pulmonary hypertension dr vidhu kohli junior resident deptt of pulmonary medicine
TRANSCRIPT

PULMONARY HYPERTENSION
DR VIDHU KOHLI JUNIOR RESIDENT DEPTT OF PULMONARY MEDICINE

INTRODUCTION
• Pulmonary hypertension is defined as mean arterial pressure of more than 25mmHg at rest or more than 30 mmHg with exercise . • Pulmonary hypertension can be due to diseases predominantly confined to
the pulmonary vasculature , as in pulmonary arterial hypertension , or can occur in association with diseases in which the primary disturbance is in the respiratory function or in left heart . • The generally accepted definition of pulmonary arterial hypertension calls for
a mean pulmonary arterial pressure more than 25mmHg at rest or more than 30mmHg with exercise , a pulmonary artery occlusion pressure or left ventricular end diastolic pressure that is less than 15mmHg, and an increase pulmonary vascular resistance greater than 3 Wood units , without significant cardiac or respiratory dysfunction .

CLASSIFICATION
• The classification of PAH has undergone several revisions since the 1960’s ; the latest and the gold standard is listed below ..• The revised classification system includes additions to WHO group 1
of chronic hemolytic anemias and schistosomiasis based on emerging evidence that these disorders produce an arteriopathy strikingly similar to other forms of PAH.

Pathology

EPIDEMIOLOGY
GROUP 1• IPAH is the prototype of group 1 pulmonary arterial hypertension . It
affects women more than men in a ratio 2:1 and may present at any age , with a mean age of onset of 37 years .• The prevalence of PAH is between 15 and 26 per million persons. • Heritable pulmonary arterial hypertension occurs in a familial context ,
most often (70%) owing to a mutation in the bone morphogenic protein receptor (BMPR2) . Other less common mutations include – mutation in activin receptor like kinase type1, or endoglin predominantly with coexistant hereditary hemorrhagic telangiectasia .

• Drug-and-toxin induced has been most clearly linked to anorexigens , observational studies also suggest amphetamines , methamphetamines and l-tryptophan to be related to development of PAH.• In the setting of connective tissue diseases , prevalence of PAH in patients
with scleroderma is in the range of 7-12%. It is less common in RA and SLE.• Prevalence in a patient with HIV is 0.5%• 2-6% of patients of portal hypertension develops PH. • A significant proportion of patients with untreated systemic-to-pulmonary
shunts , commonly owing to congenital heart disease develop PAH.

GROUP 2
• PH owing to left heart disease probably represents the most frequent cause of pulmonary hypertension seen in practice. Patients with left heart disease have an elevation of pulmonary artery pressure greater than expected based on the elevation of left heart filling pressures, with a transpulmonary gradient of >12mmHg and a pulmonary vascular resistance of more than 3 Wood units .

GROUP 3• These patients have pulmonary hypertension owing to lung diseases
or hypoxia . • COPD, ILD and sleep disordered breathing . • The pressure elevation tends to be modest with a mean pulmonary
artery pressure of 25-35mmHg.

GROUP 4• Approx. 4%of patients who have suffered an acute pulmonary
embolism progress to develop chronic thromboembolic pulmonary hypertension ( CTEPH)• CTEPH must be carefully differentiated from other groups as
treatment is quite different. • Patient with a significant clot burden can be candidates for a
pulmonary thromboendarterectomy ; which is a potentially curative procedure for this disorder.

GROUP 5Consists of forms for which etiology is unclear or multifactorial .

PATHOLOGY
• Wood in 1958 divided pulmonary hypertension into six types :
• The histologic lesions observed in the reversible kind of pulmonary hypertension consisted of medial hypertrophy of the pulmonary artey and scant intimal hyperplasia .


• Group 1: affect the distal pulmonary arteries (<500 micrometer of diameter) . They are characterized by medial hypertrophy, intimal proliferative and fibrotic changes (concentric, eccentric), adventitial thickening with moderate perivascular inflammatory infiltrates, complex lesions (plexiform, dilated lesions), and thrombotic lesions. Pulmonary veins are classically unaffected.• Group 2: enlarged and thickened pulmonary veins, pulmonary capillary
dilatation, interstitial oedema,alveolar haemorrhage, and lymphatic vessel and lymph node enlargement.

• Group 3: medial hypertrophy and intimal obstructive proliferation of the distal pulmonary arteries. A variable degree of destruction of the vascular bed in emphysematous or fibrotic areas may also be present.• Group 4: pathological lesions are characterized by organized thrombi
tightly attached to the pulmonary arterial medial layer in the elastic pulmonary arteries, replacing the normal intima. These may completely occlude the lumen or form different grades of stenosis, webs, and bands. Interestingly, in the non-occluded areas, a pulmonary arteriopathy indistinguishable from that of PAH (including plexiform lesions) can develop.


PATHOBIOLOGY
There are several lines of evidence regarding pathogenesis of IPAH ,suggesting that IPAH develops as a result of abnormal proliferation of vascular smooth muscle cells affecting all three layers of vessel wall and leading to intimal hyperplasia , medial hypertrophy and adventitial proliferation . what initiates this abnormal growth is not entirely known , but there are several clues . • Mutation in BMPR2 gene , has been associated with familial type of
IPAH , which through various messenger proteins inhibit the normal apoptosis . Hence this has been mentioned as cancer of the pulmonary artery. However , not all patients with BMPR2 mutation develops PAH.

• Defects in a specific voltage-gated potassium ion channel ,the Kv1.5 channel have been found in the pulmonary artery smooth muscle cells from patients with IPAH. • Overexpression of the serotonin transporter has also been described.• In addition to potential genetic defects , several abnormalities in
endothelial cell function have been found , many of which are likely a result of some vascular insult. The propensity of such abnormalities to produce disease appears to be determined by both the intensity and duration of these derangements, as well as an individuals genetic predisposition to abnormal vascular responses.


• The development of pulmonary arterial hypertension involves disruptions of the normal balance of vasoconstriction and vasodilatation , the control of cellular proliferation and thrombosis . Abnormalities in the expression of numerous vasoactive mediators cause , or result from changes in, endothelial, smooth muscle and platelet function and result in a thickened vessel wall and markedly narrowed or even completely obliterated lumen .

Other groups of PAH
• Patient’s with scleroderma spectrum of diseases , most notably limited scleroderma have been reported to have pulmonary hypertension in upto 40% cases. And this pulmonary involvement is associated with worse survival . Other connective tissue diseases associated with PAH , albeit less commonly , include SLE, RA and mixed connective tissue disorders . • Congenital heart disease is a well recognized “risk factor” for
development of PAH .In 1958 Wood coined the term “Eisenmenger’s complex “ to describe pulmonary hypertension at systemic level due to high pulmonary vascular resistance with reversed shunting through a large ventricular septal defect. The likelihood of developing pulmonary hypertension depends upon the size of the defect .

• In patient’s with ventricular septal defect it has beenshown that 3% of the patients with a defect of 1.5 cm or smaller will develop Eisenmenger’s syndrome whereas half of patients with larger defect will develop pulmonary hypertension , which will also appear earlier than in atrial septal defect , often in infancy . • the various studies in effects of drugs and toxins, have observed a 20
fold-increase in pulmonary hypertension after the introduction of aminorex , an appetite suppressant resembling epinephrine and amphetamine . The pulmonary lesions produced by this agent resembled plexogenic arteriopathy in every respect.

• Pulmonary hypertension has been associated with another family of appetite suppressants with similar chemical structure , fenfluramine and dexfenfluramine . In a case controlled prospective study performed in Europe , the risk of developing pulmonary hypertension was increased by 20-fold in individuals who used these drugs for periods exceeding 3months . • In a single center case control study , there has been seen a strong co-
relation between methamphetamine use and PAH .


• Portal hypertension is another disease associated with PAH and it has been speculated that multiple emboli emanating fromportocaval anastomosis were responsible for pulmonary hypertension . The exact mechanism is not known ,though there is another theory saying that portsystemic shunting allows vasoactive substances , normally cleared in liver , to reach the pulmonary vasculature .• The association between PAH and HIV is well known . In several studies the
incidence of pulmonary hypertension in HIV has been reported as high as 0.5%. The mechanism of HIV-associated pulmonary hypertension is not known ,although theories include release of cytokines and growth factors and the presence of human herpes virus 8 (HHV8), a promoter of angiogenesis. Pulmonary hypertension can occur in all stages of HIV , including patients with no detectable viral load .

PATHOPHYSIOLOGY
• The normal pulmonary vasculature bed has a remarkable capacity to dilate and recruit unused vasculature to accommodate increases in pulmonary blood flow. In pulmonary hypertension, the pulmonary artery pressure and pulmonary vascular resistance are increased at rest and further increase with exertion. Early on in the process, the right ventricle may be capable of maintaining normal cardiac output at rest, although it may fail to augment cardiac output with exercise, thereby leading to exertional dyspnea. As the disease progresses, the right ventricular dysfunction may progress to the point that resting cardiac output is impaired. Right ventricular function is a major determinant of functional capacity and prognosis in pulmonary arterial hypertension.• The two most frequent mechanisms of death are progressive right ventricular
failure and sudden death.

SYMPTOMS
• Dyspnea is the cardinal symptom of IPAH occurring in more than 95% of patients in the major clinical studies .
• Breathlessness is the presenting symptom in 60% patients and usually is first noted on exertion, but eventually occurs at rest . Its mechanism is probably complex : most likely cause being the inadequacy of cardiac output relative to metabolic requirements .Regardless of its mechanism , Packer stresses that the severity of dyspnea does not correlate with the elevation of pulmonary artery pressure in patients with IPAH .
• Closely related to dyspnea are sensations of fatigue and weakness , which are usually experienced before the general disability that is present with advanced disease . They presumably reflect impaired tissue oxygenation resulting from the depressed cardiac output in patients with IPAH.

• Substernal chest pain is another symptom which frequently occurs on exertion , radiates to left shoulder and axilla , and is relieved by rest. The pain has been attributed to coronary insufficiency in the presence of increased right ventricular work and hypoxemia ,a concept supported by the occasional relief produced by nitroglycerin. However pain may be present in young patients without coronary artery disease prompting clinicians to argue that the pain is caused by distension of the pulmonary arteries , whose afferents enter the nervous system along the same pathway as afferents from the heart

• Syncope occurs in some patients with IPAH and may be its initial manifestation . It is probably caused by decrease in cerebral blood flow that follows an increase in pulmonary artery pressure and a decrease in cardiac output . • Hemoptysis is another symptom which presumably stems from
microvascular rupture under the high pulmonary artery pressure . • Hoarseness may result from the pressure of the enlarged main
pulmonary artery on the recurrent laryngeal nerve .• Peripheral edema and ascites may develop after the onset of right
ventricular failure .

PHYSICAL FINDINGS
• Patients with early IPAH may manifest no physical abnormality . However ,signs of pulmonary hypertension and a decreased cardiac output should be evident with advanced disease . As Wood observed the hands and feet of a patient with severe pulmonary hypertension are cold, the peripheral pulse is diminished , the blood pressure is likely to be low ,and the pulse pressure is reduced . • Signs of systemic venous hypertension should be present , including a prominent
jugular venous a wave and a prominent c-v waves , which are indicative of tricuspid regurgitation . • Palpation of the chest should reveal a right ventricular lift at the left sternal
border that is sustained throughout the pressure-overloaded cardiac contraction , in contrast to unsustained parasternal impulse felt in pure volume overload .

• On auscultation of the chest , the second heart sound is closely split , and the second component is accentuated . The valvular closure sound should increase in intensity on inspiration and may become palpable as pulmonary artery pressure rises. In addition to these findings , patients with right sided heart failure usually have peripheral edema and abdominal distention due to ascites . If tricuspid regurgitation is present , the liver may become pulsatile . • Cyanosis occurs with variable frequency in patients with IPAH and is likely
to be a late phenomenon . Peripheral vasoconstriction and impaired oxygenation of arterial blood due to mixed venous hypoxemia resulting from the decreased cardiac output appear to be the most common mechanism .

DIAGNOSIS
The evaluation process of a patient with suspected PH requires a series of investigation intend to confirm the diagnosis , clarify the group of PH and the specific etiology within the PAH group , and evaluate the functional and hemodynamic impairment. 1. CLINICAL PRESENTATION : Inspiratory crackles points towards the
diagnosis of ILD . Telangiectasia , digital ulceration and sclerodactyly are seen in scleroderma . The stigmata of liver disease such as spider naevi , testicular atrophy and palmar erythema should be considered . If digital clubbing is encountered in IPAH , an alternative diagnosis such as CHD or PVOD should be sought.

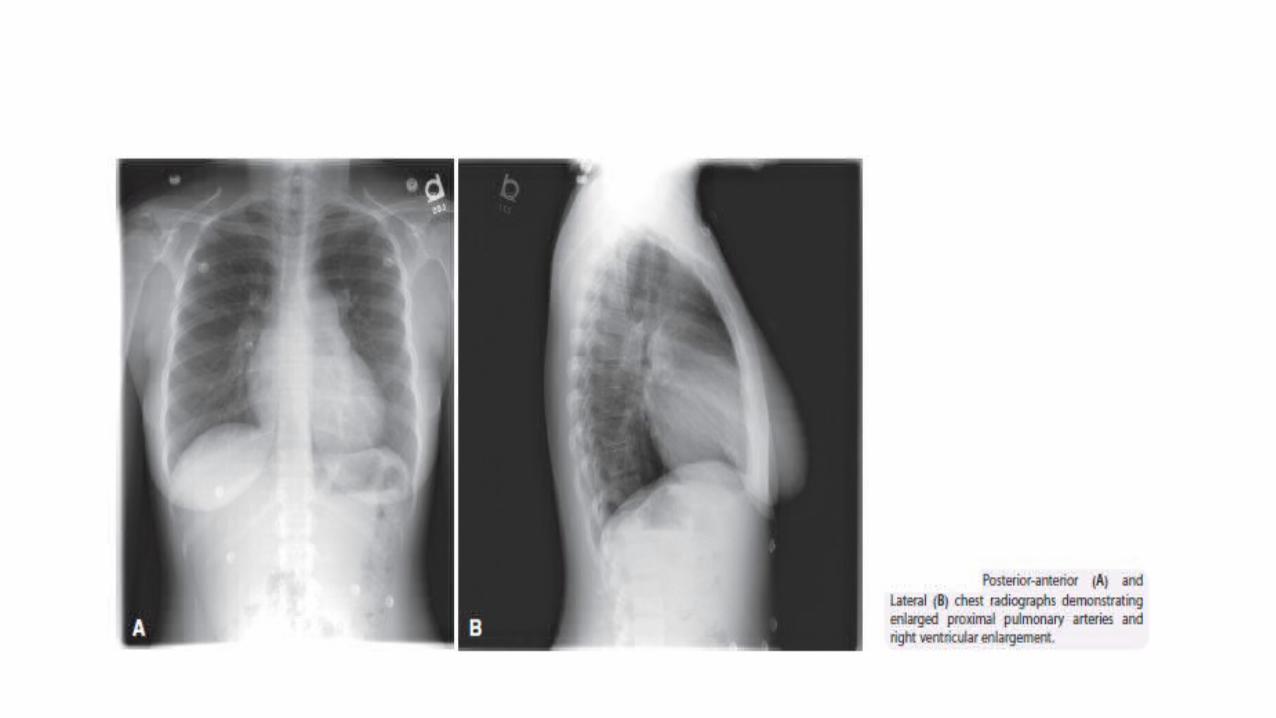
2 . ELECTROCARDIOGRAM : the ECG may provide suggestive or supportive evidence of PH by demonstrating the RV hypertrophy and strain ,and right atrial dilatation . RV hypertrophy is seen in 87% and right axis deviation in 79% of patients with IPAH . However the absence of these findings doesnot exclude the presence of PH nor does it exclude severe hemodynamic abnormalities. The ECG has insufficient sensitivity (55%) and specificity (70%) to be a screening tool for detecting significant PH. 3. CHEST RADIOGRAPH : In 90% of patients with IPAH the CXR is abnormal at the time of diagnosis . Findings include central pulmonary arterial dilatation, which contrasts with pruning of the peripheral blood vessels . Right atrium and right ventricular enlargement may be seen in more advanced cases . The chest radiograph allows associated moderate-to-severe lung diseases or PVH due to left heart disease to be reasonably excluded. Overall, the degree of PH in any given patient does not correlate with the extent of radiographic abnormalities.


4. PULMONARY FUNCTION TESTS and ABG : these will identify the contribution of underlying airway or parenchymal lung disease . Patients with PAH usually have decreased lung diffusion capacity for CO ( typically in the range of 40-80% predicted ) and mild moderate reduction of lung volumes . Arterial oxygen tension is normal or only slightly lower than normal at rest and arterial carbon dioxide is decreased because of alveolar hypertension. If clinically suspected , screening overnight oximetry or polysomnography will exclude significant obstructive sleep apnea/hypopnea.

5. EXERCISE CAPACITY : the objective assessment of exercise capacity in patients with PAH is an important instrument for evaluating disease severity and treatment effect . The commonly performed tests are • 6 –minute walk test• Cardiopulmonary exercise testing 6-minute walk test : it is technically simple , inexpensive , reproducible and standardized . In addition to distance walked , dyspnea on exertion (BORG scale ) and finger oxygen saturation are recorded . Walking distances <332m or < 250m and oxygen desaturation >10% indicate impaired prognosis in PAH. With respect to treatment effects , absolute values >380m following 3months of i.v. epoprostenol correlated with improved survival in IPAH patients .

• CPET : with this gas exchange and ventilation are continuously recorded throughout incremental exercise . In PAH, oxygen uptake at the anaerobic threshold and peak exercise are reduced in relation to disease severity ,as are peak work rate , peak heart rate , oxygen pulse and ventilator efficiency . Following multivariate analysis of clinical , hemodynamic , exercise parameter peak oxygen uptake ( < 10.4mlO2/kg/min ) and peak systolic arterial pressure during exercise ( <120mmHg) independently predicted a worse prognosis in IPAH patients .

6. ECHOCARDIOGRAPHY: transthoracic Doppler echocardiography is an excellent noninvasive screening test for the patient with suspected PH . It estimates PASP which is equivalent to RVSP in the absence of pulmonary outflow obstruction . RVSP= 4v2+RAP, where v= systolic regurgitant tricuspid flow velocity RAP= right atrial pressure (=CVP)RVSP increases with age and body mass index . Besides identification of PH, TTE can recognize left heart valvular and myocardial diseases responsible for PVH. Congenital heart diseases with systemic-to-pulmonary shunts can be easily identified.

7. VENTILATION and PERFUSION (V/Q) LUNG SCAN : In PAH, V/Q scan may be entirely normal however, it may show small peripheral non-segmental defects in perfusion. Lung V/Q scan provides a means of diagnosis of chronic thromboembolic pulmonary hypertension ( CTEPH) where perfusion defects are usually found in lobar and segmental regions , leading to segmental defects in perfusion image. V/Q scanning showed sensitivity of 90-100% with specificity of 94-100% for distinguishing between IPAH and CTEPH. However, unmatched perfusion defects are also seen in veno-occlusive disease ,such a patient requires careful further investigations.

8. HRCT : it provides the detailed views of the lung parenchyma and facilitates the diagnosis of ILD and emphysema . The presence of interstitial markings similar to those seen with advanced left ventricular failure such as diffuse central ground glass opacification and thickening of the interlobular septae suggest pulmonary veno-occlusive disease ; additional findings are lymphadenopathy , pleural shadows and effusions. Diffuse bilateral thickening of interlobular septae and the presence of small , centrilobular , poorly circumscribed nodular opacities suggest pulmonary capillary haemangiomatosis.

9. Contrast enhanced spiral CT , pulmonary angiography and cardiac MRI : Contrast enhanced spiral CT is indicated in pulmonary hypertensive patients when the V/Q lung scintigraphy shows segmental or subsegmental defects of perfusion with normal ventilation and may demonstrate central chronic pulmonary thromboemboli . CT features of chronic thromboembolic disease are complete occlusion of pulmonary arteries , eccentric filling of defects consistent with thrombi , recanalization and stenosis or webs . Traditional pulmonary angiography is still required in the work up of CTEPH to better identify patients that can benefit from the intervention of endarterectomy . Pulmonary angiograph is more accurate in the identification of distal obstruction and it is indicated also in cases of inconclusive contrast enhanced spiral CT , in patients with clinical and lung scintigraphy suspicion of CTEPH.

• MRI is increasingly used in patients with PAH for the evaluation of pathological and functional changes of both heart and pulmonary circulation. It provides direct evaluation of RV size , morphology and function, and allows non invasive assessment of blood flow including stroke volume , cardiac output , distensibility of PA and RV mass .

10. RIGHT HEART CATHETERISATION AND VASOREACTIVITY : in the evaluation of IPAH , right heart catheterization is mandatory to document the presence and severity of pulmonary hypertension , rule out cardiac causes , and determine if there is acute pulmonary vasoreactivity using pharmacologic agents . • RAP and cardiac index • PAP and the wedge pressure • Cardiac output ,and from vascular pressures, the pulmonary and systemic vascular
resistances .• Left-to-right intracardiac shunts may be excluded by the measurements of blood
oxygen conc. in various cardiac chambers .• These and other variables may be studied at rest or during exercise.

• Cardiac catheterization in patients with IPAH may reveal elevated right atrial pressure , pulmonary arterial pressures that are increased often to systemic levels , and depression of cardiac output . PVR generally is increased ,whereas systemic vascular resistance is in the normal range . • In PAH , vasoreactivity testing should be performed at the time of
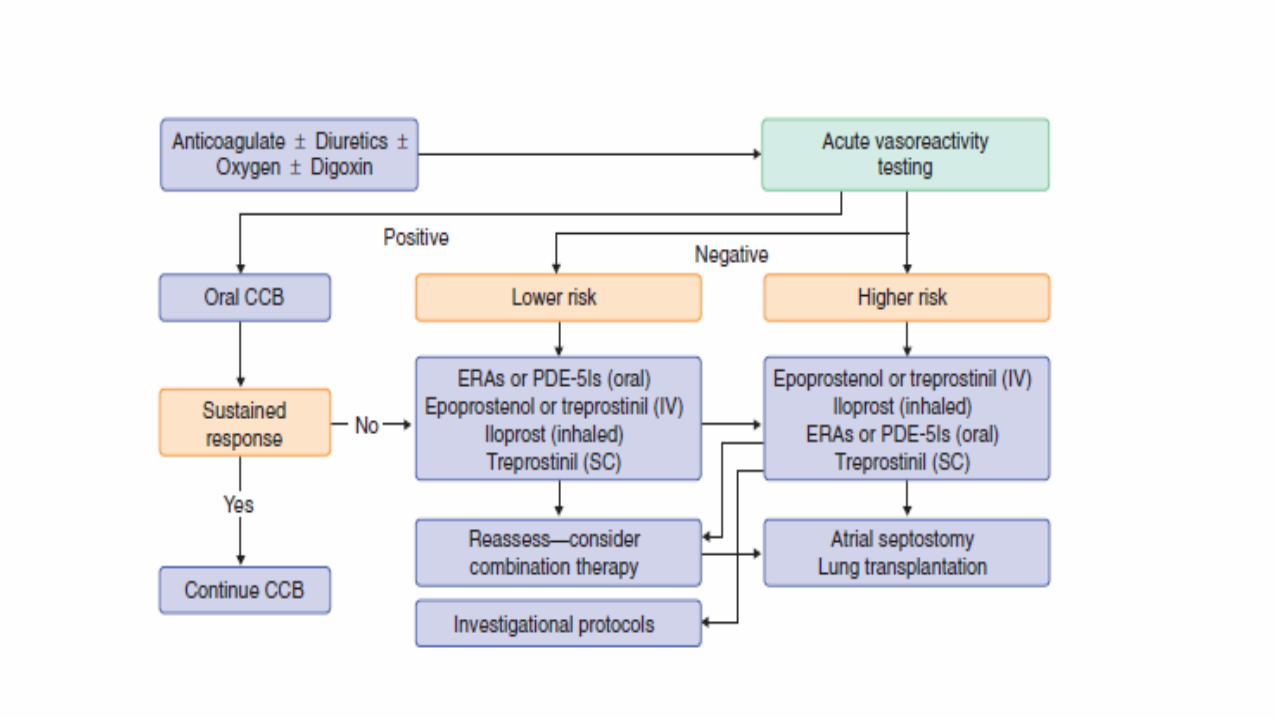
diagnostic RHC to identify patients who may benefit from long term therapy with calcium channel blockers . It should be performed with short-acting , safe and easy to administer drugs , most common being used are : nitric oxide , adenosine and prostacyclin.

• A positive acute response is defined as a reduction of mean PAP > or equal to 10mmHg to reach an absolute value of mean PAP < or equal to 40mmHg with an increased or unchanged cardiac output . Positive acute responders are most likely to show a sustained response to long term treatment with high doses of CCB’s . However ,approx. 10% of patients with IPAH will meet these criteria .

11. ABDOMINAL ULTRASOUND SCAN : Liver cirrhosis and/or hypertension can be reliablely excluded by the use of abd. ultrasound scan . The color Doppler examination also allows the differentiation between the passive portal hypertension due to RHF ,from portal hypertension caused by an increase in transhepatic venous gradient associated with liver cirrhosis .


EVALUATION OF SEVERITY
• The clinical assessment of the patient has pivotal role in the choice of the initial treatment , the evaluation of response to therapy and the possible escalation of therapy if needed. The various parameters are:
1. Clinical , haemodynamic and echocardiographic parameters. 2. Exercise evaluation.3. Biochemical markers .4. Comprehensive prognostic evaluation.5. Definition of patient status.

• BIOCHEMICAL MARKERS : biochemical markers emerged within the last decade as an attractive non-invasive tool for assessment and monitoring of RV dysfunction in patients with PH. The various markers are ..
1. Serum uric acid is a marker of impaired oxidative metabolism of ischemic peripheral tissue . High levels associated with poor survival in patients with IPAH.
2. ANP and BNP are released from myocardium in response to wall stress. During the final step of BNP synthesis from proBNP an inactive NT-proBNP and active BNP are released and it is the ratio of BNP/NT-proBNP that reflects the severity of RV dysfunction.

3. Elevated levels of cardiac troponin T and troponin I are established specific markers of myocardial damage and are prognostic indicators in acute coronary syndromes and acute pulmonary embolism. Its role in prognosis of CTEPH is under investigation. In conclusion, several circulating biomarkers convey prognostic information in patients with PAH but there value in everyday clinical practice is still not established .


TREATMENT
• IPAH is now a treatable disease. Once the diagnostic process in a patient with pulmonary hypertension is complete , and the patient is characterized as having IPAH , therapy should be initiated . Therapy is subdivided into :
1. “ Supportive /conventional” therapies 2. “Targeted therapies”

SUPPORTIVE THERAPIES
1. EXERCISE AND PHYSICAL ACTIVITY : patients should avoid activities that lead to undue symptoms such as severe dyspnea , chest pain , light headedness or syncope . However, low-to-moderate levels of exercise to prevent deconditioning and improve mental outlook are appropriate.
2. AVOIDANCE OF ALTITUDE: hypobaric hypoxia causes pulmonary vasoconstriction , and thus can worsen the pulmonary hypertension in PAH patients . It is generally recommended that patients flying on commercial airlines ( pressurized to 1500-2400m) or travelling to elevation above 5000feet use supplemental oxygen . Not surprising, patients with severe PAH residing at high elevations often improve if they move to sea level.

3. AVOIDANCE OF PREGNANCY : pregnancy and especially delivery are extremely risky for PAH and it is strongly recommended that women of childbearing potential use appropriate methods of birth control . In terms of which birth control method is preferable , no consensus exists for the type of contraceptive . It should be noted that in patients on endothelin receptor antagonists , an additional barrier method of birth control should be used. 4. WARFARIN : many of the endothelial cell abnormalities that predispose patients to pulmonary arteriopathy and increases thrombosis. Warfarin is the anticoagulant most frequently used in patients with PAH.

• However, it is important to note that none of the studies were conducted during the “modern era” of effective , targeted PAH therapies, which are already associated with better survival than that seen in these “historical” studies. Whether warfarin further improve outcome in patients on these therapies remains unclear. There may be specific cases in which warfarin would be indicated , such as in patients with advanced heart failure , indwelling central venous catheter or erythrocytosis.

5. SUPPLEMENTAL OXYGEN : most PAH patients are not hypoxemic , atleast at rest . Mild hypoxemia , when present is likely on the basis of reduced mixed venous saturation caused by low cardiac output with mild V/Q inequality . The presence of more profound hypoxemia in a PAH patient should raise suspicion for underlying parenchymal lung disease , systemic-to-pulmonary shunting , PVOD , pulmonary capillary hemangiomatosis. Although no long term studies supporting its efficacy , the consensus is that if arterial Po2 is <60mmHg or systemic arterial oxygen saturation is <90% at rest ,supplemental oxygen is indicated.

6. DIURETICS: both total body and intravascular volume overload are common in PAH patients .In addition to causing symptomatic peripheral edema , volume overload of the right ventricle can cause the compression of the left ventricle and contribute to decreased cardiac output and pre-renal azotemia . Thus , it is a common observation that in decompensated PAH patients , aggressive diuresis leads to clinical and physiologic improvement . Despite the benefits of diuretics in PAH patients ,there are no controlled studies to guide the clinicians in using these agents .

• 7. CALCIUM CHANNEL ANTAGONISTS : these diminish the vascular tone by preventing an increase in cytosolic calcium concentration by inhibiting both the influx of extracellular calcium and the release of calcium from intracellular stores. The long term prognosis is good for some IPAH patients who respond acutely to the administration of short acting pulmonary vasodilators and are treated subsequently with calcium channel antagonists. Unfortunately, only few patients demonstrate acute reactivity and of these only half experience a sustained clinical response. Non responders not only fail to benefit but are also prone to adverse side affects. Relatively high doses of calcium channel antagonists are required to promote sufficient pulmonary vasodilation. In some instances the required daily doses of Nifedipene and Diltiazem have exceeded 200mg and 700mg respectively.

TARGETED THERAPIES
• PAH-specific therapies have been available since 1995,when intravenous epoprostenol was approved by the U.S. Food and Drug Administration (FDA). These therapies were developed to offset the imbalance in endothelial derived mediators seen in PAH: excessive endothelin-1 production, deficient prostacyclin, and abnormal nitric oxide production.
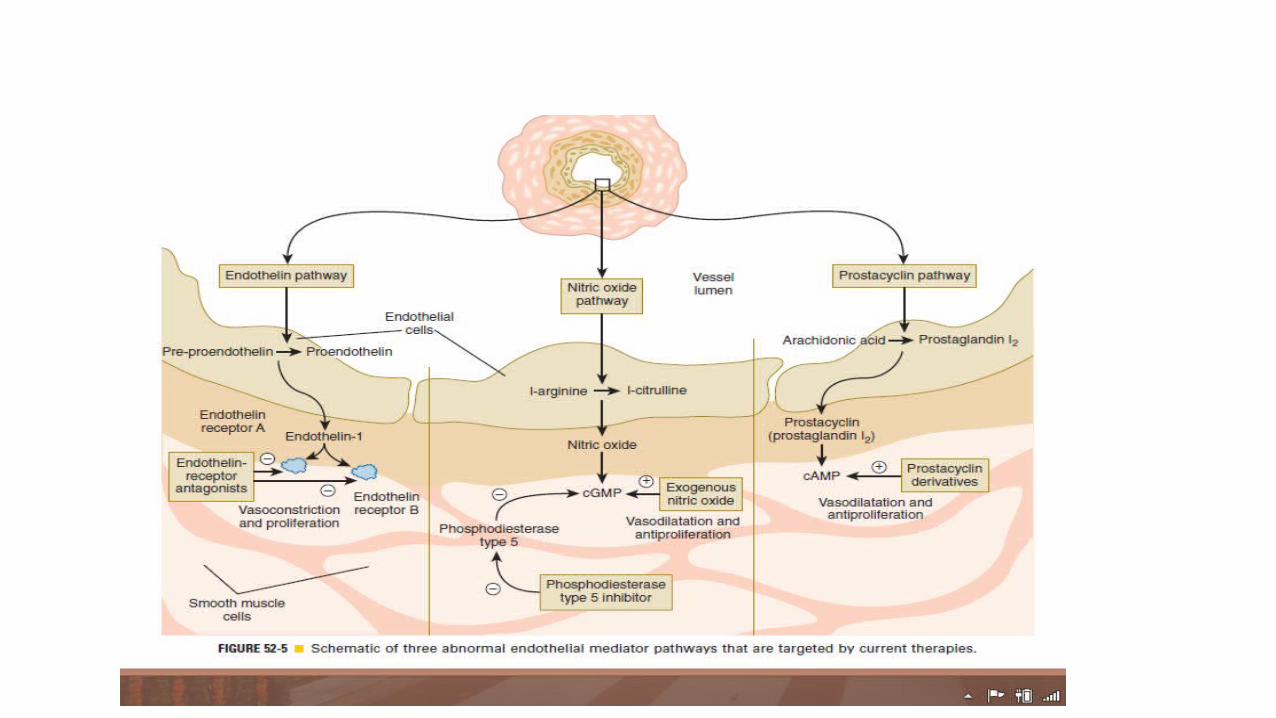

PROSTACYCLIN ANALOGUES
1.Epoprostenol :- • It has a half life of < 5 min, so requires an indwelling central venous
catheter which is connected to an infusion pump for continuous i.v. administration. • Common side effects include headache, jaw pain, flushing, nausea,
diarrhoea, skin rash, and musculoskeletal pain.• Started at a low dose of 2-4 ng/kg/min and increased gradually to 10-
16 ng/kg/min

2. Trepoprostinil sodium :-• Half life approx. 4Hrs• Used subcutaneously, can provide modest ,yet statistically significant
improvement in exercise tolerance.• The main limitation of this therapy is pain and erythema at the site of the
subcutaneous infusion, a complication that occurs in 85% of patients.• Less potent than Epoprostenol, so higher dose required.• Inhaled Trepoprostinil four times daily is effective for improving exercise
capacity. However, cough is an additional side effect with this method of administration.

3. Iloprostol :-• Inhaled by the use of ultrasonic nebulizer 6-9 times daily is also effective for improving exercise capacity.
4.Beraprost :-• Used orally, but non sustained improvement in exercise capacity, so
rarely used

ENDOTHELIN RECEPTOR ANTAGONISTS• A and B are two subtypes of ET1 receptor and antagonism of one or
both is an effective therapy .• FDA has approved two drugs 1. BOSENTAN: oral antagonist at both A and B. Initiated at a dose of 62.5mg twice daily and titrated upto 125mg twice daily after 1month.

2. AMBRISENTAN : specific ET-A antagonist 5mg or 10mg once daily
• Liver enzymes must be monitored on a monthly basis , the dose should be reduced if liver enzymes rise to greater than 3-5 times the ULN and discontinued if they rise 5 times or more the ULN .• Has teratogenic potential • Lower extremity edema , headache and nasal congestion.

PHOSPHODIESTRASE TYPE- 5 INHIBITORS1. SILDENAFIL: 20mg thrice daily . flushing, epistaxis and gastritis. use with nitrates is contraindicated.
2. TADALAFIL: 40mg once daily and has similar side effect profile.

• COMBINATION THERAPY : given the availability of therapies that target different pathologic processes , combination therapy is an attractive theoretical option in PAH . Emerging data support the incremental benefit of combining more than one targeted therapy under careful observation usually in specialized center. • INVASIVE THERAPIES: 1. ATRIAL SEPTOSTOMY : creates right-to-left interatrial shunt improved cardiac output compensates for the decreased systemic arterial oxygen saturation .

Contraindications :• Severe right ventricular failure on cardiorespiratory support • Mean right atrial pressure > 20mmHg.• Pulmonary vascular resistance index >55U/m2.• Resting oxygen saturation <90% on room air .• Left ventricular-end diastolic pressure >18mmHg.
• It is associated with high morbidity and mortality hence should be performed only by experienced operators in specialized centers.

• LUNG TRANSPLANTATION : Bilateral lung or heart lung transplantation is the final option for selected patients with PAH
indications : low or declining 6-MWT. failing therapy with i.v. epoprostenol , cardiac failure with cardiac index < 2l/min /m2 and elevated RAP > 15mmHg.
The 1-,3-,5-and 10 year survival rates are 66%, 57%,47% and 27% respectively .

• ASSESSING RESPONSE TO THERAPY : The usual recommendation is every 3-6 months in stable patients and every 1-3 months in patients with worsening or unstable symptoms or signs. • EXPERIMENTAL THERAPIES: 1. Macitentan 2. Inhaled VIP3. Riociguat4. Selexipag5. Nitric oxide synthase coupler6. Imatinib and sorafinib ( tyrosine kinase inhibitors )