pteridine dependent hydroxylases as autoantigens in autoimmune polyendocrine syndrome type 1
TRANSCRIPT

Comprehensive Summaries of Uppsala Dissertationsfrom the Faculty of Medicine 999
_____________________________ _____________________________
Pteridine Dependent Hydroxylases asAutoantigens in Autoimmune
Polyendocrine Syndrome Type 1
BY
OLOV EKWALL
ACTA UNIVERSITATIS UPSALIENSISUPPSALA 2001

Dissertation for the Degree of Doctor of Philosophy (Faculty of Medicine) in Medicine presented
at Uppsala University in 2001
ABSTRACT
Ekwall, O. 2001. Pteridine dependent hydroxylases as autoantigens in autoimmune polyendocrinesyndrome type I. Acta Universitatis Upsaliensis. Comprehensive Summaries of Uppsala
Dissertations from the Faculty of Medicine 999. 81pp. Uppsala. ISBN 91-554-4941-7.
Autoimmune polyendocrine syndrome type I (APS) is a monogenous, recessively inheriteddisease characterised by endocrine and non-endocrine autoimmune manifestations. One fifth of
APS I patients suffer from periodic intestinal dysfunction with varying degrees of malabsorbtion,
steatorrhea and constipation. Alopecia areata is found in one third of APS I patients. By
immunoscreening human cDNA libraries derived from normal human duodenum and scalp with
APS I sera, we identified tryptophan hydroxylase (TPH) as an intestinal autoantigen and tyrosinehydroxylase (TH) as a dermal autoantigen. Forty-eight percent (38/80) of the APS I patients had
TPH antibodies (Ab) and 44% (41/94) showed TH immunoreactivity. No reactivity against TPH
or TH was seen in healthy controls. TPH-Abs showed a statistically significant correlation with
gastrointestinal dysfunction (p<0.0001) and TH-Abs were significantly correlated to alopecia
(p=0.02). TPH-Ab positive APS I sera specifically immunostained TPH containing
enterochromaffin cells in normal duodenal mucosa. In affected mucosa a depletion of the TPHcontaining EC cells was seen. In enzyme inhibition experiments TPH and TH activity in vitro was
reduced by adding APS I sera. TPH and TH together with phenylalanine hydroxylase (PAH)
constitute the group of pteridine dependent hydroxylases. These are highly homologous enzymes
involved in the biosynthesis of neurotransmitters. Immunoprecipitation of PAH expressed in vitro
showed that 27% (25/94) of APS I patients had antibodies reacting with PAH, but no associationswith clinical manifestations was observed. An immunocompetition assay showed that the PAH
reactivity reflects a cross-reactivity with TPH.
In conclusion, we have identified TPH and TH as intestinal and dermal autoantigens in
APS I, coupled to gastrointestinal dysfunction and alopecia. We have also demonstrated
immunoreactivity against PAH in APS I patient sera reflecting a cross-reactivity with TPH.
Key words: APS I, alopecia, autoantigen, cDNA, malabsorbtion, phenylalanine hydroxylase,
pteridine, tryptophan hydroxylase, tyrosine hydroxylase.
Olov Ekwall, Department of Medical Sciences, University Hospital, SE-751 85 Uppsala, Sweden,
Olov Ekwall 2001
ISSN 0282-7476
ISBN 91-554-4941-7
Printed in Sweden by Eklundshofs Grafiska AB, Uppsala 2001

I n m e m o r y o f B j ö r n E k w a l l

P A P E R S
This thesis is based on the following papers, which will be referred to in the text by
their roman numerals:
I. Ekwall O, Hedstrand H, Grimelius L, Haavik J, Perheentupa J, Gustafsson J,
Husebye E, Kämpe O and Rorsman F. (1998) Identification of tryptophanhydroxylase as an intestinal autoantigen. Lancet, 1998; 352(9124): 279-283.
II. Ekwall O, Sjöberg K, Mirakian R, Rorsman F and Kämpe O. (1999)Tryptophan hydroxylase autoantibodies and intestinal disease in autoimmune
polyendocrine syndrome type 1. Lancet 1999; 354(9178): 568.
III. Hedstrand H, Ekwall O, Haavik J, Landgren E, Betterle C, Perheentupa J,Gustafsson J, Husebye E, Rorsman F and Kämpe O. (2000) Identification of
Tyrosine Hydroxylase as an Autoantigen in Autoimmune Polyendocrine
Syndrome Type I. Biochem Biophys Res Commun 2000; 267(1): 456-461.
IV. Ekwall O, Hedstrand H, Haavik J, Perheentupa J, Betterle C, Gustafsson J,Husebye E, Rorsman F and Kämpe O. (2000) Pteridine dependent
hydroxylases as autoantigens in autoimmune polyendocrine syndrome type I.J Clin Endocrinol Metab 2000; 85(8): 2944-2950.
Reprints were made with the permission of the publishers.

C O N T E N T S
ABBREVIATIONS 7
INTRODUCTION
General immunology 9
The innate immune system 9
The adaptive immune system 10
B-lymphocytes 12
Antibodies 13
T-lymphocytes 14
The T-cell receptor 15
The major histocompability complex 18
MHC class I 19
MHC class II 21
Tolerance 22
The “Danger” hypothesis 23
Autoimmunity 24
Genetic factors 25
Molecular mimicry 26
Aberrant expression of antigen 27
Defect immunoregulation through cytokines 28
Target organ defects 28
Superantigens 28
The TH1/TH2 paradigm 29
Apoptosis 29
Autoantigens, autoantibodies and autoreactive T-cells 30
Autoimmune polyendocrine syndrome type I 32
Mucocutaneous candidiasis 34

Hypoparathyroidism 35
Addison’s disease 35
Hypogonadism 36
Alopecia 36
Intestinal dysfunction 37
Vitiligo 37
Autoimmune hepatitis 38
Pernicious anaemia 38
Insulin-dependent diabetes mellitus 39
Minor components 39
Genetics 40
Autoimmune polyendocrine syndrome type II 42
Pteridine dependent hydroxylases 44
Structure 44
Function and tissue distribution 45
TPH, PAH and TH in disease 45
CURRENT INVESTIGATION
Results 47
The identification of TPH as an autoantigen in APS I (I) 47
TPH antibodies in other autoimmune intestinal diseases (II) 49
The identification of TH as an autoantigen in APS I (III) 49
Pteridine dependent hydroxylases as autoantigens in APS I (IV) 50
Discussion 52
SUMMARY 60
FUTURE PERSPECTIVES 61
ACKNOWLEDGEMENTS 63
REFERENCES 66

A B B R E V I A T I O N S
AADC Aromatic L-amino acid decarboxylase
Ab AntibodyAChR Acetylcholine receptor
AIRE Autoimmune regulator (human)Aire Autoimmune regulator (mouse)
APC Antigen presenting cellAPECED Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy
APS I Autoimmune polyendocrine syndrome type I
BH4 Tetrahydrobiopterin
CAH Chronic active hepatitisCD Cluster of differentiation
cDNA Complementary deoxyribonucleic acidCKK Cholecystokinin
CNS Central nervous system
EAE Experimental autoimmune encephalomyelitisEC Enterochromaffin
ER Endoplasmatic reticulumGAD Glutamic acid decarboxylase
HLA Human leucocyte antigenIBD Inflammatory bowel disease
IDDM Insulin dependent diabetes mellitus
IFNγ Interferon γ
Ig Immunoglobulin
ITT In vitro transcription and translationMG Myasthenia gravis
MHC Major histocompability complexPAH Phenylalanine hydroxylase
PCR Polymerase chain reaction
PKU PhenylketonureaRT-PCR Reverse transcriptase - polymerase chain reaction
SCC Side chain cleavage enzymeSLE Systemic lupus erythematosus
TCR T cell receptorTH Tyrosine hydroxylase
TPH Tryptophan hydroxylase

Olov Ekwall
9
I N T R O D U C T I O N
General immunology
The human immune system has evolved to ensure a dynamic defence against a wide
range of invading organisms. The first obstacle an invading pathogen must overcome
is a surface barrier e.g. keratinized skin or an enzyme coated mucosa. Pathogens able
to pass this first barrier are then met by two principally different but co-working
systems: the innate and adaptive immune systems. The innate immune system is
characterised by a similar response to re-invasion by the same type of invader,
irrespective of the number of previous encounters. On the other hand, the adaptive
immune system is characterised by its ability to strengthen the defence towards re-
invasion by the same type of invader.
The innate immune system
The components of the innate immune system are immunologically active cells,
complement, acute phase proteins and cytokines. The cells involved are phagocytic
cells including neutrophils, monocytes and macrophages; cells that release
inflammatory mediators including basophils, mast cells and eosinophils; and natural
killer cells. The innate immune response is rapid and does not require cell proliferation
before the intruder is attacked. The main limitation of the innate immune system is the
lack of specificity. The recognising receptors used are coded by germ line genes, and
the repertoire is limited to the hundreds, in contrast to 1014 – 1018 somatically
recombined receptors used in the adaptive immune system (110). The structures
recognised by the innate immune system are called “pathogen-associated molecular
patterns” and the receptors are referred to as “pattern-recognition receptors”. The

Pteridine dependent hydroxylases as autoantigens in APS I
10
general features of the pathogen-associated patterns are that they are specific for
microbial pathogens, often crucial for the survival of the pathogen, and shared by
whole classes of pathogens. Examples are lipopolysaccarides, peptidoglycans and
bacteria-specific DNA. The receptors are either secreted, endocytic or signalling.
Secreted circulating receptors bind to the surface of pathogens and mark them for
phagocytosis or destruction by the complement system. Endocytic receptors are
expressed on phagocytes and direct the recognised pathogen to lysosomes where it is
degraded. Signalling receptors induce the production of inflammatory mediators such
as inflammatory cytokines, when they encounter their counterpart. In contrast to the
adaptive immune system, the innate immune system cannot recognise self-structures.
This feature of the innate system is used as a control mechanism in the initiation of an
adaptive immune response. The initial activation of a T-cell requires two signals: the
recognition by the T-cell receptor of a peptide-MHC complex, and a co-stimulatory
signal, such as B7.1 or B7.2, controlled by the innate immune system. In this way the
system ensures that a peptide can activate a T-cell response only if it is derived from a
pathogen recognised by the innate immune system (111).
The adaptive immune system
The basis for the adaptive immune system is clonal proliferation of T-, and B-cells,
bearing receptors specific for the triggering antigens. A refined system of somatic gene
rearrangements allows the adaptive system to generate approximately 1015 different
receptors, each one specific to one unique epitope. T-cell receptors are expressed on T-
cells and recognise epitopes presented in complex with MHC. B-cells produce
antibodies when B-cell receptors are activated, and antibodies are the soluble forms of
B-cell receptors. Upon recognition of the specific epitope, the mature T-, or B-cell,
undergoes clonal proliferation leading to direct cell-mediated destruction,

Olov Ekwall
11
Figure 1. Overview of lymphocyte activation. T-cell receptors recognise processed
peptides presented by MHC class I or class II. A cytotoxic T lymphocyte (CTL) express
CD8, which by binding to a constant region of MHC class I restricts the CTL to
interact with MHC class I expressing cells, mainly presenting intracellular proteins of
viral, or endogenous, origin. An activated CTL kills the infected cell by inducing lysis.
A T-helper (TH) cell is in the same way restricted, by CD4 expression, to activation by
preferrably extracellular antigens presented by MHC class II. In addition to the
antigen presentation, the TH cell also requires a co-stimulatory signal, i.e. binding of
B7 to CD28, to be activated. The TH cell can be a TH1 or a TH2 cell. Activated TH1
cells produce IFNγ and IL-2, which activate CTLs or macrophages. Activated TH2
cells secrete IL-4, 5 and 6 which activate B-cells. B-cells recognise antigens directly
through membrane bound receptors and also need a second signal from an TH cell to
be activated and proliferate into antibody producing plasma cells or memory B cells
cells (From: Delves and Riott, NEJM, 2000; 343(2); 108-17).

Pteridine dependent hydroxylases as autoantigens in APS I
12
complement mediated lysis, neutralisation or phagocytosis through opsonisation by
antibodies (Figure 1) (42, 43).
B-lymphocytes
B-cells are developed throughout human life from stem cells, initially in the fetal liver,
and later in the bone marrow. During differentiation the cells go through specific
developmental stages in which they, through somatic recombination, rearrange the
variable regions of the heavy and light chain of the B-cell receptor or antibody (VDJ
rearrangement). The VDJ rearrangement is mediated by recombinases coded by the
recombination-activating genes (RAG 1 and RAG 2) and is estimated to result in the
generation of about 1015 unique variable regions (2, 169). These are checkpoints that
ensure that genetic rearrangements are no longer possible after heavy and light chain
rearrangements are completed. The end products of this development are immature B-
cells leaving the bone marrow, each one expressing a unique IgM on its surface.
Immature B-cells that bind antigens to their receptors undergo clonal deletion to
prevent the occurrence of mature and activated B-cells able of producing antibodies
against self-structures (39). The immature B-cells enter the circulation and migrate to
the secondary lymphoid tissues – mainly the spleen and the lymph nodes, where they
complete their maturation. Mature cells recirculate in the periphery and the fate of
each cell is dependent upon encountering its specific antigen. When an antigen binds
to a membrane bound IgM, the binding of the antigen sends a direct signal to the
interior of the B-cell, the antigen is then internalised and presented in a complex with
MHC class II to a CD4 positive T-cell (80). The direct signal from the receptor, and
signalling from the T-cell through cytokines, are often both required to activate the B-
cell (133). The B-cell then proliferates to become antibody-producing plasma cells,
that are predominantly located in the bone marrow or on mucosal surfaces, or as long

Olov Ekwall
13
lived memory B-cells that are mainly found in the spleen and lymph nodes. In the
proliferative stage, activated B-cells further diversify the antibody specificity by
somatic hypermutation in that cells producing antibodies with the highest affinity to
the antigen are positively selected (80).
Antibodies
Antibodies are the soluble form of the B-cell receptors. An antibody consists of two
heavy chains and two light chains held together by disulphide bonds (46). Both heavy
and light chains have hypervariable regions in their amino termini and constant
regions in their carboxy termini. The hypervariable regions from one light chain and
one heavy chain together form an antigen recognition site, thus there are two antigen
recognition sites on each antibody. All antibodies or B-cell receptors produced by a
given B-cell bear the same specificity through allelic and isotypic exclusion. There are
two types of light chains, namely λ and κ. There are five different classes of heavy
chains with different heavy-chain constant (C) domains, and the constant domains
categorise the 5 major isotypes of antibodies. The C domain can be µ, δ, γ, ε or α,
giving rise to IgM, IgD, IgG, IgE and IgA, respectively. The IgGs can be further
divided into four subtypes: IgG1, IgG2, IgG3 and IgG4, and IgAs can be either IgA1
or IgA2. The isotype switching is accomplished through alternative splicing and DNA
rearrangements (158). General characteristics of the different isotypes are summarised
in table 1. IgM is mainly produced before somatic hypermutation has occurred and
therefore has a relatively low affinity. It can rapidly be secreted into the blood as a
pentamer and acts as an effective activator of the complement system. IgD has an
obscure function in the immune system. It is present on the surface of B-cells and may
have a role in the development and/or activation of the B-cell. Very low levels of IgD
can also be detected in the plasma, but the significance of this is unknown. IgG is the

Pteridine dependent hydroxylases as autoantigens in APS I
14
predominant antibody isotype found in the circulation. It is an efficient opsonisator
and activator of complement. It can also pass the placental barrier and transmit
immunological properties to the foetus before it has started its own antibody
production. IgE is primarily found beneath the skin and mucosa where it acts as an
activator of mast cells which induce a fast and powerful immune response, mainly
through the release of histamine. IgA has the ability to associate with a J chain and
form dimers that can pass through epithelial surfaces in the gut, bronchi, mammary
glands etc. The secreted IgAs can inhibit the attachment of infectious agents to the
epithelium and form a line of defence against pathogens from the outside (80).
Function IgM IgD IgG1 IgG2 IgG3 IgG4 IgA IgE
Neutralisation + - ++ ++ ++ ++ ++ -
Opsonisation - - +++ ++/- ++ + + -
Activation of NK cells - - ++ - ++ - - -
Activation of mast cells - - + - + - - +++
Complement activation +++ - ++ + +++ - + -
Distribution
Transport across epithelium + - - - - - +++ -
Transport across placenta - - +++ + ++ +/- - -
Diff. into extravascular sites +/- - +++ +++ +++ +++ ++ +
Mean serum level (mg/ml) 1.5 0.04 9 3 1 0.5 2.1 3x10-5
Table 1. Characteristics of antibody isotypes and IgG subtypes.
T-lymphocytes
Immature T-cells migrate from the bone marrow to the thymus where they step-by-
step differentiate into mature cells. These mature cells are efficient in recognising and
reacting against non-self peptides, but also avoid attacking self structures. In this

Olov Ekwall
15
intriguing process, referred to as positive and negative selection of T-cells, the small
fraction of cells that survive are saved from apoptosis twice. First they are saved by
their T-cell receptor’s ability to recognise self-MHC, and then by the same receptor’s
inability to recognise self-peptides presented in a complex with MHC (Figure 2).
The T-cell receptor
Membrane bound receptors are expressed on the surface of T-cells to ensure the
specificity of the T-cell response. The T-cell receptor belongs to the immunoglobulin
superfamily of receptor molecules and shares many structural characteristics with B-
cell receptors, examples include subunits divided into variable and constant regions,
and genetic rearrangements responsible for antigenic variability (57). There are,
however, two major differences. Immunoglobulins recognise native antigens in
extracellular spaces while T-cell receptors only recognise processed peptides presented
in complex with MHC on cell surfaces. T-cell receptors only exist in a membrane
bound form while immunoglobulins can either be secreted antibodies or membrane
bound B-cell receptors.
The predominant T-cell receptor is an heterodimer consisting of two transmembrane
glycoprotein chains, α and β, linked by a disulphide bond. Although there are reports
of T-cells that, through incomplete allelic exclusion, express T-cell receptors with
more than one specificity (129), in principle all T-cell receptors on a given cell
recognise the same peptide-MHC complex. An alternative γ/δ-T-cell receptor is
expressed on a minority of T-cells found mainly in epithelial tissue in the epidermis
and small intestine. The γ/δ-receptor differs from the α/β-receptor in that it seems to
be able to recognise an antigen directly without the presence of a MHC molecule. The
physiological role of these γ/δ-T-cells is still unclear (24).

Pteridine dependent hydroxylases as autoantigens in APS I
16
In the same manner as B-cell receptors, the α-, and β-chains of the T-cell receptor are
coded by sets of genes which, during T-cell differentiation in the thymus, are
somatically recombined to form functional genes. The variable region of α-chain is
generated by ~70 Vα-segments rearranged to ~60 Jα-segments. The β-chain gene is a
result of the rearrangement of ~50 Vβ-, 2 Dβ-, and 13 Jβ-segments (42). The
variability is highest in the CDR3-region of the T-cell receptor, forming the centre of
the antigen binding groove, responsible for peptide recognition, and is lower in the
flanking MHC recognising parts (56). In contrast to immunoglobulins, T-cell receptors
do not undergo somatic hypermutation. This lowers the risk that T-cells, having passed
negative selection, mutate into self-reacting cells. This may also be functional, in the
sense that T-cell receptors must retain their ability to recognise MHC to be able to
stimulate an immune response.
The function of a T-cell is not only determined by the nature of the T-cell receptor
expressed, but also by the expression of CD4 or CD8 co-receptor molecules. A mature
T-cell is either expressing CD4 or CD8 associated with the T-cell receptor on the cell
surface. CD4 and CD8 exclusively bind to invariable parts of MHC class II and I,
respectively. The expression of CD4 or CD8 thus restricts the T-cell to interact with
peptides presented in a complex with either MHC class I or class II. Whether a T-cell
should express CD4 or CD8 is determined at the end of the T-cell differentiation in the
thymus (184).

Olov Ekwall
17
Figure 2. Positive and negative selection in the thymus. Immature, CD4+CD8+
T cells are predestined to apoptosis, and saved if they recognise MHC presented by
cortical epithelial cells. During this “positive selection”, 95% of lymphocytes are
eliminated. The surviving cells are challenged, in the medulla, by the presentation of
self peptides by dendritic cells, or macrophages. T cells that bind self peptides with too
high affinity are eliminated in this “negative selection”. The remaining fraction of
cells are exported to the periphery as CD4 or CD8 positive T cells (From: Delves and
Riott, NEJM, 2000; 343(1); 37-49).
Progenitor T-cells leave the bone marrow and enter the thymus at the edge of the
cortex as “double-negative” cells, lacking both CD 4 and CD 8 (CD 4- 8-) and a
rearranged T-cell receptor on the cell surface. The differentiation is initiated by the
rearrangement of the β-chain gene. When the β-chain is rearranged and expressed on

Pteridine dependent hydroxylases as autoantigens in APS I
18
the surface, the cells becomes “double positive” (TCRβ CD4+ CD 8+), and the
rearrangement of the α-chain is started. When the complete T-cell receptor is
expressed on “double positive” cells (TCRαβ CD4+ CD 8+), the cells are destined to
apoptosis if they are not saved in the process of positive selection, by the binding of
the T-cell receptor to MHC expressed on cortical epithelial cells (173). Ninety-five
percent of pre T-cells die in the thymus at this stage. Dendritic cells and macrophages
in the medulla then challenge the remaining cells for self-antigens bound to MHC. The
cells with T-cell receptors with high affinity to these self-antigens are directed towards
apoptosis (124). The remaining small fraction of cells then, depending on the
preference for MHC class I or II, cease to express either CD4 or CD8. The final result,
after approximately three weeks of development in the thymus, is the export to the
periphery, of single positive CD 4 or CD 8 expressing cells. These cells have a
rearranged T-cell receptor able to recognise MHC class I or II, but unable to interact
with self peptides in complex with MHC (Figure 2).
The major histocompability complex
All nucleated cells in the body express the major histocompability complex (MHC)
antigens on their surface. MHC antigens present processed pathogen-derived, or
endogenous, peptides for T-cells (87, 88). There are two classes of MHC: class I and
class II. MHC class I present peptides for CD 8 positive T-cells and MHC class II
present peptides for CD 4 positive T-cells. MHC class I consists of two subunits, one
membrane bound α-chain with three domains, α1, α2 and α3, and β2-microglobulin.
MHC class II molecules have one α, and one β chain, each with two domains called
α1/α2 and β1/β2, respectively. Human MHC antigens are also called human leukocyte
antigens (HLA). All human MHC, with the exception of β2-microglobulin, are coded

Olov Ekwall
19
by genes clustered on chromosome 6. A number of other proteins engaged in
processing and presentation of peptides are also encoded by genes in the MHC region.
HLA is polygenic and polymorhpic in that there are multiple genes for each HLA
locus, and there are up to as many as 200 possible alleles for each locus. Three genes
encode MHC class I (A, B and C), and four sets of genes encode MHC class II (DP,
DQ, DR1 and DR2). In addition, most individuals are heterozygous at HLA loci,
because the alleles on both chromosomes are seldom identical. This results in a total of
six different class I molecules and eight different class II molecules expressed in one
individual. The diversity is even higher if class II α and β chains from different
chromosomes are taken into account. The combination of HLA alleles expressed is
called the HLA haplotype. The HLA haplotype determines the range of peptides that
can be presented in complex with MHC and this is probably the basis for the
association between HLA haplotype and increased risk for different autoimmune
diseases (142).
MHC class I
MHC class I is expressed on all nucleated cells, and presents peptides generated within
the cell (Figure 3) (87). Endogenous or viral proteins are degraded in proteasomes and
are loaded into MHC class I in the endoplasmatic reticulum by two proteins:
“Transporters associated with Antigen Processing” -1 and –2 (TAP-1 and TAP-2). The
MHC class I-peptide complex is then transported through the Golgi apparatus to the
cell surface and presented for CD 8 positive T-cells (60). The α1 and α2 domain of the
α-chain form a peptide-binding groove where the peptide is presented. The groove is
closed at the ends restricting the length of the presented peptide to 8 – 10 amino acids
(106). Different allelic variants of MHC class I have different preferences to the
peptide bound in the cleft, and two or three residues in the peptide sequence are crucial

Pteridine dependent hydroxylases as autoantigens in APS I
20
Figure 3. Antigen processing. The upper part of the figure illustrates the processing of
antigens presented by MHC class I. Viral or endogenous proteins are degraded in
proteasomes into peptides. Peptides are transported to ER where they are loaded into
MHC class I, and the complexes are transported through the Golgi apparatus to the
surface of the cell. The lower part shows the processing of antigens presented by MHC
class II. Extracellular proteins enter the cell through endocytosis, or phagocytosis,
into early endosomes (a). MHC class II are synthesised in ER and transported through
Golgi into primary lysosomes (b), which fuse with endosomes and MHC class II
compartments (c) are formed. In MHC class II compartments the proteins are
degraded, peptides are loaded into MHC class II, and the complexes are exported to
the surface (From: Klein and Sato, NEJM, 2000; 343(10); 702-709).
for the binding. These so called anchor residues are similar in all peptides bound to a
given MHC class I molecule. The MHC class I-peptide complex is finally presented to
CD 8 positive cytotoxic T-cells resulting in cell death through the release of cytotoxic
effector proteins e.g. pore forming perforin and apoptosis inducing gramzymes from

Olov Ekwall
21
the cytotoxic T-cell. The mechanism is primarily designed to serve as a defence
against viruses and intracellular bacteria.
MHC class II
MHC class II is expressed on a subgroup of immune cells including B-cells, activated
T-cells, macrophages, dendritic cells and thymic epithelial cells. Other cells have also
been found to present MHC class II in the presence of interferon-γ (87). While class I
molecules present peptides derived from cytosolic proteins, MHC class II present
peptides from extracellular pathogens, degraded in endocytic vesicles (Figure 3).
MHC class II chains are synthesised separately in the endoplasmatic reticulum where
they are brought together and stabilised by the invariant chain protein, which binds the
peptide binding groove and thereby blocks premature peptide binding. The MHC
class II-invariant chain complex is transported in vesicles that eventually fuse with
endocytic vesicles, containing degraded extracellular proteins, where the invariant
chain is released and a peptide is loaded into the groove (26). The peptide binding cleft
of MHC class II shares many features with the corresponding cleft on MHC class I,
but one important difference is that the cleft is open in its ends, allowing longer
peptides to be presented. In addition, the MHC class II groove has pockets where
anchor residues determine whether a peptide is allowed to be presented by a specific
allelic variant of MHC class II (28). The result of the presentation of the MHC class I-
peptide complex for CD 4 positive T-cells is dependent on whether the T-cell is an
TH1 or TH2 cell (117). Mainly through the secretion of interferon-γ, IL-2, TNF-α and
TNF-β, TH 1 cells activate macrophages to destroy intracellular micro-organisms,
initiate T-cell mediated cytotoxicity, and activate B-cells to produce opsonizing
antibodies. The TH1 response is classically demonstrated by the delayed type

Pteridine dependent hydroxylases as autoantigens in APS I
22
hypersensitivity reaction. Activated TH2 produce IL-4, IL-5, IL-10 and IL-13, which
in turn, activate B-cells to produce immunoglobulins of different isotypes (145). TH2
cells also activate mast cells and eosinophils. TH1 and TH2 cells are thought to arise
from a common precursor. The course of differentiation is determined by the genetic
background, the physical form of the immunogen, the type of adjuvant, the dose of
antigen and the co-stimulatory factors present at the time of antigen presentation (154).
Tolerance
The combination of a potent immune system with an ingenious capability to generate
an enormous range of B- and T-cell reactivity along with thousands of self peptides
displayed requires the immune system to have a number of mechanisms to prevent
reaction with self structures. Central tolerance develops in the fetus and is based on the
negative selection of developing T-, or B-cells in the thymus or bone marrow, where
cells reacting with self antigens undergo apoptosis (113). There is a need for a second
line of defence against self reactivity. This is called the peripheral tolerance and it is
needed as cells can escape central tolerance mechanisms. These cells are reactive to
self antigens that are of low affinity, or expressed at low concentration, or not at all in
the thymus or bone marrow. Peripheral tolerance is developed postnatally and the
general principles are the need for dual signals to activate T- and B-cells, and low or
absense of exposure to the antigens (172). B-cells require co-stimulation by T-helper
cells to become activated and thus rely on T-cell tolerance, or a “hole” in T-cell
reactivity, for their own tolerance. An autoreactive B-cell that does not receive a co-
stimulatory signal from a TH-cell becomes anergic. There are also Fas ligand
expressing T-cells with specificity for self peptides that induce apoptosis in self
reactive T-, or B-cells (172). Ignorance of self antigens by autoreactive T-cells can
occur if:

Olov Ekwall
23
1. The antigen is anatomically sequestered behind an endothelial barrier e.g. the
blood-brain barrier.
2. The antigen is presented in amounts too low to be detected.
3. The antigen is presented on cells with few or no MHC molecules.
T-cells can also become anergic if the antigen is presented by a non-professional APC
lacking the ability to produce a co-stimulatory signal e.g. B7.1 or B7.2. The central
and peripheral mechanisms of tolerance are believed to be our main protection against
autoimmune disease (103).
The “Danger” hypothesis
An alternative model of viewing immune regulation and initiation of immune
responses is called the danger hypothesis (7, 108). This model recognises the deletion
of self reactive lymphocytes in the thymus, but does not see the self/non-self
discrimination as the on/off-button. Instead it stresses the importance of APCs in the
periphery as gatekeepers in the immune system. Autoreactive T-cells need a second
signal from an activated APC to be activated. The APC, according to this model,
becomes activated by endogenous alarm signals if the tissue is in danger. These alarm
signals can be of two principal types: “pre-packaged” as the extracellular exposition of
intracellular cell components e.g. DNA, RNA, mitochondria, or inducible as e.g. heat
shock proteins and IFNα. The type of alarm signal determines which immunological
effector response a given stimuli will provoke.

Pteridine dependent hydroxylases as autoantigens in APS I
24
Autoimmunity
In a normal physiological state a number of autoreactive T-, and B-cells circulate in
the body without causing disease. Transient autoimmune reactions are also common
especially in connection with infectious disease or trauma. Autoimmune diseases arise
when the immune system turns itself against self structures in a sustained and
uncontrolled way, resulting in tissue damage or direct pathogenic action by
autoantibodies. The classical definition of an autoimmune disease, formulated in 1957
(181), includes four criteria:
1. The existence of an autoantibody or cell mediated immunity.
2. The identification of the corresponding antigen.
3. The induction of disease in an experimental animal by immunisation with the
antigen.
4. The transfer of disease to a healthy individual by transfer of T-cells, B-cells or
autoantibodies.
Revised criteria have been formulated, but in principle these original criteria are still
valid (149). Autoimmune diseases are common, affecting around 3 percent of the
population, women are more frequently affected (3:1 sex ratio) (79). The autoimmune
diseases can be divided into three classes: systemic disease e.g. systemic lupus
erythematosus (SLE), organ specific destructive disease e.g. insulin-dependent
diabetes mellitus (IDDM) and organ specific non-destructive disease e.g. myasthenia
gravis. Systemic autoimmune diseases are characterised by autoreactivity against cell
constituents present in most cells in the body, e.g. antinuclear antibodies, and a
heterogeneous clinical presentation often mediated through immune complexes. Organ

Olov Ekwall
25
specific destructive diseases often display tissue specific intracellular enzymes as
autoantigens, e.g. glutamic acid decarboxylase (GAD) or 21-hydroxylase. The disease
leads to a destruction of target cells which is probably cell mediated. Organ specific
non-destructive diseases show autoantibodies against tissue specific cell surface
receptors, e.g. acetylcholine receptor antibodies, directly driving the
pathophysiological process, and the target tissue is not destroyed. The disease
discussed in this thesis, APS I, is a typical organ specific destructive disease, with a
number of target organs. The mechanisms underlying autoimmune diseases are largely
unknown, but some possible mechanisms and risk factors will be discussed below.
Genetic factors
Many autoimmune diseases, especially organ specific diseases show a familial
clustering. Any given individual affected by one autoimmune disease has an increased
risk of being affected by a second autoimmune disease. Different HLA alleles can be
protective or increase the susceptibility to different autoimmune diseases. For instance
the DRB1*0401 allele increases the risk of getting rheumatoid arthritis sevenfold (96),
and the DQB1*0302 allele increases the risk of getting IDDM. With the exception of
the strong association between HLA-B27 and ankylosing spondylitis, most disease
associations have been found with different class II loci (162). It is difficult to
determine exactly which gene is responsible for a particular disease association as
many genes in the MHC region are tightly linked and combinations of alleles are
needed to increase susceptibility (121). The HLA associations are, in principle,
understandable as different HLA alleles have varying preferences when it comes to
“choosing” what peptides to present for T-cells, both in the development of central
tolerance in the thymus and in the presentation for effector T-cells in the periphery.
Substitution of one single amino acid in the peptide binding groove has been shown to

Pteridine dependent hydroxylases as autoantigens in APS I
26
have major influence on peptide binding (95). Other genes encoded in the MHC
complex that have been shown to increase the risk of autoimmunity are genes
encoding complement components, especially C1, C2 and C4, in SLE (51, 120). Genes
outside the MHC complex are also thought to contribute to the genetic component in
autoimmune disease. Association studies in human IDDM have resulted in a number
of candidate genes, among them the gene encoding insulin (82). The genetic
components in most autoimmune diseases seem, however, to be very complex and
gene polymorphisms coupled to disease are to be seen as permissive rather than
causing the disease. One exception is APS I, which is a monogenous autosomal
recessive disease with total penetrance and as such, can be of value to study
mechanisms underlying autoimmune disease in general.
Molecular mimicry
The basis for molecular mimicry in autoimmune disease is that a foreign infectious
particle, or food constituent, has a linear or conformational epitope that is homologous
with a self antigen. The foreign particle must be structurally similar enough to share
determinants, but dissimilar enough to provoke an immune response. Several
mechanisms for the autoimmune reaction are possible (6, 103):
1. A viral antigen is presented in complex with MHC class I to a cytotoxic T-cell.
The cytotoxic T-cell is then activated, clonally expanded and can attack other
cells presenting a self-peptide that is homologous to the viral epitope.
2. An ignorant or anergic TH cell with self specificity is activated by a cross reactive
foreign peptide presented in complex with class II and co-stimulatory factors by a
professional APC.

Olov Ekwall
27
3. The foreign antigen activates a B-cell which proliferates and cross reacts with a
homologous self antigen.
4 . The foreign antigen activates a B-cell which proliferates, undergoes somatic
hypermutation leading to epitope spreading and cross reacts with a homologous
self antigen through epitope spreading.
Once the tolerance is broken by molecular mimicry, the autoimmune process can
continue without the presence of the foreign particle, as tissue damage leads to a non-
physiological exposition of self peptides. A number of homologies between viral or
bacterial epitopes and human autoantigens have been described (126). For example:
the homology between the AChR α subunit (the predominant autoantigen in
myasthenia gravis) and herpes simplex virus glycoprotein D (152), and the shared
determinants of GAD and the P2-C protein of coxackie B virus (11).
Aberrant expression of antigen
Intracellular self-antigens, or antigens normally presented in immunologically
privileged compartments e.g. brain or testis, can under special circumstances be
accessible to the immune system. Infections, trauma or inflammation can break down
vascular or cellular barriers. In sympathetic ophthalmia, trauma against one eye
releases intraocular proteins and an autoimmune reaction against both eyes follows
(159). Another example is that mice infected with coxsackie virus with a tropism for
pancreatic islets develop a chronic autoimmune inflammation against islet cells which
is not related to molecular mimicry (72). Aberrant MHC class II expression in target
cells normally only expressing MHC class I, e.g. due to stimulation by inflammatory
cytokines, may lead to the presentation of intracellular self peptides for CD 4 positive
TH cells.

Pteridine dependent hydroxylases as autoantigens in APS I
28
Defect immunoregulation through cytokines
The peripheral tolerance relies on a number immunoregulatory mechanisms leading to
ignorance of self epitopes. Potentially self reactive TH-cells are kept ignorant by TS-
cells, IL-10, TGF-β and prostaglandins. Cytokine imbalance has also been shown to
promote autoimmunity. An experimentally deleted or mutated IFNγ gene in pancreatic
β-cells decrease the autoimmune destruction of the β-cells (175), and in IDDM
patients an increased expression of IFNγ has been demonstrated (73). SLE patients, as
well as their healthy relatives, have a defect IL-10 regulation of TS-cells (64).
Target organ defects
Pre-morbid abnormal target organ function or aberrant MHC expression are
considered to be related to an increased risk of developing an autoimmune disorder.
Animal strains with high susceptibility for autoimmune disease, e.g. EAE receptive
animals, have been shown to upregulate MHC class II expression in target organs
(156). Obese strain chickens, prone to autoimmune thyroiditis, have a defective iodine
uptake in their thyroid gland. This has been experimentally shown to be a permissive
trait to develop thyroiditis (160).
Superantigens
Superantigens are proteins produced by pathogens that have the ability to activate
CD 4 positive T-cells by binding both the outer faces of MHC class II and the Vβ
domain of the TCR. A superantigen has a relative specificity for certain Vβ chains,
and is capable of polyclonally activating 2 – 20 % of all T-cells. It has been proposed
that bacterial or viral superantigens may have a role in the development of rheumatoid
arthritis by activating anergic self reactive T-cells (130).

Olov Ekwall
29
The TH1/TH2 paradigm
Studies, mainly those involving different animal models, have shown that organ
specific destructive autoimmune diseases have a predominant TH1 cytokine profile,
while systemic diseases show a bias towards TH2 (146). Manipulation of TH
differentiation has resulted in a changed cause of the disease, e.g. in EAE where
addition of an anti-B7.1 antibody reduced the incidence, while anti-B7.2 increased the
severity of the disease (94). It has been proposed that an imbalance in the TH1/TH2
response may be a prerequisite for autoimmune diseases to evolve. This can open the
way for new therapeutic strategies by modulating the TH1/TH2 balance (146).
Apoptosis
Apoptosis, or programmed cell death, is a mechanism for controlled cell destruction
which, in contrast to necrosis, occurs without inflammatory responses. Apoptosis is
mediated via Fas and its’ ligand, Fas L, in the immune system and has an important
role in the modulation of the immune response. Defect regulation of apoptosis could
have a possible role in autoimmunity by several mechanisms (33), (30):
1. Defect elimination of autoreactive lymphocytes.
2. Aberrant cleavage of intracellular autoantigens can result in the presentation of
new, not tolerated, epitopes.
3. In contrast to apoptosis, defective apoptosis can lead to a bias towards perforin
mediated cell destruction which promotes autoimmunity through an uncontrolled
exposition of intracellular material.

Pteridine dependent hydroxylases as autoantigens in APS I
30
The existence of Fas mutations in patients with autoimmune lymphoproliferative
syndrome (52) supports the role of apoptosis in autoimmune disease. Experimental
support also exists, e.g. the very low incidence of diabetes in perforin-negative NOD
mice (83).
Autoantigens, autoantibodies and autoreactive T-cells
Self structures identified through reactivity with autoantibodies or self reactive T-cells,
associated with autoimmune diseases, are called autoantigens. A large number of B-
cell autoantigens have been identified through immunohistochemistry,
immunoblotting and screening of cDNA libraries. T-cell autoantigens are harder to
identify and it is difficult to verify the association with a disorder in a large patient
sample, mainly due to the MHC restriction of T-cell reactivity. The effector
mechanisms in autoimmune diseases are mediated both through autoantibodies and
through T-cells. They can be classified in analogy with type II, III and IV
hypersensitivity reactions (38). Type II reactions are antibody mediated with cell
surface antigens resulting in phagocytosis or complement mediated lysis, as in
autoimmune haemolytic anaemia (45), receptor mediated stimulation as in Graves’
disease (14) or receptor blockade as in myasthenia gravis (101). Type III reactions are
immune complex mediated with extracellular antigens, matrix derived or soluble, and
can be exemplified by SLE (92). Type IV reactions are T-cell mediated, organ specific
destructive diseases and the antigens proposed are often intracellular as GAD in
IDDM (12) and 21-OH in Addison’s disease (180).
The role of autoantigens in the aetiology of autoimmune diseases, and the value of
their identification, is not obvious. B-cell activation and subsequent autoantibody
production requires activated TH-cells. The specificity of these TH-cells and the

Olov Ekwall
31
specificity of the autoantibody do not necessarily have to be the same, even if spatial
and time factors suggest a shared antigen. In myasthenia gravis, both autoantibody
specificity and T-cell epitopes are predominantly found within the same region of the
α subunit of AChR (140). Insulin and GAD65 are both reported as B-, and T-cell
autoantigens in IDDM (13, 119, 131, 132).
The value of a number of antibodies as disease-specific diagnostic markers is
undisputed. The occurrence of autoantibodies often precedes the clinical onset of the
disease (23), and can thus be used to screen persons at risk of developing disease. The
titres of some autoantibodies, e.g. anti-dsDNA antibodies in SLE (167), are correlated
to disease activity, while others are of less value for monitoring the disease.
Pteridine dependent hydroxylases as autoantigens in APS I
32
Autoimmune polyendocrine syndrome type I
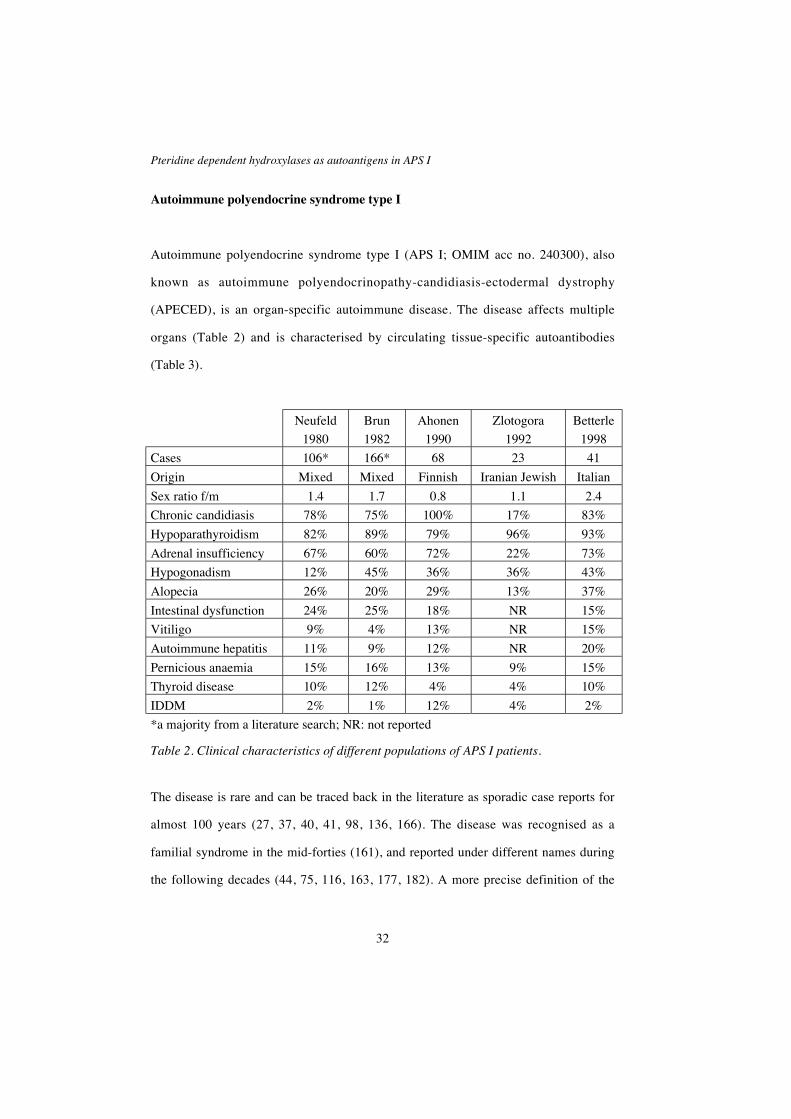
Autoimmune polyendocrine syndrome type I (APS I; OMIM acc no. 240300), also
known as autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy
(APECED), is an organ-specific autoimmune disease. The disease affects multiple
organs (Table 2) and is characterised by circulating tissue-specific autoantibodies
(Table 3).
Neufeld
1980
Brun
1982
Ahonen
1990
Zlotogora
1992
Betterle
1998
Cases 106* 166* 68 23 41
Origin Mixed Mixed Finnish Iranian Jewish Italian
Sex ratio f/m 1.4 1.7 0.8 1.1 2.4
Chronic candidiasis 78% 75% 100% 17% 83%
Hypoparathyroidism 82% 89% 79% 96% 93%
Adrenal insufficiency 67% 60% 72% 22% 73%
Hypogonadism 12% 45% 36% 36% 43%
Alopecia 26% 20% 29% 13% 37%
Intestinal dysfunction 24% 25% 18% NR 15%
Vitiligo 9% 4% 13% NR 15%
Autoimmune hepatitis 11% 9% 12% NR 20%
Pernicious anaemia 15% 16% 13% 9% 15%
Thyroid disease 10% 12% 4% 4% 10%
IDDM 2% 1% 12% 4% 2%
*a majority from a literature search; NR: not reported
Table 2. Clinical characteristics of different populations of APS I patients.
The disease is rare and can be traced back in the literature as sporadic case reports for
almost 100 years (27, 37, 40, 41, 98, 136, 166). The disease was recognised as a
familial syndrome in the mid-forties (161), and reported under different names during
the following decades (44, 75, 116, 163, 177, 182). A more precise definition of the

Olov Ekwall
33
syndrome, based on a large clinical review, was made in 1980 (122). APS I was then
defined as the occurrence of two out of three cardinal symptoms: chronic
mucocutaneous candidiasis, hypoparathyroidism and autoimmune Addison’s disease,
or the occurrence of one of these three symptoms in siblings to APS I patients. APS I
was separated from other polyglandular syndromes such as APS II and APS III. The
wide clinical variation in APS I referring to the number of, type of and age at onset for
different components, was thoroughly described in a large Finnish study of 68 patients
in 1990 (5).
As will be discussed later, APS I is enriched in some genetic isolates e.g. Finland,
Sardinia and among Iranian Jews. The clinical components vary in frequency between
different populations (5, 17, 185). Although the clinical course of the disease is highly
variable, the three main components usually develop in a specific order with
candidiasis occurring before the age of 5, hypoparathyroidism before the age of 10 and
finally Addison’s disease usually before the age of 15 (5, 17). Note that all patients do
not develop all three main components, and that only two out of three are required for
the diagnosis. In general, the earlier the age at onset of the first symptom indicates a
predisposition to developing a higher number of components, and conversely, patients
who have a late first manifestation of the disease are more likely to have fewer
components (5).
In addition to the three main components, APS I patients develop other endocrine and
non-endocrine symptoms in varying frequencies (Table 2). The different components
of APS I will be discussed in detail below.
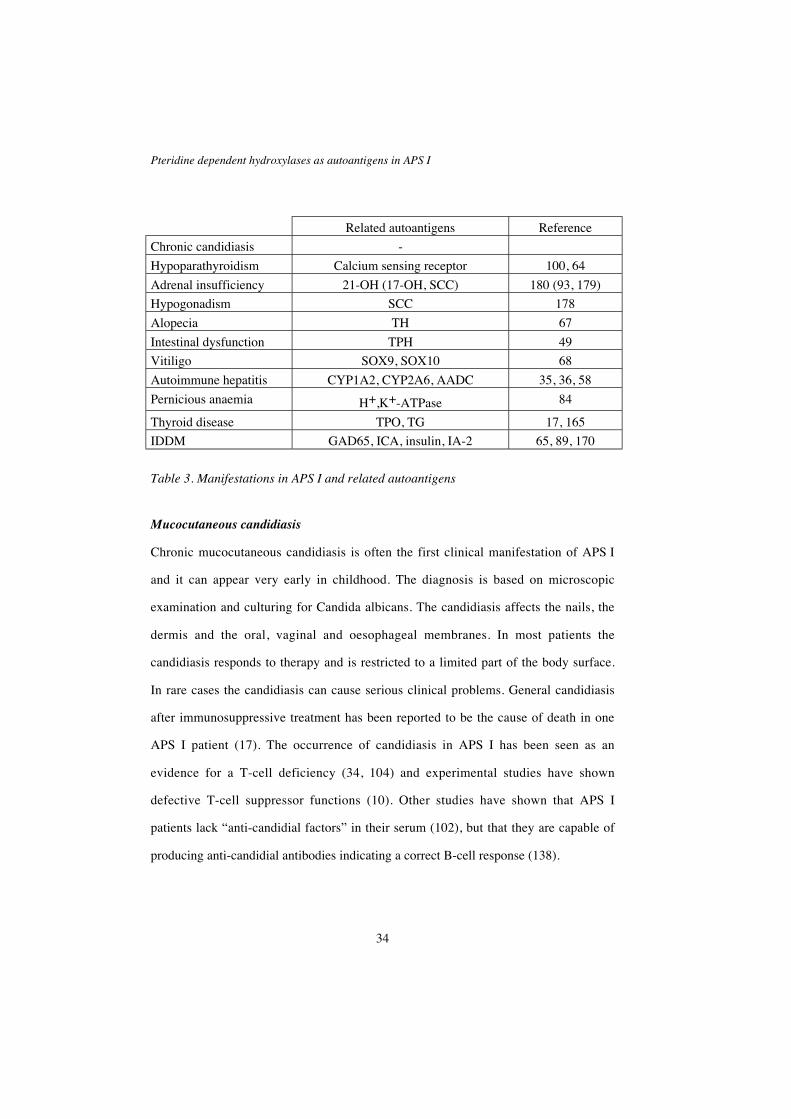
Pteridine dependent hydroxylases as autoantigens in APS I
34
Related autoantigens Reference
Chronic candidiasis -
Hypoparathyroidism Calcium sensing receptor 100, 64
Adrenal insufficiency 21-OH (17-OH, SCC) 180 (93, 179)
Hypogonadism SCC 178
Alopecia TH 67
Intestinal dysfunction TPH 49
Vitiligo SOX9, SOX10 68
Autoimmune hepatitis CYP1A2, CYP2A6, AADC 35, 36, 58
Pernicious anaemia H+,K+-ATPase 84
Thyroid disease TPO, TG 17, 165
IDDM GAD65, ICA, insulin, IA-2 65, 89, 170
Table 3. Manifestations in APS I and related autoantigens
Mucocutaneous candidiasis
Chronic mucocutaneous candidiasis is often the first clinical manifestation of APS I
and it can appear very early in childhood. The diagnosis is based on microscopic
examination and culturing for Candida albicans. The candidiasis affects the nails, the
dermis and the oral, vaginal and oesophageal membranes. In most patients the
candidiasis responds to therapy and is restricted to a limited part of the body surface.
In rare cases the candidiasis can cause serious clinical problems. General candidiasis
after immunosuppressive treatment has been reported to be the cause of death in one
APS I patient (17). The occurrence of candidiasis in APS I has been seen as an
evidence for a T-cell deficiency (34, 104) and experimental studies have shown
defective T-cell suppressor functions (10). Other studies have shown that APS I
patients lack “anti-candidial factors” in their serum (102), but that they are capable of
producing anti-candidial antibodies indicating a correct B-cell response (138).

Olov Ekwall
35
Hypoparathyroidism
Hypoparathyroidism is often the first endocrine component of APS I and usually
appears before the age of 10 (mean 7.5 yr.) (5, 17). Patients with subnormal plasma
calcium levels, supranormal plasma phosphate levels and normal renal function are
diagnosed as having hypoparathyroidism (5). Parathyroid autoantibodies by
immunofluorescence (22), and autoantibodies reacting with the extracellular domain
of the calcium sensing receptor (100) have been reported. These results have not been
confirmed in other studies. On the contrary, a recent study indicates that we are still
lacking a good serologic test for hypoparathyroidism (164).
Addison’s disease
There are a number of causes to Addison’s disease, or adrenal insufficiency.
Historically, and world-wide, tuberculosis is probably the major cause of Addison’s
disease. Other causes are metastatic malignancies, haemorrhage, sarcoidosis or rare
hereditary diseases such as adrenoleucodysdrophy. In the western world, adrenal
insufficiency is most often caused by an autoimmune destruction of the suprarenal
cortex. The term “Addison’s disease” refers to the autoimmune variant of the disease
in this paper. Addison’s disease most often occurs as an isolated disease, but can also
be one component of APS I, or other syndromes. The laboratory diagnosis of
Addison’s disease is defined as: low plasma cortisol, and/or low urinary cortisol,
and/or high plasma ACTH concentrations, and/or defect response to acute and/or
prolonged ACTH stimulation. Autoantibodies against the adrenal cortex in idiopathic
Addison’s disease have been described since the 1950s (8, 21). 21-OH has been
identified as the major autoantigen in Addison’s disease (180). 17α-OH and side-chain
cleavage enzyme (SCC) are presented as an adrenal autoantigen in APS I patients with
Addison’s disease (93) (179). A recent multivariate analysis of the diagnostic values of

Pteridine dependent hydroxylases as autoantigens in APS I
36
different autoantigens in APS I points out 21-OH and SCC as associated with
Addison’s disease in APS I (164). In APS I, the disease has an earlier age at onset
(peak 10-12 yr.) compared to isolated Addison’s disease or Addison’s disease in APS
II (peak 20-30 yr.) (122, 123). Historically, Addison’s disease has been a major cause
of death in APS I, but with modern substitution therapy the clinical management is
satisfactory. As in isolated Addison’s disease, future management will probably
include substitution with androgens to women with Addison’s disease, in addition to
the usual treatment with glucocorticoid and mineralocorticoid supplementation (9, 59).
Hypogonadism
Hypogonadism in APS I shows a female predominance and is mainly diagnosed at
puberty, but can also be detected as secondary amenorrhea before the age of 40. The
diagnosis of ovarian failure is based on amenorrhea, high levels of FSH or LH, and
slow, absent or regressive pubertal development. Testicular failure is diagnosed
through high base-line, or post-GRH stimulatory, levels of FSH or LH, and low
testosterone levels (5). Both 17α-OH and SCC have been proposed as autoantigens
associated with gonadal failure (18, 171, 178). A recent multivariate analysis of ten
different autoantibodies in a large sample of APS I patients shows that SCC alone is
statistically associated with hypogonadism (164).
Alopecia
Alopecia is present in APS I both as alopecia areata and as the more severe form
alopecia totalis. Alopecia areata is defined as a transient patchy hair loss on the scalp.
When the hair loss covers the whole scalp and becomes permanent the condition has
developed into an alopecia totalis. Alopecia totalis is twice as common as alopecia

Olov Ekwall
37
areata in APS I (69). Autoantibodies against hair follicles have been shown, by
indirect immunofluorescence, to be associated with alopecia totalis in APS I (69).
Intestinal dysfunction
Different descriptions and names such as malabsorbtion (5, 17), celiac syndrome (37,
44), constipation (29), steatorrhea and diarrhoea (41, 78, 116) have been used in the
literature to characterise the intestinal dysfunction seen in APS I. This illustrates the
great variations of the clinical manifestations of the gastrointestinal dysfunction seen
in APS I. We define the intestinal dysfunction as periodic steatorrhea, diarrhoea or
severe constipation. The dysfunction is often therapy resistant and results in marked
weight loss and/or growth failure. APS I patients with intestinal dysfunction as their
first manifestation of the disease generally have a high number of disease components
(5). Although the malabsorbtion often is severe no specific shortages are seen. Faecal
fat is often elevated. Exocrine pancreas insufficiency (176) and low levels of
cholecystokinin (71, 77) have been reported. Some reports have described a
covariation between intestinal symptoms and hypocalcemia (5, 71, 135). The intestinal
symptoms have been explained in the literature as being secondary to hypocalcemia
(71), candidial overgrowth (153), exocrine pancreas defect, or intestinal
lymphangiectasia (16). Substitution with pancreatic enzymes, normalisation of serum
calcium (71) and immunosuppressive treatment with cyclosporine (176) or
methylprednisolone (128), has been proposed for treatment.
Vitiligo
Vitiligo is characterised by loss of pigment formation by melanocytes in the skin.
Different patterns of distribution are acro-facial, focal, segmental and generalised
vitiligo. In APS I vitiligo often occurs as symmetrical depigmented patches on the

Pteridine dependent hydroxylases as autoantigens in APS I
38
face, neck, extensor surfaces of hands, wrists and legs and in the axillae. Complement-
fixing autoantibodies have been described as associated with vitiligo in APS I (137).
Recently, the transcription factors SOX 9 and SOX 10, expressed in melanocytes, have
been identified as autoantigens associated with vitiligo in APS I (68).
Autoimmune hepatitis
Autoimmune chronic active hepatitis is probably the most feared component of APS I.
The clinical course is highly variable, ranging from asymptomatic changes of liver
enzymes to fulminant hepatitis with a fatal outcome (5, 112). The histopathologic
picture resembles that of idiopathic autoimmune chronic active hepatitis and markers
of viral hepatitis are not present. Autoantibodies against cytochromes P4501A2,
P4502A6 and aromatic L-amino acid decarboxylase (AADC) have been associated
with autoimmune hepatitis in APS I (35, 36, 58). Treatment is based upon
immunosuppressive therapy in combination with corticosteroids (112). The
determination of liver enzymes should be done with regularity to promote early
detection and intervention.
Pernicious anaemia
Pernicious anaemia is secondary to vitamin B12 malabsorbtion due to a shortage of
intrinsic factor caused by a disturbed parietal cell function, coupled to autoimmune
gastritis. Clinical features are megaloblastic anaemia and achlorhydria. Lymphocytic
infiltrates and autoantibodies directed towards parietal cells have been found (116,
122). H+K+-ATPase and intrinsic factor have been described as autoantigens
associated with autoimmune gastritis (84, 150).

Olov Ekwall
39
Insulin-dependent diabetes mellitus
Insulin-dependent diabetes mellitus (IDDM) shows a great variation in occurrence
between different populations of APS I patients, with frequencies ranging from 1 to 12
percent (5, 17, 122, 143). Autoantibodies against glutamic acid decarboxylase
(GAD65), tyrosine phosphatase IA-2 (IA-2), insulin and/or islet cell antibodies (ICA)
are present in APS I patients with IDDM (65, 89, 170). In a multivariate analysis of
the clinical associations of a panel of autoantigens in APS I, only IA-2 was shown to
have a predictive value (164). AADC which has been cloned from a β-cell cDNA
library as an autoantigen in APS I does not seem to correlate to IDDM (76, 147), but
surprisingly, it is statistically associated with autoimmune hepatitis (58).
Minor components
Ectodermal dystrophy with hypoplastic enamel of the teeth, pitted nail dystrophy and
tympanic membrane dystrophy affects 77, 52 and 33 percent of APS I patients,
respectively (5). Keratoconjunctivitis is seen in 8 - 41% of APS I patients as corneal
opacities, bulbar conjunctival injection and corneal neovascularisation (5, 17, 109).
Autoimmune thyroid disease, mainly Hashimoto’s thyroiditis, has been reported in
2 – 13% of APS I patients (5, 17, 85). Thyroid autoantibodies are present in a high
proportion of APS I patients with and without thyroid disease (17, 165). The
prevalence of acquired asplenia, due to a progressive destruction of the spleen, has not
been systematically examined, but has been described in a high proportion in small
sample of APS I patients (55, 134). Cholelithiasis is also reported as over-represented
in APS I patients (55).

Pteridine dependent hydroxylases as autoantigens in APS I
40

Olov Ekwall
41
identified by positional cloning and named autoimmune regulator (AIRE) (118, 168).
The AIRE gene encodes a 545 aa, 57.5 kDa protein with a conserved nuclear
localisation signal, two plant homeodomain (PHD)-type zinc fingers, four LXXLL
motifs, one SAND and one HSR domain (Figure 4). These features are all markers for
nuclear, DNA-binding proteins suggesting a role for AIRE in transcriptional control.
AIRE has also been shown in vitro to interact with transcriptional coactivators and
activate transcription (19, 139).
AIRE is expressed mainly in thymic epithelial cells and to a lesser degree also in the
spleen, lymph nodes, fetal liver and mononuclear leukocytes (20, 118, 186). Recent
studies in mice also demonstrate Aire expression in a variety of tissues outside the
immune system e.g. kidney, testis, adrenal glands, liver and ovary (66). The sub-
cellular localisation of wild type AIRE has been described mainly as restricted to the
nucleus, but a cytoplasmatic expression has also been described (20, 70, 144). In the
nucleus AIRE is found in dots resembling, but not co-localising with, the nuclear
bodies formed by e.g. promyelotic leukaemia protein (PML), Sp 100 or Sp140.
The function of AIRE is not yet understood. The expression pattern in man and
experimental studies using murine Aire, show that AIRE is likely to be involved in the
thymic selection process and so indicates a role for AIRE in the induction of self-
tolerance (186).
To date, 29 different disease causing mutations of AIRE have been described which
are clustered in four mutational hotspots. Although 5 – 10 % of patients who fulfil the
clinical criteria for APS I lack mutations in AIRE exons, or only have one allele
mutated in the exonic parts, the general agreement is that both AIRE alleles must be

Pteridine dependent hydroxylases as autoantigens in APS I
42
mutated to cause the disease (19). A possible explanation for these APS I patients with
no detectable mutations is probably that mutations do exist in promoter regions or
intronic parts of the AIRE gene which have not been examined by sequencing. In
Finland, Sardinia and among Iranian Jews marked founder effects are seen. 89% of
Finnish APS I chromosomes have the major Finnish mutation, R257X, 92% of
Sardinian APS I chromosomes have the common Sardinian mutation, R139X, and
100% of APS I patients among Iranian Jews are homozygous for a single amino acid
change, Y85C (19, 148). Most mutations are, mainly through frame shifts, resulting in
truncated forms of AIRE, lacking DNA-binding domains or nuclear receptor binding
domains.
In experimental studies of sub-cellular localisation of mutated AIRE variants, an
altered intracellular distribution pattern of the protein was found (19, 144). Almost all
mutations examined, except the common Iranian missense mutation, resulted in a
cytoplasmatic location of the mutated protein. In transcription activation assays
truncated mutated AIRE constructs showed no, or markedly lowered, activation of
transcription. The transcriptional activation ability of the AIRE construct with the
common Iranian mutation was the same as wild type AIRE. The fact that the
experimental behaviour of the Iranian mutation does not differ much from wild type
AIRE may explain the milder clinical phenotype that can be noticed among Iranian
Jewish APS I patients.
Autoimmune polyendocrine syndrome type II
The diagnosis of autoimmune polyendocrine syndrome type II (APS II) requires the
presence of Addison’s disease plus autoimmune thyroid disease and/or insulin-

Olov Ekwall
43
dependent diabetes mellitus (IDDM) (122). Other endocrinopathies and associated
autoimmune diseases are also present, at low frequencies. APS II shows a female
predominance (123, 165), is inherited in a dominant fashion (47), and has been shown
to be associated with HLA-DR3 and -DR4 (105, 125). The age at onset for Addison’s
disease is higher in APS II (peak at age 20 – 30) compared to APS I (peak at age 10 –
12) (123).

Pteridine dependent hydroxylases as autoantigens in APS I
44
Pteridine dependent hydroxylases
Tryptophan hydroxylase (TPH; EC 1.14.16.4), tyrosine hydroxylase (TH; EC
1.14.16.2) and phenylalanine hydroxylase (PAH; 1.14.16.4) together constitute the
enzyme family of tetrahydrobiopterin dependent hydroxylases. The three enzymes are
closely related, share functional and structional characteristics and are thought to have
evolved from a common progenitor gene (63).
Structure
The functional structures of the enzymes are homologous with an N-terminal
regulatory domain, a central catalytic domain and a C-terminal tetramerization
domain. The regulatory domain is specific for each enzyme with a 20% linear
homology between the three enzymes. It determines substrate specificity and thus
reflects the unique properties of each enzyme. The central catalytic domain, containing
the active site, is highly conserved and has an overall homology of around 65%. TPH
is a 230 kDa tetramer consisting of identical subunits, each with a molecular mass of
58 kDa. TH consists of four 55-59 kDa subunits, as four isoforms (TH 1 - 4) due to
alternative splicing, forming tetramers with molecular masses of 204-217 kDa. PAH is
found in an equilibrium between tetrameric and a dimeric forms composed of 50 kDa
subunits (74). The crystal structures for the catalytic domain of TH and complete PAH
have been determined (50, 91). The structures are essentially identical, and the TPH
structure can be predicted, based on the structures of TH and PAH (174). The crystal
structure determinations are not only useful in understanding the regulation of
enzymatic activity, but may also be of great value in determining the possible

Olov Ekwall
45
conformational epitopes involved in autoimmunity directed against these enzymes
(62).
Function and tissue distribution
TPH, TH and PAH are monooxygenases, incorporating one atom of oxygen into the
substrate and reducing the other atom to water, and thereby catalyze the hydroxylation
of different amino acids. Tetrahydrobiopterin (BH4) supplies the two electrons
required. Molecular oxygen and ferrous iron are also needed in the reaction (Figure 7).
The regulatory properties differ in detail between the enzymes, but in general they are
regulated by their substrates, BH4 and phosphorylation of serines (53). TPH and TH
have central roles in the biosynthesis of the neurotransmitters serotonin and dopamine
(Figure 7). TPH catalyzes the hydroxylation of tryptophan into 5-OH tryptophan and is
the rate-limiting enzyme in the synthesis of serotonin. It is expressed in serotonergic
cells in the central nervous system and the intestine (183). TH is the rate limiting
enzyme in the biosynthesis of catecholamines where it converts tyrosine into L-Dopa,
and is mainly found in the adrenal medulla and catecholaminergic neurons throughout
the body. PAH is primarily produced in the liver where it catalyses the conversion of
phenylalanine to tyrosine. This serves two purposes: it provides an endogenous supply
of tyrosine, making tyrosine a non-essential dietary component, and it is also rate
limiting in the catabolism of phenylalanine (74).
TPH, PAH and TH in disease
Mutations in PAH are the most common cause of phenylketonurea (PKU). In PKU,
serum levels of phenylalanine are elevated to toxic levels causing brain damage
leading to mental retardation. A second result of PAH dysfunction is that tyrosine
becomes an essential dietary amino acid. The treatment is based on a diet low in

Pteridine dependent hydroxylases as autoantigens in APS I
46
phenylalanine and supplemented with tyrosine (48). Defects in the production, or
recycling, of BH4 also leads to hyperphenylalaninaemia in combination with TPH and
TH deficiencies. These conditions have a more severe clinical presentation than
isolated PAH dysfunction. TH defects and polymorphisms have been coupled to
bipolar affective disorders, schizophrenia and parkinsonism (61). Hitherto, no disease
couplings have been made with TPH dysfunction, although theoretically TPH defects
could very well be involved in affective disorders, schizophrenia, migraine, drug abuse
and intestinal movement disorders (115).

Olov Ekwall
47
C U R R E N T I N V E S T I G A T I O N
Results
The identification of TPH as an autoantigen in APS I (paper I)
Periodic gastrointestinal dysfunction represents a significant management problem in
APS I, affecting 25-30% of the patients. Before this study, the pathogenesis of the
gastrointestinal dysfunction was largely unknown, but explained in the literature as
being secondary to hypocalcemia (71), candidial overgrowth (153), exocrine pancreas
defect or intestinal lymphangiectasia (16). We undertook this study with a belief that
the intestinal dysfunction, as well as the majority of the other manifestations of APS I,
was of autoimmune origin. The aim of the study was to identify a potential intestinal
autoantigen associated with APS I and possibly other autoimmune intestinal diseases.
A commercially available, human duodenal cDNA library was immunoscreened with
sera from seven APS I patients. A positive clone was identified as tryptophan
hydroxylase (TPH, EC 1.14.16.4) and was used for in vitro transcription/translation
(ITT) followed by immunoprecipitation with sera from 80 APS I patients. Forty-eight
percent (38/80) of the APS I patients had TPH antibodies (Ab), whereas in a large
number of sera from patients with other autoimmune diseases (n=372) and healthy
blood donors (n=70) no reactivity against TPH was detected. A correlation with
clinical symptoms showed that 89% (17/19) of APS I patients with gastrointestinal
dysfunction were TPH-AB positive, compared to 34% (21/61) without (p < 0.0001).
Immunostainings of normal human small intestine with patient sera and specific
antibodies were performed. These showed that TPH-Ab positive APS I sera

Pteridine dependent hydroxylases as autoantigens in APS I
48
specifically immunostain TPH containing enterochromaffin cells in normal duodenal
mucosa. We also observed that 3/8 APS I patient sera stained goblet cells and that 3/8
patient sera reacted with Paneth cells (Figure 5). When immunostaining duodenal
biopsies from APS I patients with gastrointestinal dysfunction and TPH-Abs, a total
absence of enterochromaffin cells was seen. Finally, enzyme inhibition assays
demonstrated that TPH-Ab positive APS I sera almost completely inhibited TPH
activity at a dilution of 1:100.
Figure 5. Immunostainings of normal human duodenum with APS I showing
immunoreactivity with goblet cells (left), EC cells and Paneth cells (right).
In this study we identified TPH as an endogenous intestinal autoantigen in APS I and
established a statistical association between TPH-Ab and gastrointestinal dysfunction.
We also confirmed the reactivity against TPH with enzyme inhibition assays and
immunohistochemical stainings of normal and affected tissues.

Olov Ekwall
49
TPH antibodies in other autoimmune intestinal diseases (paper II)
We reported the identification of TPH as an intestinal autoantigen in APS I in paper I.
The gastrointestinal symptoms in APS I are in some respects related to both the
clinical presentation and the suggested autoimmune background of more common
conditions such as inflammatory bowel disease, irritable bowel syndrome and celiac
disease. Autoimmune enteropathy in children and paraneoplastic pseudoobstruction of
the gut are two rare disaeses with clinical or pathogenic features related to the
intestinal disease in APS I. In paper II, we wanted to find out if TPH-antibodies are
present in these other intestinal diseases with a possible autoimmune pathogenesis.
Sera from 22 patients with ulcerative colitis, 36 patients with Crohn’s disease, 47
patients with irritable bowel syndrome, 25 patients with celiac disease, 3 children with
autoimmune enteropathy and two patients with ANNA-1 seropositive paraneoplastic
pseudoobstruction of the gut were screened for the presence of TPH-antibodies using
ITT and immunoprecipitation of TPH (97, 114, 155). Immunoreactivity against TPH
could not be detected in sera from these patient groups. This illustrates the high
specificity of TPH-antibodies as a marker for gastrointestinal disease in APS I.
The identification of TH as an autoantigen in APS I (paper III)
Ectodermal manifestations in APS I such as alopecia, vitiligo, nail and enamel
dystrophy are frequent. Alopecia areata, characterized by sudden patchy hair-loss, has
been reported in frequencies varying from 13 to 37% in different populations of APS I
patients (5, 17, 185). In an earlier study we showed that APS I patients with alopecia
have autoantibodies directed against hair follicles (69). In this investigation, we set out
to identify the antigen responsible for this reactivity.

Pteridine dependent hydroxylases as autoantigens in APS I
50
A λ-ZAP EXPRESS cDNA library was constructed from normal human scalp and
immunoscreened with serum from an APS I patient in a 1:3000 dilution. Ten positive
clones were identified, and one of them coded for TH isoform 2. The TH clone was
used for ITT followed by immunoprecipitation with sera from 94 APS I patients from
Finland, Sweden, Norway and Italy. Forty-four percent (41/94) of the APS I patients
showed TH reactivity, whereas in a large number of sera from patients with IDDM
(n=224), Addison’s disease (n=20), and healthy blood donors (n=65) no reactivity
against TH was detected. A correlation with clinical symptoms showed that 62%
(18/29) of APS I patients with alopecia were TH-Ab positive, compared to 35%
(23/65) without (p = 0.02). No correlation with any of the other APS I components
was found. Western blot with all four TH isoforms (TH1-4) showed equal reactivity
with all isoforms. In analogy with the findings regarding TPH antibodies, although
less pronounced, TH reactive APS I sera inhibited TH enzyme activity in vitro in an
enzyme activity assay. Although TH expression in keratinocytes and hair follicles has
been described using RT-PCR (31, 32), we were not able to stain keratinocytes or hair
follicles with APS I sera or specific TH antibodies.
In conclusion, TH was identified as a dermal autoantigen in APS I, significantly
associated with alopecia. We also demonstrated that TH-Ab positive sera from APS I
patients inhibit TH activity.
Pteridine dependent hydroxylases as autoantigens in APS I (paper IV)
In papers I and III we identified TPH and TH as autoantigens in APS I associated with
intestinal dysfunction and alopecia, respectively. These two enzymes, together with
phenylalanine hydroxylase (PAH), constitute the group of biopterin dependent

Olov Ekwall
51
hydroxylases, which all are involved in the biosynthesis of neurotransmitters. Since
TPH, TH and PAH display a high linear and conformational homology (74, 174), we
wanted to investigate if APS I patients also have antibodies against PAH. We were
also interested in to what extent cross-reactivity occurred between the three enzymes.
A clone encoding PAH was used for ITT followed by immunoprecipitation with sera
from 94 APS I patients and 70 healthy controls. Twenty-seven percent of the APS I
patients had PAH antibodies. No reactivity was detected in the controls. No
statistically significant associations with the main clinical components of APS I were
found with PAH antibodies. A weak correlation with CAH (p=0.065) was observed.
Altogether 59 sera from the 94 APS I patients reacted with at least one of TPH, TH or
PAH, while 35 showed no reactivity. Nineteen of the sera contained antibodies
towards all enzymes, 12 to TPH only and 12 to TH only. No sera showed antibodies
that reacted solely to PAH. An immunocompetition assay was designed in which
[35S]-methionine labeled and non-labeled TPH, TH and PAH were expressed by ITT.
A fixed amount of labeled protein was mixed with increasing proportions of non-
labeled proteins, and an immunoprecipitation was performed. In this way the ability of
the non-labeled protein to compete with the labeled protein for antibody binding could
be determined. We demonstrated that the reactivity against PAH represents a cross
reactivity with TPH while antibodies against TPH and TH are directed towards
epitopes unique for the two enzymes.

Pteridine dependent hydroxylases as autoantigens in APS I
52
Discussion
The identification of TPH as an intestinal autoantigen in paper I and the subsequent
finding of the absence of TPH containing EC cells in patient biopsies strongly suggests
that the gastrointestinal dysfunction seen in APS I is not secondary to other disease
components. On the contrary, the deletion of TPH containing EC cells in an otherwise
intact mucosa suggests a very specific immune attack against these cells. The
possibility of a congenital EC cell aplasia, or specific EC cell apoptosis, leading to a
loss of tolerance against TPH cannot be completely excluded. However, this is in
conflict with preliminary observations in an APS I patient with severe gastrointestinal
dysfunction and loss of TPH containing EC cells. In this patient Cyclosporin A
treatment resulted in clinical remission as well as EC cell reappearance (176) (Cheri L
Deal, Montreal, personal communication).
In the gut, serotonin is classically believed to regulate motility and serotonin depletion
in mice has been shown to result in diarrhoea and decreased glucose uptake (90).
Serotonin receptors have also been found in the pancreas, and an enteropancreatic
communication mediated by serotonin has been proposed (86). One study has shown a
role for EC cell derived serotonin in mediating cholecystokinin (CKK) independent
stimulation of pancreatic secretion in response to luminal stimuli (99). The clinical
presentation of the gastrointestinal dysfunction in APS I is heterogeneous and
characterised by weight loss, fat malabsorbtion, diarrhoea, constipation, abdominal
cramps, growth failure and exocrine pancreas insufficiency in varying frequencies.
The loss of serotonin producing EC cells in APS I may explain both a dysfunction of
the exocrine pancreas and a dysregulation of intestinal motility. You can also speculate

Olov Ekwall
53
that the EC cells and/or serotonin have an as yet unknown, regulatory function for
energy absorption as some patients only suffer from marked weight loss, without
specific shortages. An interesting clinical observation is that an APS I patient with
pronounced malabsorbtion, frequent diarrhoea and marked weight loss regained
weight and normalised her defecation habits when treated with citalopram, a serotonin
reuptake inhibitor (O Kämpe, personal communication).
A recent case report describes an APS I patient with severe malabsorbtion in whom a
deficient CKK secretion, due to a transient loss of CKK producing EC cells, was
observed (77). Analysis of TPH antibodies or specific immunostaining for serotonin
producing cells was not performed. CKK deficiency has been reported before (71),
and treatment with CKK substitution has given varying clinical results (71, 176). One
can not exclude the possibility that CKK and serotonin are produced by the same EC
cells and that TPH autoreactivity can induce a loss of both serotonin and CKK
production.
A large group of autoantigens are the intracellular enzymes, e.g. GAD65 and 21-OH.
It is a common feature of the corresponding autoantibodies to inhibit the activity of the
autoantigen. The ability of APS I patient sera to decrease the enzymatic activity of
both TPH and TH in vitro is striking (Papers I and III) and it is tempting to believe that
the autoantibodies execute their action in vivo by directly affecting enzyme activity.
However, only anecdotal reports exist of autoantibodies having intracellular activity
(141) and the in vitro results can not be extrapolated to the in vivo situation without
further experimental evidence (107). Still, the observed enzyme inhibition patterns
strengthens the role for TPH and TH as autoantigens of importance in APS I. It is also

Pteridine dependent hydroxylases as autoantigens in APS I
54
noteworthy that PAH-Ab positive patient sera, possibly reflecting a cross-reaction with
TPH, does not affect PAH activity (Paper IV).
In immunostainings, three out of eight APS I patient sera showed immunoreactivity
against intestinal goblet cells (Figure 5). We do not know the significance of this
finding, but it is also interesting that sera from patients with ulcerative colitis and
Crohn’s disease are reported to have antibodies against goblet cells (54). The identity
and pathogenic role of the goblet cell antigen in inflammatory bowel disease (IBD) has
not been determined. If postulating that APS I patients and IBD patients recognise the
same goblet cell structure, APS I sera may be used to identify the antigen also of
interest for IBD. APS I sera, with high titre, high affinity, autoantibodies have been
proved suitable for immunoscreening cDNA libraries (49, 67, 147). Another finding
with unknown significance is that Paneth cells were also recognised by three out of
eight APS I patient sera (Figure 5). Paneth cells secrete small antimicrobial peptides
called cryptdins into the lumen and thus participate in the innate immune system
(127). An impaired Paneth cell function could, hypothetically, lead to a disturbed
microbial environment in the lumen.
We have used a strategy based on immunoscreening of cDNA libraries in the
identification of TPH and TH as autoantigens. Both these antigens are expressed in a
small fraction of cells in duodenum and scalp, respectively. Initial immunoblotting
experiments using whole tissues failed to show reactivity, probably because the
expression levels of these infrequently expressed proteins were below the detection
level. This illustrates the suitability of the immunoscreening methodology for
identifying rare proteins.

Olov Ekwall
55
Approximately 10 percent of APS I patients present with gastrointestinal dysfunction
as their first manifestation of the syndrome and they usually have a more severe form
of APS I (5). Since TPH antibodies are not found in other diseases with a similar
clinical presentation (Paper II), measurement of TPH-antibodies can be a valuable
diagnostic tool to identify APS I patients among young children presenting with
atypical gastrointestinal disease. An early diagnosis is desirable as it enables an early
treatment of other components of the syndrome as soon as they appear.
In paper III, TH is identified as an APS I autoantigen associated with alopecia. TH
exists in four alternatively spliced isoforms, TH1-4, which are structurally almost
identical with only minor differences in the N-terminal regulatory domain. APS I sera
do not discriminate between the four isoforms. TH is mainly expressed in the central
nervous system (CNS), the sympathetic nervous system and the adrenal medulla where
it converts tyrosine into L-dopa. Interestingly, TH expression has also been found, by
RT-PCR, in human hair follicles and keratinocytes (31, 32), and a role for TH in
keratinocyte differentiation has been suggested (151). We were unable to immunostain
hair follicles or keratinocytes by use of specific TH antibodies or APS I sera, but nerve
endings adjacent to hair follicles were clearly TH positive. The number and density of
TH containing nerve fibres has been reported to fluctuate in a hair cycle dependent
manner (25) and an autoimmune attack against TH containing cells may affect hair
growth.
Twenty-seven percent of APS I patients (25/94) had antibodies reacting with PAH
(Paper IV). All these patients also reacted with TPH, and some of them reacted with
TH as well. In immunocompetition experiments PAH reactivity could always be
inhibited by adding TPH, while adding TH did not affect PAH reactivity. Nor did

Pteridine dependent hydroxylases as autoantigens in APS I
56
PAH or TPH inhibit TH reactivity. No inhibition of PAH activity in vitro was seen
when adding PAH-Ab positive APS I sera. TPH and TH reactivities acted
independently of each other, both in the general reactivity pattern as well as in
immunocompetition experiments. These observations together strongly suggest that
the PAH reactivity seen is reflecting a cross-reactivity with TPH and that TPH and TH
antibodies are specific for each enzyme. The autoantibodies in APS I are not
monoclonal, but possibly oligoclonal. The partial inhibition of TPH reactivity seen
when adding PAH possibly reflects that some epitopes are shared and some are unique
for TPH. In a theoretical model, visualised in figure 6, three subsets of antibodies
exist. One only reacting with TH, one only reacting with TPH, and one primarily
reacting with TPH but also cross reacting with PAH through shared epitopes. These
shared epitopes are presumably located in the catalytic domain as the most widespread
homologies are found there and because cross reactivity with the regulatory domain
probably would have resulted in an inhibiting action of the cross-reacting antibodies.
It is tempting to interpret the weak, not statistically significant, correlation between
PAH-Abs and chronic active hepatitis (CAH) seen in paper IV as a possible
pathogenic mechanism considering that PAH is mainly expressed in the liver. The
possibility that these cross-reacting antibodies may have an effect in vivo, if PAH is
displayed on the cell surface, cannot be ruled out. When screening a large number of
patient sera from patients with both autoimmune and infectious hepatitis we were not
been able to find PAH antibodies (unpublished data).
The possibility of both linear and conformational epitopes makes the determination of
epitopes responsible for initiation of autoimmune responses difficult. There are still
only limited numbers of proteins for which we know the three dimensional structure.

Olov Ekwall
57
The crystal structures for TH and PAH have been determined and the TPH structure is
soon to be revealed (61, 91). The combination of this knowledge and the intriguing
patterns of reactivity against TPH, TH and PAH in APS I patients, should make it
possible to perform epitope mappings, taking both linear and conformational epitopes
in account. Chimeric proteins with regions of interest substituted between the
enzymes, could be constructed and expressed in vitro to search for structural as well as
linear epitopes.
Figure 6. Cross reactivity hypothesis. Immunisation with TPH can either result in the
generation of TPH specific antibodies only (I), or antibodies reacting with both TPH
and PAH through shared epitopes (II). TH immunisation only seems to induce TH
specific antibodies (III).

Pteridine dependent hydroxylases as autoantigens in APS I
58
Why does the immune system target intracellular enzymes? It is apparent that enzymes
of vital importance for the function of the target organ, or target cell, are chosen as
autoantigens in APS I and in other tissue specific autoimmune diseases (15, 157).
Figure 7. The figure outlining the biosynthesis of serotonin, dopamine and GABA
illustrates that key enzymes in the synthesis of neurotransmitters are autoantigens in
APS I. Cofactors required for the reactions mediated by TPH, TH and PAH are
marked.
In this case, a whole family of enzymes, all involved in the generation of
neurotransmitters, are targeted by the immune system (Figure 7). Speculations can be
made about explanations for this preference by the immune system:
1. Key enzymes are evolutionary conserved and often very homologous between
species. This increases the risks of autoimmunity induced by cross-reactivity with
infectious agents.
2. The central tolerance for enzymes is based on the presentation of the enzyme by
itself in the thymus or bone marrow. In the periphery, the enzymes are displayed

Olov Ekwall
59
bound to their substrates, and may then have a changed conformation leading to
the exposition of new, not tolerated, epitopes for the immune system.
3. The tolerance of intracellular enzymes that are normally expressed at low levels in
subpopulations of cells is primarily based on ignorance. This means that there are
potentially self reactive T-cells under normal conditions, but they are ignorant
since the number of self epitopes presented at a given time do not reach the
threshold of activating these cells. If, for instance, due to trauma, infection or
target organ stress, the amount of enzyme presented for the self reactive T-cells
increases over the threshold, T-cells are activated and the autoimmune process
starts.
In some autoimmune diseases, e.g. IDDM and myasthenia gravis, it has been shown
that autoantibodies and T-cells are directed against the same autoantigen (119, 131,
140). In APS I, no such T-cell studies have been reported, so it is still difficult to
conclude if B-cell autoantigens such as TPH, TH and PAH are just representing an
epiphenomenon, or if they represent structures that initially trigger the immune
response. The role of the autoantibodies as diagnostic markers in APS I is undisputed.
By mutational analysis of the AIRE gene approximately 90% of APS I patients are
identified (19) and by analysing a battery of APS I specific autoantibodies the
sensitivity is also around 90% (164). Since a genotype–phenotype correlation has not
yet been described in APS I, autoantibody analysis is still, by giving important clinical
information on the risk of developing different components of APS I, a valuable
complement to mutational analysis.

Pteridine dependent hydroxylases as autoantigens in APS I
60
S U M M A R Y
In this thesis we present the original identification of TPH as an endogenous intestinal
autoantigen in APS I associated with gastrointestinal dysfunction (Paper I). We have
shown that TPH autoantibodies are specific for APS I, and thereby a good diagnostic
tool to identify APS I patients among young children presenting with atypical
gastrointestinal disease (Paper II). TH is originally identified as a dermal autoantigen
in APS I associated with alopecia (Paper III). Autoantibodies reacting with PAH are
demonstrated in sera from APS I patients, probably reflecting a cross-reactivity with
TPH (Paper IV).

Olov Ekwall
61
F U T U R E P E R S P E C T I V E S
The physiological role of serotonin, and serotonin producing EC cells, in the gut
requires further study. APS I patients, and hopefully also Aire deficient mice, with a
total loss of serotonin producing EC cells in an otherwise intact mucosa represent
suitable models. Does serotonin, or serotonin producing EC cells through other
mediators affect absorption in the gut?
The epitope mapping of TPH, TH and PAH, as outlined in the discussion, should be
reasonably easy to carry out. This might give us information that makes it easier to
understand the preferences of the immune system.
TPH and TH are both rate limiting in the synthesis of neurotransmitters thought to be
involved in the pathogenesis of different diseases e.g. affective disorders,
schizophrenia and Parkinson’s disease. They ought to be suitable as targets for
pharmaceutical intervention aiming at an increased or decreased production of
serotonin or dopamine. APS I sera have been shown to markedly inhibit TPH and TH
activity, and in other studies both inhibitory and stimulatory antibodies have been
identified (81). If an epitope mapping study identifies binding sites responsible for
inhibition and possibly stimulation of TPH or TH, then a suitable drug could be
engineered to mimic this binding.
The same strategy as we have used in identifying TPH and TH as autoantigens could
be applied to other conditions. In IBD, we have already seen a clear staining pattern of
Goblet cells, similar to that given by a subgroup of APS I patients. The identification

Pteridine dependent hydroxylases as autoantigens in APS I
62
of the antigen responsible for this Goblet cell staining could supply important
information regarding the pathogenesis of IBD.
Hopefully the ongoing studies of Aire knock-out mice will provide us with new
insights into the pathogenesis of APS I, and autoimmune disease in general. The report
of Aire being expressed in thymic epithelial cells involved in negative selection
suggest that Aire is participating in the generation of central tolerance, and that
mutations may lead to loss of self tolerance and subsequent autoimmune
manifestations (186). We will have the perfect animal model to investigate organ
specific autoimmune disease if the Aire depleted mice develop autoimmune diseases.
Target organs, and infiltrating T-cells, can be studied at the time of the initial attack,
and not as in humans when the organ destruction has already taken place.

Olov Ekwall
63
A C K N O W L E D G E M E N T S
The work was performed at the Department of Medical Sciences, Uppsala University,Sweden. It has been made possible through the generous financial support from the
Swedish Medical Research Council, the Torsten and Ragnar Söderberg Foundation,the Swedish Society of Medicine, the Claes Groschinsky Memorial Foundation, the
Förenade Liv Mutual Group Life Insurance Company, the Lennander Foundation, theAgnes and Mac Rudberg Foundation, the Swedish Diabetes Association, the Torsten
Nilsson Foundation, the Åke Wiberg Foundation, the Ernfors Family Foundation,
AstraZeneca and the Swedish Society for Medical Research, which is hereby fullyacknowledged.
Scientific research is never a “one-mans work”, and many people have contributed tothis thesis. I would like to take this opportunity to thank them all, and especially:
Sverker Ljunghall, the former head of the department, who not only talked aboutbringing molecular biology into the clinical wards, but also took practical measures by
accepting a number of us “clinical molecular biologists” as PhD students.
Kjell Öberg, the present head of the department, for supplying good workingconditions and for signing all kinds of papers - just before the DHL-guy comes.
Olle Kämpe, my supervisor, for creating an inspiring atmosphere in the group byalways sharing scientific visions and knowledge. For understanding that skiing, good
food and laughter also brings science forward and, of course, for being the only one in
the group who knows how the pH-meter really works.
Fredrik Rorsman, my second supervisor, for sharing a room (with a poor view), for
your knowledge in molecular biology and for always, even in times of hard clinicalwork, having a couple of minutes to discuss experimental data. Thank you also for
letting me inherit your apartment, your childrens clothes, skates, skis and………
Eystein Husebye, “our man in Norway”, for not only supplying valuable patient
material, but also for valuable moments in front of arcade games around the world,
and for always, in an almost magical way, being first in line at check-in desks, cashiersetc.
Håkan Hedstrand, for being a good friend, for travel companionship, for amazinginventions, for thinking differently and for always contributing with new aspects and
different approaches to scientific problems.
Gennet Gebre-Medhin, for great friendship, for always being generously helpful, for
sharing a desk and my taste in “smågodis” and for always having time for a chat.

Pteridine dependent hydroxylases as autoantigens in APS I
64
Annika Söderbergh, who has left us for Arboga, for friendship and for always being
honest. Even though sometimes…….
Filip Sköldberg, for friendship, for critical thinking and for always having the latest
lab protocol – directly from some obscure web site.
Thomas Nilsson, for never hesitating to offer help and for late nights under the elk,listening to old 78 rpm records.
Eva Landgren, for your interest in GI-autoimmunity, for strengthening ourcompetence in basic science and for trying to get the lab a little bit more organised.
Jakob Skov, our “one-man brat pack”, for allowing the group to have futureperspectives.
Åsa Hallgren and Katrin Österlund, for being genuinely nice and friendly and for
expert technical help whenever needed.
Ola Winqvist, for always having an answer, and for tacos and Jacuzzi in La Jolla.
Gunnel Nordmark, for a positive attitude, and for adding a rheumatological, clinicalperspective into discussions.
Sophie Bensing, Lars Rönnblom, Anders Rönnblom, Roger Nilsson, Per Maritsand Patrik Larsson for contributing with knowledge and good spirits at our weekly
Monday seminars.
My co-authors Jan Gustafsson, for supplying patient sera, and for showingcontinuous interest, Jan Haavik, for knowing everything worth knowing about “our
enzymes”, Lars Grimelius, for invaluable help with immunohistochemistry, JaakkoPerheentupa, for generously sharing your patient sera and clinical knowledge of
APS I and Vanda A. Lennon, Klas Sjöberg and Corrado Betterle for patient sera.
Ismo Ulmanen, Petra Eskelin, Maria Halonen and Meelis Kolmer, “The Helsinki
connection”, and Cheri Deal, Leanne Ward and Patricia Crock, “The Canadian-
Australian connection”, for fruitful collaborations in the past, present and future.
Kerstin Westermark, Therese Dahlman and Majstin Wik for first introducing me
to the group, and for guiding me in my first fumbling efforts to unravel the mysteriesof thyroid cancer.
Peter Stålberg for being a good friend, for Hurricanes in New Orleans, pike-fishing,and my daily dose of irony.

Olov Ekwall
65
Håkan Melhus, Andreas Kindmark, Maria Branting, Fredrik Stiger, SaraJohansson and other members of the Osteoporosis group for laughters and lateralthinking in the coffee corner.
Anna Schölin, Mikael Kullin, Jan Melin, Britt Skogseid, Ann-Christin Syvänenand all other friends and members of various research groups at the Centre forClinical Research for small talk, sharing instruments, advice, etc.
Cindy Wong, Maud Marsden and Charlotte Sweeney for expert linguistic revision,and Cindy also for always being so nice and caring.
Elisabet Dew, Anna Carlbom-Härd, Elizabeth Dehlin and Lena Hultström forhelp with practical matters at the department throughout the years.
The pike-fishers for giving me something to look forward to each autumn.
My mother Barbro for continuous love and support. My late father Björn, who left usmuch too early, for having been the best of fathers. He will always continue to be an
inspiration for me in my research.
Axel and Erland, my two bold boys, for giving me an excuse to play with LEGO and
RC-cars.
Anna-Karin, my princess, for being the love of my life as well as an inspiring
colleague in research and in clinical work – and everything in between.

Pteridine dependent hydroxylases as autoantigens in APS I
66
R E F E R E N C E S
1. Aaltonen J, Björses P, Sandkuijl L, Perheentupa J, Peltonen L. An autosomal
locus causing autoimmune disease: autoimmune polyglandular disease type Iassigned to chromosome 21. Nat Genet 1994; 8:83-7.
2. Agrawal A, Schatz DG. RAG1 and RAG2 form a stable postcleavage synapticcomplex with DNA containing signal ends in V(D)J recombination. Cell 1997;
89:43-53.
3. Ahonen P. Autoimmune polyendocrinopathy-candidosis-ectodermal dystrophy
(APECED): autosomal recessive inheritance. Clin Genet 1985; 27:535-42.
4. Ahonen P, Koskimies S, Lokki ML, Tiilikainen A, Perheentupa J. Theexpression of autoimmune polyglandular disease type I appears associated with
several HLA-A antigens but not with HLA-DR. J Clin Endocrinol Metab 1988;66:1152-7.
5. Ahonen P, Myllarniemi S, Sipila I, Perheentupa J. Clinical variation ofautoimmune polyendocrinopathy-candidiasis- ectodermal dystrophy
(APECED) in a series of 68 patients. N Engl J Med 1990; 322:1829-36.
6. Albert LJ, Zhao Y-X, Inman RD. Molecular mimicry. In: Lahita RG, ChiorazziN, Reeves WH, eds. Textbook of the autoimmune diseases. Vol. 1.
Philadelphia: Lippincott Williams & Wilkins, 2000:153-174.
7. Anderson CC, Matzinger P. Danger: the view from the bottom of the cliff.
Semin Immunol 2000; 12:231-8.
8. Anderson JR, Goudie RB, Gray KG, Timbury GC. Auto-antibodies in
Addison's disease. Lancet 1957; 1:1123.
9. Arlt W, Callies F, van Vlijmen JC, Koehler I, Reincke M, Bidlingmaier M,Huebler D, Oettel M, Ernst M, Schulte HM, Allolio B. Dehydroepiandrosterone
replacement in women with adrenal insufficiency. N Engl J Med 1999;341:1013-20.
10. Arulanantham K, Dwyer JM, Genel M. Evidence for defectiveimmunoregulation in the syndrome of familial candidiasis endocrinopathy. N
Engl J Med 1979; 300:164-8.

Olov Ekwall
67
11. Atkinson MA, Bowman MA, Campbell L, Darrow BL, Kaufman DL, Maclaren
NK. Cellular immunity to a determinant common to glutamate decarboxylaseand coxsackie virus in insulin-dependent diabetes. J Clin Invest 1994; 94:2125-
9.
12. Bach JF. Insulin-dependent diabetes mellitus as an autoimmune disease.Endocr Rev 1994; 15:516-42.
13. Baekkeskov S, Aanstoot HJ, Christgau S, Reetz A, Solimena M, Cascalho M,Folli F, Richter-Olesen H, DeCamilli P, Camilli PD. Identification of the 64K
autoantigen in insulin-dependent diabetes as the GABA-synthesizing enzymeglutamic acid decarboxylase. Nature 1990; 347:151-6.
14. Bahn RS, Heufelder AE. Pathogenesis of Graves' ophthalmopathy. N Engl J
Med 1993; 329:1468-75.
15. Banga JP, McGregor AM. Enzymes as targets for autoantibodies in human
autoimmune disease: relevance to pathogenesis? Autoimmunity 1991; 9:177-82.
16. Bereket A, Lowenheim M, Blethen SL, Kane P, Wilson TA. Intestinal
lymphangiectasia in a patient with autoimmune polyglandular disease type Iand steatorrhea. J Clin Endocrinol Metab 1995; 80:933-5.
17. Betterle C, Greggio NA, Volpato M. Clinical review 93: Autoimmune
polyglandular syndrome type 1. J Clin Endocrinol Metab 1998; 83:1049-55.
18. Betterle C, Volpato M. Adrenal and ovarian autoimmunity. Eur J Endocrinol
1998; 138:16-25.
19. Björses P, Halonen M, Palvimo JJ, Kolmer M, Aaltonen J, Ellonen P,
Perheentupa J, Ulmanen I, Peltonen L. Mutations in the AIRE gene: effects onsubcellular location and transactivation function of the autoimmune
polyendocrinopathy- candidiasis-ectodermal dystrophy protein. Am J Hum
Genet 2000; 66:378-92.
20. Björses P, Pelto-Huikko M, Kaukonen J, Aaltonen J, Peltonen L, Ulmanen I.
Localization of the APECED protein in distinct nuclear structures. Hum Mol
Genet 1999; 8:259-66.
21. Blizzard RM, Chandler RW, Kyle MA, Hung W. Adrenal antibodies inAddison's disease. Lancet 1962; 2:901-903.
22. Blizzard RM, Chee D, Davis W. The incidence of parathyroid and other
antibodies in the sera of patients with idiopathic hypoparathyroidism. Clin Exp
Immunol 1966; 1:119-28.

Pteridine dependent hydroxylases as autoantigens in APS I
68
23. Bonifacio E, Bingley PJ, Shattock M, Dean BM, Dunger D, Gale EA, Bottazzo
GF. Quantification of islet-cell antibodies and prediction of insulin- dependentdiabetes. Lancet 1990; 335:147-9.
24. Born W, Cady C, Jones-Carson J, Mukasa A, Lahn M, O'Brien R.
Immunoregulatory functions of gamma delta T cells. Adv Immunol 1999;71:77-144.
25. Botchkarev VA, Eichmuller S, Peters EM, Pietsch P, Johansson O, Maurer M,Paus R. A simple immunofluorescence technique for simultaneous visualization
of mast cells and nerve fibers reveals selectivity and hair cycle-- dependentchanges in mast cell--nerve fiber contacts in murine skin. Arch Dermatol Res
1997; 289:292-302.
26. Brachet V, Raposo G, Amigorena S, Mellman I. Ii chain controls the transportof major histocompatibility complex class II molecules to and from lysosomes.
J Cell Biol 1997; 137:51-65.
27. Briggs JN, Goodwin JF, Wilson A. Addison's disease occurring in two
brothers. Brit Med J 1951:115-117.
28. Brown JH, Jardetzky TS, Gorga JC, Stern LJ, Urban RG, Strominger JL, Wiley
DC. Three-dimensional structure of the human class II histocompatibility
antigen HLA-DR1. Nature 1993; 364:33-9.
29. Carter AC, Kaplan SA, DeMayo AP, Rosenblum DJ. An unusual case of
idiopathic hypoparathyroidism, adrenal insufficiency, hypothyroidism andmetastatic calcification. J Clin Endocrinol 1959; 19:1633-1641.
30. Casiano CA, Tan EM. Apoptosis, autoantigens and autoimmunity. In: RoseNR, Mackay IR, eds. The autoimmune diseases. Vol. 1. San Diego: Academic
Press, 1998:193-210.
31. Chang YT, Hyland K, Mues G, Marsh JL. Human hair follicles as a peripheralsource of tyrosine hydroxylase and aromatic L-amino acid decarboxylase
mRNA. Neurosci Lett 1997; 222:210-2.
32. Chang YT, Mues G, Pittelkow MR, Hyland K. Cultured human keratinocytes
as a peripheral source of mRNA for tyrosine hydroxylase and aromatic L-amino acid decarboxylase. J Inherit Metab Dis 1996; 19:239-42.
33. Chervonsky AV. Apoptotic and effector pathways in autoimmunity. Curr Opin
Immunol 1999; 11:684-8.

Olov Ekwall
69
34. Chilgren RA, Meuwissen HJ, Quie PG, Good RA, Hong R. The cellular
immune defect in chronic mucocutaneous candidiasis. Lancet 1969; 1:1286-8.
35. Clemente MG, Meloni A, Obermayer-Straub P, Frau F, Manns MP, De
Virgiliis S. Two cytochromes P450 are major hepatocellular autoantigens in
autoimmune polyglandular syndrome type 1. Gastroenterology 1998; 114:324-8.
36. Clemente MG, Obermayer-Straub P, Meloni A, Strassburg CP, Arangino V,Tukey RH, De Virgiliis S, Manns MP. Cytochrome P450 1A2 is a hepatic
autoantigen in autoimmune polyglandular syndrome type 1. J Clin Endocrinol
Metab 1997; 82:1353-61.
37. Collins-Williams C. Idiopathic hypoparathyridism with papilledema in a boy
six years of age. Pediatrics 1950; 5:998-1007.
38. Coombs RR. Immunopathology. Br Med J 1968; 1:597-602.
39. Cornall RJ, Goodnow CC, Cyster JG. The regulation of self-reactive B cells.Curr Opin Immunol 1995; 7:804-11.
40. Craig JM, Schiff LH, Boone JE. Chronic moniliasis associated with Addison'sdisease. Am J Dis Child 1955; 89:669-684.
41. Croom DH. Addisonism as a family disease. Lancet 1909; 1:603-604.
42. Delves PJ, Roitt IM. The immune system. First of two parts. N Engl J Med
2000; 343:37-49.
43. Delves PJ, Roitt IM. The immune system. Second of two parts. N Engl J Med
2000; 343:108-17.
44. DiGeorge AM, Paschkis K. The syndrome of Addison's disease,hypoparathyroidism and superficial moniliasis. Am J Dis Child 1957; 94:476-
478.
45. Domen RE. An overview of immune hemolytic anemias. Cleve Clin J Med
1998; 65:89-99.
46. Edelman GM. Antibody structure and molecular immunology. Science 1973;180:830-40.
47. Eisenbarth GS, Wilson PW, Ward F, Buckley C, Lebovita H. Thepolyglandular failure syndrome: disease inheritance, HLA type, and immune
function. Ann Intern Med 1979; 91:528-33.

Pteridine dependent hydroxylases as autoantigens in APS I
70
48. Eisensmith RC, Woo SL. Phenylketonuria and the phenylalanine hydroxylase
gene. Mol Biol Med 1991; 8:3-18.
49. Ekwall O, Hedstrand H, Grimelius L, Haavik J, Perheentupa J, Gustafsson J,
Husebye E, Kämpe O, Rorsman F. Identification of tryptophan hydroxylase as
an intestinal autoantigen. Lancet 1998; 352:279-83.
50. Erlandsen H, Fusetti F, Martinez A, Hough E, Flatmark T, Stevens RC. Crystal
structure of the catalytic domain of human phenylalanine hydroxylase revealsthe structural basis for phenylketonuria. Nat Struct Biol 1997; 4:995-1000.
51. Fielder AH, Walport MJ, Batchelor JR, Rynes RI, Black CM, Dodi IA, HughesGR. Family study of the major histocompatibility complex in patients with
systemic lupus erythematosus: importance of null alleles of C4A and C4B in
determining disease susceptibility. Br Med J (Clin Res Ed) 1983; 286:425-8.
52. Fisher GH, Rosenberg FJ, Straus SE, Dale JK, Middleton LA, Lin AY, Strober
W, Lenardo MJ, Puck JM. Dominant interfering Fas gene mutations impairapoptosis in a human autoimmune lymphoproliferative syndrome. Cell 1995;
81:935-46.
53. Fitzpatrick PF. Tetrahydropterin-dependent amino acid hydroxylases. Annu Rev
Biochem 1999; 68:355-81.
54. Folwaczny C, Noehl N, Tschop K, Endres SP, Heldwein W, Loeschke K,Fricke H. Goblet cell autoantibodies in patients with inflammatory bowel
disease and their first-degree relatives. Gastroenterology 1997; 113:101-6.
55. Friedman TC, Thomas PM, Fleisher TA, Feuillan P, Parker RI, Cassorla F,
Chrousos GP. Frequent occurrence of asplenism and cholelithiasis in patientswith autoimmune polyglandular disease type I. Am J Med 1991; 91:625-30.
56. Garcia KC, Degano M, Pease LR, Huang M, Peterson PA, Teyton L, Wilson
IA. Structural basis of plasticity in T cell receptor recognition of a self peptide-MHC antigen. Science 1998; 279:1166-72.
57. Garcia KC, Degano M, Stanfield RL, Brunmark A, Jackson MR, Peterson PA,Teyton L, Wilson IA. An alphabeta T cell receptor structure at 2.5 A and its
orientation in the TCR-MHC complex. Science 1996; 274:209-19.
58. Gebre-Medhin G, Husebye ES, Gustafsson J, Winqvist O, Goksoyr A, Rorsman
F, Kämpe O. Cytochrome P450IA2 and aromatic L-amino acid decarboxylase
are hepatic autoantigens in autoimmune polyendocrine syndrome type I. FEBS
Lett 1997; 412:439-45.

Olov Ekwall
71
59. Gebre-Medhin G, Husebye ES, Mallmin H, Helström L, Berne C, Karlsson FA,
Kämpe O. Oral dehydroepiandrosterone (DHEA) replacement therapy inwomen with Addison's disease. Clin Endocrinol (Oxf) 2000; 52:775-80.
60. Germain RN. MHC-dependent antigen processing and peptide presentation:
providing ligands for T lymphocyte activation. Cell 1994; 76:287-99.
61. Goodwill KE, Sabatier C, Marks C, Raag R, Fitzpatrick PF, Stevens RC.
Crystal structure of tyrosine hydroxylase at 2.3 A and its implications forinherited neurodegenerative diseases. Nat Struct Biol 1997; 4:578-85.
62. Greenspan NS, Di Cera E. Defining epitopes: It's not as easy as it seems. Nat
Biotechnol 1999; 17:936-7.
63. Grenett HE, Ledley FD, Reed LL, Woo SL. Full-length cDNA for rabbit
tryptophan hydroxylase: functional domains and evolution of aromatic aminoacid hydroxylases. Proc Natl Acad Sci U S A 1987; 84:5530-4.
64. Grondal G, Kristjansdottir H, Gunnlaugsdottir B, Arnason A, Lundberg I,Klareskog L, Steinsson K. Increased number of interleukin-10-producing cells
in systemic lupus erythematosus patients and their first-degree relatives andspouses in Icelandic multicase families. Arthritis Rheum 1999; 42:1649-54.
65. Gylling M, Tuomi T, Bjorses P, Kontiainen S, Partanen J, Christie MR, Knip
M, Perheentupa J, Miettinen A. ss-Cell Autoantibodies, Human LeukocyteAntigen II Alleles, and Type 1 Diabetes in Autoimmune Polyendocrinopathy-
Candidiasis-Ectodermal Dystrophy. J Clin Endocrinol Metab 2000; 85:4434-4440.
66. Halonen M, Pelto-Huikko M, Eskelin P, Peltonen L, Ulmanen I, Kolmer M.Subcellular Location and Expression Pattern of Autoimmune Regulator (Aire),
the Mouse Orthologue for Human Gene Defective in Autoimmune
Polyendocrinopathy Candidiasis Ectodermal Dystrophy (APECED). J
Histochem Cytochem 2001; 49:197-208.
67. Hedstrand H, Ekwall O, Haavik J, Landgren E, Betterle C, Perheentupa J,Gustafsson J, Husebye E, Rorsman F, O Km. Identification of Tyrosine
Hydroxylase as an Autoantigen in Autoimmune Polyendocrine Syndrome TypeI. Biochem Biophys Res Commun 2000; 267:456-461.
68. Hedstrand H, Ekwall O, Olsson M, et al. The transcription factors SOX9 and
SOX10 are melanocyte autoantigens related to vitiligo in autoimmunepolyendocrine syndrome type I. Submitted 2000.

Pteridine dependent hydroxylases as autoantigens in APS I
72
69. Hedstrand H, Perheentupa J, Ekwall O, Gustafsson J, Michaelsson G, Husebye
E, Rorsman F, Kämpe O. Antibodies against hair follicles are associated withalopecia totalis in autoimmune polyendocrine syndrome type I. J Invest
Dermatol 1999; 113:1054-8.
70. Heino M, Peterson P, Kudoh J, et al. Autoimmune regulator is expressed in thecells regulating immune tolerance in thymus medulla. Biochem Biophys Res
Commun 1999; 257:821-5.
71. Heubi JE, Partin JC, Schubert WK. Hypocalcemia and steatorrhea--clues to
etiology. Dig Dis Sci 1983; 28:124-8.
72. Horwitz MS, Bradley LM, Harbertson J, Krahl T, Lee J, Sarvetnick N. Diabetes
induced by Coxsackie virus: initiation by bystander damage and not molecular
mimicry. Nat Med 1998; 4:781-5.
73. Huang X, Yuang J, Goddard A, Foulis A, James RF, Lernmark A, Pujol-Borrell
R, Rabinovitch A, Somoza N, Stewart TA. Interferon expression in thepancreases of patients with type I diabetes. Diabetes 1995; 44:658-64.
74. Hufton SE, Jennings IG, Cotton RG. Structure and function of the aromaticamino acid hydroxylases. Biochem J 1995; 311:353-66.
75. Hung W, Migeon CJ, Parrott RH. A possible autoimmune basis for Addison's
disease in three siblings, one with idiopathic hypoparathyroidism, perniciousanemia and superficial moniliasis. N Engl J Med 1963; 269:658-663.
76. Husebye ES, Gebre-Medhin G, Tuomi T, Perheentupa J, Landin-Olsson M,Gustafsson J, Rorsman F, Kämpe O. Autoantibodies against aromatic L-amino
acid decarboxylase in autoimmune polyendocrine syndrome type I. J Clin
Endocrinol Metab 1997; 82:147-50.
77. Högenauer C, Meyer RL, Netto GJ, Bell D, Little KH, Ferries L, Santa Ana
CA, Porter JL, Fordtran JS. Malabsorption due to cholecystokinin deficiency ina patient with autoimmune polyendocrine syndrome type I. N Engl J Med 2001;
344:270-74.
78. Jackson WPU. Steatorrhoea and hypoparathyroidism. Lancet 1957:1086-1087.
79. Jacobson DL, Gange SJ, Rose NR, Graham NM. Epidemiology and estimatedpopulation burden of selected autoimmune diseases in the United States. Clin
Immunol Immunopathol 1997; 84:223-43.
80. Janeway CA, Travers P, Walport M, Capra JD. Immunobiology - The immunesystem in health and disease. London: Elsevier, 1999:635.

Olov Ekwall
73
81. Jennings IG, Russell RG, Armarego WL, Cotton RG. Functional analysis of the
effect of monoclonal antibodies on monkey liver phenylalanine hydroxylase.Biochem J 1986; 235:133-8.
82. Julier C, Hyer RN, Davies J, Merlin F, Soularue P, Briant L, Cathelineau G,
Deschamps I, Rotter JI, Froguel P, et al. Insulin-IGF2 region on chromosome11p encodes a gene implicated in HLA- DR4-dependent diabetes susceptibility.
Nature 1991; 354:155-9.
83. Kagi D, Odermatt B, Seiler P, Zinkernagel RM, Mak TW, Hengartner H.
Reduced incidence and delayed onset of diabetes in perforin-deficient nonobesediabetic mice. J Exp Med 1997; 186:989-97.
84. Karlsson FA, Burman P, Loof L, Mardh S. Major parietal cell antigen in
autoimmune gastritis with pernicious anemia is the acid-producing H+,K+-adenosine triphosphatase of the stomach. J Clin Invest 1988; 81:475-9.
85. Kenny FM, Holliday MA. Hypoparathyroidism, moniliasis, Addison's andHashimoto's diseases. N Engl J Med 1964; 271:708-713.
86. Kirchgessner AL, Liu MT, Raymond JR, Gershon MD. Identification of cellsthat express 5-hydroxytryptamine1A receptors in the nervous systems of the
bowel and pancreas. J Comp Neurol 1996; 364:439-455.
87. Klein J, Sato A. The HLA system. First of two parts. N Engl J Med 2000;343:702-9.
88. Klein J, Sato A. The HLA system. Second of two parts. N Engl J Med 2000;343:782-6.
89. Klemetti P, Bjorses P, Tuomi T, Perheentupa J, Partanen J, Rautonen N,Hinkkanen A, Ilonen J, Vaarala O. Autoimmunity to glutamic acid
decarboxylase in patients with autoimmune polyendocrinopathy-candidiasis-
ectodermal dystrophy (APECED). Clin Exp Immunol 2000; 119:419-25.
90. Kobayashi T, Hasegawa H, Kaneko E, Ichiyama A. Gastrointestinal serotonin:
depletion due to tetrahydrobiopterin deficiency induced by 2,4-diamino-6-hydroxypyrimidine administration. J Pharmacol Exp Ther 1991; 256:773-9.
91. Kobe B, Jennings IG, House CM, Michell BJ, Goodwill KE, Santarsiero BD,Stevens RC, Cotton RG, Kemp BE. Structural basis of autoregulation of
phenylalanine hydroxylase. Nat Struct Biol 1999; 6:442-8.
92. Kotzin BL. Systemic lupus erythematosus. Cell 1996; 85:303-6.

Pteridine dependent hydroxylases as autoantigens in APS I
74
93. Krohn K, Uibo R, Aavik E, Peterson P, Savilahti K. Identification by molecular
cloning of an autoantigen associated with Addison's disease as steroid 17 alpha-hydroxylase. Lancet 1992; 339:770-3.
94. Kuchroo VK, Das MP, Brown JA, Ranger AM, Zamvil SS, Sobel RA, Weiner
HL, Nabavi N, Glimcher LH. B7-1 and B7-2 costimulatory molecules activatedifferentially the Th1/Th2 developmental pathways: application to autoimmune
disease therapy. Cell 1995; 80:707-18.
95. Kwok WW, Domeier ME, Johnson ML, Nepom GT, Koelle DM. HLA-DQB1
codon 57 is critical for peptide binding and recognition. J Exp Med 1996;183:1253-8.
96. Kwok WW, Nepom GT. Genetic influences: Major histocompability complex.
In: Rose NR, Mackay IR, eds. The autoimmune diseases. Vol. 1. San Diego:Academic Press, 1998:75-83.
97. Lennon VA, Sas DF, Busk MF, Scheithauer B, Malagelada JR, Camilleri M,Miller LJ. Enteric neuronal autoantibodies in pseudoobstruction with small-cell
lung carcinoma. Gastroenterology 1991; 100:137-42.
98. Leonard MF. Chronic idiopathic hypoparathyroidism with superimposed
Addison's disease in a child. J Clin Endocrinol 1946; 6:493-506.
99. Li Y, Hao Y, Zhu J, Owyang C. Serotonin released from intestinalenterochromaffin cells mediates luminal non-cholecystokinin-stimulated
pancreatic secretion in rats. Gastroenterology 2000; 118:1197-207.
100. Li Y, Song YH, Rais N, Connor E, Schatz D, Muir A, Maclaren N.
Autoantibodies to the extracellular domain of the calcium sensing receptor inpatients with acquired hypoparathyroidism. J Clin Invest 1996; 97:910-4.
101. Lindstrom J. Immunobiology of myasthenia gravis, experimental autoimmune
myasthenia gravis, and Lambert-Eaton syndrome. Annu Rev Immunol 1985;3:109-31.
102. Louria DB, Shannon D, Johnson G, Caroline L, Okas A, Taschdjian C. Thesusceptibility to moniliasis in children with endocrine hypofunction. Trans
Assoc Am Physicians 1967; 80:236-49.
103. Mackay IR. Science, medicine, and the future: Tolerance and autoimmunity.
Bmj 2000; 321:93-6.

Olov Ekwall
75
104. MacLaren NK, Blizzard RM. Adrenal autoimmunity and autoimmune
polyglandular syndromes. The autoimmune diseases: Academic Press,1985:201-223.
105. Maclaren NK, Riley WJ. Inherited susceptibility to autoimmune Addison's
disease is linked to human leukocyte antigens-DR3 and/or DR4, except whenassociated with type I autoimmune polyglandular syndrome. J Clin Endocrinol
Metab 1986; 62:455-9.
106. Madden DR, Gorga JC, Strominger JL, Wiley DC. The three-dimensional
structure of HLA-B27 at 2.1 A resolution suggests a general mechanism fortight peptide binding to MHC. Cell 1992; 70:1035-48.
107. Manns M, Zanger U, Gerken G, Sullivan KF, Meyer zum Buschenfelde KH,
Meyer UA, Eichelbaum M. Patients with type II autoimmune hepatitis expressfunctionally intact cytochrome P-450 db1 that is inhibited by LKM-1
autoantibodies in vitro but not in vivo. Hepatology 1990; 12:127-32.
108. Matzinger P. An innate sense of danger. Semin Immunol 1998; 10:399-415.
109. McIntyre Gass JD. The syndrome of keratokonjunctivitis, superficialmoniliasis, idiopathic hypoparathyroidism and Addison's disease. Am J
Ophthalmol 1962; 54:660-74.
110. Medzhitov R, Janeway C, Jr. Innate immunity. N Engl J Med 2000; 343:338-44.
111. Medzhitov R, Preston-Hurlburt P, Janeway CA, Jr. A human homologue of theDrosophila Toll protein signals activation of adaptive immunity. Nature 1997;
388:394-7.
112. Michele TM, Fleckenstein J, Sgrignoli AR, Thuluvath PJ. Chronic active
hepatitis in the type I polyglandular autoimmune syndrome. Postgrad Med J
1994; 70:128-31.
113. Miller JF, Basten A. Mechanisms of tolerance to self. Curr Opin Immunol
1996; 8:815-21.
114. Mirakian R, Richardson A, Milla PJ, Walker-Smith JA, Unsworth J, Savage
MO, Bottazzo GF. Protracted diarrhoea of infancy: evidence in support of anautoimmune variant. Br Med J (Clin Res Ed) 1986; 293:1132-6.
115. Mockus SM, Vrana KE. Advances in the molecular characterization of
tryptophan hydroxylase. J Mol Neurosci 1998; 10:163-79.

Pteridine dependent hydroxylases as autoantigens in APS I
76
116. Morse WI, Cochrane WA, Landrigan PL. Familial hypoparathyroidism with
pernicious anemia, ateatorrhea and adrenocortical insufficiency. N Engl J Med
1961; 264:1021-1026.
117. Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two types
of murine helper T cell clone. I. Definition according to profiles of lymphokineactivities and secreted proteins. J Immunol 1986; 136:2348-57.
118. Nagamine K, Peterson P, Scott HS, et al. Positional cloning of the APECEDgene. Nat Genet 1997; 17:393-8.
119. Naquet P, Ellis J, Tibensky D, Kenshole A, Singh B, Hodges R, Delovitch TL.T cell autoreactivity to insulin in diabetic and related non-diabetic individuals.
J Immunol 1988; 140:2569-78.
120. Navratil JS, Korb LC, Ahearn JM. Systemic lupus erythematosus andcomplement deficiency: clues to a novel role for the classical complement
pathway in the maintenance of immune tolerance. Immunopharmacology 1999;42:47-52.
121. Nepom GT, Erlich H. MHC class-II molecules and autoimmunity. Annu Rev
Immunol 1991; 9:493-525.
122. Neufeld M, Maclaren N, Blizzard R. Autoimmune polyglandular syndromes.
Pediatr Ann 1980; 9:154-62.
123. Neufeld M, Maclaren NK, Blizzard RM. Two types of autoimmune Addison's
disease associated with different polyglandular autoimmune (PGA) syndromes.Medicine (Baltimore) 1981; 60:355-62.
124. Nossal GJ. Negative selection of lymphocytes. Cell 1994; 76:229-39.
125. Obermayer-Straub P, Manns MP. Autoimmune polyglandular syndromes.
Baillieres Clin Gastroenterol 1998; 12:293-315.
126. Oldstone MB. Molecular mimicry and immune-mediated diseases. Faseb J
1998; 12:1255-65.
127. Ouellette AJ. Paneth cells and innate immunity in the crypt microenvironment.Gastroenterology 1997; 113:1779-84.
128. Padeh S, Theodor R, Jonas A, Passwell JH. Severe malabsorption inautoimmune polyendocrinopathy-candidosis- ectodermal dystrophy syndrome
successfully treated with immunosuppression. Arch Dis Child 1997; 76:532-4.

Olov Ekwall
77
129. Padovan E, Casorati G, Dellabona P, Meyer S, Brockhaus M, Lanzavecchia A.
Expression of two T cell receptor alpha chains: dual receptor T cells. Science
1993; 262:422-4.
130. Paliard X, West SG, Lafferty JA, Clements JR, Kappler JW, Marrack P, Kotzin
BL. Evidence for the effects of a superantigen in rheumatoid arthritis. Science
1991; 253:325-9.
131. Palmer JP, Asplin CM, Clemons P, Lyen K, Tatpati O, Raghu PK, Paquette TL.Insulin antibodies in insulin-dependent diabetics before insulin treatment.
Science 1983; 222:1337-9.
132. Panina-Bordignon P, Lang R, van Endert PM, Benazzi E, Felix AM, Pastore
RM, Spinas GA, Sinigaglia F. Cytotoxic T cells specific for glutamic acid
decarboxylase in autoimmune diabetes. J Exp Med 1995; 181:1923-7.
133. Parker DC. T cell-dependent B cell activation. Annu Rev Immunol 1993;
11:331-60.
134. Parker RI, O'Shea P, Forman EN. Acquired splenic atrophy in a sibship with
the autoimmune polyendocrinopathy-candidiasis syndrome. J Pediatr 1990;117:591-3.
135. Perheentupa J, Miettinen A. Type 1 autoimmune polyglandular disease. Ann
Med Interne (Paris) 1999; 150:313-25.
136. Perlmutter M, Ellison RR, Norsa L, Kantrowitz AR. Idiopathic
hypoparathyroidism and Addison's disease. Am J Med 1956; 21:634-643.
137. Peserico A, Rigon F, Semenzato G, Caretto A, Pasini CV, Betterle C. Vitiligo
and polyglandular autoimmune disease with autoantibodies to melanin-producing cells. A new syndrome? Arch Dermatol 1981; 117:751-2.
138. Peterson P, Perheentupa J, Krohn KJ. Detection of candidal antigens in
autoimmune polyglandular syndrome type I. Clin Diagn Lab Immunol 1996;3:290-4.
139. Pitkanen J, Doucas V, Sternsdorf T, et al. The autoimmune regulator proteinhas transcriptional transactivating properties and interacts with the common
coactivator CREB-binding protein. J Biol Chem 2000; 275:16802-9.
140. Protti MP, Manfredi AA, Horton RM, Bellone M, Conti-Tronconi BM.
Myasthenia gravis: recognition of a human autoantigen at the molecular level.
Immunol Today 1993; 14:363-8.

Pteridine dependent hydroxylases as autoantigens in APS I
78
141. Reichlin M. Cellular dysfunction induced by penetration of autoantibodies into
living cells: cellular damage and dysfunction mediated by antibodies to dsDNAand ribosomal P proteins. J Autoimmun 1998; 11:557-61.
142. Ridgway WM, Fathman CG. MHC structure and autoimmune T cell repertoire
development. Curr Opin Immunol 1999; 11:638-42.
143. Riley WJ. Autoimmune polyglandular syndromes. Horm Res 1992; 38 Suppl
2:9-15.
144. Rinderle C, Christensen HM, Schweiger S, Lehrach H, Yaspo ML. AIRE
encodes a nuclear protein co-localizing with cytoskeletal filaments: altered sub-cellular distribution of mutants lacking the PHD zinc fingers. Hum Mol Genet
1999; 8:277-90.
145. Romagnani S. Biology of human TH1 and TH2 cells. J Clin Immunol 1995;15:121-9.
146. Romagnani S. T-cell subsets (Th1, Th2) and cytokines in autoimmunity. In:Rose NR, Mackay IR, eds. The autoimmune diseases. Vol. 1. San Diego:
Academic Press, 1998:163-191.
147. Rorsman F, Husebye ES, Winqvist O, Björk E, Karlsson FA, Kämpe O.
Aromatic-L-amino-acid decarboxylase, a pyridoxal phosphate-dependent
enzyme, is a beta-cell autoantigen. Proc Natl Acad Sci U S A 1995; 92:8626-9.
148. Rosatelli MC, Meloni A, Devoto M, et al. A common mutation in Sardinian
autoimmune polyendocrinopathy- candidiasis-ectodermal dystrophy patients.Hum Genet 1998; 103:428-34.
149. Rose NR, Bona C. Defining criteria for autoimmune diseases (Witebsky'spostulates revisited). Immunol Today 1993; 14:426-30.
150. Samloff IM, Kleinman MS, Turner MD, Sobel MV, Jeffries GH. Blocking and
binding of antibodies to intrinsic factor and parietal cell antibodies inpernicious anemia. Gastroenterology 1968; 55:575-83.
151. Schallreuter KU, Lemke KR, Pittelkow MR, Wood JM, Korner C, Malik R.Catecholamines in human keratinocyte differentiation. J Invest Dermatol 1995;
104:953-7.
152. Schwimmbeck PL, Dyrberg T, Drachman DB, Oldstone MB. Molecular
mimicry and myasthenia gravis. An autoantigenic site of the acetylcholine
receptor alpha-subunit that has biologic activity and reacts immunochemicallywith herpes simplex virus. J Clin Invest 1989; 84:1174-80.

Olov Ekwall
79
153. Scire G, Magliocca FM, Cianfarani S, Scalamandre A, Petrozza V, Bonamico
M. Autoimmune polyendocrine candidiasis syndrome with associated chronicdiarrhea caused by intestinal infection and pancreas insufficiency. J Pediatr
Gastroenterol Nutr 1991; 13:224-7.
154. Seder RA, Paul WE. Acquisition of lymphokine-producing phenotype byCD4+ T cells. Annu Rev Immunol 1994; 12:635-73.
155. Sjöberg K, Alm R, Ivarsson SA, Lindström C, Eriksson S. Prevalence andclinical significance of gliadin antibodies in healthy children and adults. Scand
J Gastroenterol 1994; 29:248-54.
156. Sobel RA, Colvin RB. The immunopathology of experimental allergic
encephalomyelitis (EAE). III. Differential in situ expression of strain 13 Ia on
endothelial and inflammatory cells of (strain 2 x strain 13)F1 guinea pigs withEAE. J Immunol 1985; 134:2333-7.
157. Song YH, Li Y, Maclaren NK. The nature of autoantigens targeted inautoimmune endocrine diseases. Immunol Today 1996; 17:232-38.
158. Stavnezer J. Immunoglobulin class switching. Curr Opin Immunol 1996; 8:199-205.
159. Streilein JW, Ksander BR, Taylor AW. Immune deviation in relation to ocular
immune privilege. J Immunol 1997; 158:3557-60.
160. Sundick RS, Herdegen D, Brown TR, Dhar A, Bagchi N. Thyroidal iodine
metabolism in obese-strain chickens before immune- mediated damage. J
Endocrinol 1991; 128:239-44.
161. Sutphin A, Albright F, McCune DJ. Five cases (three in siblings) of idiopathichypoparathyroidism associated with moniliasis. J Clin Endocrinol 1943; 3:625-
34.
162. Svejgaard A, Platz P, Ryder LP. HLA and disease 1982--a survey. Immunol
Rev 1983; 70:193-218.
163. Sweetnam WP. Juvenile familial endocrinopathy. Lancet 1966; 1:463-5.
164. Söderbergh A, Mietinen A, Eskelin P, et al. Prevalence and clinical associations
of ten defined autoantibodies in autoimmune polyendocrine syndrome type I.manuscript 2000.
165. Söderbergh A, Winqvist O, Norheim I, Rorsman F, Husebye ES, Dolva O,
Karlsson FA, Kämpe O. Adrenal autoantibodies and organ-specific

Pteridine dependent hydroxylases as autoantigens in APS I
80
autoimmunity in patients with Addison's disease. Clin Endocrinol (Oxf) 1996;
45:453-60.
166. Talbot NB, Butler AM, MacLachlan EA. The effect of testosterone and allied
compounds on the mineral, nitrogen, and carbohydrate metabolism of a girl
with Addison's disease. J Clin Invest 1943; 22:583-93.
167. ter Borg EJ, Horst G, Hummel EJ, Limburg PC, Kallenberg CG. Measurement
of increases in anti-double-stranded DNA antibody levels as a predictor ofdisease exacerbation in systemic lupus erythematosus. A long-term, prospective
study. Arthritis Rheum 1990; 33:634-43.
168. The Finnish-German APECED Consortium. An autoimmune disease,
APECED, caused by mutations in a novel gene featuring two PHD-type zinc-
finger domains. Nat Genet 1997; 17:399-403.
169. Tonegawa S. Somatic generation of antibody diversity. Nature 1983; 302:575-
81.
170. Tuomi T, Bjorses P, Falorni A, Partanen J, Perheentupa J, Lernmark A,
Miettinen A. Antibodies to glutamic acid decarboxylase and insulin-dependentdiabetes in patients with autoimmune polyendocrine syndrome type I. J Clin
Endocrinol Metab 1996; 81:1488-94.
171. Uibo R, Aavik E, Peterson P, Perheentupa J, Aranko S, Pelkonen R, Krohn KJ.Autoantibodies to cytochrome P450 enzymes P450scc, P450c17, and P450c21
in autoimmune polyglandular disease types I and II and in isolated Addison'sdisease. J Clin Endocrinol Metab 1994; 78:323-8.
172. Van Parijs L, Abbas AK. Homeostasis and self-tolerance in the immunesystem: turning lymphocytes off. Science 1998; 280:243-8.
173. von Boehmer H. Positive selection of lymphocytes. Cell 1994; 76:219-28.
174. Vrana KE. How the regulatory and catalytic domains get together. Nat Struct
Biol 1999; 6:401-2.
175. Wang B, Andre I, Gonzalez A, Katz JD, Aguet M, Benoist C, Mathis D.Interferon-gamma impacts at multiple points during the progression of
autoimmune diabetes. Proc Natl Acad Sci U S A 1997; 94:13844-9.
176. Ward L, Paquette J, Seidman E, Huot C, Alvarez F, Crock P, Delvin E, Kampe
O, Deal C. Severe autoimmune polyendocrinopathy-candidiasis-ectodermal
dystrophy in an adolescent girl with a novel AIRE mutation: response toimmunosuppressive therapy. J Clin Endocrinol Metab 1999; 84:844-52.

Olov Ekwall
81
177. Whitaker J, Landing BH, Esselborn VM, Williams RR. The syndrome of
familial juvenile hypoadrenocorticism, hypoparathyroidism and superficialmoniliasis. J Clin Endocrinol 1956; 16:1374-87.
178. Winqvist O, Gebre-Medhin G, Gustafsson J, Ritzen EM, Lundkvist O, Karlsson
FA, Kämpe O. Identification of the main gonadal autoantigens in patients withadrenal insufficiency and associated ovarian failure. J Clin Endocrinol Metab
1995; 80:1717-23.
179. Winqvist O, Gustafsson J, Rorsman F, Karlsson FA, Kämpe O. Two different
cytochrome P450 enzymes are the adrenal antigens in autoimmunepolyendocrine syndrome type I and Addison's disease. J Clin Invest 1993;
92:2377-85.
180. Winqvist O, Karlsson FA, Kämpe O. 21-Hydroxylase, a major autoantigen inidiopathic Addison's disease. Lancet 1992; 339:1559-62.
181. Witebsky E, Rose NR, Terplan K, Paine JR, Egan RW. Chronic thyroiditis andautoimmunization. J. Am. Med. Assoc. 1957; 164:1439-1447.
182. Wuepper KD, Fudenberg HH. Moniliasis, 'autoimmune' polyendocrinopathy,and immunologic family study. Clin Exp Immunol 1967; 2:71-82.
183. Yu PL, Fujimura M, Okumiya K, Kinoshita M, Hasegawa H, Fujimiya M.
Immunohistochemical localization of tryptophan hydroxylase in the human andrat gastrointestinal tracts. J Comp Neurol 1999; 411:654-65.
184. Zamoyska R. CD4 and CD8: modulators of T-cell receptor recognition ofantigen and of immune responses? Curr Opin Immunol 1998; 10:82-7.
185. Zlotogora J, Shapiro MS. Polyglandular autoimmune syndrome type I amongIranian Jews. J Med Genet 1992; 29:824-6.
186. Zuklys S, Balciunaite G, Agarwal A, Fasler-Kan E, Palmer E, Hollander GA.
Normal thymic architecture and negative selection are associated with aireexpression, the gene defective in the autoimmune- polyendocrinopathy-
candidiasis-ectodermal dystrophy (APECED). J Immunol 2000; 165:1976-83.