psi-blast, prosite, ucsc genome browser lecture 3
Post on 18-Dec-2015
220 views
TRANSCRIPT

Psi-BLAST,Psi-BLAST,Prosite, Prosite,
UCSC Genome UCSC Genome BrowserBrowser
Lecture 3Lecture 3

Searching for remote homologsSearching for remote homologs
Sometimes BLAST isn’t enoughSometimes BLAST isn’t enough Large protein family, and BLAST only finds Large protein family, and BLAST only finds
close members. We want more distant close members. We want more distant members members

PSI-BLASTPSI-BLAST
PPosition osition SSpecific pecific IIterative terative BLASTBLAST
Regular blast
Construct profile from blast results
Blast profile search
Final results

Consensus, Pattern, PSSMConsensus, Pattern, PSSM
AATTCCTTTTGG
AAAACCTTTTGG
AAAACCTTTTCC
1 2 3 4 5 6
Seq1
Seq2
Seq3
Consensus:
the most frequent character in the column is chosen
A A C T T G A-[TA]-C-T-T-[GC]
Pattern:
represents the alignment as a regular expression Pos
Nuc112233445566
AA11.67.6700000000
CC0000110000.33.33
GG0000000000.67.67
TT00.33.3300111100
Profile = PSSM:
Position Specific Score Matrix

S(AACCAA)=1*0.67*1*1*.25*.33S(GACCAA)=0Sequences with higher scores -> higher chance of being related to the PSSM
PosNuc 112233445566
AA11..67670000..2525..3333
CC00..33331111..252500
GG00000000..2525..3333
TT00000000..2525..3333

PSI-BLASTPSI-BLAST
PPosition osition SSpecific pecific IIterative terative BLASTBLAST
Regular blast
Construct profile from blast results
Blast profile search
Final results
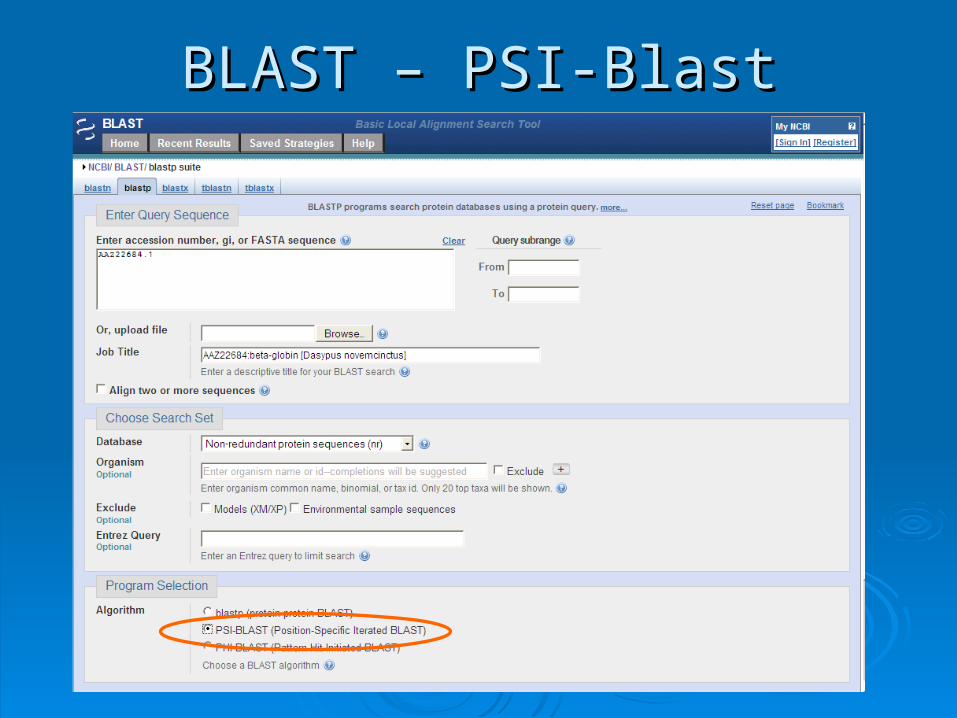
BLAST – PSI-BlastBLAST – PSI-Blast

PSI-Blast - resultsPSI-Blast - results

PSI-BLASTPSI-BLAST
AdvantageAdvantage: PSI-BLAST looks for seq’s : PSI-BLAST looks for seq’s that are close to the query, and learns that are close to the query, and learns from them to extend the circle of friendsfrom them to extend the circle of friends
DisadvantageDisadvantage: if we obtained a WRONG : if we obtained a WRONG hit, we will get to unrelated sequences hit, we will get to unrelated sequences (contamination). This gets worse and (contamination). This gets worse and worse each iterationworse each iteration

PSI-BLASTPSI-BLAST
Which of the following is/are correct?Which of the following is/are correct?
1.1. PSI-BLAST is expected to give more hits PSI-BLAST is expected to give more hits than BLASTthan BLAST
2.2. PSI-BLAST is an iterative search methodPSI-BLAST is an iterative search method
3.3. PSI-BLAST is faster than BLASTPSI-BLAST is faster than BLAST
4.4. Each iteration of PSI-BLAST can only Each iteration of PSI-BLAST can only improve the results of the previous improve the results of the previous iterationiteration

Turning information into knowledgeTurning information into knowledge
The outcome of a sequencing project are The outcome of a sequencing project are masses of raw datamasses of raw data
The challenge is to turn these The challenge is to turn these raw data raw data into biological knowledgeinto biological knowledge
A valuable tool for this challenge is an A valuable tool for this challenge is an automated diagnostic pipe through which automated diagnostic pipe through which newly determined sequences can be newly determined sequences can be streamlinedstreamlined

From sequence to functionFrom sequence to function Nature tends to innovate rather than inventNature tends to innovate rather than invent Proteins are composed of functional Proteins are composed of functional
elements: domains and motifselements: domains and motifs DomainsDomains are structural are structural
units that carry out a units that carry out a certain function. They are certain function. They are shared between different shared between different proteinsproteins
MotifsMotifs are shorter are shorter and are usually criticaland are usually criticalfor the biological activityfor the biological activity

http://www.expasy.ch/http://www.expasy.ch/prositeprosite

PrositeProsite
From analyzing conserved regions in From analyzing conserved regions in protein sequences it is possible to derive protein sequences it is possible to derive signatures of motifs and domainssignatures of motifs and domains
Prosite consists of annotated Prosite consists of annotated sites/motifs/signatures/fingerprints sites/motifs/signatures/fingerprints
Given an uncharacterized translated Given an uncharacterized translated protein sequence, prosite tries to predict protein sequence, prosite tries to predict which motifs and domains make up the which motifs and domains make up the protein and thus identify the family to protein and thus identify the family to which it belongswhich it belongs

PrositePrositeProsite represents entries with Prosite represents entries with patternspatterns or or profilesprofiles
A A C T T C
A T C T T G
A A C T T G
profile
A-[TA]-C-T-T-[GC]
Profiles are used in prosite when the motif is relatively Profiles are used in prosite when the motif is relatively divergent, and is difficult to represent as a patterndivergent, and is difficult to represent as a pattern Profiles also characterize domains over their entire length, not Profiles also characterize domains over their entire length, not just the motifjust the motif
pattern
1122334455 66
AA110.670.6700000000
TT000.330.3300111100
CC00001100000.330.33
GG00000000000.670.67
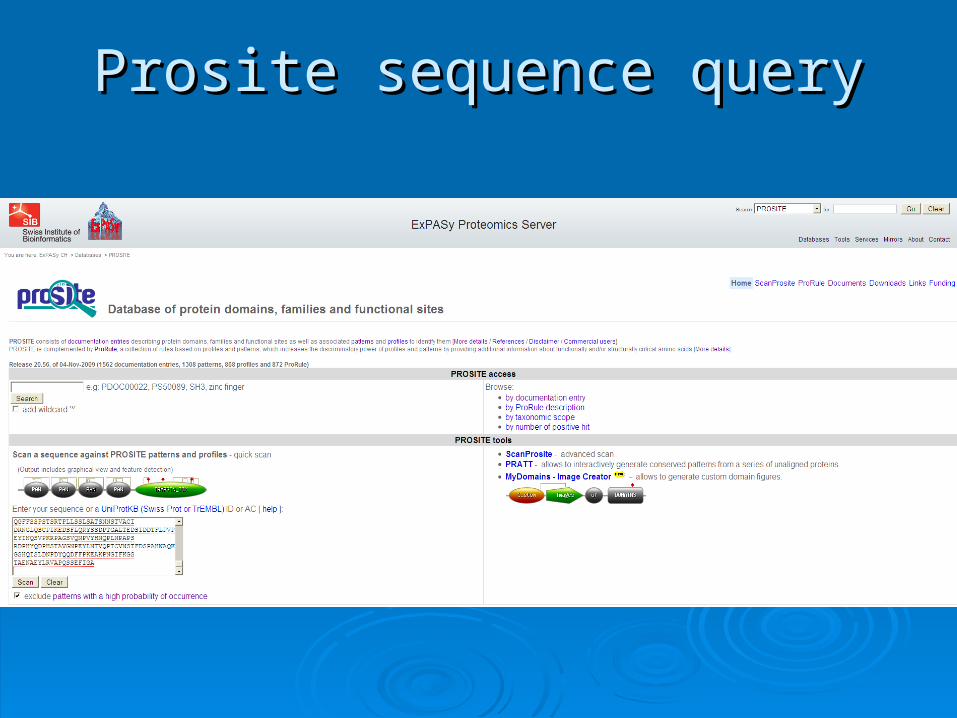
Prosite sequence queryProsite sequence query
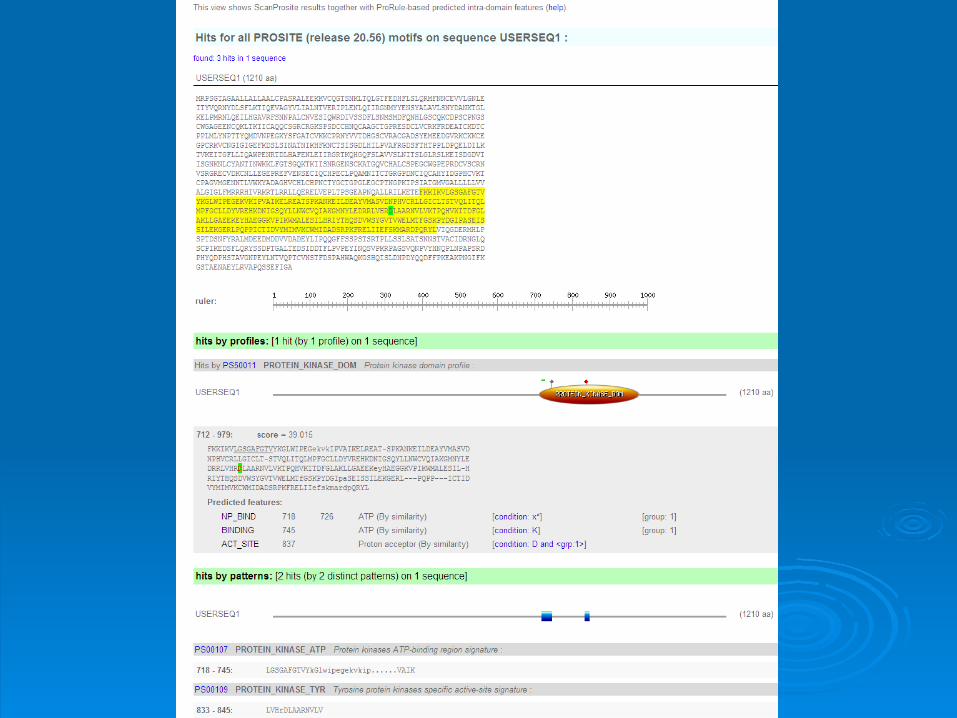

Patterns with a high probability of Patterns with a high probability of occurrenceoccurrence
Entries describing commonly found postEntries describing commonly found post--translational modifications or compositionally translational modifications or compositionally biased regionsbiased regions
Found in the majority of known protein Found in the majority of known protein sequences sequences
High probability of occurrenceHigh probability of occurrence Prosite filters them by defaultProsite filters them by default

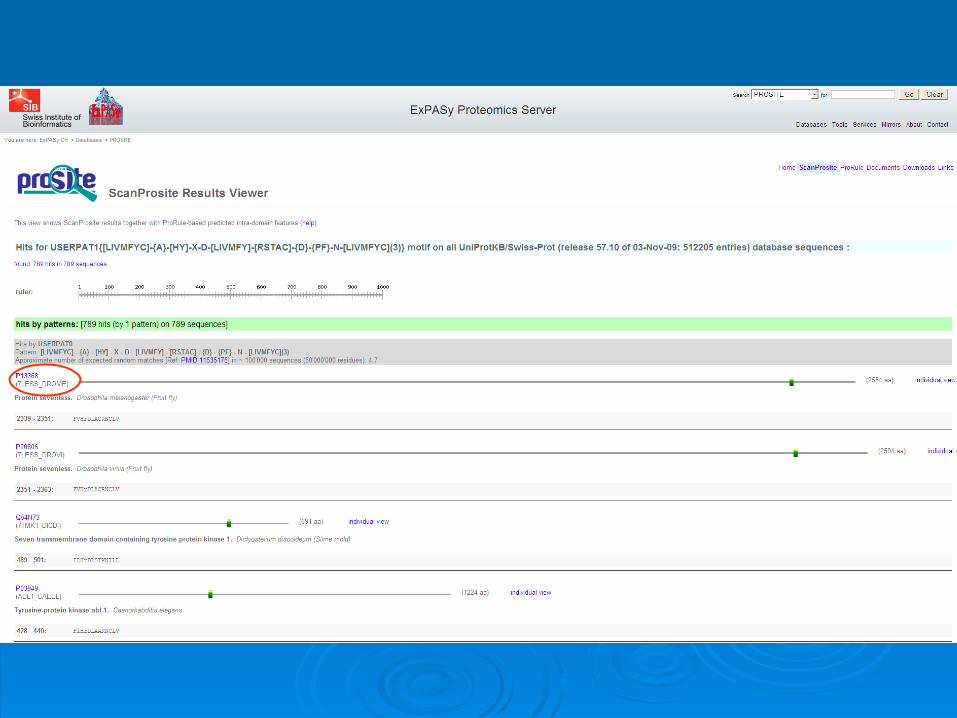
Scanning PrositeScanning Prosite
Query: sequence
Query: pattern
Result: all patterns found in the sequence
Result: all sequences which adhere to this pattern
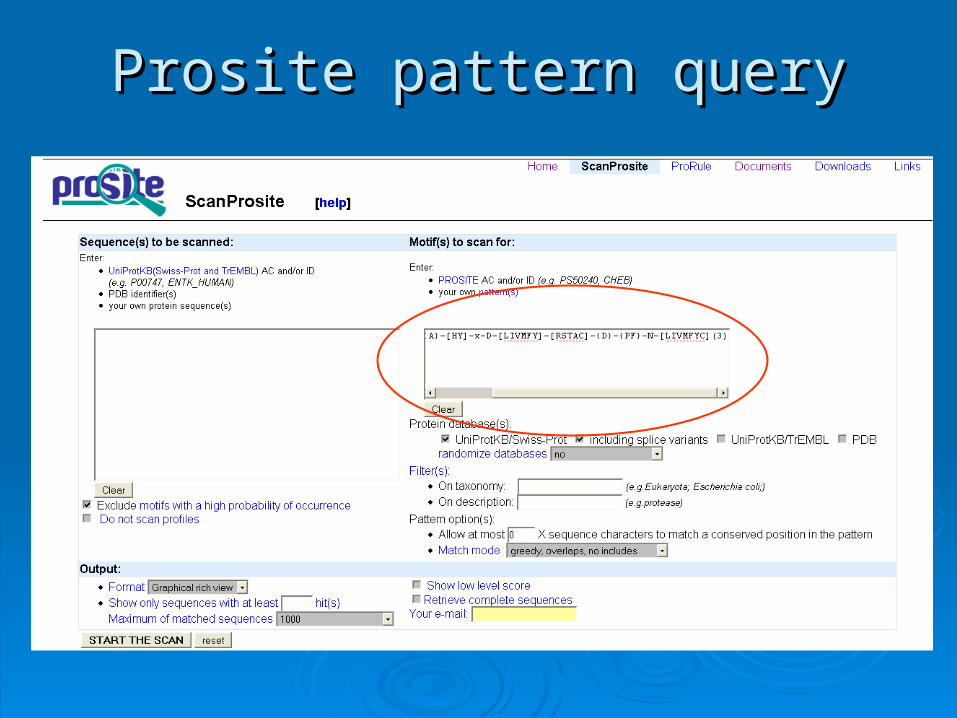
Prosite pattern queryProsite pattern query

UCSC Genome BrowserUCSC Genome Browser

UCSC Genome Browser - GatewayUCSC Genome Browser - Gateway
Reset all settings of
previous uses

UCSC Genome Browser - GatewayUCSC Genome Browser - Gateway

ResultsResults

Annotation tracksAnnotation tracks
Mammal conservation
mRNAs (GenBank)
RefSeq Genes
Base position
Species alignment
SNPs
Repeats
GeneDirection
Coding
Intron
UTRUCSC Genes

UCSC GeneUCSC Gene

UCSC Genome Browser - movementUCSC Genome Browser - movement
Zoom x3 + Center

ControllingControllingannotationannotation
trackstracks

Malariadistr.
Sickle-cell anemia distr.

BLATBLAT
BLAT = BBLAT = Blast-last-LLike ike AAlignment lignment TTool ool BLAT is designed to find similarity of BLAT is designed to find similarity of >95% on >95% on
DNADNA, , >80% for protein>80% for protein Rapid search by indexing entire genomeRapid search by indexing entire genome
Good for:Good for:
1.1. Finding genomic coordinates of cDNAFinding genomic coordinates of cDNA
2.2. Determining exons/intronsDetermining exons/introns
3.3. Finding human (or chimp, dog, cow…) Finding human (or chimp, dog, cow…) homologs of another vertebrate sequencehomologs of another vertebrate sequence

BLAT on UCSC Genome BrowserBLAT on UCSC Genome Browser
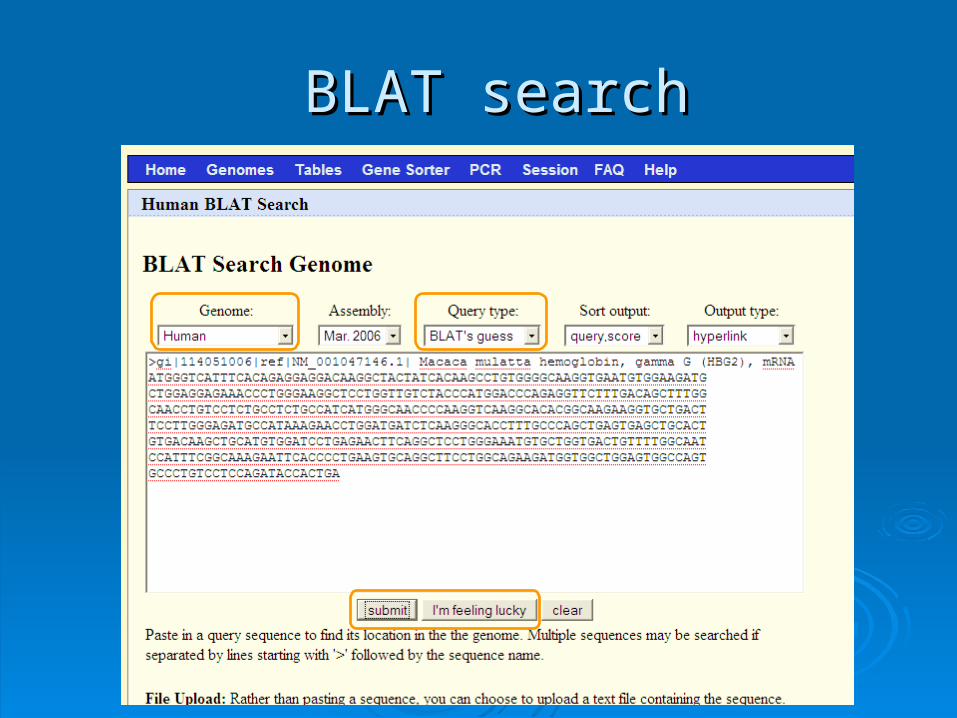
BLAT searchBLAT search

BLAT ResultsBLAT Results

BLAT ResultsBLAT Results
Match
Non-Match(mismatch/indel)
Indel boundaries
query
hit