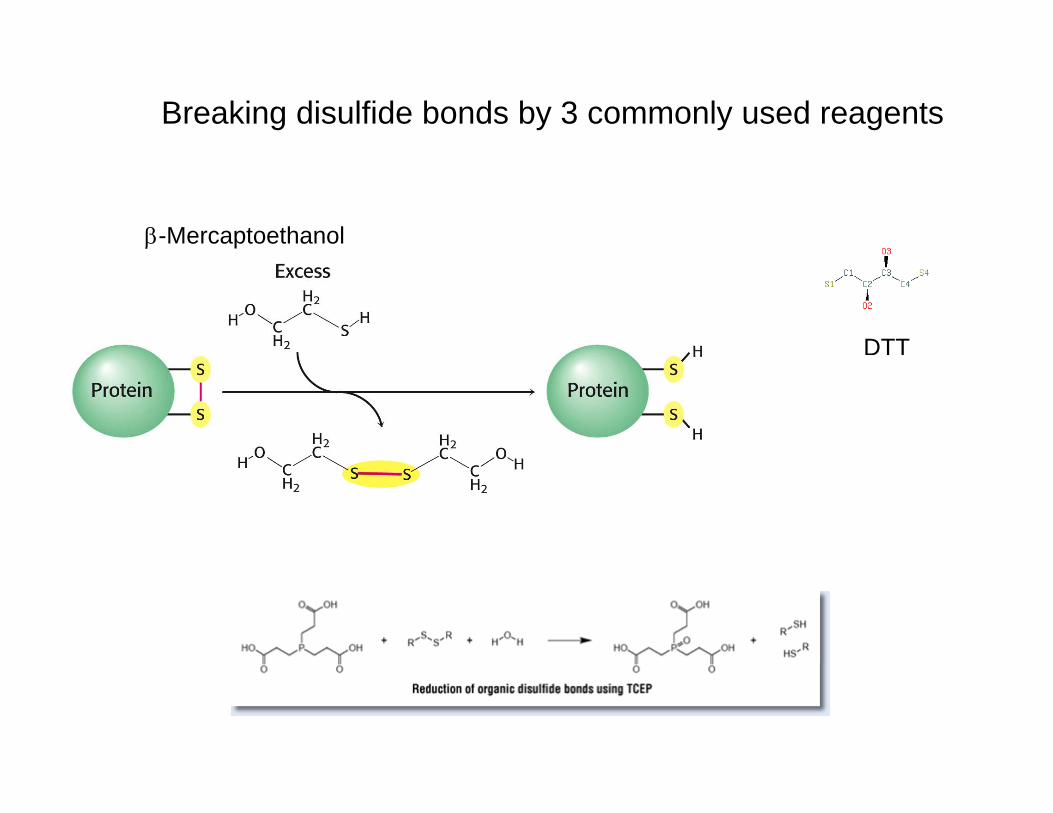
Breaking disulfide bonds by 3 commonly used reagents
β-Mercaptoethanol
DTT
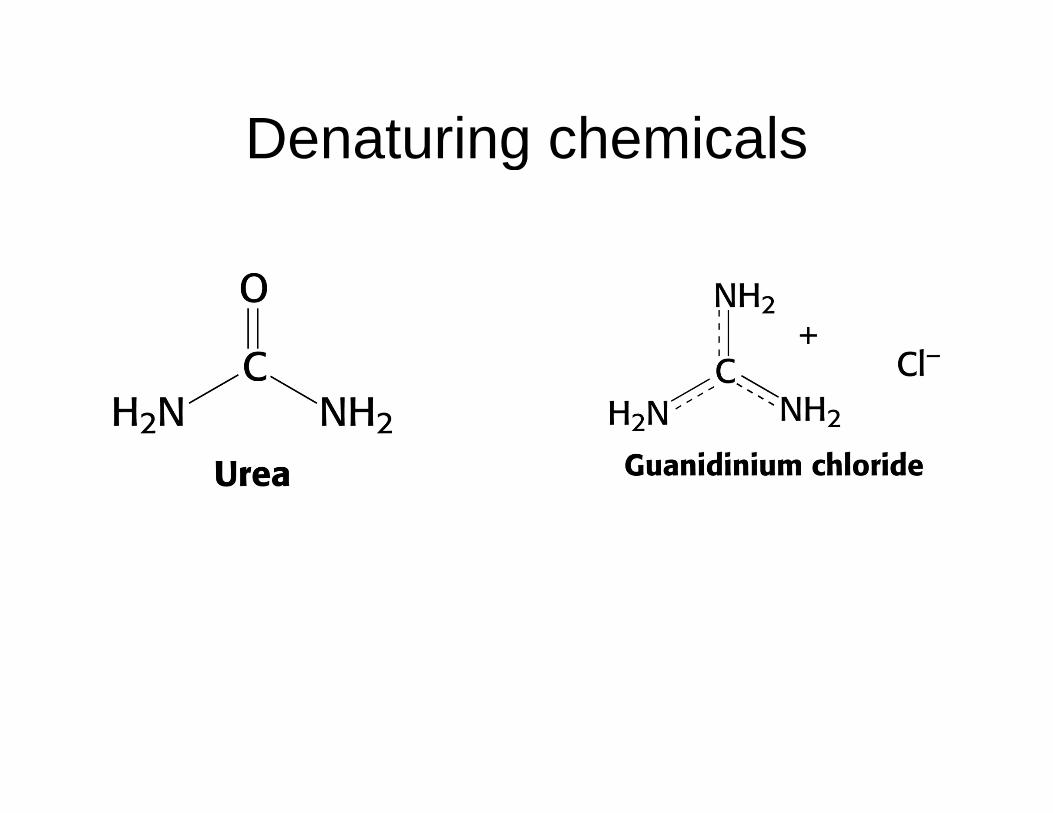
Denaturing chemicalsDenaturing chemicals
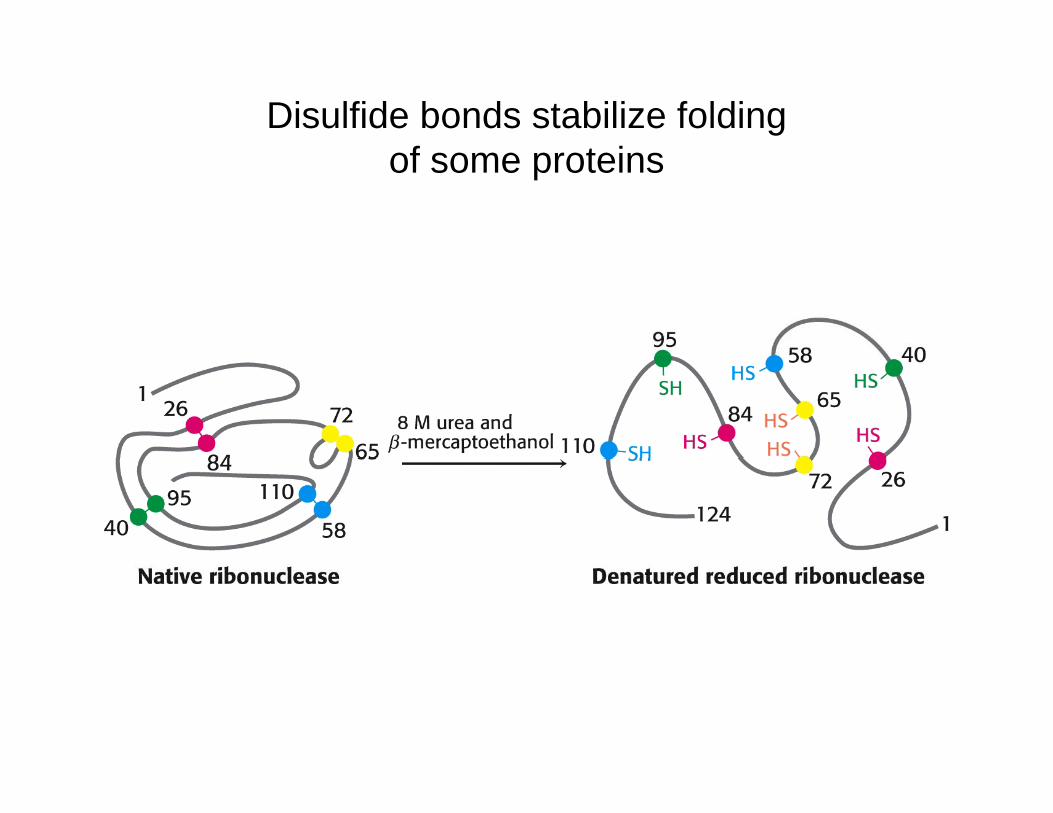
Disulfide bonds stabilize foldingf t iof some proteins
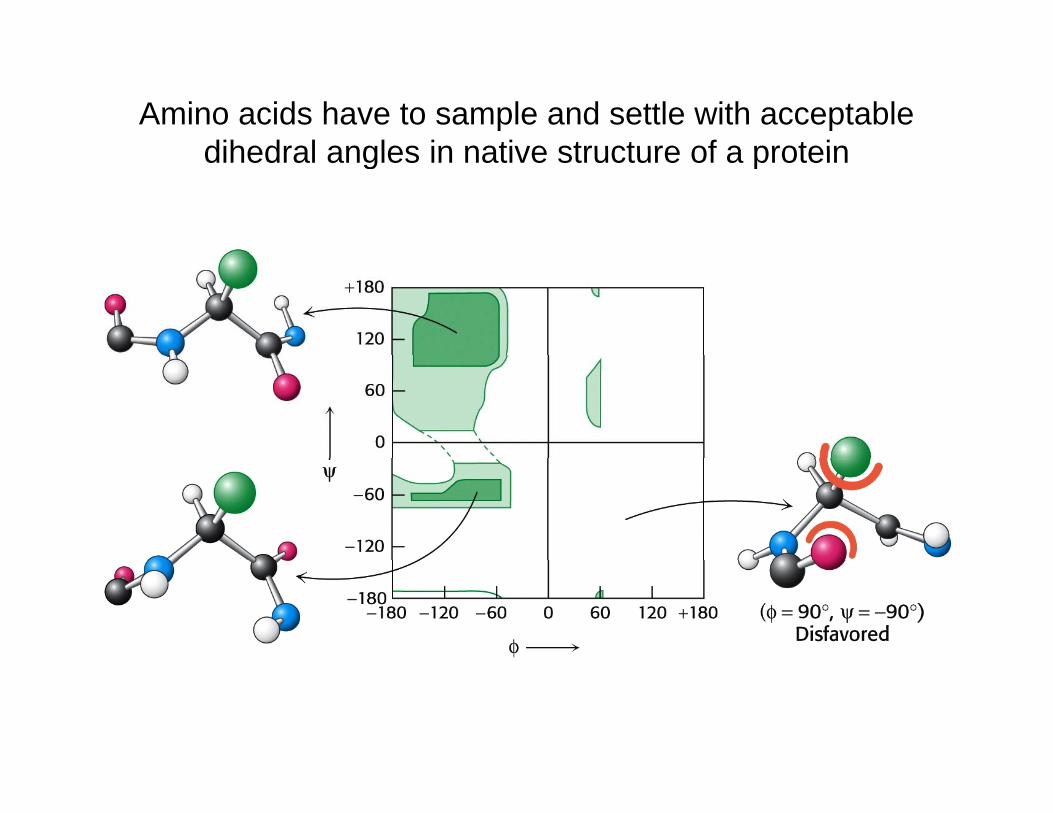
Amino acids have to sample and settle with acceptable dihedral angles in native structure of a proteindihedral angles in native structure of a protein
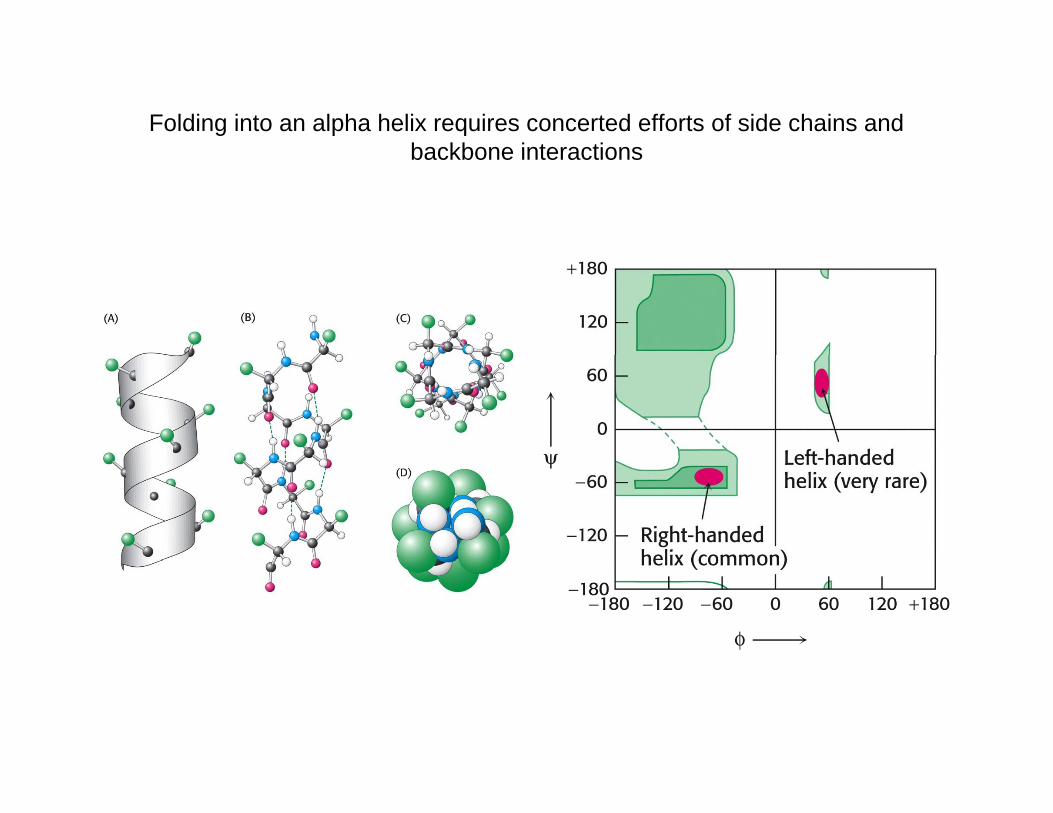
Folding into an alpha helix requires concerted efforts of side chains and backbone interactionsbackbone interactions
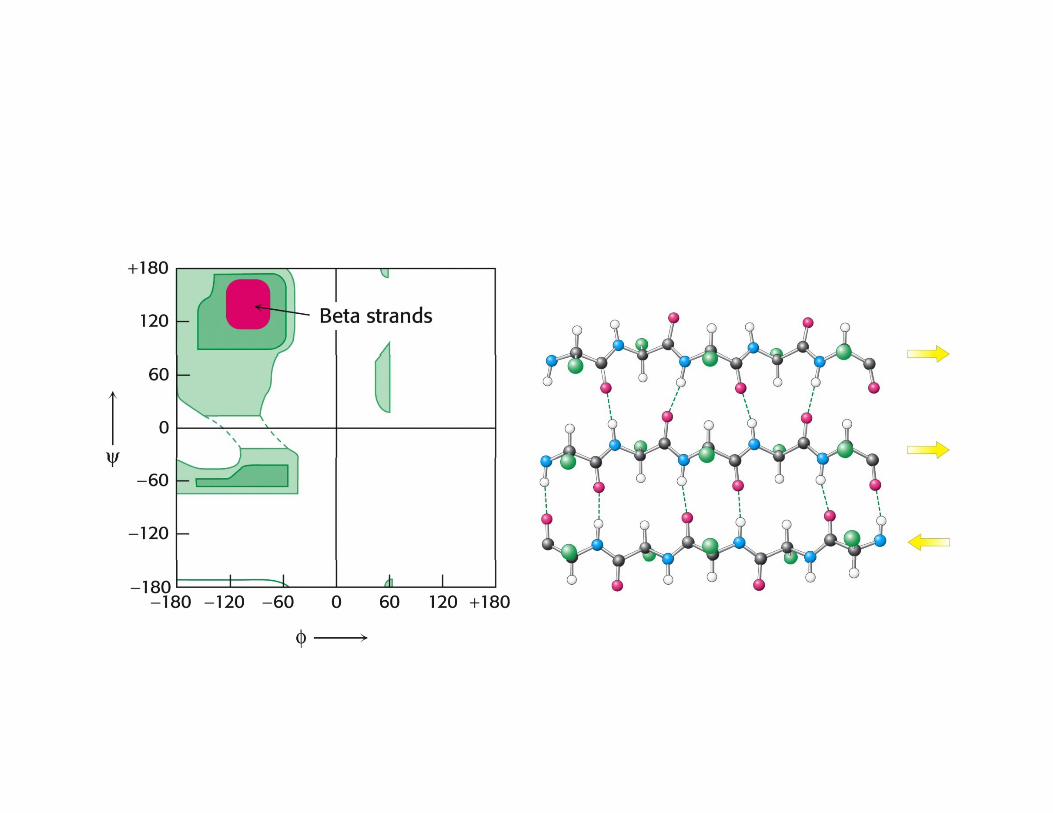
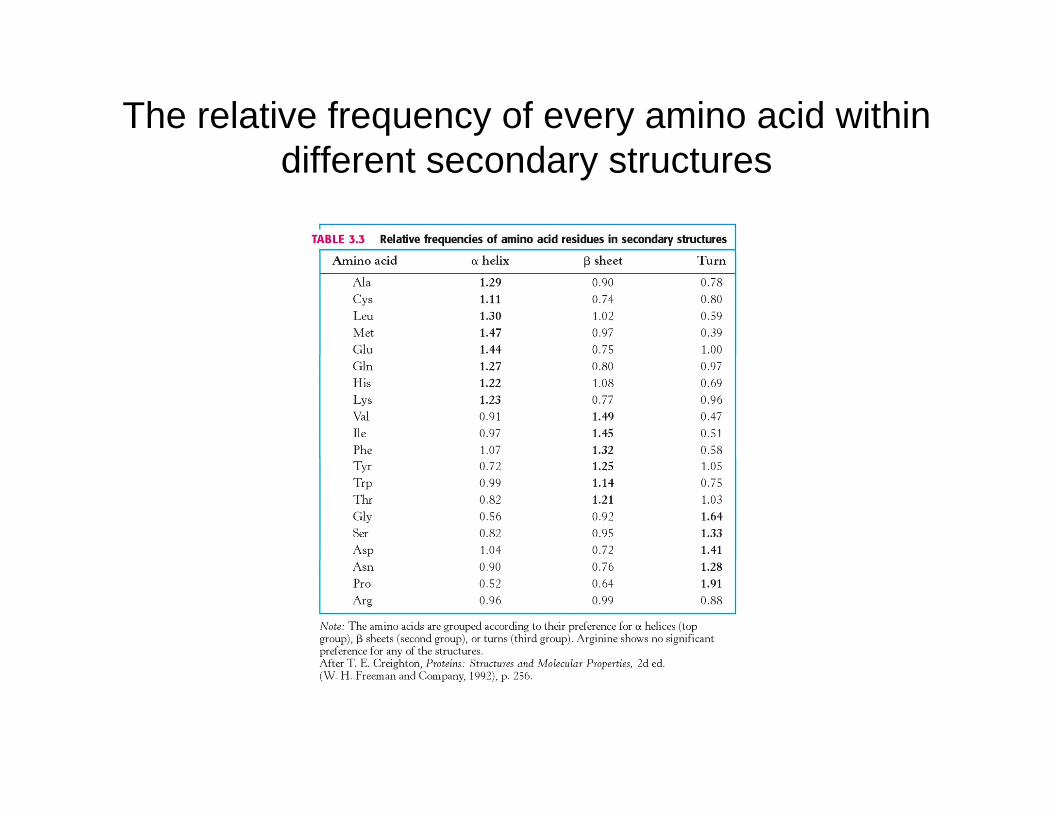
The relative frequency of every amino acid within diff t d t tdifferent secondary structures
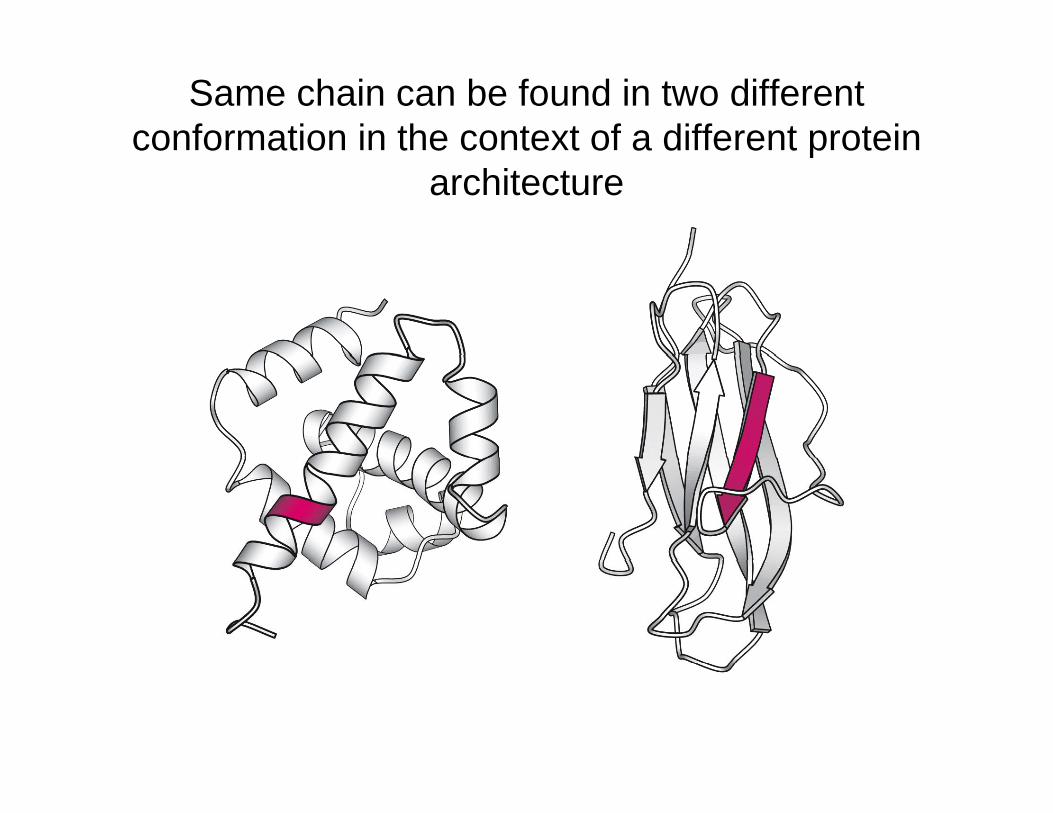
Same chain can be found in two different conformation in the context of a different protein co o at o t e co te t o a d e e t p ote
architecture
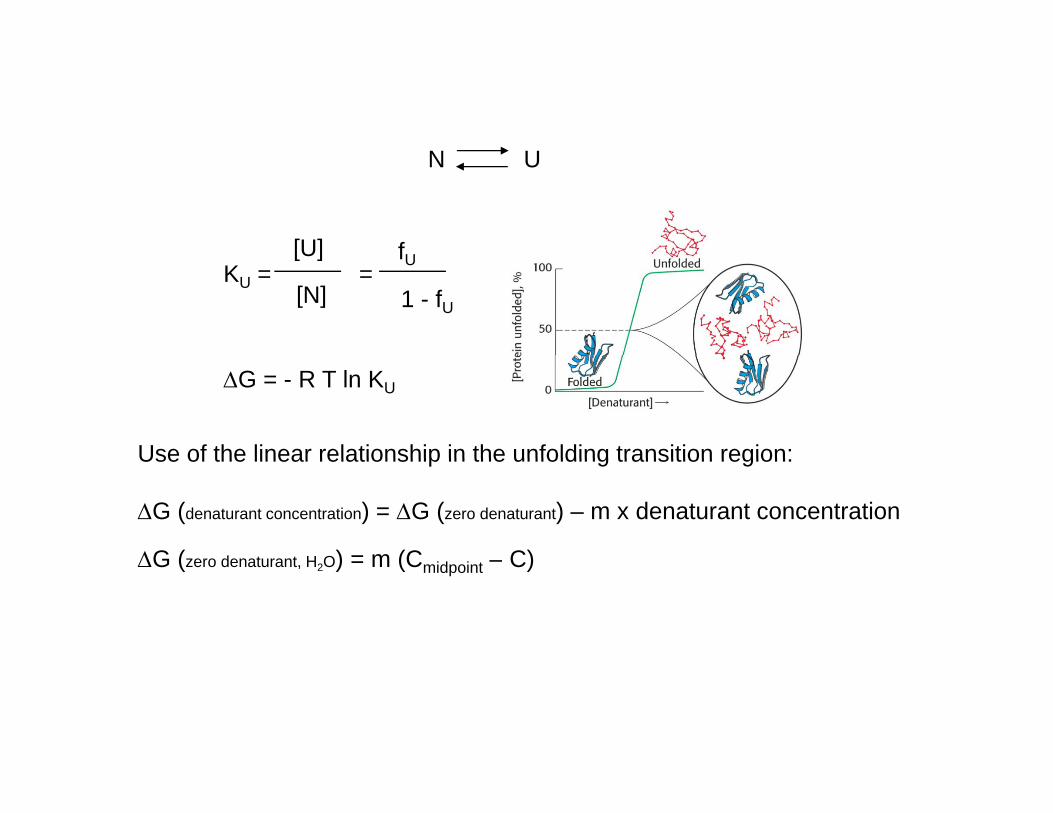
N UN U
[U] fKU = =
[U]
[N]
fU
1 - fU
ΔG = - R T ln KU
Use of the linear relationship in the unfolding transition region:Use of the linear relationship in the unfolding transition region:
ΔG (denaturant concentration) = ΔG (zero denaturant) – m x denaturant concentration
ΔG (zero denaturant H O) = m (C – C)ΔG (zero denaturant, H2O) = m (Cmidpoint – C)

Folding Paradox - Levinthal's paradox states that there are approximately 1050 possible conformationsthere are approximately 1050 possible conformations for a protein, such as ribonuclease (124 residues). If one new conformation could be attempted every 10-13
seconds, it would still take over 1030 years to randomly test all of the possibilities, yet ribonuclease can ycompletely fold in about a minute. Thus, folding must not be a completely random phenomenon.
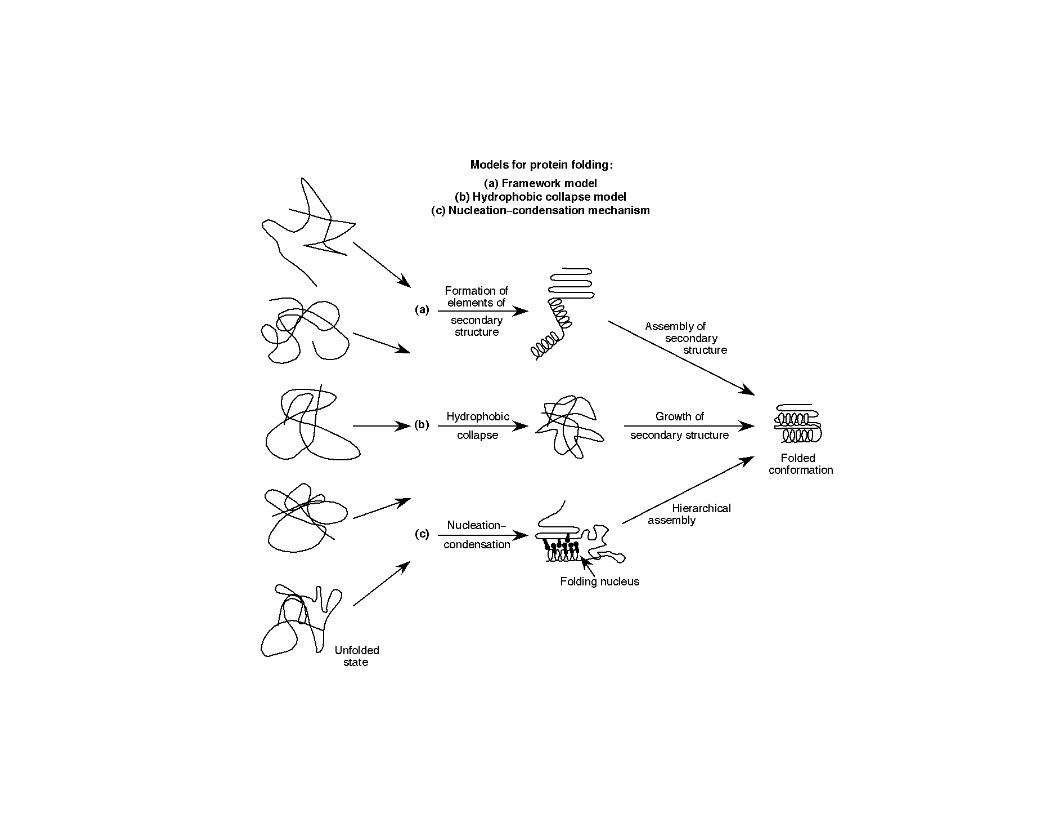
Pathway Model - The "pathway" model of protein folding is depicted at the left Nucleation is criticalfolding is depicted at the left. Nucleation is critical because it is much more difficult to begin an helix than to extend it. Nucleation may start at a number of points and all of these partially folded structures can be "funneled" by energy minimizations toward the final state. Thus, Levinthal's paradox is averted.
Matthews, Ahern, and van Holde, 3rd ed.
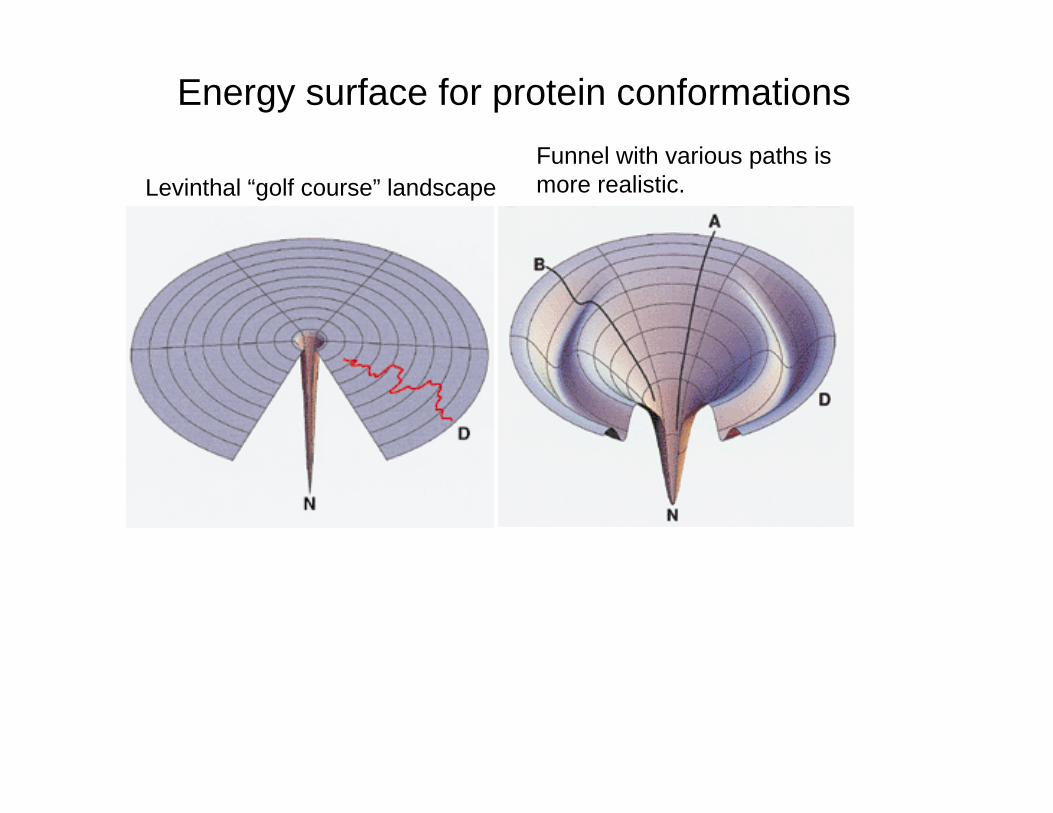
Energy surface for protein conformationsF l ith i th i
Levinthal “golf course” landscapeFunnel with various paths ismore realistic.
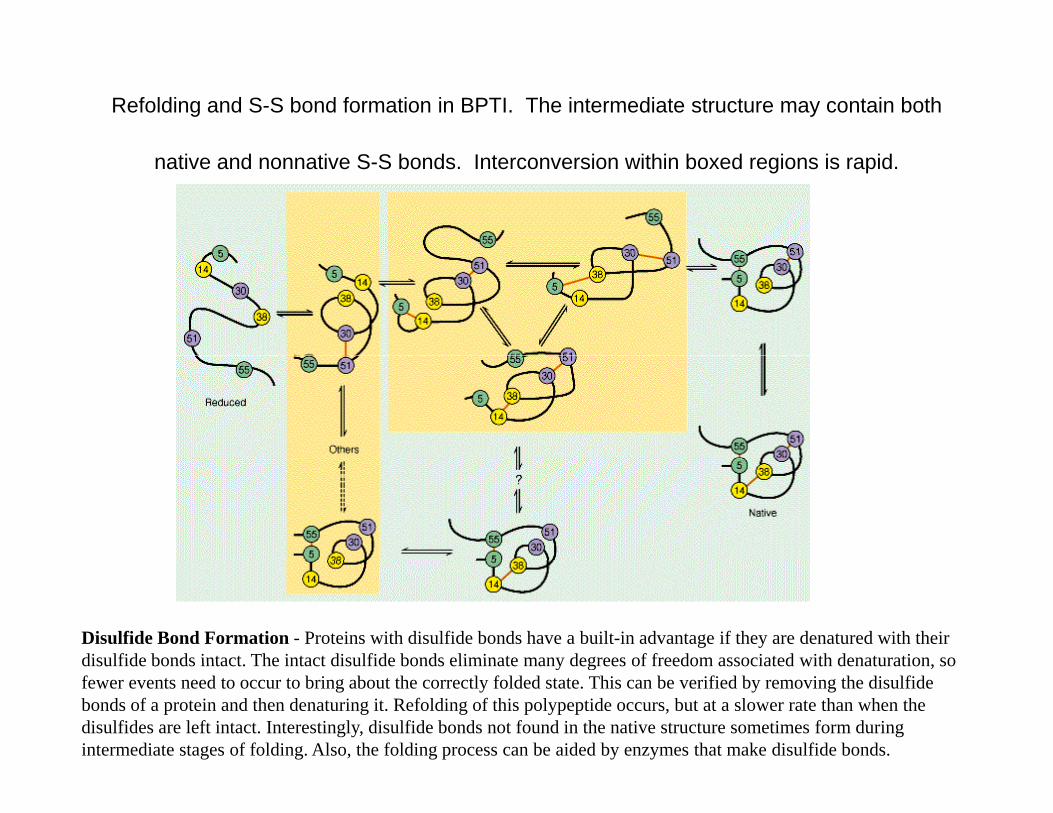
Refolding and S-S bond formation in BPTI. The intermediate structure may contain both
native and nonnative S-S bonds. Interconversion within boxed regions is rapid.
Disulfide Bond Formation - Proteins with disulfide bonds have a built-in advantage if they are denatured with their disulfide bonds intact The intact disulfide bonds eliminate many degrees of freedom associated with denaturation sodisulfide bonds intact. The intact disulfide bonds eliminate many degrees of freedom associated with denaturation, so fewer events need to occur to bring about the correctly folded state. This can be verified by removing the disulfide bonds of a protein and then denaturing it. Refolding of this polypeptide occurs, but at a slower rate than when the disulfides are left intact. Interestingly, disulfide bonds not found in the native structure sometimes form during intermediate stages of folding. Also, the folding process can be aided by enzymes that make disulfide bonds.
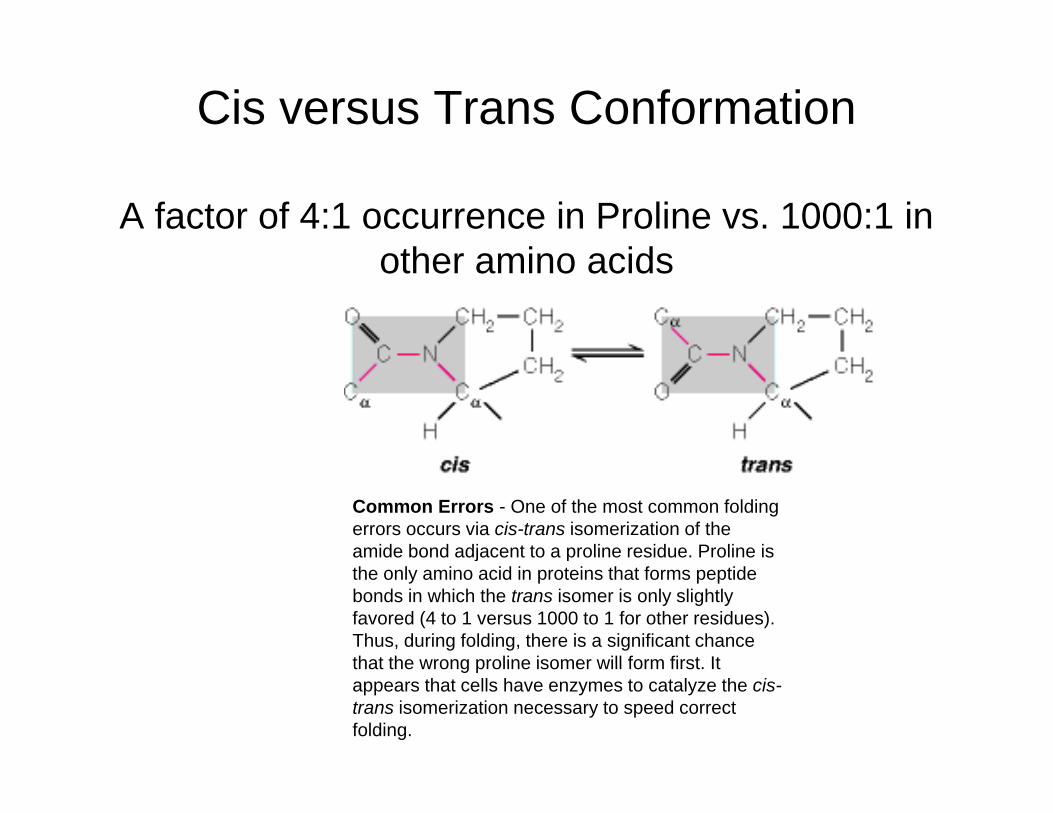
Cis versus Trans Conformation
A factor of 4:1 occurrence in Proline vs. 1000:1 in th i idother amino acids
Common Errors - One of the most common folding errors occurs via cis-trans isomerization of the amide bond adjacent to a proline residue. Proline is j pthe only amino acid in proteins that forms peptide bonds in which the trans isomer is only slightly favored (4 to 1 versus 1000 to 1 for other residues). Thus, during folding, there is a significant chance that the wrong proline isomer will form first Itthat the wrong proline isomer will form first. It appears that cells have enzymes to catalyze the cis-trans isomerization necessary to speed correct folding.

Time scale of protein motions
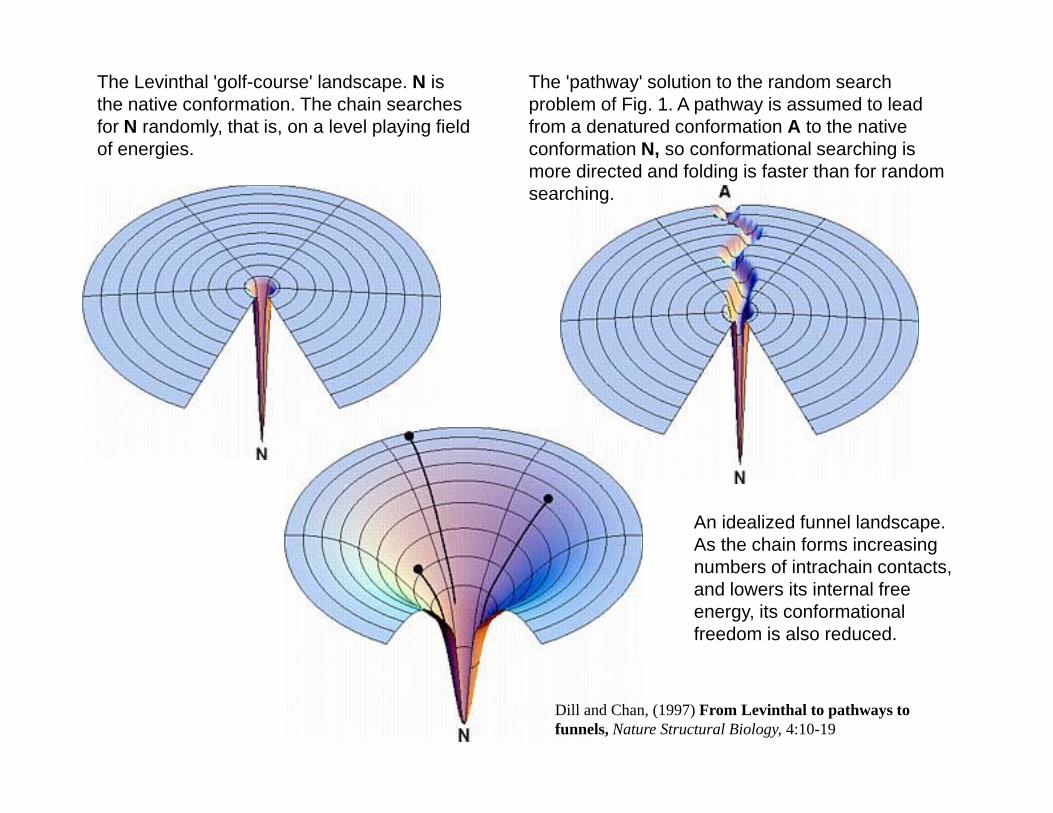
The Levinthal 'golf-course' landscape. N is the native conformation. The chain searches for N randomly, that is, on a level playing field of energies
The 'pathway' solution to the random search problem of Fig. 1. A pathway is assumed to lead from a denatured conformation A to the native conformation N so conformational searching isof energies. conformation N, so conformational searching is more directed and folding is faster than for random searching.
An idealized funnel landscape. As the chain forms increasing
b f i t h i t tnumbers of intrachain contacts, and lowers its internal free energy, its conformational freedom is also reduced.
Dill and Chan, (1997) From Levinthal to pathways to funnels, Nature Structural Biology, 4:10-19
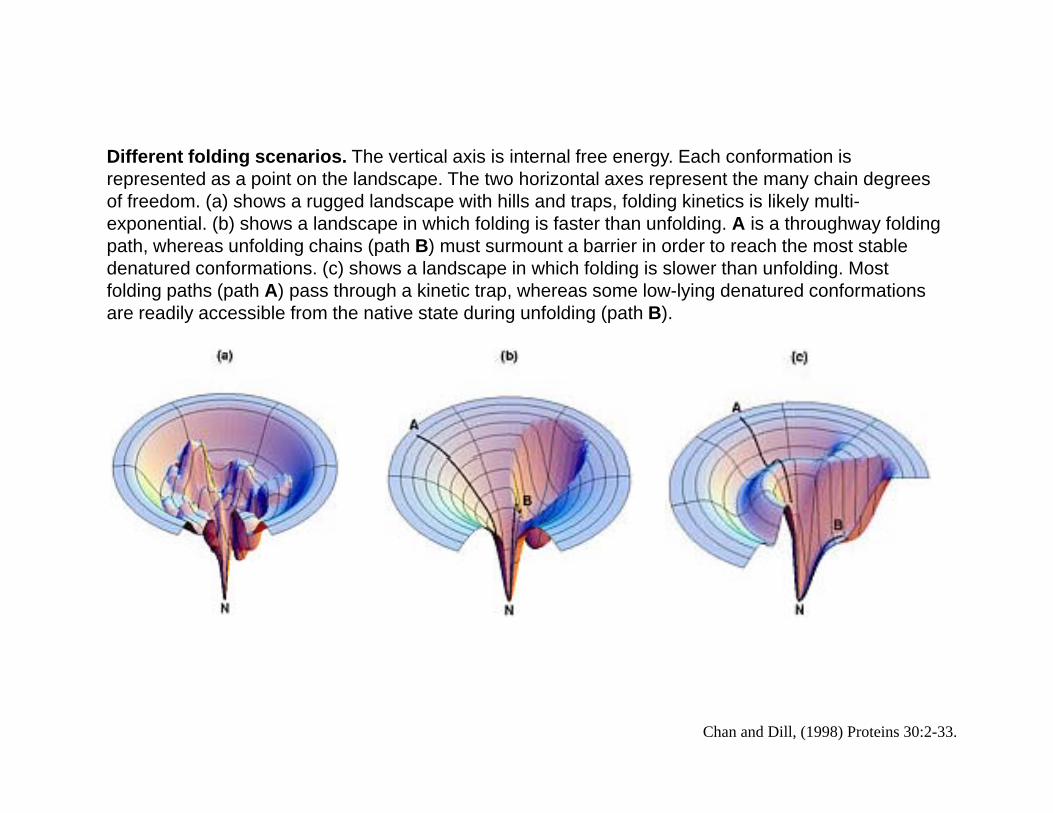
Diff t f ldi i Th ti l i i i t l f E h f ti iDifferent folding scenarios. The vertical axis is internal free energy. Each conformation is represented as a point on the landscape. The two horizontal axes represent the many chain degrees of freedom. (a) shows a rugged landscape with hills and traps, folding kinetics is likely multi-exponential. (b) shows a landscape in which folding is faster than unfolding. A is a throughway folding path, whereas unfolding chains (path B) must surmount a barrier in order to reach the most stable p , g (p )denatured conformations. (c) shows a landscape in which folding is slower than unfolding. Most folding paths (path A) pass through a kinetic trap, whereas some low-lying denatured conformations are readily accessible from the native state during unfolding (path B).
Chan and Dill, (1998) Proteins 30:2-33.
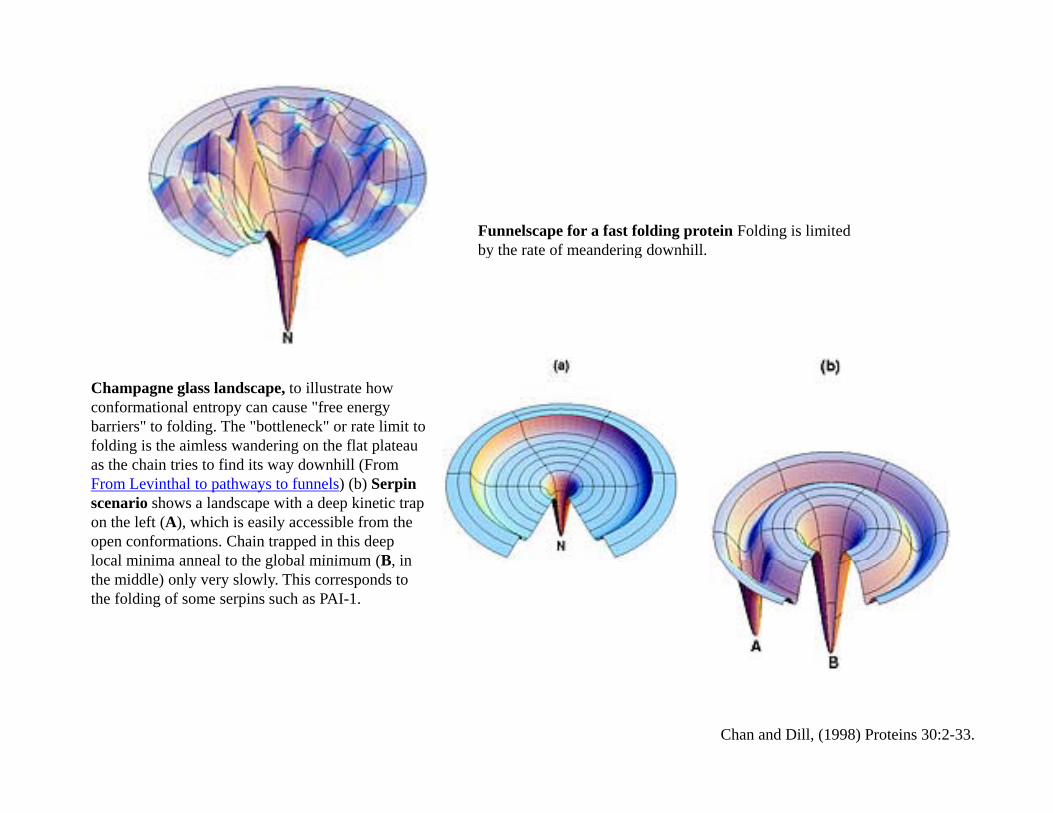
Funnelscape for a fast folding protein Folding is limited by the rate of meandering downhillby the rate of meandering downhill.
Champagne glass landscape, to illustrate how conformational entropy can cause "free energy barriers" to folding. The "bottleneck" or rate limit to folding is the aimless wandering on the flat plateau as the chain tries to find its way downhill (From From Levinthal to pathways to funnels) (b) Serpin scenario shows a landscape with a deep kinetic trap on the left (A), which is easily accessible from the open conformations. Chain trapped in this deep local minima anneal to the global minimum (B inlocal minima anneal to the global minimum (B, in the middle) only very slowly. This corresponds to the folding of some serpins such as PAI-1.
Chan and Dill, (1998) Proteins 30:2-33.
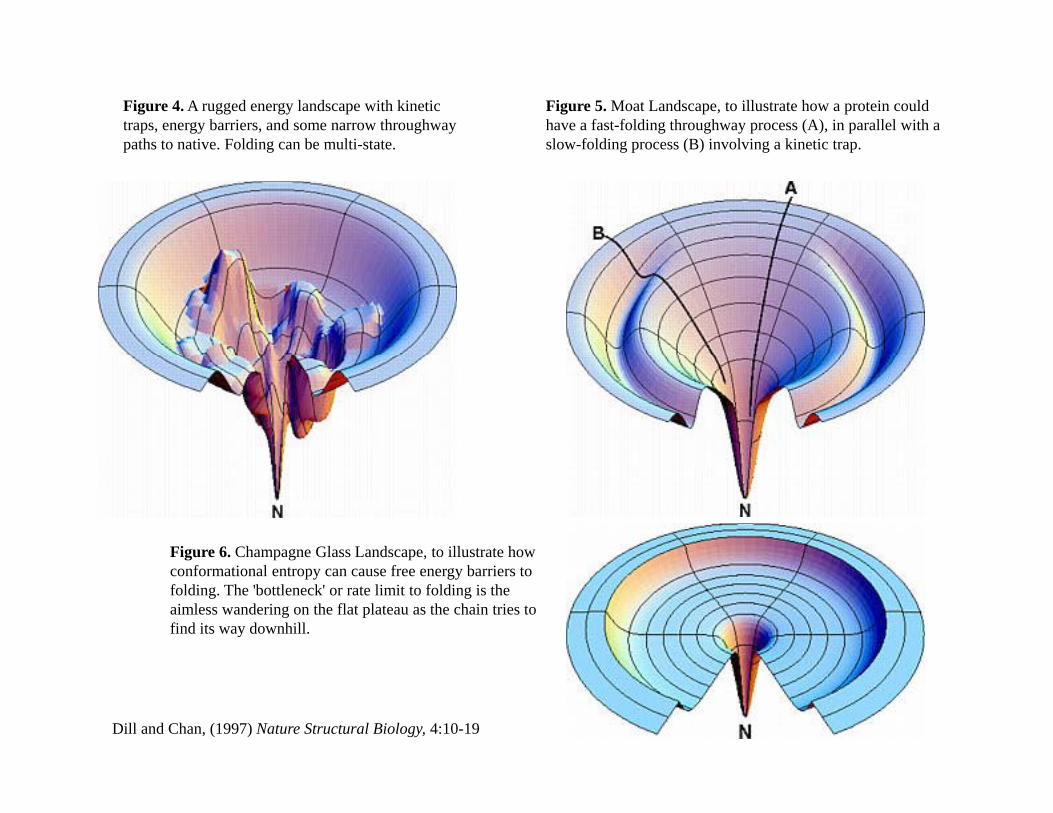
Figure 4. A rugged energy landscape with kinetic traps, energy barriers, and some narrow throughway paths to native. Folding can be multi-state.
Figure 5. Moat Landscape, to illustrate how a protein could have a fast-folding throughway process (A), in parallel with a slow-folding process (B) involving a kinetic trap.p g g p ( ) g p
Figure 6. Champagne Glass Landscape, to illustrate how conformational entropy can cause free energy barriers to folding. The 'bottleneck' or rate limit to folding is the aimless wandering on the flat plateau as the chain tries to find its way downhill.
Dill and Chan, (1997) Nature Structural Biology, 4:10-19
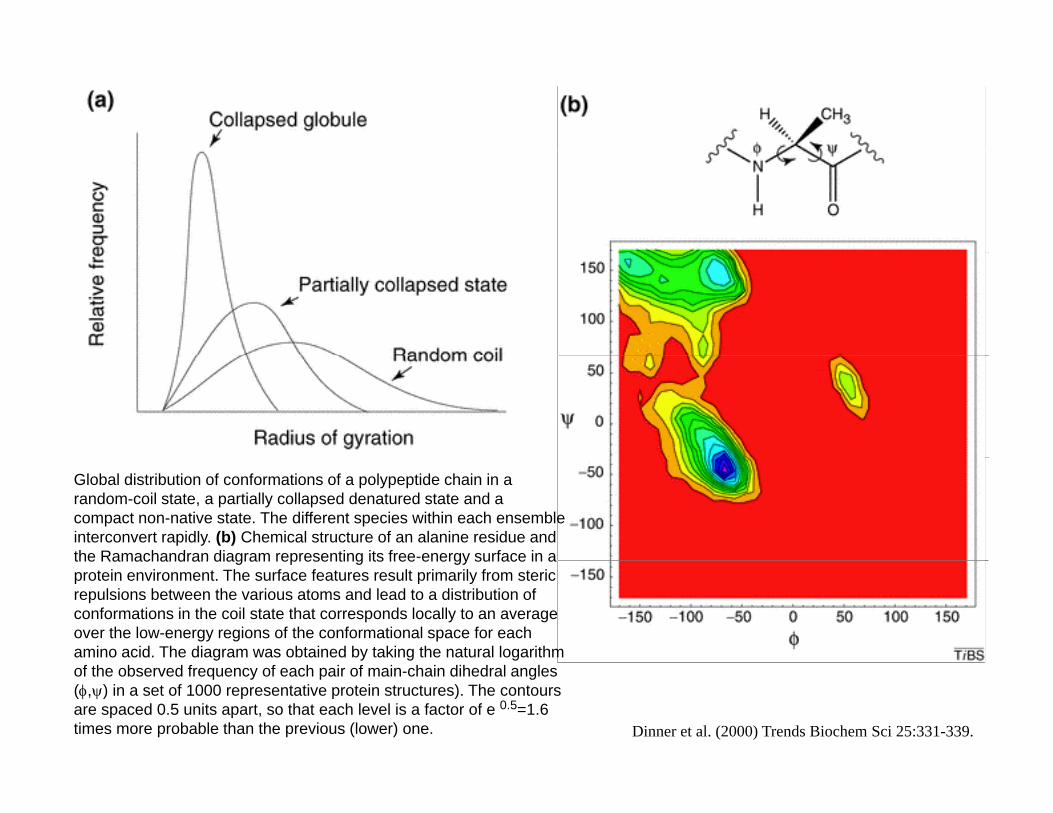
Global distribution of conformations of a polypeptide chain in a random-coil state, a partially collapsed denatured state and a compact non-native state. The different species within each ensemble interconvert rapidly. (b) Chemical structure of an alanine residue and the Ramachandran diagram representing its free-energy surface in athe Ramachandran diagram representing its free energy surface in a protein environment. The surface features result primarily from steric repulsions between the various atoms and lead to a distribution of conformations in the coil state that corresponds locally to an average over the low-energy regions of the conformational space for each amino acid. The diagram was obtained by taking the natural logarithm of the observed frequency of each pair of main-chain dihedral angles (φ,ψ) in a set of 1000 representative protein structures). The contours are spaced 0.5 units apart, so that each level is a factor of e 0.5=1.6 times more probable than the previous (lower) one. Dinner et al. (2000) Trends Biochem Sci 25:331-339.

Atomic structures of amyloid cross- spines reveal varied steric zippers
Nature 447, 453-457 (2007)
Sawaya et al.http://www ncbi nlm nih gov/entrez/query fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list uids=17468747http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=17468747
Amyloid fibrils formed from different proteins, each associated with a particular disease, contain a common cross- spine. The atomic architecture of a spine, from the fibril-forming segment GNNQQNY of the yeast prion protein Sup35, was recently revealed by X-raysegment GNNQQNY of the yeast prion protein Sup35, was recently revealed by X ray microcrystallography. It is a pair of -sheets, with the facing side chains of the two sheets interdigitated in a dry 'steric zipper'. Here we report some 30 other segments from fibril-forming proteins that form amyloid-like fibrils, microcrystals, or usually both. These include segments from the Alzheimer's amyloid- and tau proteins, the PrPThese include segments from the Alzheimer s amyloid and tau proteins, the PrPprion protein, insulin, islet amyloid polypeptide (IAPP), lysozyme, myoglobin, -synucleinand 2-microglobulin, suggesting that common structural features are shared by amyloiddiseases at the molecular level. Structures of 13 of these microcrystals all reveal stericzippers, but with variations that expand the range of atomic architectures for amyloid-likezippers, but with variations that expand the range of atomic architectures for amyloid like fibrils and offer an atomic-level hypothesis for the basis of prion strains.
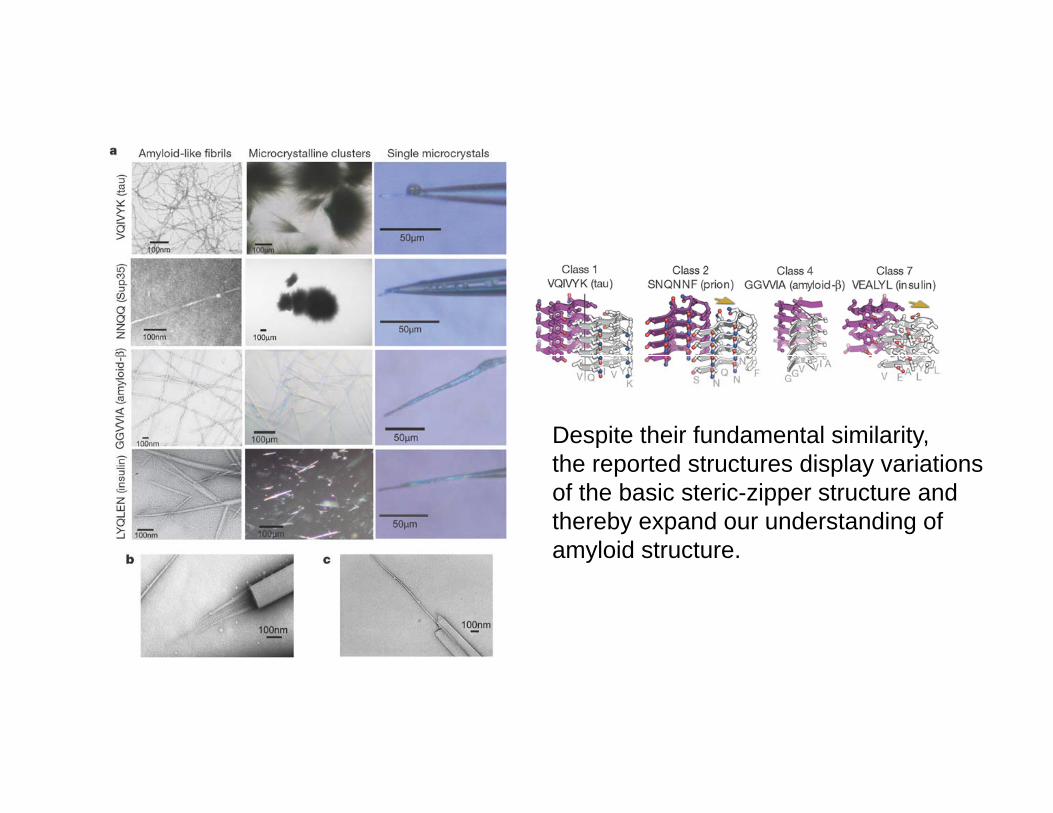
Despite their fundamental similarity, th t d t t di l i tithe reported structures display variations of the basic steric-zipper structure and thereby expand our understanding of amyloid structure. y

Th 8 l f t i iThe 8 classes of steric zippers

Mechanism of coupled folding and binding of an intrinsically disordered proteinSugase et al.g
Nature 447, 1021-1025 (2007)http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=17522630
P t i f ldi d bi di l i hi h th t i ' h ' f f blProtein folding and binding are analogous processes, in which the protein 'searches' for favourableintramolecular or intermolecular interactions on a funnelled energy landscape1, 2. Many eukaryotic proteins are disordered under physiological conditions, and fold into ordered structures only on binding to their cellular targets. The mechanism by which folding is coupled to binding is
l d t d b t it h b h th i d th ti l d th t th bi di ki tipoorly understood, but it has been hypothesized on theoretical grounds that the binding kinetics may be enhanced by a 'fly-casting' effect, where the disordered protein binds weakly and non-specifically to its target and folds as it approaches the cognate binding site7. Here we show, using NMR titrations and 15N relaxation dispersion, that the phosphorylated kinase inducible acti ation domain (pKID) of the transcription factor CREB forms an ensemble of transient enco nteractivation domain (pKID) of the transcription factor CREB forms an ensemble of transient encounter complexes on binding to the KIX domain of the CREB binding protein. The encounter complexes are stabilized primarily by non-specific hydrophobic contacts, and evolve by way of an intermediate to the fully bound state without dissociation from KIX. The carboxy-terminal helix of pKID is only partially folded in the intermediate and becomes stabilized by intermolecular interactions formedpartially folded in the intermediate, and becomes stabilized by intermolecular interactions formed in the final bound state. Future applications of our method will provide new understanding of themolecular mechanisms by which intrinsically disordered proteins perform their diverse biological functions.
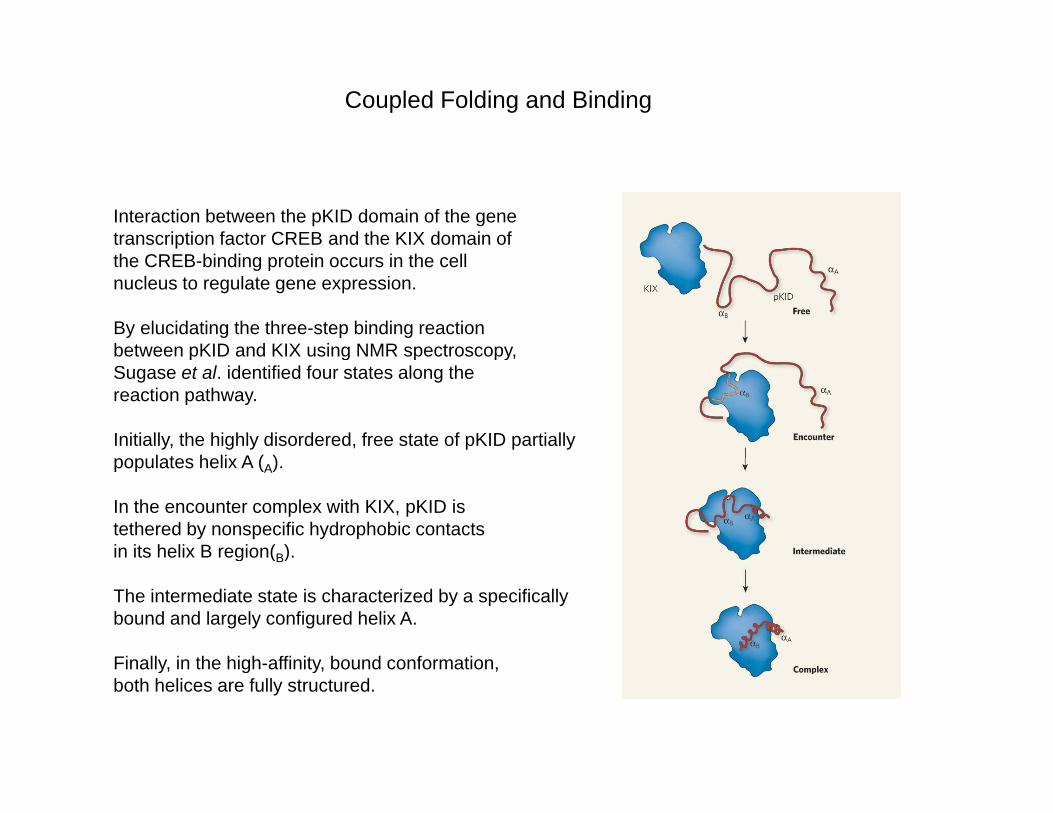
Coupled Folding and Binding
Interaction between the pKID domain of the gene transcription factor CREB and the KIX domain of the CREB-binding protein occurs in the cell nucleus to regulate gene expression.
By elucidating the three-step binding reaction between pKID and KIX using NMR spectroscopy,between pKID and KIX using NMR spectroscopy, Sugase et al. identified four states along the reaction pathway.
Initially, the highly disordered, free state of pKID partially l t h li A ( )populates helix A (A).
In the encounter complex with KIX, pKID is tethered by nonspecific hydrophobic contacts in its helix B region(B). g (B)
The intermediate state is characterized by a specifically bound and largely configured helix A.
Finally in the high affinity bound conformationFinally, in the high-affinity, bound conformation, both helices are fully structured.
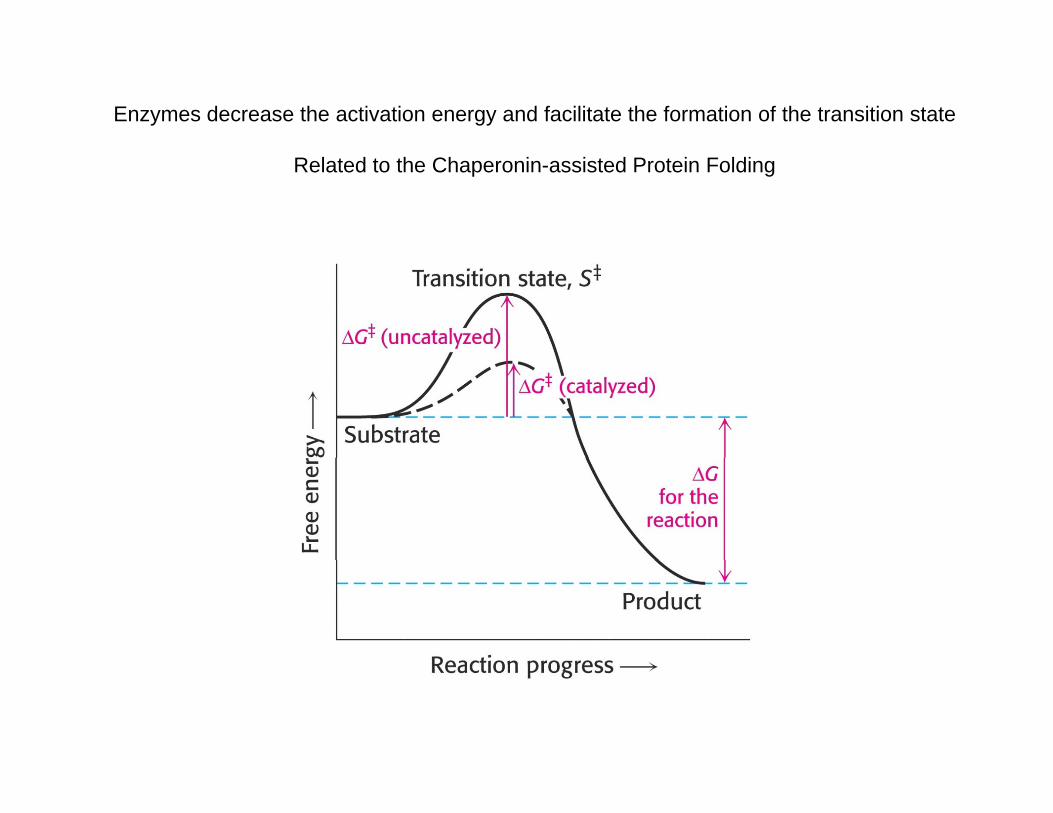
Enzymes decrease the activation energy and facilitate the formation of the transition state
Related to the Chaperonin-assisted Protein Folding
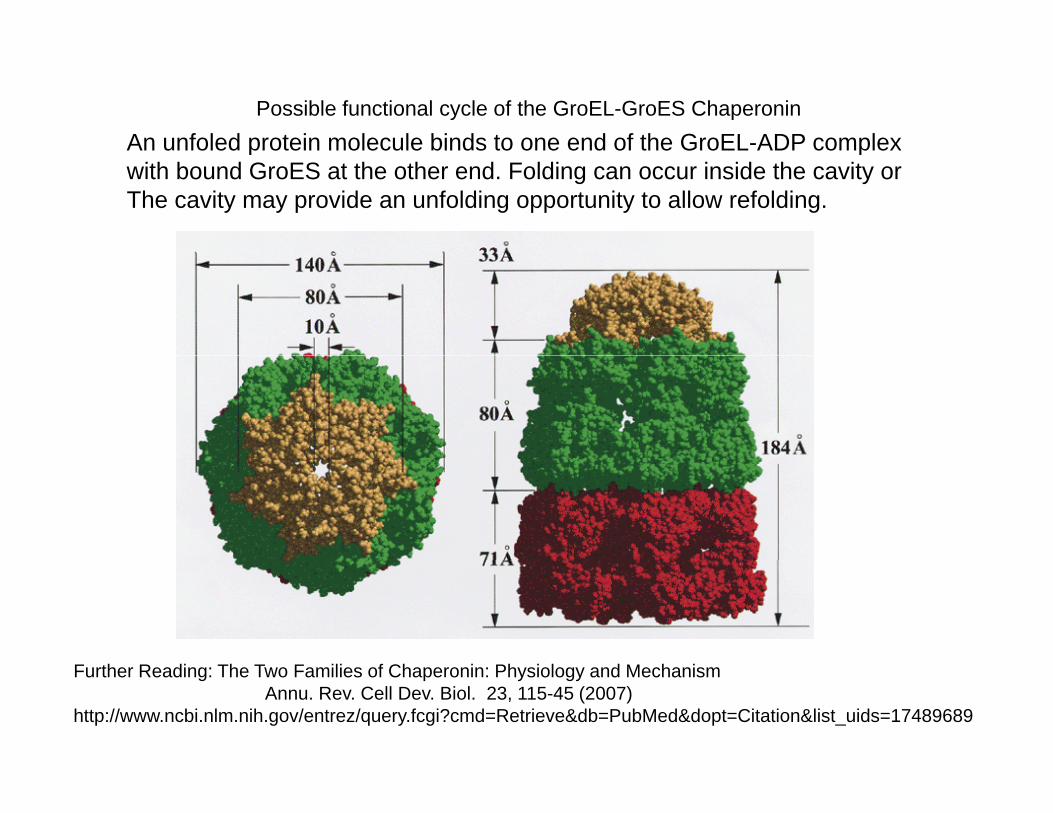
Possible functional cycle of the GroEL-GroES ChaperoninAn unfoled protein molecule binds to one end of the GroEL-ADP complexAn unfoled protein molecule binds to one end of the GroEL ADP complexwith bound GroES at the other end. Folding can occur inside the cavity orThe cavity may provide an unfolding opportunity to allow refolding.
Further Reading: The Two Families of Chaperonin: Physiology and MechanismAnnu. Rev. Cell Dev. Biol. 23, 115-45 (2007)
http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=17489689